

Abstract
Altered enteric microorganisms in concert with host genetics shape inflammatory bowel disease (IBD) phenotypes. However, insight is limited to bacteria and fungi. We found that eukaryotic viruses and bacteriophages (collectively, the virome), enriched from non-IBD, non-inflamed human colon resections, actively elicited atypical anti-inflammatory innate immune programs. Conversely, ulcerative colitis or Crohn’s disease colon resection viromes provoked inflammation, which was successfully dampened by non-IBD viromes. The IBD colon tissue virome was perturbed, including an increase in the Enterovirus B species of eukaryotic picornaviruses, not previously detected in fecal virome studies. Mice humanized with non-IBD colon tissue viromes were protected from intestinal inflammation while IBD-virome mice exhibited exacerbated inflammation in a nucleic acid sensing-dependent fashion. Furthermore, there were detrimental consequences for IBD patient-derived intestinal epithelial cells bearing loss-of-function mutations within virus sensor MDA5 when exposed to viromes. Our results demonstrate that innate recognition of IBD or non-IBD human viromes autonomously influences intestinal homeostasis and disease phenotypes. Thus, perturbations in the intestinal virome, or an altered ability to sense the virome due to genetic variation, contribute to the induction of IBD. Harnessing the virome may offer therapeutic and biomarker potential.
One Sentence Summary:
Human enteric viromes divergently shape host immunity and disease.
Introduction:
Viruses are the most abundant biological entity on earth and infect all types of life. Canonically considered obligate pathogens, viruses are a constant and emerging threat to human health, as exemplified by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) pandemic. Yet, many viruses also reside perpetually in human hosts without causing disease. These include both eukaryotic viruses and bacteriophages (1–4), existing both within their host cells, as well as free virions embedded within mucus layers of the intestine (5). The human intestinal virome is established at birth and is dominated by bacteriophages, while eukaryotic viruses gradually emerge after birth, as measured in feces that contains ~109 virus like particles (VLPs) per gram (2, 6). Despite hurdles in identifying the constituents of a normal human virome (7), robust fluctuations of the virome have been reported in IBD (1, 8–10), colorectal cancer (11, 12), type I diabetes (13), nonalcoholic fatty liver disease (14), cystic fibrosis (15), graft-versus-host-disease (16) and HIV infection (17) and a range of other diseases (18). However, the rapid cataloging of viral genomes in human tissues by sequencing and computational methods has vastly outpaced any mechanistic understanding we have of virome biology and whether these fluctuations contribute to disease.
Our understanding of the host innate immune response to virus stems predominantly from studies of resolving acute infection. This mammalian antiviral paradigm centers on the detection of virus moieties by cytosolic or endosomal receptors and production of type I or III interferon (IFNs) and hundreds of IFN stimulated genes (ISGs) that aim to eliminate virus, or the cell that it infects (19). Thus, how innate immune cells chronically respond to persistent eukaryotic or prokaryotic viruses in the intestine, as well as in other systemic niches (20), and how this shapes host physiology, are still poorly understood. Viruses inhabiting a healthy host (21, 22), and host innate sensing of viruses by gut-resident Toll-like Receptors (TLRs) or RIG-I like Receptors (RLRs) (21, 23–28) maintain homeostasis in the mouse intestine. Furthermore, loss-of-function variants in the gene IFIH1 that encodes the host virus receptor melanoma differentiation associated protein 5 (MDA5) were shown to significantly associate with IBD by genome wide-association studies, GWAS (29). However, functional understanding of human host and healthy or diseased enteric virome interactions are currently lacking.
In this study, we sought to define protective or inflammatory potential of healthy and diseased human enteric viromes recovered directly from fresh patient colon resections or ileostomy fluid in vitro and in vivo. Our results reveal a surprising autonomous functionality of the human enteric virome in educating host immunity and contributing to IBD phenotype, which could potentially be leveraged for therapeutic benefit. Finally, we identify specific eukaryotic and bacterial viruses in colon tissue and ileostomy fluid that are associated with healthy and disease phenotypes. This revealed unique virus populations that associate with IBD, including eukaryotic Enteroviruses, not previously detected in fecal viromes analyses. Taken together, our results indicate the enteric virome exerts autonomous immunoregulatory potential, akin to the microbiota.
Results:
Divergent innate immunomodulation of human macrophages by colon resection-derived healthy and IBD viromes
To determine whether the human enteric virome has homeostatic or pathophysiological properties, we enriched virus-like particles (VLPs) from fresh colon resections taken from non-inflamed/non-IBD, Ulcerative Colitis (UC) or Crohn’s disease (CD) patients, post-surgery (Fig 1A, Table S1). We reasoned that since viruses are obligate intracellular pathogens that rely on a host organism for successful replication, and bacteriophages reside within mucosal layers (5) and are readily taken up by eukaryotic cells (30–32) certain viruses may be better represented in upper intestinal tissue than in feces. Tissue from non-IBD individuals was confirmed to be not inflamed and UC and CD tissue was confirmed to be inflamed by a blinded clinical pathologist (Table S1). There are multiple protocols for isolating VLPs (33, 34), each with their own biases and limitations. We did not use a cesium chloride (CsCl) density gradient centrifugation, since it was previously shown to be detrimental to virus activity (35). Instead, we used 0.22 μm size filtration, removal of bacterial cell walls, a chloroform step that disrupts lipid membranes and endotoxin blocking. We considered that chloroform could remove enveloped viruses from our preparations but since its impact is greater on bacterial species (36), and previous reports suggested human stool contains limited enveloped viruses (34, 37), we deemed this step essential to avoid contaminating bacteria that would confound subsequent immunomodulatory experiments. We quantified our VLP preparations by confocal microscopy using nucleic acid-binding SYBR Gold and found an average of 2.8x107 VLPs per mg of colon tissue from non-IBD, UC or CD individuals (Fig 1B). Electron microscopy images of VLP preparations resembled eukaryotic and prokaryotic viruses (Fig S1A), and importantly, both endotoxin and cytokines were below the limits of detection (Fig S1B–F).
Fig 1. Human colon tissue resident viruses trigger anti-inflammatory while IBD viromes divergently provoke pro-inflammatory macrophage responses.
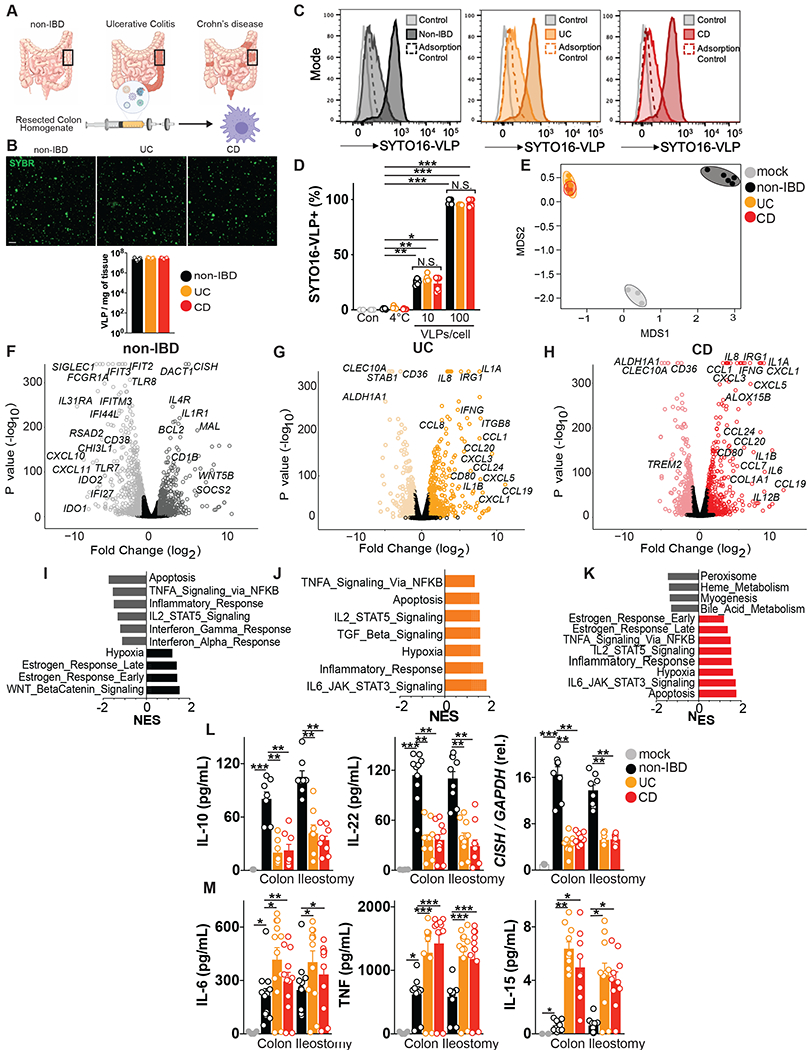
(A) Virus-like particles (VLPs) isolated from colon resections post-surgery from non-IBD (n=5), ulcerative colitis (UC, n=5) or Crohn’s disease (CD, n=5) patients. (B) Visualization and quantification of VLP isolates per mg of colon tissue using confocal microscopy for RNA/DNA using SYBR Gold. Scale bar is 1 μm. (C, D) Flow cytometric quantification of uptake of SYTO-16 labeled human colon-resection derived VLPs by human peripheral blood-derived macrophages (1 or 10 VLPs per cell). Adsorption control is fluorescently labeled VLPs delivered to human macrophages at 4°C. (E) Multidimensional scaling (MDS) plot of human macrophage transcriptional profiles induced by non-IBD or UC or CD derived VLPs (10 VLPs/cell, 24h). (F-H) Volcano plots showing differentially expressed genes comparing F) non-IBD delivery to mock adsorption, (G) UC delivery to mock adsorption or (H) CD delivery to mock adsorption. (I-K) Hallmark gene sets significantly (P value< 0.05, FDR <0.20) represented in (I) non-IBD, (J) UC or, (K) CD VLP induced transcriptional programs as assessed by Gene Set Enrichment Analysis (GSEA). NES is normalized enrichment score. (L) Production of anti-inflammatory mediators interleukin (IL)-10, IL-22 or CISH or (M) pro-inflammatory mediators TNF, IL-6, IL-15 as measured by ELISA or qPCR following delivery of VLPs (10 VLPs/cell, 24h) extracted from non-IBD, ulcerative colitis (UC) or Crohn’s disease (CD) colon resections (n=8-12 per group) or ileostomy content (n=8 per group) to primary human peripheral blood-derived macrophages. Data are mean ± s.e.m. of biological replicates. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as determined by one-way ANOVA with Tukey’s multiple comparison test.
