

Abstract
A one-step, three-component reaction between α-hydroxyketones, oxoacetonitriles, and primary amines gives N-substituted 2,3,5-functionalized 3-cyanopyrroles with complete selectivity in up to 90% isolated yields. The reaction worked on a wide substrate scope under mild reaction conditions (AcOH as a catalyst, EtOH, 70 °C, 3 h). The reaction proceeded with very high atom efficiency as water is the only molecule lost during the reaction. The practicality of the reaction was demonstrated on a large gram scale. The structures of the 3-cyanopyrroles were confirmed by single-crystal X-ray diffraction and NMR; this work provides a general and practical entry to pyrrole scaffolds suitably decorated for the synthesis of various bioactive pyrroles in a concise manner.
Keywords: pyrroles, carbohydrates, sustainable synthesis, three-component reactions, one-pot reactions
1. Introduction
Pyrroles are a class of nitrogen heterocycles with important applications in the pharmaceutical and material science industries [1,2,3]. Owing to their diverse bioactivities such as anti-inflammatory [4], anti-bacterial [5], anti-tumor [6], and anti-oxidative bioactivities [7], several pyrrole-based drugs have been successfully marketed to treat various conditions (Figure 1). Additionally, many pyrrole-based drug candidates have shown great promise in treating various conditions (Figure 1). Therefore, continuous efforts have been invested in designing simple, efficient, and sustainable routes to synthesize suitably functionalized pyrroles with specific “handles” for easy transformation to the final dug compounds. In this regard, special attention is given to synthesizing 3-cyanopyrroles since the cyano handle can easily be converted to -CXN functionality (e.g., X = H, H2, O, or simply nothing); this -CXN functionality appears in many drugs and drug candidates (Figure 1) [5,8,9,10,11,12].
Figure 1.

Examples of bioactive pyrrole-based compounds having a -CXN moiety (in blue).
Previous efforts to synthesize 3-cyanopyrroles include (i) the nickel(II) bis(acetylacetonate)-catalyzed reaction between azirines and active methylene compounds reported in 1977 (Figure 2, (i)) [13]; (ii) the reaction between 1-nitro-1-cyclopropyl ketones and primary amines followed by oxidizing the dihydropyrrole products using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give the desired 3-cyanopyrroles (Figure 2, (ii) [14]; and (iii)) the reaction between α-hydroxyketones, malononitrile and primary amines (Figure 2, (iii)) [15]; however, the first two reactions require extra steps to prepare the complex starting materials, use metals to catalyze the reactions, and are multistep syntheses. The last reaction works when the substituents at the α-hydroxyketones are similar (R1 and R1, both either methyl or phenyl, Figure 1, (iii)) and the starting material itself is not considered sustainable. In this reaction, when the substituents are different, several products are formed, which reduces the yields and complicates separation [16]; moreover, these reactions are conducted at high temperatures > 90 °C in toluene. Additionally, other inefficient multistep reactions introduced the cyano handle on a preformed pyrrole and usually used heavy metals or toxic reagents [17]. These disadvantages encourage the search for a more robust, practical, and sustainable methodology.
Figure 2.

Previous and proposed work for the synthesis of 3-cyanopyrroles.
Earlier, we developed a cascade reaction between aldoses, oxoacetonitriles, and NH4OAc to synthesize N-unsubstituted 3-cyanopyrroles (Figure 2, (iv)) [18]. Based on our previous experience [18,19], we envisioned that ketoses should also be ideal substrates for the synthesis of functionalized 3-cyanopyrroles (Figure 1, (iii)). Ketoses are cheap, sustainable materials and can introduce chirality in the pyrrole framework; moreover, their polyhydroxyalkyl chain can easily be converted into several functional groups; it is also beneficial to establish general reaction conditions that work for other α-hydroxyketones (ketoses are, in fact, masked α-hydroxyketones) such as phenacyl alcohols to add diversity to the substituents on the core 3-cyanopyrrole structure (Figure 1, (v)). Another important objective in this work is to achieve complete selectivity while using differently substituted α-hydroxyketones (R1 ≠ R1, Figure 1, (iii)) which has proven to be challenging in previous reports [16].
Herein, we report an AcOH-catalyzed selective one-pot three-component reaction between α-hydroxyketones (ketoses and phenacyl alcohols), oxoacetonitriles, and primary amines to synthesize densely functionalized 3-cyanopyrroles; this reaction gave the desired 3-cyanopyrroles in 53–90% yields and worked successfully on a gram scale. The structures of the 3-cyanopyrroles were confirmed using single-crystal X-ray analysis, and a plausible reaction mechanism was also provided.
2. Results and Discussion
2.1. Reaction Optimization
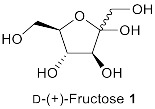
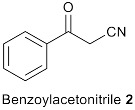
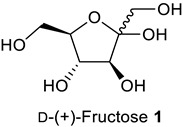
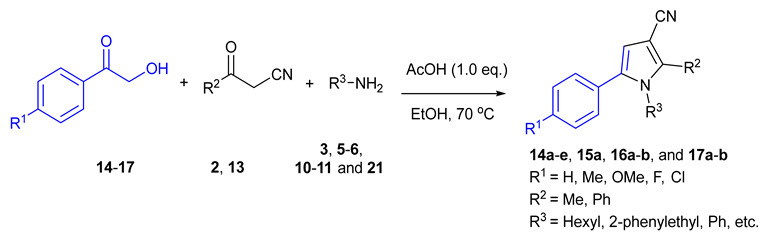
We started the investigation by stirring D-(+)-fructose 1 (1 mmol, 1.0 eq.), benzoylacetonitrile 2 (1.0 eq.), and benzylamine 3 (1.1 eq.) in one pot in the presence of AcOH (1.0 eq.) in EtOH (3 mL) at 70 °C for 3 h (Table 1, entry 1). The reaction gave 1-benzyl-2-phenyl-5-((1R,2S,3R)-1,2,3,4-tetrahydroxybutyl)-1H-pyrrole-3-carbonitrile 1a (N-substituted 2,3,5-functionalized 3-cyanopyrrole) in 77% yield (Table 1, entry 1). Under these conditions, complete selectivity was obtained and N-substituted 2,3,5-functionalized 3-cyanopyrrole 1a was the only product recovered. Attempts to increase the yield of 1a by increasing the temperature or reaction time were unsuccessful and led to more decomposition products indicated by a darkening of the reaction mixture. Next, several acidic and basic catalysts were examined. Except for ZnCl2, which gave 1a in 46% yield, other acids, including p-toluenesulfonic acid (PTSA) and camphor sulfonic acid (CSA), and bases including triethylamine (Et3N), sodium hydroxide (NaOH), potassium carbonate (K2CO3) and sodium methoxide (NaOMe) did not give the desired product, and the starting materials were recovered back (Table 1, entry 2–8). Changing EtOH solvent to acetonitrile (ACN) or dimethylformamide (DMF) did not improve the yield of 1a (Table 1, entry 1 vs. 9 vs. 10). Further, attempts to reduce the equivalents of AcOH from 1 to 0.5 reduced the yield of 1a dramatically (Table 1, entry 1 vs. 11). Attempts to reduce reaction temperature to 40 °C gave 1a in trace amount (Table 1, entry 12). Therefore, we concluded the optimum conditions to be 1 equivalent of AcOH in EtOH at 70 °C for 3 h (Table 1, entry 1).
Table 1.
Optimization of the reaction conditions for the three-component synthesis of 3-cyanopyrrole 1a.

