

Abstract
In order to improve the catalytic efficiency of recombinant Photobacterium sp. JT-ISH-224 α2–6-sialyltransferase Psp2,6ST(15–501)-His6 in sialylating α-GalNAc-containing acceptors for the synthesis of tumor-associated carbohydrate antigens sialyl Tn (STn), protein crystal structure-based mutagenesis studies were carried out. Among several mutants obtained by altering the residues close to the acceptor substrate binding pocket, mutant A366G was shown to improve the sialyltransferase activity of Psp2,6ST(15-501)-His6 toward α-GalNAc-containing acceptors by 21–115% without significantly affecting its sialylation activity to β-galactosides. Furthermore, the expression level was improved from 18–40 mg L−1 for the wild-type enzyme to 72–110 mg L−1 for the A366G mutant. In situ generation of CMP-sialic acid in a one-pot two-enzyme system was shown effective in overcoming the high donor hydrolysis of the enzyme. Mutant A366G performed better than the wild-type Psp2,6ST(15-501)-His6 for synthesizing Neu5Acα2–6GalNAcαSer/Thr STn antigens.
Keywords: Improved protein expression, mutagenesis, sialyltransferase, sialyltransferase mutant, STn antigen
Graphical Abstract
1. Introduction
Sialic acid (Sia)-containing structures in eukaryotic systems play important roles in a variety of physiological and pathological processes, including cell-cell interactions, inflammation, fertilization, viral infection, differentiation, malignancies, and cell signaling etc.1–4 Among more than 50 different sialic acid structures that have been identified in nature, N-acetylneuraminic acid (Neu5Ac) is the most common and the most abundant sialic acid form. Sialyltransferases (EC 2.4.99.X) are key enzymes involved in the biosynthesis of these sialic acid-containing oligosaccharides and glycoconjugates.5 They catalyze the transfer of a sialic acid residue from its activated sugar nucleotide donor cytidine 5’-monophosphate sialic acid (CMP-sialic acid) to an acceptor, usually a structure with a galactose (Gal), an N-acetylgalactosamine (GalNAc), an N-acetylglucosamine (GlcNAc), or another sialic acid residue. Various linkages including Siaα2–3Gal, Siaα2–6Gal/GalNAc/GlcNAc, Siaα2–8Sia, and Siaα2–9Sia can be formed. Bifunctional glycosyltransferases (SiaD)6 that are responsible for the formation of Neu5Ac-containing Neisseria meningitidis serogroups W-135 and Y capsular polysaccharides (CPSs) [–6Gal/Glcα1–4Neu5Acα2–]n have been grouped together with other glycosyltransferases in glycosyltransferase 4 (GT4) family in the Carbohydrate Activated enZyme (CAZy, http://www.cazy.org) database7,8 based on protein sequence homology. All other sialyltransferases reported to date have been grouped into five CAZy glycosyltransferase (GT) families (GT29, GT38, GT42, GT52, and GT80). All known eukaryotic sialyltransferases belong to a single CAZy GT29 family, while bacterial sialyltransferases are more spread out among CAZy GT families GT38, GT42, GT52, and GT80.9,10
Since bacterial sialyltransferases can be produced more easily as active forms in larger amounts in Escherichia coli expression systems and many of them have broader substrate specificities than their mammalian counterparts,11,12 they have been used as efficient catalysts in preparative and large scale synthesis of biological important sialosides. For example, multifunctional Pasteurella multocida α2–3-sialyltransferase 1 (PmST1) has been used as a powerful catalyst in chemoenzymatic synthesis of diverse α2–3-linked sialosides,13 Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST) has been applied in the synthesis of α2–6-linked sialosides and glycopeptides,12,14–17 Campylobacter jejuni OH4384 α2–3/8-sialyltransferase (CstII) has been used for the synthesis of GD3 and GT1a ganglioside oligosaccharides.18–22
Among sialic acid-containing biologically important sialosides, sialyl Tn antigen (Siaα2–6GalNAcα1-O-Ser/Thr) has been reported to correlate with the invasive and metastatic growth of carcinoma cells and is considered as a tumor-associated antigen for cancer vaccination development.23 In addition to chemical synthetic methods developed, sialyltransferase-catalyzed glycosylation has been shown as a highly efficient approach for the synthesis of sialyl Tn (STn) antigen.24 We previous identified recombinant Photobacterium sp. JT-ISH-224 α2–6-sialyltransferase Psp2,6ST(15–501)-His6 as a more suitable α2–6-sialyltransferase than Pd2,6ST for catalyzing the formation of STn antigens from N-acetylgalactosamine (GalNAc)-containing glycosides such as GalNAcα2AA, GalNAcαSer, and GalNAcαThr as acceptor substrates.24 Nevertheless, the efficiency of Psp2,6ST(15–501)-His6 in sialylating α-GalNAc-terminated glycosides (Tn-antigens) is still much lower than sialylating β-galactosides. On the other hand, the expression level of soluble Psp2,6ST(15–501)-His6 (25 mg L−1)24 is not as high as Pd2,6ST (36 mg L−1).25 Protein crystal structure-based mutagenesis studies were carried out to improve the catalytic efficiency of Psp2,6ST(15–501)-His6 for the formation of STn antigens. Among several mutants obtained by altering the residues close to the acceptor substrate binding pocket, a mutant A366G with a better expression level (72–110 mg L−1) and improved activities in catalyzing the formation of STn antigens from α-GalNAc-terminated glycosides was identified. The mutant is an improved catalyst in one-pot multienzyme (OPME) synthesis of STn sialosides.
2. Results and discussion
2.1. Crystal structure-based design of Psp2,6ST(15–501)-His6 mutants
The tertiary crystal structure of Δ16Psp2,6ST in complex with CMP and lactose (pdb: 2Z4T)26 showed that residues Arg153, Trp365, and Ala366 help to define a relatively narrow pocket for the acceptor substrate (lactose) of the enzyme (Fig. 1), leading to its preference towards β-linked galactosides as acceptor substrates.24 In order to accommodate acceptors terminated with an α-linked N-acetylgalactosamine residue such as Tn antigens, mutating Arg153, Trp365, and Ala366 to smaller amino acid residues was proposed. For this purpose, R153G, W365A, W365G, W365S, and A366G were designed.
Figure 1.