Macrophages are one of the most abundant leukocytes in the intestinal mucosa where they are essential for maintaining homeostasis but are also implicated in the pathogenesis of IBD (38). Macrophages are also critical sensors of virus, including endocytosed bacteriophages (30). Therefore, to identify the precise innate immune response initiated by eukaryotic or prokaryotic viruses residing in healthy or diseased human colon, we exposed primary human peripheral blood derived macrophages to collective VLPs isolated from non-IBD, UC or CD patient colon resections (Fig 1A). Fluorescently labeled colon resection-derived VLPs demonstrated dose-dependent internalization by primary human macrophages that was equal for non-IBD, UC or CD-derived viromes (Fig 1C, D). Furthermore, virome uptake was dependent on endocytosis as treatment with brefeldin A, a vesicular transport inhibitor, greatly reduced intracellular accumulation (Fig. S2). Thus, the human virome, that includes both eukaryotic viruses and bacteriophages, is readily internalized by human immune cells via endocytosis for pathogen associated molecular pattern (PAMP) recognition, consistent with other reports (30, 31). To assess the transcriptional pathways induced by the human colon tissue virome in macrophages, we performed unbiased RNA sequencing. Multi-dimensional scaling (MDS) analysis found non-IBD colon-resident viromes initiated a distinct transcriptional program, that was highly reproducible between 5 different patient VLP sources (Fig 1E). Conversely, UC or CD human enteric viromes from multiple patients induced macrophage transcriptional programs that were distinct from non-IBD virome-induced programs but almost indistinguishable from one another (Fig 1E). Interestingly, viruses resident to normal colon tissue predominantly suppressed the host innate immune response as more transcripts were down-regulated in macrophages exposed to non-IBD virus than were up-regulated (Fig S3). On the other hand, both UC and CD viromes triggered a prominent upregulation in gene expression of macrophages and these transcriptional programs were almost identical (Fig S3). A striking feature of the non-IBD virome induced macrophage transcriptional signature was a broad downregulation of the classical antiviral response. Non-IBD viromes significantly downregulated an array of virus receptors, signalling adapters and IFN stimuated genes (ISGs) such as TLR7, TLR8, IFIT2, IFIT3, RSAD2 (also known as Viperin) and IFI44L (Fig 1F and Fig S3D). This atypical host response to non-IBD viromes was also exemplified by the prominent induction of anti-inflammatory and pro-survival genes such as IL4R, CISH, BCL2, SOCS2 (Fig 1F) and multiple genes that define a homeostatic/resolving macrophage state (39, 40) (Fig S3C). In stark contrast, UC and CD viromes both robustly promoted inflammatory pathways in macrophages including many cytokines (IL6, IL1B, IL8, IFNG) and chemokines (CCL1, CCL20, CCL24, CXCL3, CXCL5, Fig 1G, H). Gene-set enrichment analysis defined the significant pathways downregulated by non-IBD viromes to be apoptosis, inflammatory and antiviral (Fig 1I) while those pathways that were upregulated were pro-survival and pathways known to polarize macrophages toward homeostatic/resolving states such as WNT-β Catenin signaling (Fig 1I). In constrast, both UC and CD viromes robustly promoted inflammatory and apoptotic pathways (Fig 1J,K). We next confirmed the divergent macrophage activation states induced by non-IBD and IBD viromes using ELISA and qPCR with VLPs isolated from additional patient colon resections. Cytokines IL-10, IL-22, TGFβ or negative regulators of inflammation CISH or SOCS2, hallmark genes of anti-inflammatory and tissue remodeling type macrophages (39, 40) were all consistently and significantly elevated in macrophages exposed to VLPs isolated from 12 different non-IBD patient colon resections (Fig 1L, Fig S4). Notably, IL-22 induction in macrophages by non-IBD VLPs is consistent with recent reports of an increased abundance of IL-22-expressing CD45+ cells in the intestinal mucosa of mice mono-associated with apathogenic eukaryotic enteric viruses (41). Conversely, macrophages that internalized viromes derived from UC or CD intestinal resections from 8-12 separate patients, repeatedly displayed significantly enhanced production of cytokines TNF, IL-6, Type I and II IFNs, IL-1β and IL-12, that define pro-inflammatory type macrophages, as well as other cytokines such as IL-15, IL-8 and IL-31 with designated roles in intestinal inflammation and IBD (42, 43) (Fig 1M, Fig S4). Importantly, inhibition of endocytosis or heat inactivation of VLPs, failed to induce macrophage cytokine responses (Fig S5). However, crosslinking of VLP preparations, at a dose that is predicted to inactivate replication of the majority of viruses, did not significantly reduce innate responses indicating that virus replication is not an absolute requirement for virome stimulatory capacity (Fig S5). Moreover, we observed similar divergent macrophage responses with virus isolated from non-IBD, UC and CD ileum lumen contents (Fig 1L,M, Fig S4, S6, Table S2). Taken together, these results demonstrate that human enteric viruses residing in colon tissue or ileum content autonomously invoke host innate immune responses. The macrophage response to enteric viruses is atypical to the classical antiviral response normally characterized by the induction of type I IFNs and ISGs. Enteric viruses from a healthy intestine instead dampened the classical antiviral response and promoted anti-inflammatory and pro-survival programs. Conversely, enteric viruses existing within an inflamed UC or CD intestine independently and robustly promoted a state of inflammation.
IECs sense and divergently respond to non-IBD and IBD enteric viruses
Intestinal epithelial cells (IECs) express innate immune receptors that recognize and respond to commensal microorganisms. Furthermore, bacteriophages are readily internalized by IECs (31, 32). We therefore also tested the ability of healthy or IBD enteric viruses to affect epithelial barrier function and the immune response of IECs. We utilized a Transwell Caco2 cell monolayer system and exposed IEC monolayers with viruses isolated from non-IBD, UC or CD colon resections and observed significantly more barrier integrity damage induced by both UC and CD-derived enteric viruses in a TLR4-independent manner, as measured by transepithelial electrical resistance (TEER, Fig S7A,B). Disrupted barrier integrity elicited by IBD viruses was also associated with a greater downregulation of transcripts for tight junction proteins zonula occludens-1 (ZO-1) and occludin (Fig S7C,D). Importantly, IFNλ which controls both acute and persistent viral infections in the intestine (44, 45) and confers protection from intestinal inflammation (22) was preferentially induced by non-IBD viruses (Fig S7E). In contrast, viruses isolated from either UC or CD colons promoted production of pro-inflammatory cytokines such as TNF and IL-15 (Fig S7F,G) that play crucial roles in intestinal tissue destruction and the pathogenesis of IBD (42, 43, 46). Of note, IL-15 was also recently shown to be induced by the murine enteric virome in dendritic cells to promote expansion of innate epithelial lymphocytes (IELs) (25). Thus, similar to macrophages, IECs sense and divergently respond to non-IBD and IBD enteric viruses whereby IBD viromes promote both inflammatory responses and disruption of epithelial barrier function.
Non-IBD viromes suppress inflammation driven by IBD viromes.
Fecal microbiota transplants (FMT) to restore disrupted microbiota are proving beneficial in both experimental and clinical settings (47) but the virome content of these preparations is currently not considered. We wondered if isolated non-IBD enteric viruses could overcome the inflammatory response induced by IBD viruses. To this end, we mixed increasing ratios of non-IBD colon resection derived viruses with IBD viruses while maintaining the same overall number of VLPs and exposed human macrophages to them. Increasing amounts of non-IBD viruses were capable of suppressing the ability of IBD-derived viruses to induce pro-inflammatory cytokines such as IL-6, TNF and IL-15 in a dose-dependent manner (Fig 2A–C). Moreover, increasing the ratio of non-IBD viruses could partially restore the ability of macrophages to produce anti-inflammatory cytokines such as IL-22 (Fig 2D). To test if the product of the immune response to healthy viruses in the form of secreted soluble factors could also suppress the IBD virome responses, we performed supernatant transfer experiments. In brief, human macrophages were exposed to non-IBD VLPs for 24 hours, then supernatant was collected, clarified, and placed on fresh cells in the presence of IBD colon-derived VLPs. Supernatant from non-IBD VLP treated macrophages was capable of suppressing the production of IL-6, TNF, IL-15 by IBD VLPs (Fig 2E–G). Furthermore, heat inactivation ameliorated suppressive activity of non-IBD virome-induced macrophage supernatant but UV crosslinking did not (Fig 2E–G), demonstrating soluble proteins, but not residual viruses in the supernatant, were eliciting anti-inflammatory activity. Thus, this data demonstrates the potential utility of viruses in a healthy intestine, or the anti-inflammatory immune response to it, to suppress the inflammatory capacity of viruses in a diseased intestine.
Fig 2. Suppression of IBD virome-induced inflammation by healthy human enteric viromes.
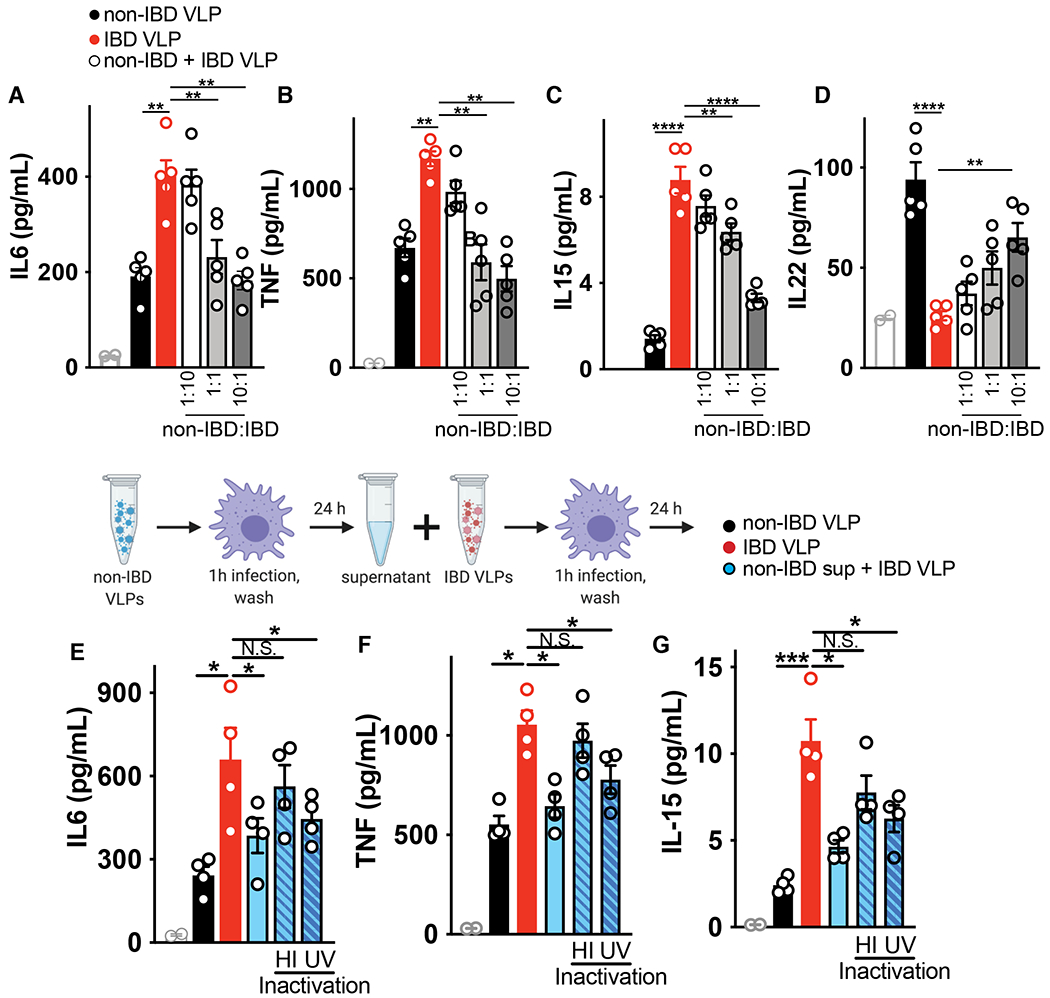
Primary human peripheral blood-derived macrophages were delivered colon resection-derived non-IBD VLPs (10 VLPs/cell), IBD VLPs (10 VLPs/cell) or indicated ratios of non IBD to IBD VLPs (total of 10 VLPs/cell) and (A) IL-6, (B) TNF, (C) IL-15 and (D) IL-22 levels were measured at 24h by ELISA. Schematic for incubation of human primary macrophages with IBD VLPs and 24h supernatant from non-IBD VLP-exposed macrophages. (E) IL-6, (F) TNF and (G) IL-15 levels were measured by ELISA following IBD VLP delivery to macrophages with the addition of non-IBD macrophage supernatant that was heat inactivated (95°C for 10 min) or UV crosslinked (three times 200 mJ/cm2 for 15 minutes) as controls. Data are mean ± s.e.m. of 5 biological replicates. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as determined by one-way ANOVA with Tukey’s multiple comparison test.
