

Abstract
This mini-review provides an update regarding the substantial progress that has been made in using single-particle cryo-EM to obtain high-resolution structures for proteins and other macromolecules whose particle sizes are smaller than 100 kDa. We point out that establishing the limits of what can be accomplished, both in terms of particle size and attainable resolution, serves as a guide for what might be expected when attempting to improve the resolution of small flexible portions of a larger structure using focused refinement approaches. These approaches, which involve computationally ignoring all but a specific, targeted region of interest on the macromolecules, is known as ‘masking and refining,’ and it thus is the computational equivalent of the ‘divide and conquer’ approach that has been used so successfully in X-ray crystallography. The benefit of masked refinement, however, is that one is able to determine structures in their native architectural context, without physically separating them from the biological connections that they require for their function. This mini-review also compares where experimental achievements currently stand relative to various theoretical estimates for the smallest particle size that can be successfully reconstructed to high resolution. Since it is clear that a substantial gap still remains between the two, we briefly recap the areas in which further improvement seems possible, both in equipment and in methods.
Introduction
One of the many appealing features of single-particle electron cryo-microscopy (cryo-EM) [1] is that biological macromolecules do not need to be constrained into specific conformational states in order to obtain high-resolution structures. Instead, as part of the normal, cryo-EM data-processing pipeline, it is often possible to correctly categorize each individual particle image into one or another of the ensemble of conformational species that are naturally present in solution [2]. The further introduction of a Bayesian method for ‘3D classification’ of individual images [3] made it possible to obtain high-resolution maps for two or more of the most highly populated conformational states that are in equilibrium with one another, under the same solution conditions.
In a subsequent development, computational tools were introduced to focus the 3D classification and reconstruction process on specific regions within a larger, flexible macromolecule (Figure 1A) [4–6]. Often, this approach begins with a ‘consensus’ reconstruction of the major, larger component of the macromolecule, followed by the generation of a 3D mask that encompasses the region of interest. The alignment parameters assigned to the larger component usually serve as starting points for ‘focused refinement’ of the smaller flexible component(s). Alignment of the targeted region can be improved by computationally removing density corresponding to the macromolecule outside of the 3D mask through a method known as ‘signal subtraction’ [6]. Such mask-and-align methods are the computational equivalent of the ‘divide and conquer’ approach [7] that has been used successfully in macromolecular crystallography. The benefit of masking and aligning, of course, is that one is no longer required to biochemically isolate the individual, smaller portions of a larger structure, thereby removing the pieces from the context in which they function.
Figure 1. Some examples of recent structures that have been determined by cryo-EM.
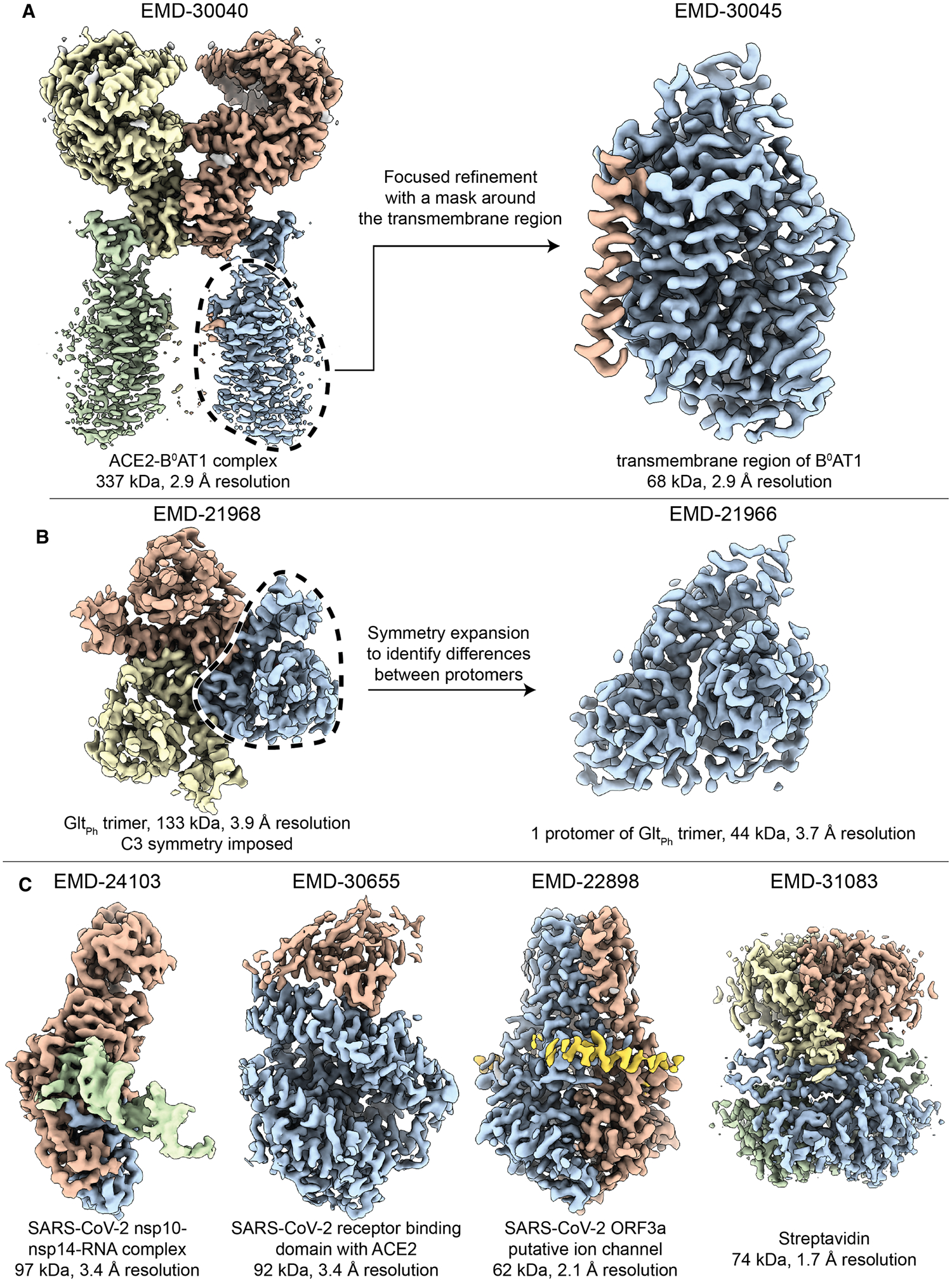
Accession numbers for each of these illustrations are indicated above the respective maps shown here. (A) The two-fold symmetric structure of the angiotensin-converting enzyme 2 (ACE2) bound to the membrane protein B0AT1, was determined at a reported resolution of better than 3 Å [73]. However, the transmembrane regions of B0AT1 were not well-resolved in this map. The mask-and-align approach was used to substantially improve the resolution of this 68 kDa region. (B) The three-fold symmetric structure of the archaeal glutamate transporter GltPh was determined with a reported resolution of 3.9 Å resolution. A specialized type of mask-and-align approach, which enables asymmetric units within a symmetric complex to be structurally analyzed in isolation (reviewed in [74]), was used to reveal the presence of distinct conformers among the protomers [75]. (C) Four recent single particle reconstructions of <100 kDa complexes at increasing resolution. Shown from left to right are: The SARS-CoV-2 exoribonuclease (nsp10–nsp14) bound to RNA [17], the pangolin coronavirus receptor binding domain bound to human ACE2 [19], the SARS-CoV-2 open reading frame 3a putative ion channel [22], and streptavidin from Streptomyces avidinii acquired using a Krios G4 electron microscope equipped with a cold FEG, Selectris energy filter and Falcon IV direct detector (personal communication with Dr. Tomohiro Nishizawa).
