

Abstract
Graphene oxide (GO) is one of the most popular materials applied in different research areas thanks to its unique properties. The application of GO requires well-designed protocols to introduce different functionalities on its surface, exploiting the oxygenated groups already present. Due to the complex and unstable chemical environment on the GO surface, it is recommended to perform the functionalization under mild conditions. The carboxylation of GO is a widely used method to introduce additional carboxylic acids, which could be further modified through amidation or esterification reactions. The strategy already reported in the literature requires harsh conditions (excess amount of sodium hydroxide). GO is readily reduced under basic conditions, but the reduction of GO during the carboxylation is barely studied. In this work, we performed the carboxylation using chloroacetic acid with different amounts of sodium hydroxide and characterized the functionalized GO with various techniques. The carboxylated GO was exploited to develop a double functionalization approach combining an epoxide ring opening reaction and an amidation. The results showed that strong basic conditions were necessary to derivatize GO. Nevertheless, these conditions resulted in a partial reduction of GO and some functionalities on GO were removed during the reaction, thus reducing the total efficiency of the functionalization in comparison to an epoxide ring opening reaction, indicating that carboxylation is not an efficient approach for the functionalization of GO.
We investigated the carboxylation of graphene oxide using chloroacetic acid and different amounts of NaOH. Increase of carboxyl groups was observed only at high amounts of NaOH, but partial reduction attenuates the yield of further functionalization.
Introduction
Over the past two decades, GO has attracted large interest and is one of the most popular nanomaterials with great potential applications in different fields covering energy storage,1,2 antibacterial agents,3,4 biosensing,5 diagnosis and therapy,6–9 and artificial muscles.10 GO is the oxidized form of graphene, usually prepared by the oxidation of graphite using strong acid treatments.11,12 It consists of a hexagonal ring-based carbon network with abundant oxygen-containing functional groups such as epoxides and hydroxyls on the basal plane, as well as carboxyl and carbonyl groups at the edges, along with limited localized graphitic domains.13 These oxygenated functional groups endow GO with unique properties including high dispersibility in water,14 good biodegradability,15,16 optical transparency and photoluminescence quenching.17,18 The high surface area of GO allows adsorption of a large amount of drugs,6,19 RNA20,21 or nanoparticles,22 while bioactive molecules can also be covalently conjugated via the oxygenated groups.23 The possibility of grafting functionalities onto the surface of GO greatly extends its application to different areas.24–27 Compared to noncovalent complexation, covalent functionalization enables us to prepare more stable conjugates and minimize the release of molecules adsorbed onto the GO surface, especially for biological applications. Several strategies have been reported to functionalize GO covalently by targeting different functional groups such as carboxylic acids,28 epoxide rings29 and hydroxyl groups30–33 allowing the development of approaches for double functionalization.31,32,34 However, the derivatization of the carboxylic acids leads to very low loading efficiency compared to the functionalization of the hydroxyl and epoxide moieties, due to the limited amount of COOH on GO.29 Therefore, a higher amount of carboxylic acids would broaden the possibilities for multi-functionalization of GO with a high loading efficiency. Because of their large surface area, two-dimensional materials have a limited amount of functional groups at the edges in comparison with the basal plane. In order to increase the amount of carboxyl groups on GO, a method of carboxylation was developed through the derivatization of hydroxyls with chloroacetic acid in the presence of sodium hydroxide.35–37 The prepared carboxylated GO was further derivatized with amine-containing functionalities by amidation.34,36,38 However, it was reported that the use of a strong base often causes deoxygenation resulting in the partial reduction of GO,39,40 thus changing its properties and influencing the efficiency of the functionalization. Although some articles already reported the partial reduction of GO during carboxylation using chloroacetic acid and sodium hydroxide,35,41 the effect of this deoxygenation on the functionalization efficiency of GO is still not clear. Under most conditions, a wide concentration range of sodium hydroxide solutions was used from 123 mM to 4 M, with 3 M being the most commonly used.
To clarify the impact of a strong alkaline solution on GO, we prepared carboxylated GO using different molarities of sodium hydroxide and performed characterization by X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), transmission electron microscopy (TEM), magic angle spinning (MAS) NMR and FT-IR spectroscopy. The introduction of the carboxylates was observed along with a partial reduction of GO. The efficiency of multifunctionalization of the carboxylated GO was evaluated by the epoxide ring opening and amidation reaction.
