

Abstract
Advances in synthetic carbohydrate chemistry have dramatically improved access to common glycans. However, many novel methods still fail to adequately address challenges associated with chemical glycosylation and glycan synthesis. Since a challenge of glycosylation has remained, scientists have been frequently returning to the traditional glycosyl donors. This review is dedicated to glycosyl halides that have played crucial roles in shaping the field of glycosciences and continue to pave the way toward our understanding of chemical glycosylation.
Graphical Abstract

1. INTRODUCTION: GLYCOSYLATION AS A CENTRAL REACTION OF TRADITIONAL AND MODERN GLYCOSCIENCES
From the building blocks of nature to disease-battling pharmaceuticals, carbohydrates have had a broad impact on many scientific and industrial fields. Numerous applications of these essential biomolecules line up at the frontier of pharmaceutical, diagnostic, and functional food development. Consequently, carbohydrates have been extensively studied by biological and medical communities. Earth-abundant and renewable sources of chirally pure materials, monosaccharides remain popular building blocks for synthetic chemists. Oligomers, glycans, or oligosaccharides, wherein multiple monosaccharides are connected to each other via glycosidic linkages, have also been popular targets for chemists. However, complex carbohydrates remain challenging targets due to the requirement for elaborate protecting group manipulations and functionalization at all stages of the synthesis. Many glycosylation methods have been developed, but performing glycosylation reactions with complete chemo-, regio-, and stereoselectivity represents a notable challenge.
Nature flawlessly executes the glycosylation reaction enzymatically,1,2 but chemical glycosylation remains cumber-some.3 The first chemical glycosylation reactions were performed by Michael,4 Fischer,5,6 and Koenigs/Knorr7 at the turn of the 20th century. With the exception of the Fischer glycosylation that relies on unprotected sugars as glycosyl donors,5 all other early methods relied on glycosyl halides, chlorides or bromides.4,6,7 Already those initial studies led to the development of glycosylation methodologies that paved the way to obtaining simple alkyl/aryl glycosides for decades. However, glycosylations of sugar acceptors were less efficient, and the synthesis of complex oligosaccharide targets was deemed practically impossible. Terminology used herein refers to “glycosylation of the acceptor with the donor” or “glycosidation of the donor with the acceptor.” One of the main directions to improve synthetic capabilities has been the investigation of leaving groups beyond the original chlorides/bromides and hemiacetals. These studies lead to the discovery of other glycosyl halides, fluorides,8 and iodides.9 However, many other classes of alternative glycosyl donors including glycosyl esters,10–13 thioglycosides,14–17 1,2-orthoesters,18,19 O-imidates,20–23 thioimidates,24–29 alkenyl glycosides,30–32 sulfones,33 thiocyanates,34 diazirines,35 glycals,36–40 sulfoxides,41 xanthates,42 selenium glycosides,43 phosphites,44,45 tellurium glycosides,46 sulfonylcarbamates,47 heteroaryl glycosides,48 phosphates,49 disulfides,50 2-(hydroxycarbonyl)benzyl glycosides,51 and compounds equipped with alkynyl-based leaving groups52–61 have been developed.
Regardless of the glycosylation reaction conditions and mechanism, unimolecular SN1-like or bimolecular SN2-like, the following steps are typically identified (Scheme 1A).62 The first step involves the formation of the activated donor as a result of the interaction of the leaving group (LG) and the promoter (A-B); this step can be reversible or irreversible depending on the leaving group and the method of activation.63 There are a few reports indicating that the glycosyl acceptor attack may be directed to the activated donor. This pure SN2 displacement pathway would be quite desirable (THE GOOD) in terms of potential stereocontrol because it would allow for the stereospecific inversion of the leaving group: equatorial LG will produce axial glycoside whereas axial LG will lead to equatorial glycoside. However, examples of such reactions remain rare.64–71 Examples wherein double SN2-like inversion would lead to the retention of the original LG configuration are also known. In a majority of cases, the second step involves the dissociation of the glycosyl donor, a typically irreversible expulsion of the activated leaving group (LGA), which is the rate-determining step (RDS) of the glycosylation reaction.
Scheme 1.

Outline of General Glycosylation Mechanisms
This process leads to the formation of the respective glycosyl carbocation and/or its stabilized form, oxacarbenium ion, along with the associated counteranion (B−). The oxacarbenium intermediate is often blamed for scrambling the stereoselectivity of the reaction (THE BAD). This is due to the existence of the oxacarbenium ion in a flattened half-chair conformation because of the sp2-hybridization of the anomeric carbon. It should be noted that other conformations of oxacarbenium intermediates are possible depending on the nature of substrates, protecting groups, and various steric and/or electronic factors. The next step involves a nucleophilic attack on the oxacarbenium ion, which is possible from either the bottom face or the top face of the ring. As a result, uncontrolled glycosylations often lead to the formation of a mixture of α/β-diastereomers. Other intermediates, the existence of which is often ignored (THE UGLY), or their impact on the reaction is underestimated, may also form at this stage with or without covalently attached B. However, there are some extended studies related to such intermediates, examples of which include tosylates, triflates, intermediates of dehydrative glycosylations among others, which can lead to unexpected side products and/or be responsible for scrambling the stereoselectivity of glycosylations. On the other hand, is the involvement of these intermediates is understood, this could lead to excellent stereocontrol how it has been proven by Schuerch,72,73 Crich,74–77 Gin,78–82 Woerpel,83–87 Bennett,69,88,89 and others.90 The last step, the existence of which is also often overlooked, involves the proton transfer. This is the termination step, after which the formation of the glycosidic bond becomes irreversible.91
The simplified mechanistic outline of glycosylation presented in Scheme 1A implies that the RDS is unimolecular and is independent of the glycosyl acceptor. However, the donor–acceptor mismatch concept by Paulsen92 and Fraser-Reid and Lopez,93–97 the double stereodifferentiation phenomenon,98 and other studies99–102 present a strong counterargument. In fact, a number of kinetic studies that have emerged in the past decade indicate that a majority of glycosylations follow a mixed mono- and bimolecular displacement mechanism,71,103–106 and the reaction order differs between various sugar series, and can even depend on the leaving group and/or covalently attached counterion type and orientation.107–111 As stated by Crich et al, the exact mechanism of a leaving group departure in a typical glycosylation reaction generally falls at a certain position on a continuum of mechanisms spanning from ideal SN1 extreme to ideal SN2 extreme (Scheme 1B).71,105–112
The goal of controlling glycosylation has been pursued in many other ways with much effort dedicated to the optimization of the reaction conditions, suppressing side reactions,113,114 studying stereoelectronics and conformation of the starting material and key reaction intermediates.62,80–84,86,87,91,115–129 Fraser-Reid’s seminal work on the armed-disarmed approach showed that the building block reactivity can be modulated through the choice of protecting groups.130,131 The scope of the original armed-disarmed concept has been expanded and a number of efforts to quantify or even predict the reactivity of building blocks have been reported by Fraser-Reid,132 Ley133,134 and Wong.135,136 Wong’s study also revealed a number of building blocks that extend beyond the traditional armed-disarmed boundary. This discovery opened a new avenue for studying building block reactivity,137 and Boons138 and, subsequently, Demchenko139 reported superdisarmed building blocks.
Two concepts for superarming glycosyl donors have also emerged. Bols showed that superarming can be achieved by changing the equatorial-rich4C1 conformation.124–127 Demchenko then reported building blocks wherein the electronic superarming was achieved via the O2/O5 cooperative effect.140–142 While the stereoelectronic and conformational effects on reactivity have been studied extensively, the impact on stereoselectivity (beyond building blocks equipped with 2-O-participating group) remain elusive. Although some model studies helped to establish general trends,83–86,115,116,128,129,143 practical applications of the stereoelectronic and conformational factors to stereocontrolling glycosylation reactions are still lacking.
Despite all these recent improvements, the challenge of glycosylation has remained, and scientists have turned their attention to reinvestigating the original glycosyl donors, hemiacetals, and halides. More recent work with hemiacetals69,81,144 and glycosyl halides70,145–147 brought these glycosylation reactions to an entirely different level of flexibility and versatility. These simple donors are typically easily accessible from a variety of precursors, can be readily activated, and offer superior atom economy. This review, dedicated to glycosyl halides, guides the reader from the first known glycosylation reactions to recent advances in the field that helped to navigate glycosciences forward. The first glycosylations performed by Michael involved glycosyl chlorides. However, in subsequent years, glycosyl chlorides were largely outshadowed by glycosyl bromides. Because of the ease of the synthesis and their higher reactivity profile, glycosyl bromides have traditionally been considered advantageous over their chloride counterparts. As a result, most of the efforts were focused on reactions of glycosyl bromides that have become prevalent glycosyl donors in the first 100 years of synthetic carbohydrate chemistry. Therefore, we will open our review with a summary of the early studies (Section 2) and then move directly to the discussion of glycosyl bromides (Section 3). Many methods developed for the activation of glycosyl bromides are also effective for the activation of glycosyl chlorides. Hence, Section 4, dedicated to glycosyl chlorides, will mainly focus on the reagent-specific or condition-specific activations of glycosyl chlorides rather than repeating the same data presented for glycosyl bromides. We will then turn our attention to glycosyl iodides (Section 5), which were outshadowed by both glycosyl bromides and glycosyl chlorides, and only recently found some notable synthetic utility. Finally, we will discuss glycosyl fluorides (Section 6), which became very prominent glycosyl donors in recent decades.
2. KEY ACCOMPLISHMENTS OF (AND LESSONS FROM) EARLY GLYCOSIDATIONS OF GLYCOSYL HALIDES AT THE TURN OF THE 20TH CENTURY
The first glycosylation was reported by Arthur Michael in 1879, about a decade before the famous addition reaction that carries his name, the Michael addition, was discovered. This reaction is depicted in Scheme 2A, wherein glycosidation of chloride α-1 with alkoxide produced glycoside 2, which was deprotected under these basic reaction conditions to produce glycoside 3. In Michael’s own words, the synthesis was “…starting from the interesting compound discovered by A. Colley…known under the name of acetochlorhydrose. This compound…, I have allowed to act on potassium phenate and potassium salicylite, and have obtained compounds which possess all the characteristic properties of the glucosides. After numerous experiments, I found the following conditions to yield the most satisfactory results: 27.5 gr. of acetochlorhydrose were mixed with about twice its volume of absolute alcohol, and the solution added to a cold alcoholic solution of 10 gr. of potassium phenate. After a few minutes, a crystalline precipitate began to separate from the solution, and at the same time a strong odor of acetic ether was noticed. The reaction proceeded very rapidly, and after four or five hours no further separation of the crystalline substance was observed… The most interesting property of this substance is its behavior towards dilute acids and emulsin. A dilute solution of sulfuric or chlorhydric acid decomposes it on gently warming very readily in glucose and phenol…”4
Scheme 2.

First Glycosylation Reactions Reported by Michael (A), Koenigs and Knorr (B), and Fischer (C)
In 1901, Koenigs and Knorr introduced silver salt-promoted activation of glycosyl bromides for the synthesis of glycosides.7 This approach is considered as a major milestone in glycochemistry because the outcome of this reaction was much easier to control or predict than that of the Fischer glycosylation with unprotected donors. It was also shown that alcohols rather than charged nucleophiles used in the Michael approach can be glycosylated directly. The first reactions involved glycosidation of acetylated glucosyl bromide β-4 with simple alcohols (methanol or ethanol) in the presence of silver carbonate (Ag2CO3) or silver nitrate (AgNO3) as shown in Scheme 2B. It should be noted that current understanding of the reaction and its mechanisms implies that the formation of pure β-anomer of acetobromoglucose is very unlikely. It is possible that during those times, the anomeric configuration of glycosyl halides was simply unknown. To preserve integrity of the discussion, we will be using configurations that were originally proposed by the authors. Contrary to the Michael glycosylation approach4 wherein neutral KCl was generated, in the Koenigs–Knorr reaction the formation of HBr was inevitable. The exact role of the silver salt was not known at that moment, but it was assumed that it acted as the acid scavenger. The synthesis of methyl glucoside β-5 was also achieved by the treatment of glycosyl bromide β-4 in the presence of either barium carbonate (BaCO3) or pyridine. It was also demonstrated that methyl glycoside can be generated in the presence of an excess of methanol without activators or additives. On the other hand, the treatment of glycosyl bromide 4 with silver acetate (CH3COOAg) in acetic acid afforded pentaacetate 6, and a similar treatment with fuming nitric acid afforded tetra-acetylated glycosyl nitrate 7. An interesting observation was made that methyl glycoside 5 and pentaacetate 6 had the same anomeric configuration as that of the starting material, β-glycosyl bromide 4. This methodology was also extended to per-acetylated galactosyl bromides. Again, it is quite possible that the authors were dealing with α-bromides that were mistakenly presented as their β-counterparts.
Independently from Koenigs and Knorr, Fischer and co-workers also synthesized methyl glucoside from α-acetochloroglucose 1 in the presence of methanol and silver carbonate, and the product was deprotected with barium hydroxide.6 Differently from the results reported by Koenigs and Knorr (vide supra), glycosidation of α-chloride 1 resulted in the formation of methyl α-glucoside 8 (Scheme 2C). Additionally, Fischer and co-workers synthesized tetra-acetylated methyl galactoside from galactosyl chloride. Apart from monosaccharides, the authors were able to synthesize hepta-acetylated maltosyl chloride 9 that was glycosidated with methanol in the presence of silver carbonate. In this case β-linked methyl maltoside 10 was obtained.6 Similarly to studies by Koenigs and Knorr, Fischer also considered silver acetate AgOAc, but noticed a significant level of the acetate transfer.6
Following these early efforts dedicated to glycosylation of simple alcohols, Fischer and co-workers synthesized a variety of relatively complex β-glycosides including hepta-acetyl menthol maltoside.148,149 The authors also tried to synthesize a 1→1-linked tetrasaccharide assuming that hepta-acetyl lactosyl bromide will self-condense with the hemiacetal produced in situ in the presence of Ag2CO3.148 Since the role of silver salt was considered to be as acid scavenger, Fischer thought of replacing it with an organic base, such as quinoline. This study showed unprecedented formation of significant amounts of α-phenyl glucoside when per-acetylated glycosyl bromide was heated with phenol in the presence of quinoline.150 The continuation of this work led to the implementation of silver oxide as an alternate acid scavenger.151 In addition to acetylated bromides, glycosidation of per-benzoylated glucosyl bromide β-11 with MeOH was proven possible in the presence of Ag2O to afford methyl glycoside 12 (Scheme 3A).
Scheme 3.
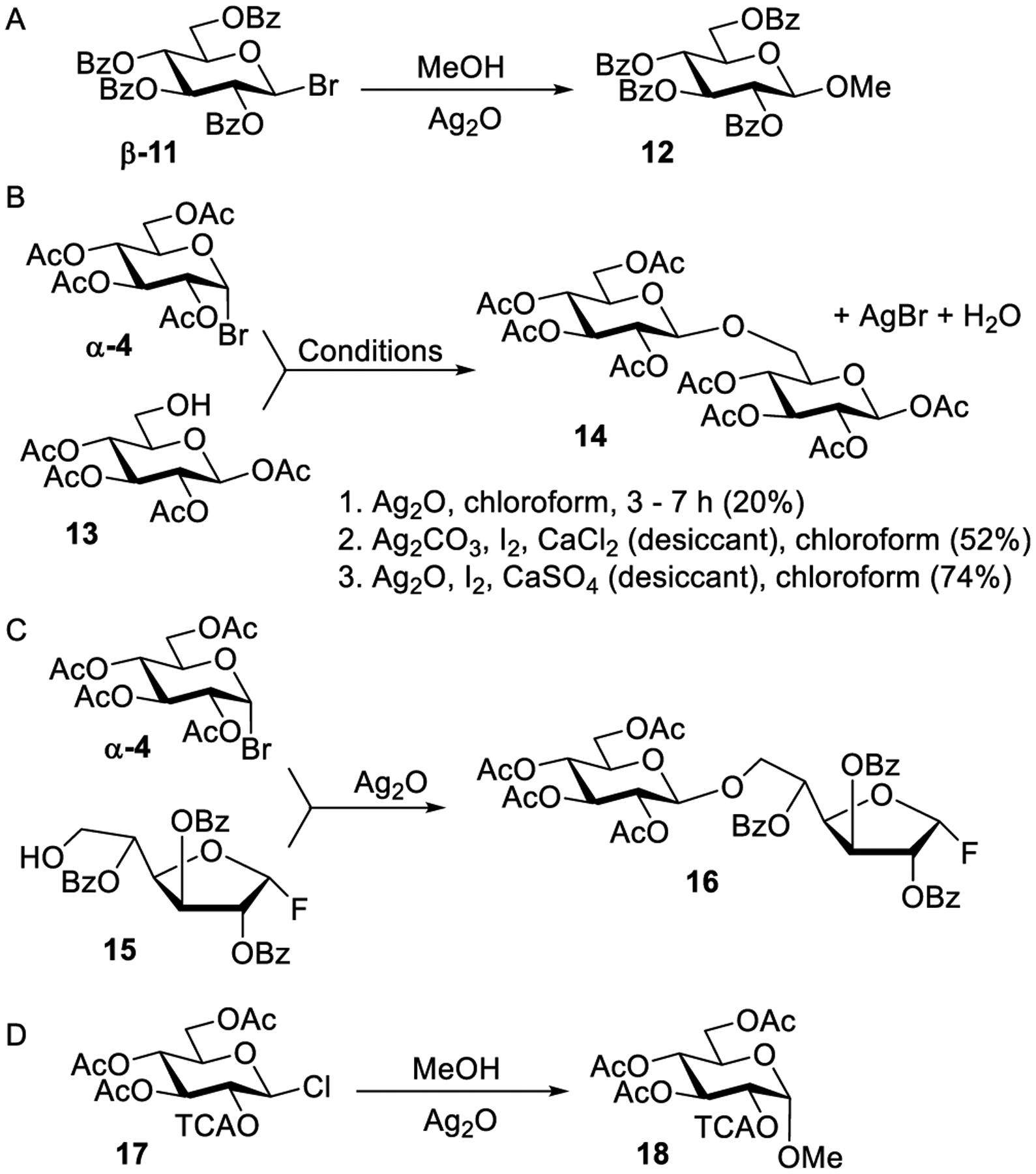
First Attempts to Enhance Utility of the Koenigs–Knorr Reaction
Helferich and co-workers were the first to synthesize a disaccharide by coupling two monosaccharide building blocks. As shown in Scheme 3B, this was achieved by treating glucosyl bromide α-4 with 1,2,3,4-tetra-O-acetyl-β-d-glucopyranose acceptor 13 under classical Koenigs–Knorr glycosylation conditions to afford per-acetylated β-gentiobiose 14 (conditions 1).152 However, only a modest yield of 20% was achieved, which was attributed to the presence of water in the reaction mixture. It was suggested that water molecules interfere with the reaction by continuously consuming glycosyl bromide α-4 and producing hydrolyzed byproducts. Helferich and co-workers then attempted to remove water by adding finely powdered calcium chloride as a desiccant. However, this led to a significant drop in the rate of glycosylation.153 It was then discovered that the reaction can be accelerated when molecular iodine was added to a silver carbonate-promoted glycosylation reaction in the presence of a desiccant (conditions 2). In this case, β-gentiobiose 14 was obtained in a significantly improved yield of 52%. Reynolds and Evans also developed reaction conditions that helped to achieve complete exclusion of water, both present in the reactants from the beginning and also that generated during the reaction.154 These reaction conditions involved preliminary stirring of glycosyl acceptor 13 and silver oxide with Drierite (CaSO4) in chloroform for 1 h. Iodine was then added followed by a slow addition of a solution of glycosyl bromide α-4 in chloroform. As a result of these improvements, a very high yield of 74% for the synthesis of gentiobiose 14 was achieved (conditions 3, Scheme 3B).
Helferich and co-workers were also the first to achieve a selective activation of one leaving group over another. Some 60–70 years later, this selective activation strategy became a very useful tool for streamlining glycan assembly because the products can be used as glycosyl donors directly, without any modification (vide infra).155 In this application, selective activation of glycosyl bromide donor α-4 over glycosyl fluoride acceptor 15 was achieved in the presence of silver oxide to produce disaccharide 16 (Scheme 3C).152 The deprotection of acyl groups furnished gentiobiosyl fluoride which was then treated with calcium carbonate to produce a fully unprotected disaccharide.
Formation of the α-phenyl glucoside in the quinoline-promoted reaction at a high temperature by Fischer (vide supra),150 demonstrated that 1,2-cis glycosides can also be obtained.156 Following this lead, Brigl and co-workers described the stereoselective synthesis of α-glycosides from β-chloride 17 protected with the 2-O-trichloroacetyl group shown in Scheme 3D.157 The authors assumed that the Walden inversion occurs during the glycosylation of methanol in the presence of silver carbonate to afford α-glycoside 18. The α/β-ratio of glycosides was found to depend on the reaction time and temperature. Similar to Brigl’s investigation, Schlubach and Schroter158 and also Hickinbottom159 investigated the synthesis of α-glycosides from β-acetochloroglucose. Over the course of this study, the authors anticipated that the synthesis of α-glycosides from β-glycosyl chlorides can be achieved only when the experimental conditions are maintained in the way that the rate of glycosylation is greater than that of the β-chloride anomerization to its α-counterpart. The authors also anticipated that the anomerization can be suppressed by using a suitable solvent. It was further reinforced that higher α-selectivity of glycosylations is achieved at lower glycosyl acceptor concentrations. To further advance the classical Koenigs–Knorr glycosylation approach, Zemplen and co-workers switched from silver salts and showed that an efficient activation of cellobiosyl bromide can be achieved with mercuric acetate.160,161 Over the course of this investigation, anomeric mixtures have been obtained, and a dedicated study revealed that the ratio of α/β-glycosides mainly depends on the amount and types of alcohol used.160,162,163
During those times, many reactions were hampered by the formation of side products. One of the most exciting observations was Fischer’s discovery of the existence of an additional isomeric glycoside that was observed to exist besides α- and β-glycosides.164 The authors coined the term of the γ-form (or third form) and suggested that it might have a different ring structure than pyranose as a result of the ring-opening, acyl migration, and subsequent ring closure during the glycosylation reaction. Also reported by Dale,165 and puzzled many others,166–170 it was not until studies by Freudenberg171,172 and, independently, Haworth et al.173,174 who suggested the γ-form to be 1,2-orthoester 19 shown in Scheme 4. To understand mechanistic details for the formation of glycosides and orthoesters, Isbell investigated various factors affecting Koenigs and Knorr reactions and byproduct formation, such as the orientation of the substituent at C-2, solvents, temperature, and the presence of water. Apart from these, the authors conducted an expansive study of physical and chemical properties of products to understand chemical composition, structure, and conformation of products.175–179
Scheme 4.
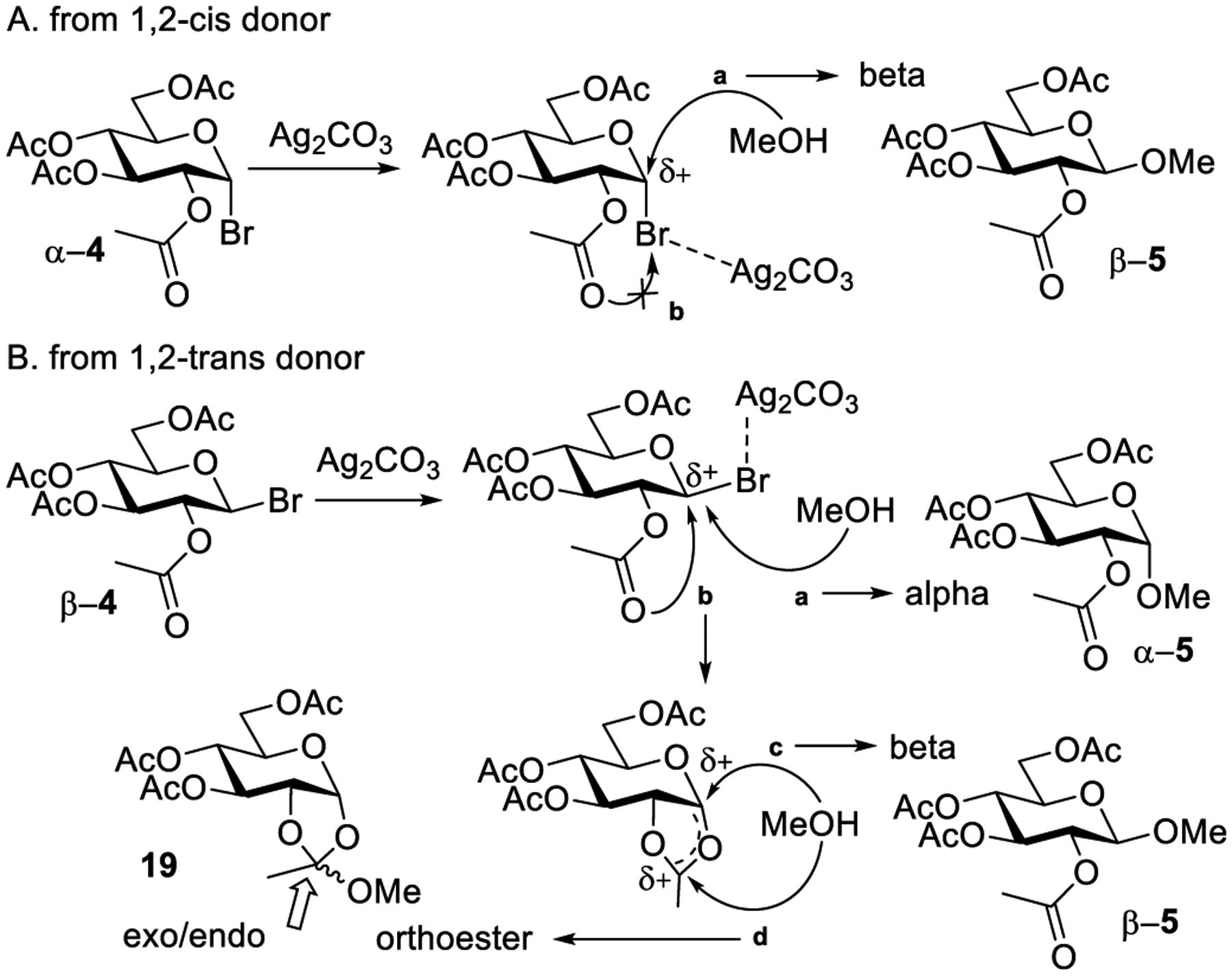
Glycosidation of 1,2-cis and 1,2-trans Glycosyl Halides and the Formation of 1,2-Orthoesters
On the basis of these studies, Isbell and co-workers concluded that 1,2-trans glycosylation occurs when there is a participating ester group present at the C-2 position. Taking into consideration the neighboring group participation, Isbell proposed two distinct pathways for glycosidation of 1,2-cis and 1,2-trans glycosyl halides, α-4 and β-4, respectively.176 The activation process wherein the anomeric bromide complexes with the silver salt is the same for both glycosyl donor configurations (Scheme 4). This decreases the electron density at the anomeric center, making it more susceptible to nucleophilic attack. In the case of the 1,2-cis glycosyl halide α-4, only the expected inversion product β-5 can be obtained (Scheme 4A, pathway a). In this case, the 1,2-orthoester does not form (pathway b) because the approach of the 2-O-acetyl group is blocked by the halide. Conversely, the 1,2-trans bromide β-4 yielded two anomeric glycosides α/β-5 and the orthoesters exo/endo-19 (Scheme 4B). Following the activation, the 1,2-cis product α-5 was obtained via direct nucleophilic displacement from the bottom face of the ring (pathway a). Additionally, the intramolecular attack from the adjacent carbonyl oxygen leads to the formation of a reactive acyloxonium intermediate (pathway b). Depending on the site of nucleophilic attack, the latter can produce a 1,2-trans glycoside β-5 (pathway c) and a 1,2-orthoester exo/endo-19 (pathway d). In this case, the neighboring group participation may or may not offer anchimeric assistance and hence accelerate the leaving group departure. It should be noted that although sometimes (inaccurately) used interchangeably, “neighboring group participation” and “anchimeric assistance” are not the same: while the former refers to the effect on stereoselectivity, the latter refers to the acceleration of the reaction rate. It was originally believed that that the formation of orthoesters and participation is only possible when the leaving halogen (bromide) is present in the 1,2-trans position to the participating group on the neighboring carbon atom. However, later it was clearly demonstrated are orthoesters can also form from 1,2-cis glycosyl halides.180–190 Subsequent studies of orthoesters were extended to sugar alcohols191 and led to observation of diastereomeric exo/endo-orthoesters the existence of which was later proven by nuclear magnetic resonance (NMR).192
Over the years, Koenigs–Knorr glycosylation reactions found broad application in glycosylation of simple alcohols. Glycosylations of sugar acceptors were less efficient and the synthesis of complex oligosaccharide targets was deemed practically impossible. One of the main directions to improve synthetic capabilities has been the investigation of different activation conditions. It should be particularly emphasized that it is seminal studies of glycosyl halide donors discussed in this section that have enabled our understanding of many basic aspects and mechanisms behind glycosylation reactions. This, in turn, helped to resolve challenges in synthesizing many targets ranging from simple glycosides to complex glycans and glycoconjugates.
3. GLYCOSYL BROMIDES
Since first silver salt-promoted activations of glucosyl bromides, innumerable reagents for the activation of glycosyl bromides have emerged. Not only glycosidation but also the synthesis of glycosyl bromides underwent significant improvements over the years with the introduction of mild reagents and developing efficient reaction conditions.
3.1. Synthesis of Glycosyl Bromides
Synthesis of glycosyl bromides is very crucial since many compounds of this class are not crystalline and their stability differs drastically based on their structure and protecting group pattern. Numerous methods have been developed for the synthesis of glycosyl bromides that are categorized below by the type of the starting material (anomeric protecting/leaving group) used for the introduction of the anomeric bromide moiety.
3.1.1. Preparation from Unprotected Sugars with Concomitant Per-acetylation.
As aforementioned, synthesis of glycosyl chlorides was known since studies by Colley and then Michael. These syntheses were accomplished from the corresponding free sugars that were treated with acetyl chloride. Following this general concept, Koenigs and Knorr synthesized acetobromoglucose by treating glucose with neat acetyl bromide. The authors were able to isolate β-bromide in a good yield after crystallization (Scheme 5A).7 For many years, this method remained popular for the synthesis of acetylated glycosyl bromides.7,148,193 Decades later, Koto and co-workers showed that the synthesis of glycosyl bromides can be achieved by treating the corresponding free sugars with acetyl bromide that was diluted with acetic acid.194,195 The authors demonstrated that acetic acid helps to prevent sudden evolution of toxic HBr gas compared to previously known methods for the formation of glycosyl bromide donor. Kartha and Jennings reported a one-pot synthesis of per-acetylated glycosyl bromide wherein free sugars were first treated with acetic anhydride followed by the addition of HBr in AcOH.196 Another method for the synthesis of glycosyl bromides involves the treatment of free sugars with acetic anhydride in the presence of catalytic HClO4 in acetic acid followed by bromination with AcBr-MeOH with or without sonication.197,198 A reversal in the order of the reagent addition wherein sugar and acetic anhydride were added to a mixture of acetyl bromide and methanol in acetic acid also produced glycosyl bromides in excellent yields.167 Lin et al. treated unprotected sugars with LiClO4-Ac2O followed by the reaction with HBr-AcOH.199 Recently, Bennett, Pohl, and co-workers developed a continuous flow platform for the synthesis of orthogonally protected monosaccharide building blocks.200 One relevant example includes acetylation and bromination of unprotected sugars performed in a single step. This transformation was rapidly achieved by employing acetyl bromide as acetylating and brominating reagent in a continuous flow manner. Saturated aq. NaHCO3 was then used for in-line quench of excess HBr.
Scheme 5.
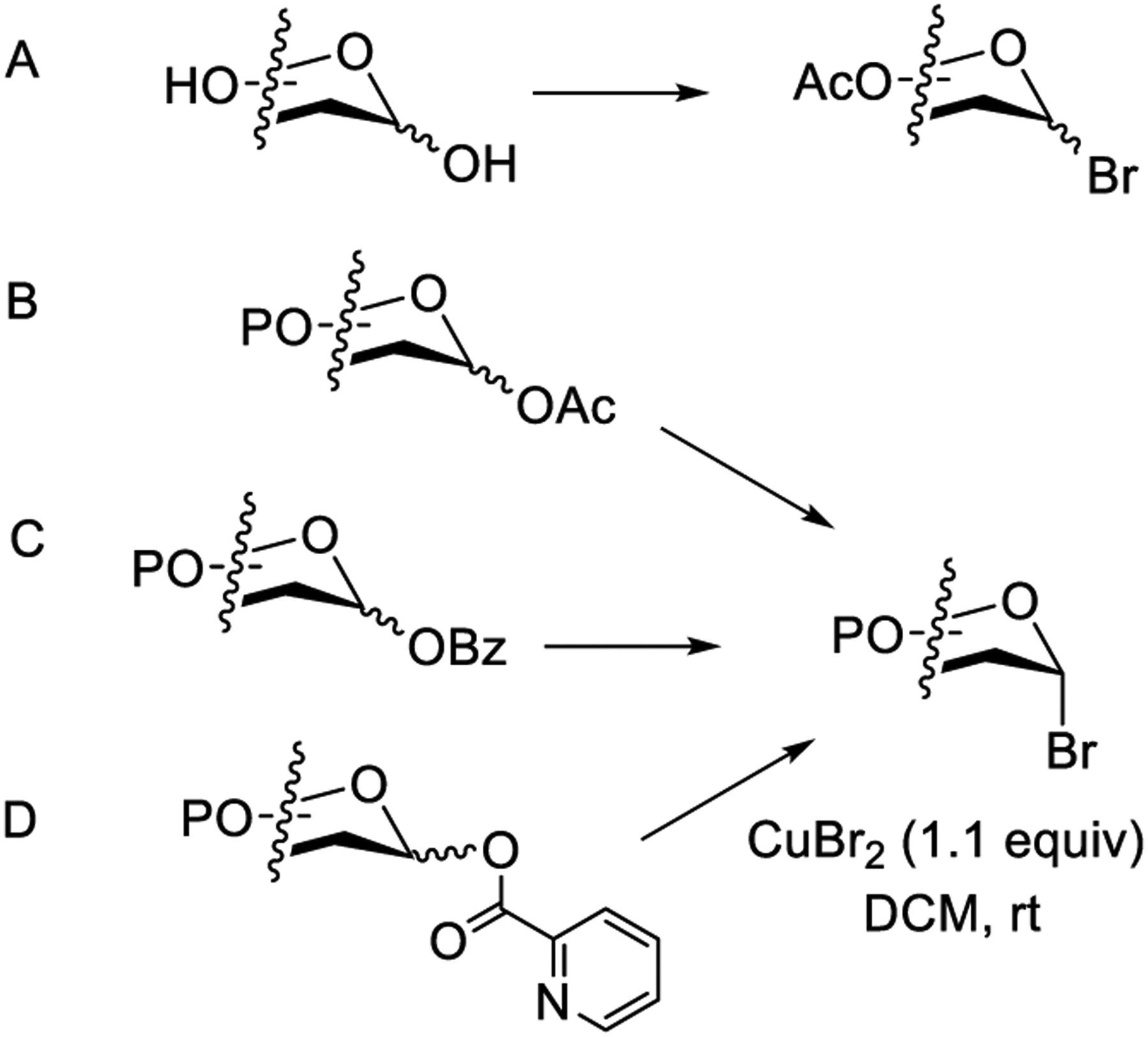
Synthesis of Glycosyl Bromides from Unprotected Sugars and Glycosyl Esters
3.1.2. Preparation from Glycosyl Esters of Differentially Protected Sugars.
The synthesis of acetylated glycosyl bromides can also be performed starting from presynthesized per-acetates (Scheme 5B). As described by Fischer, the treatment of glucose pentaacetate with HBr neat or in glacial acetic acid produced per-acetylated β-glucosyl bromide.149 In early days, a few other methods such as red phosphorus/bromine or PBr3 were commonly used for the synthesis of bromides.201 Zemplen and co-worker employed titanium tetrabromide for the synthesis of glycosyl bromide wherein a solution of titanium tetrabromide in chloroform was added to a solution of tetra-acetylated rhamnose in chloroform to produce acetobromo-α-l-rhamnose in an excellent yield and in crystalline form.202 This protocol works well for the synthesis of 2-azido-2-deoxy bromides.203
More recent methods were predominantly focused on the development of milder reaction conditions for the formation of anomeric bromides. One such procedure for bromination involves a dilute solution of HBr in dichloromethane (DCM).204–206 A convenient method to generate HBr in situ is by reaction of MeOH and AcBr; this protocol is particularly advantageous for the synthesis of highly reactive bromides and/or those equipped with acid-sensitive protecting groups.64 Hunsen and co-workers utilized this protocol also for the synthesis of glycosyl bromides from the corresponding free sugars in the presence of Ac2O.197 Thiem and co-workers discovered that anomeric acetates can be efficiently converted to corresponding anomeric bromides upon treatment with trimethylsilyl bromide (TMSBr) in chloroform or benzene.207 The side product, trimethylsilyl acetate can be removed in vacuo owing to its low boiling point. Around the same time, a similar observation was independently made by Gillard and co-worker who converted different anomeric acetates to the corresponding bromides.208 The use of TMSBr is attractive since it eliminated the aqueous work up, which is a common operation used in many other methods. Apart from this, many common protecting groups are compatible with TMSBr. Later, Montero and co-workers reported a milder approach for halogenation by employing bismuth(III)-based reagents complexed with halosilanes.209 The treatment of per-acetylated sugars with BiBr3-Me3SiBr produced corresponding glycosyl bromides in excellent yields. Mizuno and co-workers reported light-induced (352 nm, 15 W) conversion of per-acetylated sugars to the corresponding glycosyl bromides in the presence of bromine.210
Preparation of glycosyl bromides from per-benzoylated sugars has also been reported (Scheme 5C). Fischer and Helferich treated per-benzoylated glucose with a saturated a solution of HBr in glacial acetic acid.151 This method was further modified by Hudson and co-workers.211,212 Another efficient method for the synthesis of benzoylated glycosyl bromides involves the treatment of per-benzoylated sugars with AcBr-MeOH using ultrasound irradiation.198 All the previous methods for the synthesis of glycosyl bromides from the corresponding 1-O-acyl derivatives rely on highly reactive, toxic, sensitive, and corrosive acids. To address these issues, Tang and co-workers described a milder reaction conditions for an efficient synthesis of glycosyl bromides.213 In this approach, 1-O-picoloyl (Pico) derivatives were treated with copper(II) bromide in DCM at ambient temperature (Scheme 5D). This reaction produced the corresponding α-glycosyl bromides in good to excellent yields, and the method was found to be compatible with several protecting groups including acid-sensitive groups.
3.1.3. Preparation from Thioglycosides or Other Substrates.
One of the most convenient methods for the formation of glycosyl bromides involves treatment of thioglycosides with bromine to produce the corresponding glycosyl bromides (Scheme 6A). This reaction typically completes in minutes, does not require any work up, and the glycosyl bromides can be used immediately after solvent evaporation.214–217 Because of mild reaction conditions, a broad range of protecting groups including acyl, alkyl, silyl, acetal, and ketal are tolerated. A few other methods have been developed. Kobayashi and co-workers reported the conversion of 2-O-benzylated hemiacetals to corresponding glycosyl bromides by the treatment with PPh3-CBr4, commonly known as Appel conditions (Scheme 6B).218 A one-pot conversion of benzyl-protected methyl or p-methoxyphenyl glycosides to per-benzoylated glycosyl bromides by treating with zinc triflate and benzoyl bromide, is another useful approach to the synthesis of per-benzoylated glycosyl bromides (Scheme 6C).219
Scheme 6.