The development of the mask-and-align approach in single-particle cryo-EM raises the question of how small a region can be targeted and still be large enough to assign accurate alignment parameters for high-resolution structure determination. This question is not as easily answered as is the simpler one — how small an isolated single particle can be, which was addressed previously and continues to be an active area of study [8–11]. Theoretical estimation for the case of mask-and-align is made more difficult because of residual errors and artifacts that remain after computationally subtracting information from surrounding parts of the larger structure, which add a confounding background that is both particle- and view- (orientation) specific. Nevertheless, results achieved with isolated macromolecules at least provide a best-case limit for what might be achieved with the mask-and-align approach.
We first update previous reviews on the progress made in determining the structures of isolated, smaller macromolecules by cryo-EM, pointing out that the current state-of-the-art still falls well short of the estimated theoretical limits. Further improvement in the capabilities of cryo-EM is sure to follow, however. We thus conclude this review by offering our perspective on what it may take to further narrow the gap between theory and what is possible in practice.
Cryo-EM of proteins smaller than 100 kDa continues to make rapid progress
The high level of noise in single-particle cryo-EM images, and the resulting ambiguity of particle alignment that this causes, initially led the cryo-EM community to underestimate its ability to resolve the structures of smaller targets. Even after the advent of direct detector cameras, it still was widely believed that any biological targets smaller than 200 kDa would fail to reconstruct to high resolution [12]. Then, after high resolution reconstructions were achieved of complexes smaller than 200 kDa using conventional single-particle cryoEM methods, it was initially thought that targets smaller than 100 kDa, would require the use of phase plate technology [11]. Even this assumption proved to be wrong [13], however, and the number of high-resolution structures of biological targets smaller than 100 kDa that is determined using conventional cryo-EM methods, including many membrane proteins, rises every year. A few representative examples of the density maps are shown in Figure 1C and in Figure 2B.
Figure 2. Illustration of the rapid growth in the ability of single-particle cryo-EM to produce high-resolution structures of small macromolecules.
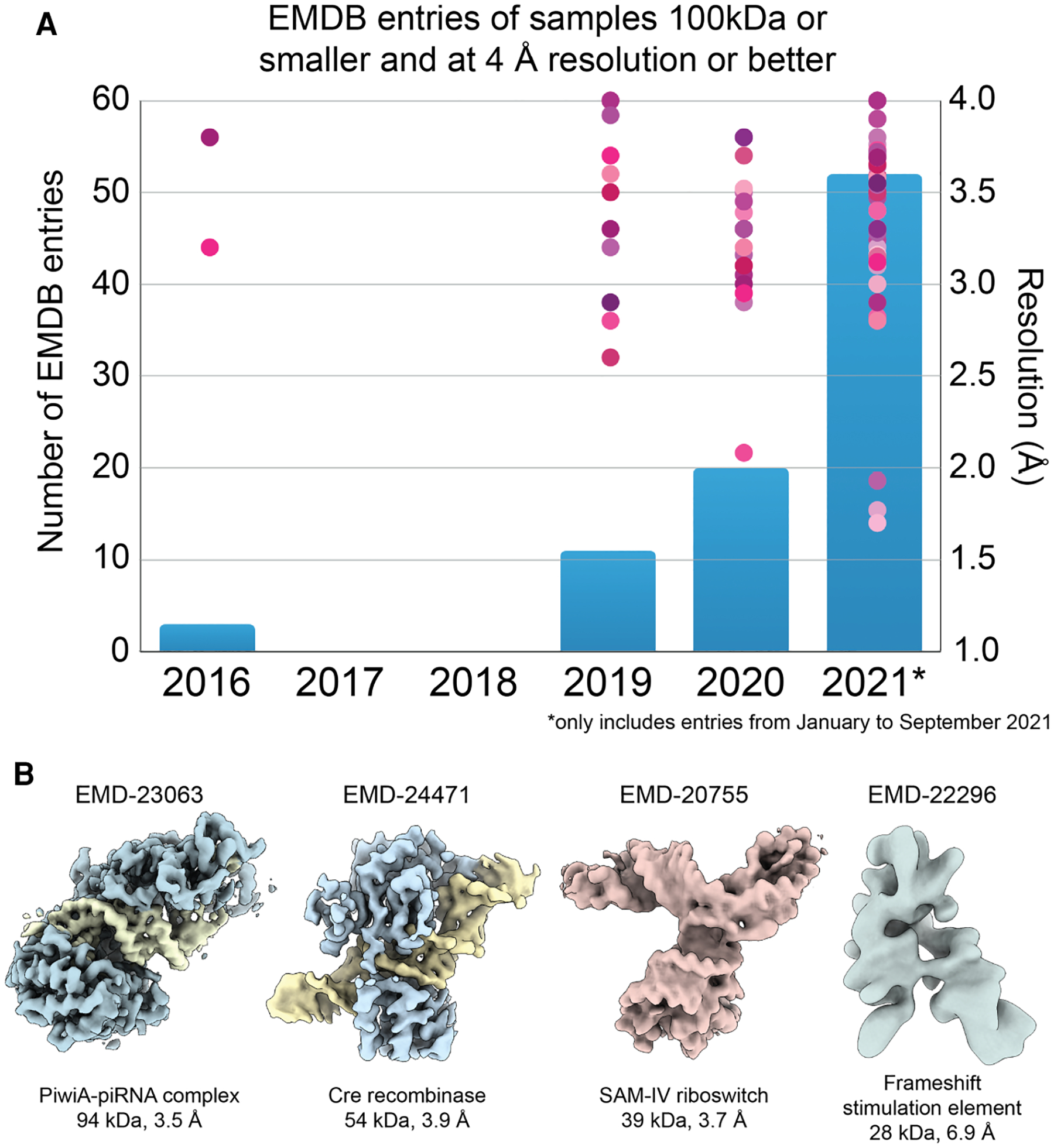
(A) The number of <100 kDa entries released by the EMDB (blue bars) has begun to increase substantially as more groups recognize the capacity of existing cryoEM technology to resolve high resolution structures of molecular targets in this size range. Simultaneously, the resolutions attained by single particle cryoEM continues to improve. (B) Four nucleic-acid-containing complexes smaller than 100 kDa in size. Shown from left to right are: the reconstructed densities of the PiwiA-piRNA complex [32], Cre recombinase bound to a DNA substrate [31], the SAM-IV Riboswitch [33], and Frameshift Stimulation Element (FSE) from the SARS-CoV-2 RNA Genome [34]. The riboswitch and FSE structures are purely nucleic acid, and demonstrate the potential for nucleic acids, which have a higher density than protein, to aid in attaining high resolution structures at the theoretical size limit of cryo-EM.
