

Abstract
Background and Objectives:
High dose melphalan is an integral part of conditioning chemotherapy prior to both autologous and allogeneic hematopoietic cell transplantation (HCT). While underexposure may lead to relapse, overexposure may lead to toxicities include mucositis, diarrhea, bone marrow suppression, and rarely sinusoidal obstruction syndrome. In this study, we describe the population pharmacokinetics (popPK) of high dose melphalan as a first step towards individualized dosing.
Methods:
Melphalan samples were collected in patients receiving an allogeneic or autologous HCT between August 2016 and August 2020 at Memorial Sloan Kettering Cancer Center. A population pharmacokinetic model was developed using NONMEM.
Results:
Based on a total of 3418 samples from 452 patients receiving a median cumulative dose of 140 mg/m2, a two-compartment popPK model was developed. Fat free mass was a covariate for clearance, central volume of distribution and intercompartmental clearance, while glomerular filtration rate predicted clearance. Simulation studies showed that based on fixed body surface area-based dosing, renal impairment has a higher impact in increasing melphalan exposure compared to obesity.
Conclusion:
The proposed model adequately describes the popPK of melphalan in adult patients receiving a hematopoietic cell transplantation. This model can be used to define the therapeutic window of melphalan, and subsequently to develop individualized dosing regimens aiming for that therapeutic window in all patients.
Introduction
Melphalan, a nitrogen mustard and potent alkylating agent, has been used since the 1950s to treat a variety of malignancies,1 including multiple myeloma, ovarian cancer, sarcomas, breast cancer, retinoblastoma, and neuroblastoma 2–5. Melphalan is also frequently used in high dosages as a conditioning agent for both autologous and allogeneic hematopoietic cell transplantation (HCT)6–8. Oral and intravenous formulations of melphalan are available; with the intravenous formulation with propylene glycol (PG) as the solvent (Alkeran®) or a PG-free formulation (Evomela®) which are suggested to be bio-equivalent9,10.
The lower limit of the therapeutic window of melphalan in the setting of HCT is mainly determined by relapse and graft-failure. For those patients receiving an autologous HCT, early recurrence could be a result of underexposure. High dose melphalan has been associated with severe gastro-intestinal side effects including diarrhea and mucositis, in addition to bone marrow suppression that at doses higher than 140 mg/m2 require stem cell support. Sinusoidal obstruction syndrome (SOS) incidence is low when used as a single alkylator but can be higher when used in regimens with two or more alkylators.
High dose melphalan (HD-MEL) is usually dosed between 100mg/m2 and 200mg/m2 11. For patients receiving an autologous HCT for multiple myeloma, melphalan is given as monotherapy, while in lymphoma, melphalan is often given in combination with carmustine, etoposide and cytarabine (BEAM)12. Most allogeneic HCT-regimens incorporating melphalan give the drug for one or two days, mostly in combination with fludarabine, busulfan, and/or thiotepa13.
Patients usually receive a fixed dose of melphalan in mg/m2. Due to variable pharmacokinetics (PK) in a population, this fixed dosing may lead to differences in exposure. Part of this variability is caused by patient characteristics such as kidney function14 and body weight including obesity15. However, these characteristics however are not routinely taken into account when dosing patients. As a result, melphalan exposure varies between patients, with a part of the population that is potentially underdosed, and another part that is overdosed. This in turn may lead to decreased efficacy (relapse, non-engraftment) in case of underexposure or increased toxicity in case of over-exposure. To attain optimal and predictable melphalan exposure in all patients, an individualized dosing regimen is needed that accounts for known factors that impact PK. Some population PK-studies are available in literature16–21. Of these studies, 3 focus on HD-MEL in the context of HCT, 2 on low dose melphalan, and one pediatric study on HD-MEL. These PK-models for HD-MEL however are based on relatively small cohorts of patients.
The aim of this study was to develop a population PK model to describe the PK of HD-MEL in a large cohort of patients receiving either an allogeneic of autologous HCT as a first step towards individualized dosing.
Methods
Patients and Treatments
We included patients aged ≥18 years who had consented to research sample collection and were treated between August 2016 and August 2020 at the Memorial Sloan Kettering Cancer Center (MSKCC) with an autologous or allogeneic HCT with intravenous propylene glycol free melphalan used as part of the conditioning. All HCT’s were eligible for sample collection in cases of patients receiving multiple transplantations during that time period. All data including concentration samples were collected after informed consent was obtained in accordance with the Declaration of Helsinki. This study was approved by the Institutional Review Board of MSKCC.
Most of patients received a cumulative melphalan dose of 140–200 mg/m2 depending on the regimen. Most patients received the full melphalan dose in one infusion, while some patients received the cumulative dose divided over 2 consecutive days. Body surface area was calculated using the Mosteller equation, where the patient’s weight was imputed either as actual body weight or adjusted body weight depending on treatment regimen. Another part of the cohort received a test dose of either 20 mg/m2 or 100 mg/m2 with therapeutic drug monitoring, followed by an adjusted dose to reach an area under the curve of 13.5 mg*h/L based on a previous study22. Melphalan was administered as a 30–45 minute infusion in all patients. Melphalan concentration samples were drawn at 5, 15, 30, 40, 75, and 150 minutes after the end of each infusion. These time points were chosen based on prior studies; no formal optimal sampling strategy was performed using modelling and simulation21,23.
Melphalan assay
Melphalan was measured using a previously described mass spectrometry assay24. Briefly, the assay utilized turbulent flow liquid chromatography (TFLC) and tandem mass spectrometry to simultaneously measure the concentration of busulfan (Bu) and melphalan (Mel) in human plasma. The method involved precipitating proteins in the specimen with an organic solvent containing deuterated internal standards. Following centrifugation, an aliquot of the supernatant was injected into the TFLC mass spectrometry system operated in the positive ion mode. The analytical measurement range for both compounds was 10–5000 ng/mL. Intra-day and inter-day (n=20 day) precision studies showed a coefficient of variation (CV) of <7% at several concentrations spanning the measurement range. Accuracy was determined via recovery studies performed at several concentrations spanning the measurement range; all recoveries for both compounds were between 98 and 103%.