| Entry | Catalyst (eq.) | Solvent | Temp. | Yield (%) [a] |
|---|---|---|---|---|
| 1 | AcOH (1.0) | EtOH | 70 ° C | 77 |
| 2 | ZnCl2 (1.0) | EtOH | 70 °C | 46 |
| 3 | PTSA (1.0) | EtOH | 70 °C | ND [b] |
| 4 | CSA (1.0) | EtOH | 70 °C | ND |
| 5 | Et3N (1.0) | EtOH | 70 °C | Trace |
| 6 | NaOH (1.0) | EtOH | 70 °C | ND |
| 7 | K2CO3 (1.0) | EtOH | 70 °C | ND |
| 8 | NaOMe (1.0) | EtOH | 70 °C | ND |
| 9 | AcOH (1.0) | ACN | 70 °C | 69 |
| 10 | AcOH (1.0) | DMF | 70 °C | 76 |
| 11 | AcOH (0.5) | EtOH | 70 °C | 58 |
| 12 | AcOH (1.0) | EtOH | 40 °C | Trace |
[a] Isolated yields. All reactions were conducted at 70 °C for 3 h using 3 mL of the solvent. [b] ND: Not detected.
2.2. Substrate Scope
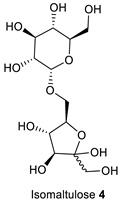
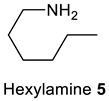
Initially, the scope of the reaction was investigated by reacting different ketoses, including D-(+)-fructose 1 and isomaltulose (also known as palatinose) 4 with primary amines 3 and 5–12 and oxoacetonitriles 2 and 13, (Table 2) under the optimized condition (Table 1, entry 1). The selected ketoses were representative of mono- and disaccharides, the amines were representative of primary, secondary, unsaturated, aliphatic, aromatic, and heterocyclic amines, while the oxoacetonitriles were representative of aliphatic and aromatic nitriles. Ketoses 1 and 4 reacted selectively to give the N-substituted 2,3,5-functionalized 3-cyanopyrroles 1a–k and 4a–c in 55–86% isolated yields. In particular, 3-cyanopyrroles 4a–c are interesting structures since they combine both a pyrrole moiety and a sugar moiety and are water-soluble [20]. The yield of the 3-cyanopyrroles 1a–k and 4a–c was affected by the type of sugar and the type of the primary amine. Hence, D-(+)-fructose/amines provide higher yields than isomaltulose/amines (Table 2, entries 1–6 vs. 12–14), and aliphatic amines provided higher yields than aromatic amines due to their higher nucleophilicity (Table 2, entries 1–6 vs. 7–9). All the 3-cyanopyrroles 1a–k and 4a–c were purified by column chromatography using a mixture of MeOH/DCM (5–10% MeOH/DCM for 1a–k, and 15–20% MeOH/DCM for 4a–c). The high polarity of the 3-cyanopyrroles is attributed to the extensive hydrogen bonding in the polyhydroxyalkyl chains.
Table 2.
Substrate scope of the AcOH-catalyzed three-component reaction between ketoses 1, and 4, primary amines 3 and 5–12, and oxoacetonitriles 2 and 13 for the selective synthesis of 3-cyanopyrroles 1a–k and 4a–c [a].

| No. | Ketose | Oxoacetonitrile | Amine | 3-Cyanopyrrole | Yield [b] |
|---|---|---|---|---|---|
| 1 |

|

|

|

|
77 |
| 2 |

|

|

|

|
86 |
| 3 |

|

|

|

|
84 |
| 4 |

|

|

|

|
73 |
| 5 |
|
|

|

|
75 |
| 6 |

|

|

|

|
68 |
| 7 |

|

|

|
|
72 |
| 8 |

|

|

|

|
61 |
| 9 |

|

|

|

|
60 |
| 10 |

|

|

|

|
71 |
| 11 |
|

|

|

|
67 |
| 12 |

|

|

|

|
68 |
| 13 |

|

|

|

|
55 |
| 14 |
|

|
|

|
57 |
[a] A mixture of ketoses 1 or 4 (1.0 mmol), oxoacetonitriles 2 or 13 (1.0 mmol), primary amines 3 or 5–12 (1.1 mmol) and AcOH (1.0 mmol) were stirred in EtOH (3 mL) at 70 °C for 3 h. [b] Isolated yields.
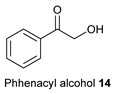
Next, the reactivity of phenacyl alcohols 14–17 as the α-hydroxyketone was tested under the optimum conditions (Table 1, entry 1) to further extend the scope of this reaction and examine the selectivity. Additionally, phenacyl alcohols would also give needed diversity to the substituents on the 3-cyanopyrrole core. The reaction between phenacyl alcohols 14–17 [21] oxoacetonitriles 2, 13, and 18–20, and primary amines 3, 5, 6, 10, 11, and 21 selectively gave the desired N-substituted 2,3,5-functionalized 3-cyanopyrroles 14a–e, 15a, 16a–b, and 17a–b in excellent 70–90% isolated yields (Table 3). Again, aromatic amines generally gave lower yields than aliphatic amines due to their lower nucleophilicity. 3-Cyanopyrroles 14a–e, 15a, 16a-b, and 17a–b were purified using silica gel column chromatography using a mixture of 5–35% EtOAc/Hexane. These 3-cyanopyrroles showed the expected lower polarity on silica gel TLC and column chromatography compared to 3-cyanopyrroles 1a–k and 4a–c due to lower hydrogen bonding.
Table 3.
The substrate scope of the AcOH-catalyzed three-component reaction between phenacyl alcohols 14–17, oxoacetonitriles 2, 13, and 18–20, and primary amines 3, 5, 6, 10, 11, and 21 for the selective synthesis of 3-cyanopyrroles 14a–e, 15a, 16a–b, and 17a–b [a].
| No. | α-Hydroxyketon | Oxoacetonitrile | Amine | 3-Cyanopyrrole | Yield [b] |
|---|---|---|---|---|---|
| 1 |
|

|

|
|
90 |
| 2 |

|

|

|

|
88 |
| 3 |

|
|

|

|
86 |
| 4 |

|

|

|

|
80 |
| 5 |

|

|

|

|
77 |
| 6 |

|

|

|

|
74 |
| 7 |

|

|

|

|
82 |
| 8 |

|

|

|

|
70 |
| 9 |

|

|

|

|
84 |
| 10 |

|

|

|
|
70 |
[a] A mixture of phenacyl alcohols 14–17 (1.0 mmol), oxoacetonitriles 2, 13 and 18–20 (1.0 mmol), and primary amines 3, 5, 6, 10, 11 and 21 (1.1 mmol) were stirred in EtOH (3 mL) at 70 °C for 3 h. [b] Isolated yields.
The structures of 3-cyanopyrroles 1c and 14c and the substitution pattern on the pyrrole ring were unambiguously confirmed by single-crystal X-ray diffraction analysis. The crystal structures of 3-cyanopyrroles 1c (CCDC 2154839) and 14c (CCDC 2154829) (Figure 3) combined with the NMR data (see Supplementary Materials) also confirmed the structures of pyrroles 14a–e, 15a, 16a–b, and 17a–b.
Figure 3.

Single-crystal XRD of 3-cyanopyrroles 1c and 14c (displacement ellipsoids are drawn at the 50% probability level).
2.3. Large-Scale Reactions
To examine the practicality of this one-step synthesis, the reactions were conducted using 8–10 mmol of the α-hydroxyketones. D-(+)-fructose 1 (1.80 g, 10 mmol), benzoylacetonitrile 2, and benzylamine 3 reacted to give 3-cyanopyrrole 1c in 78% isolated yield (Scheme 1, (i)). Similarly, isomaltulose 4 (3.42 g, 10 mmol), benzoylacetonitrile 2, and hexylamine 5 reacted to give 3-cyanopyrrole 4a in 66% isolated yield (Scheme 1, (ii)). Additionally, the reaction between 1-(4-fluorophenyl)-2-hydroxyethan-1-one 16 (1.23 g, 8 mmol), 3-oxobutanenitrile 13 and aniline 11 gave 3-cyanopyrrole 16b in 64% isolated yield (Scheme 1, (iii)). All large-scale reactions worked smoothly and selectively to give the desired 3-cyanopyrroles, which demonstrated the potential of this route even on a larger scale.
Scheme 1.

Large-scale reactions for the synthesis of 3-cyanopyrroles 1c, 4a, and 16b.
2.4. Structural Modification of the Synthesized Pyrroles
The polyhydroxyalkyl chains of 3-cyanopyrroles 1a–k are useful handles for easy modifications into other functional groups. For example, the polyhydroxyalkyl chain of pyrrole 1c was oxidized effectively using sodium periodate to an aldehyde moiety, thus generating 5-formyl 3-cyanopyrroles 22 in 98% yield (Scheme 2) [22,23]. Further, the reduction of the formyl group of 22 using sodium borohydride gave 5-hydroxymethyl 3-cyanopyrroles 23 in 95% yield (Scheme 2) [24]. Both the aldehyde and hydroxyl groups can be used for homologation reactions or transformations to other functional groups. The 5-formyl and 5-hydroxymethyl groups are important functional substitutions that enable further modifications of 3-cyanopyrroles to become bioactive molecules or natural products [25,26].
Scheme 2.