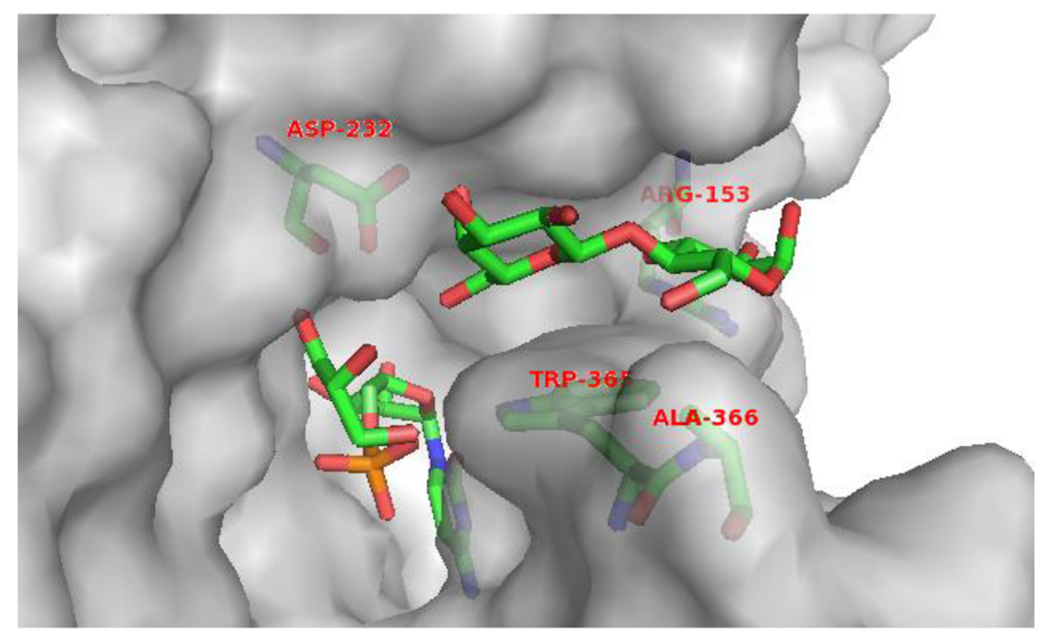
The amino acid residues in the lactose-binding pocket of Psp2,6ST(15-501)-His6. The protein structure was modeled based on the reported crystal structure of Δ16pspST6 (pdb: 2Z4T) and analyzed using PyMOL (carbon, nitrogen, oxygen, and phosphorus atoms in lactose and the amino acid residues of interests (stick model) are shown in green, blue, red, and orange colors, respectively).
2.2. Donor hydrolysis activity of Psp2,6ST(15–501)-His6 and design of Psp2,6ST(15–501)-His6 mutants with potentially decreased donor hydrolysis activity
We showed previously that donor hydrolysis activity of glycosyltransferases competed with glycosylation process and led to low glycosylation yields if a poor glycosyltransferase acceptor substrate was used.27 Indeed, when GalNAcα2AA was used as an acceptor for Psp2,6ST(15–501)-His6, adding one equivalent of CMP-Neu5Ac led to the formation of only 7.3% of sialylated product Neu5Acα2–6GalNAcα2AA and the reaction was completed in 15 min (Fig. 2A). A longer incubation time for up to 60 min (solid line marked with white diamonds in Fig. 2A) or 240 min (dashed line marked with black circles in Fig. 2A) did not improve the sialylation yield. Adding additional two equivalents of CMP-Neu5Ac, the sialylation yield was improved to 17% in 15 min (solid line marked with white diamonds in Fig. 2A). Similarly, adding the third and the fourth doses (2 equiv at each time) of CMP-Neu5Ac pushed the reaction yields to 25% and 33%, respectively (solid line marked with white diamonds in Fig. 2A). These provided evidence that the donor hydrolysis activity of Psp2,6ST(15-501)-His6 where water molecules competing with the GalNAcα2AA acceptor molecules (Fig. 2B) caused the relatively low yields of GalNAcα2AA α2–6-sialylation reactions.
Figure 2.
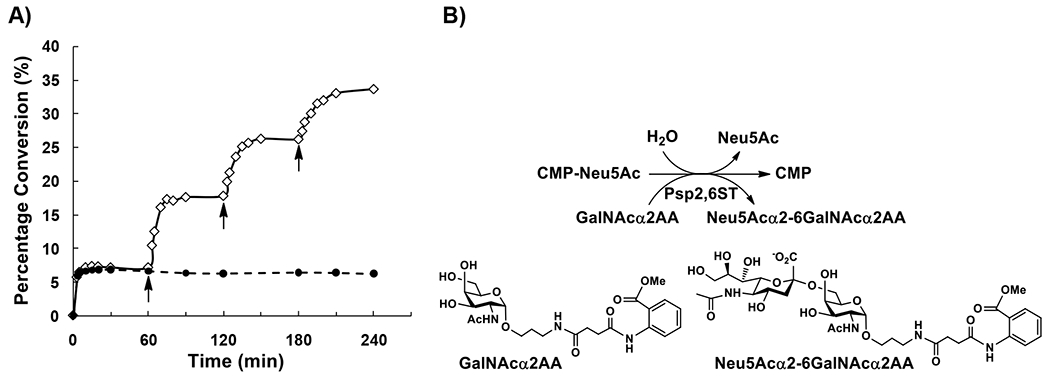
A) Time course study for the donor hydrolysis activity of Psp2,6ST(15-501)-His6 where GalNAcα2AA was used as an acceptor and with periodic addition of 2 equiv of CMP-Neu5Ac (shown by arrows). B) Schematic illustration of donor hydrolysis of Psp2,6ST(15-501)-His6 where water competes with the GalNAcα2AA acceptor for Psp2,6ST(15-501)-His6-catalyzed sialylation reaction.
Previous crystal structure-based mutagenesis studies of another CAZy GT80 family member PmST1 which shares 36% amino acid sequence identity with the sialyltransferase domain of Psp2,6ST showed that mutating a neutral amino acid residue (Met144) in a close proximity to the catalytic base Asp141 of PmST1 generated a mutant (PmST1 M144D) with decreased donor hydrolysis activity.27 The corresponding catalytic base is Asp232 and the corresponding residue for mutation is Ala235 in Psp2,6ST(15-501)-His6 (Fig. 3). Therefore, Psp2,6ST(15-501)-His6 A235D mutant was designed in the attempt to reduce the donor hydrolysis activity and to enhance the sialylation activity of the enzyme. Furthermore, another neutral amino acid residue Ala124 close to the catalytic Asp232 of Psp2,6ST(15-501)-His6 as shown in the crystal structure26 was identified (Fig. 3) and the A124D mutant was also designed to test enhanced sialylation activity of the enzyme.