Synthesis of Glycosyl Bromides from Thioglycosides (A), Hemiacetals (B), and O-Glycosides (C)
3.2. Glycosidation of Glycosyl Bromides
Since the first silver salt-promoted activation of glucosyl bromides (vide supra), innumerable reagents for the activation of glycosyl bromides have emerged. To streamline the discussion, we chose to divide the activating reagents into different categories based on their halophilic nature (Table 1). The discussion begins from silver salts that were very instrumental for understanding the metal salt involvement in splitting the anomeric C–Br bond and investigating the effects of the counteranion of silver salts. We will then focus the discussion on how those studies enabled scientists to better understand the glycosylation reaction, helped develop improved methods, and how the improved methods enhanced our synthetic capabilities.
Table 1.
Activation of Glycosyl Bromide Donors
| promoter | additive |
|---|---|
| silver and copper salts | |
| Ag2CO36,7 | I2,153,220 Lewis acid221 |
| AgNO37,151 | crown ether222,223 |
| AgOAc6,7 | |
| Ag2O151 | I2,154 borinic acid,224 HOFox/Lewis acid,217,225 TMSOTf,146,221,226 TfOH221 |
| AgClO4227–229 | |
| AgBF4227,230 | |
| AgPF6230 | |
| AgOTf230 | TMU231 |
| other organic Ag salts221,232–234 | TfOH221 |
| Ag silicate-alumina235 | |
| Ag zeolite236 | |
| Ag silica-alumina237 | |
| silver imidazole | ZnCl2238 |
| Ag2SO4 | TfOH,221,226 |
| CuI + BPhen + Xantphos147 |
DTBMP147 |
| mercury, zinc, and cadmium salts | |
| Hg(OCOCH3)2160,161 | |
| Hg(CN)2239 | HgBr2239 |
| HgO239 | HgBr2240 |
| HgBr2241,242 | |
| Hg(PhCOO)2243 | |
| Hg(NpCOO)2243 | |
| HgI2244 | |
| ZnO239 | |
| ZnCl2245,246 | TrCl;245 TMSCl247 |
| ZnBr2245,246 | TrBr;245 TMSBr246,247 |
| Zn(OTf)2 | TMSBr248 |
| CdO239 | |
| CdCO3249–251 | |
| CdS249 | |
| indium, tin, and bismuth salts | |
| InCl3252,253 | |
| InBr3253 | |
| InI3254 | |
| In(OTf)3254 | |
| In(NTf2)3254 | |
| SnCl4255 | |
| Sn(OTf)2 | base256 |
| PbCO3251 | |
| non-nucleophilic bases | |
| pyridine7,148,182,257 | |
| quinoline150,156 | |
| collidine180 | |
| phenanthroline derivatives257 | |
| 2,2′-bipyridine257 | |
| halogens or halide ions | |
| NR4Br258,259 | |
| I2260,261 | DDQ260 |
| IBr262,263 | DABCO262 |
| ICl263 | |
| NIS261,263 | I2;263 protic acids261 |
| other methods | |
| NR3264–266 | |
| PR3265,266 | |
| SR2265,266 | |
| solvolysis7,230,267,268 | NR4Br;267,268 silver salts230 |
3.2.1. Activation with Silver Salts or Other Group 11 Metal Salts (Copper).
As mentioned previously, glycosylation reactions in the presence of insoluble silver salts such as Ag2O and Ag2CO3 proceed slowly and may result in inefficient glycosylations despite using large excess of reagents and, sometimes, excess reactants. This ultimately affects the yield of glycosides that typically fall within 30 to 70%. To improve the glycoside synthesis, several soluble silver salts were introduced as promoters for the activation of glycosyl bromides. These salts include silver acetate,6,7 which led to the predominant 1-O-acetylation, silver nitrate first studied by Koenigs and Knorr,7 and in a greater detail investigated by Knöchel et al.222,223 Several other soluble silver salts were explored including perchlorate,227,229 tetrafluoroborate,227,230 hexafluorophosphate,230 and trifluoromethanesulfonate (triflate).230 While being efficient activators, these silver salts required multiple equivalents of the acid scavenger to be added in these reactions. Wulff and co-workers studied glycosylation of steroidal alcohols in the presence of several silver salts of hydroxyacids and dicarboxylic acids.232–234,269,270 In a majority of cases, glycosides were observed as major products, but in some cases significant amounts of 1-O-acyl and 1,2-orthoester derivatives were also present. A direct correlation was paved between the formation of products and the distance between the hydroxy and carboxy groups in the organo-silver salts. As depicted in Figure 1, interactions between OH or OAg and the carbonyl group in 2-, 3-, and 4-hydroxycarboxylate salts (A), and 1,2-, 1,3-, 1,4-dicarboxylate salts (B) may have a role in suppressing the nucleophilic attack of the carboxylate ion. Stronger interactions help to lower the formation of 1-O-acyl derivatives. For hydroxy acids, silver 3-hydroxypentanoate and salicylate provided the highest yields, whereas disilver maleate outperformed all other diacids investigated.
Figure 1.
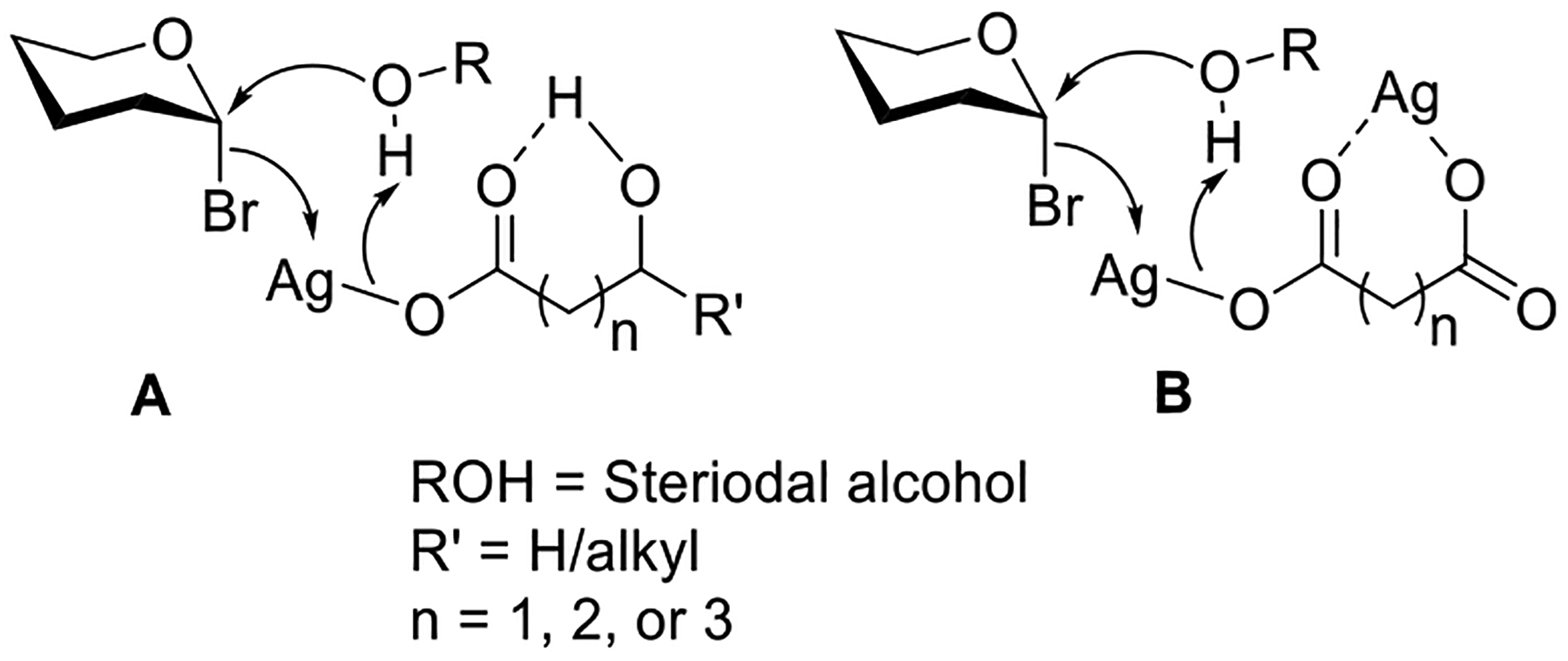
Mechanistic interpretation of action of hydroxyl carboxylate and dicarboxylate silver salts.
Apart from the above-mentioned silver salts, numerous other silver-based promoters were reported for the activation of glycosyl bromides such as silver imidazole-ZnCl2,238 silver silicate,235 silver silicate-alumina,235 silver zeolite,236 silver silica-alumina.237 As discovered by Paulsen and co-workers, silver silicates are very advantageous for the synthesis of β-linked glycosides.235 This study was recently extended by Herzon and co-worker to the stereoselective synthesis of 2-deoxy and 2,6-dideoxy β-glycosides, from corresponding anomeric bromides upon activation with silver silicate.271 The authors employed in situ formation of glycosyl bromides α-22 and α-23 from the corresponding anomeric acetates in the presence of TMSBr in dichloromethane as shown in Scheme 7. The synthesized glycosyl bromides α-22 and α-23 were then reacted with (−)-menthol acceptor 24 in the presence of silver silicate to produce the respective β-linked glycosides in good yields and with high stereoselectivity: 25 (81%, α/β = 1:18) and 26 (74%, α/β = 1:22). The authors pointed out that the nature of protecting groups of the glycosyl bromide donors may play an important role in enhancing or reducing the stereoselectivity. It was also observed that acid sensitive and azide protecting groups remained unaffected during these reaction conditions.
Scheme 7.

Silver Silicate-Promoted Glycosidation of 2-Deoxy- and 2,6-Dideoxyglycosyl Bromides
Borinic acid-catalyzed regioselective glycosidation of glycosyl bromide (and chloride) was developed by Taylor and co-workers224 in accordance with their approach, complexation of partially protected cis-diol glycosyl acceptor 27 with borinic acid 28 to afford a borinate complex takes place first (Scheme 8).272 The nucleophilicity of sugar hydroxyl groups is predicted by Fukui index calculation,273–275 which states that the boron-bound oxygen to be more nucleophilic than the free hydroxyl group. The borinate complex subsequently attacks glycosyl bromide α-4 to furnish disaccharide 28. The outcome of the glycosylation in terms of yields and stereoselectivity mainly depends on the stereochemistry of the glycosyl halide and reactivity of the acceptor. Exclusive 1,2-trans selectivity was observed in borinic acid-catalyzed glycosylation wherein α-glycosyl halides were used.
Scheme 8.
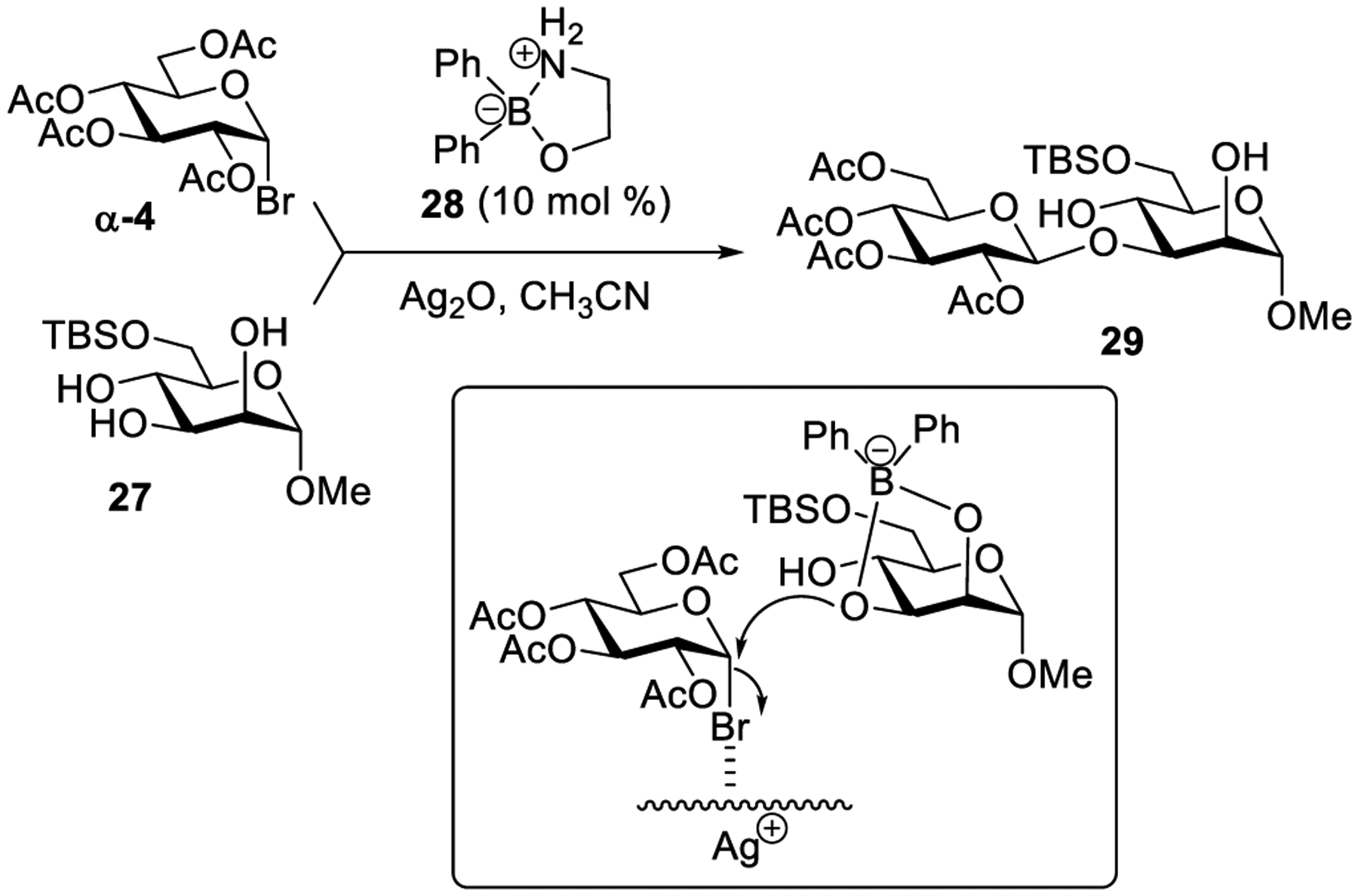
Borinic Acid-Catalyzed Glycosidation of Glycosyl Bromide
Demchenko and co-workers have developed a regenerative strategy to activate glycosyl bromides.217,225 As shown in Scheme 9, ethylthio glycoside 30 was first treated with a stoichiometric amount of bromine to afford glycosyl bromide 31. The latter was dried in vacuo and used without purification for the regenerative glycosylation cycle wherein both the synthesis of the reactive OFox imidate intermediate 33 and its glycosidation were performed in a catalytic, regenerative fashion. First, HOFox aglycone 32 used in catalytic amounts was reacted with glycosyl bromide 31 in the presence of silver oxide to afford OFox imidate 33.276 Second, glycosyl acceptor and a Lewis acid (TMSOTf) were added afford the corresponding glycosides 34. The leaving group from the OFox intermediate departs as HOFox aglycone 32 that can be used to generate the next batch of OFox imidate 33. It was also shown that the rate of glycosylation reaction can be increased by increasing the amount of HOFox used. As illustrated in Scheme 8, only 0.25 equiv of HOFox additive was very effective at enhancing glycosidations of glycosyl bromides of the gluco, galacto, and manno series. More recently, a streamlined procedure that allows for bypassing the intermediacy of glycosyl bromides was reported.277
Scheme 9.