Perturbed UC and CD human colon tissue virome
To gain a mechanistic understanding of the divergent immunomodulation by non-IBD or IBD derived human colon tissue or ileostomy fluid-derived viromes, we profiled the virus types in each cohort since only fecal VLPs have been examined to date. DNA and RNA were purified from preparations of VLPs from surgical colon resections or ileostomy fluid, RNA was reverse transcribed, followed by second strand synthesis of total nucleic acid, and random amplification and characterized by metagenomic sequencing. After filtering out human and other contaminant (primers and adapters) and low-quality data, sequences were assigned a eukaryotic viral or bacteriophage taxonomic lineage. Specific eukaryotic viral families were selected for subsequent analysis based on the alignment length and percent identity for each family that separated the high-quality alignments from potentially spurious alignments (Table S3). Total reads aligning to eukaryotic or prokaryotic viruses revealed a significant increase in eukaryotic virus reads in CD patients compared to non-IBD individuals and a significant increase in bacteriophage reads in CD patients compared to non-IBD or UC patients (Fig 3A). Of the high-quality reads for subsequent analysis, we found Papillomaviridae, Picornaviridae, Coronaviridae, Orthomyxoviridae and Anelloviridae animal viruses, Virgaviridae plant viruses and a number of unknown eukaryotic virus families in colon tissue (Fig 3B). Furthermore, we found the majority of VLP classifications in human colon tissue to be bacteriophage families consisting of dsDNA Caudovirales order including Siphoviridae, Myoviridae, Podoviridae, Herelleviridae, Ackermannviridae and unknown; Ligamenvirales order member Lipothrixviridae, as well as ssDNA phage family Microviridae. Quantification of viral species richness showed no significant difference between groups (Fig S8A) but differential analysis showed a significant elevation in the Picornaviridae family member Enterovirus B, in both UC and CD colon tissue compared to non-IBD controls (Fig 3C). Enterovirus B has not been reported in fecal metagenomic studies performed to date, but has been previously detected in resected terminal ileum of immunocompetent pediatric patients with Crohn’s disease (48). This further highlights the need to catalog viruses and other microorganisms (49) in intestinal tissue that is not fully captured in fecal material. Human enteroviruses are extremely common RNA viruses that spread mainly through the fecal-oral route. Species B enteroviruses consist of coxsackieviruses B1–B6 (CVB1–6), coxsackievirus A9 (CVA9), over 30 serotypes of echoviruses and more than 20 EV-B serotypes that all share the same overall structure found in picornaviruses, a single stranded ~7500 bp RNA genome. We searched for specific Enterovirus B serotypes in our data and found high identity mapping to Echovirus B75, Echovirus B5 and Echovirus 26, that were significantly elevated in both UC and CD colon resection samples. The wide prevalence and persistence of enteroviruses within humans has suggested a potential role for these viruses in complex human disease, including type I diabetes (50). Here we now identify Enterovirus B, and particularly Echovirus serotypes, as a potential pathogenic factor in UC and CD.
Fig 3. Detection and characterization of VLPs in colon surgical resections from non-IBD, Ulcerative Colitis and Crohn’s disease patients.
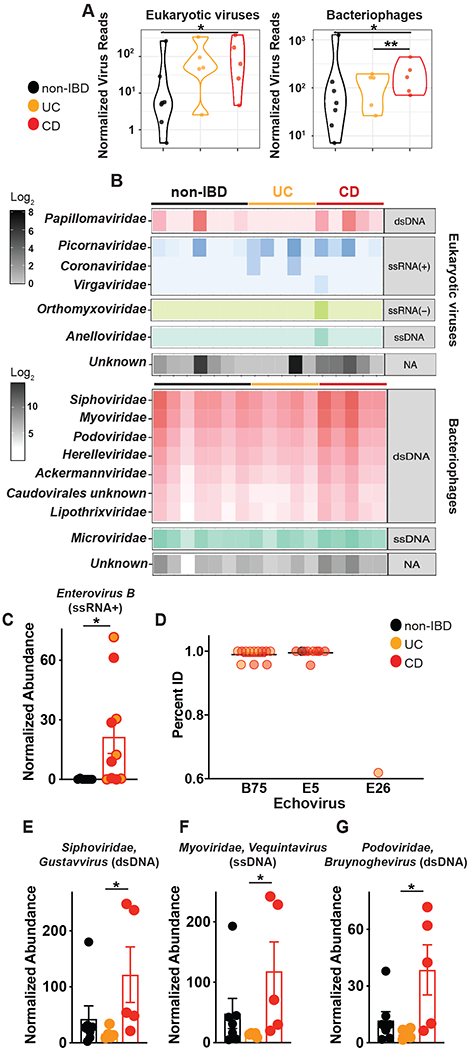
VLPs were assessed using VLP DNA and RNA metagenomic sequence data. Sequences were assigned a eukaryotic viral or phage taxonomic lineage using primary searches against virus sequence databases and subsequently confirmed in secondary searches using reference databases with additional non-viral taxonomic lineages. Specific eukaryotic viral families were selected for subsequent analysis by gating on the alignment length and percent identity for each family that separated the high-quality alignments from potentially spurious alignments (Table S3). (A) Violin plots represent the distribution of individual datasets for normalized reads aligning to eukaryotic viruses or bacteriophages from non-IBD, UC or CD cohorts; samples were compared using two-sided Wilcoxon rank sum tests, *P<0.05. (B) Taxonomic assignments of VLP sequences in non-IBD, UC or CD colon tissue. (C) Significantly elevated abundance of eukaryotic virus, Picornaviridae>Enterovirus B in UC and CD colon resections. (D) Identification of Echovirus B75, Echovirus E5 and Echovirus E26 Enterovirus B serotypes in UC or CD colon tissue by bbmap. All potential Picornavirales reads were re-mapped one at a time to six Enterovirus B genomes (KF874627.1, NC_001472.1, AF083069.1, AY302539.1, D00627.1, NC_038307.1), using the bbmap tool, mapPacBio.sh with the parameters recommended for remote homologies (vslow k=8 maxindel=200 minratio=0.1). The subset of these reads that mapped to Enterovirus B, along with the previously identified Enterovirus B reads, were plotted, split into Serotypes on the x-axis and percent identity on the y-axis. Each read found is plotted and grouped and colored by the type of sample in which it was found (Non-IBD, UC or CD). Non-IBD versus UC and non-IBD versus CD were significantly different (p-adj = 0.029, 0.001). (E) Top significantly increased genera of Siphoviridae, (F) Myoviridae or (G) Podoviridae bacteriophages in CD colon resections. Data are the mean of 5-7 biological replicates. *P<0.05, Kruskal–Wallis test with a Dunn’s post-hoc test.
For bacteriophages, CD colon tissue exclusively demonstrated a significant expansion, consistent with reports of CD fecal viromes (1). We found significant elevations in total phage aligned reads (Fig 3A), a distinct separation of CD from non-IBD and UC by NMDS analysis (Fig S8B) and a multitude of significantly increased genus of Caudovirales members Siphoviridae, Myoviridae and Podoviridae (Fig 3D–F, Fig S8C). Interestingly, ileostomy fluid VLPs showed significantly reduced viral diversity compared to colon derived VLPs (Fig S9C) underscoring tissue rather than luminal contents to be the abundant source of virus in the intestine. Although, NMDS analysis did indicate a distinct separation in ileostomy fluid virus populations between non-IBD, UC and CD samples (Fig S9D), despite UC pathology being confined to colon. Detectable eukaryotic families in ileostomy fluid were limited to Anelloviridae, Papillomaviridae, Picornaviridae and Virgaviridae eukaryotic viruses and the bacteriophage families were Caudovirales family members Siphoviridae, Myoviridae, Podoviridae, Herelleviridae, Ackermannviridae and unknown; Ligamenvirales order member Lipothrixviridae; as well as ssDNA phage family Microviridae (Fig S9E). Interestingly, differential analysis of ileostomy fluid viruses revealed a significant reduction in the dsDNA bacteriophage Ackermannviridae Kuttevirus and a significant elevation in Helleviridae Okubovirus, Siphoviridae Beetrevirus and Siphoviridae Vegasvirus in IBD patients (Fig S9F) indicating distinct virome fluctuations in different niches of the intestine.