Figure 3.
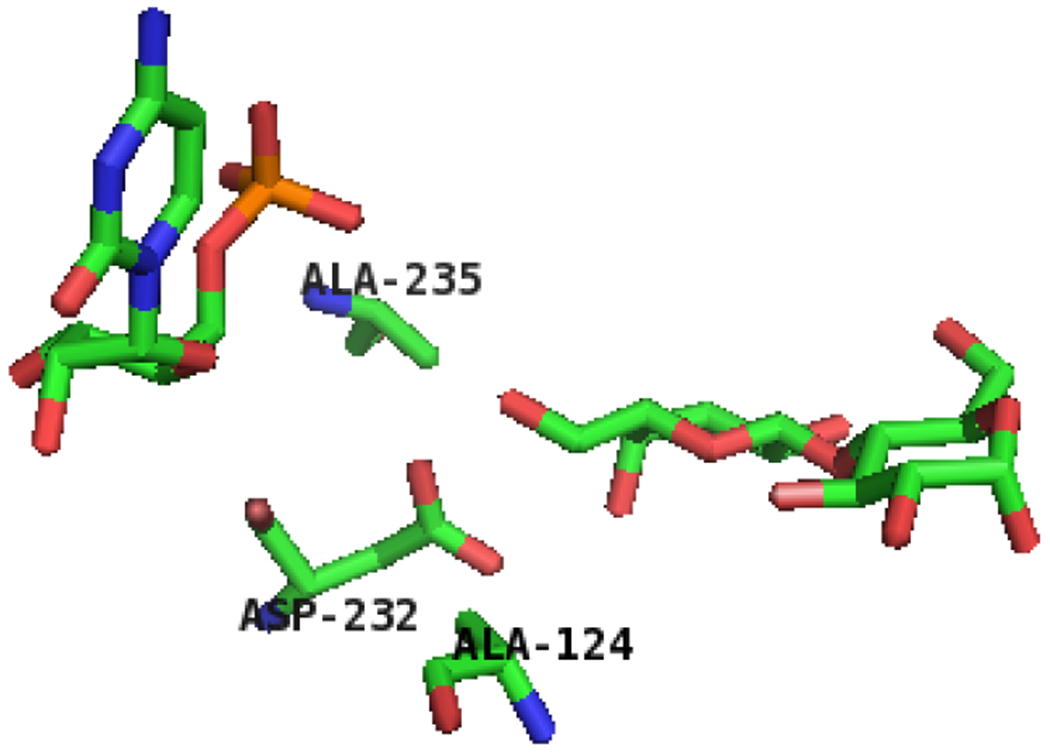
The amino acid residues in close proximity to the catalytic base (Asp232) of Psp2,6ST(15-501)-His6. The protein structure was modeled based on the reported crystal structure of Δ16pspST6 (pdb: 2Z4T) and analyzed using PyMOL (carbon, nitrogen, oxygen, and phosphorus atoms in lactose and the amino acid residues of interests (stick model) are shown in green, blue, red, and orange colors, respectively).
2.3. Psp2,6ST(15–501)-His6 mutants expression and purification
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the lysates (Fig. 4) showed that all of the obtained Psp2,6ST(15–501)-His6 mutants A124D, R153G, A235D, W365A, W365G, W365S, and A366G were expressed well as soluble proteins with a molecular weight similar to that of the wild-type enzyme of 56 kDa. Quite interestingly as obviously seen in the SDS-PAGE (Fig. 4), A366G (lane 2), W365S (lane 5), W365G (lane 6), W365A (lane 7), and R153G (lane 8) mutants had significantly improved expression level of soluble recombinant proteins in the cell lysates compared to the wild-type enzyme (lane 1) as well as A235D (lane 3) and A124D (lane 4) mutants. It was unclear how a single site mutation close to the substrate binding pocket can improve the expression level of the soluble protein in the Escherichia coli expression system. Repeated expression in duplicates or triplicates and more detailed quantitation showed that compared to wild-type Psp2,6ST(15–501)-His6 whose expression level fell in a range of 18–40 mg L−1, A366G mutant was able to be expressed and purified by nickel-nitrilotriacetic acid (Ni2+-NTA) column chromatography in a range of 72–110 mg per liter of Escherichia coli culture.
Figure 4.
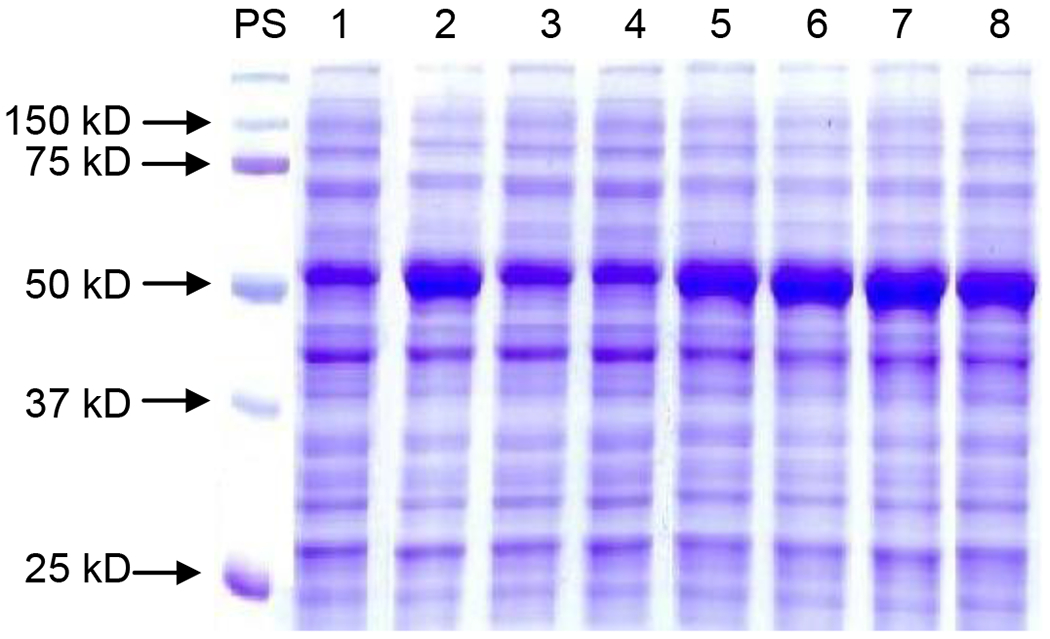
SDS-PAGE analysis of the expression of Psp2,6ST(15–501)-His6 and its mutants in cell lysates. Lanes: PS, protein standards; 1, wild-type; 2, A366G mutant; 3, A235D mutant; 4, A124D mutant, 5, W365S mutant; 6, W365G mutant; 7, W365A mutant; 8, R153G mutant.
