

Abstract
The apoptotic antiproliferative actions of our previously reported CB1 allosteric modulators 5-chlorobenzofuran-2-carboxamide derivatives VIIa–j prompted us to develop and synthesise a novel series of indole-2-carboxamide derivatives 5a–k, 6a–c, and 7. Different spectroscopic methods of analysis were used to validate the novel compounds. Using the MTT assay method, the novel compounds were examined for antiproliferative activity against four distinct cancer cell lines. Compounds 5a–k, 6a–c, and 7 demonstrated greater antiproliferative activity against the breast cancer cell line (MCF-7) than other tested cancer cell lines, and 5a–k (which contain the phenethyl moiety in their backbone structure) demonstrated greater potency than 6a–c and 7, indicating the importance of the phenethyl moiety for antiproliferative action. Compared to reference doxorubicin (GI50 = 1.10 µM), compounds 5d, 5e, 5h, 5i, 5j, and 5k were the most effective of the synthesised derivatives, with GI50 ranging from 0.95 µM to 1.50 µM. Compounds 5d, 5e, 5h, 5i, 5j, and 5k were tested for their inhibitory impact on EGFR and CDK2, and the results indicated that the compounds tested had strong antiproliferative activity and are effective at suppressing both CDK2 and EGFR. Moreover, the studied compounds induced apoptosis with high potency, as evidenced by their effects on apoptotic markers such as Caspases 3, 8, 9, Cytochrome C, Bax, Bcl2, and p53.
Keywords: indole, carboxamide, apoptosis, antiproliferative, multi-target
1. Introduction
In response to increased global morbidity and mortality rates from so-called incurable diseases, medication research and development has never been static but has grown increasingly dynamic. Traditionally, therapeutic drug discovery has relied on the design of highly selective chemical entities that target a single biological entity assumed to play a dominant role in a particular disease [1,2]. By means of this method, researchers hoped to eliminate any unwanted side effects and ensure that drug candidates had more drug-like properties. Highly selective or specific therapeutic medicines focused on single molecular targets, on the other hand, have shown to be ineffective, particularly in the treatment of complicated disorders. Drug resistance has been linked to the use of highly selective therapeutic agents.
However, due to the low efficacy of single target medications against multifactorial disorders whose aetiology is based on a collection of biochemical processes and many bioreceptors acting concurrently, drug design methodologies have to be reconsidered. Over the last few years, medicinal chemistry has been exploring new tools and alternatives to attain more agility, security, and efficiency in the synthesis and prospection of drug candidates.
Because of the ineffectiveness of certain single-drug therapies, the hunt for improved clinical outcomes has prompted the introduction of polypharmacology as a novel therapeutical technique. Polypharmacology is the development or application of pharmacological drugs that operate on several molecular targets or metabolic pathways. This multi-target strategy can take several forms, such as drug associations, drug combinations, or a single agent with numerous ligands, all of which are aimed at multiple targets. HIV/AIDS treatment, cancer treatment, TB treatment, and hypertension medication are all examples of combination therapy [3,4,5]. However, the inefficiency of combination therapy, as well as the negative consequences of drug–drug interactions, different pharmacokinetics, toxicity, and costs, has fuelled the development of new drug discovery methodologies. This current technique promotes combining diverse structural components in a single scaffold to allow molecular recognition by more than one bioreceptor, operating in many targets associated with biochemical networks responsible for multifactorial disease pathophysiology [6,7,8].
A breakdown of balance between cell proliferation and apoptosis is a symptom that enhances the inability of damaged cells to be removed by apoptosis. Activating apoptotic pathways in tumour cells is a critical practise for cancer treatment [9]. Apoptosis is triggered by extracellular or intracellular cues, which initiate a signalling cascade with characteristics such as nuclear condensation and DNA fragmentation [10]. Furthermore, the deregulations responsible for cancer genesis and progression involve hundreds of genes or signalling cascades [11]
Caspase, a highly specialized family of cysteine proteases, is known to mediate an important stage of the apoptotic process [12]. Numerous in vitro and in vivo research demonstrated that aberrant caspase activation control is critical to avoiding cancer cell death [13]. Furthermore, other genes, including Bcl-2 and p53, are known to be involved in apoptotic pathways. Overexpression of anti-apoptotic Bcl-2 has been linked to a variety of cancers [14]. The suppression of caspase proteins is thought to be the mechanism by which Bcl-2 prevents apoptosis [15]. p53 has been discovered to be required for cellular senescence caused by mutations in genes involved in mitosis and chromosomal segregation [16]. To maintain genomic integrity, the p53 gene can activate cell cycle checkpoints, DNA repair, and apoptosis (Ahmad et al., 2012). Most malignancies are caused by p53 mutations or deletions [17].
Antiproliferative actions have been documented for cannabinoids such as THC (I) and the CB1 allosteric modulator CBD (II) [18,19,20,21,22], but no data for other CB1 allosteric modulators such as the 5-chloroindole-2-carboxamide derivatives III and IV, and their furan congeners V and VI have been reported (Figure 1). Recently, we reported on the antiproliferative action of 5-chlorobenzofuran-2-carboxamide CB1 allosteric modulators V and VI for the first time [23]. Based on this, we designed and synthesised a novel series of 5-chlorobenzofuran-2-carboxamide derivatives VIIa–j (Figure 1). The newly synthesized compounds were tested for their antiproliferative effects in A549 lung, MCF-7 breast, Panc-1 Pancreatic, and HT-29 colon cancer cell lines. VIIa–j compounds showed significant antiproliferative action, the most potent derivative of VIIa–j had a GI50 value of 1.35 µM against the four examined cell lines, being equipotent to the reference doxorubicin (mean GI50 = 1.13 µM) and even more potent than doxorubicin in MCF-7. The compounds examined had a strong apoptotic effect, with significant increases in caspase 3, 8, and 9, as well as Cytochrome C levels. Furthermore, compared to doxorubicin, the investigated compounds triggered a significant rise in Bax levels and a decrease in anti-apoptotic Bcl-2 protein levels in MCF-7 cells [23].
Figure 1.

Structures of compounds I–VI and VIIa–j.
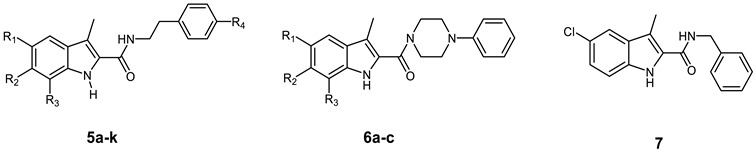
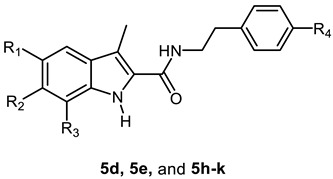
In the present work, the apoptotic antiproliferative actions of our previously reported 5-chlorobenzofuran-2-carboxamide derivatives VIIa–j (Figure 1) prompted us to develop and synthesise a novel series of indole-2-carboxamide derivatives 5a–k, 6a–c, and 7 (Figure 2). A small library of fifteen new compounds in which the methyl group was kept as a substituent at C3. To investigate the effect of substituent modification, the para positions of the phenethyl tails in the newly synthesised compounds were left unsubstituted or substituted with 4-dimethylamino, morpholin-4-yl, piperidin-1-yl, or 2-methylpyrrolidine-1-yl. To investigate the impact of the linker nature on anticancer activity, the phenethyl amino carbonyl moieties were modified to 4 phenylpiperazin-1-yl carbonyl as in compounds 6a–c or benzyl carbonyl as in compound 7. The position and number of halogen atoms on the indole moiety’s phenyl ring were also investigated. The antiproliferative activity of compounds 5a–k, 6a–c, and 7 against a panel of cancer cell lines were investigated. The most active compounds were evaluated for mechanistic activity as multi-targeted kinase inhibitors such as EGFR and CDK2. Furthermore, the compounds were evaluated for apoptotic activity against caspases 3, 8, and 9, as well as Cytochrome C, Bax, Bcl2, and p53.