Results and discussion
(1) Carboxylation of GO with high amount of sodium hydroxide
Large GO flakes (30 μm) were prepared following a modified Hummers method. By using a jet mill with a 0.1 mm nozzle, GO with an average lateral size of 1 μm was obtained.31 We first derivatized GO with chloroacetic acid under strongly basic conditions following the protocol published by other groups to increase the amount of carboxylic acids.37 Briefly, the pristine GO was dispersed in 3 M sodium hydroxide solution followed by the immediate addition of chloroacetic acid (Scheme 1). After washing and dialysis, the resulting GO 1 was characterized by XPS, TGA, FT-IR and TEM.
Scheme 1. Carboxylation reaction on GO. For the sake of clarity, only one hydroxyl group is derivatized and the epoxide rings are closed. The epoxide ring opening by sodium hydroxide is under an equilibrium and some of the C–O− groups can close again to form a ring.42,43.

XPS provides information on the chemical composition of the surface of GO (Fig. 1). The atomic percentage compositions of the pristine GO and GO 1 were measured (Fig. 1a and c, respectively), presenting a C/O ratio of 2.6 for the starting GO and 3.0 for GO 1. The increase of the C/O ratio after carboxylation was certainly due to a partial reduction of GO,44 indicating that some of the oxygen-containing functional groups were removed during the reaction. The reduction of GO was also supported by the analysis of the high resolution C 1s spectra. The C 1s spectrum of the pristine GO exhibited two major peaks assigned to the C–C bond at 284.8 eV and the C–O bond from alcohols and epoxides at 286.6 eV (Fig. 1b). The shoulder in the 288.1 to 289.1 eV region could be deconvoluted into two components, the carboxyl groups in the region from 288.7 eV to 289.1 eV and the carbonyl groups from 288.1 eV to 288.3 eV. The C 1s spectrum changed significantly after the reaction (Fig. 1d). The carboxyl groups on GO increased from 3.9% to 6.2%, proving the successful modification of GO with carboxylic acids. However, a significant decrease of the C–O component from 44.0% to 28.7% was also observed, confirming the partial reduction of GO during the reaction.45 Some labile oxygenated groups and oxidative debris46 were likely removed from the GO surface during the treatment with sodium hydroxide. Some of the epoxide rings were probably opened by OH− and the hydroxyl groups could be further reduced by OH− with the assistance of Na+ and water molecules, resulting in the formation of CO2, vacancy defects and extension of conjugation, as previously reported.39,47 The partial reduction of GO affects its intrinsic properties and may decrease the loading of molecules conjugated to the COOH groups. The high resolution O 1s peak was deconvoluted into three components: C O (531.4–530.3 eV), C–O (533.0–532.0 eV) and H2O (535.2–534.8 eV). The increase of the C O component from 5.0% to 5.7% after carboxylation was not very significant, probably due to the contribution of the chloroacetic acid moiety to both peaks (Fig. S1†).
Fig. 1. XPS survey spectra (a and c) and high resolution C 1s (b and d) spectra of pristine GO (top row) and GO 1 (bottom row).

The reduction of GO was corroborated by thermogravimetric analysis under an inert atmosphere (Fig. 2a). The TGA curve of the pristine GO showed two main weight losses due to (i) physisorbed water (30–100 °C) and (ii) decomposition of labile oxygenated groups (∼150–200 °C). As evident in Fig. 2a, the TGA curve of GO 1 displayed a higher thermal stability in the 150–200 °C region and the total weight loss was lower.47 The increase of the thermal stability could be attributed to the partial reduction of GO by removal of some labile oxygen-containing groups.
Fig. 2. (a) TGA of the pristine GO and GO 1 performed in an inert atmosphere. (b) FT-IR spectra of the pristine GO and GO 1.

The GO samples were characterized by attenuated total reflectance (ATR) FT-IR spectroscopy (Fig. 2b). The spectrum of the pristine GO displayed a broad peak around 3400 cm−1 assigned to the O–H stretching of the adsorbed water and the hydroxyl functionalities of GO.48 The band at 1723 cm−1 was attributed to the stretching of C O groups. The peak at 1619 cm−1 corresponded to the H–O–H bending vibration of water molecules and the skeletal C C bond vibrations of the graphitic domains. The band at 1371 cm−1 was assigned to the O–H bending vibration. The C–O–C vibration band of the epoxides was located at 1232 cm−1 and the peak at 1143 cm−1 was assigned to the C–O stretching. After the carboxylation, the stretching band of C O at 1723 cm−1 was still present as well as the O–H stretching at ∼3400 cm−1.