Regenerative Glycosylation
Very recently, Singh and Demchenko discovered an acid-catalyzed silver salt-promoted glycosyl halide activation. It was observed that the addition of a catalytic amount of TMSOTf to a silver oxide-promoted glycosylation dramatically accelerates the reaction and rapidly affords glycosides in very high yields.146 As depicted in Scheme 10, TMSOTf-catalyzed glycosidation of mannosyl bromide 35 with glycosyl acceptor 36 afforded disaccharide 37 in 99% yield in 10 min. This new reaction was applied to the synthesis of a variety of glycosides of different series. A tentative mechanism was proposed, wherein it was assumed that silver oxide coordinates with anomeric bromide (Intermediate A) and the oxide oxygen gets silylated with TMSOTf (intermediate B). This helps to shift the equilibrium of the reaction, reinforces the Ag–Br bond formation, and results in the release of AgBr that precipitates from the reaction mixture making the reaction irreversible (Scheme 10).
Scheme 10.

TMSOTf-Catalyzed Koenigs–Knorr Glycosylation
Depending on the C-2 protection group, either an oxacarbenium or acyloxonimun ion is formed as a reactive intermediate (intermediate C), which then reacts with the glycosyl acceptor. After deprotonation and TMS exchange, TMSOTf along with silver hydroxide are formed. TMSOTf gets cycled back into the reaction, and AgOH is decomposed to silver oxide by losing a water molecule to the desiccant (molecular sieves). The authors eliminated a possibility of the in situ formation of AgOTf by showing that these conditions are ineffective for the activation of glycosyl STaz29 and SBox27,28 glycosyl donors that are readily activated with AgOTf. While no further mechanistic evidence has been presented, it is certainly possible that the intermediate B may undergo an SN2-like displacement instead of leading to the oxacarbenium ion. Modest stereoselectivity observed in reactions with glycosyl donors equipped with a nonparticipating group at C-2 is indicative of the intermediacy of the oxacarbenium ion.
From the proposed mechanistic pathway, it became clear that as little as 0.50 equiv of silver oxide (stoichiometric silver) should be sufficient for the complete consumption of glycosyl bromide donor. Further investigation of roles of different silver salts, Lewis/Bronsted acid additives, and solvents revealed further particulars of this cooperatively catalyzed reaction.221 Thus, it was noted that reactions in polar solvents proceed much slower and are prone to side processes. Silver(I) oxide and acid-catalyzed glycosylation in nonpolar solvents proceed as explained in Scheme 10, and the identical reaction pathway was envisaged for both Lewis acid and protic acid cocatalysis. In polar solvents, however, it was assumed that upon addition of Lewis or Bronsted acids, strongly ionized species get solvated in polar solvents as shown for intermediate D in Scheme 11. The solvation reduces the effective interaction with the anomeric bromide E thus resulting in longer reaction times or incomplete consumption of glycosyl bromide or formation of side products. Ultimately, this investigation led to a successful activation of glycosyl bromides in the presence of 0.50 equiv of silver oxide and 0.35 equiv of TfOH in toluene. One of the most interesting aspects of the reaction was that the progress and completion of the reaction could be monitored by eye due to stark visual changes, when the glycosidation proceeds from dark brown-black appearance due the presence of Ag2O of the reaction mixture to complete decolorization when 0.50 equiv of Ag2O has been entirely consumed.
Scheme 11.
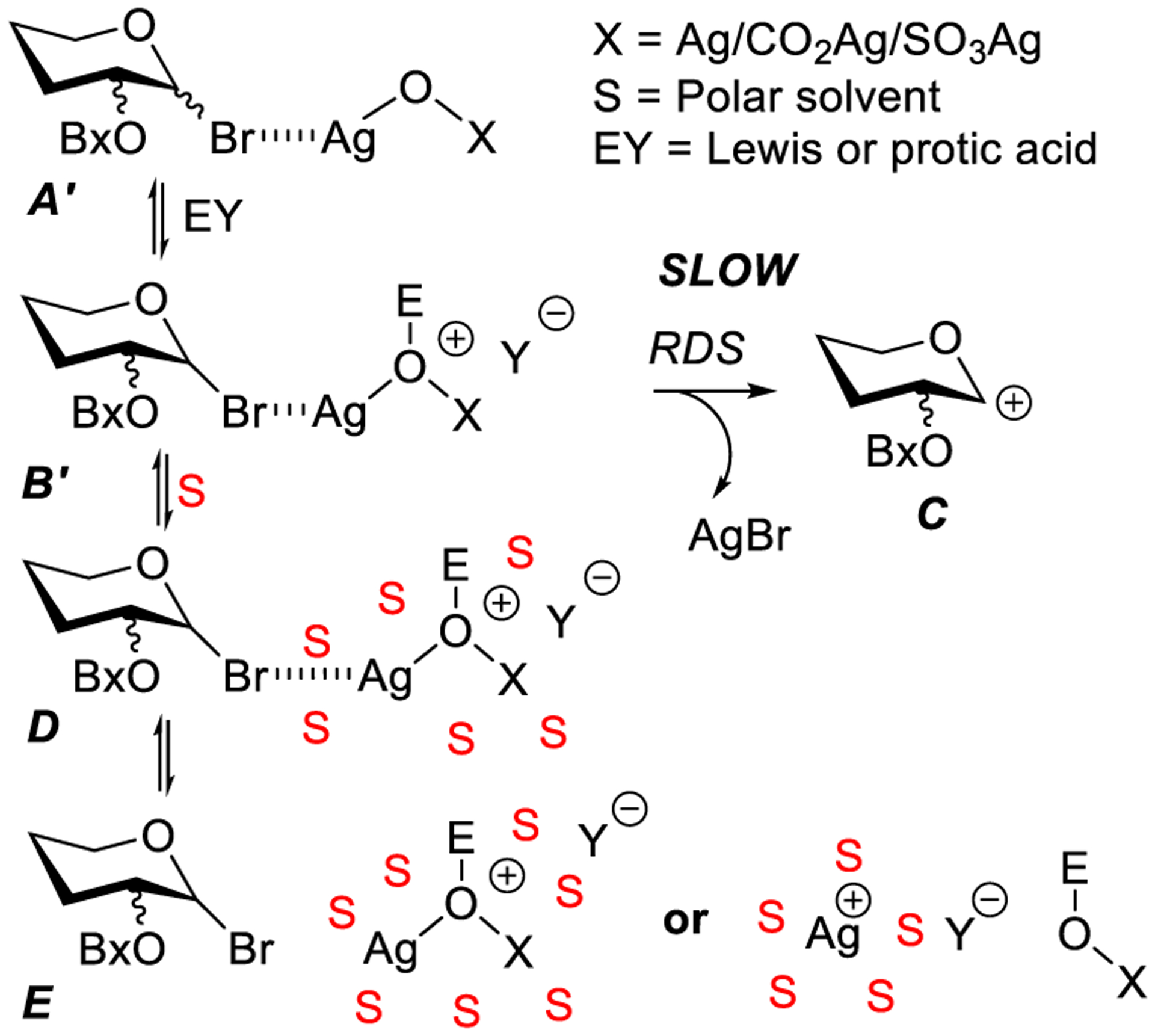
Cooperatively Catalyzed Koenigs–Knorr Glycosylation in Polar and Nonpolar Solvents
An unusual reactivity trend has been unveiled in these cooperatively catalyzed glycosylation conditions where benzoylated α-bromides 35 and 11 turned out to be much more reactive compared to their benzylated counterparts 39 and 38 (Figure 2). The higher reactivity of benzoylated α-bromides compared to their benzylated counterparts strikingly contradicts the armed-disarmed theory proposed by Fraser-Reid.278 This was found to be consistent irrespective of silver salts, acids, and sugar series employed with a less pronounced effect in highly reactive galactosyl bromide donors 40 and 41.
Figure 2.
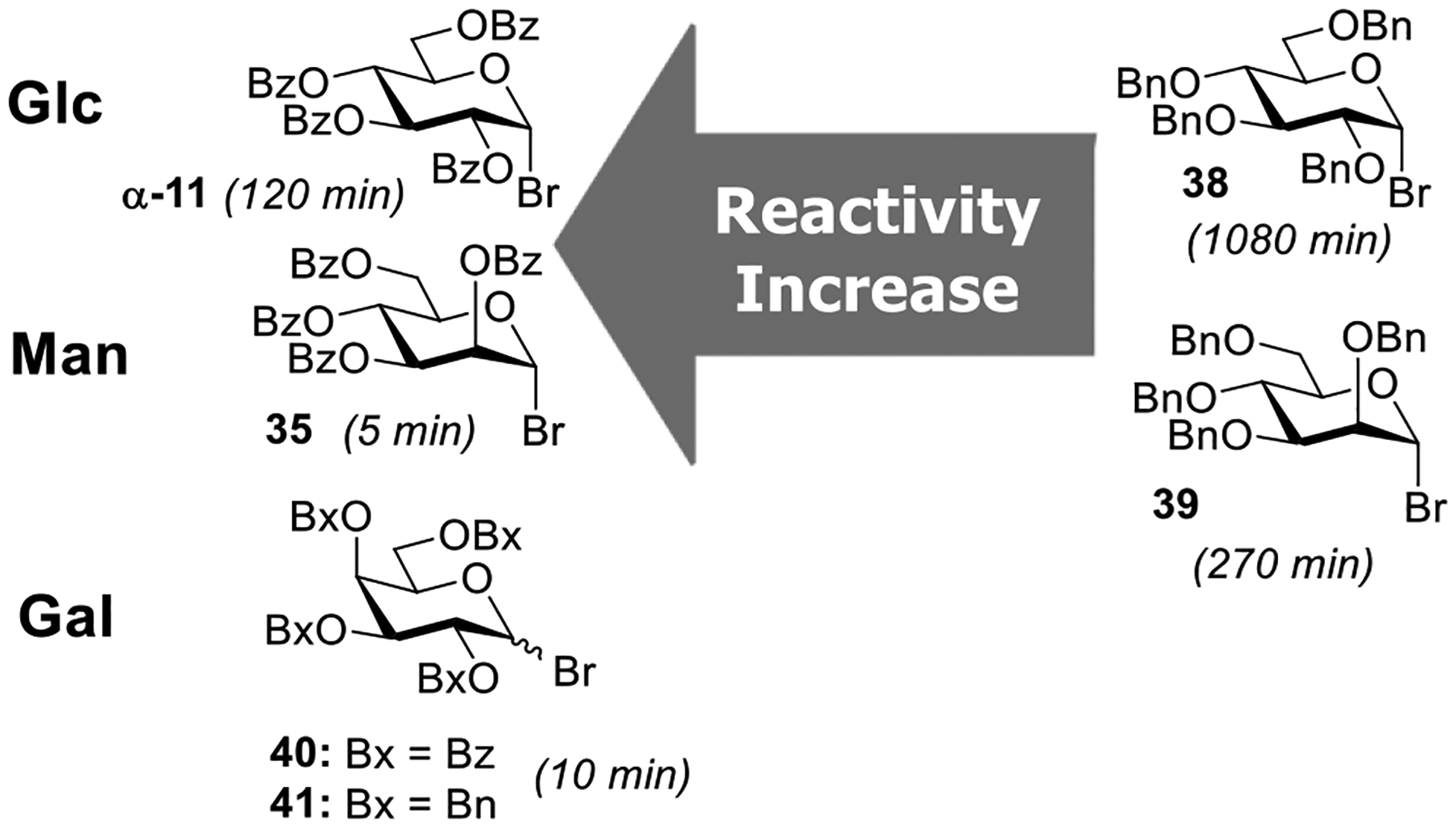
Contradicting reactivity trends in the cooperatively catalyzed Koenigs–Knorr glycosylations.
The glycosidation of glycosyl bromide donors bearing a nonparticipating (benzyl) group at C-2 under cooperative catalysis produce glycosides in good yield albeit no stereoselectivity. To address this issue, Demchenko and co-workers performed a preliminary investigation of 1,2-cis selective galactosylation reaction by optimizing the reaction conditions in combination with switching the location of common protecting groups OBn and OBz across different position of galactosyl bromides.226 For this purpose, galactosyl bromide 43, obtained from bromination thioglycoside 42, was used as glycosyl donor and methyl 2,3,4-tri-O-benzyl-α-d-glucopyranoside acceptor 36 in the presence of Ag2SO4 and TfOH (Scheme 12). Disaccharide 44 was obtained in 87% yield with predominant α-selectivity (α/β = 6.0:1). From this study, the authors concluded that the 4-O-acyl group is strictly necessary, as previously discovered by Boons,279 but it may be insufficient to drive α-selective galactosylation under cooperative catalysis of Ag2SO4 (1.5 equiv) and TfOH (0.20 equiv) in dichloromethane.
Scheme 12.
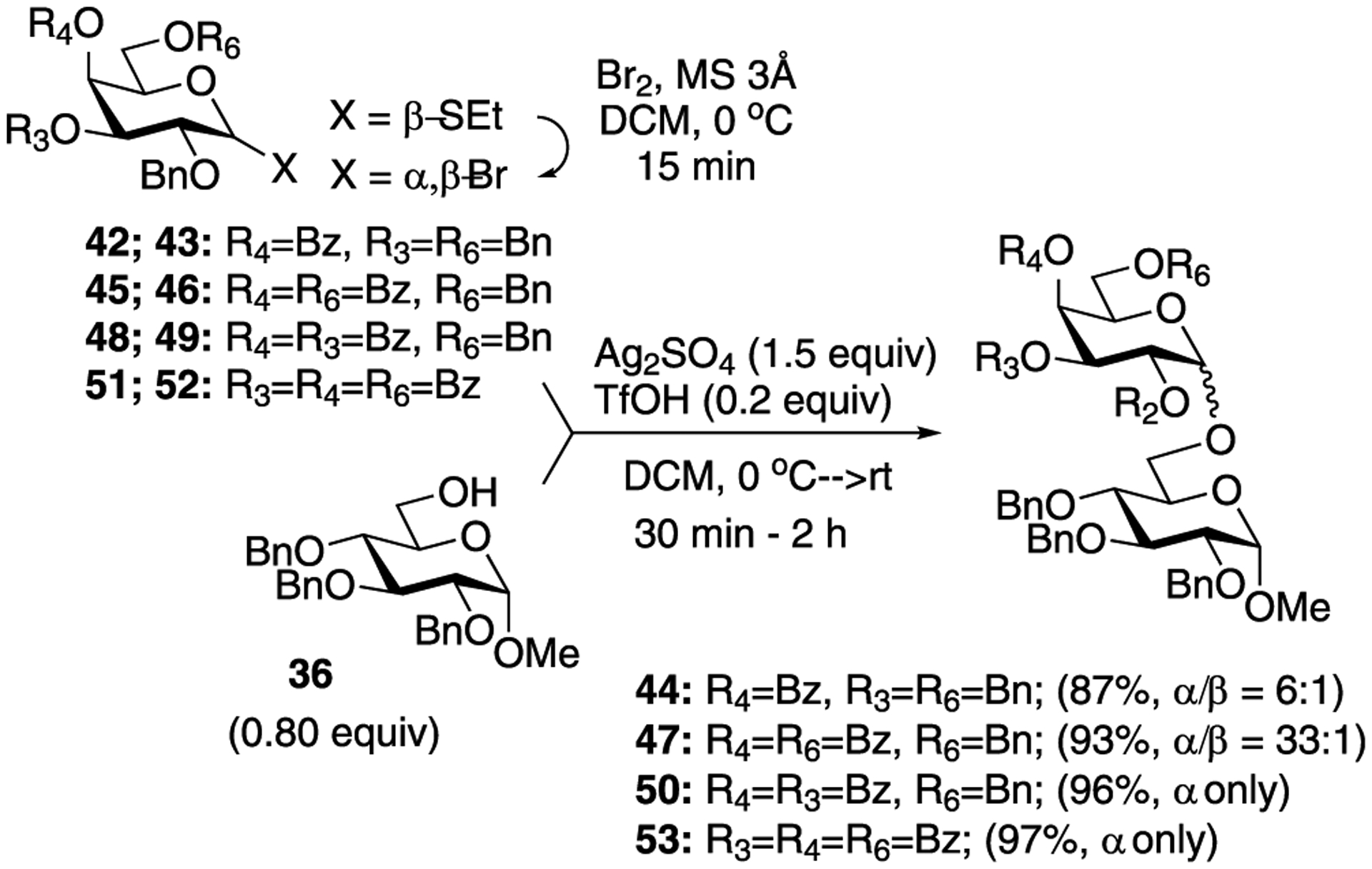
Acyl Group-Directed α-Stereoselective Galactosylation in Cooperatively Catalyzed Glycosylations
Subsequently, the authors achieved an excellent 1,2-cis α-galactosylation from galactosyl bromide 46 having 4- and 6-OBz groups. The bromide donor 46 was obtained by bromination of thiogalactoside 45. Glycosidation of donor 46 with glycosyl acceptor 36 produced disaccharide 47 in 93% yield (α/β = 33.0:1) as shown in Scheme 12. Following this general line of thought, thiogalactoside 48 having 3- and 4-OBz groups and 51 having 3-, 4-, and 6-OBz groups were synthesized, which were later brominated to produce galactosyl bromides 49 and 52, respectively. Glycosidation of both galactosyl bromides 49 and 52 with glycosyl acceptor 36 produced the respective disaccharides 50 and 53 in excellent yields and with exclusive α-selectivity. Construction of different types of 1,2-cis galactosidic linkages was then extended to a variety of differently protected glycosyl acceptors.
Very recently, Nguyen and co-workers developed a method for copper-catalyzed activation of glycosyl bromides in the presence of visible light.147 Reaction optimization revealed that copper iodide (CuI), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (XantPhos), 4,7-diphenyl-1,10-phenanthroline (BPhen), and di-tert-butylmethylpyridine (DTBMP) are strictly necessary to induce an efficient coupling between glycosyl bromide 54 and glycosyl acceptor 55 in acetonitrile affording disaccharide 56 in 72% yield (Scheme 13). The comparison of chemical shift value (31P NMR, δ = −11.99 ppm) obtained from in situ generated complex was identical with the isolated complex [Cu(BPhen)(Xantphos)]BF4 (31P-NMR, δ = −11.87 ppm) and the glycosylation outcome from the isolated catalyst, [Cu(BPhen)(Xantphos)]BF4 was similar to the aforementioned conditions, indicating [Cu(BPhen)-(Xantphos)]+ as an active catalyst for this transformation. The optimized reaction conditions facilitated the construction of glycosidic linkages using coupling partners of different sugar series.
Scheme 13.

Visible Light-Mediated Cu(I)-Catalyzed 1,2-cis α-Selective Glycosylation
In addition to the outcome of glycosylations, data from absorption and emission spectroscopies and electrochemistry experiments suggested the following mechanistic pathway for the visible light-mediated copper-catalyzed activation of glycosyl bromide. The initial step includes the reaction of Cu(I) catalyst A with alcohol to produce Cu(I)-oxygen complex B (Scheme 13).280,281 Upon photoirradiation (blue light) the latter is converted to excited Cu(I)-complex C that enables electron transfer to glycosyl bromide D to afford glycosyl radical E and Cu(II) complex F. This can potentially lead to either Cu(III) complex G282–285 via Path I or oxacarbenium ion intermediate H via Path II.286 Control experiments were performed by replacing oxygen at C-2 with fluorine/hydrogen atom. Fluorine is known to interact with the copper center;287 indeed, 2-fluoro-2-deoxy-derivative favored α-1,2-cis selectivity. Conversely, the 2-deoxy derivative led to the formation of an α/β mixture, which could be due to the formation of a 2-deoxy cation. These observations strongly suggest that α-1,2-cis selectivity does not arise from the oxacarbenium ion intermediate H generated either via Path II or from the collapse of Cu(III) complex G. This concludes the Path I is more likely to be operative wherein reductive elimination of Cu(III) complex G affords glycoside I and regenerates Cu (I) catalysts to endure the catalytic glycosylation cycle.
Herein, we are describing a representative example to construct C-glycosidic linkages by activating glycosyl bromides with a combined effect of visible light and [Ru(bpy)3]2+.288 Thus, as described by Gagné and co-workers, these reactions produce a glycosyl radical from glycosyl bromide, which gets intermolecularly trapped with electron-deficient alkene to afford C-glycosides in a stereoselective manner. For example, per-benzoylated glycosyl bromide α-11 produced the corresponding glycosyl radical upon visible light-mediated activation in the presence of [Ru(bpy)3](BF4)2 (5 mol %) and a stoichiometric amount of N,N-diisopropylethylamine (DIPEA) in DCM. Once formed, the glycosyl radical gets immediately consumed by methyl acrylate to produce the corresponding α-C-glycoside 57 (Scheme 14). The authors observed the formation of an overconjugate addition product 58. This side reaction could be suppressed by the addition of Hantzsch ester 59, which also was found to accelerate the reaction. This observation allowed to obtain α-C-glycoside 57 in a very impressive 92% yield and led to an improved understanding of the mechanistic pathway. As shown in Scheme 14, as electron transfer occurs from the photogenerated species [Ru(bpy)3]+ to anomeric bromide A producing glycosyl radical B. The latter can get reduced to produce C or can be trapped by an electron-deficient alkene to produce C-glycoside radical D. Subsequent step depends on relative rates for the formation of overconjugate product E versus that of the formation of expected C-glycoside F. As it was mentioned, Hantzsch ester 59 accelerates the reduction of D into F.
Scheme 14.