Population PK analysis
A population PK model was developed using non-linear mixed effects modeling. First-order conditional estimation (FOCE) with interaction was used throughout the modelling process. Melphalan concentrations were logarithmically transformed and fitted simultaneously. Very few concentrations were reported below the limit of quantification (BLQ) (n=2), and only occurred at the tail-end of the concentration-time curve. The first sample BLQ was set at half the BLQ, subsequent samples were removed in accordance with method M6 as reported in literature25. We assumed interindividual variability on PK parameters to have a log-normal distribution, and thus were implemented in the model according to equation 1:
| (1) |
where Pi is the individual value of the parameter in the ith individual, Ppop the value of the population mean for this parameter, and ηi the interindividual variability of the ith individual, sampled from a normal distribution with mean of 0 and a variance of ω2. We used an additive error model, which due to the logarithmic transformation is de facto an additive error. Observations were described as equation 2:
| (2) |
where Yi,j is the observed concentration for the jth observation in the ith individual, Cpred,i,j the predicted concentration for the jth observation in the ith individual and ϵ the error sampled from a normal distribution with a mean of 0 and a variance of σ2.
We tested inter-occasion variability, there the pharmacokinetics changes with time within the same individual. This was described using equation 3:
| (3) |
Where compared to equation 1, the term km depicts the interoccasion variability k on the mth occasion.
Model building and selection was evaluated using several steps. First, a degree in objective function value (OFV) of 3.84 points between nested models was considered a significant improvement; this correlated with p<0.05 based on a Chi-square distribution with 1 degree of freedom. Goodness-of-fit (GOF) plots were evaluated, including observed versus individual- and population predicted concentrations, conditional weighted residuals (CWRES) versus time, and versus observed concentrations. Lastly, parameter uncertainty and eta-shrinkage are evaluated.
Individual PK-parameters (post-hocs) were estimated using the POSTHOC function in the software. NONMEM 7.3.0 (Icon Development Solutions, Hanover, MD, USA) will be used for modelling with R version 3.6.1 and Pirana26 for preparations and visualization of the data.
Covariate model
Patient characteristics were evaluated to assess their predictive power for PK-parameters. These included body size parameters such as actual body weight, ideal body weight, and fat-free mass. Fat free mass was calculated according to equations 4 and 5 as proposed by Janmahasatian et al27:
Other patient parameters such as height, sex, age were also included. Transplant- and disease-related parameters such as underlying disease (categorical with 4 levels, Table 1), stem cell source, conditioning intensity, number of HCT’s received, kidney- and liver function (AST, ALT and bilirubin), albumin concentrations, and hematocrit were evaluated. As kidney function is a covariate in most population PK models for melphalan16–21, we included estimated glomerular filtration rate (eGFR) using the CKD-EPI formula and actual creatine concentration estimated shortly before the first dose, but maximum 48h before, as covariates. We also evaluated changes in eGFR from the day of the first melphalan dose to the next day, and implement this as a time-varying covariate. As such, changes of eGFR were only implemented in patients who received melphalan over 2 days, and was available in all of these patients. Missing data regarding the change in eGFR was imputed as no change.
Table 1:
Patient characteristics
| Auto | Allo | Total | |
|---|---|---|---|
|
| |||
| Number of transplants (n) | 321 | 138 | 459 |
| Transplant number (n) | |||
| First transplant | 319 | 133 | 452 |
| Second transplant | 2 | 3 | 5 |
| Third transplant | 0 | 2 | 2 |
| Male sex (%) | 59 | 62 | 60 |
| Age (years) | 61.0 (50.4–67) | 62.0 (55.2–67) | 61 (52.1–67) |
| Weight (kg) | 81.0 (69.0–93) | 80.0 (69.0–90) | 80.4 (69–92) |
| Fat free mass (kg) | 56.0 (44.5–65) | 57.0 (43.4–63) | 56.8 (43.9–64) |
| Number of samples [n (mean per patient)] | 2194 (6.8) | 1224 (8.9) | 3418 (7.4) |
| Cumulative dose (mg) | 300.0 (260–370) | 260.0 (210–280) | 280 (240–340) |
| Cumulative dose (mg/m2) | 143.0 (139–188) | 131.0 (120.2–138) | 140 (133–182) |
| Albumin concentration (g/L) | 39 (36.00–41) | 37 (34.00–39) | 38.00 (36.00–41.0) |
| Creatinin concentration (mmol/L) | 71 (62.00–88) | 71 (62.00–80) | 71.00 (62.00–84.0) |
| eGFR (CKD-EPI; ml/min/1.73 m2) | 93.5 (76.70–105) | 92 (81.20–97.6) | 93.10 (78.30–102.7 |
| Change in eGFR from first to second day* | 0 (−8.00–0) | 0 (0.00–0) | 0.00 (−3.35–0.0) |
| Diagnosis (%) | |||
| Multiple Myeloma | 58 | 10 | 44 |
| Amyloidisis | 6 | 0 | 4 |
| Lymphoma | 36 | 1 | 25 |
| Acute leukemia | 0 | 89 | 27 |
| Conditioning regimen (%) # | |||
| Melphalan monotherapy 140mg/m2 | 16 | 0 | 11 |
| Melphalan monotherapy 200mg/m2 | 36 | 0 | 25 |
| Melphalan monotherapy other dose# | 11 | 0 | 8 |
| BEAM (140mg/m2) | 33 | 0 | 23 |
| BuMelFlu (140 mg/m2) | 0 | 48 | 14 |
| MelFlu (120 mg/m2) | 0 | 52 | 16 |
| BendaMel (140 mg/m2) | 4 | 0 | 3 |
Shown as median (quantiles) unless otherwise specified
only in patients receiving melphalan during >1 day
Melphalan monotherapy (140 and 200 mg/m2) given in 1 day
Melphalan monotherapy other dose: 2 days melphalan with TDM
BEAM: Carmustine, etoposide, cytarabine, 1 day melphalan
BuMelFlu: Busulfan, 2 days melphalan, fludarabine
MelFlu: 1 day melphalan, fludarabine
BendaMel: Bendamustine, 1 day melphalan
We included only potential covariates where a physiological or pharmacological mechanism could be identified. Covariates were evaluated for predictive power by plotting interindividual variability, CWRES, and posthocs against the covariate. Stepwise covariate modelling was performed in parallel.
Continuous covariates were tested in a linear and power function relationship according to equations 6 and 7:
where Pi is the individual value of the parameter in the ith individual, Ppop the value of the population mean for this parameter, Covi the covariate value for the ith individual, and Covmedian the median value for the covariate in the population. In the linear parameter-covariate relationship, the slope is depicted by l, while k depicts the scaling factor in the power relationship.