The morphology of GO was characterized by TEM (Fig. S2†). The GO sheets have an average lateral dimension of around 1 μm and a wavy shape with folded edges. After carboxylation, the morphology of GO was not affected.
Although the carboxylation method using chloroacetic acid and sodium hydroxide has been widely used for GO surface modification, the partial reduction of the material was never clearly mentioned. On the other hand, the reduction of GO in strongly alkaline solution at room temperature has been reported by several groups,39,40,46,47 and it has been proved as an efficient way to produce reduced GO.38
To better understand the effect of the partial reduction on GO functionalization, we combined an epoxide ring opening reaction and the carboxylation of GO followed by an amidation using O-(2-aminoethyl)-O′-[2-(Boc-amino)ethyl]decaethylene glycol (BocNH-PEG10-NH2) in the presence of activating agents (EDC/NHS) to achieve the double functionalization (Scheme 2). The colorimetric Kaiser test was used to determine the amount of amino-PEG groups grafted onto GO after Boc deprotection.30,49,50 The epoxide ring opening reaction was first performed since the epoxides easily react with amines through a nucleophilic addition forming C–N bonds and new hydroxyl groups,29 which can be further derivatized with chloroacetic acid. For this purpose, the pristine GO was mixed with BocNH-PEG10-NH2 in water and stirred for 3 days at room temperature (Scheme 2). After washing and dialysis, GO 2 was obtained. The amount of primary amines on GO 2 was assessed by the Kaiser test after Boc deprotection and it was estimated to be 59 μmol g−1, proving that the PEG chain was successfully grafted onto GO (Fig. 3a). For the carboxylation procedure, GO 2 was mixed with chloroacetic acid in the presence of 3 M sodium hydroxide under bath sonication following the protocol described above, leading to GO 3 (Scheme 2). We cannot exclude that chloroacetic acid may also react with the secondary amine of the PEG chain introduced after the epoxide ring opening. However, the close proximity of this secondary amine with the surface of GO likely hampers such a derivatization due to steric inaccessibility. The Kaiser test of GO 3 after Boc deprotection revealed that the amount of amines decreased to 14 μmol g−1 after the carboxylation (Fig. 3a), indicating that the PEG chain may be partially removed during the reaction due to the partial reduction of GO during the carboxylation step. GO 3 was further reacted with BocNH-PEG10-NH2 through amidation in the presence of EDC/NHS to give the double functionalized GO 4. An increase of the amount of primary amines was measured by the Kaiser test (27 μmol g−1), confirming that the PEG chain was linked to GO through amidation of the carboxyl groups (Fig. 3a). Meanwhile, a control reaction was performed by directly grafting BocNH-PEG10-NH2 on GO 2 through amidation obtaining GO 4-CTR (Scheme S1†). The Kaiser test value (62 μmol g−1) showed a little increase compared to GO 2 (Fig. 3a), thus confirming that there is only a limited amount of carboxyl groups at the edges of GO and that the reaction with chloroacetic acid allows more COOH moieties to be introduced on GO.
Scheme 2. Double functionalization of GO combining the epoxide ring opening reaction and the carboxylation reaction. For the sake of clarity, only one type of functional group is derivatized.

Fig. 3. (a) Kaiser test of GO 2, GO 3, GO 4 and GO 4-CTR, and (b) TGA of the pristine GO, GO 2, GO 3 and GO 4 performed in an inert atmosphere.

The removal of some functional groups during the carboxylation step due to the strongly basic conditions was confirmed by thermogravimetric analysis (Fig. 3b). Compared to the starting GO, GO 2 displayed another weight loss region located between 200 and 400 °C, corresponding to the degradation of the PEG chain bound to GO. However, GO 3 showed a higher thermal stability compared to GO 2, probably due to the deoxygenation under strongly basic conditions. After the amidation reaction, more PEG chains were grafted onto the GO surface, resulting in a higher weight loss for GO 4 between 200 and 400 °C. Overall, the TGA results confirmed the successful double functionalization of GO thanks to the presence of the additional carboxyl groups. Nevertheless, the distinct thermal properties of GO 3 compared to GO 2 revealed that some functionalities were partially removed during the carboxylation step. The XPS analysis also confirmed the removal of some functional groups during the reaction (Fig. S3†). After the carboxylation step, the ratio of nitrogen atoms on GO 3 increased from 1.0% to 1.8% compared to GO 2 due to the partial deoxygenation on GO. After the amidation reaction, the %N in GO 4 increased to 2.1%, proving that more PEG chains were grafted onto the GO surface.