Visible Light-Mediated Intermolecular Addition of Glycosyl Bromide to Methyl Acrylate
3.2.2. Activation with Mercury and Other Group 12 Metal Salts (Zinc or Cadmium).
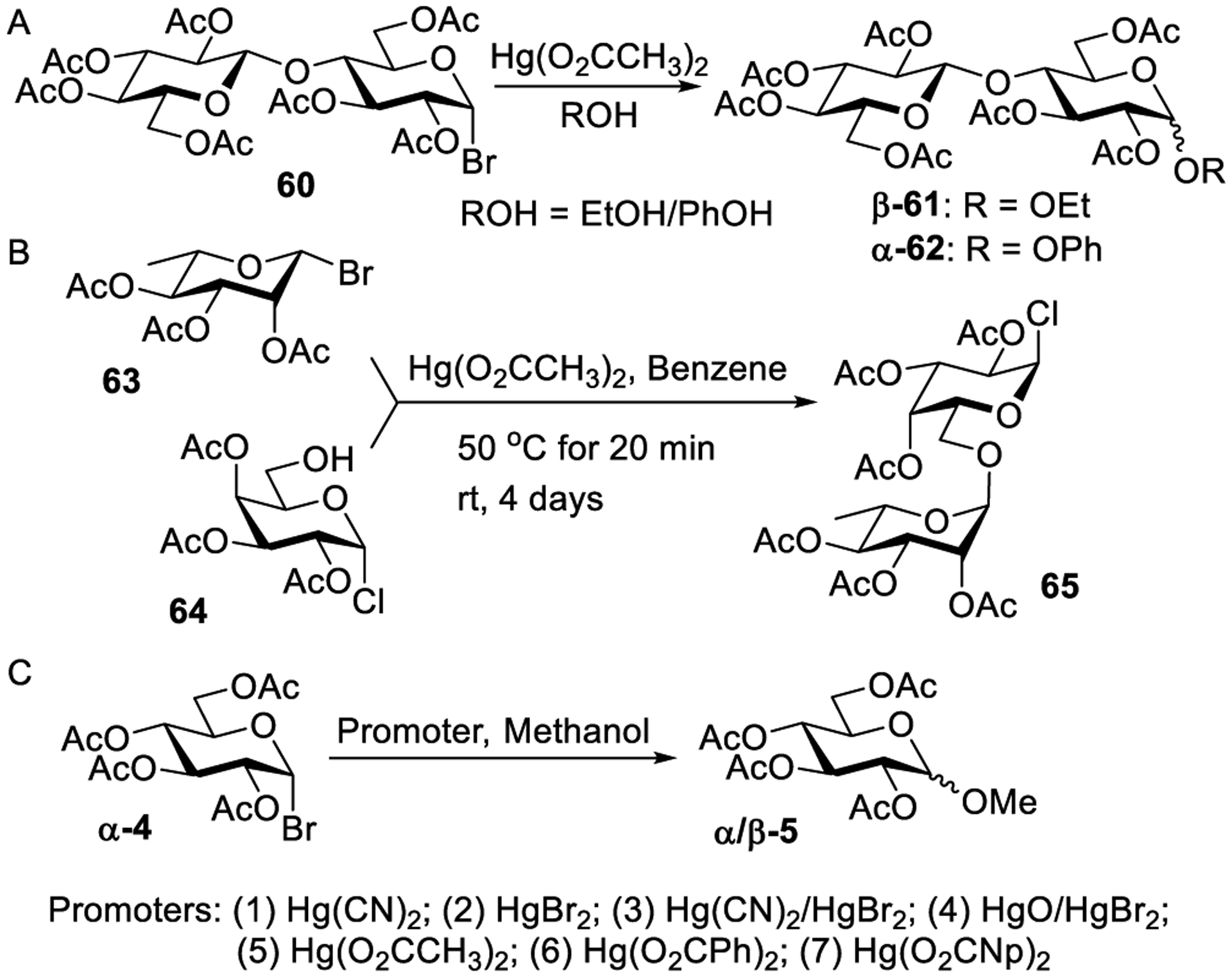
As aforementioned, Zemplen and co-workers showed an efficient activation of cellobiosyl bromide 60 in the presence of aluminum metal and mercuric acetate Hg(OAc)2. However, it was later realized that aluminum did not play an active role in splitting the C–Br bond.160,161 The anomeric distribution of glucosides was difficult to predict, and a subsequent dedicated study revealed that the ratio of α/β-linked glycosides mainly depends on the amount and types of alcohol used.160,162,163 For example, as shown in Scheme 15A, the treatment of glycosyl bromide 60 with ethanol or phenol in the presence of Hg(OAc)2 produces the respective β-glycoside 61 and α-glycoside 62. The α-anomer formation was favored when the reaction was performed with an excess of alcohol. Zemplen and co-workers applied this methodology to the synthesis of several disaccharides wherein selective activation of the leaving group was employed. For example, selective glycosidation of acetobromorhamnose 63 with partially protected galactosyl chloride acceptor 64 was performed in the presence of Hg(OAc)2 to produce disaccharide chloride 65 (Scheme 15B).289 Several other disaccharide chlorides were synthesized employing this selective activation of glycosyl bromides over chlorides in the presence of Hg(OAc)2.290 The selective activation approach helps to streamline oligosaccharide assembly because the products are already equipped with a leaving group, chloride in this case, and hence can be used as glycosyl donors in subsequent glycosylations directly.
Scheme 15.
Mercury Salt-Promoted Glycosidations of Glycosyl Bromides
Helferich and co-workers showed that a very fast activation of glycosyl bromide α-4 can take place in the presence of mercuric cyanide. This powerful improvement of the basic methodology earned a name of the Helferich modification of the classical Koenigs–Knorr method.239 By varying the amount of alcohol and mercuric cyanide, a rapid formation (30–100 min) of methyl β-glycoside 5 from glycosyl bromide α-4 and methanol has been achieved in good yields (up to 83%, Scheme 15C). It was also found that the addition of mercuric bromide to a mercuric cyanide-promoted glycosylation, further increases the rate of the reaction, whereas mercuric bromide used by itself did not produce any glycoside product. However, later it was shown that mercuric bromide can activate glycosyl bromide donors in the presence of MS 4 Å in refluxing methylene chloride. These reactions produced α-glycosides.241,242 Several other mercuric salts were tested (Scheme 15C), and some aryl mercuric salts such as phenyl mercury acetate, and naphthyl mercury acetate turned out to be effective promoters due to their better solubility in organic solvents.243 Green and co-workers have shown that mercuric oxide in combination with a small amount of mercuric bromide is also capable of an effective activation of glycosyl bromides.240 Meldal and Bock showed that mercuric iodide alone is capable of activating glycosyl bromides.244 Mercuric salt-catalyzed glycosylation method was employed for the synthesis of different types of biologically relevant glycosides and oligosaccharides.181,242,291,292
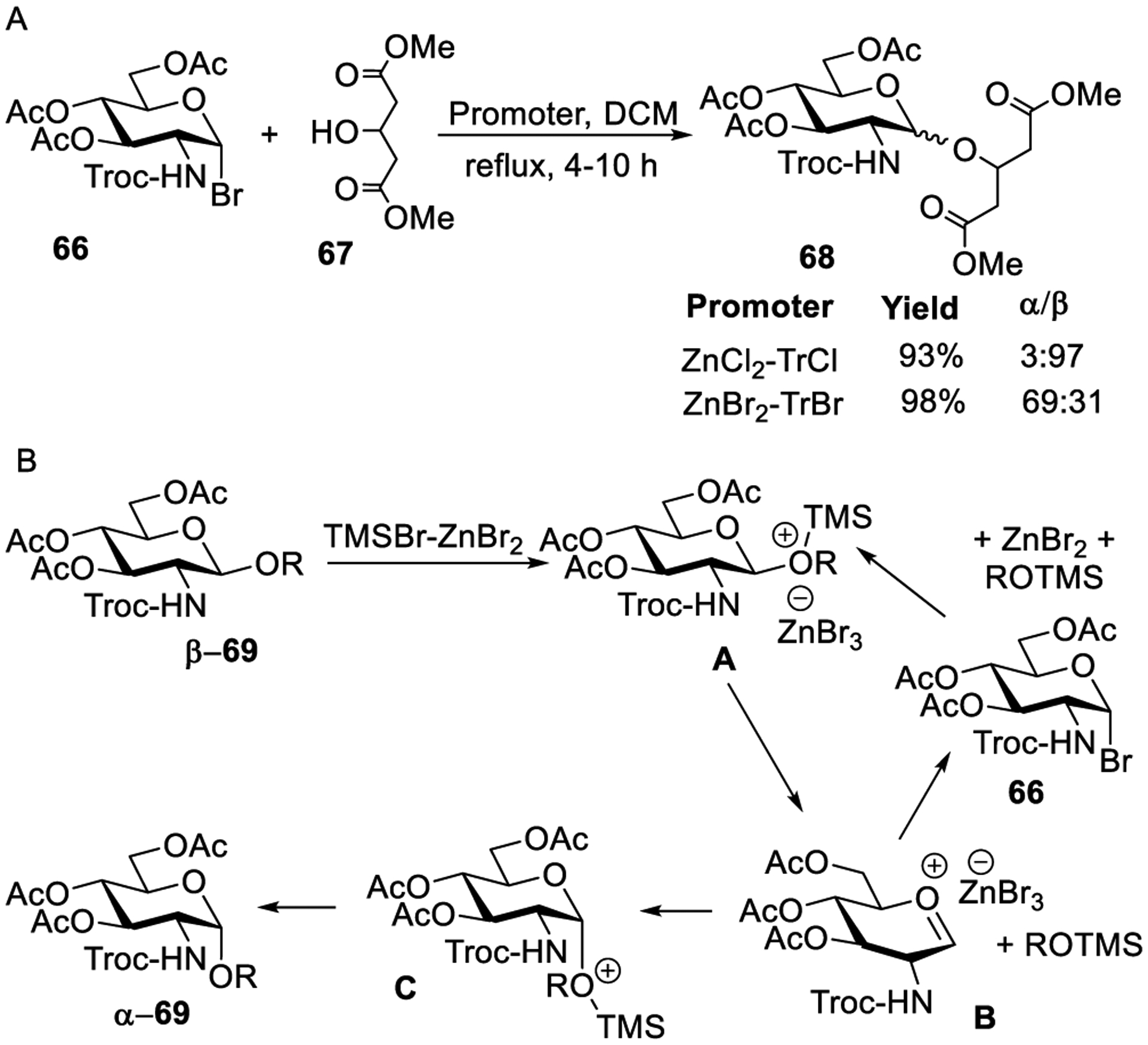
In other attempts to replace silver salts in glycosylations, Helferich and co-workers investigated zinc salts as activators of glycosyl bromides.239 For example, when glycosidation of bromide α-4 with methanol was conducted in the presence of zinc oxide methyl β-glycoside, 5 was obtained in 37% yield. Different primary alcohols were tested, and the corresponding glycosides were obtained in good yields (41–76%). Increasing the amount of zinc oxide was detrimental for the glycosylation reaction and led to decreased yields of glycosides. However, changing the reaction solvent from nonpolar to polar showed a positive effect. A slower activation was observed when zinc acetate was used instead under otherwise identical reaction conditions. Kusama and co-workers showed that glycosyl bromide 66 can be activated with equimolar amounts of zinc chloride and trityl chloride. Glycosylation of acceptor 67 produced glucoside 68 in a high yield with excellent β-stereoselectivity (Scheme 16A).245
Scheme 16.
Glycosylation and Anomerization in Zinc Salt-Promoted Glycosylations
A similar activation was also observed with ZnBr2 and trityl bromide (TrBr), but a notable loss of stereoselectivity was observed (Scheme 16A). In this case, α-glucoside 68 was obtained as the major product.247 Several other alcohols were glycosylated, and a similar stereoselectivity trend was observed. It is well-known that Lewis acids such as TiCl4, AlCl3, ZnCl2, SnCl2, and BF3-Et2O promote anomerization of glycosides.293–299 Analysis of the reaction mixture revealed that in this case the formation of β-glucoside takes place first, which is then followed by anomerization as the reaction progresses. A dedicated study confirmed that HBr generated during glycosylation induces the anomerization of the kinetic β-glucoside product to the thermodynamic α-glucoside. A plausible mechanistic pathway for anomerization in the presence of ZnBr2 and TMSBr was proposed (Scheme 16B).247,248 Reaction of β-glycoside β-69 in the presence of TMSBr-ZnBr2 produced salt A, which collapsed to oxacarbenium ion B and trimethylsilyl ether. Nucleophilic displacement by bromide ion (ZnBr3−) can produce glycosyl bromide 66. Conversely, a more thermodynamically stable product C can be formed by the nucleophilic attack of trimethylsilyl ether to form α-69. It was later discovered that adding 1.0–2.0 equiv of a glycosyl acceptor can drastically decrease the formation of glycosyl bromide and afford predominantly α-linked products. This methodology was applied to the synthesis of 1→6-linked glycolipid derivatives.300,301 Surprisingly, only β-disaccharides were obtained irrespective of the promoter system (TrCl-Zn salts or zinc salts alone). When β-linked disaccharides were exposed to the anomerization conditions (TMSBr-ZnBr2), only the starting material was recovered. The stability of β-linked disaccharide lipid derivatives was attributed to the inductive effect of the oxygen functional group at the reducing end. Shibakami and co-workers presented a similar activation protocol for per-benzoylated glucosyl bromide wherein NBS and catalytic ZnI2 or ZnBr2 were employed.246 In the case of ZnI2, the reaction was found to proceed via intermediacy of the respective glycosyl iodide formed in situ.
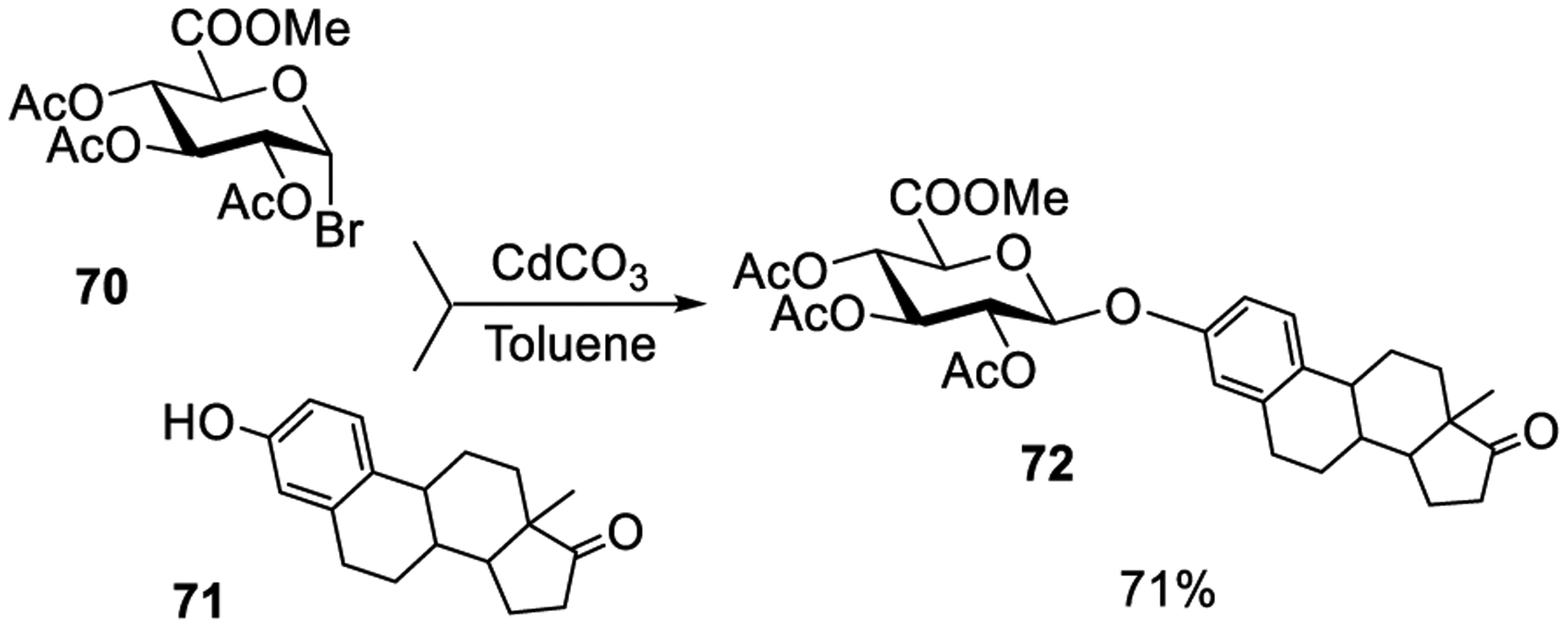
Helferich and co-workers reported a cadmium salt-promoted activation of glycosyl bromide donors.239 The authors demonstrated that reaction of per-acetylated glucosyl bromides with MeOH in the presence of cadmium oxide afforded the corresponding methyl β-glucoside in a moderate yield. Bernstein and Conrow investigated the Cd salt-promoted glycosylation in a greater detail by employing glycosyl halides of different sugar series.249 For example, glycosidation of glycosyl uronide donor 70 with estrone acceptor 71 in the presence of CdCO3 produced glucuronide 72 in 71% yield (Scheme 17). Cadmium sulfide was also investigated for the activation of donor 70; however, the yield of product 72 was only 20%. Several other steroidal phenolic glucuronides, glycosides, and acetyl glucosaminides were successfully synthesized using similar glycosylation conditions.250,302,303 In addition to β-glycosides, small amounts of the corresponding α-glucosides and/or C-glycosides were also isolated, and the ratio of products was found to depend on the coupling partners and the reaction conditions employed. It was suggested that the generated cadmium bromide was the actual activator, but cadmium bromide by itself was ineffective, similar to Helfrich’s observations made with mercuric cyanide activation.239 Later, it was also confirmed that the product composition depends on the surface area of cadmium carbonate.251
Scheme 17.
Cadmium Salt-Promoted Glycosylation
3.2.3. Activation with Group 13–15 Post-transition Metal Salts (Indium, Tin, Lead, or Bismuth).
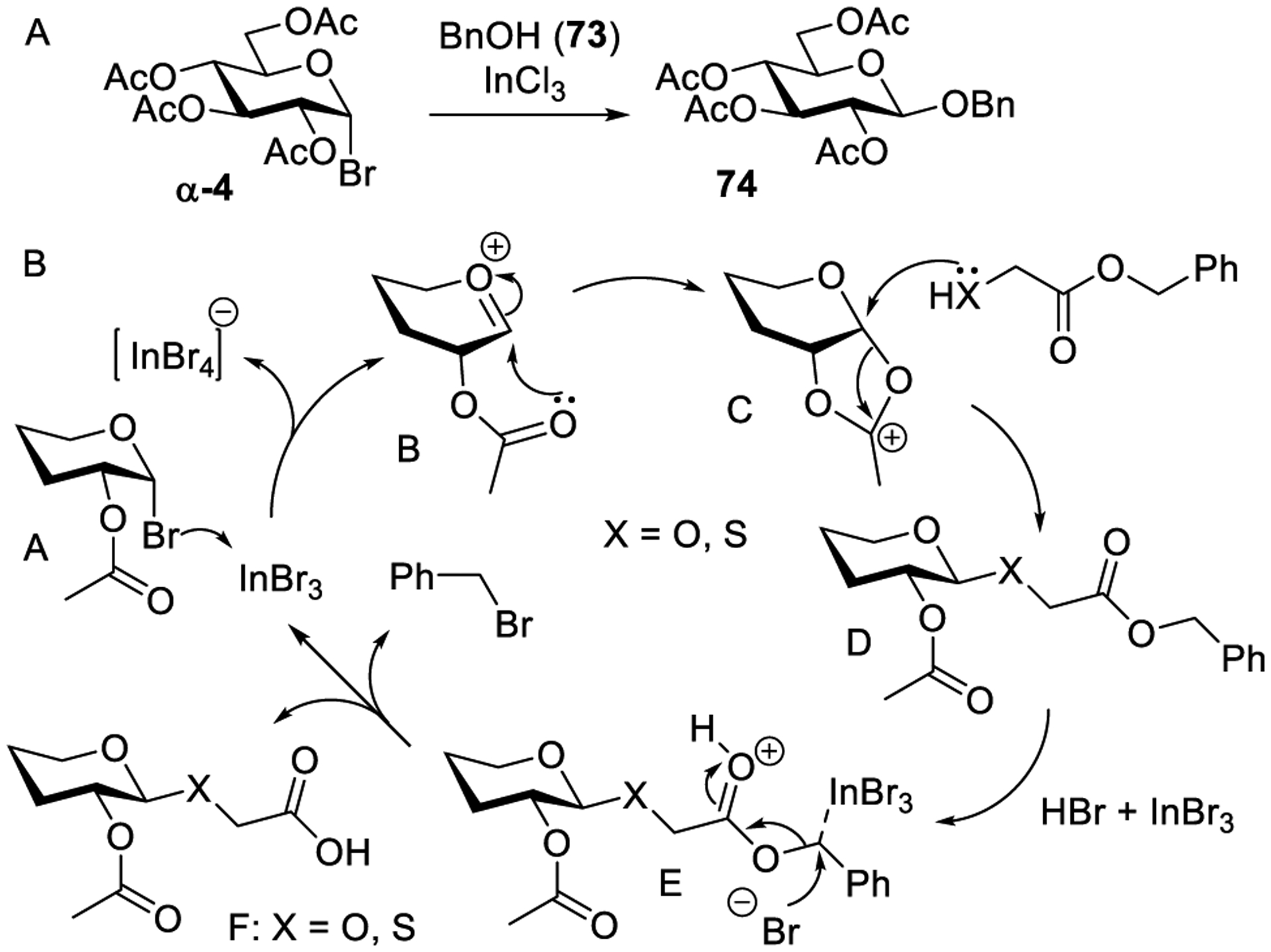
Chowdhury and co-workers developed indium chloride-promoted activation of glycosyl bromides (Scheme 18A).252 A catalytic amount of indium chloride without any additive was sufficient to promote the glycosylation. For example, when tetra-acetylated glucosyl bromide α-4 was treated with benzyl alcohol (73, 1.0 equiv) in the presence of InCl3 (0.4 equiv), glucoside 74 was obtained in 68% yield. Several other glycosides and disaccharides were successfully synthesized in good yields (62–90%). Indium chloride-catalyzed C-glycosylations of pyrrole and indoles were also described.304 In addition, various O- and S-glycosyl esters were synthesized by activation of glycosyl bromides with catalytic InCl3.253
Scheme 18.
Indium Salt-Promoted Glycosidations of Glycosyl Bromides
It was also found that glycosylation and removal of benzyl esters can be achieved in one pot in the presence of indium bromide (InBr3). A tentative mechanism involving coordination of the anomeric bromide A with indium tribromide to afford the reactive intermediate, oxacarbenium ion B, which gets stabilized as the acyloxonium ion C has been proposed (Scheme 18B). Following the nucleophilic attack and proton exchange, InBr3 and HBr were generated. The latter is capable of protonating benzyl ester D to produce intermediate E, which undergoes the nucleophilic attack by the bromide ion to produce O- or S-glycosyl acids F and benzyl bromide as a side product. To broaden the scope of indium salt-catalyzed glycosylations, Xue and co-workers investigated several other indium salts. The authors found that as low as 0.05–0.15 equiv of In(NTf2)3 without any external additive can catalyze glycosidations of glycosyl bromides with simple alcohols.254 To achieve efficient glycosylation of sugar alcohols, 2.0 equiv of glycosyl acceptors was needed.
Matsui and Ogawa showed that glycosidation of glycosyl bromides with trialkylstannyl alkoxide acceptors can be achieved in the presence of and tin(IV) chloride.255 Different alkoxide acceptors produced either α- or β-glycosides albeit with fair stereoselectivity. For example, when tetra-acetylated glucosyl bromide α-4 was reacted with tributylstannylated cyclohexyloxide 75 in the presence of SnCl4. The corresponding α-glucoside 76 was obtained in 47% yield, as shown in Scheme 19A. When similar reaction conditions were applied to glycosylation of the tributylstannyl benzyloxide acceptor, the corresponding β-glucoside was obtained in 52% yield. By employing the halide ion-catalyzed anomerization procedure in the presence of Et4NBr,305 several trilalkylstannyl alkoxide acceptors were converted to 1,2-orthoesters 77 (Scheme 19B).183
Scheme 19.

Tin Salt-Mediated Glycosidation of Glycosyl Bromides
Malleron and Lubineau developed β-stereoselective glycosidations of glycosyl bromides by employing tin triflate as an activator.256 One equivalent of a base as an acid scavenger was necessary to produce glycosides in good yields. The authors observed a competitive transesterification side reaction when primary alcohols were used as glycosyl acceptors. This led to compromised yields, for example, methyl glycoside was obtained in 33% yield. Secondary alcohols, however, did not seem to encounter this side reaction and produced glycosides in better yields. Several β-glycosides, β-disaccharides, β-aminosugars,306 and N-linked-β-disaccharides307 were successfully synthesized. In another study, the authors successfully applied tin triflate-promoted activation of glycosyl chloride donors (vide infra).306 Dick has reported activation of glycosyl bromides in the presence of lead(II) carbonate, wherein a moderate yield (37%) for the synthesis of tetra-acetylated α,β-phenyl glucosides was obtained when per-acetylated glucosyl bromide was treated with PhOH in the presence of PbCO3.251
Recently, Demchenko and co-workers have shown an efficient activation of glycosyl halides in the presence of Bi(OTf)3.308 Compared to most common metal salt activators of glycosyl halides wherein stoichiometric amount of activators is needed, a rapid activation of glycosyl bromides was achieved in the presence of 0.35 equiv of Bi(OTf)3. Due to the detrimental effect of strongly acidic TfOH produced as a byproduct, the authors have increased the amount of molecular sieves and identified nitromethane-DCM as the preferred reaction solvent. As a result, glycosidation of benzoylated and benzylated galactosyl bromides 40 and 41 with glycosyl acceptor 36 in the presence of Bi(OTf)2 (0.35 equiv) afforded respective galactosides 78 and 79 in good yields (Scheme 20). An effective glycosylation of a variety of glycosyl acceptors was achieved for sugars of the gluco, manno, rhamno, and glucosamino series equipped with different protecting groups.308
Scheme 20.

Bismuth(III) Salt-Promoted Activation of Glycosyl Halides
3.2.4. Activation with Non-nucleophilic Organic Bases.
Koenigs and Knorr were the first to report non-metal-based activation of glycosyl bromides, wherein glycosyl bromide was treated with methanol in the presence of pyridine that afforded methyl β-glycoside.7 The exact mechanistic action of pyridine was not discussed or known, rather it was assumed that pyridine acts as hydrogen bromide scavenger. A few years later, Fischer and co-workers showed the formation of anomeric mixtures of pyridinium salts α/β-80 when tetra-O-acetylated glucosyl bromide α/β-4 was treated with pyridine in the absence of an alcohol (Scheme 21A).148 This undoubtedly excluded the possibility of pyridine purpose solely as the hydrogen halide scavenger.
Scheme 21.

Reaction of Glycosyl Bromide 4 in the Presence of Pyridine
Fischer also investigated glycosylations in the presence of quinoline.150 Lemieux and Morgan also observed the formation of anomeric pyridinium salts.182 To gain mechanistic insights for this reaction, the authors performed an extensive concentration-dependence study depicted in Scheme 21B. It was found that the anomeric distribution of α- and β-pyridinium salts 80 depends on the concentration of glycosyl bromide α-4 in pyridine. At a low concentration, the formation of β-80 predominates, but at higher concentrations both α- and β-80 are produced and the ratio of anomers was found to depend on the concentration of glycosyl bromide α-4. When the equimolar amount of tetra-n-butyl ammonium bromide was added to the reaction, only α-pyridinium salt 80 was obtained, whereas reactions in the presence of tetra-n-butylammonium perchlorate produced the anomeric mixture.
These results were indicative of the intermediacy of β-bromide β-4 and acyloxonium intermediate A. In the presence of methanol this reaction led to a significant amount of orthoester 19. NMR spectroscopy studies suggested that α-pyridinium salt α-80 adopts the1C4 conformation, as shown in Scheme 21B.309 Further experiments were conducted to support the routes for the formation of α-80 and its β-linked counterpart,183,310 which ultimately led to appreciation that charged intermediates can serve as glycosyl donors. A variety of other approaches to obtain charged intermediates followed by their glycosidation have been explored. Micheel and Micheel examined the formation of positively charged anomeric salts from triethylamine. However, the authors never used these salts as a glycosyl donors.264,311 Hess and Heumann treated a glycosyl chloride with trimethylamine in the presence of ethyl alcohol and observed the formation of ethyl α-glycosides in addition to the anticipated anomeric salts.312
Expanding upon these findings, Schuerch and co-workers performed a dedicated study of the formation and glycosidation of positively charged intermediates that led to the development of a highly stereocontrolled α-glycosylation.265,266 It was shown that positively charged intermediates 81–83 depicted in Scheme 22 can be synthesized by treating glycosyl bromide 38 with triethylamine, dimethyl sulfide, or triphenylphosphine, respectively. These intermediates can be isolated and purified. The positively charged anomeric substituents would have a strong propensity to adopt the equatorial position due to the reverse anomeric effect.309 As a result, these positively charged equatorially placed leaving groups would undergo the nucleophilic attack from the α-face to give axial glycoside products.
Scheme 22.