Modification of the polyhydroxyalkyl chain of 3-cyanopyrroles 1c to 3-cyanopyrroles 22 and 23.
2.5. Mechanistic Considerations
Plausible reaction pathways are shown in Figure 4. In path A, the AcOH-catalyzed reaction between the α-hydroxyketones and amines gives the imine intermediate I which tautomerizes to intermediate II. The reaction of II with oxoacetonitriles gives enaminone intermediate III, which upon cyclization and dehydration, gives intermediate IV. Finally, the aromatization of IV gives the product (Figure 4, path A) [15,27]. In path B, oxoacetonitriles react first with primary amines to give the enamine intermediate V, which condenses with the α-hydroxyketones to give VI. Intermediate VI tautomerizes to VII, which upon intermolecular cyclization and loss of water gives VIII. Aromatization of VIII then gives the product (Figure 4, path B). In path C, the condensation between oxoacetonitriles and α-hydroxyketones gives intermediate IX, which upon condensation with the primary amines gives intermediate X, which isomerizes to intermediate XI. The product was formed from intermediate XI after intramolecular cyclization, dehydration, and aromatization (Figure 4, path C).
Figure 4.

Proposed mechanism (paths A, B and C) for the formation of functionalized 3-cyanopyrroles.
In all three pathways, the reaction proceeded with very high atom efficiency. Water was the only molecule lost during this three-component reaction.
3. Materials and Methods
All chemicals and AR grade solvents were obtained from Sigma-Aldrich (Saint Louis, MO, USA), Merck (Lebanon, NJ, USA) or Alfa Aesar (Tewksbury, MA, USA) and were used as received without further purification. IR spectra were recorded using Bruker MPA FT-IR machine (Karlsruhe, Germany). 1H NMR spectra were recorded at 300 MHz on a Bruker Avance DPX 300 or at 400 MHz Bruker Avance III 400 (BBFO 400) (Karlsruhe, Germany). 13C NMR spectra were recorded at 75.47 MHz on a Bruker Avance DPX 300 or at 101 MHz Bruker Avance III 400 (BBFO 400). Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. HRMS were measured using a hybrid Quadrupole Time-of-Flight (Q-TOF) (Waters Xevo G2-X2 MS) on Qstar XL MS/MS system (Milford, CT, USA). LCMS spectra were recorded using Agilent 6530 LCMS (Santa Clara, CA, USA). Single-crystal X-ray crystallographic analysis was done using Bruker D8 Quest (Karlsruhe, Germany). Analytical TLC was performed using Merck 60 F254 precoated silica gel plates (0.2 mm thickness) (Oakville, ON, Canada). The plates were visualized using UV radiation (254 nm) or stained in ceric ammonium sulfate solution with heating to detect the reaction spots. Flash chromatography was performed using Merck silica gel 60 (230–400 mesh) (Oakville, ON, Canada). The general procedure of phenacyl alcohols preparation, synthesis of 3-cyanopyrroles 1a–k, 4a–c, 14a–e, 15a, 16a–b, 17a–b and compounds 22 and 23 are found below.
3.1. General Procedure—Preparation of Substituted Phenacyl Alcohols 14–17
A mixture of phenacyl bromide (20 mmol) and sodium formate (16 mmol) was stirred in ethanol/water (30 mL, EtOH:H2O = 9:1) at 90 °C for 12 hours. Upon completion of the reaction (TLC), the mixture was cooled to room temperature, and ethanol was evaporated under reduced pressure. The resulting concentrated mixture was diluted with water (8 mL) and extracted using ethyl acetate (10 mL × 3 times). The organic layers were dried over Mg2SO4, filtered, and evaporated to dryness under reduced pressure. The residual compound was purified using column chromatography using an EtOAc/Hexane mixture (1:4). This general procedure was used to prepare phenacyl alcohols 14–17 (Scheme 3).
Scheme 3.