2.4. Activity comparison of Psp2,6ST(15–501)-His6 and mutants
Activity assays using LacβMU, GalNAcα2AA, GalNAcαSer, or GalNAcαThr as an acceptor for Psp2,6ST(15–501)-His6 and mutants (Fig. 5) showed that the activities of W365 mutants, including W365A, W365G, and W365S, decreased drastically in sialylating LacβMU (1–5% activity of the wild-type enzyme) and GalNAcαSer (9–16% activity of the wild-type enzyme) and the effect was more severe for LacβMU. The activities of W365 mutants also decreased in sialylating GalNAcα2AA (39–62% activity of the wild-type enzyme) and GalNAcαThr (65–82% activity of the wild-type enzyme), but to a less extent. These results indicated that the acceptor stabilizing effect by Trp365 van der Waals stacking interaction28 is more significant when LacβMU or GalNAcαSer, instead of GalNAcα2AA or GalNAcαThr, was used as the acceptor. Changing the bulkier tryptophan residue to a smaller alanine, glycine, or serine residue did not provide benefit in accommodating α-linked N-acetyl galactosides as acceptor substrates for Psp2,6ST(15–501)-His6.
Figure 5.
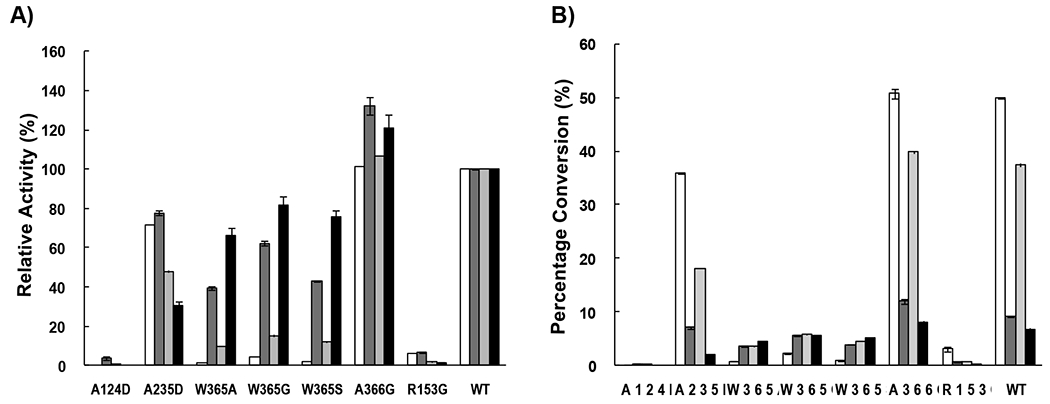
Sialyltransferase activity comparison of Psp2,6ST(15–501)-His6 and its mutants when different acceptors were used: LacβMU (while columns), GalNAcα2AA (dark grey columns), GalNAcαSer (light grey columns) and GalNAcαThr (black columns). A) Relative activities of the mutants when the yields for wild-type enzyme were assigned as 100%. B) Percentage conversions of sialylation reactions catalyzed by the wild-type enzyme and the mutants.
Mutating Arg153, a residue that is conserved among GT80 sialyltransferases characterized so far, to a glycine residue knocked out the sialylation activity completely. The corresponding Arg63 in PmST128 was shown to form ion pairs with the sialyl carboxylic acid group which explains the importance of the negative charge in the residue.
To our delight, A366G mutation successfully improved the sialyltransferase activity of Psp2,6ST(15–501)-His6 when α-linked N-acetyl galactosaminides, especially when GalNAcα2AA or GalNAcαThr, was used as the acceptor. These results indicate that a smaller glycine residue may provide a bigger acceptor binding pocket to accommodate α-linked N-acetyl galactosaminides.
Similar to that observed previously for PmST1 A35D and A35H mutants,27 the corresponding Psp2,6ST(15–501)-His6 A124D mutant lost sialylation activity. In comparison, Psp2,6ST(15–501)-His6 A235D mutant did have a decreased donor hydrolysis activity (Table 1) while maintaining most of the sialylation activity (Fig. 5). The CMP-Neu5Ac hydrolysis efficiency catalyzed by Psp2,6ST(15–501)-His6 was 17 mM−1 min−1, which was about 14-fold more efficient than its sialyltransferase activity when GalNAcα2AA was used as an acceptor (kcat/Km = 1.2 mM−1 min−1), although it was 13-fold lower than the sialyltransferase activity when LacβMU was used as acceptor (kcat/Km = 2.2×102 mM−1 min−1). This can explain the lower yield of sialylating GalNAcα2AA by Psp2,6ST(15–501)-His6. In comparison, the CMP-Neu5Ac hydrolysis efficiency catalyzed by a closely related Photobacterium damselae α2–6-sialyltransferase His6-Pd2,6ST(16–497)12,25 was 12 mM−1 min−1 (Table 1) which was very similar to that of Psp2,6ST(15–501)-His6. For Psp2,6ST(15–501)-His6 A235D mutant, the efficiency for donor hydrolysis was 6.6 mM−1 min−1. However, this decreased donor hydrolysis did not lead to the overall improvement of the sialylation efficiency of the enzyme.
Table 1.