Synthesis of α-Glycoside via Positively Charged Glycosyl Donors
Thus, glycosidation of ammonium 81 and phosphonium 83 salts produced α-glycosides 84 exclusively, whereas glycosidation of sulfonium 82 salt showed somewhat relaxed stereoselectivity (Scheme 22).265 An attempt to synthesize an α-1→3-linked disaccharide has failed, and this outcome was attributed to low reactivity of the sugar alcohol. Upon subsequent optimization of the reaction conditions wherein reaction solvents and temperatures were refined, a successful synthesis of an α-1→6-linked disaccharide has been achieved.313
Very recently, Nguyen and co-workers presented an improved approach to achieve stereoselective activation of glycosyl bromides bearing a nonparticipating group at C-2.257 It was discovered that the activation of glycosyl bromide 54 with phenanthroline catalyst 85 in the presence of isobutylene oxide (as hydrogen bromide scavenger) preferentially afforded 1,2-cis glycosides. For example, glycosylation of glycosyl acceptor 55 produced disaccharide 56 in 73% with excellent α-stereoselectivity, as shown in Scheme 23A. The mechanistic pathway involves double SN2-like displacement wherein the first step involves the formation of β-phenanthrolium intermediate A depicted in Scheme 23B. The stability of intermediate A was attributed to noncovalent interaction of anomeric α-hydrogen with nitrogen of phenanthroline. Subsequently, intermediate A reacts with a glycosyl acceptor to form an α-linked product. This stereoselective glycosylation methodology was utilized to synthesize many glycosidic linkages as well as a series of α-glycans (vide infra).
Scheme 23.
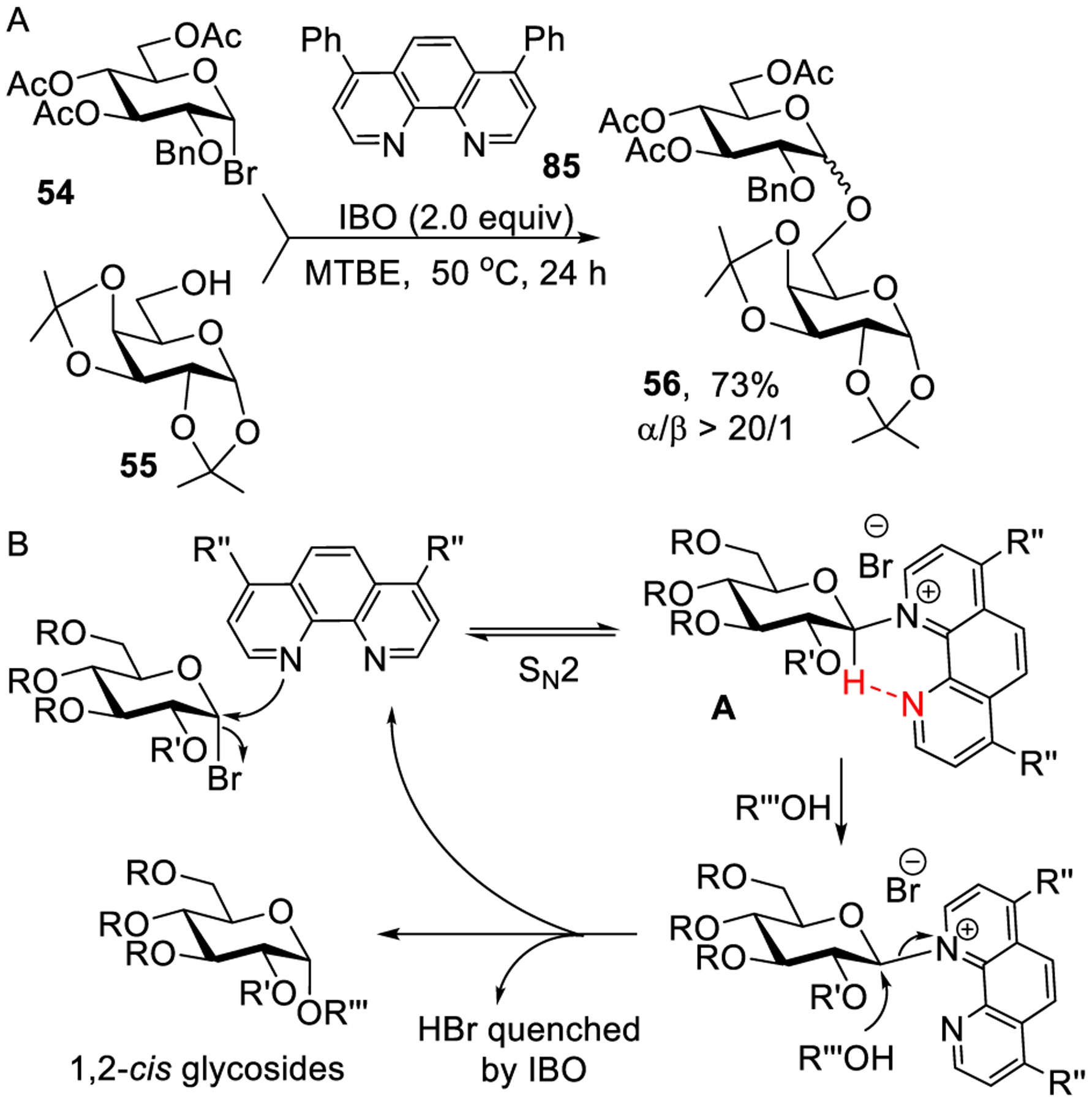
1,2-cis Glycosylation via β-Phenanthrolium Intermediates
3.2.5. Activation with Halogens, Halide Ions, or Halonium Ions.
Lemieux and co-workers performed an extensive study of a halide ion-catalyzed glycosylation with glycosyl halides. This approach has led to effective 1,2-cis stereoselective syntheses of glycosides and oligosaccharides.258 The early attempts to address the issues of stereoselectivity were primarily directed on the development of new catalytic systems, as well as optimization of the reaction conditions (solvent, temperature, pressure).314 Many valuable developments for the synthesis of 1,2-cis glycosides have been published by Schuerch and co-workers,72,230,265,315–318 However, a major breakthrough in the understanding of the principles of the α-glycosidic bond formation emerged with the discovery and thorough elaboration of the in situ anomerization concept, so-called “halide ion catalyzed glycosidation reactions” by Lemieux and co-workers (Scheme 24).258 Thus, it was observed that a rapid equilibrium could be established between a relatively stable α-halide A and its far more reactive β-counterpart F by the addition of tetraalkyl ammonium bromide (Et4NBr). Therefore, a glycosyl acceptor (ROH) would attack the more reactive intermediate in an overall SN2 fashion (via D), providing α-glycoside L. More detailed analysis of the glycosylation process showed that the energy barrier for a nucleophilic substitution F to L (formation of α-glycosides from the highly reactive β-bromide F) is somewhat lower than that for the reaction from A to I (formation of β-glycosides from the less reactive α-bromide A).
Scheme 24.

Formation of 1,2-cis Glycosides via In Situ Anomerization
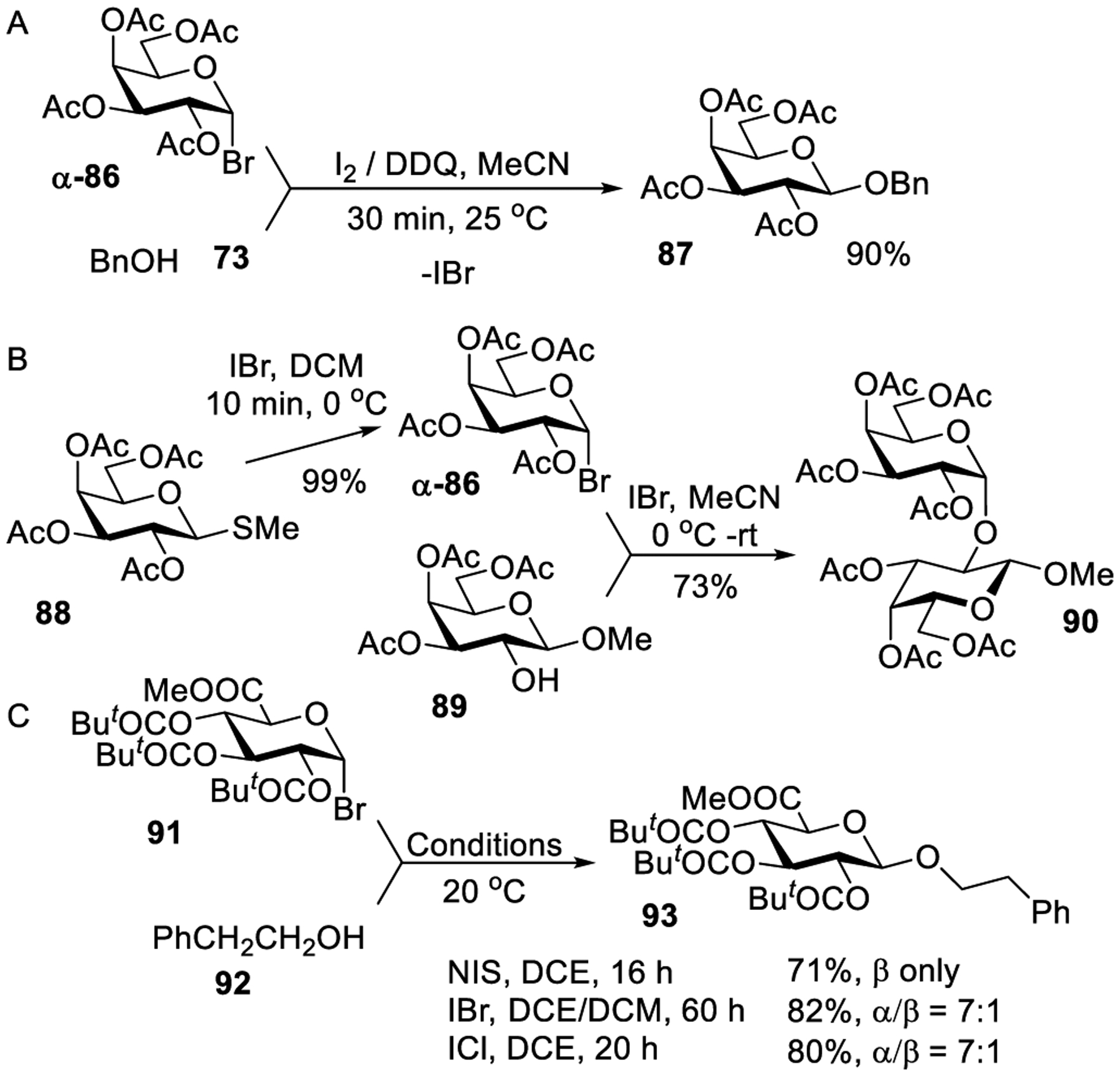
Indeed, α-glycoside L is formed faster than its β-linked counterpart I, which in combination with higher thermodynamic stability of the α-anomer makes this glycosylation process very favorable overall. If the difference in the energy barrier had been sufficient, it would be possible to direct the reaction toward the formation of α-anomers with complete stereoselectivity. Therefore, in order to achieve high stereoselectivity, the entire glycosylation process has to be performed in a highly controlled manner. In this particular case the control is achieved by the use of extremely mild catalyst (Et4NBr), although very reactive substrates and prolonged reaction times are thus required. A series of per-benzylated glycosyl halides of the gluco, galacto, and fuco series were tested. These reactions produced good yields of glycosides with excellent α-stereoselectivity. This method found practical application in synthesizing blood group determinants and other biologically relevant oligosaccharides having α-glycosidic linkages (vide infra).259,319 In another effort to find a substitute to heavy metal promoters, Field and co-workers studied the activation of per-acetylated glycosyl bromides by utilizing I2 alone or with DDQ as an additive. As shown in Scheme 25A, when galactosyl bromide α-86 was reacted with benzyl alcohol 73 in the presence of I2 and DDQ at rt, an excellent yield of galactoside 87 was achieved within 30 min.260 It was proposed that iodine acts as a halophilic reagent which, following the interaction with the anomeric bromide, fragments to give iodine monobromide and oxacarbenium or acyloxonium ion. Subsequently, the authors realized that iodine monobromide (IBr) could also be used as an efficient activator for glycosyl bromides. Indeed, IBr alone or with DABCO additive could activate glycosyl bromides to produce different disaccharides in 35–73% yields. The lower yields in this series were generally attributed to side reactions rather than promoter problems. All glycosyl bromide donors were synthesized by treating the corresponding thioglycosides with IBr.
Scheme 25.
Iodine or Iodine Monobromide/Chloride-Promoted Glycosylations
One such example is the synthesis of galactosyl bromide α-86 from the corresponding methylthio galactoside 88 in the presence of IBr depicted in Scheme 25B. The bromide donor α-86 was later glycosidated with secondary galactosyl acceptor 89 in the presence of IBr that produced disaccharide 90 in 73% yield.262 Glycosyl chloride donors were also activated under identical reaction conditions. Besides glycosyl halide activation, thioglycoside activation was also shown to occur in the presence of IBr. Iodine-promoted glycosidation of glycosyl halide was further extended to amino acid acceptors.320 Anhydrous potassium carbonate was used as a scavenger for a hydrogen iodide byproduct.
Stachulski employed N-iodosuccinimide as a source of the iodonium ion for the activation of glycosyl bromides.263 An efficient activation of pivaloyl protected bromide 91 was achieved in the presence of NIS, IBr or ICl at 20 °C. For example, glycosylation of acceptor 92 produced glucuronide 93 in good yields of 71–82% as depicted in Scheme 25C. Excellent to good yields of other glucuronides were achieved with several primary and secondary alcohols. However, the stereoselectivity was found to differ drastically depending on the type of promoter used. Thus, NIS produced the product in high β-stereoselectivity, whereas IBr- or ICl-promoted reactions were α-stereoselective.
Demchenko developed reaction conditions wherein bromine was used both as the reagent to convert thioglycosides into bromides and to activate the latter for glycosylation.321 To achieve highly stereocontrolled 1,2-cis glycosylation, it was deemed necessary to form reactive β-glycosyl bromides. It was assumed that α-thioglycoside upon bromination can produce the reactive β-bromide as shown in Scheme 26A. This reaction was monitored by NMR, showing that β-bromide is indeed the reactive intermediate, which can undergo a rapid anomerization into the α-linked counterpart. Once formed, the α-bromide was found to be totally unreactive under the established reaction conditions. The glycosylations of primary acceptors were smooth and stereoselective. For example, glycosidation of thioglycoside 94 with 6-OH glycosyl acceptor 36 in the presence of Br2 produced disaccharide 95 in 67% yield with complete 1,2-cis stereoselectivity (Scheme 26B).
Scheme 26.
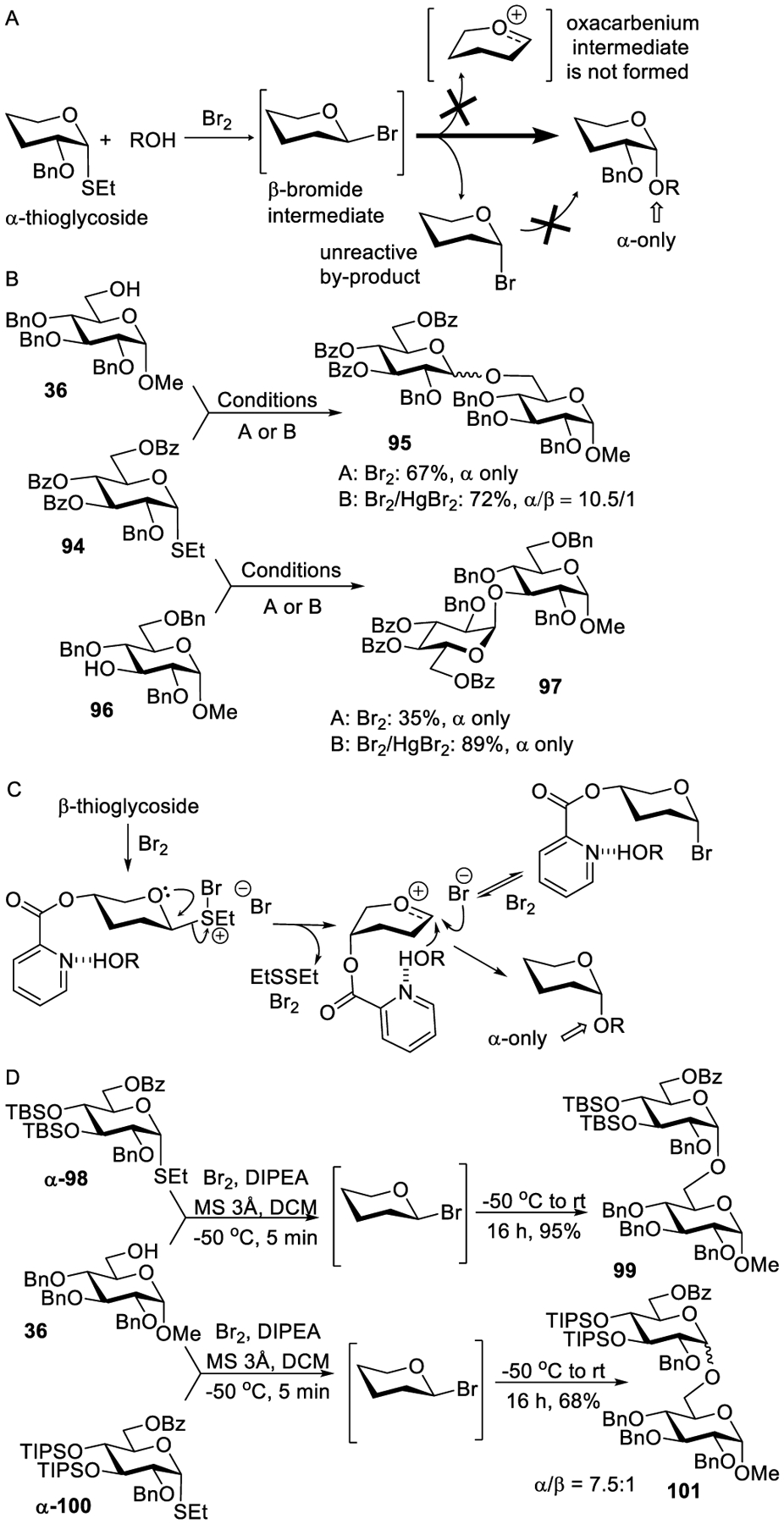
Bromine-Promoted α-Selective Glycosylations
However, slower glycosylations of secondary acceptors were less efficient due to the competing β→α halide anomerization, although all reactions were still α-stereoselective. For example, glycosidation of thioglycoside 94 with 3-OH glycosyl acceptor 96 in the presence of Br2 produced disaccharide 97 in 35% yield with complete 1,2-cis stereoselectivity. It was also shown that the α-bromide can be reactivated in the presence of mercury(II) additive. This pathway was found to be very beneficial for the glycosylation of secondary alcohols. For example, glycosidation of thioglycoside 94 with 3-OH glycosyl acceptor 96 in the presence of Br2 and HgBr2 produced disaccharide 97 in a significantly improved yield of 89% yield with complete 1,2-cis stereoselectivity. However, when applied to glycosylation of highly reactive primary acceptors, these reaction conditions can compromise α-selectivity. Thus, glycosidation of thioglycoside 94 with 6-OH glycosyl acceptor 36 in the presence of Br2 and HgBr2 produced disaccharide 95 in a slightly improved yield of 72% yield albeit reduced stereoselectivity (α/β = 10.5/1, Scheme 26B).
Bromine has also been used as a promoter in glycosylations by means of an H-bond-mediated Aglycone Delivery (HAD).322 Glucosyl donors bearing the 4-O-picoloyl group exhibited excellent stereoselectivity with a number of different glycosyl and aliphatic acceptors.323 Furthermore, bromine was also shown to activate a wide variety of leaving groups including thioglycosides, imidates, and thioimidates. A low temperature NMR monitoring of the reaction showed direct and rapid formation of the α-bromide. Differently from the benzoylated bromides (vide supra), a highly benzylated α-bromide was reactive under these reaction conditions. Upon departure of the bromide leaving group, the 4-O-picoloyl mediated HAD ensures that the nucleophile is delivered exclusively from the bottom face of the resulting oxacarbenium intermediate (Scheme 26C). Mercuric bromide could also be added to this reaction to improve the yield and shorten the reaction time.
Further efforts were made to apply the bromine-promoted glycosylation reaction to conformationally superarmed glycosyl donors.124,125 Dedicated NMR experiments and computational studies revealed that introduction TBS and TIPS groups at C-3 and C-4 positions of β- or α-thioglycosides 98 and 100 distort the ring conformation.143 Reactive β-glycosyl bromide formation was seen only with α-98, whereas β-98 afforded an anomeric mixture of bromides. Glycosidation of α-98 with primary glycosyl acceptor 36 in the presence of Br2 and DIPEA as an additive provided 1,2-cis disaccharide 99 in an excellent yield of 95% (α only) as shown in Scheme 26D. The reactivity trend of the corresponding TIPS protected glycosyl donor 100 was similar; however, the yield and stereoselectivity both decreased. For example, when glycosyl donor α-100 was glycosidated with acceptor 36 disaccharide 101 was produced in 68% yield with reduced stereoselectivity (α/β = 7.5:1). This reduced yield was attributed to competing silyl group cleavage occurring under these reaction conditions.
Madsen and co-workers used protic acid in combination with N-iodosuccinimide to achieve a facile release of the iodonium ion.261 An efficient glycosidation of per-benzoylated glucosyl bromide α-11 with reactive glycosyl acceptor 36 was achieved in the presence of NIS/TfOH to afford disaccharide 102 in yields up to 88% (Scheme 27). However, changing the sugar series and protecting groups of glycosyl donors led to reduced yields (60–80%) even in reactions with reactive glycosyl acceptors.
Scheme 27.
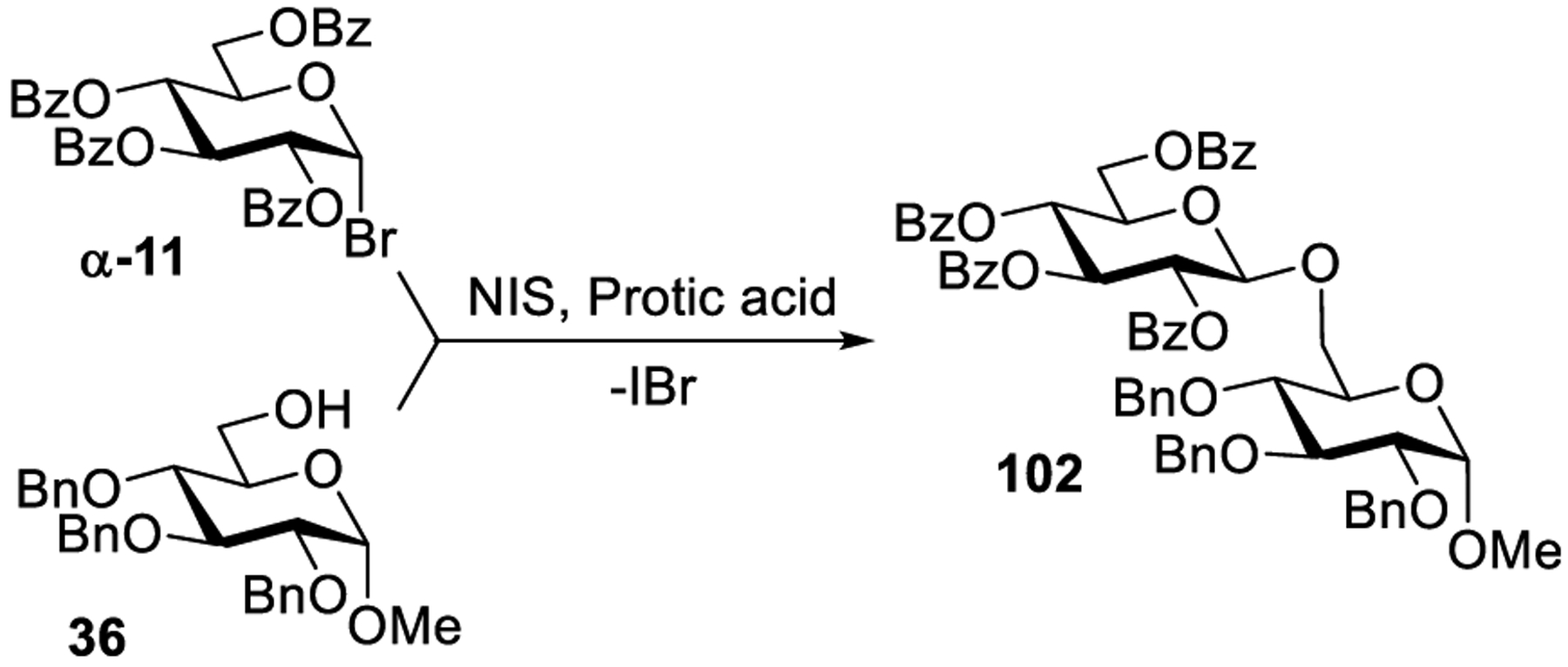
NIS-TfOH-Promoted Glycosidation of Glycosyl Bromides
3.2.6. Solvolysis.
Methanolysis of glycosyl bromides was studied both in the presence and the absence of various additives such as tetrabutylammonium bromide267 or silver salts.230 To understand the mechanistic details of solvolysis reactions, several experiments were conducted and it was determined that the concentration of alcohol plays a significant role on reaction rates and stereoselectivity.230 At lower concentrations of methanol, less reactive α-glycosyl bromides are in anomeric equilibrium with the more reactive β-counterpart, which subsequently gives methyl α-glycoside as the major product.230,267 At high concentrations of methanol, α-glycosyl bromides directly react with MeOH to preferentially produce methyl β-glycoside. A study by Frechet and Schuerch revealed variations in the rate of methanolysis or anomerization caused by steric and/or electronic effects of substituents at C-6 of glycosyl donors.268 The study concluded that the anomeric distribution of methyl glycosides from methanolysis (solvolysis) of glycosyl bromides strongly depends on various factors such as structure of glycosyl halides, concentration of methanol, and reaction conditions. Several glycosyl bromides having different acyl groups at C-6 have been investigated revealing useful trends.268 By changing the electronics of the functional groups at C-6, methyl glycosides were formed from 90% of α-anomer to over 90% of its β-counterpart. It was proposed that it is the interaction of the oxacarbenium ion with the carbonyl group of the substituent at C-6 that plays a significant role in deciding the stereochemistry of the product. For the instance, electron rich p-methoxybenzoyl group increases the electron density on the carbonyl group, stabilizes the positive charge on C-1, and facilitates the top face attack of the glycosyl acceptor (pathway a, Scheme 28A). The hydrogen bonding with acceptor could also be possible due to electron-rich carbonyl oxygen at C-6, which would also favor the formation of the β-linked product. Conversely, the p-nitrobenzoyl group with the electron deficient carbonyl group is unable to stabilize the positive charge on C-1. As a result, the attack of the glycosyl acceptor will take place from the less shielded bottom face resulting in the corresponding α-glycoside. The anomeric distribution of methyl glycosides produced from glycosyl bromides equipped with differentially p-substituted 6-O-benzoates was shown to follow the σ Hammett substituent constant value. Frechet and Schuerch extended the alcoholysis (methanolysis) of glycosyl bromide to solid phase synthesis wherein glycosyl bromides were coupled with polystyrene resin-immobilized glycosyl acceptors.324,325
Scheme 28.