Nucleic acid sensing requirement for VLP immunomodulation
We next determined the virus receptor requirement for host responses to human enteric viruses we isolated from patient colon resections. Virus-derived RNA structures are recognized by pattern recognition receptors (PRRs) such as cytosolic (RIG-I)-like receptors (RLRs), or endosomal Toll-like receptor (TLR)3, 7, 8 and viral DNA is mainly recognized by cyclic GMP-AMP (cGAMP) synthase (cGAS) or TLR9 (51). So far, endosomal TLR3, TLR7, TLR9 and cytosolic RIG-I have all been shown to be essential for sensing of the healthy murine virome (21, 25, 52). Furthermore, since VLP isolation methods only enrich for viruses, other bioactive molecules may be present so demonstrating the requirement of nucleic acid sensing for the observed immunomodulation was also an essential control. We generated bone marrow–derived macrophages from mice deficient in the RLR signaling adapter mitochondrial antiviral signaling protein (MAVS), DNA sensor cGAS, TLR adapter protein MyD88 or endotoxin sensor TLR4, and exposed them to human colon VLPs (Fig 4A,B). Indeed, MAVS, cGAS or MyD88 deletion resulted in attenuated non-IBD virome responses, as measured by pro-survival gene Cish (Fig 4A). Conversely, deletion of endotoxin receptor TLR4 had little effect (Fig 4A), underscoring recognition of virus nucleic acid in VLP-driven responses. Similarly, pro-inflammatory cytokine IL-6 production by colon tissue derived IBD VLPs was also significantly diminished in MAVS-, cGAS-, and MyD88-deficient macrophages but was unchanged in TLR4-deficient macrophages (Fig 4B). Notably, ileostomy fluid-derived VLPs, that had significantly reduced virus diversity compared to colon tissue (Fig S9) and was mostly bacteriophages and animal viruses of the Anelloviridae family (Fig S9), required DNA sensor cGAS exclusively for cytokine induction. Certainly, a dominance of viral-derived DNA was observed in the ileum content whilst the colonic tissue virome was a mix of RNA and DNA (Fig S10). This data suggests there is recognition redundancy for normal/non-IBD or IBD enteric viruses but qualitative and quantitative differences in the downstream immune responses induced by them. RLRs RIG-I (retinoic acid-inducible gene I; encoded by DDX58) and MDA5 (melanoma differentiation-associated gene 5; encoded by IFIH1) are activated by distinct viral RNA structures to differentiate between viral and cellular RNAs. MDA5 senses long double-stranded (ds)RNA, while RIG-I responds to blunt-ended dsRNA bearing a triphosphate (ppp) moiety (53). Transfection of purified RNA isolated from non-IBD VLPs, triggered more IL-10 than equal concentrations of RNA from UC or CD VLPs (Fig 4C). In contrast, UC or CD VLP RNA triggered more IL-6 production (Fig 4D) demonstrating divergent macrophage responses were not due to differences in viral tropism or levels. Importantly, digestion of single-stranded or double-stranded VLP RNA with RNase I or III, respectively, or removal of phosphates with Calf Intestinal Phosphatase (CIP), eliminated this response (Fig 4C, D) demonstrating the existence of “non-self” RNA moieties within the human enteric virome and the requirement for both RIG-I and MDA5 in their recognition. Divergent anti- or pro-inflammatory responses by non-IBD or IBD enteric viruses were also induced by transfection of VLP-derived DNA, in a cGAS-dependent manner (Fig 4E, F). Thus, altered RNA or DNA moieties that are sensed by host virus receptors RIG-I, MDA5 or cGAS (and likely other receptors), rather than an adaptive function of viral proteins on the host, contribute to the divergent activation thresholds and immunomodulation by normal/non-IBD and IBD enteric viromes. This deviation in RNA and DNA viral ligands within the diseased virome is likely attributed to the fluctuations of certain viruses we observed. However, it is also appealing to speculate that akin to some bacteria (54), normally symbiotic viruses may also become pathobionts when in an inflamed environment.
Fig 4. Host viral receptor requirement for sensing human enteric viruses.
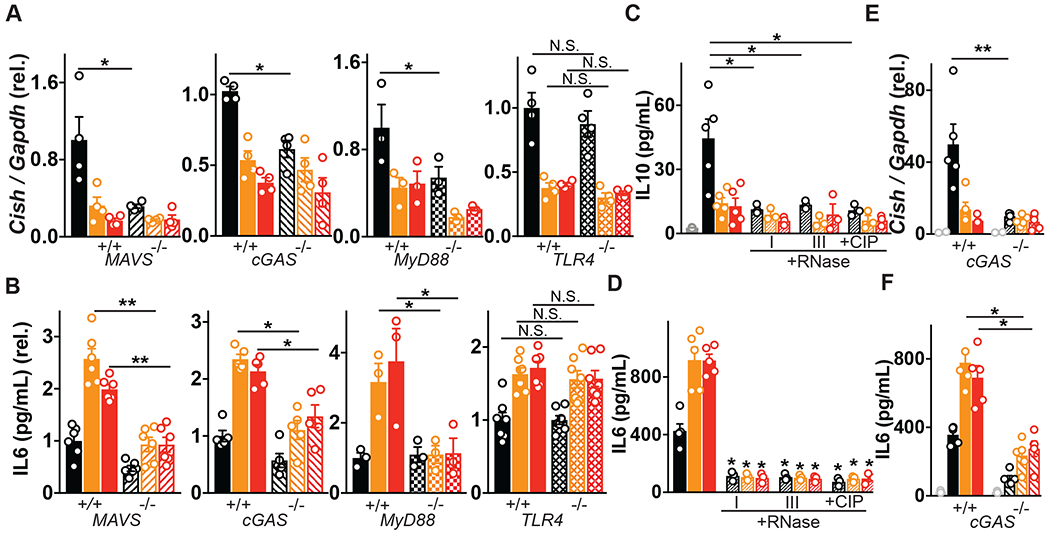
(A) Cish or (B) IL-6 levels following exposure of primary bone marrow derived macrophages (BMDMs) from wild-type, MAVS−/−, cGAS−/−, MyD88−/− or TLR4-deficient mice to non-IBD, Ulcerative Colitis (UC) or Crohn’s disease (CD) colon resection-derived VLPs as measured by qPCR and ELISA. (C) IL-10 or (D) IL-6 production 24h following transfection of THP1 cells with 2.5 μg of VLP RNA or VLP RNA pre-treated with RNase I (5 μg/mL, 1h), RNase III (5 μg/mL, 1h) or Calf Intestinal Phosphatase (CIP, 250U, 1h). (E) Cish levels or (F) IL-6 production 24h following transfection of WT or cGAS−/− BMDMs with 2.5 μg of VLP DNA as measured by qPCR or ELISA. Data are mean ± s.e.m. of 2-5 biological replicates repeated twice. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as determined by one-way ANOVA with Tukey’s multiple comparison test.
Lack of virome immunomodulation with IBD-associated MDA5 loss-of-function
Given the existence of genetic mutations within IFIH1, that codes for the innate virus sensor MDA5, were identified to associate with IBD incidence by GWAS (29) and that we observed elevated Enterovirus B of the Picornaviridae family in IBD colon tissue, that can be sensed by MDA5 (55), we examined the consequence of these mutations on responses to the human non-IBD and IBD enteric virome. Notably, IFIH1 gain-of-function has been reported as a cause of a type I interferonopathy encompassing a spectrum of autoinflammatory phenotypes including systemic lupus erythematosus and Aicardi-Goutières syndrome (56). Yet, intriguingly, all IBD-associated IFIH1 variants have predicted loss-of-function biological effects, either truncating the protein (rs35744605, E627X), affecting essential splicing positions (rs35732034 in a conserved splice donor site at position +1 in intron 14) or altering a highly conserved amino acid (rs35667974/I923V; Fig 5A) within the C-terminal domain (CTD) which recognizes and binds to RNA (57). We transfected HEK-293T cells with wild-type MDA5 or E627X or I923V MDA5 coding variants and found the wild-type and MDA5 mutants were expressed at comparable levels although the E627X truncation, that lacks a part of the helicase domain and the entire CTD in which I923 resides, showed slightly reduced MDA5 protein levels (Fig 5B). Wild-type MDA5 conferred similar responsiveness to the synthetic RNA ligand poly(I:C) and RNA isolated from healthy human colon tissue VLPs but was not responsive to self RNA (Fig 5C). However, both IBD-associated MDA5 variants exhibited a loss of function phenotype to poly(I:C) (Fig 5C) in agreement with a previous report (58). Importantly, a loss of function phenotype to human colon derived viral RNA was observed for both IBD-associated coding MDA5 variants (Fig 5C). We next examined the functional consequence of a loss of MDA5 function on intestinal health. MDA5 is predominantly expressed in epithelial cells (Fig S11) so we examined functional consequences of loss-of-function MDA5 by converting induced pluripotent stem cells (iPSCs) derived from a compound heterozygote UC patient carrying a single allele of rs35732034/IVS14+1 and a single allele of rs35744605/E627X into human intestinal epithelial cells (hIECs, Fig 5D, Fig S12). Compared to non-affected individuals, hIECs from patients carrying loss-of-function MDA5E627X/IVS14+1 displayed similar morphology and barrier integrity at steady state, as measured by confocal microscopy for F-actin, ZO1 and E-cadherin (Fig 5E). However, unlike iPSC-derived hIECs cells bearing wild-type MDA5, MDA5E627X/IVS14+1 iPSC-derived hIECs were severely impaired in their ability to maintain barrier integrity when exposed to enteric viruses derived from non-IBD colon resections (Fig 5F). Furthermore, damage to MDA5E627X/IVS14+1 hIECs was exacerbated when exposed to IBD derived viromes (Fig 5F). Moreover, IBD-risk MDA5E627X/IVS14+1 iPSC-derived hIECs displayed significantly abrogated virome-induced IFNλ production compared to wild-type cells (Fig 5G). Thus, abolished innate responses to the virome due to human genetic variation within virus receptor MDA5 has detrimental consequences for intestinal epithelial cell function and integrity. Moreover, intestinal damage is intensified in patients bearing IBD-risk MDA5 variants responding to an IBD virome, underscoring the ability of environmental cues to uniquely impact IBD phenotypes in the context of host genetics.
Fig. 5. Detrimental consequences for IBD patient intestinal epithelial cells bearing MDA5 loss-of-function variants in the context of the virome.