The current resolution record for a <100 kDa complex is that of streptavidin from Streptomyces avidinii, which forms a soluble 74 kDa tetramer that was resolved to ~1.7 Å resolution (EMD-31083, Figure 1C) using a Titan Krios G4 microscope equipped with a cold FEG and a Selectris energy filter, with images acquired using a Falcon IV direct detector (personal communication with Dr. Tomohiro Nishizawa). This reconstruction confirms the capacity for next-generation instrumentation to enable single particle approaches to surpass the 2 Å resolution barrier for stable <100 kDa complexes. Considering the ongoing technological and methodological developments discussed later in this review, as well as the current trends demonstrated in Figure 2A, we can expect that such resolutions will become more commonplace for similarly or smaller sized complexes.
Given the fact that the vast majority of the proteins that make up the mammalian proteome are less than 100 kDa [14], further development of methodologies targeting macromolecules in this size range are of significant biomedical importance. Furthermore, advances in our ability to resolve small, isolated protein complexes will likely be of benefit to mask-and-align studies of targeted regions within larger complexes. This is because the success of the mask-and-align approach in improving the resolution of a particular region of a map is directly influenced by the mass of the targeted region. A larger mass will scatter more electrons, producing a projection containing increased signal, which is needed for accurate orientation assignment by alignment algorithms. For example, although it was previously shown that individual lysozyme proteins (14 kDa) can be observed as distinct dark spots using cryo-EM [15], the projection images of these particles do not contain sufficient structural details to enable accurate orientational assignments. Computer simulations have since confirmed that solving the structure of lysozyme is a challenge for cryo-EM, but suggest that it should be achievable when a phase plate is used [16].
The realization that high-resolution structures of <100 kDa complexes could be determined using conventional single particle cryoEM methods was particularly timely in the context of the COVID-19 pandemic, as over a third of the <100 kDa depositions to the EMDB so far this year are associated with SARS-COV-2. These entries include the Coronavirus exoribonuclease, [17] the receptor-binding domain bound to human receptors or neutralizing Fabs/nanobodies [18–20], viral envelope proteins [21], and putative viral membrane channels [22]. While the trimeric spike protein, which contains the receptor-binding domains, is over 400 kDa in size, the mask-and-align approach was particularly useful in increasing resolution of binding modes of neutralizing antibodies or nanobodies, providing a detailed description of residues involved in neutralizing interactions [23–25]. The masks used for these focused image analyses often encompassed density that were smaller than 100 kDa in cumulative mass, emphasizing the utility of the mask-and-align approach in improving resolution for small, targeted regions of a map.
One of the most impressive examples in the category of <100 kDa COVID-related structures is the 2.1 Å resolution structure of the SARS-CoV-2 ORF3a ion channel embedded in lipid nanodiscs (Figure 1C) [22]. This putative viral ion channel forms a dimer amassing 62 kDa, and it is the highest resolution <100 membrane protein determined to date. Notably, this structure was determined without the incorporation of a tightly binding Fab, as is usually required for structure determination of small membrane proteins. Incorporation of size-increasing moieties into small membrane proteins is common since a substantial portion of any small membrane protein will likely be buried within detergent or lipid micelles, which obscure structural features of the membrane-embedded regions in the projection images. Increasing the apparent size of the soluble domains can be accomplished either through selection of a high affinity Fab or nanobody that stably binds an exposed region of the target protein [26,27], or by fusing the target protein to a rigid soluble domain, such as a BRIL domain, that can be targeted by a known Fab [28,29]. A recent study introduced the ‘Legobody’, wherein a target-specific nanobody is integrated into a stable 120 kDa scaffold bearing a distinct shape that aids in accurate orientation assignment. This Legobody was used to determine high resolution structures of a 23 kDa membrane protein (KDEL receptor), and the 22 kDa receptor-binding domain of the SARS-COV-2 spike protein [30].
A number of <100 kDa complexes are of nucleoprotein complexes. The higher density of nucleic acids (averaging 1.7 g/cm3 vs. an average protein density of 1.35 g/cm3) imparts greater electron scattering per cubic Å, thereby enhancing the contrast of these regions in the resulting projections. Indeed, 2D classification of small nucleoprotein complexes often show significantly brighter density corresponding to the nucleic acid components of the assembly relative to the protein components [31,32]. Thus, incorporation of nucleic acid, if well-ordered relative to the protein interactors, likely improves the accuracy of 3D orientation assignments. An excellent example of such a small nucleoprotein complex is the Cre recombinase bound to a DNA substrate, assembling into a 54 kDa complex that was determined to 3.9 Å resolution [31]. Likely owing to their higher density properties, the cryo-EM reconstructions that have come closest to the theoretical size limit of single particle analysis have been of RNA oligomers (Figure 2B). To date, the smallest isolated complex resolved to better than 4 Å resolution using single particle approaches is the 39 kDa S-adenosylmethionine-IV riboswitch RNA (~3.7 Å resolution) [33]. It was also recently demonstrated that a 28 kDa frameshift stimulation element of the SARS-CoV-2 RNA genome, which is notably smaller than the predicted theoretical 38 kDa size limit, could be resolved to ~6.9 Å resolution [34]. Such observations indicate that integration of nucleic acid into protein complexes, such as through the development of site-specific aptamers, may enable higher resolution structure determination of isolated small complexes or flexible regions through mask-and-align approaches.
To ensure success when imaging small particles, one of the more important considerations is to use a high enough magnification that the pixel size of the resulting micrographs is one fourth or even less (rather than just half) the sought-after resolution. Using a higher magnification decreases the field of view and thereby reduces the number of particles included in each image, but the improved camera performance achieved (see below) more than compensates for this. However, a field of view that is too small can decrease the accuracy of motion correction and CTF estimation algorithms, and thus it is important not to work at too high of a magnification. Other considerations that help are to record images in areas where the ice thickness is less than ~30 nm, and to minimize beam-induced motion (addressed further below) by preparing samples on holey gold films that are supported on gold grids [35]. It is also beneficial to prepare samples that have a large number of particles per unit area, not just to thereby reduce the number of images that must be recorded, but also to increase the level of signal in the contrast transfer function (CTF).