With a combined epoxide ring opening and carboxylation/amidation, a stepwise double GO functionalization was successfully achieved. But, the partial reduction of GO resulted in a lower efficiency of total functionalization and limited this method to a certain extent for further application.
(2) Carboxylation of GO using a reduced amount of sodium hydroxide
In order to limit the significant reduction of GO during the carboxylation, we repeated this reaction using a lower amount of sodium hydroxide. The carboxylation of GO was performed in a sodium hydroxide solution at different pH values, at pH 9 (10−5 M NaOH in water) and pH 13 (0.1 M aqueous NaOH), giving GO 5a and GO 5b, respectively (Scheme 3). Both samples were characterized by XPS (Fig. 4). However, based on the C 1s spectra, we could not observe an evident increase of the amount of carboxyl groups in both cases, indicating a lower efficiency of carboxylation. Moreover, the C–O component of GO 5a decreased to 39.7% compared to the pristine GO (44.0%), revealing a slight reduction of GO at pH 9. The reduction was more pronounced in GO 5b (35.0% C–O) due to the higher pH. These results are consistent with an increase of the C/O ratio calculated from the survey spectra, from 2.6 for the pristine GO to 2.8 for GO 5a and 3.0 for GO 5b.
Scheme 3. Double functionalization of GO combining the carboxylation reaction using a reduced amount of sodium hydroxide and the epoxide ring opening reaction, followed by amidation. For the sake of clarity, only one type of functional group is derivatized.

Fig. 4. XPS survey spectra (a and c) and high resolution C 1s (b and d) spectra of GO 5a (top row) and GO 5b (bottom row).

GO 5a and GO 5b were also characterized by TGA (Fig. 5). Both GO samples displayed a slightly higher thermal stability than the pristine GO at around 200 °C, which could be assigned to the removal of labile oxygen-containing groups. The TGA results are in agreement with the XPS data, showing a slight reduction of GO during the carboxylation step.
Fig. 5. TGA of the pristine GO, GO 5a and GO 5b performed in an inert atmosphere.

In order to clarify the low efficiency of the carboxylation reaction at lower pH, the pristine GO and GO 5a were characterized by quantitative 13C direct polarization (DP) solid state MAS-NMR (Fig. 6). A control reaction was also performed under the same conditions but without adding chloroacetic acid leading to GO 5a-CTR (Scheme S2†). Detailed analysis of 13C spectra required line shape fitting (Fig. S4†) with a CSA (Chemical Shift Anisotropy) model in order to get the intensities for inner components of each broad band (Table S1†). The 13C NMR spectrum of GO confirmed the presence of abundant epoxides and hydroxyl groups with a strong band at 60.1 ppm and 69.8 ppm, respectively, and C C bonds (peak at ∼130 ppm).29,51,52 The peak of the carboxyl groups around 164 ppm is negligible since the amount of carboxyl groups located at the edges of the pristine GO sheets is very low.29 After the carboxylation reaction, unfortunately, the band of the carboxyl groups did not increase, confirming the inefficiency of the reaction at a lower pH.
Fig. 6. 13C DP/MAS NMR spectra of the pristine GO, GO 5a and GO 5a-CTR.
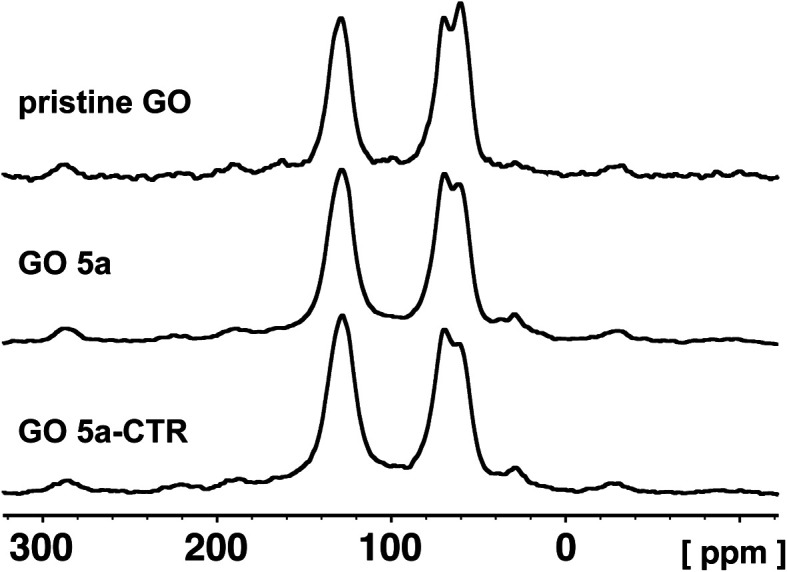
The intensity of the peaks of the epoxides decreased from 22% in the pristine GO to 16% in GO 5a and GO 5a-CTR, while hydroxyls showed a little increase from 20% in pristine GO to 23% in GO 5a and 22% in GO 5a-CTR, respectively (Table S1†). The difference in the peak areas of the epoxides and the hydroxyls could be assigned to the opening of a small proportion of the epoxides by sodium hydroxide. The fitting of the C C signal evidenced two peaks at ca. 126 and 132 ppm, which could be ascribed to graphitic localized clusters of C C and to the other C C in the proximity of oxygenated groups, respectively (Table S1†).