Figure 2.

Structures of new compounds 5a–k, 6a–c, and 7.
2. Results and Discussion
2.1. Chemistry
Scheme 1 depicts the synthesis of target compounds 5a–k, 6a–c, and 7. Derivatives of phenyl hydrazine Hydrochloride 1a–d under Fisher Indole cyclization were reacted with 2-oxopropanoic acid 2 in the presence of PTSA (p-toluenesulfonic acid) to provide 3-methylindole-2-carboxylates 3a–d [24]. The carboxylic acids 4a–d was obtained by alkaline hydrolysis of the esters 3a–d [25]. The appropriate amines were coupled with carboxylic acids 4a–d in the presence of DIPEA in DCM using BOP as a coupling reagent [23], yielding target carboxamides 5a–k, 6a–c, and 7. 1H NMR, 13C NMR, and HRESI-MS were used to identify the newly synthesised derivatives. The 1H NMR spectrum of 5d revealed the appearance of three singlet signals: one at δ 11.34 ppm (1H) consistent with indole NH, one at δ 7.89 ppm (1H) relating to amidic NH, and one at δ 2.41 ppm (3H) corresponding to the methyl group. The spectrum also indicated the existence of signals corresponding to ethylene protons at δ 3.48 (q, J = 7.1 Hz, 2H, NHCH2) and δ 2.77 (t, J = 7.4 Hz, 2H, NHCH2), in addition to the morpholine group’s distinctive signals at δ 3.70 (t, J = 4.8 Hz, 4H, morph-H) and δ 3.02 (t, J = 4.8 Hz, 4H, morph-H). HRESI-MS revealed a peak for [M + H]+ at m/z 398.1629, which corresponds to the molecular formula C22H25ClN3O2.
Scheme 1.

Synthesis of the target compounds 5a–k, 6a–c, and 7. Reagents and conditions: (a) PTSA, EtOH, reflux, 20 h, 82%; (b) 5% NaOH, EtOH, 40 °C, overnight, 95%; (c) BOP, DIPEA, DCM, rt, overnight, 75–94%.
2.2. Evaluation of Biological Activities
2.2.1. In Vitro Anticancer Activity
Cell Viability Assay
The MCF-10A (human mammary gland epithelial) cell line was used in the cell viability experiment. Compounds 5a–k, 6a–c, and 7 were incubated with MCF-10A cells for 4 days at 50 µM concentration, and the viability of cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test. [26]. All compounds had no cytotoxic effects, and the vitality of the cells was more than 83% for most of the compounds examined.
Antiproliferative Activity
Using the MTT assay with doxorubicin as the reference drug, the antiproliferative activities of 5a–k, 6a–c, and 7 against four human cancer cell lines, including pancreas cancer cell line (Panc-1), breast cancer cell line (MCF-7), colon cancer cell line (HT-29), and epithelial cancer cell line (A-549) were investigated [27]. Table 1 shows the results of calculating the median inhibitory concentration (IC50) for all derivatives. Generally, compounds 5a–k, 6a–c, and 7 demonstrated greater antiproliferative activity against the breast cancer cell line (MCF-7) than other tested cancer cell lines, and 5a–k (which contain the phenethyl moiety in their backbone structure) demonstrated greater potency than 6a–c and 7, indicating the importance of the phenethyl moiety for antiproliferative action.
Table 1.
Antiproliferative activity of compounds 5a–k, 6a–c, 7, and Doxorubicin.
| ||||||
|---|---|---|---|---|---|---|
| Compd. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (µM) | ||||
| A-549 | MCF-7 | Panc-1 | HT-29 | Average | ||
| 5a | 89 | 3.70 ± 0.30 | 3.20 ± 0.30 | 3.90 ± 0.30 | 3.90 ± 0.30 | 3.70 |
| 5b | 87 | 3.20 ± 0.30 | 2.90 ± 0.30 | 3.50 ± 0.30 | 3.60 ± 0.30 | 3.30 |
| 5c | 87 | 1.70 ± 0.20 | 1.40 ± 0.20 | 1.80 ± 0.20 | 1.80 ± 0.20 | 1.70 |
| 5d | 89 | 1.05 ± 0.10 | 0.90 ± 0.10 | 1.10 ± 0.10 | 1.10 ± 0.10 | 1.05 |
| 5e | 93 | 0.95 ± 0.05 | 0.80 ± 0.05 | 1.00 ± 0.20 | 1.10 ± 0.10 | 0.95 |
| 5f | 90 | 1.90 ± 0.20 | 1.70 ± 0.20 | 2.10 ± 0.20 | 2.10 ± 0.20 | 1.95 |
| 5g | 89 | 4.90 ± 0.50 | 4.80 ± 0.50 | 5.20 ± 0.50 | 5.10 ± 0.50 | 5.00 |
| 5h | 87 | 1.00 ± 0.10 | 0.90 ± 0.10 | 1.20 ± 0.10 | 1.20 ± 0.10 | 1.10 |
| 5i | 90 | 1.55 ± 0.20 | 1.30 ± 0.10 | 1.60 ± 0.20 | 1.65 ± 0.20 | 1.50 |
| 5j | 83 | 1.20 ± 0.10 | 1.00 ± 0.10 | 1.30 ± 0.10 | 1.30 ± 0.10 | 1.20 |
| 5k | 87 | 1.40 ± 0.20 | 1.20 ± 0.10 | 1.50 ± 0.20 | 1.50 ± 0.20 | 1.40 |
| 6a | 90 | 2.90 ± 0.30 | 2.60 ± 0.20 | 2.80 ± 0.20 | 2.90 ± 0.20 | 2.80 |
| 6b | 91 | 2.50 ± 0.20 | 2.30 ± 0.20 | 2.65 ± 0.20 | 2.80 ± 0.20 | 2.60 |
| 6c | 89 | 2.20 ± 0.20 | 2.10 ± 0.20 | 2.40 ± 0.20 | 2.50 ± 0.20 | 2.30 |
| 7 | 91 | 4.10 ± 0.40 | 4.00 ± 0.40 | 4.40 ± 0.40 | 4.60 ± 0.40 | 4.30 |
| Doxorubicin | - | 1.20 ± 0.20 | 0.90 ± 0.10 | 1.40 ± 0.20 | 1.00 ± 0.10 | 1.10 |
The 2-methylpyrrolidin-4-yl phenethyl derivative 5e (R1 = Cl, R2 = R3 = H, R4 = 2-methylpyrrolidin-1-yl) was the most potent derivative, with a GI50 value of 0.95 µM against the four cell lines, being more potent than the reference doxorubicin (GI50 = 1.10 µM) and also it was more potent than doxorubicin in A-549, MCF-7, and Panc-1 cell lines (IC50 = 0.95, 0.80, and 1.00 µM, respectively, while for doxorubicin IC50 = 1.20, 0.90, and 1.40 µM, respectively). The unsubstituted derivative 5a (R1 = Cl, R2 = R3 = R4 = H) was roughly four-fold less effective than 5e, with a GI50 = 3.70 µM, whereas the 4-dimethylamino derivative 5b (R1 = Cl, R2 = R3 = H, R4 = dimethylamino) had a GI50 = 3.30 µM.
Compound 5d (R1 = Cl, R2 = R3 = H, R4 = morpholin-4-yl) rated second in activity with a GI50 of 1.05 µM against the four cancer cell lines, being somewhat less potent (1.1-fold) than 5e but equipotent to doxorubicin and even more potent than doxorubicin against A-549 and Panc-1 cell lines.
Replacement of the 2-methylpyrrolidin-4-yl moiety in compound 5e or the morpholin-4-yl in 5d by 4-piperidin-1-yl in compound 5c resulted in at least 1.8- and 1.6-fold reduction of the mean GI50 values, respectively, signifying the importance of the 2-methylpyrrolidin-4-yl and 4-morpholinophenethyl moieties for the antiproliferative activity.