Preparation of Substituted Phenacyl Alcohols 14–17.
3.2. General Procedure—Synthesis of 3-Cyanopyrroles 1a–k, 4a–c, 14a–e, 15a, 16a–b, 17a–b
AcOH (1.0 eq.) was added to a stirred solution of the α-hydroxyketones 1, 4, or 14-17 (1.0 eq.), oxoacetonitriles 2, 13, or 18–20 (1.0 eq.), and primary amines 3, 5–12, or 21 (1.1 eq.) in EtOH (3 mL) at room temperature. The reaction mixture was then heated at 70 °C for 3 h. Upon completion (TLC), the reaction mixture was evaporated to dryness under vacuum to produce the crude product as a foam. The foam was purified using silica gel column chromatography using eluent MeOH/CH2Cl2 (5:95 for D-(+)-fructose and 20:80 for isomaltulose) or EtOAc/Hexane (5–35:95–65) as eluants for phenacyl alcohols 14–17 to produce pure 3-cyanopyrroles. All reactions were conducted using 1.0 mmol of the substrates 1, 4, and 14–17. This general procedure was used to prepare 3-cyanopyrroles 1a–k, 4a–c, 14a–e, 15a, 16a–b, and 17a–b.
1-Benzyl-2-Phenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Arbonitrile (1a)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), benzylamine 3 (120 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1a (291 mg, 77%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3416, 2221, 1637, 1398, 1077 cm−1. 1H NMR (300 MHz, MeOD) δ 7.43–7.38 (m, 3H), 7.34 (d, J = 5.2 Hz, 2H), 7.29–7.20 (m, 3H), 6.87 (d, J = 7.5 Hz, 2H), 6.79 (s, 1H), 5.40 (d, J = 12.0 Hz, 1H), 5.24 (d, J = 12.0 Hz, 1H), 4.88 (s, 1H), 3.79 – 3.69 (m, 3H), 3.61 (dd, J = 10.0, 4.3 Hz, 1H). 13C NMR (75 MHz, MeOD) δ 143.8, 138.9, 137.6, 131.0, 130.9, 130.4, 129.9, 129.9, 128.5, 126.9, 118.4, 112.0, 92.8, 74.4, 72.9, 66.1, 64.8, 49.4. DEPT135 13C NMR (75 MHz, MeOD) δ 130.9, 130.4, 129.9 (2 × CH signals), 128.5, 126.9, 112.0, 74.4, 72.9, 66.1, 64.8, 49.4. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H23N2O4+ 379.1658; found 379.1644.
1-Hexyl-2-Phenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1b)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), hexylamine 5 (144 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1b (320 mg, 86%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3293, 2957, 2928, 2220, 1447, 1090 cm−1. 1H NMR (300 MHz, MeOD) δ 7.45–7.37 (m, 3H), 7.35–7.29 (m, 2H), 6.56 (s, 1H), 4.95 (s, 1H), 4.06–3.83 (m, 2H), 3.75–3.63 (m, 3H), 3.58–3.49 (m, 1H), 1.48–1.34 (m, 2H), 1.02-0.95 (m, 6H), 0.68 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, MeOD) δ 144.5, 138.5, 133.1, 132.5, 131.7, 131.5, 119.9, 113.1, 93.8, 76.2, 74.4, 67.2, 66.3, 47.6, 33.6, 33.4, 28.6, 24.9, 15.7. DEPT135 13C NMR (75 MHz, MeOD) δ 132.5, 131.7, 131.5, 113.1, 76.2, 74.4, 67.2, 66.3, 47.6, 33.6, 33.4, 28.6, 24.9, 15.7. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C21H28N2O4Na+ 395.1947; found 395.2000.
1-Phenethyl-2-Phenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1c)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), 2-phenylethylamine 6 (138 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1c (329 mg, 84%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3427, 3341, 2959, 2224, 1434, 1077 cm−1. 1H NMR (300 MHz, MeOD) δ 7.37–7.33 (m, 3H), 7.17–7.1 (m, 2H), 7.05-7.00 (m, 3H), 6.70 (dd, J = 6.6, 2.9 Hz, 2H), 6.58 (s, 1H), 4.99 (d, J = 1.4 Hz, 1H), 4.16–4.07 (m, 2H), 3.77–3.64 (m, 3H), 3.55 (dd, J = 10.2, 4.5 Hz, 1H), 2.69 (t, J = 7.5 Hz, 2H). 13C NMR (75 MHz, MeOD) δ 144.9, 140.5, 138.5, 132.8, 132.4, 131.6, 131.4, 131.3, 131.1, 129.2, 119.9, 113.2, 93.9, 76.3, 74.5, 67.3, 66.3, 49.3, 39.6. DEPT135 13C NMR (75 MHz, MeOD) δ 132.4, 131.6, 131.4, 131.3, 131.1, 129.2, 113.2, 76.3, 74.5, 67.3, 66.3, 49.3, 39.6. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C23H25N2O4+ 393.1814; found 393.1834.
1-(2-(1H-Imidazol-1-yl)ethyl)-2-Phenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1d)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), 2-(1H-imidazol-1-yl)ethanamine 7 (107 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1d (279 mg, 73%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3407, 2220, 1651, 1400, 1077 cm−1. 1H NMR (300 MHz, MeOD) δ 7.32 (dd, J = 6.2, 2.7 Hz, 3H), 7.05–6.95 (m, 3H), 6.69 (s, 1H), 6.61 (s, 1H), 6.44 (s, 1H), 4.88 (d, J = 2.5 Hz, 1H), 4.48–4.27 (m, 2H), 4.24–4.04 (m, 2H), 3.75–3.62 (m, 3H), 3.55 (dd, J = 10.8, 5.1 Hz, 1H). 13C NMR (75 MHz, MeOD) δ 145.4, 139.6, 138.7, 132.3, 131.8, 131.8, 131.4, 130.7, 122.2, 119.4, 113.8, 94.5, 76.4, 74.6, 67.4, 66.1, 49.3, 48.4. DEPT135 13C NMR (75 MHz, MeOD) δ 139.6, 132.3, 131.8, 131.4, 130.7, 122.2, 113.8, 76.4, 74.6, 67.4, 66.1, 49.3, 48.4. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H22BrN2O3+ 441.0814; found 441.0795.
2-Phenyl-1-(Prop-2-yn-1-yl)-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1e)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), propargylamine 8 (70 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1e (245 mg, 75%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3290, 2219, 1699, 1400, 1089 cm−1. 1H NMR (300 MHz, MeOD) δ 7.49–7.39 (m, 5H), 6.58 (d, J = 0.4 Hz, 1H), 5.17–5.09 (m, 1H), 4.83 (dd, J = 18.2, 2.5 Hz, 1H), 4.60 (dd, J = 18.2, 2.5 Hz, 1H), 3.79–3.65 (m, 3H), 3.57 (dd, J = 10.7, 5.4 Hz, 1H), 2.78 (t, J = 2.5 Hz, 1H). 13C NMR (75 MHz, MeOD) δ 144.6, 138.5, 132.4, 132.0, 131.5, 119.6, 131.5, 94.1, 81.1, 76.9, 75.9, 75.5, 67.6, 66.3, 37.4. DEPT135 13C NMR (75 MHz, MeOD) δ 132.4, 132.0, 131.5, 113.5, 75.9, 74.5, 67.5, 66.3, 37.4. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H22BrN2O3+ 441.0814; found 441.0795.
1-(Naphthalen-1-ylmethyl)-2-Phenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1f)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), 1-naphthylmethylamine 9 (162 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1f (291 mg, 68%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3400, 2922, 2221, 1653, 1479, 1076 cm−1. 1H NMR (400 MHz, MeOD) δ 7.91–7.86 (m, 2H), 7.79 (d, J = 8.3 Hz, 1H), 7.53–7.48 (m, 2H), 7.42 – 7.38 (m, 1H), 7.34 (dd, J = 7.6, 1.8 Hz, 2H), 7.31 – 7.24 (m, 3H), 6.91 (s, 1H), 6.63 (d, J = 7.0 Hz, 1H), 5.79 (dd, J = 84.3, 17.7 Hz, 2H), 4.81 (d, J = 1.9 Hz, 1H), 3.81 (dd, J = 7.8, 2.1 Hz, 1H), 3.72 (dd, J = 10.9, 3.2 Hz, 1H), 3.67–3.62 (m, 1H), 3.58 (dd, J = 10.9, 5.7 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 142.5, 136.4, 133.6, 133.2, 129.7, 129.3, 129.2, 129.0, 128.4 (2 × CH), 127.7, 126.2, 125.8, 125.2, 122.1, 121.9, 117.0, 110.8, 91.5, 73.1, 71.4, 64.7, 63.4, 46.3. DEPT135 13C NMR (101 MHz, MeOD) δ 129.2, 129.0, 128.4 (2 × CH), 127.7, 126.2, 125.8, 125.2, 122.1, 121.9, 110.8, 73.1, 71.4, 64.7, 63.4, 46.3. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C26H24N2O4Na+ 451.1634; found 451.1610.
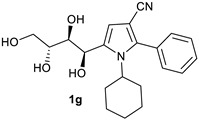