Examples of Alcoholysis Reactions
Recently, Csuk and co-workers synthesized several β-glycosides by treating acetylated glycopyranosyl bromides with excess of n-alkyl alcohol without any additives.326 Glycosides were produced on gram scale in good to moderate yields in 4–7 days at room temperature. Matheu and co-workers discovered a green approach to construct glycosidic linkages.327 For that, glycosylation was conducted employing glycosyl halide donors in super critical CO2 without any volatile organic solvent and metal catalysts. The authors demonstrated an efficient activation of tetra-acetylated galactosyl bromide α-86 with benzyl alcohol 73 in the presence of scCO2 at 1500 Psi and 60 °C to afford galactoside 87 in 63% yield (α/β = 1:3.8, Scheme 28B). To broaden the scope, differently protected galactosyl bromide donors in combination with various glycosyl acceptors were tested to afford the corresponding galactosides in moderate to good yields. Interestingly, pivaloyl-protected galactosyl bromide donor produced far better results in terms of yields and stereoselectivity in comparison to those achieved with its acetyl-, benzoyl-, or benzyl-protected counterparts. Reactions performed in the presence of 2,6-lutidine led to the formation of orthoester 104 in a good yield (81%, Scheme 28B). Glycosyl chlorides were investigated alongside providing comparable results; however, the activation of glycosyl chloride turned out to be slower and required high temperatures (90 °C).
3.3. Glycosyl Bromides in Glycan and Glycoconjugate Synthesis
3.3.1. Linear Synthesis.
After understanding the mechanistic details for the formation of side products, strategies were developed for the synthesis of higher oligosaccharides. As depicted in Figure 3A, linear oligosaccharide synthesis consists of the glycosylation of a monosaccharide donor with a monosaccharide acceptor in the presence of a promoter to give the desired disaccharide. Most commonly, the latter is then converted into the second-generation glycosyl acceptor via liberation a specific hydroxyl group. The acceptor is then allowed to react with a glycosyl donor, resulting in the formation of trisaccharide, and this deprotection–glycosylation sequence can be then reiterated to yield a glycan of the desired length.
Figure 3.
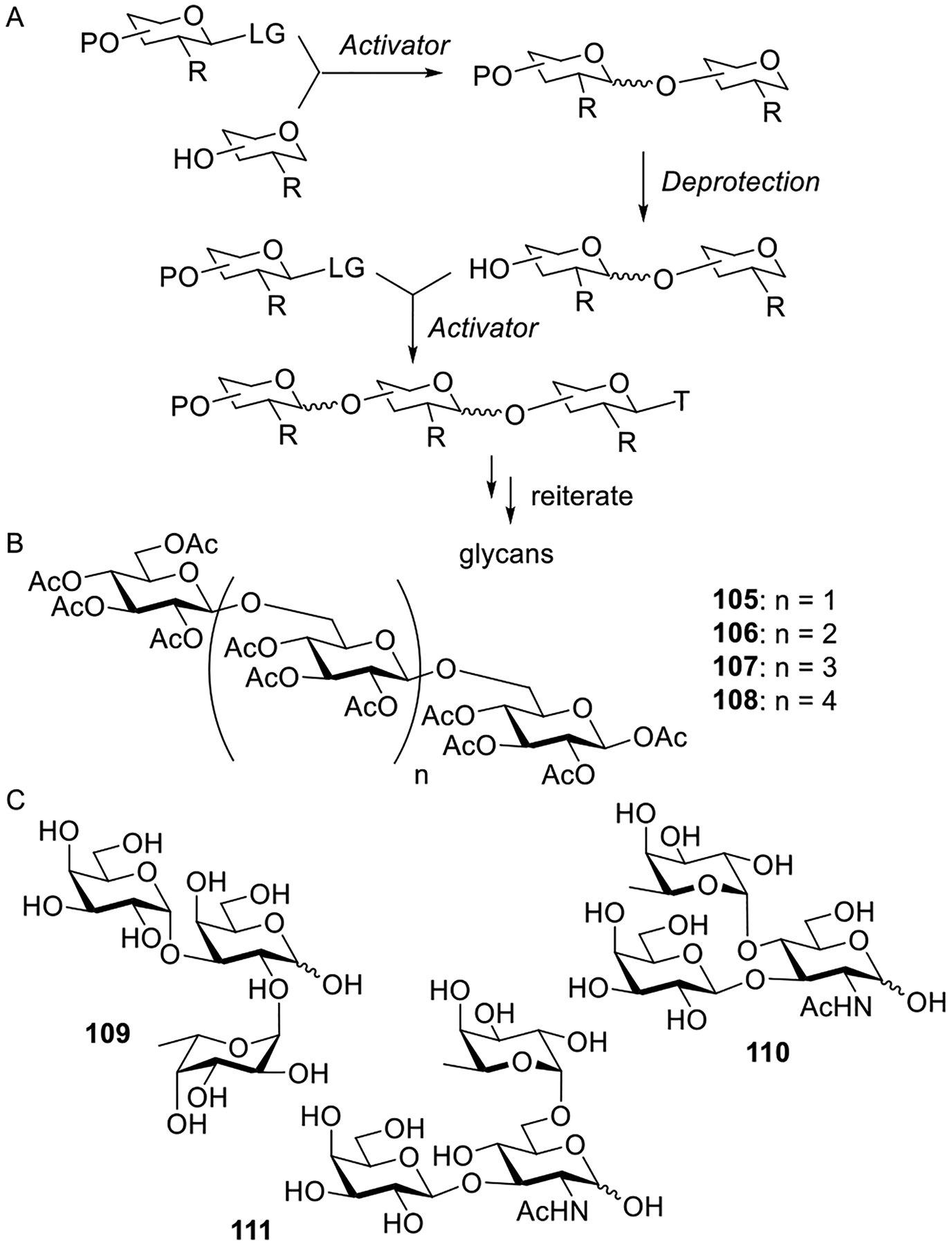
Linear oligosaccharides syntheses using Koenigs–Knorr (A) and halide-catalyzed (B) approaches
Alternatively, the intermediate oligosaccharides can be converted into a glycosyl donor via the introduction of a suitable leaving group. Many glycans have been synthesized using linear approaches; the only approach that was known in early days of oligosaccharide synthesis. Successful stepwise linear syntheses of gentiotriose 105 and gentiotetraose 106 were successfully demonstrated employing the Koenigs–Knorr glycosylation method in the presence of silver salt, iodine, and Drierite (Figure 3B).220,228 Following the same strategy, Takiura et al. synthesized gentio-oligosaccharides 105–108.328 The halide ion-catalyzed glycosylation method was applied to the synthesis of blood group determinants 109–111 containing challenging α-glycosidic linkages (Figure 3C).259,319
3.3.2. Convergent Building Block Assembly.
Pioneering studies by Zen,329 Paulsen,330 and Ogawa331 paved the way to what is currently known as a convergent building-block strategy for oligosaccharide synthesis. According to this strategy, two di- (or oligo) saccharides, a glycosyl donor and a glycosyl acceptor, are constructed individually and then coupled together (converged) to afford a larger oligosaccharide. One benefit of such block synthesis is the decreased number of steps required for the overall assembly. One relevant example is a 2+3+2 assembly of a biantennary heptasaccharide 117 as depicted in Scheme 29.332 According to the developed strategy, lactosamine bromide donor 112 was glycosidated with trimannosyl acceptor 113 in the presence of AgOTf and 2,6-lutidine to produce pentasaccharide 114 in 71% yield. The latter was subjected to the allyloxycarbonyl (Alloc) group removal with Pd(PPh3)4 to provide glycosyl acceptor 115 in 88% yield, which was then glycosylated with glycosyl donor 116 in the presence of AgOTf and 2,6-lutidine to produce the heptasaccharide 117 in 76% yield.
Scheme 29.
Convergent Synthesis of Heptasaccharide 117
A conceptually related convergent approach has been proven particularly advantageous if the synthesis of two or more repeating units is required. In this case, an intermediate di- (or oligo) saccharide is divided into two portions: one portion is converted into a glycosyl donor and another into a glycosyl acceptor. Subsequently, the two reactants are coupled to afford a dimer. This manipulation can be then repeated for the synthesis of larger structures. A relevant recent example of such an approach was described by Nguyen and co-workers. The authors employed their phenanthroline-catalyzed glycosylation (vide supra) to assemble 1,2-cis-linked oligosaccharides as shown in Scheme 30.
Scheme 30.

Phenanthroline-Catalyzed Synthesis of 1,2-cis Glycans
Glycosyl bromide 118 was glycosidated with glycosyl acceptor 29 in the presence of 5% of phenanthroline 85 to obtain disaccharide 119 in 89% yield (α/β > 20:1). The latter was converted to glycosyl bromide donor 120 via sequential acetolysis-bromination. Intermediate 119 was also converted to glycosyl acceptor 121 by removing acetyl in the presence of sodium methoxide in methanol. Further, glycosyl bromide donor 120 was glycosidated with glycosyl acceptor 121 in the presence of phenanthroline 85 to obtain tetrasaccharide 122 in 86% yield (α/β > 20:1). The latter was the converted into tetrasaccharide bromide donor 123 and tetrasaccharide acceptor 124, and the resulting counterparts were coupled in the presence of phenanthroline 85 to produce octasaccharide 125 in a good yield and excellent stereocontrol (77%, α/β > 20:1).
3.3.3. Selective Activation.
When the arsenal of the glycosylation techniques was limited to Fischer and Koenigs–Knorr approaches (or their variations), selective activation of one leaving group over another was rare. A few relevant examples wherein more reactive glycosyl bromides were activated over less reactive glycosyl halides (chlorides, fluorides) that could act as glycosyl acceptors were discussed in previous subsections. However, when stable glycosyl donors such as thio- or selenoglycosides have emerged, the ability to activate one leaving group over another dramatically enhanced. Pioneering research of Zen,329 Nicolaou,333,334 Lonn,335,336 Garegg,216,337 and others338,339 involved the activation of alkyl halides over S-alkyl/aryl glycosides. In addition to a few relevant examples discussed previously, an example of such a synthesis is presented in Scheme 31.340 First, glycosyl bromide donor 126 was activated over the phenylselenide acceptor 127 in the presence of AgOTf/γ-collidine to give the desired disaccharide 128 in 60% yield. The latter was then directly activated over the SEt acceptor 129 in the presence of AgOTf/K2CO3 to produce the desired trisaccharide 130 in 81% yield. Apparently, since the latter is equipped with the anomeric SEt leaving group, it can be directly activated for subsequent glycosylations under appropriate reaction conditions. A similar activation sequence was reported by the Ley group.341
Scheme 31.
Selective Activation Utilizing Three Leaving Groups
3.3.4. Two-Step Activation and Regenerative Glycosylation Concept.
Two-step activation of thioglycosides with bromine via glycosyl bromides has several potential applications for oligosaccharide synthesis. It can range from simple glycosylation to sophisticated iterative approaches. As shown in Scheme 32, the initial step involves the treatment of thioglycoside precursor 131 with bromine. The resulting glycosyl bromide is then directly reacted with glycosyl acceptor 132 in the presence of silver triflate to afford trisaccharide 133.231 Since the acceptor is not carrying a leaving group, it can be present in the reaction mixture from the beginning. The two-stage activation approach can also be applied to situations when both glycosyl donor and glycosyl acceptor initially bear the same type of a leaving group (thioglycoside).329 In this case, thioglycoside precursor of the donor is first converted to glycosyl bromide. The acceptor equipped with the SR leaving group is then added, and the glycosyl bromide is selectively activated in the presence of a suitable activator. Since the resulting product is a thioglycoside, this two-step activation sequence can be reiterated.
Scheme 32.

In Situ Activation of Thioglycosides via Bromides
Demchenko and co-workers have developed a regenerative strategy to activate glycosyl bromides (vide supra).217,225 In accordance with this approach, a thioglycosides precursor is first treated with bromine to afford the corresponding glycosyl bromide. The latter is then activated with HOFox/TMSOTf for the regenerative glycosylation cycle wherein both the synthesis of the reactive OFox imidate intermediate and its glycosidation is performed in a catalytic, regenerative fashion. Further, the regenerative glycosylation was applied to oligosaccharide synthesis. For this purpose, per-benzoylated galactosyl bromide 40, generated from the corresponding thiogalactoside 134, was glycosidated with thiogalactoside acceptor 135 in the presence of Ag2O, HOFox, and TMSOTf, as depicted in Scheme 33. As a result, disaccharide 136 was obtained in 84% yield. Subsequently, thioglycoside 136 was converted to bromide donor 137 and coupled with acceptor 135 under the regenerative reaction conditions to produce trisaccharide 138 in 87% yield. Finally, the latter was converted into bromide 139, which was then coupled with acceptor 135 to afford tetrasaccharide 140 in 76% yield.
Scheme 33.
Oligosaccharide Synthesis via Regenerative Glycosylation
4. GLYCOSYL CHLORIDES
First glycosylations performed by Michael (vide supra) involved glycosyl chlorides.4 However, in subsequent years, glycosyl chlorides were largely outshadowed by glycosyl bromides that have traditionally been considered advantageous over their chloride counterparts. After the thorough discussion of glycosyl bromides (Section 3), we note that many methods developed for the activation of glycosyl bromides are also effective for the activation of glycosyl chlorides. Hence, this section will mainly focus on the reagent-specific or condition-specific synthesis, activation, and application of glycosyl chlorides rather than repeating the same data presented for glycosyl bromides.
4.1. Synthesis of Glycosyl Chlorides
For the first time, glycosyl chlorides were synthesized from free glucose by treatment with acetyl chloride by Colley in 1870.342 Expanding on this early work, others have also used AcCl for the synthesis of glycosyl chlorides of other sugar series.343,344 Subsequently, more efficient methods, surveyed in this Section, have been developed for the synthesis of glycosyl chlorides from a variety of precursors.
4.1.1. Preparation from Hemiacetals or Glycosyl Esters.
Most commonly, glycosyl chlorides are prepared from two anomeric groups, either an ester such as acetate or a hemiacetal (Scheme 34). Selected reagents suitable for converting anomeric esters into glycosyl chlorides include TiCl4,345 SOCl2,346 and PCl5.347 Representative reagents used in the synthesis of chlorides from hemiacetals include CHCl2OMe,348,349 SOCl2,350 oxalyl chloride,351–353 n-BuLi and ClPO(OPh)2,354 chloroenamine,355 and triphosgene.356 Evidently, many if not all of these reactions use harsh conditions and/or toxic reagents. Finding new methods to avoid some of these harsh conditions, toxic, or heavy metal reagents has been a vibrant area of study in recent years.
Scheme 34.