The 5-substitution impact was also investigated. As indicated in Table 1, compound 5g (R2 = Cl, R1 = R3 = H, R4 = 4-piperidin-1-yl) had much lower antiproliferative efficacy (3 times) than compound 5c (R1 = Cl, R2 = R3 = H, R4 = 4-piperidin-1-yl). Compound 5f (R2 = Cl, R1 = R3 = H = R4 = H) was, on the other hand, more potent (1.9 times) than compound 5a (R1 = Cl, R2 = R3 = R4 = H).
Furthermore, we attempt to explore the effect of increasing the number of halogen atoms on antiproliferative activity. For instance, the dihalo derivatives 5h (R1 = R3 = Cl, R2 = H, R4 = 4-piperidin-1-yl) and 5k (R1 = R3 = F, R2 = H, R4 = 4-piperidin-1-yl) had higher antiproliferative activity than the monohalo derivative 5c (R1 = Cl, R2 = R3 = H, R4 = 4-piperidin-1-yl) with GI50 values of 1.10 µM and 1.40 µM, respectively, compared to 5c (GI50 = 1.70 µM), indicating the relevance of dihalo atoms for antiproliferative activity and that the chlorine atom is better tolerated than the fluorine one. The same is true for 5j (R1 = R3 = F, R2 = R4 = H), which has higher potency (GI50 = 1.20 µM) than 5a (R1 = Cl, R2 = R3 = R4 = H, GI50 = 3.7 µM) and 5f (R2 = Cl, R1 = R3 = H = R4 = H, GI50 = 1.95 µM). The situation is somewhat different in the case of 5,7-dichloro derivative 5i (R1 = R3 = Cl, R2 = H, R4 = morpholin-4-yl), which demonstrated lower potency (GI50 = 1.50 µM) than the 5-chloro derivative 5d (R1 = Cl, R2 = R3 = R4 = morpholin-4-yl) with a GI50 value of 1.05 µM.
Furthermore, among the tested compounds, the 4-benzyl carbonyl derivative 7 and the 4-phenylpiperazin-1-yl carbonyl derivatives 6a–c had the lowest mean GI50 values, implying that the N-phenethyl carboxamide architecture is important for antiproliferative action and correlating with previous SAR studies [23].
2.2.2. EGFR Inhibitory Activity
The inhibitory efficacy of 5d, 5e, and 5h–k against EGFR was evaluated using the EGFR-TK assay [28], and the findings are presented in Table 2. The results of this test supplement the findings of the cancer-cell-based investigation. All the tested derivatives (5d, 5e, and 5h–k) inhibited EGFR significantly, with IC50 values ranging from 89 to 137 nM. Based on the findings given, three derivatives 5d, 5e, and 5j were found to be the most potent, with EGFR inhibitory effects (IC50 = 89 ± 6 nM and 93 ± 8 nM, and 98 ± 8 nM, respectively) comparable to the positive erlotinib (IC50 = 80 ± 5 nM). Again, the 2-methylpyrrolidin-1-yl phenethyl derivative 5e (R1 = Cl, R2 = R3 = H, R4 = 2-methylpyrrolidin-1-yl) and the 4-morpholin-4-yl phenethyl 5d (R1 = Cl, R2 = R3 = H, R4 = morpholin-4-yl) were the most potent of all synthesized derivatives, with IC50 value of 93 nM and 89 nM being equipotent to the reference erlotinib.
Table 2.
Effects of compounds 5d, 5e, 5h–k, and Erlotinib on EGFR and Dinaciclib.
| ||||||
|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | R3 | R4 | EGFR Inhibition IC50 ± SEM (nM) |
CDK2 Inhibition IC50 ± SEM (nM) |
| 5d | Cl | H | H |
|
89 ± 6 | 23 ± 2 |
| 5e | Cl | H | H |
|
93 ± 8 | 13 ± 1 |
| 5h | Cl | H | Cl |
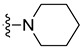
|
118 ± 10 | 11 ± 1 |
| 5i | Cl | H | Cl |
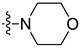
|
137 ± 12 | 27 ± 3 |
| 5j | F | H | F | H | 98 ± 8 | 34 ± 3 |
| 5k | F | H | F |
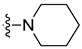
|
129 ± 11 | 19 ± 2 |
| Erlotinib | -- | -- | -- | -- | 80 ± 5 | ND |
| Dinaciclib | -- | -- | -- | -- | ND | 20 ± 2 |
2.2.3. CDK2 Inhibitory Assay
Compounds 5d, 5e, and 5h–k were further examines for their ability to inhibit the CDK2 enzyme [29]. The IC50 values are shown in Table 2. In comparison to the reference dinaciclib (IC50 = 20 nM), all investigated derivatives inhibited CDK2 effectively, with IC50 values ranging from 11 nM to 34 nM. Three derivatives, 5e, 5h, and 5k, were shown to be superior to the standard dinaciclib as CDK2 inhibitors, with IC50 values of 13, 11, and 19 nM, respectively. Compound 5e, the most potent antiproliferative derivative, displayed significant anti-CDK2 activity with an IC50 value of 13 nM, which is 1.5-fold more active than the reference dinaciclib. On the other hand, compounds 5d, 5i, 5j, and 5k exhibited significant activity against CDK2 (IC50 = 23, 27, 34, and 19 µM) comparable to dinaciclib. The findings of the EGFR and CDK2 tests revealed that the examined compounds exhibit significant antiproliferative activity and are efficient at suppressing both CDK2 and EGFR.
2.2.4. Apoptosis Assay
A previous report has shown that CBD (2), a CB1 allosteric modulator, can trigger apoptosis [30]. Therefore, to assess the proapoptotic potential of our target compounds, we evaluated the most active compounds 5d, 5e, and 5h for their capacity to initiate the apoptosis cascade in the breast cancer (MCF-7) cell line.
Activation of Proteolytic Caspases Cascade
Caspases play a crucial role in the initiation and completion of the apoptotic process [31]. Caspase-3 is a crucial caspase that cleaves a variety of proteins in cells, causing apoptosis [32]. The effects of compounds 5d, 5e, and 5h on caspase 3 were assessed and compared to doxorubicin, which was used as a control [33]. The results showed that when compared to control cells, the tested compounds increased the level of active caspase 3 by 8–10 folds and that 5d, 5e, and 5h induce outstanding overexpression of caspase-3 protein level (570.00 ± 5.00, 635.50 ± 5.50 and 537.50 ± 5.00 pg/mL, respectively) compared to doxorubicin (503.50 ± 4.50 pg/mL). In comparison to the control untreated cells, the most active antiproliferative derivative 5e increases caspase 3 levels by 9.70 times.
The impact of compounds 5d, 5e, and 5h on caspases 8 and 9 was also investigated to highlight the involvement of the intrinsic and extrinsic apoptotic pathways in the antiproliferative actions of these compounds, Table 3. When compared to control cells, compound 5e increased caspase 8 and 9 levels by 10.90 and 18.15 folds, respectively, while compound 5d increased caspase 8 and 9 levels by 9.70 and 17.80 folds, respectively, indicating activation of both intrinsic and extrinsic pathways with a stronger effect on the intrinsic pathway because caspase 9 levels were higher [34].
Table 3.