Apparent kinetic parameters for the CMP-Neu5Ac hydrolysis activities of Psp2,6ST(15–501)-His6 and A235D mutant.
| Enzymes | Km (mM) | kcat (min−1) | kcat/Km (mM−1 min−1) |
|---|---|---|---|
| His6-Pd2,6ST(16–497) | (1.8±0.4)×10 | (2.2±0.2)×102 | 1.2×10 |
| Psp2,6ST(15–501)-His6 | (1.7±0.4)×10 | (2.9±0.3)×102 | 1.7×10 |
| Psp2,6ST(15–501)-His6 A235D mutant | (3.8±0.5)×10 | (2.5±0.2)×102 | 6.6 |
2.5. Kinetic study for Psp2,6ST(15–501)-His6 A366G mutant
Among the mutants obtained, A366G mutant which has an enhanced expression level and improved sialylation activities was characterized further. Kinetics studies showed that its catalytic efficiencies (kcat/Km) were 4.4, 29, and 5.7 mM−1 min−1, respectively, when GalNAcα2AA, GalNAcαSer, and GalNAcαThr were used as the acceptor substrates, which were 1.3, 1.2 and 2.1-fold of the wild-type enzyme (Table 2). For acceptor GalNAcα2AA, the decreased Km value contributed to the improved catalytic efficiency of the mutant. For acceptors GalNAcαSer and GalNAcαThr, higher kcat values contributed to the enhanced catalytic efficiencies of the mutant. For acceptor LacβMU, the kcat and the Km value of the mutant remained almost the same as the wild-type enzyme, leading to similar kcat/Km values for the mutant and the wild-type enzyme. Overall, compared to the wild-type Psp2,6ST(15–501)-His6, the A366G mutant has improved catalytic efficiency towards α-linked N-acetylgalactosaminides without changing its efficiency in sialylating β-linked galactosides.
Table 2.
Apparent kinetic parameters of Psp2,6ST(15–501)-His6 and A366G mutant.
| Enzymes | Substrates | Km (mM) | kcat (min−1) | kcat/Km (mM−1 min−1) |
|---|---|---|---|---|
| Psp2,6ST(15–501)-His6a | CMP-Neu5Ac | (6.2±0.4)×10−1 | (1.4±0.1)×102 | 2.2×102 |
| LacβMU | (3.6±0.7)×10−1 | (1.0±0.1)×102 | 2.9×102 | |
| CMP-Neu5Ac | 3.9±0.4 | 4.8±0.2 | 1.2 | |
| GalNAcα2AA | 9.5±2.7 | (3.1±0.5)×10 | 3.3 | |
| GalNAcαSer | 1.4±0.1 | (3.4±0.3) ×10 | 2.4×10 | |
| GalNAcαThr | 5.9±1.6 | (1.6±0.1) ×10 | 2.7 | |
| Psp2,6ST(15–501)-His6 A366G mutant | CMP-Neu5Ac | (6.3±0.5)×10−1 | (1.3±0.1)×102 | 2.1×102 |
| LacβMU | (4.1±0.4)×10−1 | (1.0±0.1)×102 | 2.4×102 | |
| CMP-Neu5Ac | 3.3±0.4 | 5.9±0.2 | 1.8 | |
| GalNAcα2AA | 5.5±1.0 | (2.4±0.1)×10 | 4.4 | |
| GalNAcαSer | 1.7±0.7 | (4.9±1.2) ×10 | 2.9×10 | |
| GalNAcαThr | 7.2±3.0 | (4.2±0.5) ×10 | 5.8 |
The kinetic parameters for the wild-type enzyme were cited from reference 24.24
2.6. Efficiency of a one-pot two-enzyme system for synthesizing STn antigens using Psp2,6ST(15–501)-His6 and mutants
Psp2,6ST(15-501)-His6 mutants A366G, W365G, W365A, and W365S which showed high expression levels and good or reasonable α2–6-sialyltransferase activities for sialylating α-linked N-acetylgalactosaminides were used with Neisseria meningitidis CMP-sialic acid synthetase (NmCSS)29 in a one-pot two-enzyme system for synthesizing STn antigens from Tn-antigens GalNAcαSer (white and black bars in Fig. 6) and GalNAcαThr (Grey bars in Fig. 6) in the presence of Neu5Ac and CTP. In this system, CTP and Neu5Ac were used by NmCSS for the formation of CMP-Neu5Ac in situ, which was used as the donor substrate for Psp2,6ST(15-501)-His6 or its mutants. As shown in Figure 6, the application of the one-pot multienzyme (OPME) approach effectively improved the yields of STn formation compared to the reactions catalyzed by Psp2,6ST(15-501)-His6 or its mutants alone (Fig. 5B). For example, using 1 equiv of Neu5Ac and 1.5 equiv of CTP in the one-pot two-enzyme (OP2E) reaction (white bars in Fig. 6) improved the GalNAcαSer α2–6-sialylation reaction yields from less than 6% (1 equiv CMP-Neu5Ac was used) (Fig. 5B) to 32–51% for W365A, W365G, W365S mutants. Similarly, the OP2E reaction improved the GalNAcαSer α2–6-sialylation reaction yields from 40% and 37% to 75% and 67% for A366G mutant and the wild-type enzyme, respectively. Increasing the concentrations of Neu5Ac and CTP to 2.5 and 5.0 equiv further improved the reaction yields to more than 67% for Psp2,6ST(15-501)-His6 and its mutants (black bars in Fig. 6). Quite significantly, the yields for synthesizing Neu5Acα2–6GalNAcαThr from GalNAcαThr, a poorer acceptor for Psp2,6ST(15-501)-His6 and mutants other than A366G, were improved to more than 65% for all enzymes and reached 85%, 91%, 93%, and 88% for W365A, W365G, A366G mutants and the wild-type enzyme, respectively (gray bars in Fig. 6). Therefore, in situ generation of CMP-Neu5Ac, the sugar nucleotide donor for sialyltransferases, was proven an efficient method to enhance the yields for sialylation reactions catalyzed not only by the wild-type enzymes but also sialyltransferase mutants.
Figure 6.