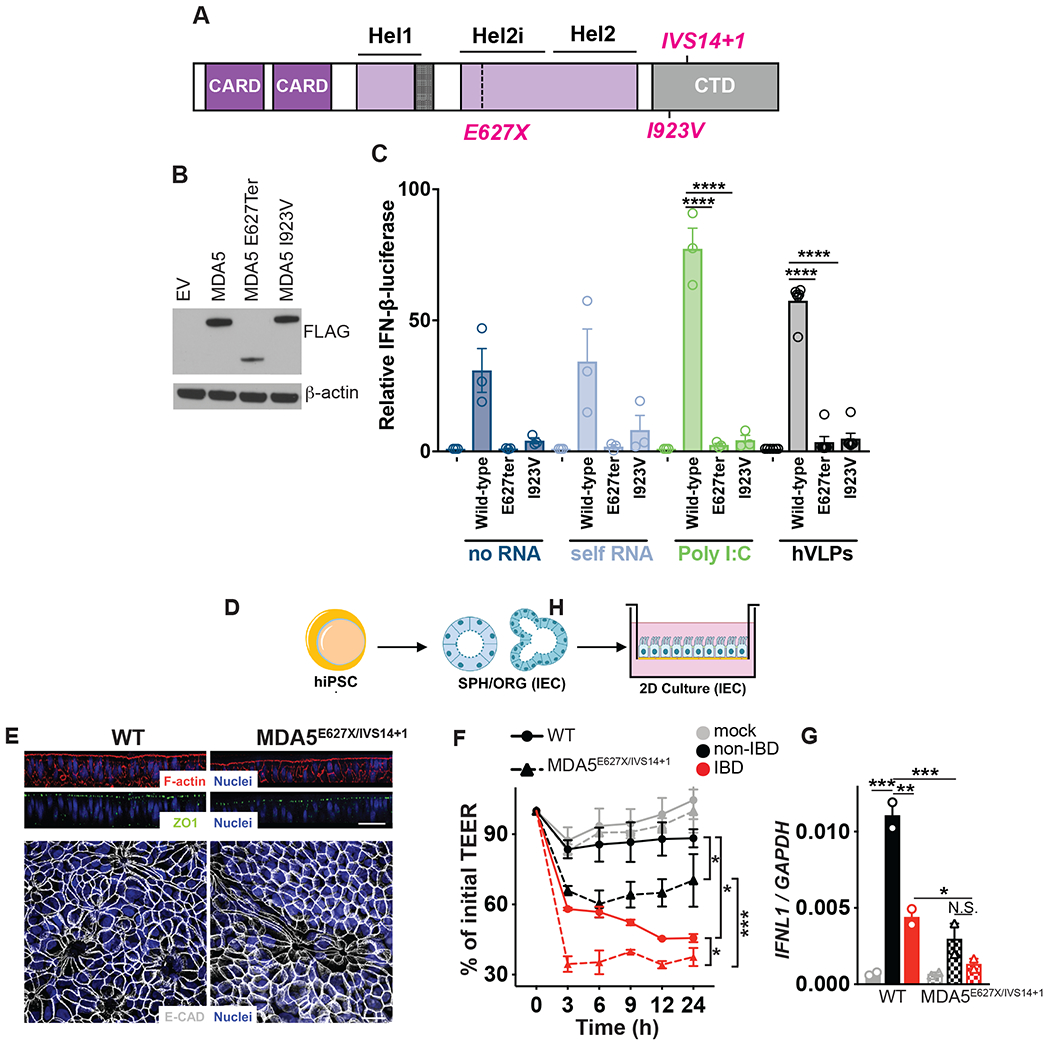
(A) Schematic of human MDA5 and mapped IBD-associated MDA5 variants rs35744605/E627X, rs35667974/I923V and rs35732034/IVS14+1. (B) Western Blot of ectopically expressed, FLAG-tagged MDA5 in HEK293T cells. (C) Relative activation of IFNβ luciferase reporter in HEK293T cells transfected with Empty vector (EV), FLAG-tagged WT MDA5, MDA5E627Ter or MDA5I923V and transfected with 1μg/mL RNA isolated from the cytoplasm of HEK293T cells (self RNA), poly I:C or RNA isolated from human colon resection virus like particles (VLPs) for 24 hours. (D) Schematic of patient lymphoblastoid cell line (LBL) derived human induced pluripotent stem cells (hiPSCs) transdifferentiated into intestinal epithelial cell monolayers. (E) Differentiated mature monolayers evaluated for structural integrity markers. Upper panel, orthogonal view of F-actin (phalloidin, red), tight junction protein ZO-1 (green) and nuclei (DAPI, blue) by confocal microscopy. Scale bar represents 50μm. Lower panel, maximal intensity projection images of epithelial adherens junction marker E-cadherin (gray) and nuclei (DAPI, blue). Scale bar is 20 μm. (F) Transepithelial electrical resistance (TEER) over indicated time or (G) IFNL1 levels at 24h as measured by qPCR of WT or MDA5 E627Ter/IVS14+1 patient hiPSC-derived intestinal epithelial cell monolayers and exposed to VLPs (10 VLPs/cell) extracted from non-IBD or IBD colon resections. Data are mean ± s.e.m. of 2-5 biological replicates repeated twice. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as determined by one-way ANOVA with Tukey’s multiple comparison test.
Spontaneous expansion of inflammatory phagocytes in humanized IBD virome mice
We next assessed the ability of non-IBD or IBD human colon-resident viruses to shape local immunity and intestinal disease in vivo. To achieve this, we first diminished the murine enteric virome of C57BL/6 mice using an antiviral (AV) cocktail by oral gavage for 10 days (21) and then administered human colon resection-derived VLPs 3 times intragastrically (Fig 6A). To complement our functional data in macrophages, we first assessed the influence of healthy or diseased enteric viromes on gut resident mononuclear phagocytes (MNPs), especially those bearing the fractalkine receptor CX3CR1 (CX3CR1+ MNPs), since they have established roles in the induction of immune responses toward enteric bacteria (59) and more recently intestinal fungi (60). In parallel, we examined the influence of human enteric viromes on subsets of dendritic cells (DCs) differentially expressing integrins CD11b and CD103 that initiate immune tolerance (61, 62). Mice with a healthy humanized virome showed no changes in lamina propria CX3CR1+ MNPs compared to wild-type or antiviral treated mice (Fig 6B,C) but had elevated tolerizing CD11b+CD11c+ phagocytes expressing CD103 (Fig 6B,C, Fig S13). In contrast, mice with an IBD humanized virome displayed a significant accumulation of MNPs expressing CX3CR1 and a reduction in CD11b+CD11c+ MNPs expressing CD103 in the colonic lamina propria, both in number and as a fraction of the total CD11b+ phagocyte compartment (Fig 6B,C, Fig S13). Thus, IBD-associated enteric viruses can promote an inflammatory state in the intestine in vivo by spontaneously influencing the ratio of CX3CR1+ and CD103+ lamina propria MNPs.
Fig 6. Mice with a “humanized” non-IBD virome had attenuated intestinal inflammation while those with a “humanized” IBD-derived virome exhibited intestinal inflammation.
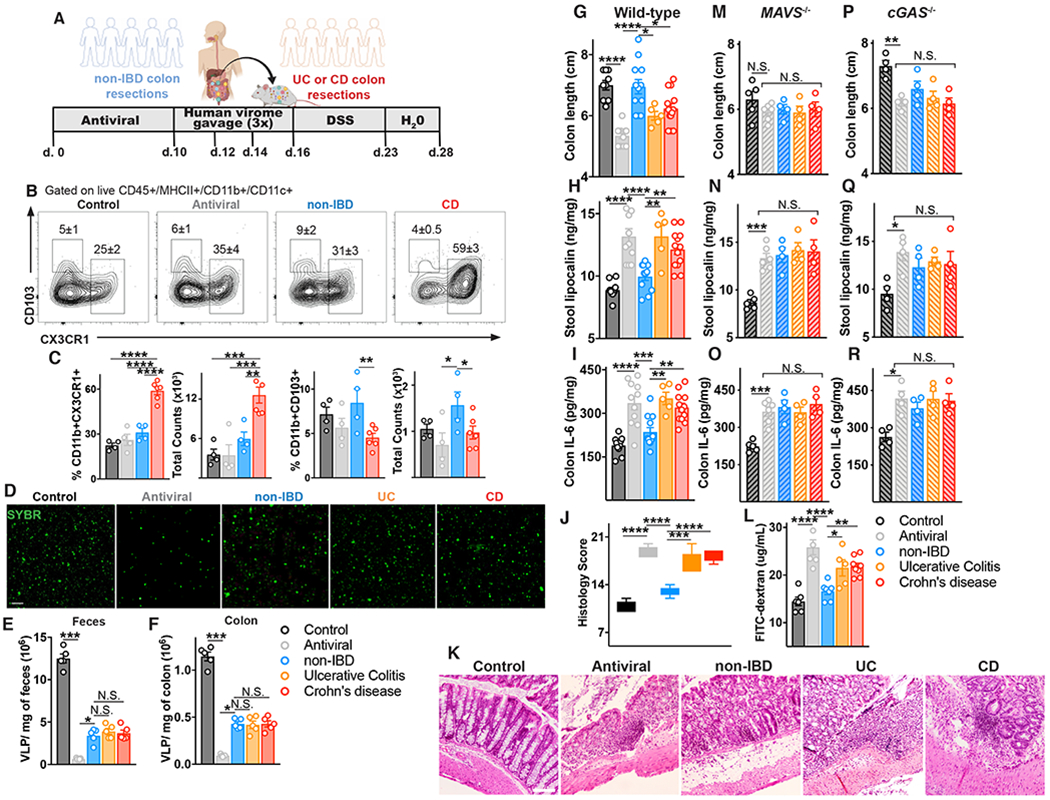
(A) Schematic of mouse gut virome depletion using an antiviral cocktail (acyclovir 20 mg/kg, lamivudine 10 mg/kg, ribavirin 30 mg/kg, oseltamivir 10 mg/kg, gray, daily gavage for 10 days), and reconstitution with 200 μL of human non-IBD or IBD VLPs pool (4×108 VLPs per mouse) by gavage on day 10, 12 and 14. 2.5% DSS colitis model was commenced on day 16. (B) Flow cytometry plots of CD45+MHCII+CD11c+CD11b+ cells from the colonic lamina propria, and percentages and counts of (C) CX3CR1+ or CD103+ mononuclear phagocytes in control, antiviral depleted (AV), non-IBD or IBD humanized virome mice on day 17. (D-F) Confocal images and quantification of viral-like particles (VLPs) using SYBR Gold in feces or colon tissue of humanized virome mice. (G) Colon length, (H) stool lipocalin, (I) IL-6 levels in colonic explant supernatants cultured for 24 hours following 12 days of induced DSS colitis measured by ELISA. (J,K) Representative hematoxylin and eosin–stained sections and blinded histologic scores in mouse colon tissue (scale bar, 50 μm). (L) FITC-dextran level in serum 4 hours after oral gavage following 7 days of DSS. (M-O) Colon length, stool lipocalin or colonic explant IL-6 levels following induction of DSS colitis in antiviral-treated MAVS−/− mice or (P-R) cGAS−/− mice administered human non-IBD, UC or CD colon-derived viromes. Data are mean ± s.e.m. of 4-8 animals. Representative of 2 independent experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, two-tailed unpaired t-test or one-way ANOVA with Tukey’s multiple comparison test.
Human non-IBD viromes protect while IBD viromes exacerbate DSS-colitis
The ability of the human enteric virome to contribute to disease phenotype in a dextran sulfate sodium (DSS)-induced colitis model was then assessed. Since this model is more closely associated with UC, mice diminished of their murine virome were administered human enteric viruses derived from colon resections of non-IBD or UC or CD patients 3 times intragastrically (Fig 6D). We assessed the abundance of isolated virus-like particles (VLPs) in each treatment group by staining fecal or colon tissue derived VLPs with SYBR. We found a mean of 1.1x107 VLPs/mg of feces and 1.1x106 VLPs/mg of colon tissue in naïve C57BL/6 mice (Fig 6E,F). After AV cocktail treatment, fecal VLPs declined ~20-fold to a mean of 6x105 VLPs/mg in accordance with previous publications in Balb/c mice (21) and VLPs in colon tissue declined ~12 fold to 9x104 VLP/mg. Quantification of viruses in feces or colon tissue of humanized virome mice revealed an equal abundance of ~4x106 VLPs/mg of feces and ~4x105 VLPs/mg of colon tissue from mice humanized with healthy, UC or CD viromes (Fig 6E, F). Consistent with recent publications (21, 22), depletion of viruses in healthy mice lead to significantly exacerbated DSS induced intestinal damage (Fig 6G–I). Remarkably, mice repopulated with a healthy humanized virome conferred robust protection against colitis, including less shortening of the colon (Fig. 6G), rescued stool lipocalin levels (Fig. 6H), rescued colon IL-6 levels (Fig 6I). Thus, normal/non-IBD human enteric viromes actively immunodulate and incite protection from intestinal inflammation in vivo, consistent with our in vitro results. In contrast, both UC or CD human colonic viromes were incapable of rescuing antiviral treated mice and promoted intestinal disease in vivo (Fig 6G–I), results that were confirmed by histological analysis (Fig 6J,K). Moreover, confirming our in vitro data, both UC or CD intestinal viromes resulted in reduced intestinal barrier integrity in vivo, as measured by gavaged FITC-dextran in the serum (Fig 6L).