Synthesis of Glycosyl Chlorides from Ethers or Hemiacetals
In 2017, Iadonisi developed a solvent-free method for the synthesis of a variety of glycosyl chlorides (Scheme 35A).357 Using triphenyl phosphine and hexachloroacetone at 70 °C, glycosyl chloride 142 was obtained from the hemiacetal precursor 141 in 45 min. These conditions were applied to various sugar series including mannose, galactose, and fucose giving high yields (80%+) in most cases. This method was less effective when applied to nitrogen-containing sugar series. For example, the glycosyl chloride from glucosamine was obtained in only 44% yield.
Scheme 35.
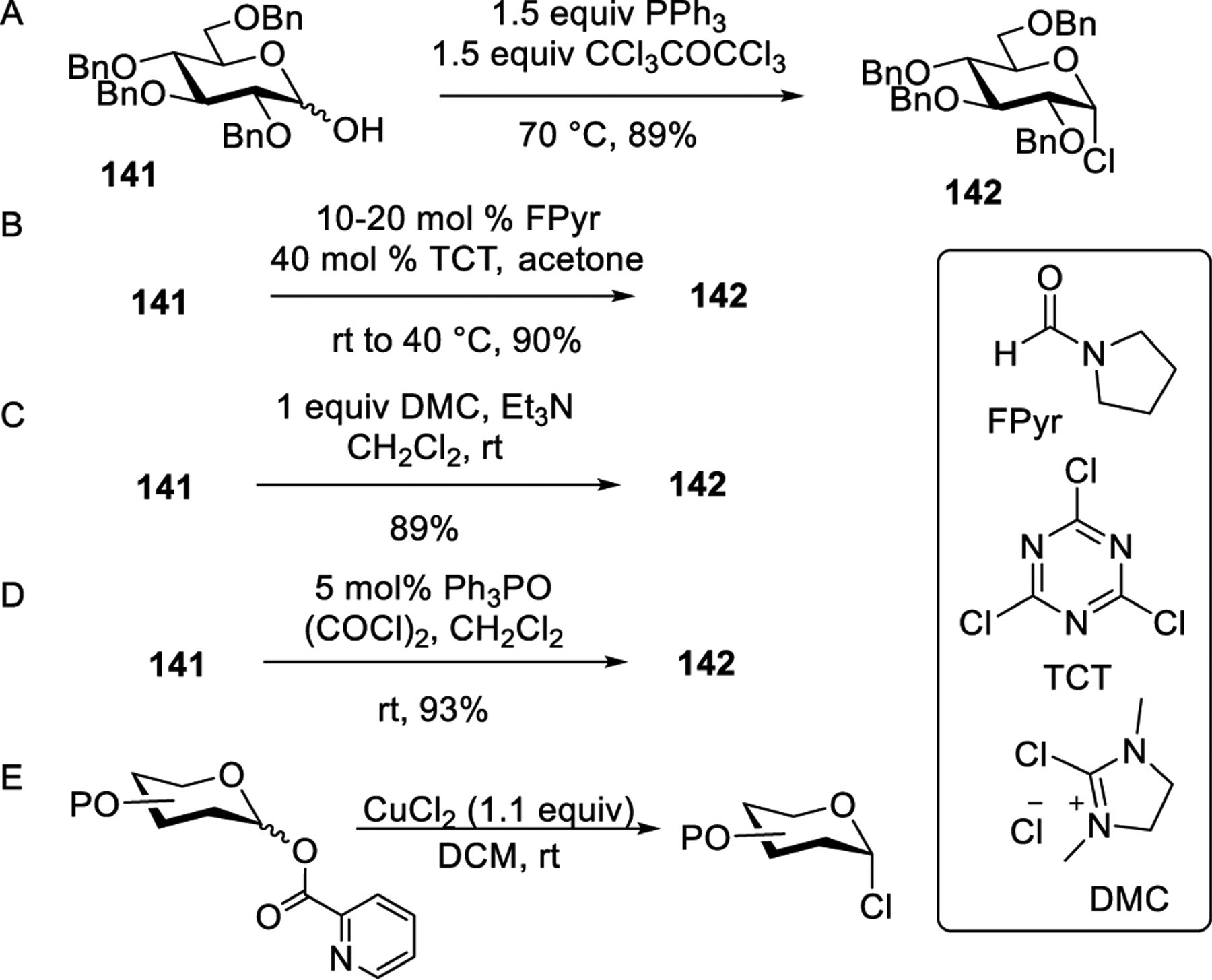
New Methods for the Synthesis of Glycosyl Chlorides
Huy and Filbrich358 have recently developed a method for the synthesis of glycosyl chlorides from the corresponding hemiacetal derivatives using as little as 34 mol % of trichlorotriazine (TCT) as a source of the stoichiometric chlorine. Thus, reaction of hemiacetal 141 with TCT in the presence of 10–20 mol % of N-formylpyrrolidine (FPyr) at 40 °C produced glycosyl chloride 142 in 90% yield (Scheme 35B). These conditions were shown to work both with sugar substrates such as glucose and fructose and with aliphatic alcohols. In 2018, Judeh et al.359 introduced a chlorinating reagent 2-chloro-1,3-dimethylimidazolinum chloride (DMC) that was applied to the synthesis of glycosyl chlorides. Using stoichiometric DMC in the presence of triethylamine, hemiacetal 141 was converted into the corresponding glycosyl halide 142 in 15–30 min in 89% yield (Scheme 35C). This reaction worked well for a variety of sugars (glucose, mannose, galactose) giving 80–95% yield in most cases. The developed conditions were found to be compatible with many commonly used protecting groups such as acetates, silyl ethers, and acetals. McGarrigle et al.360 found that catalytic Appel conditions using 5 mol % Ph3PO and 1.5 equiv (COCl)2 were also capable of chlorinating hemiacetals (Scheme 35D). Using these conditions, chloride 142 was synthesized from hemiacetal 141 in 93% yield. While this protocol worked well for glucose, most other sugars such as mannose, galactose, and 2-deoxy glucose gave lower yields between 67 and 79%. Glucosamine and galactosamine derivatives performed the worst under these reaction conditions, giving mixtures of products.
Recently, Tang and co-workers developed a milder reaction conditions for the synthesis of glycosyl chloride donors.213 In accordance with their approach, 1-O-picoloyl (Pico) derivatives were treated with copper(II) chloride in DCM at ambient temperature (Scheme 35E). An interesting insight of this method is that 1-O-picoloyl (Pico) derivatives exclusively produced the corresponding α-glycosyl chlorides, and the method was found to be compatible with many commonly used protecting groups.
4.1.2. Preparation from Thioglycosides.
Thioglycosides have many advantages such as being stable, allowing for protecting group manipulations, and can be stored for long periods of time. Hence, most modern building block syntheses today involve thioglycosides. Conversion from thioglycosides to glycosyl chlorides typically involved a two-step protocol with the first step being hydrolysis of the anomeric thioglycoside to the hemiacetal. This represented a notable disadvantage, and the effort has been made to develop methods for the direct conversion of thioglycosides to glycosyl chlorides. The first method was reported by Sugiyama and Diakur,361 wherein 4-chlorophenylthio glycoside 143 was treated with oxalyl chloride in dichloromethane. The reaction was found to proceed through the intermediacy of a glycosyl halosulfonium salt shown in Scheme 36A. The latter is unstable and falls apart to form the corresponding glycosyl chloride 144. The resulting glycosyl chlorides were isolated in high yields (90%+) or crude chlorides could be used for subsequent glycosylations directly. While this protocol allowed for the one-step conversion, it still used harsh reaction conditions and the scope of this reaction was limited to 4-chlorophenylthio glycosides.
Scheme 36.
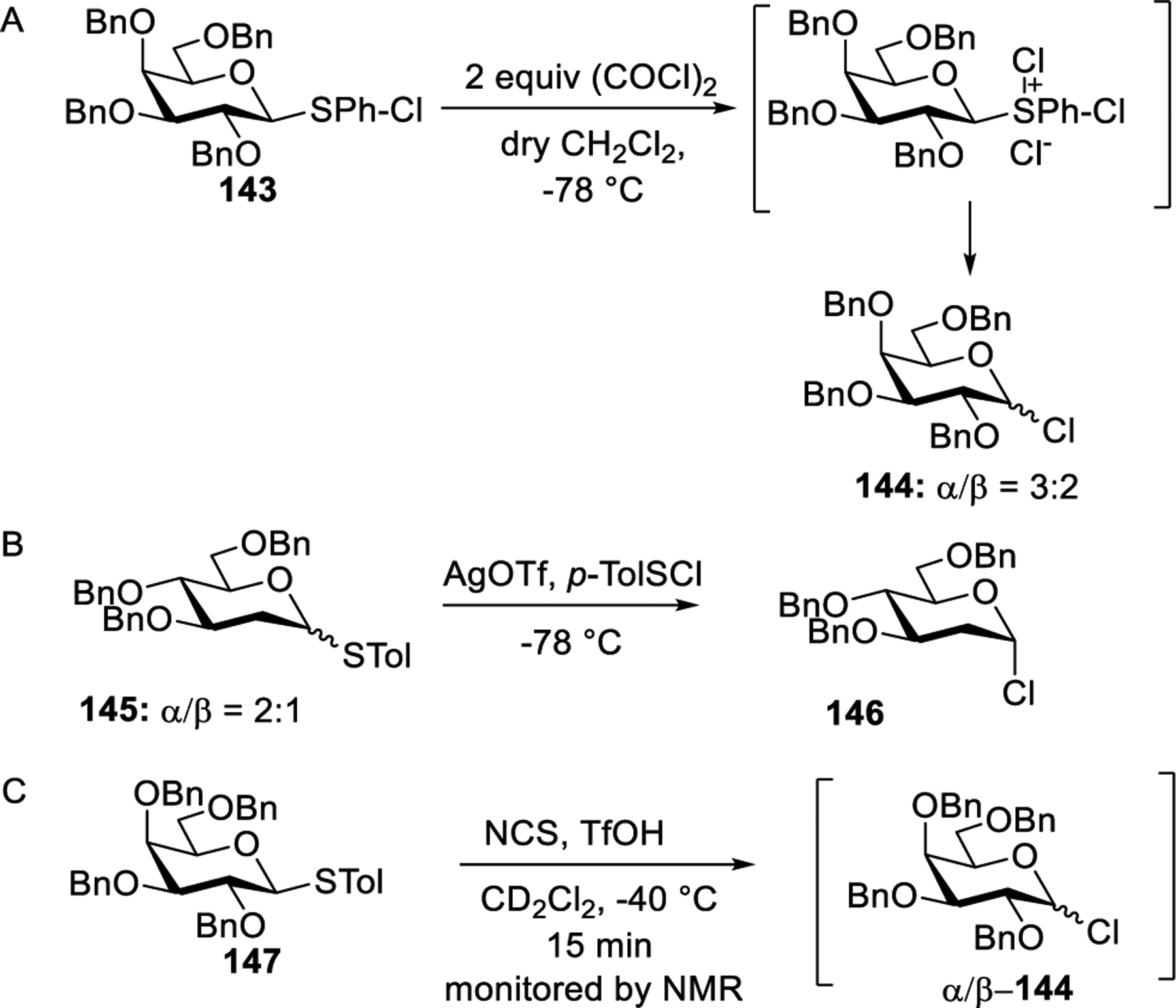
Direct Synthesis of Glycosyl Chlorides from Thioglycosides
Verma and Wang362 employed stoichiometric p-TolSCl in the presence of catalytic AgOTf to successfully convert p-tolylthio (STol) 2-deoxyglycoside 145 into the corresponding chloride 146 (Scheme 36B). Following this conversion, the corresponding chloride could be used for glycosylation without further purification. Subsequently, the authors discovered that the formation of anomeric chlorides can be accompanied by anomeric triflates, and the product distribution was dependent on the relative reactivity value (RRV)135 of the tolylthio glycoside precursor. For example, when armed tolylthio galactoside 147 (RRV = 17 000) was treated with N-chlorosuccinimide NCS (1.0 equiv) and TfOH (0.3 equiv) in CD2Cl2 at −40 °C, exclusive formation of anomeric chloride 144 was observed by means of the NMR spectroscopy monitoring (Scheme 36C).363 Regardless of the reaction intermediates, tolylthio glycosides could be glycosidated in the presence of a suitable glycosyl acceptor, NCS, and triflic acid. Reactions proceeding via the intermediacy of glycosyl chlorides were highly α-stereoselective.
4.2. Glycosidation of Glycosyl Chlorides
Michael in 1879 was the first to perform a glycosylation reaction with glycosyl chlorides (vide supra).4 Per-acetylated glucosyl chloride was reacted with potassium phenoxide giving phenyl glucoside as the product. Then in 1901, the activation of glycosyl chlorides was performed by Koenigs and Knorr.7 These reactions used insoluble silver(I) salts such as silver oxide or silver carbonate which were thought to act as an acid scavenger. Little was done to improve the activation of chlorides until 1949 when Helferich364 introduced mercury salts as active promoters. While these methods can be used to synthesize oligosaccharides,365,366 mercury salts are very toxic and the environmental considerations of the 21st century called for further quest of better activation conditions. Nevertheless, both the Koenigs-Knorr method and the Helferich modification have found broad utility in the synthesis of simple glycosides to date.
Some early work included the activation of glycosyl chlorides with cadmium(II) salts.249 For example, glycosidation of glycosyl uronide donor 148 with estrone acceptor 71 in the presence of CdCO3 produced glucuronide 72 in 71% yield (Scheme 37A). Cadmium sulfide was also investigated as the activator for the glycosidation of the anomeric chloride; however, the yield of the glucuronide was below the preparative value. Several other steroidal phenolic glucoronides, glycosides, and acetyl glucosaminides were successfully synthesized.250,302,303 In addition to β-glycosides, a small amount of the corresponding α-glucosides and/or C-glycosides were also isolated, and the ratio of reagents was found to depend on coupling partners and reaction conditions employed. An interesting aspect of these reactions was the development of color as the reaction progresses. It was suggested that the generated cadmium chloride was the actual catalyst of activation. However, cadmium chloride by itself was ineffective, similar to Helferich’s explanation for mercuric cyanide activation.239 As discussed in Section 3.2.2, the product composition also in this case depends on the surface area of cadmium carbonate.251
Scheme 37.
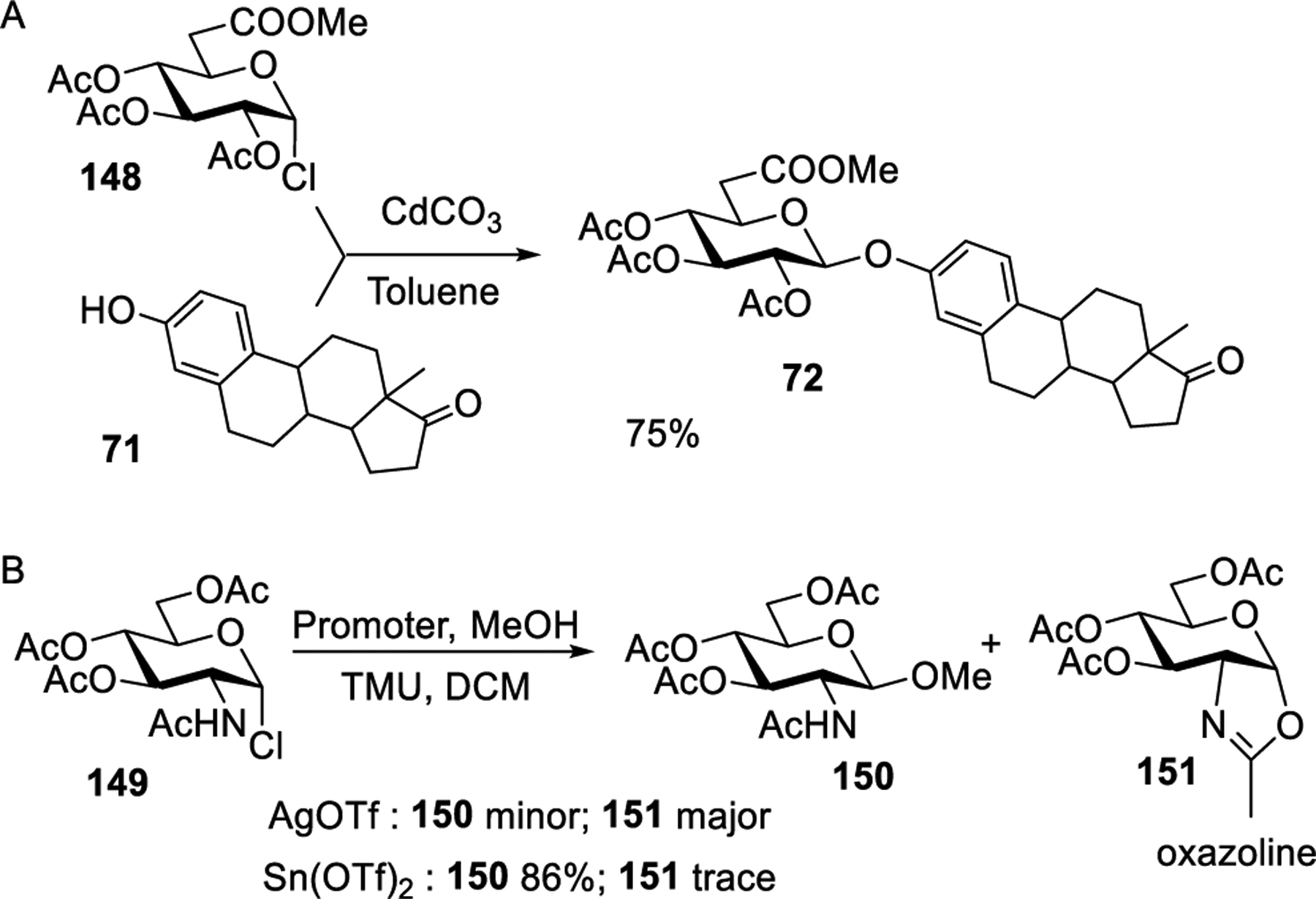
Cadmium or Tin Salt-Promoted Glycosidations of Glycosyl Chlorides
In another study, the authors successfully applied the stannous triflate-promoted glycosylation for the activation of glycosyl chloride donors.306 When glycosyl chloride 149 bearing a 2-acetamido (NHAc) group was employed as glycosyl donor in the presence of AgOTf, oxazoline 151 was observed as a major product (Scheme 37B). Changing AgOTf with Sn(OTf)2 under otherwise identical reaction conditions suppressed the formation of oxazoline 151 while producing glycoside 150 as the major product. The control experiments showed that oxazoline does not serve as a reaction intermediate under these reaction conditions.
4.2.1. Activation with Silver Salts.
Being partially soluble, silver(I) triflate is typically an effective promoter for the activation of glycosyl chlorides. However, these conditions were somewhat ineffective in cases of relatively unreactive glycosyl donors such as sialic acid chlorides.367,368 Thus, glycosidation of chloride 152 with glycosyl acceptor 153 in the presence of AgOTf produced glycoside 154 in a poor yield of 22% without any preferred stereoselectivity (Scheme 38). While investigating a participating moiety at C-1, Gin and co-workers introduced sialyl chloride donor 155 bearing an N,N-dimethylglycolamido ester functionality.369 A commendable result was obtained with galactosyl acceptor 153 for the formation of disaccharide 156 in 56% yield and with high α-stereoselectivity α/β = 13/1. Glycosylation of the primary glycosyl acceptor β-36 was nearly as effective with both sialyl donors 152 and 155 in terms of the yields of the respective products 158 and 157 that were isolated in 60–61% yields. However, the activation of the standard sialyl chloride donor 152 was nonstereoselective, and disaccharide 158 was isolated as a racemic mixture.
Scheme 38.
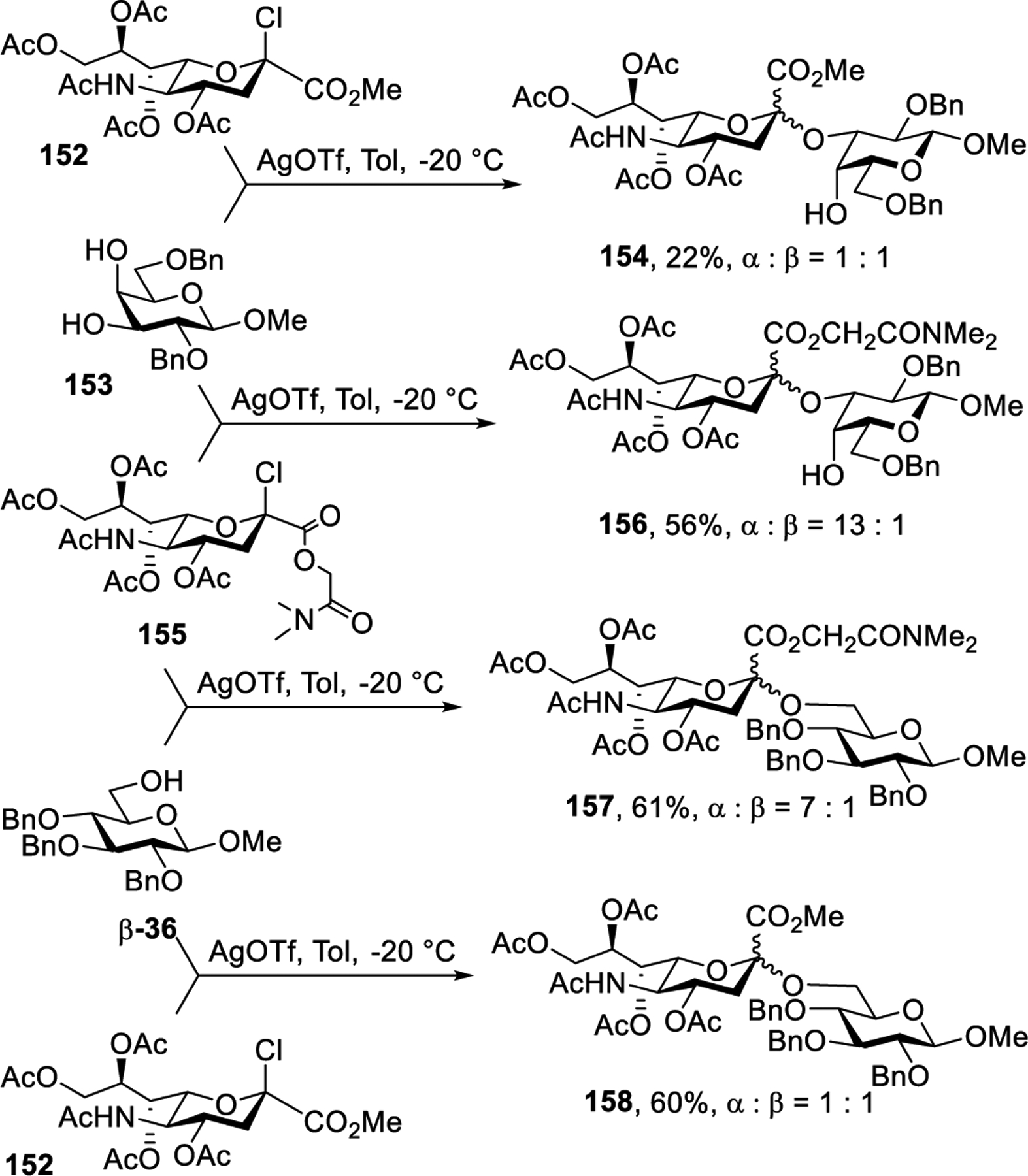
Silver Triflate-Promoted Activation of Sialic Acid Chlorides
As described in Section 3.2.1, Taylor and co-workers developed borinic acid-catalyzed regioselective activation of glycosyl bromides.224,272 A similar approach was later applied by the authors to the β-selective synthesis of 2-deoxy and 2,6-dideoxyglycosides.370 Anomeric chlorides as glycosyl donors and partially protected cis-1,2- and 1,3-diols as glycosyl acceptors were found to be very effective. As shown in Scheme 39, per-acetylated 2-deoxyglycosyl chloride 159, synthesized by treating with BCl3, was reacted with glycosyl acceptor 160 in the presence of aminoethyl diphenylborinate 28 and silver oxide to produce 2-deoxy disaccharide 161 in 77% yield with high β-selectivity (α/β = 1:16). The authors also synthesized challenging β-2,6-dideoxy glycosides commonly found in bioactive natural products. For example, reaction of 2-deoxy-L-rhamnopyranosyl chloride 162 with glycosyl acceptor 163 under similar reaction conditions produced disaccharide 164 in a good yield of 60% with high β-selectivity (α/β = 1:10). Changing the protecting group from OAc to OBn turned out to be detrimental for both β-selectivity and yields. Similarly to that discussed in Section 3.2.1, the enhanced nucleophilicity of the hydroxyl group, as predicted by Fukui index calculation, in the borinate complex resulted in an SN2-like attack on the glycosyl chloride donor.
Scheme 39.

Organoboron-Catalyzed Glycosidation of 2-Deoxy and 2,6-Dideoxy Glycosyl Chlorides
Following the initial success with catalytic glycosidation of glycosyl chlorides, the Demchenko group investigated acid-catalyzed silver(I) oxide-mediated glycosylations that were initially developed for glycosyl bromides (vide supra).221 Using Ag2O combined with TfOH allows one to drastically increase the glycosylation reaction rate while reducing the amount of silver needed to 0.50 equiv. This reaction worked well with benzylated glucosyl chloride 142 and disaccharide 165 was obtained in 97% yield in the presence of just 0.50 equiv of Ag2O and 0.25 equiv of TfOH in 30 min (Scheme 40).371 Glycosylations of secondary glycosyl acceptors provided similar results. Benzylated mannosyl and galactosyl chlorides required a higher amount of TfOH (0.50 equiv). These reaction conditions provided high yields (98%+) and fast reaction times.
Scheme 40.

Glycosyl Chloride Activation Using Ag2O and TfOH Promoter System
Nitrogen-containing sugar derivatives such as glucosamine and sialic acid could also be readily activated under these reaction conditions, but required higher amounts of activators. Thus, primary glycosyl acceptor 36 was coupled with phthalimide-protected glycosyl chloride donor 166 in the presence of 1.5 equiv of Ag2O and 0.5 equiv of TfOH afforded disaccharide 167 in 97% yield. However, upon switching to less reactive secondary acceptors the yields dropped to 68–76%. Sialic acid chloride 168 was glycosidated with the primary glycosyl acceptor 55 allowing disaccharide 169 in an excellent yield of 97% yield, albeit with poor stereoselectivity.
4.2.2. Organocatalytic Activation.
These classical methods were state-of-the-art until recently, when new methods for the activation of glycosyl chlorides have emerged. The first new method was introduced by the Ye group in 2016.145 Using a variety of benzylated glycosyl chlorides, 20 mol % of Schreiner’s catalyst 170 and 2.0 equiv K2CO3 in benzene at 80 °C the respective disaccharides were produced in high yields (80%+). At first, these reactions were rather slow (24 h), and the stereoselectivity was poor. Thus, glycosylation between glycosyl chloride donor 142 and primary acceptor 36 gave disaccharide 165 in 95% yield as an anomeric mixture (α/β = 1:1, Scheme 41A). When the same reaction was carried out in the presence of 1.5 equiv of tris(2,4,6-trimethoxyphenyl)-phosphine (TTMPP) as an additive, the anomeric stereoselectivity could be improved to a commendable α/β = 12.6:1. The improvement of stereoselectivity with TTMPP was seen throughout a series of glycosyl acceptors. On the basis of the NMR data, the authors theorized that TTMPP noncovalently interacted with the anomeric carbon from the β-face. This interaction directed the acceptor attack from the opposite face giving rise to α-linked glycosides.
Scheme 41.

Glycosyl Chloride Activation Using Thioureas
In 2019, McGarrigle360 applied the Appel conditions (vide supra) to the synthesis of glycosyl chlorides followed by their glycosidation in one pot. The treatment of hemiacetal 141 with Ph3PO and oxalyl chloride in dichloromethane produced glycosyl chloride 142. Then glycosyl acceptor 36 was added in situ along with 20 mol % of Schreiner’s catalyst 170, 2.2 equiv of K2CO3, and 20 mol % of TTMPP (Ye’s conditions, Scheme 41B). This one-pot approach led to a decrease in yield of disaccharide 165 that was obtained in 71% yield (vs 95% reported by Ye) with an anomeric ratio of α/β = 88:12.
The Jacobsen group developed thiourea catalyst 171,70 which cooperatively activates both the glycosyl chloride donor and the glycosyl acceptor. The glycosyl chloride donor hydrogen bonds with the thiourea portion of catalyst 171, which enhances its leaving group ability. At the same time, the incoming nucleophile is also activated by the catalyst via Lewis basic interactions with the carbonyl oxygen of the amide of the catalyst. The combination of these two activations by the catalyst leads to an SN2-like displacement. These reactions were conducted in the presence of 5 mol % of 171, 2 equiv of isobutylene oxide (IBO), which acts as an electrophilic trap for the released HCl, in o-dichlorobenzene. Reacting donor 142 with acceptor 172 using the conditions above gave disaccharide 173 in 77% yield (α/β = 7:93, Scheme 41C). Other sugars showed similar yields and stereoselectivity. To prove that these glycosylations undergo an SN2-like displacement, the authors also studied the glycosyl chloride configuration at the anomeric center. It was determined that an α-chloride leaving group gave primarily the β-linked product whereas a β-chloride gave predominantly the α-linked product. This trend was seen throughout a series of glycosyl chlorides, showing that these reactions follow an SN2-like displacement of the leaving group, without the formation of an oxacarbenium ion intermediate.
Codee and co-workers developed a halogen bond-mediated activation method for glycosyl chlorides. For that purpose iodoimidazolium compounds 174 and 175 was employed as halogen-bond donors.372 NMR experiments were performed to establish the activation of 2,3,4,6-tetra-O-benzyl α,β-glucosyl chloride 142 that was treated with bis(iodoimidazolium) compound 174 (1.0 equiv) in CD3CN in an NMR tube (Scheme 42A). Based on NMR signals and LC-MS data it was suggested that more reactive β-glycosyl chloride was completely consumed and furnished anomeric acetamide 176 by a Ritter type process373 while the α-anomer did not react. Later, it was realized that the stable α-anomer requires longer duration (several weeks) for activation under these reaction conditions. Several control experiments were performed to confirm that halogen-bond activation was essential for activating the anomeric C–Cl bond. In a separate NMR experiment, it was shown that glucosyl bromide is more reactive than corresponding chloride however, its use is restricted by lower stability and spontaneous decomposition. To explore the ability of the dicationic halogen-bond donors to promote the synthesis of O-glycosidic linkages, several attempts were made. When the reactive l-oleandrosyl chloride 177 was treated with isopropyl alcohol in the presence of a lipophilic halogen bond activator 175 and 2,4,6-tritert-butyl pyrimidine as an acid scavenger a satisfactory conversion to corresponding glycoside 178 was achieved as shown in Scheme 42B. This method was largely limited to highly reactive glycosyl chlorides.
Scheme 42.

Halogen Bond-Mediated Activation of Glycosyl Chloride
4.2.3. Catalytic Activation with Iron, Bismuth, or Palladium Salts.
In 2018, Geringer and Demchenko introduced the first catalytic glycosidation of glycosyl chlorides.374 Thus, benzylated glucosyl chlorides could be readily glycosidated in the presence of only 20 mol % of iron(III) chloride (FeCl3) and molecular sieves (4 Å) in dichloromethane. These glycosylations led to the corresponding disaccharides in moderate to good yields (47–80%). For example, glycosidation of chloride 142 with primary acceptor 36 afforded disaccharide 165 in 67% yield (α/β = 1.1:1, Scheme 43). Somewhat modest yields in these glycosylations were partially attributed to the formation of an undesirable side product, 1,6-anhydro-2,3,4-tri-O-benzyl-β-d-glucopyranose. Notably, switching to benzoylated glucosyl chlorides or either mannosyl or galactosyl chlorides, resulted in an increase in yields to 52–98% depending on the acceptor. For example, the reaction of benzoylated chloride 179 with acceptor 36 afforded disaccharide 102 in 98% yield as shown in Scheme 43. In these cases, no 1,6-anhydro derivative formation was noticed. The proposed reaction mechanism follows a traditional Lewis acid-mediated reaction according to which FeCl3 associates with the corresponding glycosyl chloride, causing the activated glycosyl donor to form. The chloride then leaves resulting in the formation of the oxacarbenium ion, which was the cause of the poor stereoselectivity observed in most glycosylations with benzylated glycosyl bromides due to the ability of the acceptor to attack from either side of the flattened chair. Besides being catalytic, the ability to glycosylate the disarmed, benzoylated sugars was advantageous over previously described methods.
Scheme 43.