To confirm whether more carboxyl groups were covalently linked onto the GO surface, the carboxylated GO samples prepared at pH 9 (GO 5a) and pH 13 (GO 5b) were derivatized with N-Boc-2,2′-(ethylenedioxy)diethylamine (Boc-TEG-NH2) (Scheme 3). Since the reduction of GO was not severe at pH 9 and pH 13, we assumed it was possible to perform the carboxylation keeping the epoxide group which can be susceptible to the following ring opening reaction. The carboxylated GO 5a and GO 5b reacted first with Boc-TEG-NH2 through a nucleophilic epoxide ring opening, giving GO 6a and GO 6b, respectively. Then, Boc-TEG-NH2 was coupled to GO 6a and GO 6b by amidation with the carboxyl groups of GO, obtaining GO 7a and GO 7b.
The GO samples were characterized by XPS (Fig. S5†). A significant increase of the %N values was observed for GO 6a and GO 6b, indicating the successful functionalization of GO through epoxide ring opening (Table 1). However, the %N did not increase significantly after the amidation reaction, revealing that the amount of carboxyl groups on the GO surface was very low. These results were supported by the Kaiser test. After Boc deprotection, the amounts of primary amines in GO 6a and GO 6b were calculated as 105 and 99 μmol g−1, respectively (Table 1). Both double functionalized GO 7a and GO 7b did not present a higher amount of amines due to the low efficiency of the carboxylation step at pH 9 and 13. The slight decrease of the amine level after the amidation was likely caused by the removal of some PEG chains physisorbed onto GO by DMF.
Percentage of nitrogen element and amount of amines on GO samples assessed by XPS and the Kaiser test, respectively.
| Sample | %N | Amine (μmol g−1) |
|---|---|---|
| 5a | 0.1 | — |
| 6a | 1.3 | 105 |
| 7a | 1.4 | 75 |
| 5b | 0.2 | — |
| 6b | 1.3 | 99 |
| 7b | 1.4 | 75 |
By applying this alternative strategy using a lower amount of sodium hydroxide, the reduction of GO was limited. Nevertheless, the carboxylation was not efficient, thus limiting the scope of the double functionalization strategy based on carboxylation followed by epoxide ring opening.