Potential covariates were evaluated using forward inclusion and backwards elimination, with a significance level of p<0.005 (−7.9 points in OFV) for forward inclusion and p<0.001 (−10.8 points in OFV) for backwards elimination. Building of the covariate model was comparable to the development of the structural model in terms of goodness-of-fit plots, parameter uncertainty and eta-shrinkage. In addition, a decrease in interindividual variability had to be accomplished by introduction of a covariate.
Model evaluation
The developed final model was analyzed for robustness. Internal validation was performed using prediction-corrected visual predictive check (pcVPC). Model robustness and parameter precision were evaluated using the sampling importance resampling (SIR) method28.
Model-based Simulation studies
Since body surface is not the most frequently included body size parameter in PK-models, simulation studies with different dosing levels are useful to give insight in melphalan exposures. For this reason, we simulated typical patients using only population means of PK-parameters and covariate effects and the most frequently used doses of melphalan in HCT (140 and 200 mg/m2) infused over 30 minutes.
Software
Population PK analysis was performed using NONMEM version 7.5.0 (Icon, Hanover, MD, USA) with Pirana version 2.9.9 (Certara, Princeton, NJ, USA) as modelling workbench. R version 1.2.5001 was used for database building and visualization of data.
Results
Patients and Data
A total of 452 patients were included with a median age of 61 years (range 19.2–79.9 years), who underwent a total of 459 HCTs (Table 1). Diagnosis included multiple myeloma (44%), acute leukemia (27%), lymphoma (25%), and other hematologic malignancies; 321 patients underwent an autologous and 138 an allogeneic HCT. The median cumulative dose of melphalan was somewhat higher in patients undergoing an autologous HCT (143 mg/m2 in autologous versus 131 mg/m2 in allogeneic HCT). Most patients received the full dose of melphalan in one infusion (76%), a lower initial dose of melphalan with area-under-the-curve targeted therapeutic drug monitoring was used in 8% of patients. We collected 3418 blood samples for melphalan concentrations, a mean of 7.4 per patient. Among all patients, 144 patients (32%) had an impaired kidney function of whom 122 (26%), 34 (8%), 4 (1%) and 6 (1%) patients with stage 2, 3, 4 and 5 chronic kidney disease scores, respectively.
Structural model
Melphalan pharmacokinetics were well described using a two-compartment model (Table 2; Figure 1). A two-compartment model was superior over a one-compartment model with a decrease in OFV of 1459 (p<0.001) and improvement in GOF-plots. The variance between IIV on CL and V1 was implemented using an omega block. Due to the absence of samples taken during infusion, the estimation of the distribution was not optimal. Inclusion of inter-individual variability on the peripheral volume of distribution led to substantial eta shrinkage and high dependency on initial values and was therefore omitted. We also tried to estimate the peripheral volume as a factor or the individual central volume; this was inferior to the previous model in terms of OFV and GOF-plots. A three-compartment model was unstable with inaccurate parameter estimations. We tested IOV on clearance, this yielded no improvement in the model in terms of GOF-plots and OFV, inclusion of IOV led residual standard errors and shrinkage on these parameters. There was no evidence of non-linear elimination looking at the individual concentration-time plots, which could be suggestive for saturation of elimination pathways. Inclusion of a non-linear clearance pathway also did not improve the model. A proportional error was implemented adding an additive error that did not improve the model.
Table 2:
Parameter estimates and SIR results
| Dataset [estimate (RSE)] | Shrinkage | SIR [estimate (RSE)] | |
|---|---|---|---|
|
| |||
| Structural model | |||
| CLpop (L/h) | 12.60 (9%) | 12.62 (6%) | |
| k | 0.73 (9%) | 0.73 (7%) | |
| l | 1.15 (17%) | 1.15 (11%) | |
| m | −0.012 (19%) | −0.01 (13%) | |
| V1,pop (L) | 24.2 (2%) | 24.17 (2%) | |
| n | 0.58 (13%) | 0.58 (11%) | |
| V2,pop (L) | 14.8 (6%) | 14.71 (4%) | |
| Qpop (L/h) | 15 (5%) | 15.01 (4%) | |
| p | 1.19 (13%) | 1.2 (11%) | |
| Random variability | |||
| Inter-individual variability on CL (%) | 25 (5%) | 4% | 25 (7%) |
| Inter-individual variability on V1 (%) | 18 (16%) | 13% | 18 (19%) |