1. -Cyclohexyl-2-Phenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1g)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), cyclohexylamine 10 (127 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1f (266 mg, 72%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3425, 2931, 2224, 1439, 1077 cm−1. 1H NMR (300 MHz, MeOD) δ 7.41–7.39 (m, 3H), 7.32–7.22 (m, 2H), 6.61 (s, 1H), 5.14 (s, 1H), 4.09 (s, 1H), 3.76–3.52 (m, 4H), 1.77 (s, 4H), 1.65 (d, J = 13.0 Hz, 2H), 1.48 (d, J = 12.4 Hz, 1H), 1.20–1.03 (m, 2H), 0.93 (s, 1H). 13C NMR (75 MHz, MeOD) δ 144.5, 138.9, 134.2, 133.2, 131.9, 131.1, 119.8, 113.7, 94.9, 76.5, 74.5, 67.0, 66.4, 61.5, 36.1, 35.9, 28.9 (2 × CH2), 27.8. DEPT135 13C NMR (75 MHz, MeOD) δ 133.2, 131.9, 131.1, 76.5, 74.5, 67.0, 66.4, 61.5, 36.1, 35.9, 28.9 (2xCH2), 27.8. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C21H27N2O4+ 371.1971; found 371.1972.
1,2-Diphenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1h)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), aniline 11 (100 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1g (222 mg, 61%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3408, 2219, 1635, 1497, 1095 cm−1. 1H NMR (300 MHz, MeOD) δ 7.30–7.25 (br s, 4H), 7.17–7.12 (m, 3H), 7.11–7.07 (m, 2H), 7.02 (s, 1H), 6.77 (d, J = 0.5 Hz, 1H), 4.57 (d, J = 2.0 Hz, 1H), 3.56–3.46 (m, 3H), 3.41 (dt, J = 5.5, 2.4 Hz, 1H). 13C NMR (75 MHz, MeOD) δ 144.4, 140.2, 139.8, 132.4, 132.3, 131.8, 131.7, 131.5, 131.0, 130.8, 119.8, 113.6, 94.6, 76.4, 74.4, 67.3, 66.1. DEPT135 13C NMR (75 MHz, MeOD) δ 132.3, 31.8, 131.5, 131.0, 130.8, 113.6, 76.4, 74.4, 67.3, 66.1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C21H21N2O4+ 365.1501; found 365.1494.
1-(4-Methoxyphenyl)-2-Phenyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1i)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), p-anisidine 12 (126 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1h (236 mg, 60%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3402, 2942, 2223, 1514, 1400, 1251, 1078 cm−1. 1H NMR (300 MHz, MeOD) δ 7.23–7.08 (m, 6H), 6.98–6.74 (m, 3H), 6.73 (s, 1H), 4.55 (d, J = 1.9 Hz, 1H), 3.68 (s, 3H), 3.59–3.47 (m, 3H), 3.45–3.36 (m, 1H). 13C NMR (75 MHz, MeOD) δ 162.8, 144.6, 140.4, 132.7, 132.6, 132.3, 131.0, 130.8, 119.9, 116.8 (CH), 116.8 (Cq), 113.4, 94.3, 76.5, 74.5, 67.4, 66.1, 57.5. DEPT135 13C NMR (75 MHz, MeOD) δ 132.7, 132.3, 131.0, 130.8, 116.8, 113.4, 76.4, 74.4, 67.3, 66.1, 57.5. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C22H22N2O5Na+ 417.1426; found 417.1478.
2-Methyl-1-Phenethyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1j)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), 3-oxobutanenitrile 13 (83 mg, 1.0 mmol), 2-phenylethylamine 6 (138 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1i (234 mg, 71%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3350, 2944, 2218, 1638, 1429, 1077 cm−1. 1H NMR (400 MHz, MeOD) δ 7.33–7.23 (m, 3H), 7.14 (d, J = 7.6 Hz, 2H), 6.51 (s, 1H), 5.00 (d, J = 1.9 Hz, 1H), 4.32–4.12 (m, 2H), 3.86–3.70 (m, 3H), 3.64 (dd, J = 11.0, 5.1 Hz, 1H), 3.03 (t, J = 7.3 Hz, 2H), 2.04 (s, 3H). 13C NMR (101 MHz, MeOD) δ 138.6, 138.1, 134.3, 128.7, 128.3, 126.5, 117.2, 108.9, 89.2, 73.4, 71.7, 64.2, 63.3, 45.9, 36.6, 9.8. DEPT135 13C NMR (101 MHz, MeOD) δ 128.7, 128.3, 126.5, 108.9, 73.4, 71.7, 64.2, 63.3, 45.9, 36.6, 9.8. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C18H23N2O4+ 331.1658; found 331.1620.
1-Benzyl-2-Methyl-5-((1R,2S,3R)-1,2,3,4-Tetrahydroxybutyl)-1H-Pyrrole-3-Carbonitrile (1k)
Obtained by general procedure 2 using D-(+)-fructose 1 (180 mg, 1.0 mmol), 3-oxobutanenitrile 13 (83 mg, 1.0 mmol), benzylamine 3 (120 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 1j (212 mg, 67%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3411, 2217 1636, 1428, 1078 cm−1. 1H NMR (400 MHz, MeOD) δ 7.36–7.24 (m, 3H), 6.98 (d, J = 7.4 Hz, 2H), 6.56 (s, 1H), 5.41 (d, J = 16.0 Hz, 1H), 5.27 (d, J = 16.0 Hz, 1H), 4.88 (s, 1H), 3.76–3.64 (m, 3H), 3.58 (dd, J = 11.0, 5.5 Hz, 1H), 2.25 (s, 3H). 13C NMR (101 MHz, MeOD) δ 138.8, 137.1, 135.0, 128.5, 127.1, 125.5, 117.0, 108.8, 89.9, 73.1, 71.4, 64.5, 63.3, 47.4, 10.0. DEPT135 13C NMR (101 MHz, MeOD) δ 128.5, 127.1, 125.5, 108.8, 73.1, 71.4, 64.5, 63.3, 47.4, 10.0. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C17H21N2O4+ 317.1501; found 317.1503.
1-Hexyl-2-Phenyl-5-((1R,2S,3R)-1,2,3-Trihydroxy-4-(((2S,3R,4S,5S,6R)-3,4,5-Trihydroxy-6-(Hydroxymethyl)tetrahydro-2H-Pyran-2-yl)oxy)butyl)-1H-Pyrrole-3-Carbonitrile (4a)
Obtained by general procedure 2 using isomaltulose 4 (342 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), hexylamine 5 (144 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 4a (363 mg, 68%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3391, 2928, 2221, 1653, 1457, 1078 cm−1. 1H NMR (300 MHz, MeOD) δ 7.45–7.37 (m, 3H), 7.33 (dd, J = 7.6, 1.8 Hz, 2H), 6.58 (s, 1H), 5.01 (s, 1H), 4.74 (d, J = 3.7 Hz, 1H), 4.04–3.76 (m, 4H), 3.71 (dd, J = 13.4, 3.2 Hz, 2H), 3.61–3.49 (m, 4H), 3.33 (dd, J = 9.6, 3.6 Hz, 1H), 3.27–3.21 (m, 1H), 1.42 (d, J = 5.9 Hz, 2H), 1.1–0.9 (m, 6H), 0.69 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, MeOD) δ 144.5, 138.8, 133.1, 132.5, 131.7, 131.5, 120.0, 113.2, 101.7, 93.9, 76.7, 75.4, 75.2, 75.1, 73.2, 72.8, 71.6, 67.0, 64.1, 47.6, 33.6, 33.4, 28.6, 24.9, 15.7. DEPT135 13C NMR (75 MHz, MeOD) δ 132.5, 131.7, 131.5, 113.2, 101.7, 76.7, 75.4, 75.2, 75.1, 73.1, 72.8, 71.6, 67.0, 64.1, 47.6, 33.6, 33.4, 28.6, 24.9, 15.7. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C27H38N2O9Na+ 557.2475; found 557.2474.
1-Benzyl-2-Phenyl-5-((1R,2S,3R)-1,2,3-Trihydroxy-4-(((2S,3R,4S,5S,6R)-3,4,5-Trihydroxy-6-(Hydroxymethyl)tetrahydro-2H-Pyran-2-yl)oxy)butyl)-1H-Pyrrole-3-Carbonitrile (4b)
Obtained by general procedure 2 using isomaltulose 4 (342 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), benzylamine 3 (120 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 4b (297 mg, 55%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3402, 2938, 2221, 1636, 1400, 1077, 1028 cm−1. 1H NMR (300 MHz, MeOD) δ 7.30 (dd, J = 6.6, 2.7 Hz, 3H), 7.24–7.20 (m, 2H), 7.13 (dd, J = 10.5, 7.1 Hz, 3H), 6.76 (d, J = 6.7 Hz, 2H), 6.68 (s, 1H), 5.29 (d, J = 16.0 Hz, 1H), 5.11 (d, J = 17.0 Hz, 1H), 4.82 (s, 1H), 4.70 (d, J = 3.6 Hz, 1H), 3.94–3.86 (m, 1H), 3.72–3.68 (m, 3H), 3.60–3.56 (m, 3H), 3.46–3.38 (m, 1H), 3.32 (dd, J = 9.7, 3.7 Hz, 1H), 3.25 (d, J = 9.1 Hz, 1H). 13C NMR (75 MHz, MeOD) δ 145.2, 140.4, 139.3, 132.4, 132.4, 131.8, 131.4 (CH), 131.4 (Cq), 131.3, 130.0, 128.3, 119.9, 113.5, 101.7, 94.2, 76.7, 75.2, 75.1, 73.2, 72.6, 71.4, 67.4, 64.1, 50.9. DEPT135 13C NMR (75 MHz, MeOD) δ 132.4, 131.8, 131.4, 130.0, 128.3, 113.5, 101.7, 76.7, 75.2, 75.0, 73.1, 72.6, 71.4, 67.4, 64.0, 50.9. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C28H32N2O9Na+ 563.2006; found 563.2002.
1-Hexyl-2-Methyl-5-((1R,2S,3R)-1,2,3-Trihydroxy-4-(((2S,3R,4S,5S,6R)-3,4,5-Trihydroxy-6-(hydroxymethyl)tetrahydro-2H-Pyran-2-yl)oxy)butyl)-1H-Pyrrole-3-Carbonitrile (4c)