Conclusion
In summary, we investigated the influence of different amounts of sodium hydroxide on the carboxylation of GO aiming to prepare a multifunctional platform using controlled chemical approaches. The significant increase of the carboxyl groups on GO was observed only under strongly basic conditions, but a concomitant partial reduction of GO also occurred. Combining an epoxide ring opening, carboxylation and amidation reaction, GO was doubly functionalized, provoking, however, a significant reduction of the material accompanied by the removal of some functionalities during the carboxylation step. With a lower amount of sodium hydroxide, GO was not significantly reduced. Only a slight dehydration was observed due to a small proportion of the epoxides being opened by sodium hydroxide. Nevertheless, under these conditions, the introduction of carboxylates was negligible. Although it was possible to attach two distinct molecules by combining carboxylation with the epoxide ring opening reaction, the total level of functionalization remained lower than that of a direct epoxide ring opening. Overall, our results demonstrate that carboxylation is not an efficient approach for the functionalization of GO due to the partial reduction of GO and it can be hardly extended to achieve an efficient double functionalization of GO for further modifications. This work shows that it is very important to consider side reactions that may not be mentioned in the previous literature. The control of the derivatization of GO is crucial, in particular for biological purposes. The surface modification would tune the intrinsic properties of GO, affecting the loading efficiency and other factors which are important for its application in biosensing and disease treatment as drug nanocarriers.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of the Centre National de la Recherche Scientifique (CNRS) through the International Research Project MULTIDIM between the I2CT Unit and Okayama University, the International Center for Frontier Research in Chemistry (icFRC), and financial support from the Agence Nationale de la Recherche (ANR) through the LabEx project Chemistry of Complex Systems (ANR-10-LABX-0026_CSC). SG is indebted to the Chinese Scholarship Council for supporting his PhD internship. We wish to thank Cathy Royer and Valérie Demais for help with TEM analyses at the “Plateforme Imagerie in vitro” at the Center of Neurochemistry (INCI, Strasbourg, France).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00561d
References
- Zhang L. Ding Y. Zhang C. Zhou Y. Zhou X. Liu Z. Yu G. Chem. 2018;4:1035–1046. [Google Scholar]
- Farooqui U. R. Ahmad A. L. Hamid N. A. Renewable Sustainable Energy Rev. 2018;82:714–733. doi: 10.1016/j.rser.2017.09.081. [DOI] [Google Scholar]
- Lu X. Feng X. Werber J. R. Chu C. Zucker I. Kim J. H. Osuji C. O. Elimelech M. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E9793–E9801. doi: 10.1073/pnas.1710996114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M. Y. Xie Y. Yu C. H. Chen G. Y. Li Y. H. Zhang T. Peng Q. J. Controlled Release. 2019;307:16–31. doi: 10.1016/j.jconrel.2019.06.011. [DOI] [PubMed] [Google Scholar]
- Vulcano F. Kovtun A. Bettini C. Xia Z. Liscio A. Terzi F. Heras A. Colina A. Zanfrognini B. Melucci M. Palermo V. Zanardi C. 2D Mater. 2020;7:024007. doi: 10.1088/2053-1583/ab734f. [DOI] [Google Scholar]
- Ji D. K. Ménard-Moyon C. Bianco A. Adv. Drug Delivery Rev. 2019;138:211–232. doi: 10.1016/j.addr.2018.08.010. [DOI] [PubMed] [Google Scholar]
- Reina G. González-Dominguez J. M. Criado A. Vázquez E. Bianco A. Prato M. Chem. Soc. Rev. 2017;46:4400–4416. doi: 10.1039/C7CS00363C. [DOI] [PubMed] [Google Scholar]