| Inter-individual variability on Q (%) | 32 (20%) | 28% | 32 (23%) |
| Proportional residual error (%) | 16 (5%) | 11% | 16 (3%) |
RSE relative standard error, SIR sample importance resampling, CL linear clearance, FFM fat free mass, FFMmean mean fat free mass (56.8 kg), eGFR glomerular filtration rate (CKD-EPI), eGFRmean mean eGFR (93.3 ml/min/1.73 m2), eGFRdelta change in eGFR from the first day of melphalan dosing to the second day of melphalan dosing in those patients receiving 2 doses, V1 central volume of distribution, V2 peripheral volume of distribution, Q intercompartmental clearance
Figure 1:
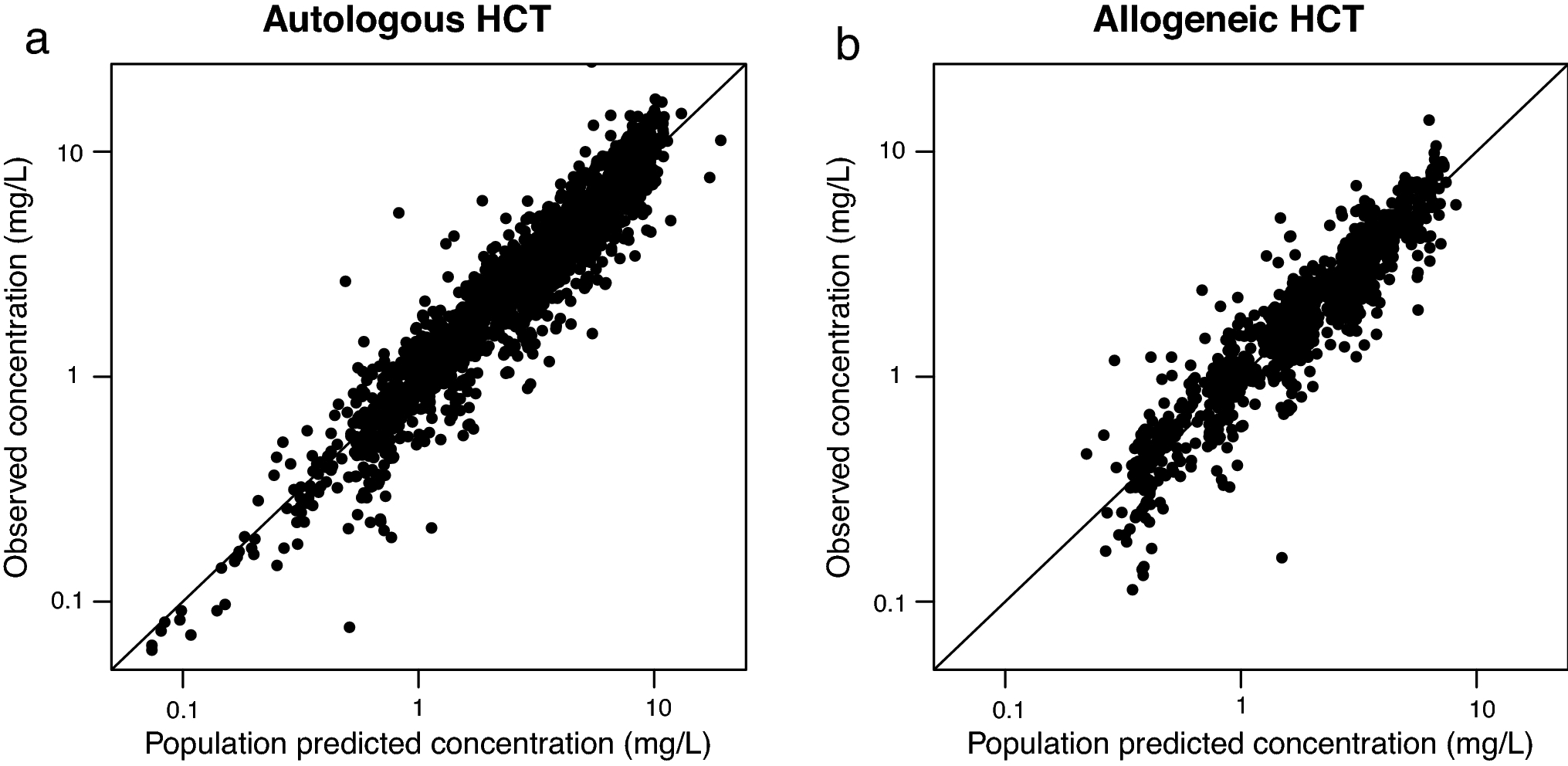
Model performance in patients receiving an autologous (panel a) and allogeneic (panel b) cell transplantation. Dots indicate the individual concentration versus population predicted value; the lines depict x=y.
Covariate model
The covariate analysis showed that all body size related variables were predictive for clearance, central volume of distribution, and intercompartmental clearance. Out of actual body weight, fat free mass, and body surface area, fat free mass was most predictive for all parameters with a decrease in OFV of 226, 219 and 196 when including FFM, BSA and weight, respectively, as a covariate on CL, V1 and Q (Supplemental Figure S1). Inclusion in FFM over BSA and weight led to a superior improvement in GOF and decrease in IIV. Furthermore, an exponential relationship between FFM and PK-parameters yielded a further drop in OFV compared to a linear relationship with comparable GOF plots. Inclusion of fat free mass, a parameter based on body weight, length, and sex, resulted in a drop in OFV of 92, 73, and 60 points in clearance, central volume of distribution, and intercompartmental clearance, respectively. While conditioning regimen was led to a significant decrease in OFV, GOF and covariate-IIV plots did not improve. Therefore, conditioning regimen was not included in the final model. Next, we evaluated renal function as a covariate on clearance. We evaluated two parallel linear elimination pathways, where only the renal pathway was impacted by a renal function parameter20,21. This approach was unsuccessful due to high dependencies on initial estimates on both clearance parameters with a tendency to shrink non-renal clearance to zero. Renal function was evaluated in terms of eGFR and actual creatinine concentration, of which eGFR was superior in terms of GOF and OFV (13 versus 105 points decrease in creatinine clearance and eGFR, respectively, p<0.001). Furthermore, combining a baseline eGFR with a time-varying change in eGFR according to Wahlby et al29 resulted in a further 99 points decrease in OFV, despite the fact that data on time-varying eGFR was available in part of the population (Figure S5). Finally, we tested other covariates suggested in literature including hematocrit18,21 and sex19 on clearance. No correlation between the covariate and IIV could be identified in the IIV versus covariate plots for both covariates (Supplemental Figure S2). Inclusion of haematocrit on clearance did decrease OFV (13.8 points), however based on the IIV-covariate plot, the GOF-plots, the absence of reduction in IIV on clearance and the relatively high RSE, we did not include haematocrit in the final model. Inclusion of sex on clearance did not improve any of the criteria for covariate inclusion. Moreover, given the heterogeneous cohort with both allogeneic and autologous transplants, we tested stem cell source as a covariate. None were impacted by the stem cell source (IIV on clearance versus stem cell source in Figure S2). Therefore, the final model included fat free mass on clearance, volume of distribution and intercompartmental clearance, with baseline eGFR and delta eGFR on clearance (Supplemental figure S3). Inclusion of these covariates led to an absolute decrease in random interindividual variability of 10%, 5% and 12% in clearance, central volume of distribution, and intercompartmental clearance, respectively (Table 2, Table S2). Actual exposure data (area under the curve) for different dosing levels is included in the supplements (Supplemental Table S3)
Model evaluation
Sampling importance resampling with 10.000 samples and an M/m ratio of 5 was performed, with satisfying SIR diagnostic plots (Supplemental figure S4). The results of the SIR were in line with the model in terms of parameter estimation and precision (Table 2). A prediction corrected visual predictive check yielded overlapping observations and predictions (Figure 2).