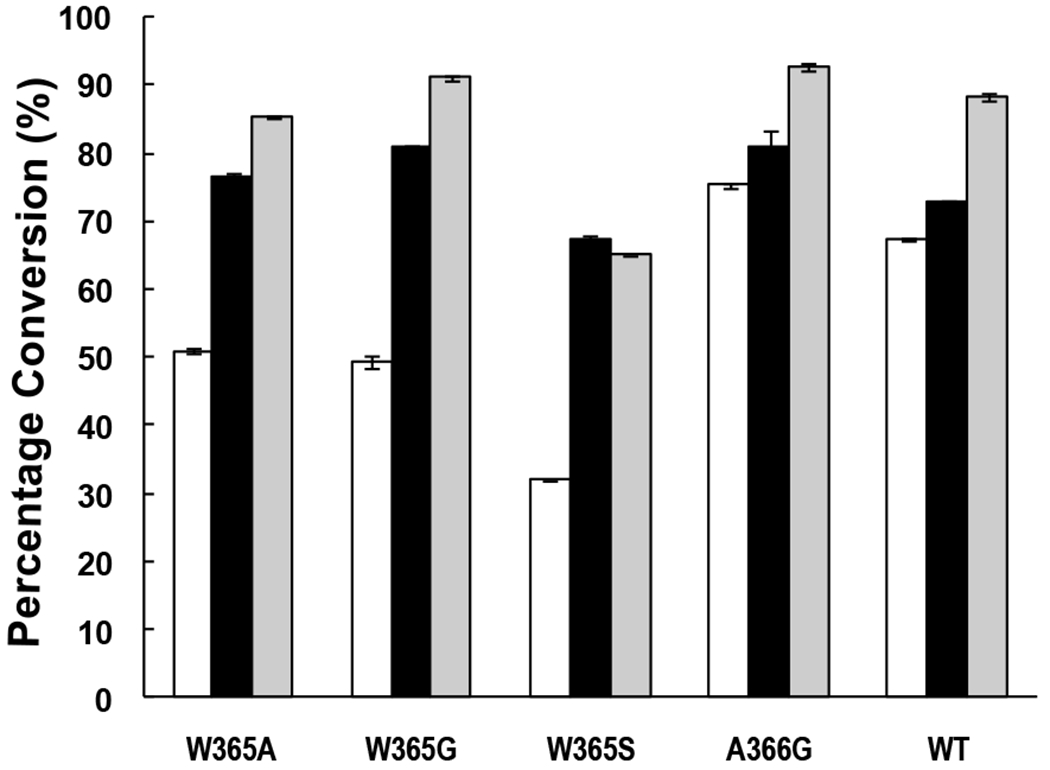
Reaction yields from one-pot two-enzyme (OP2E) systems containing Neisseria meningitidis CMP-sialic acid synthetase and Psp2,6ST(15–501)-His6 or its mutants. White bars, GalNAcαSer was used as an acceptor with 1 equiv of Neu5Ac and 1.5 equiv of CTP; Black bars, GalNAcαSer was used as an acceptor with 2.5 equiv of Neu5Ac and 5.0 equiv of CTP; Gray bars, GalNAcαThr was used as an acceptor with 2.5 equiv of Neu5Ac and 5.0 equiv of CTP.
3. Conclusion
In conclusion, a Psp2,6ST(15-501)-His6 mutant (A366G) with an enhanced expression level (>2-fold) and moderately improved activities in catalyzing the formation of Neu5Acα2–6GalNAcαSer/Thr STn antigens was generated and characterized. Protein crystal structure-based site-directed mutagenesis was demonstrated again a practical approach to obtain sialyltransferase mutants with improved function. In situ generation of CMP-Neu5Ac by one-pot multienzyme (OPME) system was also confirmed to be an efficient approach for high-yield enzymatic and chemoenzymatic synthesis of sialosides. With an improved expression level and enhanced activities, the Psp2,6ST(15-501)-His6 A366G mutant is a powerful catalyst for enzymatic and chemoenzymatic synthesis of α2–6-linked sialosides.
4. Experimental section
4.1. Material
Escherichia coli BL21 (DE3) was from Invitrogen (Carlsbad, CA, USA). Nickel-nitrilotriacetic acid agarose (Ni2+-NTA agarose) and QIAprep spin miniprep kit were from Qiagen (Valencia, CA, USA). Bicinchoninic acid (BCA) protein assay kit was from Pierce Biotechnology, Inc. (Rockford, IL). QuikChange Multi Site-Directed Mutagenesis Kit was from Agilent Technologies company/Stratagene (Santa Clara, CA).
4.2. Site-directed mutagenesis
Site-directed mutagenesis was carried out using the QuikChange Multi Site-Directed Mutagenesis Kit from Stratagene according to the protocol from the manufacturer. The primers (the sites for mutations are underlined) used are shown in Table 3.
Table 3.
Primers used for site-directed mutagenesis. Mutation sites are underlined.
| Mutant | Primer |
|---|---|
| A124D | 5’ GAAGTTTATGTTGATCATGATAGCCTGCCGACCCTGCAG 3’ |
| R153G | 5’ CGTTATATTGCATGGGGTGGTATTGTTCCGACCGATGAG 3’ |
| A235D | 5’ TCTGTATGACGATGGCAGCGATGAGTACGTGAATCTGTATAAT 3’ |
| W365G | 5’ TTACCGGCACCACCGTTGGCGCAGGTAATCATGAACG 3’ |
| W365A | 5’ TTACCGGCACCACCGTTGCGGCAGGTAATCATGAACG 3’ |
| W365S | 5’ TTACCGGCACCACCGTTAGCGCAGGTAATCATGAACG 3’ |
| A366G | 5’ GGCACCACCGTTTGGGGTGGTAATCATGAACG 3 |
4.3. Protein expression and purification of Psp2,6ST(15-501)-His6 and mutants
The plasmids containing mutant genes were transformed into Escherichia coli BL21 (DE3). The Escherichia coli cells were cultured in LB-rich media (10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl) supplemented with ampicillin (100 μg mL−1). Overexpression of the mutants was achieved by adding 0.3 mM of isopropyl-1-thio-β-D-galactopyranoside (IPTG) to the Escherichia coli culture when its OD600 nm reached 0.8. The induced culture was incubated at 20 °C for 20 h with vigorous shaking at 250 rpm in a C25KC incubator shaker (New Brunswick Scientific, Edison, NJ). His6-tagged mutant proteins were purified from the cell lysate. To obtain cell lysate, the cell pellet harvested by centrifugation at 4000 rpm for 2 h was resuspended in 20 mL (for cells obtained from one-liter culture) of lysis buffer (pH 8.0, 100 mM Tris-HCl containing 0.1% Triton X-100). Lysozyme (50 μg mL−1) and DNaseI (3 μg mL−1) were added to the resuspended cells followed by shaking at 37 °C for 60 min. The lysate was obtained as the supernatant after centrifugation at 11,000 rpm for 20 min. Purification of His6-tagged proteins from the lysate was achieved using 10 mL column packed with Ni2+-NTA agarose. The column was pre-equilibrated with 8 column volumes of binding buffer (5 mM imidazole, 0.5 M NaCl, 50 mM Tris-HCl pH 7.5). After loading the sample, the column was washed with 8 column volumes of the binding and 8 column volumes of washing buffer (20 mM imidazole, 0.5 M NaCl, 50 mM Tris-HCl pH 7.5). Protein was eluted using 8 column volumes of the elute buffer (200 mM imidazole, 0.5 M NaCl, 50 mM Tris-HCl pH 7.5). The fractions containing the purified enzyme were collected and stored at 4 °C. Protein concentrations were quantified by bicinchoninic acid (BCA) protein assay kit according to manufacturer’s instruction using bovine serum albumin (BSA) as the protein standard. The wild-type enzyme and A366G mutant were expressed, purified, and quantified in duplicates or triplets for at least three times.