Estimated theoretical limits suggest that room for improvement still remains
There are fundamental limitations on what can be accomplished by any experimental technique. In cryo-EM of small particles, the factors that fundamentally limit what is possible include (A) the limited scattering of electrons from individual atoms, (B) the size of the particle, i.e. the total number of atoms present in the targeted particle, and (C) structural homogeneity of the particles, i.e. the percentage of atoms that are in identical locations from one particle to the next.
In addition, there is the equally important, practical factor that high-energy electrons are ionizing radiation, and biological structures thus become damaged beyond the point of recognition if the exposures are too high [8,36–38]. The values of electron exposure that may be safely used are, in fact, much smaller than what is needed to visualize features at atomic resolution, as will be further explained below. As a consequence, cryo-EM images, when taken with ‘safe’ exposures, are quite noisy due to the random distribution of just a limited number of electrons per unit area in the images.
Both the values of the atomic scattering cross sections — and thus the amount of signal that is available for a particle of a given size, and the tolerated electron exposures — and thus the level of shot-noise in the image, are dependent on the energy of the electrons used to record images [39]. The voltage-dependent increase in one of these factors is largely offset, however, by a voltage-dependent decrease in the other. As a result, we believe that comparable results can be expected for images recorded at either 200 keV or at 300 keV. On the other hand, instrumental factors such as reduced performance of the camera, and the reduced temporal coherence of the electron beam, currently have a notably adverse effect on images recorded at 100 keV, although it has also been pointed out that the information that is obtained, for a given amount of radiation damage, is slightly higher at 100 keV if the samples are thin enough [39].
Since images of single biological molecules are too noisy to be interpretable at atomic resolution [8], data must be merged from images of a large number of identical particles, i.e. asymmetric units in the case of symmetric oligomers, no matter what energy of electrons is used. Doing so improves the signal-to-noise ratio, thereby allowing three-dimensional density maps to be correctly interpreted in terms of the chemical structure of the particle. Theoretically, the required number of asymmetric units might be as small as ~1000 identical particles, or even less depending upon the sought-after resolution and the value of the so-called B factor (a term used to represent the sum of resolution-limiting factors associated with an experimental setup) [10]. The required number of asymmetric units is independent of the size of the particle, provided that the particle’s size is above a threshold value, which has been estimated to be as small as 20 kDa [9], although a higher value was estimated in the section titled ‘Averaging particle images’ in [10]. In current practice, however, the required number of asymmetric units used to obtain an interpretable map at ~3.5 Å resolution generally remains above 15,000, as it was for subtomogram averaging of ribosomes [40], and at least eight million asymmetric units are needed to obtain a map at better than 1.3 Å resolution [41,42].
A large gap thus remains between the current requirements for obtaining high-resolution maps and the estimated theoretical limits. This brings us to the important question of what was assumed, when making theoretical estimates, that has not yet been realized in current experiments.
Prospects for narrowing the gap between experiment and theory
The ideal-case assumptions that were made in theoretical estimates include (A) there is no loss of signal due to imperfect performance of the camera, (B) there is no loss of signal due to the chromatic aberration of the microscope, (C) there is no loss of signal due to uncorrected, beam-induced motion, and (D) there is no loss of signal due to oscillations in the CTF. The available technology for biological cryo-EM still falls well short of realizing all four of these assumptions. Instead, these practical issues still need much improvement in order to close the gap between what can be achieved at present and what have been estimated to be the theoretical limits of cryo-EM.
Camera performance
All cameras that are currently used in cryo-EM consist of pixelated silicon sensors, regardless of the particular technology on which they are based, such as CCD, CMOS, or ‘hybrid’ devices [43]. Although the improved performance introduced by direct electron detection revolutionized cryo-EM [44,45], pixelation unavoidably produces a significant loss of signal as one goes to higher and higher resolutions. As a result, the spectral signal-to-noise ratio (SNR) progressively falls off in the Fourier transform of a pixelated image, relative to what it is in the image intensity itself.
This unwanted falloff is usually quantified by measuring the spectral detective quantum efficiency, DQE(s), of a camera device. The falloff in DQE is the result of the inability to distinguish precisely which single pixel on the detector is initially hit by an incident electron. In effect, the recorded image intensity is a 2D array of samples of the convolution product between (A) the image intensity, as it hits the camera, and (B) a function whose value is 1.0 inside a single pixel and 0.0 outside. Figure 3 shows an early comparison of the theoretical of DQE(s) for a pixelated camera and some experimental curves [43]. While the performance levels of some newer cameras now approach the theoretical limit, shown by the dotted curve in Figure 3, the falloff in DQE(s) incurred by pixelization will always introduce a large gap relative to the limiting value of DQE(s) = 1.0, which has been assumed when making ideal-case estimates.
Figure 3. Curves comparing the experimentally determined DQE(s) values for three older, commercially available direct-detection cameras; the curve for photographic film; and the theoretically best-possible curve for a pixelated camera.