Obtained by general procedure 2 using isomaltulose 4 (342 mg, 1.0 mmol), 3-oxobutanenitrile 13 (83 mg, 1.0 mmol), hexylamine 5 (144 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 4c (269 mg, 57%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 3402, 2931, 2217, 1651, 1429, 1078, 1027 cm−1. 1H NMR (400 MHz, MeOD) δ 6.50 (s, 1H), 5.03 (s, 1H), 4.84 (d, J = 3.7 Hz, 1H), 4.11–3.99 (m, 2H), 3.98–3.89 (m, 1H), 3.87–3.76 (m, 3H), 3.71–3.60 (m, 4H), 3.44 (dd, J = 9.6, 3.7 Hz, 1H), 3.38–3.33 (m, 1H), 2.39 (s, 3H), 1.79–1.67 (m, 2H), 1.38 (s, 6H), 0.94 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 137.8, 134.4, 117.3, 108.7, 98.8, 89.3, 73.8, 72.5, 72.3, 72.1, 70.3, 69.9, 68.7, 63.9, 61.2, 44.4, 31.2, 30.5, 26.2, 22.3, 13.0, 10.0. DEPT135 13C NMR (101 MHz, MeOD) δ 108.7, 98.8, 73.8, 72.5, 72.3, 72.1, 70.2, 69.8, 68.7, 63.9, 61.2, 44.4, 31.2, 30.5, 26.2, 22.3, 13.0, 10.0. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C22H36N2O9Na+ 495.2319; found 495.2320.
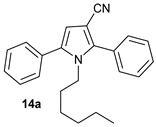
1-Hexyl-2,5-Diphenyl-1H-Pyrrole-3-Carbonitrile (14a)
Obtained by general procedure 2 using phenacyl alcohol 14 (136 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), hexylamine 5 (144 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 14a (295 mg, 90%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2956, 2929, 2857, 2220, 1604, 1474, 1346 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.53–7.40 (m, 10H), 6.49 (s, 1H), 4.06–3.94 (m, 2H), 1.25–1.13 (m, 2H), 1.07–0.96 (m, 2H), 0.92–0.79 (m, 4H), 0.71 (t, J = 7.2 Hz, 3H). 13C NMR (75 MHz, MeOD) δ 142.4, 136.2, 132.1, 130.1, 129.7, 129.3, 129.0 (2xCH), 128.8, 128.3, 117.4, 111.8, 92.7, 45.6, 30.7, 30.1, 25.6, 22.1, 13.8. DEPT135 13C NMR (75 MHz, MeOD) δ 129.7, 129.3, 129.0, 129.0, 128.8, 128.3, 111.8, 45.6, 30.7, 301., 25.6, 22.1, 13.8. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C23H24N2Na+ 351.1837; found 351.1845.
1-Benzyl-2,5-Diphenyl-1H-Pyrrole-3-Carbonitrile (14b)
Obtained by general procedure 2 using phenacyl alcohol 14 (136 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), benzylamine 3 (120 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 14b (294 mg, 88%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2220, 1473, 1452, 1415, 1346 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.42 (br s, 5H), 7.39–7.30 (m, 5H), 7.21–7.11 (m, 3H), 6.72–6.54 (m, 3H), 5.19 (s, 2H). 13C NMR (75 MHz, MeOD) δ 142.8, 137.4, 136.7, 131.6, 129.7 (CH), 129.7 (Cq), 129.5, 129.2, 128.9, 128.6, 128.5, 128.4, 127.4, 125.9, 117.2, 112.0, 93.3, 49.1. DEPT135 13C NMR (75 MHz, MeOD) δ 129.7, 129.5, 129.2, 128.9, 128.6, 128.5, 128.4, 127.4, 125.9, 112.0, 49.1. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C24H18N2Na+ 357.1368; found 357.1360.
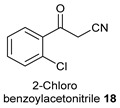
2-(2-Chlorophenyl)-1-Cyclohexyl-5-Phenyl-1H-Pyrrole-3-Carbonitrile (14c)
Obtained by general procedure 2 using phenacyl alcohol 14 (136 mg, 1.0 mmol), 2-chloro benzoylacetonitrile 18 (152 mg, 1.0 mmol), cyclohexylamine 10 (127 µL, 1.0 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 14c (310 mg, 86%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2943, 2928, 2847, 2220, 1458, 1431, 1338 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.0 Hz, 1H), 7.49-7.40 (m, 8H), 6.44 (s, 1H), 3.91–3.82 (m, 1H), 1.95 – 1.83 (m, 2H), 1.63 (d, J = 14.0 Hz, 2H), 1.56-1.34 (m, 3H), 1.09 – 0.93 (m, 2H), 0.77-0.71 (m, 1H). 13C NMR (101 MHz, MeOD) δ 138.0, 135.9, 135.8, 133.4, 132.7, 131.1, 130.4, 129.9, 128.5, 128.3, 126.6, 116.5, 111.9, 94.4, 59.6, 33.9, 32.8, 26.2 (2xCH2), 25.0. DEPT135 13C NMR (101 MHz, MeOD) δ 133.4, 131.1, 130.5, 129.9, 128.5, 128.3, 126.6, 111.8, 59.6, 33.9, 32.8, 26.2, 26.2, 25.0. HRMS (ESI-TOF) m/z: [M+Na]+ calcd for C23H21N2ClNa+ 383.1291; found 383.1295.
1,2,5-Triphenyl-1H-Pyrrole-3-Carbonitrile (14d)
Obtained by general procedure 2 using phenacyl alcohol 14 (136 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), aniline 11 (100 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 14d (256 mg, 80%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2229, 1597, 1493, 1085 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.21–7.18 (m, 5H), 7.17–7.10 (m, 6H), 7.00–6.94 (m, 2H), 6.92–6.86 (m, 2H), 6.64 (s, 1H). 13C NMR (75 MHz, MeOD) δ 141.9, 137.3, 136.1, 131.2, 129.7, 129.4, 129.1, 129.0, 128.6, 128.4, 128.3 (2xCH), 128.2, 127.6, 117.2, 112.0, 93.6. DEPT135 13C NMR (75 MHz, MeOD) δ 129.7, 129.1, 129.0, 128.6, 128.4, 128.3 (2 × CH), 128.2, 127.6, 112.4. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C23H17N2+ 321.1392; found 321.1379.
1-(4-Fluorophenyl)-2,5-Diphenyl-1H-Pyrrole-3-Carbonitrile (14e)
Obtained by general procedure 2 using phenacyl alcohol 14 (136 mg, 1.0 mmol), benzoylacetonitrile 2 (145 mg, 1.0 mmol), 4-fluoroaniline 21 (104 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 14e (260 mg, 77%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2227, 1602, 1514, 1489, 1232 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.23–7.09 (m, 8H), 6.97 (dd, J = 6.6, 2.9 Hz, 2H), 6.95-6.80 (m, 4H), 6.63 (d, J = 0.6 Hz, 1H). 13C NMR (75 MHz, MeOD) δ 162.0 (d, J = 247.5 Hz, C-F), 141.9, 136.2, 133.3, 131.0, 130.3, 130.2, 129.8, 129.2, 129.0, 128.6, 128.5, 128.3, 127.7, 117.0, 116.2 (d, J = 22.5 Hz, C-H), 112.1, 93.7. DEPT135 13C NMR (75 MHz, MeOD) δ 130.3, 130.2, 129.8, 129.0, 128.6, 128.5, 128.3, 127.8, 116.3, 116.0, 112.1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C23H16N2F+ 339.1298; found 339.1283.
1-Benzyl-2-Methyl-5-(p-tolyl)-1H-Pyrrole-3-Carbonitrile (15a)
Obtained by general procedure 2 using 2-hydroxy-1-(p-tolyl)ethan-1-one 15 (150 mg, 1.0 mmol), 3-oxobutanenitrile 13 (83 mg, 1.0 mmol), benzylamine 3 (120 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 15a (212 mg, 74%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2218, 1700, 1559, 1496, 1353, 1155 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.35–7.28 (m, 3H), 7.15 (s, 4H), 6.90 (d, J = 7.1 Hz, 2H), 6.42 (s, 1H), 5.11 (s, 2H), 2.36 (s, 3H), 2.30 (s, 3H). 13C NMR (101 MHz, MeOD) δ 138.7, 138.1, 136.9, 135.6, 129.3, 129.1, 129.0, 128.6, 127.6, 125.5, 117.3, 109.8, 92.0, 48.2, 21.2, 11.8. DEPT135 13C NMR (101 MHz, MeOD) δ 129.3, 129.1, 129.0, 127.6, 125.5, 48.2, 21.2, 11.8. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C20H19N2+ 287.1548; found 287.1542.
2-(4-Bromophenyl)-5-(4-Fluorophenyl)-1-Phenethyl-1H-Pyrrole-3-Carbonitrile (16a)