- Jana B. Mondal G. Biswas A. Chakraborty I. Saha A. Kurkute P. Ghosh S. Macromol. Biosci. 2013;13:1478–1484. doi: 10.1002/mabi.201300129. [DOI] [PubMed] [Google Scholar]
- Jana B. Biswas A. Mohapatra S. Saha A. Ghosh S. Chem. Commun. 2014;50:11595–11598. doi: 10.1039/C4CC04924A. [DOI] [PubMed] [Google Scholar]
- Han B. Zhang Y. L. Zhu L. Li Y. Ma Z. C. Liu Y. Q. Zhang X. L. Cao X. W. Chen Q. D. Qiu C. W. Sun H. B. Adv. Mater. 2019;31:1806386. doi: 10.1002/adma.201806386. [DOI] [PubMed] [Google Scholar]
- Marcano D. C. Kosynkin D. V. Berlin J. M. Sinitskii A. Sun Z. Slesarev A. Alemany L. B. Lu W. Tour J. M. ACS Nano. 2010;4:4806–4814. doi: 10.1021/nn1006368. [DOI] [PubMed] [Google Scholar]
- Hummers W. S. Offeman R. E. J. Am. Chem. Soc. 1958;80:1339. doi: 10.1021/ja01539a017. [DOI] [Google Scholar]
- Dreyer D. R. Park S. Bielawski C. W. Ruoff R. S. Chem. Soc. Rev. 2010;39:228–240. doi: 10.1039/B917103G. [DOI] [PubMed] [Google Scholar]
- Compton O. C. Nguyen S. T. Small. 2010;6:711–723. doi: 10.1002/smll.200901934. [DOI] [PubMed] [Google Scholar]
- Kotchey G. P. Allen B. L. Vedala H. Yanamala N. Kapralov A. A. Tyurina Y. Y. Klein-Seetharaman J. Kagan V. E. Star A. ACS Nano. 2011;5:2098–2108. doi: 10.1021/nn103265h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurapati R. Russier J. Squillaci M. A. Treossi E. Ménard-Moyon C. Del Rio-Castillo A. E. Vazquez E. Samori P. Palermo V. Bianco A. Small. 2015;11:3985–3994. doi: 10.1002/smll.201500038. [DOI] [PubMed] [Google Scholar]
- Golmohammadi H. Morales-Narváez E. Naghdi T. Merkoçi A. Chem. Mater. 2017;29:5426–5446. doi: 10.1021/acs.chemmater.7b01170. [DOI] [Google Scholar]
- Cheeveewattanagul N. Morales-Narváez E. Hassan A.-R. H. A. Bergua J. F. Surareungchai W. Somasundrum M. Merkoçi A. Adv. Funct. Mater. 2017;27:1702741. doi: 10.1002/adfm.201702741. [DOI] [Google Scholar]
- Chai D. Hao B. Hu R. Zhang F. Yan J. Sun Y. Huang X. Zhang Q. Jiang H. ACS Appl. Mater. Interfaces. 2019;11:22915–22924. doi: 10.1021/acsami.9b03983. [DOI] [PubMed] [Google Scholar]
- Reina G. Chau N. D. Q. Nishina Y. Bianco A. Nanoscale. 2018;10:5965–5974. doi: 10.1039/C8NR00333E. [DOI] [PubMed] [Google Scholar]
- Chau N. D. Q. Reina G. Raya J. Vacchi I. A. Ménard-Moyon C. Nishina Y. Bianco A. Carbon. 2017;122:643–652. doi: 10.1016/j.carbon.2017.07.016. [DOI] [Google Scholar]
- Bhosale S. S. Jokar E. Fathi A. Tsai C.-M. Wang C.-Y. Diau E. W.-G. Adv. Funct. Mater. 2018;28:1803200. doi: 10.1002/adfm.201803200. [DOI] [Google Scholar]
- Cao C. Jin R. Wei H. Yang W. Goldys E. M. Hutchinson M. R. Liu S. Chen X. Yang G. Liu G. ACS Appl. Mater. Interfaces. 2018;10:33078–33087. doi: 10.1021/acsami.8b13518. [DOI] [PubMed] [Google Scholar]
- Ji P. Zhang W. Ai S. Zhang Y. Liu J. Liu J. He P. Li Y. Nanotechnology. 2019;30:115701–115710. doi: 10.1088/1361-6528/aaf8e4. [DOI] [PubMed] [Google Scholar]
- Wang M. Jamal R. Wang Y. Yang L. Liu F. Abdiryim T. Nanoscale Res. Lett. 2015;10:370–381. doi: 10.1186/s11671-015-1078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakilas V. Tiwari J. N. Kemp K. C. Perman J. A. Bourlinos A. B. Kim K. S. Zboril R. Chem. Rev. 2016;116:5464–5519. doi: 10.1021/acs.chemrev.5b00620. [DOI] [PubMed] [Google Scholar]
- Li F. Jiang X. Zhao J. Zhang S. Nano Energy. 2015;16:488–515. doi: 10.1016/j.nanoen.2015.07.014. [DOI] [Google Scholar]
- Mei K. C. Rubio N. Costa P. M. Kafa H. Abbate V. Festy F. Bansal S. S. Hider R. C. Al-Jamal K. T. Chem. Commun. 2015;51:14981–14984. doi: 10.1039/C5CC05412E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacchi I. A. Spinato C. Raya J. Bianco A. Ménard-Moyon C. Nanoscale. 2016;8:13714–13721. doi: 10.1039/C6NR03846H. [DOI] [PubMed] [Google Scholar]