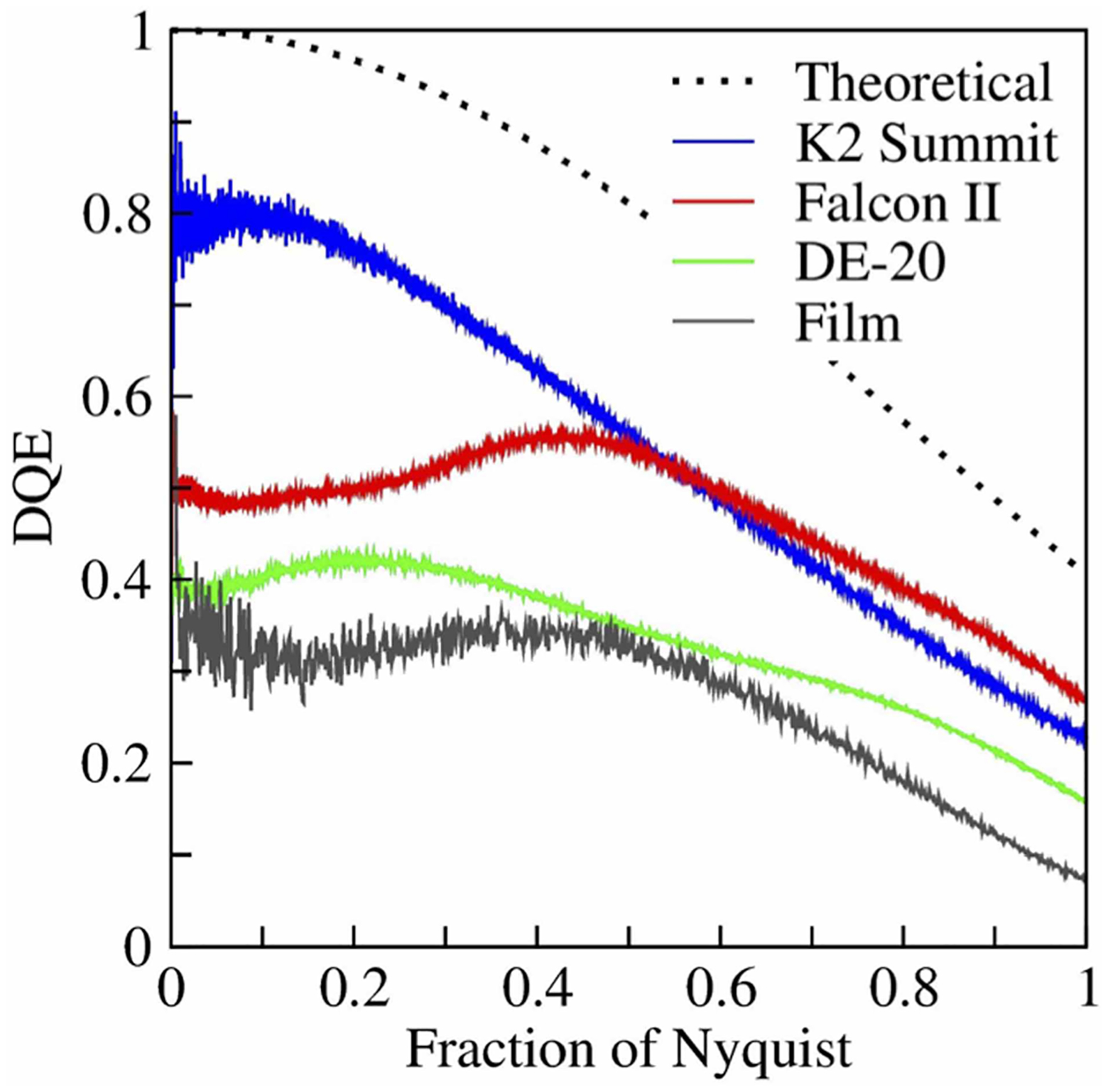
Although more recent developments in camera performance have brought the values for all commercially available cameras closer to the theoretical limit, this figure nonetheless emphasizes the limiting way in which pixilation causes the DQE to fall off as the resolution increases. As is explained in the text, one can collect images at higher magnification in order that the sought-after (best) spatial frequency is no more than 0.4 or 0.5 of the Nyquist frequency, thus incurring a much smaller amount of falloff. In the future, it might be hoped that technology to interpolate the location of single-electron events (i.e. super-localization technology) might become more effective than what is currently available, thus overcoming the limitation set by pixelization. This is a redrawn figure, modified from one presented as Figure 4 in [76], which was prepared for us courtesy of Dr. Greg McMullan.
One way to increase the values of DQE (s) at high resolution is, in principle, to fit a model of the point-spread function of a camera to its measured responses to single-electron events, and assume that electron counts should be assigned to the centers of the fitted point-spread functions. Mathematically analogous to super-resolution (super-localization) approaches that have successfully improved the resolution of fluorescence-based light microscopy [46], this approach has so far given only a marginal improvement in the performance of direct-detection cameras. An alternative approach, which was recommended above, is to record images at two, or even three times higher magnification than is needed to place the desired resolution at the Nyquist limit. The approach of using ‘empty’ magnification is now used quite frequently, and it greatly improves the resolution achieved in reconstructions, but it comes at the heavy cost of increasing the number of images that must be recorded.
Further development of camera performance thus remains an essential priority. Improvements may be achieved (A) by increasing the number of pixels in the camera, in order to increase the imaged field of view at a given magnification, (B) by improving the technology used for super-localization of single-electron events, or (C) by a combination of the two. The final result, however achieved, is likely to have significant impact on what is achieved by cryo-EM.
Chromatic aberration and temporal coherence
The finite temporal coherence of the electrons that pass through the electron microscope give rises to an ‘envelope function’ [47], the exponential falloff of which depends upon the product of the chromatic aberration coefficient of the objective lens and the energy spread of the beam. This envelope function significantly reduces the signal in Fourier transforms of images at high resolution, relative to the signal in the electron beam as it exits the specimen. This issue is best addressed by reducing the energy spread of the beam, as has been done for some time in materials-science applications. As mentioned above, it has recently been shown that using either a gun monochromator [41] or a cold field-emission gun [42] makes it possible to extend the resolution achieved by single-particle cryo-EM to below 1.3 Å, and likely aided in breaking the 2 Å resolution barrier for a <100 kDa complex. A current challenge, however, is that data must still be merged from up to 24 million structurally identical asymmetric units to achieve such high resolutions, something that is impractical unless the protein molecules are assembled into highly symmetrical oligomers, such as apoferritin. Nevertheless, it can be expected that technology to improve the temporal coherence will become increasingly important as the other limiting factors begin to be mitigated, and thus the number of asymmetric units is reduced significantly.
Beam-induced motion
Any sample motion that occurs while recording an image will clearly result in blurring of the image, resulting in a loss of signal at high resolution. Just being irradiated by the electron beam is enough, unfortunately, to cause significant specimen motion [48], and deflection of the electron beam also occurs when steps are not taken to prevent electrostatic charging of the specimen while being illuminated [49]. Although the problem of beam-induced motion is mitigated by recording images as a series of frames that can be subsequently aligned to one another [50,51], motion that occurs during the exposure of a single frame still results in unrecoverable loss of signal. Unfortunately, the amount of beam-induced motion is greatest during the first few frames of a movie, thus substantially reducing the high-resolution signal in images at the time when it is still at its highest level within the specimen, i.e. prior to it being lost due to radiation damage [52].
The use of holey gold specimen-support films, themselves supported on gold grids, reduces beam-induced motion to a considerable extent [53]. A recent modification, in which the holey gold support films are mounted on molybdenum grids, seems to further reduce ‘cryo-crinkling’ and its associated contribution to beam-induced motion, even for grids tilted by up to 45 degrees [54]. A further improvement can also be had by greatly reducing the hole size of the thin gold specimen support film [55], but — if the hole size is extremely small — the improvement comes at the expense of reducing the amount of imageable sample within the hole. Fractionation of the exposure into a greater number of frames, especially at the beginning of the exposure, might be an alternative way to reduce the impact of beam-induced motion. The use of electron event representation (EER) [56], or the equivalent, is an especially efficient strategy in this regard, although shorter frames carry less signal and may not be aligned accurately. This low-signal issue associated with ‘super-fractionated’ frames could be overcome with the incorporation of high-contrast fiducials, or increasing the contrast of the specimen itself through the use of a phase plate.