Human virome immunomodulation in vivo is dependent on nucleic acid sensing
We had demonstrated an autonomous immunomodulatory capacity of intestine-derived viromes in cells, however, to assess any virome-microbiome community interactions vivo, we performed 16S sequencing of host microbiota. Bacterial richness were not significantly affected by antiviral treatment or gavage administration of human healthy or IBD viromes (Fig S14A). However, in UniFrac-based PCoA analysis, groups were clustered separately dependent on virus depletion or human virome source (P<0.05, PERMANOVA, Fig S14B). By proportion (Fig S14C) and LEfSe analysis, the bacterial groups most changed by AV cocktail treatment was a reduction in the phyla Firmicutes and an elevation certain genera of the Ruminococcaceae family (Fig S14D). Both healthy and UC humanized virome mice displayed similar changes in murine microbiota (Fig S14E,F) suggesting microbiome changes are unlikely to be the driver of divergent human virome-induced phenotypes. Furthermore, CD humanized virome mice had unique changes in the murine microbiome (Fig S14G) reflecting the distinct bacteriophage alterations we had observed in CD colon tissue. Importantly however, virus nucleic acid sensing by the host immune system was an absolute requirement for immunomodulation by healthy or IBD human viromes in vivo. Mice deficient in MAVS, the adapter protein of RNA sensors RIG-I and MDA5, or mice deficient in the DNA sensor cGAS, displayed no change in colon length, lipocalin or colon IL-6 production following administration of non-IBD, UC or CD human enteric viromes, compared to antiviral-treated controls (Fig 6M–R). Thus, an ability of human enteric viromes to directly shape intestinal disease phenotype in vivo was via host innate immune recognition of viruses. Moreover, it demonstrates that fluctuations in the virome are not correlative with disease but rather there are direct functional consequences on host immune and intestinal state due to active host sensing of an altered virome in IBD (1, 8, 10).
Discussion
Inflammatory bowel diseases (IBD) affect more than 3.5 million people, with incidence increasing worldwide. These diseases, the most prevalent forms of which are Crohn’s disease (CD) and ulcerative colitis (UC), are characterized by debilitating and chronic relapsing and remitting inflammation of the gastrointestinal tract (for CD) or the colon (in UC). These conditions result from a complex interplay between host, microbial, and environmental factors. While disruption of the intestinal microbiome is an established contributor to IBD, the virome alterations observed in IBD by metagenomic analyses have been correlative findings. Indeed, the interplay of the microbiota and virome is unsurprising given the dominance of bacteriophages. However, whether there is an autonomous functional role for the human intestinal virome in educating host immunity and IBD disease phenotype has remained elusive. To address this deficiency in knowledge of virome biology, in this study we enriched viruses directly from colon surgical resections or ileostomy fluid of non-IBD, UC or CD patients and assessed their immunomodulation by delivering them to human macrophages, intestinal epithelial cells and creating mice with a humanized virome in vivo. We also identified the viruses that are present in colon tissue or ileostomy fluid of non-IBD, UC or CD individuals by metagenomic analysis since only the fecal virome has been elucidated to date. Moreover, we established how genetic variation within IFIH1 that results in loss-of-function of the virus sensor MDA5 in IBD may contribute to disease in the context of the virome.
Our data demonstrate that viruses resident to normal human colon tissue are readily endocytosed and can autonomously elicit an immunomodulatory transcriptional program in macrophages and intestinal epithelial cells, consistently promote production of anti-inflammatory cytokines, and can maintain homeostasis and protect against induced colitis in vivo. Conversely, viromes from inflamed colon tissue, and specifically UC and CD, that are perturbed, including significantly elevated eukaryotic Enteroviruses and bacteriophages of the Caudovirales order, triggered pro-inflammatory macrophage and IEC barrier loss phenotypes, and intestinal inflammation in vivo upon transfer. Moreover, viruses isolated from ileostomy fluid of non-IBD or IBD patients, which had reduced diversity compared to colon tissue highlighting specific niches of the virome, also triggered anti- and pro-inflammatory innate immune responses, respectively. Importantly, these functional differences of non-IBD and IBD viromes were dependent on innate viral sensors and known RNA virus moieties, indicating active and unique recognition of the intestinal virome by the host. Thus, human enteric viruses residing in colon tissue or ileum content autonomously invoke host innate immune responses and this response is divergent for viruses resident to colon tissue of non-IBD or IBD individuals. Notably, our data also show that viruses isolated from healthy colon tissues were capable of suppressing inflammatory responses invoked by UC or CD patient colon tissue viromes. Therefore, collective virus populations isolated from healthy individuals, and particularly from colon tissue, should be considered as a novel therapeutic approach to suppress inflammation in IBD. Indeed, it is appealing to speculate that viruses already play a role in the protection mediated by fecal transplants, as well as in the pathology of intestinal inflammation. Certainly, filtered feces (removing the bacterial component) have similar efficacy to unfiltered feces in treating the Clostridium difficile patients (63).
To progress beyond virome transfers to more specific virus-based therapeutic strategies in IBD, individual prokaryotic or eukaryotic viral strains in the intestinal microbiota that contribute to or prevent disease will need to be elucidated. Certainly, bacteriophages are the most abundant viral community in the human gastrointestinal tract, but eukaryotic viruses are also a critically important virome component. Specifically, we found a significant elevation in Enterovirus B, of the Echovirus subtype, in IBD patient colon tissue which was missed in previous fecal virome analyses. This further emphasizes that viruses and other microorganisms (49) may be uniquely found within intestinal tissue, and not feces. Enterovirus B infection has already been shown to associate with type 1 diabetes (50) indicating that prolonged infections with these viruses, with a fecal-oral route, might contribute to development of chronic inflammatory disorders more broadly. Certainly, based on our new data presented here and previous findings (48), the role of enteroviruses in triggering IBD and therapeutic strategies to suppress these viruses, or the immune response to them, warrants exploration in greater detail and with larger cohorts. As for the contribution of Caudovirales phage expansion in IBD to human innate immunomodulation, much evidence demonstrates that bacteriophages are readily internalized by human cells (31, 64) and directly influence human immunity, independent of their effects on their bacterial hosts (30, 32, 52). Efforts to both better characterize bacteriophage populations, especially RNA phage, and to cultivate different phages will be necessary to yield culturable phage communities for future experimental application in IBD. Moreover, improvements on virome experimental models, such as standardized techniques for high purity VLP preparations and improved methods to deplete murine viromes with antiviral treatments, or ideally the development of “virus-free” mice, for colonization with defined viral species will be advantageous. Finally, understanding the fundamental mechanisms of innate recognition and divergent transcriptional responses to so-called commensal (e.g. Anelloviruses) versus pathogenic (e.g. Enteroviruses) viruses will be invaluable and a large advance for the innate immunology field.
Altogether, our findings provide some of the first functional evidence that the collective viruses in a normal/non-inflamed intestine can help build gut immunity in humans - a contrast to the misconception that all viruses are harmful. Hence, an alteration in the intestine-resident prokaryotic or eukaryotic virome, or disrupted virome sensing due to genetic variation in the virus receptor MDA5, has detrimental consequences for the human intestine and can contribute directly to IBD phenotype. Therapeutic modulation of the virome through targeted elimination or replacement of disease- or health-driving intestinal viruses presents an novel strategy to treat intestinal and immunological diseases more broadly.
Materials and Methods:
Study design
The purpose of this study was to directly compare the autonomous immunomodulatory capacity of viruses enriched from colon resections or ileostomy fluid obtained from ulcerative colitis or Crohn’s disease patients, or non-IBD controls, both in vitro and in vivo. Fresh colon resections were obtained post-surgery from 8-12 individuals per group and confirmed to be inflamed or non-inflamed by a surgical pathologist. The human study population were de-identified adult patients enrolled in the Prospective Registry in IBD Study at Massachusetts General Hospital (PRISM), a single-center ongoing patient registry that aims to understand the causes of IBD and factors affecting disease progression. Ileostomy fluid was collected from patients enrolled in the “Quantifying the Pro-Inflammatory Nature of Human Fluid Collection Study”. Study research coordinators obtained consent, and medical history was obtained and confirmed by review of the electronic medical record. Animal studies were conducted under protocols approved by the MGH Institutional Animal Care and Use Committee (IACUC), and in compliance with appropriate ethical regulations. All in vitro experiments with primary mouse, primary human macrophages or intestinal epithelial cells included three to twelve biological replicates performed in triplicate, as indicated in the figure legends. To avoid donor variation in primary human macrophage experiments, individual blood donors were pooled. All in vivo experiments were performed independently two times, with five to seven animals per group. Descriptions of some of the methods used are located in a Supplementary Methods section within the Supplementary Materials.
Patient intestinal resections and ileostomy fluid
All human samples were collected under Institutional Review Board (IRB)-approved protocols by Massachusetts General Hospital (MGH) and Research Blood Components, including informed consent obtained in accordance with relevant ethical regulations. Fresh colon resections were collected from patients that were enrolled in the “Prospective Registry in IBD Study at Massachusetts General Hospital (PRISM, IRB# FWA00003136, Table S1). Study research coordinators obtained consent, and medical history was obtained and confirmed by review of the electronic medical record. Inflammation in UC or CD colon resection samples was confirmed grossly and microscopically by a clinical pathologist. Non-IBD controls included individuals undergoing colon resection surgeries for colorectal cancer, familial adenomatous polyposis (FAP) or diverticulitis. Lack of inflammation in non-IBD control resection samples was confirmed by a pathologist. Ileostomy fluid was collected from patients enrolled in the “Quantifying the Pro-Inflammatory Nature of Human Fluid Collection Study” that was approved by the Committee for the Protection of Human Subjects at MGH (IRB#2011P002755, Table S2).
Virus-like particle (VLP) isolation
100 mg of colon tissue or centrifuged ileostomy fluid was resuspended in 500 μL of saline magnesium buffer (SM) buffer (8 mM MgSO4, 100 mM NaCl, 50 mM Tris-HCl pH 7.4, and 0.002% (w/v) gelatin, passed through a 0.02μm Whatman filter), homogenized using sterile metal beads, and centrifuged. The supernatant was then passed through a 0.45 μm pore-sized membrane, followed by 0.22 μm pore-sized membrane (Millipore). The filtrates were treated with lysozyme (10 μg/mL; Sigma) for 30 min at 37 °C, followed by incubation with 0.2 volumes of chloroform for 10 min to degrade any remaining bacterial and host cell membranes and then centrifuged at 2,500 × g for 5 min at room temperature. The aqueous phase was collected and incubated with DNase I (3U/200μL, Sigma) for 1 h at 37 °C to remove any non-virus protected DNA. Enzyme activity was inactivated by incubation at 65°C for 15 min as described (1). To remove any potential residual endotoxin, samples were treated with Polymyxin B (10 μg/mL, Sigma) for 30 minutes. Undetectable endotoxin was confirmed using Limulus Amebocyte Lysate (LAL) assay kit (GenScript) following the manufacturer’s instructions.