Effects of compounds 5d, 5e, 5h and doxorubicin on active Caspases 3, 8, 9 and Cytochrome C in MCF-7 breast cancer cell line.
| Compound Number | Caspase-3 | Caspase-8 | Caspase-9 | Cytochrome C | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (pg/mL) |
Fold Change |
Conc (ng/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (ng/mL) | Fold Change | |
| 5d | 570.00 ± 5.00 | 8.70 | 1.94 | 9.70 | 16.90 | 17.80 | 0.70 | 14 |
| 5e | 635.50 ± 5.50 | 9.70 | 2.17 | 10.90 | 17.25 | 18.15 | 0.80 | 16 |
| 5h | 537.50 ± 5.00 | 8.20 | 1.88 | 9.50 | 16.65 | 17.50 | 0.65 | 13 |
| Doxorubicin | 503.50 ± 4.50 | 7.70 | 1.80 | 9.00 | 16.25 | 17.00 | 0.60 | 12 |
| Control | 65.50 | 1 | 0.20 | 1 | 0.95 | 1 | 0.05 | 1 |
Cytochrome C Assay
The quantity of cytochrome C within the cell is important for activating caspases and commencing the intrinsic apoptosis process [35]. Table 3 shows the findings of testing indole-2-carboxamide derivatives 5d, 5e, and 5h as Cytochrome C activators in the MCF-7 human breast cancer cell line. Compounds 5d, 5e, and 5h increased Cytochrome C levels in the MCF-7 human breast cancer cell line by 14, 16, and 13 times, respectively, compared to untreated control cells. The findings add to the evidence that apoptosis can be attributed to Cytochrome C overexpression and activation of the intrinsic apoptotic pathway triggered by the investigated compounds.
Bax and Bcl-2 Levels Assay
The most potent caspase activators, 5d and 5e, were investigated further for their influence on Bax and Bacl-2 levels in a breast cancer cell line (MCF-7) using doxorubicin as a control [36]. Table 4 shows that 5d and 5e caused a significant increase in Bax levels when compared to doxorubicin. Compound 5e demonstrated a comparable induction of Bax (296.50 pg/mL) compared to doxorubicin (276 pg/mL) with a 36-fold increase over control untreated breast cancer cells, followed by compound 5d (290 pg/mL and 35-fold rise). Finally, compound 5e reduced the anti-apoptotic Bcl-2 protein levels to 0.87 ng/mL in MCF-7 cells, followed by compound 5d (0.89 ng/mL) in comparison to doxorubicin (0.98 ng/mL).
Table 4.
Effects of compounds 5d, 5e, and doxorubicin on Bax and Bcl-2.
| Compd. No. | Bax | Bcl-2 | ||
|---|---|---|---|---|
| Conc (pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | |
| 5d | 289.70 ± 2.50 | 35 | 0.89 | 5.70 |
| 5e | 296.50 ± 2.50 | 36 | 0.87 | 5.90 |
| Doxorubicin | 275.80 ± 2.50 | 33 | 0.98 | 5.20 |
| Cont. | 8.25 | 1 | 5.10 | 1.00 |
Effect of Compounds 5d and 5e on p53 Transcription in MCF-7
p53 is a unique protein that participates in several physiological processes such as cell metabolism [37], stem cell maintenance [38], and cell adhesion [39]. Because p53 is frequently inactivated in cancer cells, the cells are unable to undergo apoptosis [40,41]. Similarly, activating, or stabilizing p53 aids cancer cells in normalizing p53-controlled physiological processes and increasing apoptotic activity [42]. The effects of 5d and 5e on p53 transcription were evaluated and compared to doxorubicin as a control [43], Table 5. The results revealed an increase of at least 27-folds in p53 level compared to the test cells and that the p53 protein level of 5d and 5e was significantly inductive (1375 and 1435 pg/mL, respectively) in relation to doxorubicin (1265 pg/mL).
Table 5.
Effects of compounds 5d, 5e, and doxorubicin on p53.
| Compd. No. | p53 | |
|---|---|---|
| Conc (pg/mL) | Fold Change | |
| 5d | 1375 ± 15 | 27 |
| 5e | 1435 ± 15 | 28 |
| Doxorubicin | 1265 ± 10 | 25 |
| Cont. | 51.50 | 1 |
2.3. Docking Study
Interestingly, running docking simulations of compounds 5d and 5e within EGFR active site revealed docking scores (S; −6.90 and −6.79 kcal/mol; respectively), so much close to that of co-crystallized ligand, erlotinib (−7.30 kcal/mol), which co-insides with what obtained in-vitro against EGFR enzyme (as shown in Table 2). Moreover, visual inspection of the best docking poses of compounds 5d and 5e showed their close distance to key amino acids lining EGFR active site. Additionally, compounds 5d and 5e showed a number of H-bonding and pi–H interactions with LEU 694 and THR 766 amino acid residues (as listed in Table 6 and shown in Figure 3).
Table 6.
MD of compounds 5d and 5e within EGFR and CDK2 active sites.
| Compd. | EFGR (PDB ID: 1M17) | CDK2 (PDB ID: 1PYE) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S a | RMSD (Å) |
Binding Interactions | S | RMSD (Å) |
Binding Interactions | |||||
| a.a. Residue |
Type | Distance (Å) |
a.a. Residue |
Type | Distance (Å) |
|||||
| 5d | −6.90 | 1.49 | THR 766 | H-acceptor | 2.91 | −6.03 | 1.77 | GLN 131 | H-donor | 3.60 |
| LEU 694 | pi-H | 3.99 | ||||||||
| 5e | −6.79 | 1.51 | LEU 694 | pi-H | 3.70 | −6.99 | 1.68 | LYS 33 | pi-cation | 4.65 |
| Ref | −7.3 b | 1.28 | GLN 767 | H-donor | 3.15 | −5.89 c | 1.84 | GLU 81 | H-donor | 3.05 |
| MET 769 | H-acceptor | 2.70 | LEU 83 | H-acceptor | 3.07 | |||||
a S: docking score (kcal/mol); b Ref: co-crystallized ligand (Erlotinib); c Ref: co-crystallized ligand (Dinaciclib).
Figure 3.

Schematic 2D representation of best docking poses of 5d (left) and 5e (right) within EGFR (PDB ID: 1M17) active site showing pi-H (green-dotted line) and H-acceptor interactions (green arrow).
Both compounds 5d and 5e showed a common settling profile within EGFR active represented by U-shaped bending of the whole molecule, so its indole ring interacts with LEU 694 (as shown in Figure 3). On the other hand, compound 5d showed additional H-acceptor bonding with THR 766 that resulted in its better docking score over its congener, compound 5e.
Additionally, and as shown in Table 6, MDs of compound 5e within the CDK2 active site revealed its better docking score (S = −6.99 kcal/mol) over its congener 5d (S = −6.03 kcal/mol), although its inability to have strong H-bonding with amino acid residues lining active site, its close proximity to key amino acid residues (revealed by its proximity contour as shown in Figure 4) could explain its better scoring over 5d.
Figure 4.

Schematic 2D representation of best docking poses of 5d (left) and 5e (right) within CDK2 (PDB ID: 1PYE) active site showing pi-cation (green-dotted line) and H-donor interactions (blue arrow).
3. Materials and Methods
3.1. Chemistry
3-Methylindole-2-carboxylates 3a–d [24], carboxylic acids 4a–d [25], and carboxamides 5a–k, 6a–c, and 7 [23] were synthesized according to previously reported procedures.
3.1.1. 5-Chloro-3-methyl-N-phenethyl-1H-indole-2-carboxamide (5a)
Yield % 91, m.p 182–184 °C, 1H NMR (400 MHz, CDCl3) δ 9.21 (s, 1H, indole NH), 7.53 (d, J = 2.2 Hz, 1H, Ar-H), 7.39–7.17 (m, 7H, Ar-H), 5.97 (s, 1H, amide NH), 3.81 (q, J = 6.4 Hz, 2H, NHCH2), 2.97 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.26 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 162.10 (C=O), 138.50, 133.32, 128.85, 128.81, 128.53, 126.84, 125.53, 125.00, 119.38, 112.79, 110.80, 40.83, 35.51, 9.88. HRESI-MS m/z calcd for [M + H]+ C18H18ClN2O: 313.1102, found: 313.1104.