Figure 2:
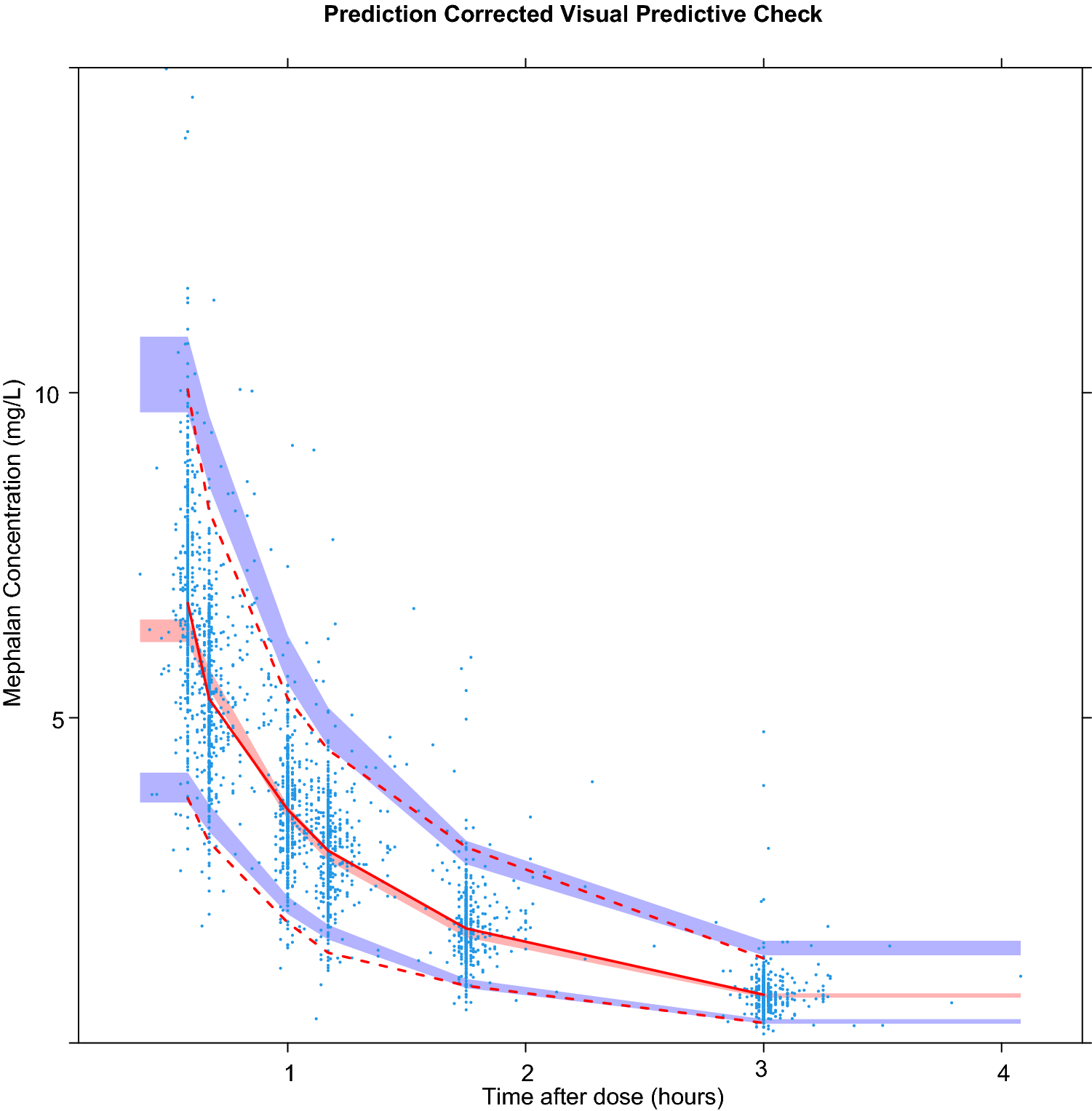
Prediction corrected visual predictive check (PC-VPC) with 1000 simulations. Dots: observed concentration data. Solid line: median observed concentration over time; dashed lines: 2.5 and 97.5% quartiles of observed concentration of time. Red shaded area: 95% confidence interval (CI) of median predictions; Blue shaded area: 95% CI of 2.5 and 97.5% predictions.
Simulation studies
We simulated lean and obese male patients with a length of 180 cm and a body weight of 70 and 150 kg. This corresponds to an FFM of 56.8 and 83.7 kg in lean and obese simulated patients, respectively. A glomerular filtration rate of 30 and 90 ml/min/1.73m2 was chosen for the simulations. The simulation studies showed that there was a substantial difference in AUC; a 60% decrease in area under the curve (AUC) between obese patients with impaired renal function compared to lean patients with normal eGFR after a dose of 200mg/m2 (Figure 3). Therefore, to equalize AUC between patients, dosing requires adjustment based on the covariates FFM and eGFR.