Quantification of VLPs
Virus-like particles (VLPs) were diluted 10-fold serially, stained for 30 minutes with 10× SYBR Gold (Thermo Fisher Scientific) for nucleic acid or the dialkylcarbocyanine DiI (Thermo Fisher Scientific) for lipid bilayers of enveloped viruses, and imaged using a Zeiss LSM510 laser scanning confocal microscope. Images were captured using Zeiss software (ZEN). Particles <0.5 μm in diameter were regarded as VLPs. SM buffer was used as a negative control. 10 VLP images were captured per sample and VLPs were counted using an image analyzer (InnerView™).
VLP sequencing
Total RNA and DNA was extracted from VLPs on a MagNA Pure 24 instrument (Roche), according to the manufacturer’s instructions. In order to evaluate samples for both RNA and DNA viruses, the total nucleic acids were randomly amplified as described previously (65, 66) using barcoded primers consisting of a base-balanced 16-nucleotide-specific sequence upstream of a random 15-mer and used for NEBNext library construction (New England Biolabs). The libraries were multiplexed on an Illumina NextSeq (Washington University Center for Genome Sciences) using the paired-end 2 × 150 protocol.
Identification and analysis of viral-like sequences
Unprocessed paired-end reads were processed through a multistage quality-control procedure to remove primers and adapters, human and other contaminant and low-quality sequence data. Exact duplicate sequences were removed reserving a single copy and all remaining sequence dereplicated allowing for 4 substitutions. These high-quality / low-redundancy sequences were systematically queried against protein or genomic reference databases using MMseqs2 (67) translated and untranslated search strategies. Sequences assigned a eukaryotic viral or phage taxonomic lineage were first identified using primary searches against virus sequence databases and subsequently confirmed in secondary searches using reference databases with additional non-viral taxonomic lineages. Specific eukaryotic viral families were selected for subsequent analysis by gating on the alignment length and percent identity for each family that separated the high-quality alignments from potentially spurious alignments (Table S3). Only high-quality reads were used for subsequent analysis. Specific phage reads were selected for subsequent analysis by gating on an alignment length of 75 nt, to account for the current state of phage taxonomy. In order to determine which phage and eukaryotic viral reads are differentially abundant in the three cohorts, we analyzed the reads at the family, genus and species levels. The non-parametric Kruskal–Wallis test was used to assess the difference in median abundance of eukaryotic viral and phage taxa in the Non-IBD, UC and CD cohorts. The Dunn’s post-hoc test was used to correct for multiple comparisons and only those taxa with an adjusted p-value < 0.05 were considered significant. The mean abundance of viral or phage reads, scaled by library size, was determined for each sample in the colon and ileostomy samples. The non-parametric Welch’s paired t-test was used to determine which cohorts had significantly different numbers of viral or phage reads.
Human peripheral blood derived macrophages
Peripheral Blood Mononuclear Cells (PBMCs) were isolated from 20-30 mL blood buffy coats from healthy human volunteers (MGH blood components lab). Briefly, mononuclear cells were isolated by density gradient centrifugation of PBS-diluted buffy coat/blood (1:2) over Ficoll-Paque Plus (GE Healthcare). The PBMC layer was carefully removed and washed 3 times with PBS, mononuclear cells were collected as PBMC. In order to obtain macrophages, PBMCs were re-suspended in X-VIVO medium (Lonza) containing 1% penicillin/streptomycin (Gibco) and incubated at 37°C, 5% CO2 for 1h to adhere to the tissue culture dish. After 1h, adherent cells were washed 3 times with PBS and differentiated in complete X-VIVO medium containing 100 ng/mL human M-CSF (PeproTech) for 7 days at 37°C, 5% CO2. On day 4, cultures were supplemented with one volume of complete X-VIVO medium containing 100 ng/mL human M-CSF.
Delivery of VLPs to macrophages
VLPs or SM buffer (8 mM MgSO4, 100 mM NaCl, 50 mM Tris-HCl, pH 7.4, and 0.002%, w/v, gelatin) were pre-treated for 30 minutes with Polymyxin B (Sigma) to ensure endotoxin-free conditions. Media was removed from 1×106 adherent macrophages and VLPs (10 VLPs per cell) or SM buffer (“mock”) was added drop-wise onto the cells at a fixed volume sufficient to cover the well surface. VLPs were allowed to adsorb to adherent cells for 1 hour at 37°C with occasional rocking. After 1 hour, VLPs were aspirated, cells were gently washed with PBS, and media was replaced (this was considered time = 0). For heat inactivation experiments, VLPs were first inactivated for 1 hour at 60°C. For UV crosslinking of VLPs, VLPs were placed in a UV Stratalinker 2400 (Stratagene) on ice to UVB-irradiate three times with 200 mJ/cm2 for 15 minutes. For in vitro ratio experiments, non-IBD colon resection-derived VLPs were mixed with CD colon resection-derived VLPs at increasing ratios to the final concentration of 10 VLPs per cell and VLPs were delivered to macrophages as described above. For supernatant in vitro experiments, the supernatant from macrophages that were exposed to the non-IBD VLP 24 hours prior was mixed with IBD VLPs and added dropwise onto the cells. VLPs and supernatant mix were allowed to adsorb to adherent cells for 1 hour at 37°C with occasional rocking. Heat inactivated (95°C for 10 min) or UV crosslinked (three times 200 mJ/cm2 for 15 minutes) supernatant were used as controls.
Fluorescent labeling of VLPs and uptake assays
Purified, quantified colon resection derived VLP preparations were labeled with 0.01 μM SYTO™ 16 Green Fluorescent Nucleic Acid Stain (Thermo Fisher Scientific, #S7578) according to manufacturer’s instructions. Labeled VLPs were then separated from unincorporated dye using PD10 Sephadex G-25 desalting columns (GE Healthcare, #17085101) according to manufacturer’s instructions. Uptake assays of SYTO16-labeled VLPs were performed with human peripheral blood monocyte derived macrophages. Cells were seeded at a density of 105 cells/well in a 24-well plate. Cells were delivered 106 or 107 non-IBD, UC or CD VLPs or SM buffer control by adsorption in 100 μL for 1h, washed and left for 3 hours at 37°C for uptake analysis or 4°C for adsorption analysis. Cells were removed from the culture plate surface with cold PBS, washed in flow cytometry buffer twice, stained for live–dead discrimination and acquired on an LSR II flow cytometer (BD Biosciences). For the inhibition assays, cells were treated with 3.0 mg/mL of Brefeldin A (Invitrogen, #00-4506) for 30 min at 37°C before addition of labeled VLPs and during the adsorption. Data was analyzed using FlowJo v10 software (TreeStar).
VLP RNA or DNA isolation
For VLP RNA or DNA transfection experiments, VLP preparations were mixed with TRIzol LS (Invitrogen) and RNA and DNA fractions isolated. DNA was further purified by ethanol precipitation. The RNA fraction was subjected to RNeasy Mini Kit (Qiagen) with on-column DNase I digest for clean-up and removal of residual DNA. For enzyme treatments of nucleic acids, 2.5 μg of RNA was treated with RNase I (5 μg/mL, Ambion) or RNase III (5 μg/mL, Ambion) or with Calf Intestinal Phosphatase (CIP, 250U, Roche) at 37 °C for 1 h. Enzyme-treated RNA was purified with RNeasy Mini Kit (Qiagen) before transfection. Human macrophages were transfected with 2.5 μg of RNA or DNA using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol.
ELISA
Cell culture supernatants were removed from human macrophages and centrifuged to remove debris and non-adherent cells. Secreted cytokines were measured using a human Cytokine 25-Plex Magnetic Panel for Luminex® Platform (ThermoFisher, EPX250-12166-901) and read on a Luminex FLEXMAP 3D® instrument. Human and mouse TNF, IL-6, IFN-β, IL-8, IL-15, IL-10 and IL-22 ELISA kits (R&D) were also used. Supernatants and kit-supplied cytokine standards were processed in triplicate according to the manufacturer’s instructions.
Quantitative PCR
RNA was extracted using the RNeasy Mini Kit (Qiagen) with on-column DNase digest (Qiagen) according to manufacturer’s instruction. 100ng-1μg RNA was used to synthesize cDNA by reverse transcription using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR reactions were run with cDNA template in the presence of 0.625 μM forward and reverse primer and 1x solution of iTaq Universal SYBR Green Supermix (Bio-Rad). Quantification of transcript was normalized to the indicated housekeeping gene. A complete list of primer sequences is provided in Supplementary Table 4.
Mice
Five- to six-week-old female C57BL/6J, B6;129-Mavstm1Zjc/J, B6(C)-Cgastm1d(EUCOMM)Hmgu/J or TLR4-deficient C3H/HeJ mice were purchased from The Jackson Laboratory. B6;129-Mavstm1Zjc/J mice were bred as heterozygotes and littermate controls were used. All mice were housed in specific pathogen-free conditions according to the National Institutes of Health (NIH), and all animal experiments were conducted under protocols approved by the MGH Institutional Animal Care and Use Committee (IACUC), and in compliance with appropriate ethical regulations. For all experiments, age-matched mice were randomized and allocated to experimental group, with 5-8 mice per group, and repeated three independent times. No statistical method was used to determine sample size. For “humanized” virome mice, mice were first depleted of viruses using an antiviral cocktail (acyclovir 20 mg/kg, lamivudine 10 mg/kg, ribavirin 30 mg/kg, oseltamivir 10 mg/kg) by daily gavage for 10 days as described (21). AV-treated mice were then gavaged with 200 μL of SM buffer as a control or 4×108 VLPs isolated from non-IBD, UC or CD patient fresh colon resections. Mice received the human VLP gavage every other day for three times in total. To ensure that the gastric pH would not affect the viability of the gavaged VLPs, each mouse was gavaged with 100 μL of 1M NaHCO3 15 min prior to VLP gavage to keep gastric pH at 7.4. Quantification of viruses in different treatment groups was performed by confocal imaging of VLPs isolated from feces and colon tissue.