3.1.2. 5-Chloro-N-(4-(dimethylamino)phenethyl)-3-methyl-1H-indole-2-carboxamide (5b)
Yield % 89, m.p 202–204 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1H, indole NH), 7.87 (t, J = 5.6 Hz, 1H, amide NH), 7.62 (d, J = 2.0 Hz, 1H, Ar-H), 7.37 (d, J = 8.6 Hz, 1H, Ar-H), 7.16 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 7.06 (d, J = 8.6 Hz, 2H, Ar-H), 6.66 (d, J = 8.7 Hz, 2H, Ar-H), 3.46 (q, J = 6.7 Hz, 2H, NHCH2), 2.82 (s, 6H, N(CH3)2), 2.74 (t, J = 6.6 Hz, 2H, NHCH2CH2), 2.41 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 162.01 (C=O), 149.54, 134.07, 129.94, 129.59, 129.55, 127.31, 124.06, 123.97, 119.35, 113.92, 113.13, 112.93, 41.47, 40.79, 34.69, 9.98. HRESI-MS m/z calcd for [M + H]+ C20H23ClN3O: 356.1524, found: 356.1522.
3.1.3. 5-Chloro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5c)
Yield % 92, m.p 216–218 °C, 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H, indole NH), 7.53 (s, 1H, Ar-H), 7.29 (d, J = 8.6 Hz, 1H, Ar-H), 7.20 (dd, J = 8.6, 0.7 Hz, 1H, Ar-H), 7.13 (d, J = 8.6 Hz, 2H, Ar-H), 6.92 (d, J = 8.6 Hz, 2H, Ar-H), 5.97 (s, 1H, amide NH), 3.76 (q, J = 6.6 Hz, 2H, NHCH2), 3.17–3.09 (m, 4H, piperidin-H), 2.87 (t, J = 6.7 Hz, 2H, NHCH2CH2), 2.27 (s, 3H, CH3), 1.74–168 (m, 4H, piperidin-H), 1.63–1.53 (m, 2H, piperidin-H). 13C NMR (101 MHz, CDCl3) δ 162.03 (C=O), 151.21, 133.26, 129.74, 129.37, 128.77, 128.68, 125.48, 124.90, 119.37, 116.95, 112.75, 110.71, 50.82, 40.92, 34.48, 25.79, 24.25, 9.94. HRESI-MS m/z calcd for [M + H]+ C23H27ClN3O: 396.1837, found: 396.1837.
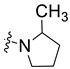
3.1.4. 5-Chloro-3-methyl-N-(4-morpholinophenethyl)-1H-indole-2-carboxamide (5d)
Yield % 91, m.p 210–212 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1H, indole NH), 7.89 (t, J = 5.6 Hz, 1H, amide NH), 7.62 (s, 1H, Ar-H), 7.38 (d, J = 8.6 Hz, 1H, Ar-H), 7.21–7.08 (m, 3H, Ar-H), 6.85 (d, J = 8.3 Hz, 2H, Ar-H), 3.70 (t, J = 4.8 Hz, 4H, morph-H), 3.48 (q, J = 7.1 Hz, 2H, NHCH2), 3.02 (t, J = 4.8 Hz, 4H, morph-H), 2.77 (t, J = 7.4 Hz, 2H, NHCH2), 2.41 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 162.05 (C=O), 149.99, 134.08, 130.44, 129.91, 129.62, 129.59, 124.08, 124.00, 119.36, 115.72, 113.94, 112.98, 66.57, 49.17, 41.29, 34.71, 9.98. HRESI-MS m/z calcd for [M + H]+ C22H25ClN3O2: 398.1630, found: 398.1629.
3.1.5. 5-Chloro-3-methyl-N-(4-(2-methylpyrrolidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5e)
Yield % 85, m.p 186–188 °C. 1H NMR (400 MHz, CDCl3) δ 9.67 (s, 1H, indole NH), 7.53 (d, J = 2.0 Hz, 1H, Ar-H), 7.32 (d, J = 8.6 Hz, 1H, Ar-H), 7.20 (dd, J = 8.7, 2.0 Hz, 1H, Ar-H), 7.10 (d, J = 8.5 Hz, 2H, Ar-H), 6.57 (d, J = 8.5 Hz, 2H, Ar-H), 6.07 (t, J = 5.5 Hz, 1H, amide NH), 3.91–3.71 (m, 3H, pyrrolidin-H, NHCH2), 3.47–3.37 (m, 1H, pyrrolidin-H), 3.18–3.12 (m, 1H, pyrrolidin-H), 2.87 (t, J = 6.7 Hz, 2H, NHCH2CH2), 2.31 (s, 3H, CH3), 2.15–1.94 (m, 3H, pyrrolidin-H), 1.73–1.70 (m, 1H, pyrrolidin-H), 1.17 (d, J = 6.2 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3) δ 162.23 (C=O), 146.18, 133.48, 129.68, 129.54, 128.78, 125.34, 124.78, 124.45, 119.29, 112.92, 112.16, 110.77, 53.67, 48.26, 41.25, 34.42, 33.09, 23.28, 19.29, 10.01. HRESI-MS m/z calcd for [M + H]+ C23H27ClN3O: 396.1837, found: 396.1832.
3.1.6. 6-Chloro-3-methyl-N-phenethyl-1H-indole-2-carboxamide (5f)
Yield % 80, m.p 170–172 °C, 1H NMR (400 MHz, CDCl3) δ 9.67 (s, 1H, indole NH), 7.46 (d, J = 8.6 Hz, 1H, Ar-H), 7.42–7.22 (m, 6H, Ar-H), 7.07 (dd, J = 8.6, 1.9 Hz, 1H, Ar-H), 6.00 (s, 1H, amide NH), 3.82 (q, J = 6.4 Hz, 2H, NHCH2), 2.98 (t, J = 6.7 Hz, 2H, NHCH2CH2), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 162.39 (C=O), 138.53, 135.49, 130.42, 128.84, 128.83, 127.88, 127.20, 126.81, 120.92, 120.67, 111.61, 40.89, 35.53, 9.94. HRESI-MS m/z calcd for [M + H]+ C18H18ClN2O: 313.1102, found: 313.1101.
3.1.7. 6-Chloro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5g)
Yield % 75, m.p 218–220 °C, 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H, indole NH), 7.47 (d, J = 8.6 Hz, 1H, Ar-H), 7.37 (d, J = 1.8 Hz, 1H, Ar-H), 7.13 (d, J = 8.6 Hz, 2H, Ar-H), 7.08 (dd, J = 8.6, 1.8 Hz, 1H, Ar-H), 6.92 (d, J = 8.6 Hz, 2H, Ar-H), 5.97 (s, 1H, amide NH), 3.76 (q, J = 6.6 Hz, 2H, NHCH2), 3.17–3.09 (m, 4H, piperidin-H), 2.88 (t, J = 6.6 Hz, 2H, NHCH2CH2), 2.29 (s, 3H, CH3), 1.76–1.66 (m, 4H, piperidin-H), 1.64–1.54 (m, 2H, piperidin-H). 13C NMR (101 MHz, CDCl3) δ 162.12 (C=O), 151.17, 135.22, 130.41, 129.39, 128.87, 128.04, 127.32, 120.95, 120.70, 116.97, 111.44, 111.39, 50.86, 40.93, 34.51, 25.79, 24.24, 9.97. HRESI-MS m/z calcd for [M + H]+ C23H27ClN3O: 396.1837, found: 396.1837.