4.4. Sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
SDS-PAGE was performed in a 12% Tris–glycine gel using Bio-Rad Mini-protein III cell gel electrophoresis unit (Bio-Rad, Hercules, CA) at DC = 150V. Bio-Rad Precision Plus Protein Standards (10–250 kD) were used as molecular weight standards. Gels were stained with Coomassie Blue. Cell lysates from the wild-type enzyme and the mutants were prepared exactly the same and the same volume (~30 mL) of the lysate was obtained for each construct. The same volume of the lysate was used to prepare samples for SDS-PAGE and the same volume (10 μL) was loaded to the gel.
4.5. Psp2,6ST(15-501)-His6 sialylation assay with periodic addition of CMP-Neu5Ac.
Two reactions were set up at 20 °C in MES buffer (200 mM, pH 5.0) containing Psp2,6ST(15-501)-His6 (6.0 μM), CMP-Neu5Ac (1 mM), and GalNAcα2AA (1 mM) in a total volume of 50 μL. One of the reactions was used as a control and was incubated for 4 h. The other was incubated for 1 h, 2 equiv of CMP-Neu5Ac was then added. The same dose was added every hour for up to 4 h reaction duration. Aliquots (2 μL each) were withdrawn added to 10 μL of pre-chilled ethanol followed by centrifugation. The samples were diluted and kept on ice until aliquots of 8 μL were injected and analyzed by a Shimadzu LC-6AD system equipped with a membrane on-line degasser, a temperature control unit, and a fluorescence detector (Shimadzu RF-10AXL). A reverse-phase Premier C18 column (250 × 4.6 mm i.e., 5 μm particle size, Shimadzu) protected with a C18 guard column cartridge was used. The mobile phase was 25% acetonitrile in water. The 2-aminobenzoic acid (2AA)-labeled fluorescent acceptor and the product formed were detected with excitation at 315 nm and emission at 400 nm.
4.6. Sialyltransferase activity assays for Psp2,6ST(15-501)-His6 and mutants
When LacβMU was used as an acceptor, the assay was performed in duplicate in 10 μL of Tris-HCl buffer (100 mM, pH 8.0) containing CMP-Neu5Ac (1 mM), LacβMU (1 mM), MgCl2 (20 mM), wild-type Psp2,6ST(15-501)-His6 or its mutant (0.3 μM). When an α-GalNAc-terminated acceptor was used, the assay was performed in duplicate in 10 μL of Tris-HCl buffer (100 mM, pH 8.0 for GalNAcαSer/Thr) or NaOAc-HOAc (100 mM, pH 5.0 for GalNAcα2AA) containing CMP-Neu5Ac (1.5 mM), the acceptor (1 mM), MgCl2 (20 mM), wild-type Psp2,6ST(15-501)-His6 or its mutant (3.0 μM). Reactions were allowed to proceed at 20 °C for 20 min and stopped by adding 10 μL of pre-chilled ethanol. The analysis of sialylated product conversion was performed using the HPLC system as described above for sialyltransferase activity assay. The 4-methylumbelliferone (MU)-labeled fluorescent acceptor and the product formed were detected with excitation at 325 nm and emission at 372 nm. The 2-aminobenzoic acid (2AA)-labeled fluorescent acceptors and the products formed were detected with excitation at 315 nm and emission at 400 nm. The 9-fluorenylmethylcarbamate (Fmoc)-labeled fluorescent acceptors and the products formed were detected with excitation at 262 nm and emission at 313 nm.
4.7. Kinetics studies for the sialyltransferase activity of A366G mutant
Reactions were carried out in duplicate at 20 °C for 20 min in a total volume of 10 μL in a proper buffer at the optimal pH according to the pH profile of the wild-type enzyme.24 When LacβMU was used as an acceptor, the conditions were: Tris-HCl buffer (200 mM, pH 8.0), enzyme (0.3 μM), varied concentrations of LacβMU (0.1, 0.25, 0.4, 1.0, 2.0, 4.0, and 6.0 mM) with a fixed concentration of CMP-Neu5Ac (1.0 mM) or varied concentrations of CMP-Neu5Ac (0.1, 0.25, 0.4, 1.0, 2.0, 4.0, and 6.0 mM) with a fixed concentration of LacβMU (1.0 mM). When GalNAcα2AA was used as an acceptor, the conditions were: NaOAc-HOAc buffer (200 mM, pH 5.0), enzyme (6 μM), varied concentrations of GalNAcα2AA (0.5, 0.8, 1.0, 2.0, 4.0, 8.0, and 10.0 mM) and a fixed concentration of CMP-Neu5Ac (4.0 mM) or varied concentrations of CMP-Neu5Ac (0.5, 1.0, 2.0, 4.0, 5.0, 8.0, and 10.0 mM) with a fixed concentration of GalNAcα2AA (1.0 mM). When GalNAcαSer was used as an acceptor, the conditions were: Tris-HCl buffer (200 mM, pH 8.0), enzyme (0.6 μM), varied concentrations of GalNAcαSer (0.1, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 10.0 mM) and a fixed concentration of CMP-Neu5Ac (2.0 mM). When GalNAcαThr was used as an acceptor, the conditions were: Tris-HCl buffer (200 mM, pH 8.0), enzyme (3.0 μM), varied concentrations of GalNAcαThr (0.5, 1.0, 2.0, 4.0, 10.0, 20.0, 40.0, 60.0 mM) and a fixed concentration of CMP-Neu5Ac (4.0 mM). Results analysis was performed using the HPLC system as described above for sialyltransferase activity assays. Apparent kinetic parameters were obtained by fitting the experimental data (the average values of duplicate assay results) into the Michaelis-Menten equation using Grafit 5.0.