Figure 3:
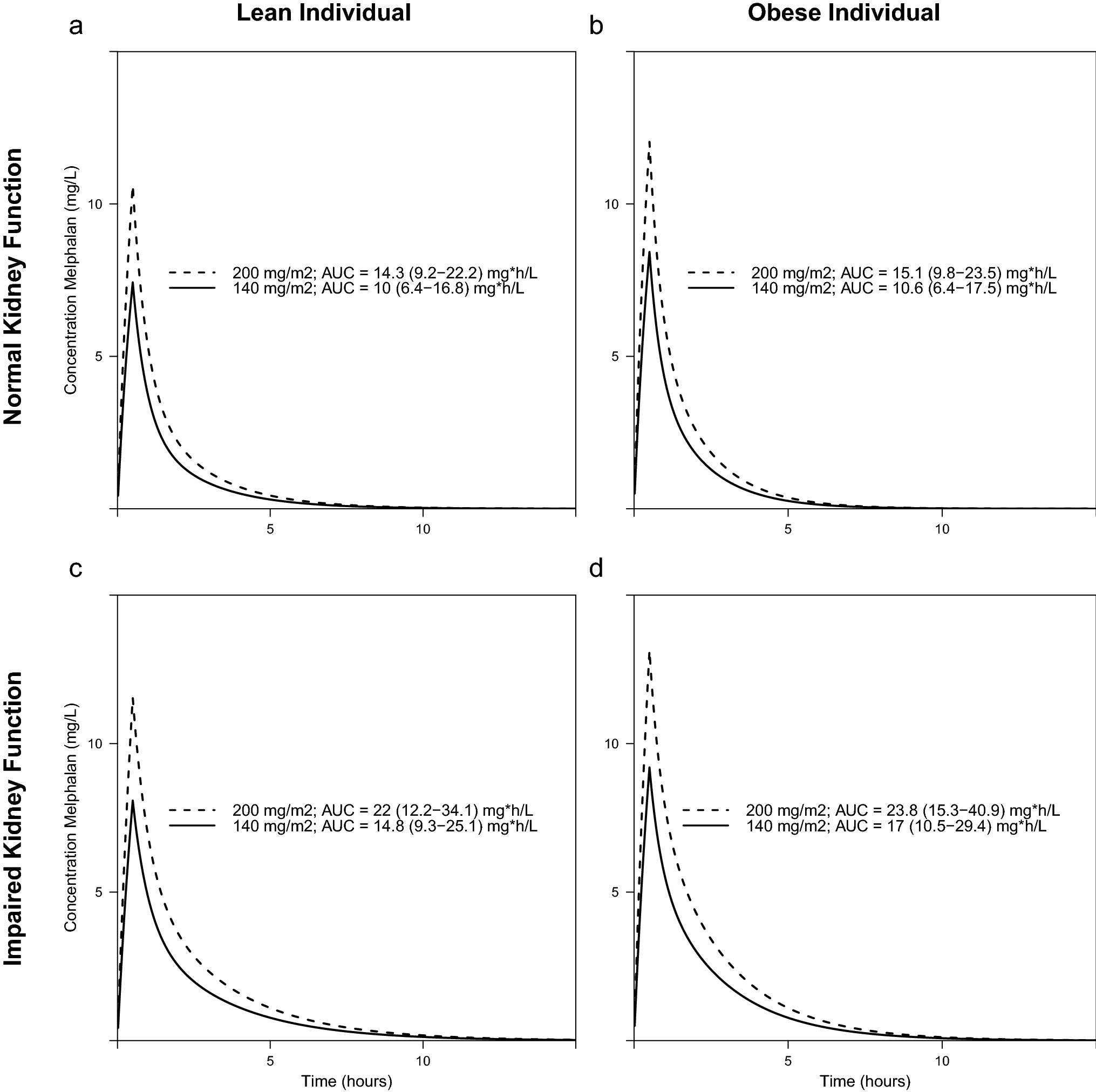
Simulation studies of patients with a body weight of 70 kg (lean patients; panels A and C) and 150 kg (obese patients; panels B and D) having a glomerular filtration rate of 90 (panels A and B) or 30 ml/min/1.73 m2 (panels C and D). Median area under the curve (AUC) with 95% confidence intervals in 200 simulations per dose-eGFR-body weight category.
Discussion
We describe the population pharmacokinetics of high-dose melphalan in the context of receiving either an autologous or allogeneic HCT in, to our knowledge, the largest cohort of adult patients in the literature. The pop-PK could be best described using a two-compartment model incorporating linear clearance. Lean body weight was a covariate on clearance, volume of distribution, and intercompartmental clearance in an exponential relationship. Furthermore, eGFR was included as a time-varying covariate on clearance reflecting the renal excretion of melphalan. Simulation studies show that impaired renal function, and to a lesser extent fat free mass, led to increased exposure after fixed body surface area-based dosing.
The developed model and parameter estimations are in line with the expected clinical pharmacology of melphalan. The central volume of distribution of 24.2 L (residual standard error 2%) is consistent with a drug with moderate protein binding30. The main route of elimination of melphalan is non-enzymatous hydrolysis to monohydroxymelphalan and dihydroxymelphalan, while 6–21% of melphalan is excreted unchanged in urine30,31. This is reflected in the moderate slope (slope parameter of 1.15 [RSE 19%]) of the relation between melphalan clearance and eGFR. Faecal excretion of unchanged melphalan is described in the literature; however, only after oral administration and thus probably reflecting non-absorption rather than biliary excretion31,32.
Some population PK models for melphalan have been described previously. Five of these models were developed in adult patients17–21. The number of patients included in these studies are substantially smaller (15–118 patients) compared to the current cohort (452 patients), making our pop-PK model more substantial. Two previous models incorporated two parallel linear clearance pathways depicted by a renal and non-renal elimination, where eGFR was included as a covariate on the renal pathway20,21. This parallel elimination pathway was not identifiable in our data, with the population mean of the non-renal clearance shrinking to zero. This difference may be due many factors, including patient characteristics such as the distribution of eGFR in the population, study design with published studies taking more tail-end samples, melphalan formulation where published studies likely administer only part of the dose due to hydrolysation (CL in those studies can conceptionally be seen as CL/F), and modelling approaches (handling of samples below the limit of quantification, identifiability of parameters. Still, actual eGFR calculated with the CKD-EPI formula33 was included on the common elimination pathway. In contrast to previous studies20,21, we could not identify IIV on the peripheral volume, which may be due to the sampling scheme in the current study. Two other previous models include haematocrit as a covariate on clearance, either on total or non-renal clearance18,21. The pharmacological rationale of including haematocrit on clearance was that a lower haematocrit would lead to a lower binding of melphalan to erythrocytes, which has been suggested to lead to higher plasma and ultrafiltrate concentrations of melphalan. Following this argumentation, the more logical parameter to include haematocrit would be volume of distribution. In the current model, we could not identify a relationship between haematocrit and both melphalan clearance and volume of distribution, and therefore it was not included in the current model. The same applied for central volume of distribution. Another major difference between the current study and the current literature is the use of PG-free melphalan. This is more stable, and is therefore expected to be more accurate in terms of the actual dose that is administered. With non-PG-free melphalan, part of the dose is actually administered, which is conceptually comparable with bioavailability. As such, the reported values population parameters such as CL and V1 should be interpreted as CL/F and V1/F. For CL and Q, this is in line with the lower estimates for these parameters in our study compared to those published previously. Volume of distribution however is estimated to be higher than that in previous studies. We hypothesize a possibility for this difference may be due to saturated protein binding: the two largest published studies dosed at 200 mg/m2 and 190 mg/2, respectively18,21. Patients in the current study received a significantly lower median dose of 140mg/m2, divided over 2 doses in 24% (111/459) transplants. The lower dose may be less prone to lead to oversaturation, which may have result in a higher estimated volume of distribution in the current study. This however is not in line with one of the previous studies reporting on both total and unbound melphalan concentrations21.