3.1.8. 5,7-Dichloro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5h)
Yield % 85, m.p 178–180 °C, 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H, indole NH), 8.36 (t, J = 5.6 Hz, 1H, amide NH), 7.66 (d, J = 1.8 Hz, 1H, Ar-H), 7.36 (d, J = 1.9 Hz, 1H, Ar-H), 7.07 (d, J = 8.0 Hz, 2H, Ar-H), 6.83 (d, J = 8.3 Hz, 2H, Ar-H), 3.45 (q, J = 7.3 Hz, 2H, NHCH2), 3.03 (t, J = 5.4 Hz, 4H, piperidin-H), 2.75 (t, J = 7.5 Hz, 2H, NHCH2CH2), 2.46 (s, 3H, CH3), 1.63–1.43 (m, 6H, piperidin-H). 13C NMR (101 MHz, DMSO-d6) δ 161.24 (C=O), 150.70, 131.41, 130.65, 130.24, 129.68, 129.53, 124.16, 123.24, 118.73, 117.38, 116.60, 116.52, 50.33, 41.27, 34.69, 25.76, 24.34, 10.19. HRESI-MS m/z calcd for [M + H]+ C23H26Cl2N3O: 430.1447, found: 430.1448.
3.1.9. 5,7-Dichloro-3-methyl-N-(4-morpholinophenethyl)-1H-indole-2-carboxamide (5i)
Yield % 84, m. p 185–187 °C. 1H NMR (400 MHz, CDCl3) δ 9.25 (s, 1H, indole NH), 7.43 (d, J = 1.7 Hz, 1H, Ar-H), 7.25 (d, J = 1.8 Hz, 1H, Ar-H), 7.15 (d, J = 8.5 Hz, 2H, Ar-H), 6.88 (d, J = 8.7 Hz, 2H, Ar-H), 6.02 (s, 1H, amide NH), 3.90–3.83 (m, 4H, morph-H), 3.77 (q, J = 6.6 Hz, 2H, NHCH2), 3.17–3.09 (m, 4H, morph-H), 2.89 (t, J = 6.7 Hz, 2H, NHCH2CH2), 2.28 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 161.59 (C=O), 150.23, 130.98, 130.33, 129.69, 129.53, 129.33, 125.41, 123.94, 118.19, 117.65, 116.11, 111.86, 66.86, 49.47, 41.01, 34.48, 10.12. HRESI-MS m/z calcd for [M + H]+ C22H24Cl2N3O2: 432.1240, found: 432.1240.
3.1.10. 5,7-Difluoro-3-methyl-N-phenethyl-1H-indole-2-carboxamide (5j)
Yield % 82, m.p 198–200 °C, 1H NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1H, indole NH), 8.45 (t, J = 5.6 Hz, 1H, amide NH), 7.69–7.50 (m, 6H, Ar-H), 7.42 (t, J = 11.6 Hz, 1H, Ar-H), 3.86 (q, J = 7.8 Hz, 2H, NHCH2), 3.20 (t, J = 7.4 Hz, 2H, NHCH2CH2), 2.75 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 161.91 (C=O), 140.15, 130.98, 129.46, 129.17, 126.95, 120.88, 116.06, 101.43, 101.15, 99.81, 99.61, 99.50, 41.29, 35.83, 10.49. HRESI-MS m/z calcd for [M + H]+ C18H17F2N2O: 315.1303, found: 315.1305.
3.1.11. 5,7-Difluoro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5k)
Yield % 88, m.p 192–194 °C, 1H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H, indole NH), 8.07 (s, 1H, amide NH), 7.25 (dd, J = 9.3, 2.2 Hz, 1H, Ar-H), 7.13–7.04 (m, 3H, Ar-H), 6.84 (d, J = 8.6 Hz, 2H, Ar-H), 3.46 (q, J = 7.2 Hz, 2H, NHCH2), 3.05 (t, J = 5.4 Hz, 4H, piperidin-H), 2.74 (t, J = 7.4 Hz, 2H, NHCH2CH2), 2.42 (s, 3H, CH3), 1.64–1.55 (m, 4H, piperidin-H), 1.51–1.48 (m, 2H, piperidin-H). 13C NMR (101 MHz, DMSO-d6) δ 161.50 (C=O), 157.34, 155.00, 150.69, 147.63, 131.03, 129.65, 120.65, 116.52, 115.77, 101.08, 99.44, 99.24, 50.33, 41.24, 34.64, 25.76, 24.35, 10.17. HRESI-MS m/z calcd for [M + H]+ C23H26F2N3O: 398.2038, found: 398.2038.
3.1.12. (5-Chloro-3-methyl-1H-indol-2-yl)(4-phenylpiperazin-1-yl)methanone (6a)
Yield % 85, m.p 165–167 °C, 1H NMR (400 MHz, CDCl3) δ 9.0 (s, 1H, indole NH), 8.25 (d, J = 8.8 Hz, 1H, Ar-H), 7.56 (d, J = 2.0 Hz, 1H, Ar-H), 7.40 (dd, J = 8.7, 2.0 Hz, 1H, Ar-H), 7.35–7.21 (m, 5H, Ar-H), 3.76 (t, J = 7.6 Hz, 4H, piperazin-H), 3.00 (t, J = 8.0 Hz, 4H, piperazin-H), 2.65 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 164.41 (C=O), 150.78, 134.46, 129.28, 128.23, 127.44, 125.47, 124.14, 120.80, 119.61, 118.01, 116.80, 112.67, 49.87, 18.26, 14.76. HRESI-MS m/z calcd for [M + H]+ C20H21ClN3O: 354.1368, found: 354.1367.
3.1.13. (6-Chloro-3-methyl-1H-indol-2-yl)(4-phenylpiperazin-1-yl)methanone (6b)
Yield % 80, m.p 180–182 °C, 1H NMR (400 MHz, CDCl3) δ 9.68 (s, 1H, indole NH), 7.47 (d, J = 8.5 Hz, 1H, Ar-H), 7.37–7.25 (m, 2H, Ar-H), 7.08 (d, J = 8.0 Hz, 1H, Ar-H), 6.95–6.92 (m, 3H, Ar-H), 3.88 (t, J = 5.1 Hz, 4H, piperazin-H), 3.22 (t, J = 5.3 Hz, 4H, piperazin-H), 2.38 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 164.69 (C=O), 150.82, 136.51, 129.76, 129.29, 127.52, 126.64, 120.73, 120.67, 120.56, 116.78, 112.10, 111.53, 49.88, 10.18. HRESI-MS m/z calcd for [M + H]+ C20H21ClN3O: 354.1368, found: 354.1368.
3.1.14. (5,7-Difluoro-3-methyl-1H-indol-2-yl)(4-phenylpiperazin-1-yl)methanone (6c)
Yield % 82, m.p 171–173 °C, 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1H, indole NH), 7.29 (t, J = 7.9 Hz, 2H, Ar-H), 7.13–6.66 (m, 5H, Ar-H), 3.89 (t, J = 5.8 Hz, 4H, piperazin-H), 3.23 (t, J = 5.7 Hz, 4H, piperazin-H), 2.34 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 163.99 (C=O), 150.78, 129.34, 129.28, 120.79, 116.82, 100.35, 100.31, 100.12, 100.08, 99.28, 99.08, 98.78, 49.94, 10.12. HRESI-MS m/z calcd for [M + H]+ C20H20F2N3O: 356.1569, found: 356.1568.
3.1.15. N-Benzyl-5-chloro-3-methyl-1H-indole-2-carboxamide (7)
Yield % 84, m.p 203–205 °C, 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H, indole NH), 8.43 (t, J = 5.9 Hz, 1H, amide NH), 7.64 (d, J = 2.1 Hz, 1H, Ar-H), 7.41–7.20 (m, 6H, Ar-H), 7.17 (dd, J = 8.7, 2.1 Hz, 1H, Ar-H), 4.50 (d, J = 5.9 Hz, 2H, NHCH2), 2.48 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 162.16 (C=O), 139.83, 134.16, 129.66, 129.56, 128.76, 127.82, 127.28, 124.10, 119.41, 113.96, 113.46, 42.92, 10.07. HRESI-MS m/z calcd for [M + H]+ C17H16ClN2O: 299.0946, found: 299.0946.