4.8. Kinetics for donor hydrolysis of His6-Pd2,6ST(16–497), Psp2,6ST(15-501)-His6 and its A235D mutant
The kinetics study for the donor hydrolysis activity of His6-Pd2,6ST(16–497) was carried out at 37 °C for 20 min in Tris-HCl buffer (200 mM, pH 8.0). The kinetics study for the donor hydrolysis activities of Psp2,6ST(15-501)-His6 and its A235D mutant was carried out at 20 °C for 10 min in Tris-HCl buffer (200 mM, pH 8.0). All reactions were performed in duplicate in a total volume of 10 μL containing varied concentrations of CMP-Neu5Ac (10, 20, 40, 60, 80 and 100 mM) and the enzyme (6 μM). Reactions were stopped by adding 10 μL of ethanol and then centrifuged. The supernatants were analyzed by a P/ACETM capillary electrophoresis (CE) system equipped with a photodiode array (PDA) detector (Beckman Coulter, Inc., Fullerton, CA). CE conditions were as follows: 75 μm i.d. capillary, 25 KV/80 μÅ, 5 s vacuum injections, monitored at 200 nm and 254 nm, the running buffer used was sodium tetraborate (25 mM, pH 9.4). Apparent kinetic parameters were obtained by fitting the experimental data (the average values of duplicate assay results) into the Michaelis-Menten equation using Grafit 5.0.
Highlights.
Several mutants of Photobacterium sp. α2–6-sialyltransferase (Psp2,6ST) were obtained.
A366G mutant has an increased expression level.
A366G mutant also has an improved catalytic activity in sialylating Tn antigens.
A one-pot two-enzyme sialylation system improved STn antigen synthetic efficiency.
Psp26ST A366G mutant is a powerful catalyst for synthesizing α2–6-sialosides.
Acknowledgments
This work was supported by Camille Dreyfus Teacher-Scholar Fund to X. C., National Institutes of Health grants R01HD065122 and R01GM094523. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NICHD, NIGMS, or NIH.
Reference:
- 1.Chen X; Varki A ACS Chem. Biol 2010, 5, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traving C; Schauer R Cell Mol. Life Sci 1998, 54, 1330–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schauer R Curr. Opin. Struct. Biol 2009, 19, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varki A Nature 2007, 446, 1023–1029. [DOI] [PubMed] [Google Scholar]
- 5.Harduin-Lepers A; Recchi MA; Delannoy P Glycobiology 1995, 5, 741–758. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharjee AK; Jennings HJ; Kenny CP; Martin A; Smith IC Can. J. Biochem 1976, 54, 1–8. [DOI] [PubMed] [Google Scholar]
- 7.Campbell JA; Davies GJ; Bulone V; Henrissat B Biochem. J 1997, 326 ( Pt 3), 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coutinho P; Deleury E; Davies G; Henrissat B J. Mol. Biol 2003, 328, 307–317. [DOI] [PubMed] [Google Scholar]
- 9.Li Y; Chen X Appl. Microbiol. Biotechnol 2012, 94, 887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Audry M; Jeanneau C; Imberty A; Harduin-Lepers A; Delannoy P; Breton C Glycobiology 2011, 21, 716–726. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T Mar. Drugs 2010, 8, 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu H; Huang S; Chokhawala H; Sun M; Zheng H; Chen X Angew. Chem. Int. Ed 2006, 45, 3938–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu H; Chokhawala H; Karpel R; Wu B; Zhang J; Zhang Y; Jia Q; Chen XJ Am. Chem. Soc 2005, 127, 17618–17619. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto T; Nagae H; Kajihara Y; Terada I Biosci. Biotechnol. Biochem 1998, 62, 210–214. [DOI] [PubMed] [Google Scholar]
- 15.Kajihara Y; Akai S; Nakagawa T; Sato R; Ebata T; Kodama H; Sato K Carbohydr. Res 1999, 315, 137–141. [DOI] [PubMed] [Google Scholar]
- 16.Teo CHT; Chen P; Hung C; Gao H; Chang L; Lin C Adv. Synth. Catal 2005, 347, 967–972. [Google Scholar]
- 17.Yu H; Chokhawala HA; Huang S; Chen X Nat. Protoc 2006, 1, 2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert M; Brisson JR; Karwaski MF; Michniewicz J; Cunningham AM; Wu Y; Young NM; Wakarchuk WW J. Biol. Chem 2000, 275, 3896–3906. [DOI] [PubMed] [Google Scholar]
- 19.Blixt O; Vasiliu D; Allin K; Jacobsen N; Warnock D; Razi N; Paulson JC; Bernatchez S; Gilbert M; Wakarchuk W Carbohydr. Res 2005, 340, 1963–1972. [DOI] [PubMed] [Google Scholar]
- 20.Antoine T; Heyraud A; Bosso C; Samain E Angew. Chem. Int. Ed 2005, 44, 1350–1352. [DOI] [PubMed] [Google Scholar]
- 21.Cheng J; Yu H; Lau K; Huang S; Chokhawala HA; Li Y; Tiwari VK; Chen X Glycobiology 2008, 18, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H; Cheng J; Ding L; Khedri Z; Chen Y; Chin S; Lau K; Tiwari VK; Chen XJ Am. Chem. Soc 2009, 131, 18467–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J; Guo Z Bioconj. Chem 2006, 17, 1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding L; Yu H; Lau K; Li Y; Muthana S; Wang J; Chen X Chem. Commun 2011, 47, 8691–8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun M; Li Y; Chokhawala HA; Henning R; Chen X Biotechnology letters 2008, 30, 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kakuta Y; Okino N; Kajiwara H; Ichikawa M; Takakura Y; Ito M; Yamamoto T Glycobiology 2008, 18, 66–73. [DOI] [PubMed] [Google Scholar]
- 27.Sugiarto G; Lau K; Qu J; Li Y; Lim S; Mu S; Ames JB; Fisher AJ; Chen X ACS Chem. Biol 2012, 7, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni L; Chokhawala HA; Cao H; Henning R; Ng L; Huang S; Yu H; Chen X; Fisher AJ Biochemistry 2007, 46, 6288–6298. [DOI] [PubMed] [Google Scholar]
- 29.Yu H; Yu H; Karpel R; Chen X Bioorg. Med. Chem 2004, 12, 6427–6435. [DOI] [PubMed] [Google Scholar]