Contrast transfer
Cryo-EM specimens exhibit very little amplitude contrast, corresponding to the fact that, as explained in Section 1.2.2 of [1], few electrons are absorbed (inelastically scattered) in thin, unstained objects. On the other hand, the same specimens exhibit strong phase contrast when the images are defocused by a sufficiently large amount. Unfortunately, using defocus to generate phase contrast results in a significant corruption of the image The resulting CTF for defocused images always remains extremely low at low resolution, and, although the CTF reaches ideal values at some intermediate spatial frequencies, it actually oscillates between values of 1.0 and −1.0 as the resolution increases, see [57,58]. The information that can be recovered, by making now-standard computational corrections, still deviates significantly from the ideal case in which the signal in the Fourier transform of the image remains identical with that of the electron wave function as it first exits the specimen.
Although obtaining nearly ideal phase contrast is a theoretical possibility in electron microscopy [59], as it is in light microscopy [60], devising an appropriate phase plate for electrons has proven to be more elusive than had been imagined [61]. A significant advance was made in this direction by taking advantage of an effect in which the local value of the surface potential of a thin, heated-carbon foil changes when the foil is irradiated by an intense electron beam [62]. While effective, the resulting, so-called ‘Volta’, phase plate suffers from being unable to produce phase shifts of the desired value for more than limited periods of time. More importantly, it also suffers from losses of signal due to electron scattering by the foil itself, as well as exhibiting a poorly understood, but significant, loss of signal at high resolution [63].
More recently, however, progress has been made towards using an intense laser beam as a phase plate for high-energy electrons [64,65]. While the theoretical CTF for such a phase plate could approach the desired value of 1.0 over a broad range of spatial frequencies, this technology is still in the early development phase.
Summary and conclusions
Year after year, single-particle cryo-EM increasingly contributes to breakthroughs in biochemistry and structural biology. This review has emphasized the rapid extension of high-resolution cryo-EM to macromolecules whose sizes are well below 100 kDa, once considered to be inaccessible by this method. Such advances greatly expand the power of cryo-EM to investigate different conformational states of flexible, dynamic macromolecules, i.e. states which normally co-exist under physiologically relevant buffer conditions. As a result, regions of a map that once were blurred to low resolution are now being refined to high resolution by masking all but the region in question, which is the computational equivalent of the ‘divide and conquer’ approach previously used so successfully in biochemistry and structural biology. By extending the masked-refinement approach to smaller and smaller subunits, it becomes possible to observe individual parts of macromolecular machines in their native structure environments, without needing to first physically separate them in order to ‘conquer’.
Given the importance of cryo-EM structure determination in the field of drug design [66] we expect that advances in these methods could be extended to unambiguously identify the binding modes of low occupancy or low affinity small molecules in their respective binding pockets to benefit structure-aided drug design. Further advances in masked refinement technologies will also benefit ongoing development of algorithms aimed at describing the complete conformational landscapes of macromolecules, enabling more detailed characterization of the motions of small components [67–72]. We anticipate that such computational developments, which are increasingly taking advantage of powerful deep-learning frameworks, will eventually do away with our need to define masks or manually guide classification and refinement. The expectation is that 3D analyses of single particle cryoEM data in the future would automatically produce conformational landscapes of the targeted molecules rather than a single structure.
Perspectives.
Advances in single-particle cryo-EM now make it possible to determine high-resolution structures of macromolecules, and even flexible regions of macromolecular complexes, whose size is significantly smaller than 100 kDa.
The lower particle weight limit of macromolecules that can be reconstructed using single-particle cryo-EM is currently below 50 kDa for proteins, and even lower for nucleic acids. The eventual limit might thus become less than ~20 kDa.
Future advances in cryo-EM technology, which can be expected to facilitate high-resolution structure determination of smaller macromolecules, include (1) better camera performance, (2) more monochromatic electron sources, (3) better recovery of high-resolution information that is lost because of beam-induced motion, and (4) the use of phase plates that can provide nearly perfect contrast transfer over the full range of spatial frequencies.
Acknowledgements
We thank Dr. Greg McMullan for providing the graphics used in Figure 3.
Funding
This work was supported in part by funds from NIH Grants R01 GM126011 to Prof. Holger Mueller and R21 AG067594 and R21 GM142196 to GCL.
Abbreviations
- ACE2
angiotensin-converting enzyme 2
- CTF
contrast transfer function
- DQE
detective quantum efficiency
- FSE
Frameshift Stimulation Element
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.2021. Single-particle Cryo-EM of biological macromolecules. (Glaeser RM, Nogales E and Chiu W, eds), IOP Publishing, Bristol [Google Scholar]
- 2.Baxter WT, Grassucci RA, Gao H and Frank J (2009) Determination of signal-to-noise ratios and spectral SNRs in cryo-EM low-dose imaging of molecules. J. Struct. Biol 166, 126–132 10.1016/j.jsb.2009.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheres SHW (2012) A Bayesian view on cryo-EM structure determination. J. Mol. Biol 415, 406–418 10.1016/j.jmb.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilca SL, Kotecha A, Sun X, Poranen MM, Stuart DI and Huiskonen JT (2015) Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun 6, 8843 10.1038/ncomms9843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Q, Huang X, Sun S, Li X, Wang H-W and Sui S-F (2015) Cryo-EM structure of SNAP-SNARE assembly in 20S particle. Cell Res. 25, 551–560 10.1038/cr.2015.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L et al. (2015) An atomic structure of human γ-secretase. Nature 525, 212–217 10.1038/nature14892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strelkov SV, Herrmann H, Geisler N, Lustig A, Ivaninskii S, Zimbelmann R et al. (2001) Divide-and-conquer crystallographic approach towards an atomic structure of intermediate filaments. J. Mol. Biol 306, 773–781 10.1006/jmbi.2001.4442 [DOI] [PubMed] [Google Scholar]
- 8.Henderson R (1995) The potential and limitations of neutrons, electrons and X-rays for atomic-resolution microscopy of unstained biological molecules. Q. Rev. Biophys 28, 171–193 10.1017/S003358350000305X [DOI] [PubMed] [Google Scholar]
- 9.Glaeser RM (1999) Review: electron crystallography: present excitement, a nod to the past, anticipating the future. J. Struct. Biol 128, 3–14 10.1006/jsbi.1999.4172 [DOI] [PubMed] [Google Scholar]