3.2. Biology
3.2.1. In Vitro Anticancer Activity
Cell Viability Assay
The MCF-10A (human mammary gland epithelial) cell line was used in the cell viability experiment. Compounds 5a–k, 6a–c, and 7 were incubated with MCF-10A cells for 4 days at 50 µM concentration, and the viability of cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test [26].
Antiproliferative Activity
Using the MTT assay with doxorubicin as the reference drug, the antiproliferative activities of 5a–k, 6a–c, and 7 against four human cancer cell lines, including pancreas cancer cell line (Panc-1), breast cancer cell line (MCF-7), colon cancer cell line (HT-29), and epithelial cancer cell line (A-549) were investigated [27].
3.2.2. EGFR Inhibitory Activity
The inhibitory efficacy of 5d, 5e, and 5h–k against EGFR was evaluated using the EGFR-TK assay [28].
3.2.3. CDK2 Inhibitory Assay
Compounds 5d, 5e, and 5h–k were further examined for their ability to inhibit the CDK2 enzyme [29].
3.2.4. Apoptosis Assay
Activation of Proteolytic Caspases Cascade
The effects of compounds 5d, 5e, and 5h on caspases 3, 8, and 9 were assessed and compared to doxorubicin, which was used as a control [33].
Cytochrome C Assay
Indole-2-carboxamide derivatives 5d, 5e, and 5h were evaluated as Cytochrome C activators in the MCF-7 human breast cancer cell line [35].
Bax and Bcl-2 Levels Assay
The most potent caspase activators, 5d and 5e, were investigated for their influence on Bax and Bacl-2 levels in a breast cancer cell line (MCF-7) using doxorubicin as a control [36].
Effect of Compounds 5d and 5e on p53 Transcription in MCF-7
The effects of 5d and 5e on p53 transcription were evaluated and compared to doxorubicin as a control [43].
4. Conclusions
A new series of EGFR/CDK2 dual inhibitors containing indole-2-carboxamides has been reported. A total of fifteen target compounds were synthesized and evaluated in vitro against four cancer cell lines as well as these two kinases. The majority of the compounds examined had promising antiproliferative activity. The most effective of these compounds were 5d, 5e, 5h, 5i, 5j, and 5k. The novel compounds induced apoptosis and increased Caspase 3, 8, 9, and Cytochrome C levels. Furthermore, the investigated compounds increased Bax and p53 levels while decreasing anti-apoptotic Bcl2 protein levels. Following optimization, these compounds form a novel class of compounds capable of acting as potent apoptotic anticancer agents for both EGFR and CDK2.
Author Contributions
Conceptualization, B.G.M.Y.; Funding acquisition, L.H.A.-W.; Investigation, L.T.; Methodology, L.H.A.-W., M.H.A., A.H.E.-B., L.T. and B.G.M.Y.; Project administration, L.H.A.-W.; Resources, L.H.A.-W. and Y.A.M.; Software, Y.A.M.; Supervision, L.T.; Validation, Y.A.M. and L.T.; Visualization, L.T.; Writing—original draft, B.G.M.Y.; Writing—review & editing, B.G.M.Y. All authors have read and agreed to the published version of the manuscript.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Funding Statement
This work was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2022R3), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Medina-Franco J.L., Giulianotti M.A., Welmaker G.S., Houghten R.A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov. Today. 2013;18:495–501. doi: 10.1016/j.drudis.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramsay R.R., Popovic-Nikolic M.R., Nikolic K., Uliassi E., Bolognesi M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018;7:3–17. doi: 10.1186/s40169-017-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Msomi N.Z., Shode F.O., Pooe O.J., Mazibuko-Mbeje S., Simelane M.B.C. Iso-Mukaadial Acetate from Warburgia salutaris Enhances Glucose Uptake in the L6 Rat Myoblast Cell Line. Biomolecules. 2019;9:520. doi: 10.3390/biom9100520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu C., Zhang M., Hu M., Guo H.-F., Li J., Yu Y.-L., Jin S., Wang X.-T., Liu L., Liu X.-D. Increased glucagon-like peptide-1 secretion may be involved in antidiabetic effects of ginsenosides. J. Endocrinol. 2013;217:185–196. doi: 10.1530/JOE-12-0502. [DOI] [PubMed] [Google Scholar]
- 5.Zhou P., Xie W., He S., Sun Y., Meng X., Sun G., Sun X. Ginsenoside Rb1 as an anti-diabetic agent and its underlying mechanism analysis. Cells. 2019;8:204. doi: 10.3390/cells8030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qaseem A., Barry M.J., Humphrey M.J., Forciea M.A., Fitterman N., Horwitch C., Kansagara D., McLean R.M., Wilt T.J. Oral pharmacologic treatment of type 2 diabetes mellitus: A clinical practice guideline update from the American college of physicians. Ann. Intern. Med. 2017;166:279–290. doi: 10.7326/M16-1860. [DOI] [PubMed] [Google Scholar]
- 7.Garber A.J., Abrahamson M.J., Barzilay J.I., Blonde L., Bloomgarden Z.T., Bush M.A., Dagogo-Jack S., Davidson M.B., Einhorn D., Garvey W.T., et al. American Association of Clinical Endocrinologists’ comprehensive diabetes management algorithm 2013 consensus statement—Executive summary. Endocr. Pract. 2013;19:536–557. doi: 10.4158/EP13176.CS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussein Z., Wentworth J.M., Nankervis A.J., Proietto J., Colman P.G. Effectiveness, and side effects of thiazolidinediones for type 2 diabetes: Real-life experience from a tertiary hospital. Med. J. Aust. 2004;181:536–539. doi: 10.5694/j.1326-5377.2004.tb06441.x. [DOI] [PubMed] [Google Scholar]
- 9.Elkady A.I., Abuzinadah O.A., Baeshen N.A., Rahmy T.R. Differential control of growth, apoptotic activity, and gene expression in human breast cancer cells by extracts derived from medicinal herbs Zingiber officinale. J. Biomed. Biotechnol. 2012;2012:614356. doi: 10.1155/2012/614356. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Ahmad J., Ahamed M., Akhtar M.J., Alrokayan S.A., Siddiqui M.A., Musarrat J., Al-Khedhairy A.A. Apoptosis induction by silica nanoparticles mediated through reactive oxygen species in human liver cell in eHepG2. Toxicol. Appl. Pharmacol. 2012;259:160–168. doi: 10.1016/j.taap.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Teiten M.H., Eifes S., Dicato M., Diederich M. Curcumin–the paradigm of a multi-target natural compound with applications in cancer prevention and treatment. Toxins. 2010;2:128–162. doi: 10.3390/toxins2010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pourhassanali N., Roshan-Milani S., Kheradmand F., Motazakker M., Bagheri M., Saboory E. Zinc attenuates ethanol-induced Sertoli cell toxicity and apoptosis through caspase-3 mediated pathways. Reprod. Toxicol. 2016;61:97–103. doi: 10.1016/j.reprotox.2016.03.041. [DOI] [PubMed] [Google Scholar]