Obtained by general procedure 2 using 1-(4-fluorophenyl)-2-hydroxyethan-1-one 16 (154 mg, 1.0 mmol), 4-bromo benzoylacetonitrile 19 (224 mg, 1.0 mmol), 2-phenylethylamine 6 (138 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 16a (365 mg, 82%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2220, 1557, 1493, 1225, 1157, 1071 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.65–7.55 (m, 2H), 7.34–7.28 (m, 2H), 7.22–7.11 (m, 7H), 6.67–6.51 (m, 2H), 6.46 (s, 1H), 4.21 (t, J = 7.0 Hz, 2H), 2.44 (t, J = 7.0 Hz, 2H). 13C NMR (101 MHz, MeOD) δ 162.8 (d, J = 242.4 Hz, C-F), 141.2, 136.7, 135.4, 132.2, 131.2, 131.1 (2xCH), 128.6, 128.4, 127.8 (2xCq), 126.9, 123.6, 116.8, 116.0, 115.8, 112.2, 93.2, 47.1, 36.4. DEPT135 13C NMR (101 MHz, MeOD) δ 132.2, 131.2, 131.1, 131.1, 128.6, 128.4, 126.9, 115.9 (d, J = 20.2 Hz, C-H), 47.1, 36.4. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C25H19N2FBr+ 445.0716; found 445.0730.
5-(4-Fluorophenyl)-2-Methyl-1-Phenyl-1H-Pyrrole-3-Carbonitrile (16b)
Obtained by general procedure 2 using 1-(4-fluorophenyl)-2-hydroxyethan-1-one 16 (154 mg, 1.0 mmol), 3-oxobutanenitrile 13 (83 mg, 1.0 mmol), aniline 11 (100 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 16b (193 mg, 70%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2221, 1700, 1559, 1496, 1225, 1160 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.47–7.42 (m, 3H), 7.16–7.11 (m, 2H), 7.04–6.98 (m, 2H), 6.88 (t, J = 8.7 Hz, 2H), 6.52 (s, 1H), 2.29 (s, 3H). 13C NMR (101 MHz, MeOD) δ 162.0 (d, J = 252.5 Hz, C-F), 139.9, 137.4, 134.2, 130.1, 130.0, 129.5, 128.8, 128.1, 127.6, 117.0, 115.3 (d, J = 20.2 Hz, C-H), 110.1, 92.5, 12.4. DEPT135 13C NMR (101 MHz, MeOD) δ 130.1, 130.0, 129.5, 128.8, 128.1, 115.4, 115.2, 110.0, 12.4. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C18H14N2F+ 277.1141; found 277.1138.
1.-Benzyl-5-(4-Chlorophenyl)-2-(3-Methoxyphenyl)-1H-Pyrrole-3-Carbonitrile (17a)
Obtained by general procedure 2 using 1-(4-chlorophenyl)-2-hydroxyethan-1-one 17 (170 mg, 1.0 mmol), 3-methoxy benzoylacetonitrile 20 (175 mg, 1.0 mmol), benzylamine 3 (120 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 17a (334 mg, 84%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2221, 1603, 1557, 1487, 1287, 1093 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.38–7.30 (m, 3H), 7.24–7.20 (m, 5H), 7.03 (d, J = 7.9 Hz, 1H), 6.96 (dd, J = 8.3, 2.5 Hz, 1H), 6.91 (d, J = 1.4 Hz, 1H), 6.76–6.66 (m, 2H), 6.60 (s, 1H), 5.17 (s, 2H), 3.69 (s, 3H). 13C NMR (101 MHz, MeOD) δ 159.7, 142.9, 137.5, 135.3, 134.5, 130.6, 130.5, 130.0, 128.8, 128.7, 127.6, 125.8, 122.1, 116.9, 115.6, 114.6, 112.4, 93.5, 55.2, 49.2. DEPT135 13C NMR (101 MHz, MeOD) δ 130.6, 130.0, 128.9, 128.7, 127.6, 125.8, 122.1, 115.6, 114.6, 112.4, 55.2, 49.2. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C25H20N2OCl+ 399.1264; found 399.1274.
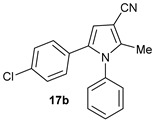
5-(4-Chlorophenyl)-2-Methyl-1-Phenyl-1H-Pyrrole-3-Carbonitrile (17b)
Obtained by general procedure 2 using 1-(4-chlorophenyl)-2-hydroxyethan-1-one 17 (170 mg, 1.0 mmol), 3-oxobutanenitrile 13 (83 mg, 1.0 mmol), aniline 11 (100 µL, 1.1 mmol), and AcOH (57 µL, 1.0 mmol). The reaction produced the desired product 17b (205 mg, 70%) as a hygroscopic pale-yellow solid. IR (KBr film) νmax = 2223, 1700, 1559, 1496, 1399, 1094 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.48–7.42 (m, 3H), 7.19–7.11 (m, 4H), 6.96 (d, J = 8.5 Hz, 2H), 6.56 (s, 1H), 2.29 (s, 3H). 13C NMR (101 MHz, MeOD) δ 140.2, 137.3, 133.9, 133.2, 129.9, 129.6, 129.4, 128.9, 128.5, 128.1, 116.9, 110.4, 92.7, 12.4. DEPT135 13C NMR (101 MHz, MeOD) δ 129.6, 129.4, 128.9, 128.5, 128.1, 110.4, 12.4. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C18H14N2Cl+ 293.0846; found 293.0843.
5-Formyl-1-Phenethyl-2-Phenyl-1H-Pyrrole-3-Carbonitrile (22)
NaIO4 (235 mg, 1.1 mmol) was slowly added to a solution of 1c (392 mg, 1.0 mmol) in MeOH (10 mL) at room temperature, and the reaction mixture was stirred at room temperature for 3 h. The reaction mixture was then concentrated under vacuum, diluted with H2O (5 mL), and extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were dried over magnesium sulfate and filtered, and the solvent was then evaporated under vacuum to produce 22 (295 mg, 98%) as a pale-yellow solid. IR (KBr film) νmax = 2228, 1674, 1457, 1404, 1363, 1142 cm−1. 1H NMR (400 MHz, CDCl3) δ 9.67 (s, 1H), 7.50-7.46 (m, 3H), 7.33 (s, 1H), 7.22–7.15 (m, 5H), 6.87 (d, J = 6.5 Hz, 2H), 4.55 (t, J = 7.2 Hz, 2H), 2.90 (t, J = 7.1 Hz, 2H). 13C NMR (101 MHz, MeOD) δ 179.4, 148.5, 137.0, 131.7, 130.2, 129.5, 129.0, 128.7, 128.7, 127.3, 127.3, 126.9, 115.2, 95.0, 48.3, 37.3. DEPT135 13C NMR (101 MHz, MeOD) δ 179.4, 130.2, 129.5, 129.0, 128.7, 128.7, 127.3, 126.9, 48.3, 37.3. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C20H17N2O+ 301.1341; found 301.1335.
5-(Hydroxymethyl)-1-Phenethyl-2-Phenyl-1H-Pyrrole-3-Carbonitrile (23)
NaBH4 (28 mg, 0.74 mmol) was added to a stirred solution of 22 (200 mg, 0.67 mmol) in MeOH (10 mL) at room temperature. Stirring was continued for 1 h (TLC), and the solvent was then evaporated under reduced vacuum. The residue was flushed via silica gel column chromatography and was eluted with a gradient of 40% EtOAc/ hexane to produce 23 (191 mg, 95% yield) as a pale-yellow solid. IR (KBr film) νmax = 3478, 2221, 1674, 1481, 1346 cm−1. 1H NMR (400 MHz, CDCl3) δ 7.51-7.37 (m, 3H), 7.41–7.35 (m, 2H), 7.22-7.19 (m, 3H), 6.84 (s, 2H), 6.45 (s, 1H), 4.51 (s, 2H), 4.27 (t, J = 7.4 Hz, 2H), 2.78 (t, J = 7.3 Hz, 2H). 13C NMR (101 MHz, MeOD) δ 142.7, 137.4, 133.3, 129.6, 129.6, 129.3, 129.0, 128.7, 128.6, 126.9, 117.0, 111.9, 92.1, 56.6, 46.6, 37.2. DEPT135 13C NMR (101 MHz, MeOD) δ 129.6, 129.3, 129.0, 128.7, 128.6, 126.9, 111.9, 56.6, 46.6, 37.2. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C20H19N2O+ 303.1497; found 303.1494.
4. Conclusions
In summary, we established an efficient and simple three-component reaction between α-hydroxyketones, oxoacetonitriles, and primary amines to selectively give densely functionalized 3-cyanopyrroles in 55–90% isolated yields. The structure and the substitution pattern of the products were unambiguously confirmed by single-crystal X-ray analysis. The practicality of this method was demonstrated on large-scale syntheses of several pyrroles. Noteworthy is the high atom economy of this reaction which makes it attractive for practical applications. Currently, we are expanding the scope of this reaction and using the process to synthesize different pyrrole-based drug candidates and intermediates such as the anti-tuberculosis drugs BM212, BM521, and BM533.
Acknowledgments
We thank Nanyang Technological University, Singapore for financial support (CoE, Start-up Grant).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27165285/s1. Copies of the 1H NMR, 13C NMR, and 2D NMR spectra of the synthesized compounds as well as single-crystal X-ray data of pyrrole 1c and 13c are available online.
Author Contributions