- 10.Rosenthal PB and Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol 333, 721–745 10.1016/j.jmb.2003.07.013 [DOI] [PubMed] [Google Scholar]
- 11.Wu M and Lander GC (2020) How low can we go? structure determination of small biological complexes using single-particle cryo-EM. Curr. Opin. Struct. Biol 64, 9–16 10.1016/j.sbi.2020.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costa TRD, Ignatiou A and Orlova EV (2017) Structural analysis of protein complexes by cryo electron microscopy. Methods Mol. Biol 1615, 377–413 10.1007/978-1-4939-7033-9_28 [DOI] [PubMed] [Google Scholar]
- 13.Herzik MA, Wu M and Lander GC (2019) High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM. Nat. Commun 10, 1032 10.1038/s41467-019-08991-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiessen A, Pérez-Rodríguez P and Delaye-Arredondo LJ (2012) Mathematical modeling and comparison of protein size distribution in different plant, animal, fungal and microbial species reveals a negative correlation between protein size and protein number, thus providing insight into the evolution of proteomes. BMC Res Notes 5, 85 10.1186/1756-0500-5-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinothkumar KR and Henderson R (2016) Single particle electron cryomicroscopy: trends, issues and future perspective. Q. Rev. Biophys 49, 1–25 10.1017/S0033583516000068 [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Tammaro R, Peters PJ and Ravelli RBG (2020) Could Egg white lysozyme be solved by single particle cryo-EM? J. Chem. Inf. Model 60, 2605–2613 10.1021/acs.jcim.9b01176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C, Shi W, Becker ST, Schatz DG, Liu B and Yang Y (2021) Structural basis of mismatch recognition by a SARS-CoV-2 proofreading enzyme. Science 373, 1142–1146 10.1126/science.abi9310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun D, Sang Z, Kim YJ, Xiang Y, Cohen T, Belford AK et al. (2021) Potent neutralizing nanobodies resist convergent circulating variants of SARS-CoV-2 by targeting diverse and conserved epitopes. Nat. Commun 12, 4676 10.1038/s41467-021-24963-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niu S, Wang J, Bai B, Wu L, Zheng A, Chen Q et al. (2021) Molecular basis of cross-species ACE2 interactions with SARS-CoV-2-like viruses of pangolin origin. EMBO J. 40, e107786 10.15252/embj.2021107786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zahradník J, Marciano S, Shemesh M, Zoler E, Harari D, Chiaravalli J et al. (2021) SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol 6, 1188–1198 10.1038/s41564-021-00954-4 [DOI] [PubMed] [Google Scholar]
- 21.Chai J, Cai Y, Pang C, Wang L, McSweeney S, Shanklin J et al. (2021) Structural basis for SARS-CoV-2 envelope protein recognition of human cell junction protein PALS1. Nat. Commun 12, 3433 10.1038/s41467-021-23533-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kern DM, Sorum B, Mali SS, Hoel CM, Sridharan S, Remis JP et al. (2021) Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol 28, 573–582 10.1038/s41594-021-00619-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koenig PA, Das H, Liu H, Kümmerer BM, Gohr FN, Jenster LM et al. (2021) Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 371, eabe6230 10.1126/science.abe6230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.VanBlargan L, Adams L, Liu Z, Chen RE, Gilchuk P, Raju S et al. (2021) A potently neutralizing SARS-CoV-2 antibody inhibits variants of concern by utilizing unique binding residues in a highly conserved epitope. Immunity 54, 2399–2416.e6 10.1016/j.immuni.2021.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song D, Wang W, Dong C, Ning Z, Liu X, Liu C et al. (2021) Structure and function analysis of a potent human neutralizing antibody CA521 (FALA) against SARS-CoV-2. Commun. Biol 4, 500 10.1038/s42003-021-02029-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bloch JS, Pesciullesi G, Boilevin J, Nosol K, Irobalieva RN, Darbre T et al. (2020) Structure and mechanism of the ER-based glucosyltransferase ALG6. Nature 579, 443–447 10.1038/s41586-020-2044-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan YZ, Rodrigues J, Keener JE, Zheng RB, Brunton R, Kloss B et al. (2020) Cryo-EM structure of arabinosyltransferase EmbB from Mycobacterium smegmatis. Nat. Commun 11, 3396 10.1038/s41467-020-17202-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukherjee S, Erramilli SK, Ammirati M, Alvarez FJD, Fennell KF, Purdy MD et al. (2020) Synthetic antibodies against BRIL as universal fiducial marks for single-particle cryoEM structure determination of membrane proteins. Nat. Commun 11, 1598 10.1038/s41467-020-15363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsutsumi N, Mukherjee S, Waghray D, Janda CY, Jude KM, Miao Y et al. (2020) Structure of human Frizzled5 by fiducial-assisted cryo-EM supports a heterodimeric mechanism of canonical Wnt signaling. eLife 9, e58464 10.7554/eLife.58464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X and Rapoport TA (2021) Cryo-EM structure determination of small proteins by nanobody-binding scaffolds (Legobodies). Proc. Natl. Acad. Sci 118, e2115001118 10.1073/pnas.2115001118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stachowski K, Norris A, Potter D, Wysocki V and Foster MP (2021) Unloxing the assembly and activation mechanism of Cre recombinase using cryo-EM. bioRxiv 10.1101/2021.07.31.454597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anzelon TA, Chowdhury S, Hughes SM, Xiao Y, Lander GC and MacRae IJ (2021) Structural basis for piRNA targeting. Nature 597, 285–289 10.1038/s41586-021-03856-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang K, Li S, Kappel K, Pintilie G, Su Z, Mou TC et al. (2019) Cryo-EM structure of a 40 kDa SAM-IV riboswitch RNA at 3.7 Å resolution. Nat. Commun 10, 5511 10.1038/s41467-019-13494-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang K, Zheludev IN, Hagey RJ, Haslecker R, Hou YJ, Kretsch R et al. (2021) Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. Nat. Struct. Mol. Biol 28, 747–754 10.1038/s41594-021-00653-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russo CJ and Passmore LA (2016) Progress towards an optimal specimen support for electron cryomicroscopy. Curr. Opin. Struct. Biol 37, 81–89 10.1016/j.sbi.2015.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker LA, Smith EA, Bueler SA and Rubinstein JL (2010) The resolution dependence of optimal exposures in liquid nitrogen temperature electron cryomicroscopy of catalase crystals. J. Struct. Biol 169, 431–437 10.1016/j.jsb.2009.11.014 [DOI] [PubMed] [Google Scholar]
- 37.Baker LA and Rubinstein JL (2010) Radiation damage in electron cryomicroscopy. Methods Enzymol. 481, 371–388 10.1016/S0076-6879(10)81015-8 [DOI] [PubMed] [Google Scholar]
- 38.Glaeser RM (2016) Specimen behavior in the electron beam. Methods Enzymol. 579, 19–50 10.1016/bs.mie.2016.04.010 [DOI] [PubMed] [Google Scholar]
- 39.Peet MJ, Henderson R and Russo CJ (2019) The energy dependence of contrast and damage in electron cryomicroscopy of biological molecules. Ultramicroscopy 203, 125–131 10.1016/j.ultramic.2019.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tegunov D, Xue L, Dienemann C, Cramer P and Mahamid J (2021) Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 Å in cells. Nat. Methods 18, 186–193 10.1038/s41592-020-01054-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yip KM, Fischer N, Paknia E, Chari A and Stark H (2020) Atomic-resolution protein structure determination by cryo-EM. Nature 587, 157–161 10.1038/s41586-020-2833-4 [DOI] [PubMed] [Google Scholar]