- Vacchi I. A. Raya J. Bianco A. Ménard-Moyon C. 2D Mater. 2018;5:035037. doi: 10.1088/2053-1583/aac8a9. [DOI] [Google Scholar]
- Guo S. Nishina Y. Bianco A. Ménard-Moyon C. Angew. Chem., Int. Ed. Engl. 2020;59:1542–1547. doi: 10.1002/anie.201913461. [DOI] [PubMed] [Google Scholar]
- Vacchi I. A. Guo S. Raya J. Bianco A. Ménard-Moyon C. Chem.–Eur. J. 2020;26:6591–6598. doi: 10.1002/chem.201905785. [DOI] [PubMed] [Google Scholar]
- He D. He X. Wang K. Zou Z. Yang X. Li X. Langmuir. 2014;30:7182–7189. doi: 10.1021/la501075c. [DOI] [PubMed] [Google Scholar]
- Imani R. Prakash S. Vali H. Faghihi S. Biomater. Sci. 2018;6:1636–1650. doi: 10.1039/C8BM00058A. [DOI] [PubMed] [Google Scholar]
- Yin T. Liu J. Zhao Z. Zhao Y. Dong L. Yang M. Zhou J. Huo M. Adv. Funct. Mater. 2017;27:1604620. doi: 10.1002/adfm.201604620. [DOI] [Google Scholar]
- Jiang T. Sun W. Zhu Q. Burns N. A. Khan S. A. Mo R. Gu Z. Adv. Mater. 2015;27:1021–1028. doi: 10.1002/adma.201404498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. Liu Z. Welsher K. Robinson J. T. Goodwin A. Zaric S. Dai H. Nano Res. 2008;1:203–212. doi: 10.1007/s12274-008-8021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Zhang Q. Chu X. Chen T. Ge J. Yu R. Angew. Chem., Int. Ed. Engl. 2011;50:7065–7069. doi: 10.1002/anie.201101351. [DOI] [PubMed] [Google Scholar]
- Chen C. Kong W. Duan H. M. Zhang J. Phys. Chem. Chem. Phys. 2014;16:12858–12864. doi: 10.1039/C4CP01031K. [DOI] [PubMed] [Google Scholar]
- Fan X. Peng W. Li Y. Li X. Wang S. Zhang G. Zhang F. Adv. Mater. 2008;20:4490–4493. doi: 10.1002/adma.200801306. [DOI] [Google Scholar]
- Park K.-W. J. Mater. Chem. A. 2014;2:4292–4298. doi: 10.1039/C3TA14223J. [DOI] [Google Scholar]
- Han J.-L. Zhang D. Jiang W. Tao Y. Liu M.-J. Haider M. R. Ren R.-Y. Wang H.-c. Jiang W.-L. Ding Y.-C. Hou Y.-N. Zhang B. Cheng H.-Y. Xia X. Wang Z. Wang A.-J. J. Membr. Sci. 2019;576:190–197. doi: 10.1016/j.memsci.2018.12.055. [DOI] [Google Scholar]
- Taniguchi T. Kurihara S. Tateishi H. Hatakeyama K. Koinuma M. Yokoi H. Hara M. Ishikawa H. Matsumoto Y. Carbon. 2015;84:560–566. doi: 10.1016/j.carbon.2014.12.054. [DOI] [Google Scholar]
- Zhang P. Li Z. Zhang S. Shao G. Energy Environ. Mater. 2018;1:5–12. doi: 10.1002/eem2.12001. [DOI] [Google Scholar]
- Shams M. Guiney L. M. Huang L. Ramesh M. Yang X. Hersam M. C. Chowdhury I. Environ. Sci.: Nano. 2019;6:2203–2214. doi: 10.1039/C9EN00355J. [DOI] [Google Scholar]
- Rourke J. P. Pandey P. A. Moore J. J. Bates M. Kinloch I. A. Young R. J. Wilson N. R. Angew. Chem., Int. Ed. Engl. 2011;50:3173–3177. doi: 10.1002/anie.201007520. [DOI] [PubMed] [Google Scholar]
- Dimiev A. M. Alemany L. B. Tour J. M. ACS Nano. 2013;7:576–588. doi: 10.1021/nn3047378. [DOI] [PubMed] [Google Scholar]
- Szabó T. Berkesi O. Dékány I. Carbon. 2005;43:3186–3189. doi: 10.1016/j.carbon.2005.07.013. [DOI] [Google Scholar]
- Lucherelli M. A. Raya J. Edelthalhammer K. F. Hauke F. Hirsch A. Abellan G. Bianco A. Chem.–Eur. J. 2019;25:13218–13223. doi: 10.1002/chem.201903165. [DOI] [PubMed] [Google Scholar]
- Kaiser E. Colescott R. L. Bossinger C. D. Cook P. I. Anal. Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- Lerf A. He H. Forster M. Klinowski J. J. Phys. Chem. B. 1998;102:4477–4482. doi: 10.1021/jp9731821. [DOI] [Google Scholar]
- Cai W. Piner R. D. Stadermann F. J. Park S. Shaibat M. A. Ishii Y. Yang D. Velamakanni A. An S. J. Stoller M. An J. Chen D. Ruoff R. S. Science. 2008;321:1815–1817. doi: 10.1126/science.1162369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.