We did, however, include FFM as a covariate on clearance, central volume of distribution, and intercompartmental clearance. From a modelling perspective, this was superior to other body size parameters in terms of GOF-plots and objective function value. From a clinical pharmacological point of view, the increase in body weight of fat tissue in obese patients compared to lean body weight patients apparently does not result in an increase in volume of distribution and clearance of melphalan. This is in line with literature, where clearance is often best described using body size parameters correcting for adiposity, and volume of distribution depends on the lipophilicity of the drug34. The logP of melphalan is −0.5, making it mildly hydrophilic, which makes the superior correlation with an adjusted body size parameter compared to actual body weight logical35.
Albeit being frequently used, the therapeutic window of melphalan has not been fully defined. However, higher exposures to melphalan are suggested to be associated to improved overall survival and event free survival, but also lead to more toxicity in terms of mucositis21,23,36,37. Considering the working mechanism, underexposure of melphalan may lead to an increased relapse probability and graft failure in an allogeneic setting. Overexposure can lead to toxicity including severe mucositis, GvHD and SOS. Further investigation is ongoing evaluating the relationship between melphalan exposure and outcomes, including toxicity (e.g. mucositis) and efficacy (e.g. disease control, engraftment) in the current cohort of patients.
In summary, we developed and evaluated a population PK model for melphalan which accurately describes melphalan PK in adult patients. Fat free mass and eGFR were identified as covariates influencing the pharmacokinetics of melphalan. Current body surface area-based dosing results in variable exposure across patients with different renal function, as well as lean versus obese patients. Once the therapeutic window has been definitely identified in various HCT settings, the current model may serve as a basis to develop an individualized dosing regimen for melphalan. In line with recent examples38–41 such as busulfan, rATG, and fludarabine, this personalized dosing may lead to an optimization of outcomes (including survival) following HCT.
Supplementary Material
Key points:
Melphalan pharmacokinetics can be predicted using a population pharmacokinetic model, being the first step towards an individualized dosing regimen
Fat free mass and glomerular filtration rate are the most important covariates predicting pharmacokinetics
Melphalan area under the curve almost doubles in obese patients with poor renal function as compared to lean non-renally impaired individuals
Acknowledgements:
The study was supported by funding from the Sawiris Family Research Fund, Melvin Berlin Family Fund for Myeloma Research, A.C. Israel Foundation, and the Donna and Patrick Martin Foundation. This research was supported in part by National Institutes of Health award number P01 CA23766 and National Institutes of Health/National Cancer Institute Cancer Center Support grant P30 CA008748. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The work was performed independently of all funders.
Footnotes
Disclosure of conflicts of Interest:
GS reports research funding from Janssen and Amgen unrelated to the current project. MS reports has served as a consultant for McKinsey & Company, Angiocrine Bioscience, Inc., Omeros Corporation; has received research funding from Angiocrine Bioscience, Inc and Omeros Corporation; has served on an advisory board for Kite – A Gilead Company; and has served as a CME speaker for i3Health, all unrelated to the current project.
JB has received honoraria for consulting for Race oncology, AvroBio, Omeros, Sanofi, Advanced Clinical, BlueRock, all unrelated to the current project.
SG has consulted for and received research funding from Amgen, Actinium, Celgene, Johnson & Johnson, and Takeda; has consulted for Jazz Pharmaceuticals, Novartis, Kite, and Spectrum Pharmaceuticals; and has received research funding from Miltenyi unrelated to the current project. MS has received research support from Angiocrine Bioscience, Inc.; Omeros Corporation, performed consultancy for Angiocrine Bioscience, Inc.; Omeros Corporation; McKinsey & Company, has advised for Kite – A Gilead Company, and received speaking engagement from i3Health. RA has received research grants from Sanofi, unrelated to the current project.
Code availability:
The model codes are available upon request
Availability of Data and Material:
The data and materials for this study are not available
References
- 1.Papac R, Galton D, Till M, Wiltshaw E. Preliminary clinical trial of p-di-2-chloroethyl-amino-L-phenylalanine (CB 3025, melphalan) and of di-2-chloroethyl methanesulfonate (CB 1506). Ann N Y Acad Sci 1958; 68: 1126–7. [DOI] [PubMed] [Google Scholar]
- 2.Barlogie B, Shaughnessy J, Tricot G, et al. Treatment of multiple myeloma. Blood 2016; 103: 20–33. [DOI] [PubMed] [Google Scholar]
- 3.Verhoef C, Wilt JHW, Grünhagen DJ, Geel AN, Hagen TLM, Eggermont AMM. Isolated limb perfusion with melphalan and TNF-α in the treatment of extremity sarcoma. Curr Treat Options Oncol 2007; 8: 417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samuels BL, Bitran JD. High-dose intravenous melphalan: A review. J Clin Oncol 1995; 13: 1786–99. [DOI] [PubMed] [Google Scholar]
- 5.Hsieh T, Liao A, Francis JH, et al. Comparison of efficacy and toxicity of intravitreal melphalan formulations for retinoblastoma. PLoS One 2020; 15: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayraktar UD, Bashir Q, Qazilbash M, Champlin RE, Ciurea SO. Fifty Years of Melphalan Use in Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant 2013; 19: 344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain T, Alahdab F, Firwana B, Sonbol MB, Almader-Douglas D, Palmer J. Choosing a Reduced-Intensity Conditioning Regimen for Allogeneic Stem Cell Transplantation, Fludarabine/Busulfan versus Fludarabine Melphalan: A Systematic Review and Meta-Analysis. Biol Blood Marrow Transplant 2019; 25: 728–33. [DOI] [PubMed] [Google Scholar]
- 8.Dahi PB, Lazarus HM, Sauter CS, Giralt SA. Strategies to improve outcomes of autologous hematopoietic cell transplant in lymphoma. Bone Marrow Transplant 2019; 54: 943–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aljitawi OS, Ganguly S, Abhyankar SH, et al. Phase IIa cross-over study of propylene glycol-free melphalan (LGD-353) and alkeran in multiple myeloma autologous transplantation. Bone Marrow Transplant 2014; 49: 1042–5. [DOI] [PubMed] [Google Scholar]