Conceptualization, M.X. and Z.M.A.J.; methodology, M.X. and Z.M.A.J.; validation, M.X. and Z.M.A.J.; formal analysis, M.X., Z.M.; investigation, M.X., Z.M.; data curation, M.X.; writing—original draft preparation, M.X.; writing—review and editing, Z.M.A.J., Z.M. and M.X.; supervision, Z.M.A.J.; project administration, Z.M.A.J.; funding acquisition, Z.M.A.J. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in supplementary materials.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
Funding Statement
This research was funded by the College of Engineering (Startup Grant), Nanyang Technological University, Singapore and by United Arab Emirates University (UAEU), Al-Ain (Grant no. G00003291/Fund no. 31S401/12S040/Project #852).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gholap S.S. Pyrrole: An emerging scaffold for construction of valuable therapeutic agents. Eur. J. Med. Chem. 2016;110:13–31. doi: 10.1016/j.ejmech.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 2.Trofimov B.A., Al’bina I.M., Schmidt E.Y., Sobenina L.N. Chemistry of Pyrroles. CRC Press; Boca Raton, FL, USA: 2014. [Google Scholar]
- 3.Kaur R., Rani V., Abbot V. Recent synthetic and medicinal perspectives of pyrroles: An overview. J. Pharm. Chem. Chem. Sci. 2017;1:17–32. [Google Scholar]
- 4.Battilocchio C., Poce G., Alfonso S., Porretta G.C., Consalvi S., Sautebin L., Pace S., Rossi A., Ghelardini C., Mannelli L.D.C. A class of pyrrole derivatives endowed with analgesic/anti-inflammatory activity. Bioorganic Med. Chem. 2013;21:3695–3701. doi: 10.1016/j.bmc.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Biava M., Porretta G., Manetti F. New Derivatives of BM212: A Class of Antimycobacterial Compounds Based on the Pyrrole Ring as a Scaffold. Mini Rev. Med. Chem. 2007;7:65–78. doi: 10.2174/138955707779317786. [DOI] [PubMed] [Google Scholar]
- 6.Gupton J.T. Pyrrole Natural Products with Antitumor Properties. Top. Heterocycl. Chem. 2006;2:53–92. [Google Scholar]
- 7.Alaiz M., Zamora R., Hidalgo F.J. Antioxidative Activity of Pyrrole, Imidazole, Dihydropyridine, and Pyridinium Salt Derivatives Produced in Oxidized Lipid/Amino Acid Browning Reactions. J. Agric. Food Chem. 1996;44:686–691. doi: 10.1021/jf950606k. [DOI] [Google Scholar]
- 8.Fujita M., Nakao Y., Matsunaga S., Seiki M., Itoh Y., Yamashita J., van Soest R.W., Fusetani N. Ageladine A: An Antiangiogenic Matrixmetalloproteinase Inhibitor from the Marine Sponge Agelas nakamurai. J. Am. Chem. Soc. 2003;125:15700–15701. doi: 10.1021/ja038025w. [DOI] [PubMed] [Google Scholar]
- 9.Colhoun H., Thomason M., Mackness M., Maton S., Betteridge D., Durrington P., Hitman G., Neil H., Fuller J., Investigators C. Design of the Collaborative AtoRvastatin Diabetes Study (CARDS) in Patients with Type 2 Diabetes. Diabet. Med. 2002;19:201–211. doi: 10.1046/j.1464-5491.2002.00643.x. [DOI] [PubMed] [Google Scholar]
- 10.Park H.Y., Kim M.K., Moon S.I., Cho Y.H., Lee C.H. Cell cycle arrest and apoptotic induction in LNCaP cells by MCS-C2, novel cyclin-dependent kinase inhibitor, through p53/p21(WAF1/CIP1) pathway. Cancer Sci. 2006;97:430–436. doi: 10.1111/j.1349-7006.2006.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao P., Knaus E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008;11:81s–110s. doi: 10.18433/J3T886. [DOI] [PubMed] [Google Scholar]
- 12.Faivre S., Demetri G., Sargent W., Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 2007;6:734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 13.Filho P.F.D.S., Schuchardt U. Nickel(II)-katalysierte Pyrrolsynthese aus 2H-Azirinen und aktivierten Ketonen. Angew. Chem. 1977;89:672–673. doi: 10.1002/ange.19770890920. [DOI] [Google Scholar]
- 14.Wurz R.P., Charette A.B. Doubly activated cyclopropanes as synthetic precursors for the preparation of 4-nitro- and 4-cyano-dihydropyrroles and pyrroles. Org. Lett. 2005;7:2313–2316. doi: 10.1021/ol050442l. [DOI] [PubMed] [Google Scholar]
- 15.Pichler H., Folkers G., Roth H.J., Eger K. Synthesis of 7-unsubstituted 7h-pyrrolo[2,3-D]pyrimidines. Liebigs Ann. Chem. 1986;9:1485–1505. doi: 10.1002/jlac.198619860901. [DOI] [Google Scholar]
- 16.Procopiou P.A., Draper C.D., Hutson J.L., Inglis G.G., Ross B.C., Watson N.S. Inhibitors of Cholesterol-Biosynthesis.2. 3,5-Dihydroxy-7-(n-Pyrrolyl)-6-Heptenoates, a Novel Series of Hmg-Coa Reductase Inhibitors. J. Med. Chem. 1993;36:3658–3662. doi: 10.1021/jm00075a021. [DOI] [PubMed] [Google Scholar]
- 17.Magnus N.A., Staszak M.A., Udodong U.E., Wepsiec J.P. Synthesis of 4-Cyano Pyrroles via Mild Knorr Reactions with β-Ketonitriles. Org. Process Res. Dev. 2006;10:899–904. doi: 10.1021/op060104f. [DOI] [Google Scholar]
- 18.Xia M., Lambu M.R., Tatina M.B., Judeh Z.M.A. A Practical Synthesis of Densely Functionalized Pyrroles via a Three-Component Cascade Reaction between Carbohydrates, Oxoacetonitriles, and Ammonium Acetate. J. Org. Chem. 2021;86:837–849. doi: 10.1021/acs.joc.0c02381. [DOI] [PubMed] [Google Scholar]
- 19.Xia M., Lambu M.R., Judeh Z.M.A. Selective one-pot cascade synthesis of N-substituted highly functionalized pyrroles from unprotected sugars, primary amines, and oxoacetonitriles. J. Org. Chem. 2022 doi: 10.1021/acs.joc.2c01270. manuscript accepted. [DOI] [PubMed] [Google Scholar]
- 20.Lichtenthaler F.W., Brust A., Cuny E. Sugar-derived building blocks. Part 26.Part 25. See ref. 1. Hydrophilic pyrroles, pyridazines and diazepinones from d-fructose and isomaltulose. Green Chem. 2001;3:201–209. doi: 10.1039/b105630c. [DOI] [Google Scholar]
- 21.Zhang Z., Jiang X. Oxidative Coupling of Terminal Alkyne with α-Hydroxy Ketone: An Expedient Approach toward Ynediones. Org. Lett. 2014;16:4400–4403. doi: 10.1021/ol502298a. [DOI] [PubMed] [Google Scholar]
- 22.Bouhadir K.H., Hausman D.S., Mooney D.J. Synthesis of cross-linked poly(aldehyde guluronate) hydrogels. Polymer. 1999;40:3575–3584. doi: 10.1016/S0032-3861(98)00550-3. [DOI] [Google Scholar]
- 23.Sudalai A., Khenkin A., Neumann R. Sodium periodate mediated oxidative transformations in organic synthesis. Org. Biomol. Chem. 2015;13:4374–4394. doi: 10.1039/C5OB00238A. [DOI] [PubMed] [Google Scholar]
- 24.Chaikin S.W., Brown W.G. Reduction of Aldehydes, Ketones and Acid Chlorides by Sodium Borohydride. J. Am. Chem. Soc. 1949;71:122–125. doi: 10.1021/ja01169a033. [DOI] [Google Scholar]
- 25.Wood J.M., Furkert D.P., Brimble M.A. 2-Formylpyrrole natural products: Origin, structural diversity, bioactivity and synthesis. Nat. Prod. Rep. 2019;36:289–306. doi: 10.1039/C8NP00051D. [DOI] [PubMed] [Google Scholar]
- 26.van Putten R.-J., van der Waal J.C., de Jong E., Rasrendra C.B., Heeres H.J., de Vries J.G. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 2013;113:1499–1597. doi: 10.1021/cr300182k. [DOI] [PubMed] [Google Scholar]
- 27.Tamaddon F., Amirpoor F. Improved Catalyst-Free Synthesis of Pyrrole Derivatives in Aqueous Media. Synlett. 2013;24:1791–1794. doi: 10.1055/s-0033-1339294. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available in supplementary materials.