Statistical analysis
Results are shown as mean ± s.e.m. Visual examination of the data distribution as well as normality testing demonstrated that all variables appeared to be normally distributed. Comparisons and statistical tests were performed as indicated in each figure legend. Briefly, for comparisons of multiple groups over time or with two variables, a two-way analysis of variance (ANOVA) was used and corrected for multiple post hoc comparisons, comparing all groups to each other, all groups to a control, or selected groups to each other. For comparisons of multiple groups with only one variable, a one-way ANOVA was performed and corrected for multiple post hoc comparisons. For comparisons of two groups, two-tailed paired or unpaired t tests were used, except where indicated. Statistical analyses were performed in the GraphPad Prism 8 software. The non-parametric Kruskal–Wallis test with a Dunn’s post-hoc test was used to assess the difference in median abundance of eukaryotic viral and phage taxa in the non-IBD, UC and CD cohorts. P values denoted throughout the manuscript highlight biologically relevant comparisons. A P value of less than 0.05 was considered significant, denoted as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 for all analyses. FDR correction was performed for all RNA-seq data.
Supplementary Material
Acknowledgments:
The authors wish to thank the clinical coordinators and patients enrolled in the Prospective Registry in IBD study at Massachusetts General Hospital (PRISM). We would also like to thank the MGH Next Gen Sequencing core, Thomas Diefenbach at the Ragon Institute Imaging Core for confocal imaging of VLPs, the Harvard Medical School electron microscopy facility, Hembly Rivas (Harvard Virology Ph.D. program) for assistance with MDA5 experiments, Laurie Pruneau and Marie-Eve Rivard (Montreal Heart Institute) for technical expertise with iPSC to human epithelial cell transdifferentiation using LCL cells lines from NIDDK Inflammatory Bowel Disease Genetics Consortium (IBDGC) and Chloé Lévesque for confocal immunofluorescence analyses of the hiPSC-derived epithelial 2D cultures. The NIDDK IBDGC was conducted by the IBDGC Investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The lymphoblastoid cell lines and related data from the IBDGC reported here were supplied by the NIDDK Central Repositories. Other than J.D.R., this manuscript was not prepared in collaboration with investigators of the IBDGC study and does not necessarily reflect the opinions or views of the IBDGC study, the NIDDK Central Repositories, or the NIDDK. We thank Amy Avery (Massachusetts General Hospital) for technical assistance with 16S sequencing and Raza Hoda (Massachusetts General Hospital) for blindly scoring intestinal pathology. We thank Corinne Maurice and Irah King (McGill University), Naama Geeva-Zatorsky (Technion) and the entire Jeffrey lab for valuable scientific discussions as well as Robert Anthony, James Moon and Ramnik Xavier for critical reading of the manuscript. A provisional patent related to this work has been filed. Kate L. Jeffrey is an employee of Moderna Inc., 200 Technology Square, Cambridge MA 02138, since November 2021.
Funding:
This study was supported by the Kenneth Rainin Foundation (Innovator and Synergy Awards to K.L.J), NIH R21AI144877 (K.L.J), NIH R01DK119996 (K.L.J), Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL 1TR002541) and financial contributions from Harvard University and its affiliated academic healthcare centers (K.L.J), Canadian Institute of Health Research (CIHR) postdoctoral fellowship (H.A.), NIH P30DK040561 (R.S.), Canada Research Chair (J.D.R), NIH DK062432 (J.D.R) and NIH RC2DK116713 (S.H.). K.L.J is a John Lawrence MGH Research Scholar 2020-2025.
Footnotes
Competing interests: A provisional patent related to this work has been filed with F.A and K.L.J as inventors. K.L.J. is an employee of Moderna Inc., 200 Technology Square, Cambridge MA 02138, since November 2021. K.L.J and S.H. are members of the scientific advisory board for Ancilia Biosciences. None of these relationships influenced the work performed in this study. The other authors declare no competing interests.
Data and materials availability: RNA sequencing raw and processed data was deposited at Gene Expression Omnibus (GEO) and is accessible using accession number GSE135223. Human colon resection and ileostomy fluid VLP sequencing were deposited at European Nucleotide Archive (ENA) under accession number PRJEB44121. 16S rRNA sequences from the feces of humanized virome mice were deposited at Sequencing Read Archive (SRA) under accession number PRJNA816891. All other data needed to evaluate the conclusions are present in the paper or the Supplementary Materials.
References and Notes
- 1.Norman JM et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang G et al. The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature 581, 470–474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards RA et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat Microbiol 4, 1727–1736 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camarillo-Guerrero LF, Almeida A, Rangel-Pineros G, Finn RD, Lawley TD, Massive expansion of human gut bacteriophage diversity. Cell 184, 1098–1109 e1099 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barr JJ et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A 110, 10771–10776 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim ES et al. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat Med 21, 1228–1234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adiliaghdam F, Jeffrey KL, Illuminating the human virome in health and disease. Genome Med 12, 66 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clooney AG et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe, (2019). [DOI] [PubMed] [Google Scholar]
- 9.Duerkop BA et al. Murine colitis reveals a disease-associated bacteriophage community. Nat Microbiol 3, 1023–1031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang G et al. The dynamics of the stool virome in very early onset inflammatory bowel disease. J Crohns Colitis, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannigan GD, Duhaime MB, Ruffin M. T. t., Koumpouras CC, Schloss PD, Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. MBio 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakatsu G et al. Alterations in Enteric Virome Are Associated With Colorectal Cancer and Survival Outcomes. Gastroenterology 155, 529–541 e525 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Zhao G et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc Natl Acad Sci U S A 114, E6166–E6175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang S et al. Intestinal Virome Signature Associated With Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willner D et al. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS One 4, e7370 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legoff J et al. The eukaryotic gut virome in hematopoietic stem cell transplantation: new clues in enteric graft-versus-host disease. Nat Med 23, 1080–1085 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Monaco CL et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 19, 311–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iliev ID, Cadwell K, Effects of Intestinal Fungi and Viruses on Immune Responses and Inflammatory Bowel Diseases. Gastroenterology, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park A, Iwasaki A, Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 27, 870–878 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Virgin HW, The virome in mammalian physiology and disease. Cell 157, 142–150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang JY et al. Enteric Viruses Ameliorate Gut Inflammation via Toll-like Receptor 3 and Toll-like Receptor 7-Mediated Interferon-beta Production. Immunity 44, 889–900 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Broggi A, Tan Y, Granucci F, Zanoni I, IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol 18, 1084–1093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price AE et al. A Map of Toll-like Receptor Expression in the Intestinal Epithelium Reveals Distinct Spatial, Cell Type-Specific, and Temporal Patterns. Immunity 49, 560–575 e566 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y et al. Rig-I−/− mice develop colitis associated with downregulation of G alpha i2. Cell Res 17, 858–868 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Liu L et al. Commensal viruses maintain intestinal intraepithelial lymphocytes via noncanonical RIG-I signaling. Nat Immunol 20, 1681–1691 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Kernbauer E, Ding Y, Cadwell K, An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neil JA et al. IFN-I and IL-22 mediate protective effects of intestinal viral infection. Nat Microbiol, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XD et al. Mitochondrial antiviral signaling protein (MAVS) monitors commensal bacteria and induces an immune response that prevents experimental colitis. Proc Natl Acad Sci U S A 108, 17390–17395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sweere JM et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 363, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen S et al. Bacteriophage Transcytosis Provides a Mechanism To Cross Epithelial Cell Layers. mBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bichet MC et al. Bacteriophage uptake by mammalian cell layers represents a potential sink that may impact phage therapy. iScience 24, 102287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleiner M, Hooper LV, Duerkop BA, Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 16, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang G, Bushman FD, The human virome: assembly, composition and host interactions. Nat Rev Microbiol 19, 514–527 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carroll-Portillo A et al. Standard Bacteriophage Purification Procedures Cause Loss in Numbers and Activity. Viruses 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conceicao-Neto N et al. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci Rep 5, 16532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bushman FD, McCormick K, Sherrill-Mix S, Virus structures constrain transmission modes. Nat Microbiol 4, 1778–1780 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zigmond E, Jung S, Intestinal macrophages: well educated exceptions from the rule. Trends Immunol 34, 162–168 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Murray PJ et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez FO et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood 121, e57–69 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Dallari S et al. Enteric viruses evoke broad host immune responses resembling those elicited by the bacterial microbiome. Cell Host Microbe, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jabri B, Abadie V, IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol 15, 771–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neurath MF, Cytokines in inflammatory bowel disease. Nat Rev Immunol 14, 329–342 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Pott J et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci U S A 108, 7944–7949 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldridge MT et al. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 347, 266–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graham DB, Xavier RJ, Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 578, 527–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giles EM, D’Adamo GL, Forster SC, The future of faecal transplants. Nat Rev Microbiol 17, 719 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Nystrom N et al. Human enterovirus species B in ileocecal Crohn’s disease. Clin Transl Gastroenterol 4, e38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain U et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science 371, 1154–1159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hober D, Sauter P, Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol 6, 279–289 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Tan X, Sun L, Chen J, Chen ZJ, Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annu Rev Microbiol 72, 447–478 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Gogokhia L et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 25, 285–299 e288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goubau D, Deddouche S, Reis e Sousa C, Cytosolic sensing of viruses. Immunity 38, 855–869 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jochum L, Stecher B, Label or Concept - What Is a Pathobiont? Trends Microbiol, (2020). [DOI] [PubMed] [Google Scholar]
- 55.Kato H et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Rice GI et al. Genetic and phenotypic spectrum associated with IFIH1 gain-of-function. Hum Mutat 41, 837–849 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ablasser A, Hur S, Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat Immunol 21, 17–29 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Shigemoto T et al. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J Biol Chem 284, 13348–13354 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim M et al. Critical Role for the Microbiota in CX3CR1(+) Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity 49, 151–163 e155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leonardi I et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coombes JL et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204, 1757–1764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinnebrew MA et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 36, 276–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ott SJ et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients With Clostridium difficile Infection. Gastroenterology 152, 799–811 e797 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Lehti TA, Pajunen MI, Skog MS, Finne J, Internalization of a polysialic acid-binding Escherichia coli bacteriophage into eukaryotic neuroblastoma cells. Nat Commun 8, 1915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Finkbeiner SR et al. Identification of a novel astrovirus (astrovirus VA1) associated with an outbreak of acute gastroenteritis. J Virol 83, 10836–10839 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang D et al. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol 1, E2 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steinegger M, Soding J, MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35, 1026–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anders S et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8, 1765–1786 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DeBoever C et al. Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nat Commun 9, 1612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar S, Curran JE, Glahn DC, Blangero J, Utility of Lymphoblastoid Cell Lines for Induced Pluripotent Stem Cell Generation. Stem Cells Int 2016, 2349261 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tamminen K et al. Intestinal Commitment and Maturation of Human Pluripotent Stem Cells Is Independent of Exogenous FGF4 and R-spondin1. PLoS One 10, e0134551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takahashi Y et al. Reciprocal Inflammatory Signaling Between Intestinal Epithelial Cells and Adipocytes in the Absence of Immune Cells. EBioMedicine 23, 34–45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Erben U et al. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int J Clin Exp Pathol 7, 4557–4576 (2014). [PMC free article] [PubMed] [Google Scholar]
- 78.Bolyen E et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37, 852–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Callahan BJ et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Katoh K, Misawa K, Kuma K, Miyata T, MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Price MN, Dehal PS, Arkin AP, FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lozupone C, Knight R, UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bokulich NA et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McDonald D et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Segata N et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.