- 13.Olsson M., Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011;8:1441–1449. doi: 10.1038/cdd.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo R., Overman M., Chatterjee D., Rashid A., Shroff S., Wang H., Katz M.H., Fleming J.B., Varadhachary G.R., Abbruzzese J.L., et al. Aberrant expression of p53, p21, cyclinD1, and BCL2 and their clinicopathological correlation in ampullary adenocarcinoma. Hum. Pathol. 2014;45:1015–1023. doi: 10.1016/j.humpath.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 15.Shi L., Teng H., Zhu M., Li C., Huang K., Chen B., Dai Y., Wang J. Paeoniflorin inhibits nucleus pulposus cell apoptosis by regulating the expression of BCL2 family proteins and caspase-9 in a rabbit model of intervertebral disc degeneration. Exp. Ther. Med. 2015;10:257–262. doi: 10.3892/etm.2015.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan Y., Lu H., An L., Wang C., Zhou Z., Feng F., Ma H., Xu Y., Zhao Q. Effect of active fraction of Eriocaulon sieboldianum on human leukemia K562 cells via proliferation inhibition, cell cycle arrest and apoptosis induction. Environ. Toxicol. Pharmacol. 2016;43:13–20. doi: 10.1016/j.etap.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Lohrum M.A.E., Vousden K.H. Regulation and function of the p53-related proteins: Same family, different rules (Electronic version) Trends Cell Biol. 2000;10:197–202. doi: 10.1016/S0962-8924(00)01736-0. [DOI] [PubMed] [Google Scholar]
- 18.Shrivastava A., Kuzontkoski P.M., Groopman J.E., Prasad A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the crosstalk between apoptosis and autophagy. Mol. Cancer Ther. 2011;10:1161–1172. doi: 10.1158/1535-7163.MCT-10-1100. [DOI] [PubMed] [Google Scholar]
- 19.McAllister S.D., Murase R., Christian R.T., Lau D., Zielinski A.J., Allison J., Almanza C., Pakdel A., Lee J., Limbad C., et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res. Treat. 2011;129:37–47. doi: 10.1007/s10549-010-1177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blazquez C., Casanova M.L., Planas A., Gomez Del Pulgar T., Villanueva C., Fernandez-Acenero M.J., Aragones J., Huffman J.W., Jorcano J.L., Guzman M. Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 2003;17:529–531. doi: 10.1096/fj.02-0795fje. [DOI] [PubMed] [Google Scholar]
- 21.Vaccani A., Massi P., Colombo A., Rubino T., Parolaro D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br. J. Pharmacol. 2005;144:1032–1036. doi: 10.1038/sj.bjp.0706134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramer R., Bublitz K., Freimuth N., Merkord J., Rohde H., Haustein M., Borchert P., Schmuhl E., Linnebacher M., Hinz B. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule 1. FASEB J. 2012;26:1535–1548. doi: 10.1096/fj.11-198184. [DOI] [PubMed] [Google Scholar]
- 23.Youssif B.G.M., Mohamed A.M., Osman E.E.A., Abou-Ghadir O.F., Elnaggar D.H., Abdelrahman M.H., Treamblu L., Gomaa H.A. 5-Chlorobenzofuran-2-carboxamides: From allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019;177:1–11. doi: 10.1016/j.ejmech.2019.05.040. [DOI] [PubMed] [Google Scholar]
- 24.Abdelrahman M.H., Aboraia A.S., Youssif B.G.M., Elsadek B.E.M. Design, synthesis and pharmacophoric model building of new 3-alkoxymethyl/3-phenyl indole-2-carboxamides with potential antiproliferative activity. Chem. Biol. Drug Des. 2017;90:64–82. doi: 10.1111/cbdd.12928. [DOI] [PubMed] [Google Scholar]
- 25.Gomaa H.A.M., Shaker M.E., Alzarea S.I., Hendawy O.M., Mohamed F.A.M., Gouda A.M., Ali A.T., Morcoss M.M., Abdelrahman M.H., Trembleau L., et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022;120:105616. doi: 10.1016/j.bioorg.2022.105616. [DOI] [PubMed] [Google Scholar]
- 26.Al-Wahaibi L.H., Gouda A.M., Abou-Ghadir O.F., Salem O.I.A., Ali A.T., Farghaly H.S., Abdelrahman M.H., Trembleau L., Abdu-Allah H.H.M., Youssif B.G.M. Design, and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020;104:104260. doi: 10.1016/j.bioorg.2020.104260. [DOI] [PubMed] [Google Scholar]
- 27.El-Sheref E.M., Elbastawesy M.A.I., Brown A.B., Shawky A.M., Gomaa H.A.M., Bräse S., Youssif B.G.M. Design and Synthesis of (2-oxo-1, 2-Dihydroquinolin-4-yl)-1,2,3-triazole derivatives via Click Reaction: Potential Apoptotic Antiproliferative Agents. Molecules. 2021;26:6798. doi: 10.3390/molecules26226798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdel-Aziz S.A., Taher E.S., Lan P., Asaad G.F., Gomaa H.A.M., El-Koussi N.A., Youssif B.G.M. Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1, 3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg. Chem. 2021;111:104890. doi: 10.1016/j.bioorg.2021.104890. [DOI] [PubMed] [Google Scholar]
- 29.Mekheimer R.A., Allam S.M.R., Al-Sheikh M.A., Moustafa M.S., Al-Mousawi S.M., Mostafa Y.A., Youssif B.G.M., Gomaa H.A.M., Hayallah A.M., Abdel Aziz M., et al. Discovery of new pyrimido [5,4-c] quinolines as potential antiproliferative agents with multitarget actions: Rapid synthesis, docking, and ADME studies. Bioorg. Chem. 2022;121:105693. doi: 10.1016/j.bioorg.2022.105693. [DOI] [PubMed] [Google Scholar]
- 30.Khurana L., Mackie K., Piomelli D., Kendall D.A. Modulation of CB1 cannabinoid receptor by allosteric ligands: Pharmacology and therapeutic opportunities. Neuropharmacology. 2017;124:3–12. doi: 10.1016/j.neuropharm.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen T., Li J.X., Thomas B.F., Wiley J.L., Kenakin T.P., Zhang Y. Allosteric Modulation: An Alternate Approach Targeting the Cannabinoid CB1 Receptor. Med. Res. Rev. 2017;37:441–474. doi: 10.1002/med.21418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997;236:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abou-Zied H.A., Youssif B.G.M., Mohamed M.F.A., Hayallah A.M., Abdel-Aziz M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity, and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019;89:102997. doi: 10.1016/j.bioorg.2019.102997. [DOI] [PubMed] [Google Scholar]
- 34.Slee E.A., Adrain C., Martin S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001;276:7320–7326. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 35.Hisham M., Youssif B.G.M., Osman E.E.A., Hayallah A.M., Abdel-Aziz M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019;176:117–128. doi: 10.1016/j.ejmech.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 36.Mitupatum T., Aree K., Kittisenachai S., Roytrakul S., Puthong S., Kangsadalampai S., Rojpibulstit P. mRNA Expression of Bax, Bcl-2, p53, Cathepsin B, Caspase-3 and Caspase-9 in the HepG2 Cell Line Following Induction by a Novel Monoclonal Ab Hep88 mAb: Cross-Talk for Paraptosis and Apoptosis. Asian Pac. J. Cancer Prev. 2016;17:703–712. doi: 10.7314/APJCP.2016.17.2.703. [DOI] [PubMed] [Google Scholar]
- 37.Gatza C., Moore L., Dumble M., Donehower L.A. Tumor suppressor dosage regulates stem cell dynamics during aging. Cell Cycle. 2007;6:52–55. doi: 10.4161/cc.6.1.3667. [DOI] [PubMed] [Google Scholar]
- 38.Godar S., Ince T.A., Bell G.W., Feldser D., Donaher J.L., Bergh J., Liu A., Miu K., Watnick R.S., Reinhardt F., et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finlay C.A., Hinds P.W., Levine A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 40.Levine A.J., Hu W., Feng Z. The p53 pathway: What questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 41.Kim N.H., Kim H.S., Kim N.-G., Lee I., Choi H.-S., Li X.-Y., Kang S.E., Cha S.Y., Ryu J.K., Na J.M., et al. p53 and MicroRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 2011;4:197. doi: 10.1126/scisignal.2001744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Junttila M.R., Evan G.I. p53 a Jack of all trades but master of none. Nat. Rev. Cancer. 2009;9:821–829. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 43.Gomaa H.A.M., El-Sherief H.A.M., Hussein S., Gouda A.M., Salem O.I.A., Alharbi K.S., Hayallah A.M., Youssif B.G.M. Novel 1, 2,4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorg. Chem. 2020;105:104369. doi: 10.1016/j.bioorg.2020.104369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data is contained within the article.