- 42.Nakane T, Kotecha A, Sente A, McMullan G, Masiulis S, Brown PMGE et al. (2020) Single-particle cryo-EM at atomic resolution. Nature 587, 152–156 10.1038/s41586-020-2829-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMullan G, Chen S, Henderson R and Faruqi AR (2009) Detective quantum efficiency of electron area detectors in electron microscopy. Ultramicroscopy 109, 1126–1143 10.1016/j.ultramic.2009.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glaeser RM (2013) Stroboscopic imaging of macromolecular complexes. Nat. Methods 10, 475 10.1038/nmeth.2486 [DOI] [PubMed] [Google Scholar]
- 45.Kühlbrandt W (2014) The resolution revolution. Science 343, 1443–1444 10.1126/science.1251652 [DOI] [PubMed] [Google Scholar]
- 46.Schermelleh L, Ferrand A, Huser T, Eggeling C, Sauer M, Biehlmaier O et al. (2019) Super-resolution microscopy demystified. Nat. Cell Biol 21, 72–84 10.1038/s41556-018-0251-8 [DOI] [PubMed] [Google Scholar]
- 47.Wade RH and Frank J (1977) Electron-microscope transfer-functions for partially coherent axial illumination and chromatic defocus spread. Optik 49, 81–92 [Google Scholar]
- 48.Brilot AF, Chen JZ, Cheng AC, Pan JH, Harrison SC, Potter CS et al. (2012) Beam-induced motion of vitrified specimen on holey carbon film. J. Struct. Biol 177, 630–637 10.1016/j.jsb.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brink J, Sherman MB, Berriman J and Chiu W (1998) Evaluation of charging on macromolecules in electron cryomicroscopy. Ultramicroscopy 72, 41–52 10.1016/S0304-3991(97)00126-5 [DOI] [PubMed] [Google Scholar]
- 50.Li XM, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S et al. (2013) Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 10.1038/nmeth.2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai XC, Fernandez IS, McMullan G and Scheres SH (2013) Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife Sci. 2, e00461 10.7554/eLife.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheres SHW (2014) Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife 3, e03665 10.7554/eLife.03665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russo CJ and Passmore LA (2014) Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380 10.1126/science.1259530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang X, Xuan S, Zuckermann RN, Glaeser RM, Downing KH and Balsara NP (2021) Minimizing crinkling of soft specimens using holey gold films on molybdenum grids for cryogenic electron microscopy. Microsc. Microanal 27, 767–775 10.1017/S1431927621000520 [DOI] [PubMed] [Google Scholar]
- 55.Naydenova K, Jia P and Russo CJ (2020) Cryo-EM with sub–1 Å specimen movement. Science 370, 223–226 10.1126/science.abb7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo H, Franken E, Deng Y, Benlekbir S, Singla Lezcano G, Janssen B et al. (2020) Electron-event representation data enable efficient cryoEM file storage with full preservation of spatial and temporal resolution. IUCrJ 7, 860–869 10.1107/S205225252000929X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rohou A and Grigorieff N (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 10.1016/j.jsb.2015.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang K (2016) Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12 10.1016/j.jsb.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boersch H (1947) Uber die Kontraste von Atomen im Electronenmikroskop Zeitschrift Fur Naturforschung Section a-a. J. Phys. Sci 2, 615–633 10.1515/zna-1947-11-1204 [DOI] [Google Scholar]
- 60.Zernike F (1955) How I discovered phase contrast. Science 121, 345–349 10.1126/science.121.3141.345 [DOI] [PubMed] [Google Scholar]
- 61.Glaeser RM (2013) Invited review article: methods for imaging weak-phase objects in electron microscopy. Rev. Sci. Instrum 84, 111101 10.1063/1.4830355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Danev R, Buijsse B, Khoshouei M, Plitzko JM and Baumeister W (2014) Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc. Natl Acad. Sci. U.S.A 111, 15635–15640 10.1073/pnas.1418377111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buijsse B, Trompenaars P, Altin V, Danev R and Glaeser RM (2020) Spectral DQE of the Volta phase plate. Ultramicroscopy 218, 113079 10.1016/j.ultramic.2020.113079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwartz O, Axelrod JJ, Campbell SL, Turnbaugh C, Glaeser RM and Muller H (2019) Laser phase plate for transmission electron microscopy. Nat. Methods 16, 1016–2020 10.1038/s41592-019-0552-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turnbaugh C, Axelrod JJ, Campbell SL, Dioquino JY, Petrov PN, Remis J et al. (2021) High-power near-concentric Fabry–Perot cavity for phase contrast electron microscopy. Rev. Sci. Instrum 92, 053005 10.1063/5.0045496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Renaud J-P, Chari A, Ciferri C, Liu WT, Rémigy H-W, Stark H et al. (2018) Cryo-EM in drug discovery: achievements, limitations and prospects. Nat. Rev. Drug Discov 17, 471–492 10.1038/nrd.2018.77 [DOI] [PubMed] [Google Scholar]
- 67.Punjani A and Fleet DJ (2021) 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol 213, 107702 10.1016/j.jsb.2021.107702 [DOI] [PubMed] [Google Scholar]
- 68.Nakane T, Kimanius D, Lindahl E and Scheres SHW (2018) Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 7, e36861 10.7554/eLife.36861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dashti A, Mashayekhi G, Shekhar M, Ben Hail D, Salah S, Schwander P et al. (2020) Retrieving functional pathways of biomolecules from single-particle snapshots. Nat. Commun 11, 4734 10.1038/s41467-020-18403-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen M and Ludtke SJ (2021) Deep learning-based mixed-dimensional Gaussian mixture model for characterizing variability in cryo-EM. Nat. Methods 18, 930–936 10.1038/s41592-021-01220-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhong ED, Bepler T, Berger B and Davis JH (2021) CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 18, 176–185 10.1038/s41592-020-01049-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsumoto S, Ishida S, Araki M, Kato T, Terayama K and Okuno Y (2021) Extraction of protein dynamics information from cryo-EM maps using deep learning. Nat. Mach. Intell 3, 153–160 10.1038/s42256-020-00290-y [DOI] [Google Scholar]
- 73.Yan R, Zhang Y, Li Y, Xia L, Guo Y and Zhou Q (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 10.1126/science.abb2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serna M (2019) Hands on methods for high resolution cryo-electron microscopy structures of heterogeneous macromolecular complexes. Front. Mol. Biosci 6, 33 10.3389/fmolb.2019.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen I, Pant S, Wu Q, Cater RJ, Sobti M, Vandenberg RJ et al. (2021) Glutamate transporters have a chloride channel with two hydrophobic gates. Nature 591, 327–331 10.1038/s41586-021-03240-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McMullan G, Faruqi AR, Clare D and Henderson R (2014) Comparison of optimal performance at 300 keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy 147, 156–163 10.1016/j.ultramic.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]