- 10.Miller KC, Gertz MA, Buadi FK, et al. Comparable Outcomes using Propylene Glycol-Free Melphalan for Autologous Stem Cell Transplantation in Multiple Myeloma. Bone Marrow Translantation 2019; 54: 587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw PJ, Nath CE, Lazarus HM. Not too little, not too much-just right (Better ways to give high dose melphalan). Bone Marrow Transplant 2014; 49: 1457–65. [DOI] [PubMed] [Google Scholar]
- 12.Choi T Is autologous stem cell transplantation still relevant for multiple myeloma? Curr Opin Hematol 2019; 26: 386–91. [DOI] [PubMed] [Google Scholar]
- 13.Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: One size does not fit all. Blood 2014; 124: 344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dimopoulos MA, Terpos E, Chanan-Khan A, et al. Renal impairment in patients with multiple myeloma: A consensus statement on behalf of the International Myeloma Working Group. J Clin Oncol 2010; 28: 4976–84. [DOI] [PubMed] [Google Scholar]
- 15.Shultes KC, Arp C, Stockerl-goldstein K, Trinkaus K, Defrates S. Transplantation Impact of Dose-Adjusted Melphalan in Obese Patients Undergoing Autologous Stem Cell Transplantation. Biol Blood Marrow Transplant 2018; 24: 687–93. [DOI] [PubMed] [Google Scholar]
- 16.Nath CE, Shaw PJ, Montgomery K, Earl JW. Population pharmacokinetics of melphalan in paediatric blood or marrow transplant recipients. Br J Clin Pharmacol 2007; 64: 151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sezer O, Heider U, Meineke I, et al. Population Pharmacokinetics of Melphalan and Glutathione S -transferase Polymorphisms in Relation to Side Effects. Clin Pharmacol Ther 2008; 83: 749–57. [DOI] [PubMed] [Google Scholar]
- 18.Cho YK, Sborov DW, Lamprecht M, et al. Associations of high-dose melphalan pharmacokinetics and outcomes in the setting of a randomized cryotherapy trial. Clin Pharmacol Ther 2017; 102: 511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mougenot P, Pinguet F, Fabbro M, et al. Population pharmacokinetics of melphalan, infused over a 24-hour period, in patients with advanced malignancies. Cancer Chemother Pharmacol 2004; 53: 503–12. [DOI] [PubMed] [Google Scholar]
- 20.Mizuno K, Dong M, Fukuda T, Chandra S. Population Pharmacokinetics and Optimal Sampling Strategy for Model-Based Precision Dosing of Melphalan in Patients Undergoing Hematopoietic Stem Cell Transplantation. Clin Pharmacokinet 2018; 57: 625–36. [DOI] [PubMed] [Google Scholar]
- 21.Nath CE, Shaw PJ, Trotman J, et al. Population pharmacokinetics of melphalan in patients with multiple myeloma undergoing high dose therapy. Br J Clin Pharmacol 2010; 69: 484–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah GL, Lin A, Kamrowski A, et al. Successful Personalization of Propylene Glycol Free Melphalan (PGF-MEL) for Multiple Myeloma (MM) and AL Amyloidosis (AL) Patients Undergoing Autologous Hematopoietic Stem Cell Transplant (AHCT) Using Pharmacokinetic (PK)-Directed Dosing. Biol Blood Marrow Transplant 2020; 26: S154–5. [Google Scholar]
- 23.Nath CE, Trotman J, Tiley C, et al. High melphalan exposure is associated with improved overall survival in myeloma patients receiving high dose melphalan and autologous transplantation. Br J Clin Pharmacol 2016; : 149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schofield RC, Landau HJ, Giralt SA, et al. Measurement of the DNA alkylating agents busulfan and melphalan in human plasma by mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 2019; 1125: 121711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 2001; 28: 481–504. [DOI] [PubMed] [Google Scholar]
- 26.Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed 2011; 101: 72–9. [DOI] [PubMed] [Google Scholar]
- 27.Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet 2005; 44: 1051–65. [DOI] [PubMed] [Google Scholar]
- 28.Dosne AG, Bergstrand M, Harling K, Karlsson MO. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J Pharmacokinet Pharmacodyn 2016; 43: 583–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wählby U, Thomson AH, Milligan PA, Karlsson MO. Models for time-varying covariates in population pharmacokinetic- pharmacodynamic analysis. Br J Clin Pharmacol 2004; 58: 367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evomela SmPc, Spectrum Pharmaceuticals. .
- 31.Alberts DS, Chang SY, Chen HS, et al. Kinetics of intravenous melphalan. Clin Pharmacol Ther 1979; 26: 73–80. [DOI] [PubMed] [Google Scholar]
- 32.Alberts DS, Chang SY, George Chen HS, Evans TL, Moon TE. Oral melphalan kinetics. Clin Pharmacol Ther 1979; 26: 737–45. [DOI] [PubMed] [Google Scholar]
- 33.Levey A, Stevens L, Schmid C, et al. A New Equation to Estimate Glomerular Filtration Rate. Ann Intern Med 2009; 150: 604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green B, Duffull SB. What is the best size descriptor to use for pharmacokinetic studies in the obese? Br J Clin Pharmacol 2004; 58: 119–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.National Library of Medicine (US): National Center for Biotechnology. PubChem Compound Summary for CID 460612, Melphalan. Available from https//pubchem.ncbi.nlm.nih.gov/. 2004.
- 36.Parmar SR, Bookout R, Shapiro JF, et al. Comparison of 1-day vs 2-day dosing of high-dose melphalan followed by autologous hematopoietic cell transplantation in patients with multiple myeloma. Bone Marrow Transplant 2014; 49: 761–6. [DOI] [PubMed] [Google Scholar]
- 37.Attal M, Harousseau JL, Stoppa AM, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. N Engl J Med 1996; 335: 91–7. [DOI] [PubMed] [Google Scholar]
- 38.Admiraal R, van Kesteren C, Jol-van Der Zijde CM, et al. Association between anti-thymocyte globulin exposure and CD4+ immune reconstitution in paediatric haematopoietic cell transplantation: a multicentre, retrospective pharmacodynamic cohort analysis. Lancet Haematol 2015; 2: e194–e203. [DOI] [PubMed] [Google Scholar]
- 39.Admiraal R, Nierkens S, de Witte MA, et al. Association between anti-thymocyte globulin exposure and survival outcomes in adult unrelated haemopoietic cell transplantation: a multicentre, retrospective, pharmacodynamic cohort analysis. Lancet Haematol 2017; 4: e183–91. [DOI] [PubMed] [Google Scholar]
- 40.Bartelink IH, Lalmohamed A, van Reij EML, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol 2016; 3: e526–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langenhorst JB, van Kesteren C, van Maarseveen EM, et al. Fludarabine exposure in the conditioning prior to allogeneic hematopoietic cell transplantation predicts outcomes. Blood Adv 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and materials for this study are not available