Glycosyl Chloride Activation Using Catalytic FeCl3
In 2021, Demchenko and co-workers have developed very mild reaction conditions for the activation glycosyl chlorides in the presence of bismuth(III) triflate.308 This reaction worked similarly to that of glycosylation with glycosyl bromides. Tetra-benzoylated and tetra-benzylated galactosyl chlorides 180 and 144 were glycosidated with glycosyl acceptor 36 in the presence of Bi(OTf)2 (0.35 equiv) to produce galactosides 78 and 79, respectively (Scheme 44A). An interesting observation was made when activation of glycosyl chlorides and bromides were compared. The activation of galactosyl chloride 180 was faster (15 min) when compared with the activation of a similarly protected galactosyl bromide 40 (30 min). Benzylated galactosyl chloride 144 produced galactoside 79 in a better yield (97%, α/β = 1/1.2) when compared to results from benzylated galactosyl bromide 34 (78%, α/β = 1/1.1).
Scheme 44.
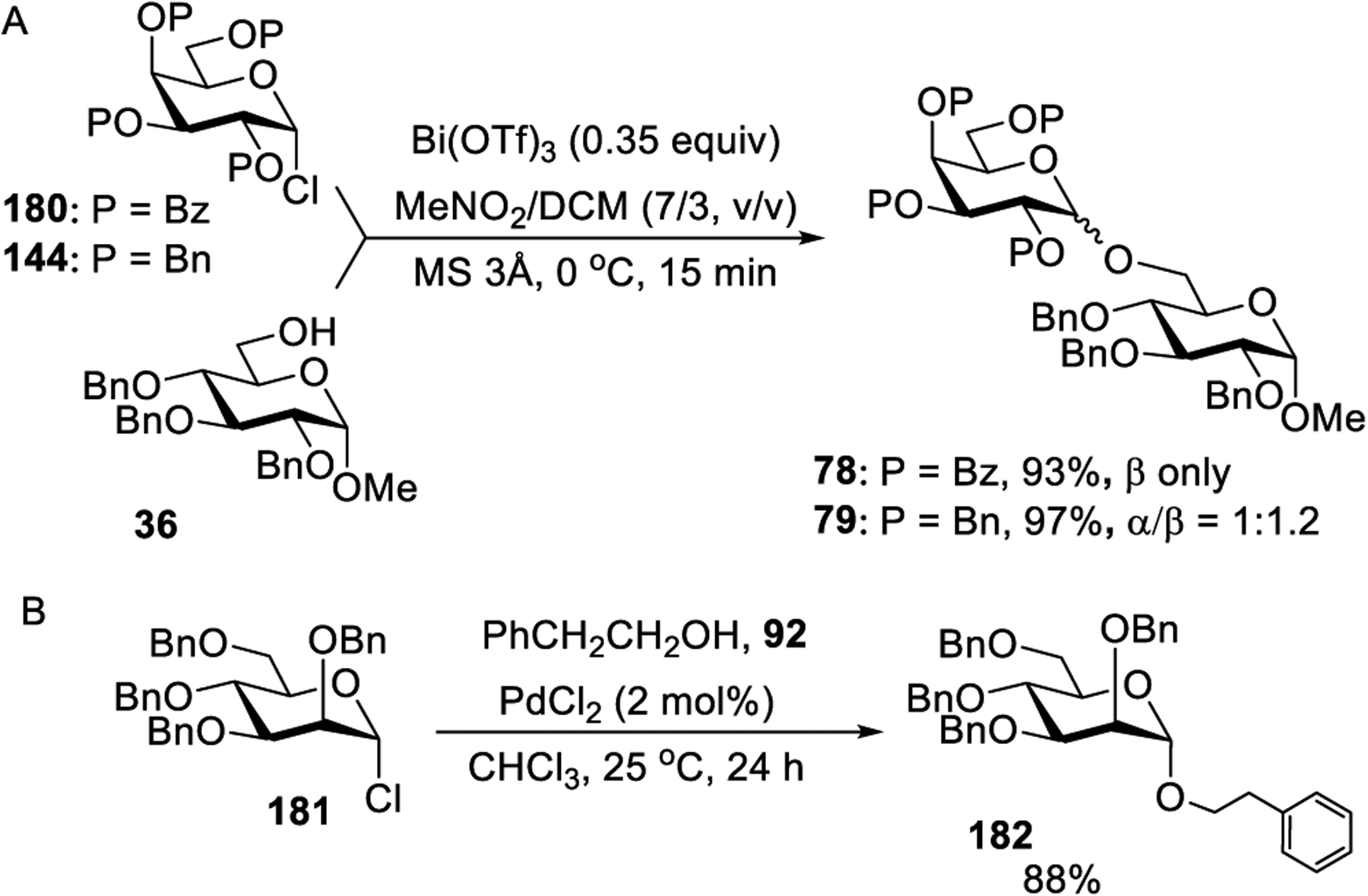
Bismuth(III)- and Palladium(II)-Catalyzed Activations of Glycosyl Chlorides
Very recently, Chen and co-workers developed a new protocol to activate glycosyl chloride donors to synthesize O-glycosides using palladium(II) reagents.375 On the basis of their previous work of palladium-catalyzed C-aryl glycoside synthesis, the authors established that palladium acetate can serve as a Lewis acid that will interact with the chloride leaving group thereby activating the anomeric C–Cl bond.376 To broaden the scope of this approach to the synthesis of O-glycosides, 2,3,4,6-tetrabenzyl mannosyl bromide 181 was treated with 2-phenylethanol 92 in the presence of 2 mol % of Pd(OAc)2 in chloroform at room temperature (Scheme 44B). The glycosylation produced glycoside 182 in 88% yield with exclusive α-stereoselectivity. By tuning the reaction temperature (25 to 110 °C) and amount of Pd(OAc)2 (2 to 5 mol %), various glycosyl acceptors were glycosylated producing the respective products in good to excellent yields with exclusive α-stereoselectivity. Glycosyl donors of different sugar series including gluco, galacto, rhamno, ribofurano, and mannofurano were also investigated. Glycoside products were obtained in good yields; however, the stereoselectivity was difficult to predict.
4.3. Glycosyl Chlorides in Glycan and Glycoconjugate Synthesis
2-Chloro and 2-fluoro sialic acid derivatives 183 and 184, both bearing a participating 3-S-phenyl auxiliary, have been employed for the preparation of tetrasaccharide 187 using a selective activation strategy.367,377 Glycosyl fluorides are stable toward the reaction conditions required for the activation of glycosyl chlorides. Thus, coupling of sialyl chloride donor 183 with 8-OH sialyl fluoride acceptor 184 in the presence of AgOTf gave (2→8)-linked dimer 185 in 49% yield with complete α-stereoselectivity (Scheme 45). The resulting dimer 185 was used in the subsequent glycosylation of thiolactosyl acceptor 186 in the presence of the AgOTf/SnCl2 promoter system. The resulting tetrasaccharide 187 was obtained in 39% yield and its anomeric thioglycoside moiety can be activated for subsequent glycosylations directly.
Scheme 45.
Selective Activation of Sialyl Chloride 142 over Sialyl Fluoride 167
5. GLYCOSYL IODIDES
5.1. Synthesis of Glycosyl Iodides
The first synthesis of glycosyl iodides was reported by Fischer, who synthesized 2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl iodide by reaction of per-O-acetylated glucose with HI.148 Fischer also noted that the glycosyl iodide quickly reacted with methanol in the presence of silver carbonate to afford the methyl glycoside. The field did not have much growth until 1974 when Kronzer and Schuerch315 discovered that the glycosidation of benzylated glucosyl bromides could be promoted by the addition of sodium iodide. These glycosylation reactions were performed under metal-free conditions and presumed to occur through the intermediacy of glycosyl iodides.
Shortly thereafter, Thiem and Meyer207 reported that glycosyl iodides could be synthesized from a variety of precursors such as anhydrosugars, methyl glycosides, and per-acetylated hexoses using TMSI. This discovery allowed for the synthesis of many glycosyl iodide donors that for the first time became readily available. However, only acetylated glycosyl iodides were sufficiently stable to be fully characterized. Benzylated iodides were deemed too unstable and had to be synthesized and used in subsequent glycosylations in situ.51 This was the case until Gervay et al. devised a technique to fully characterize benzylated glycosyl iodides by monitoring their formation in the presence of TMSI in CD2Cl2 at −100 °C in an NMR spectrometer.378 The authors found that the anomeric peaks appeared as a doublet at either 6.68 ppm for α-glycosyl iodide 189 or at 5.61 ppm for β-glycosyl iodide. The ratios of these products depended on the configuration of the anomeric acetate in the substrate, α- or β-acetate 188. Regardless of the initial configuration, the iodide displaced the acetates in an SN2-like manner. Following the displacement, anomerization of the β-iodide rapidly occurs to the thermodynamically stable α-iodide (Scheme 46). Following these first major mechanistic studies, glycosyl iodides found a much broader application in synthetic chemistry.
Scheme 46.

Anomerization of Glycosyl Iodides
5.2. Glycosidation of Glycosyl Iodides
Among the first applications of glycosyl iodides as donors was the synthesis of C-glycosides that were shown to form in an SN2-like manner with direct displacement of the glycosyl iodide with the nucleophile. Using tetrabutylammonium cyanide (TBACN) in tetrahydrofuran (THF) and α-mannosyl iodide 190, β-cyanoglycoside 191 was obtained in 55% yield (Scheme 47).379 While this reaction worked well with the mannose iodide, glucosyl iodide α-189 produced β-cyanoglycoside 193 in a modest yield of 32% along with the major side product oxyglycal 192 resulting from the competing E2 elimination reaction. Switching to glucosyl iodide 194 equipped with TMS ethers allowed for a much more successful synthesis of the corresponding β-cyanoglucoside. This was accomplishe using TBACN in toluene, followed by the cleavage of the TMS groups that was affected by addition of MeOH, and subsequent acetylation using Ac2O in pyridine to give 195 in an overall yield of 67%. Similar C-glycoside formation was accomplished by using Grignard reagents in the synthesis of the glycolipid BbGL2, as reported by Kulkarni and Gervay-Hague.380
Scheme 47.
Synthesis of C-Glycosides Using Glycosyl Iodides
The synthesis of O-glycosides has also been approached from glycosyl iodides. When small nucleophiles were used such as phenol with NaHMDS in THF, glycosyl iodide α-189 gave phenol glycoside 196 in 61% yield (Scheme 48A). Other small nucleophiles such as sodium acetate or sodium tert-butoxide worked well giving complete β-stereoselectivity following direct displacement of the α-iodide.9,381 Synthesis of the disaccharide has also proven to be straightforward, however, converting the disaccharide into the second-generation glycosyl donor was troublesome. During displacement of the O-acetyl anomeric group from anomeric acetate 197 to the iodide donor, cleavage of the interglycosidic bond has occurred (Scheme 48B). This problem was solved by Lam and Gervay-Hague382 by a simple addition of an acetate (or other electron-withdrawing) group at the C-6 position of the glycosyl donor. This modification allowed for the synthesis of oligosaccharide derivatives using both solid phase and solution phase strategies using TBAI as a promoter system. Other promoter systems used for the synthesis of oligosaccharides include AgOTf,382 tetrabutylammonium bromide/Na2CO3,383 AgNO3,384 ZnI,385 and TBAI/DIPEA.386
Scheme 48.
Synthesis of O-Glycosides Using Glycosyl Iodides
Another method to help combat the interglycosidic bond cleavage employs fully trimethylsilyl protected substrates. Introduced by Gervay-Hague and co-workers,387 this approach allowed to achieve high yields in glycosidations of per-O-silylated galactosyl iodides. This approach was successfully applied to α-stereoselective synthesis of glycolipids, however some decline in yields was seen due to the formation of silylated acceptors as side products. The reaction was not perfected until later when Gervay-Hague388 found that the formation of silylated side products can be suppressed by reducing the amount of TBAI to 1.5 equiv as opposed to 3.0 equiv used previously. Since this discovery, many research groups have used per-O-silylated sugars to synthesize a variety of natural products.389–391 Glycosyl iodides have been used in a variety of ways that were comprehensively discussed in previous reviews by Kulkarni,392 Lowary,393 and Gervay-Hague.394
More recently, Zhang and co-workers395 expanded the scope of per-silylated glycosyl donors such as 198 and improved the outcome of the reaction by supplementing TBAI-promoted glycosylations with triethylamine (Scheme 48C). Under these conditions, glycosyl donor 198 was reacted with glycosyl acceptor 199 to form disaccharide 200 in 63% yield over two steps after subsequent acetylation. These reactions worked well with a variety of sugar series such as glucosyl, galactosyl, and fucosyl donors.
Glycosyl iodides were also used in α-stereoselective ribofuranosylation of alcohols.396 Ribofuranosyl iodide could be generated using TMSI from precursor 201 (Scheme 49A). Following the addition of i-Pr2NEt and triphenylphosphine oxide, which acts as an additive to improve α-stereoselectivity, and glycosyl acceptor 36, ribofuranosylation would occur. As a result, disaccharide 202 was obtained in 77% yield with complete α-stereoselectivity. These conditions worked well for a variety of glycosyl acceptors ranging from primary aliphatic alcohols to hindered sugar alcohols with yields of 75% or higher. Ribosylation was also studied by Houston and Koreeda397 using iPrOH as the glycosyl acceptor in the presence of I2 as a promoter in THF. This reaction gave the corresponding β-ribosides in high yields. The authors also found that ribosylations performed in the presence of acetone led to the formation of a 1,2-O-isopropylidene derivative instead.
Scheme 49.
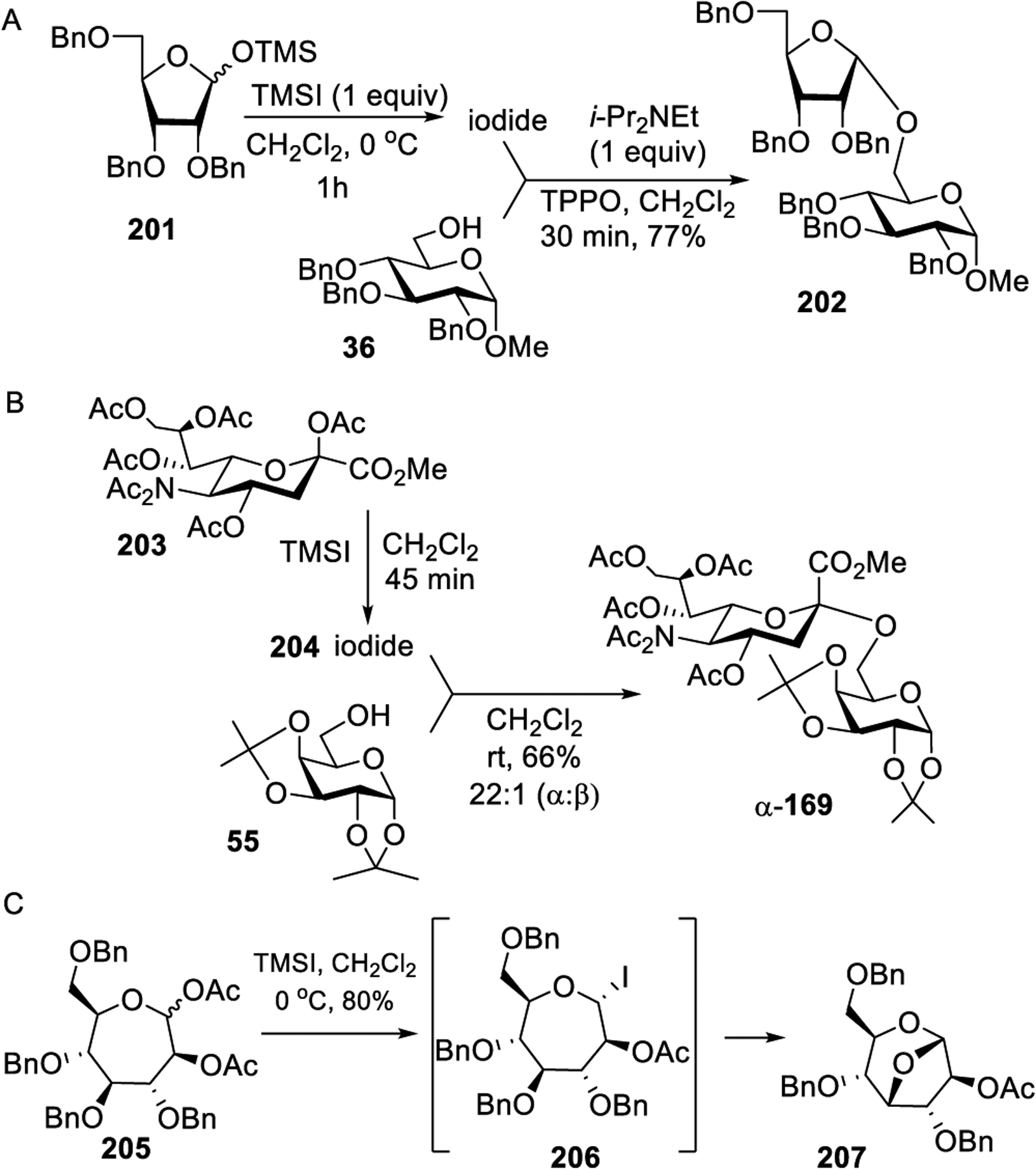
Glycosidation of Iodides of Different Series
The most recent advancement in the application of glycosyl iodides was reported by Park and Gervay-Hague.398 The authors achieved the first, promoter-free sialylations with sialyl iodides, which was applied to the synthesis of steryl β-sialosides. These glycosylations only worked with C-5 modified sialic acid donors, whereas traditional N-acetamido sialic acids underwent 2,3-elimination upon the attempt to obtain a sialyl iodide donor. However, when 5-N-acetylacetamido precursor 203 was investigated, the corresponding α-iodide donor 204 was smoothly produced (Scheme 49B). Sialylation could then be performed in a one-pot manner at rt; for example, reaction of the primary glycosyl acceptor 55 gave disaccharide 169 in 66% yield with excellent α-stereoselectivity (α:β = 22:1). Cholesterol-based acceptors provided respectable yields ranging from 52% to 85% giving sialosides with complete β-stereoselectivity.
Applications of glycosyl iodides in synthesis span beyond their use as glycosyl donors. For instance, glycosyl iodides were used as precursors in the formation of 1,4-anhydroseptanoses.399 As depicted in Scheme 49C, septanose 205 was reacted with TMSI to form septanosyl iodide 206 that readily rearranged to form 1,4-anhydroseptanose 207 in 30 min in 80% yield. This rapid cyclization was occurring in septanoses derived from glucose, mannose, xylose, and galactose.
Bennett and co-workers reported a dehydrative glycosidation of 2-deoxy and 2,6-dideoxy-sugars in the presence of 3,3-dichloro-1,2-diphenylcyclopropene 209, tetrabutylammonium iodide (TBAI), and N,N-diisopropylethylamine (DIPEA).400 The authors assumed that the treatment of benzyl protected 2-deoxy hemiacetal 210 under these reaction conditions will lead to the in situ formation of the corresponding glycosyl iodide 211. The latter will then be trapped with cholesterol 212 to produce glycoside 213 in 72% yield with preferential α-selectivity (α/β = 4:1, Scheme 50A). Increasing the amount of TBAI to 5.0 equiv increased the yield of 213 to 82%, however, there was no apparent change in stereoselectivity. Similarly, an α-selective formation 2,6-dideoxyglycosides was achieved.
Scheme 50.
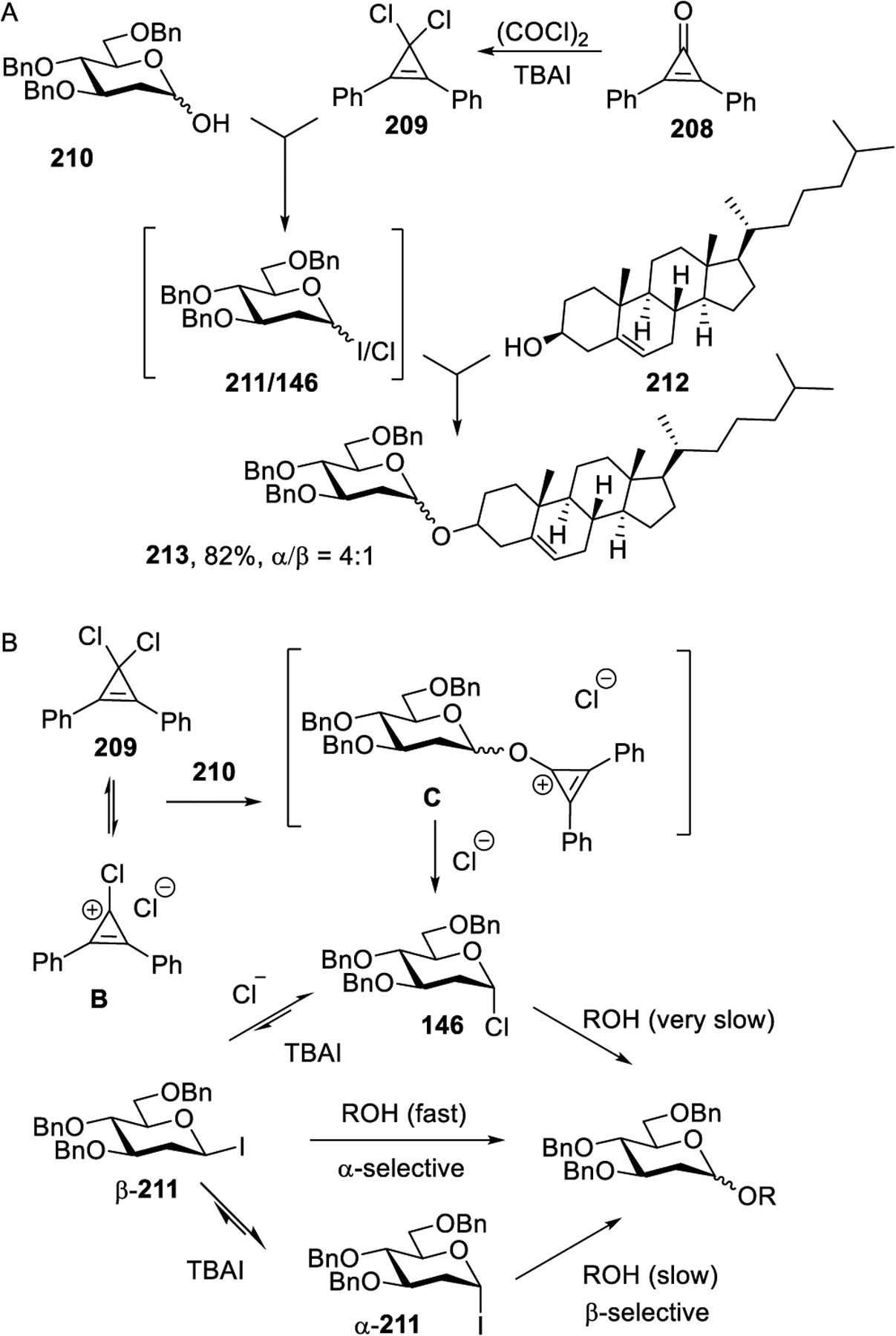
Dehydrative Glycosidation of Hemiacetals
The authors proposed a stepwise mechanistic conversion. 3,3-Dichloro-1,2-diphenylcyclopropene 209 is postulated to exist in equilibrium with intermediate B (Scheme 50B). The latter assists conversion of hemiacetal 210 to the corresponding anomeric chloride 146 through a reactive intermediate C. As evident from NMR experiments, anomeric chloride 146 exists in equilibrium with another species that is postulated to be either α-211 or β-211. Excess iodide (TBAI) in reaction mixture promotes equilibrium between stable α-211 and its reactive counterpart β-211. The fact that preferential α-selective was observed for the products suggests that glycosyl acceptor might be approaching β-iodide intermediate 211 in an SN2-like fashion.
5.3. Glycosyl Iodides in Glycan and Glycoconjugate Synthesis
Bennett used a glycosyl iodide generated in situ for the synthesis of α-glycosides without directing groups.88 Starting from stable thioglycoside 214, the corresponding anomeric triflate was generated in the presence of Ph2SO, Tf2O, and 4 Å molecular sieves in dichloromethane at −78 °C (Scheme 51). Following generation of the glycosyl triflate in situ, 5 equiv of TBAI was added to produce glycosyl iodide 189. 1,4-Dioxane was then added to improve α-stereoselectivity along with glycosyl acceptor 215. As a result, disaccharide 216 was synthesized in 65% yield in excellent α-stereoselectivity (α→β = 23/1). This reaction sequence was reiterated with in situ generated iodide 217 and glycosyl acceptor 36 allowing for the synthesis of trisaccharide 218. However, a modest yield of 42% was observed since a sterically bulky glycosyl donor was used at this stage.
Scheme 51.

Iterative Synthesis of Trisaccharide 196 Using Glycosyl Iodides
6. GLYCOSYL FLUORIDES
The early expansion of synthetic carbohydrate chemistry was made possible due to the improved understanding of the mechanistic aspects of glycosylation with glycosyl halides (mainly chlorides and bromides) as donors. The synthesis of glycosyl fluorides has been known for a long time, but their glycosidation under Koenigs-Knorr conditions is impossible due to their higher stability. This demanded further investigation, and Mukaiyama and co-workers were the first to show the activation of glycosyl fluorides in the presence of Lewis acids such as tin(II) chloride (SnCl2). The activation protocol was subsequently improved by using silver salts as additives. These early attempts led to the formation of 1,2-cis glycosides, which are difficult to synthesize. Following these early discoveries, glycosyl fluorides have become increasingly popular glycosyl donors in synthetic glycochemistry due to a unique combination of high stability and relatively mild Lewis acidic conditions required for their glycosidation.
6.1. Synthesis of Glycosyl Fluorides
Numerous methods for the synthesis of glycosyl fluorides from a variety of precursors have been developed. In the following, all known methods have been categorized by the type of the starting material used for the introduction of the anomeric fluoride moiety.
6.1.1. Preparation from Anomeric Acetates.
The very first synthesis of glycosyl fluorides comprised the treatment of per-acetylated sugars with anhydrous hydrofluoric acid. The acid was generated by heating dry potassium hydrogen fluoride allowing for various glycosyl fluorides to be synthesized in 30 min (Scheme 52A).401 Over the years, milder protocols that are compatible with acid-sensitive functional and protecting groups have been developed. One popular protocol involves Py-HF complex as a source of HF. Thus, Sharma and Eby employed the fluorination procedure developed by Olah et al.402 to convert sialic acid 219 to the corresponding β-fluoride 220 (Scheme 52B).403 Noyori and co-workers extended this approach to different sugar series for the synthesis of anomeric fluorides.404,405 It was shown that the treatment of a solution of benzyl or acetyl protected glycosyl acetates in toluene with 50 or 70% hydrogen fluoride-pyridine mixture readily produced the corresponding fluorides in good to excellent yields. This method turned out to be advantageous for the synthesis of thermodynamically favored α-fluorides. Differentially protected hemiacetals are also suitable substrates (vide infra), but their conversion to fluorides under these conditions was not as efficient as that from the anomeric acetates.406
Scheme 52.
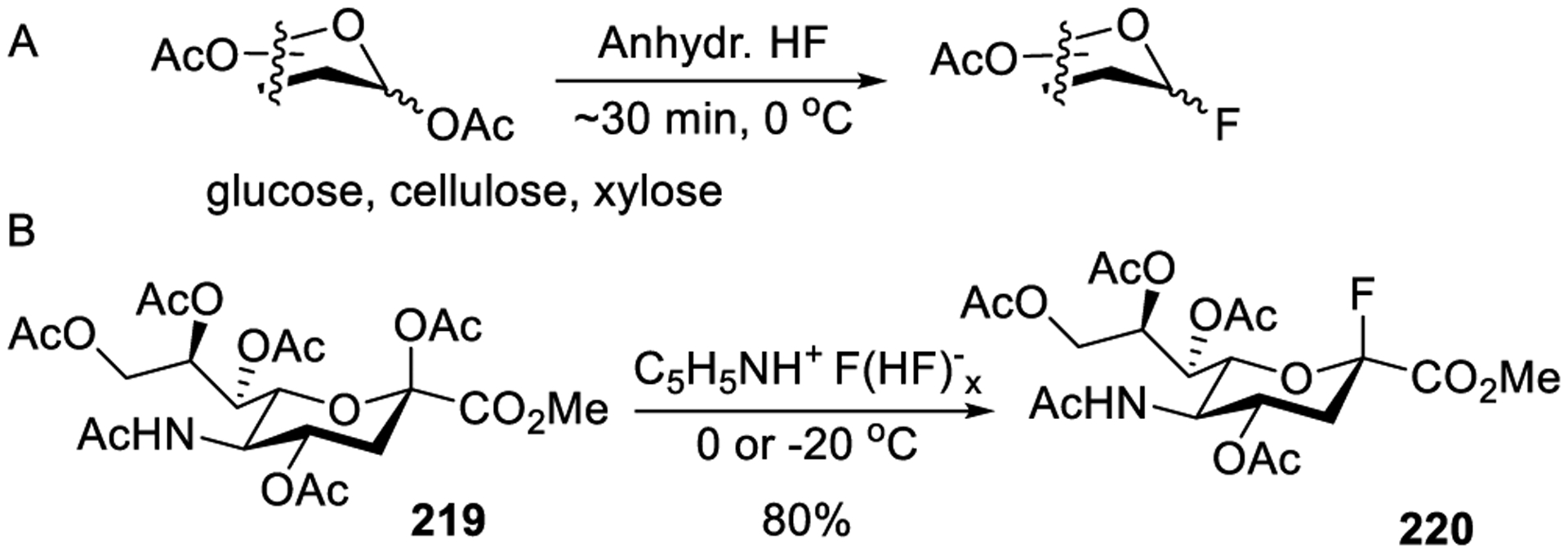
Synthesis of Glycosyl Fluorides from Acetates
6.1.2. Preparation from Glycosyl Chlorides and Bromides.
Helferich and Gootz performed a halogen exchange reaction to synthesize glucosyl fluorides from the corresponding glucosyl bromides.407 For example, when 2,3,4,6-tetra-O-acetyl-α-d-glucosyl bromide α-4 was treated with silver fluoride (AgF) in acetonitrile the corresponding fluoride β-221 was afforded in 54% yield after recrystallization (Scheme 53A). Klemer and Micheel extended this approach to the synthesis of anomeric fluorides of other sugar series from the corresponding glycosyl bromides and chlorides.408,409 Hall and co-workers employed a similar approach to obtain glycosyl fluorides to study their conformational properties.410 Igarashi and co-workers reported the synthesis of glycosyl fluorides from the corresponding chlorides in the presence of AgBF4 in ether or toluene.411
Scheme 53.
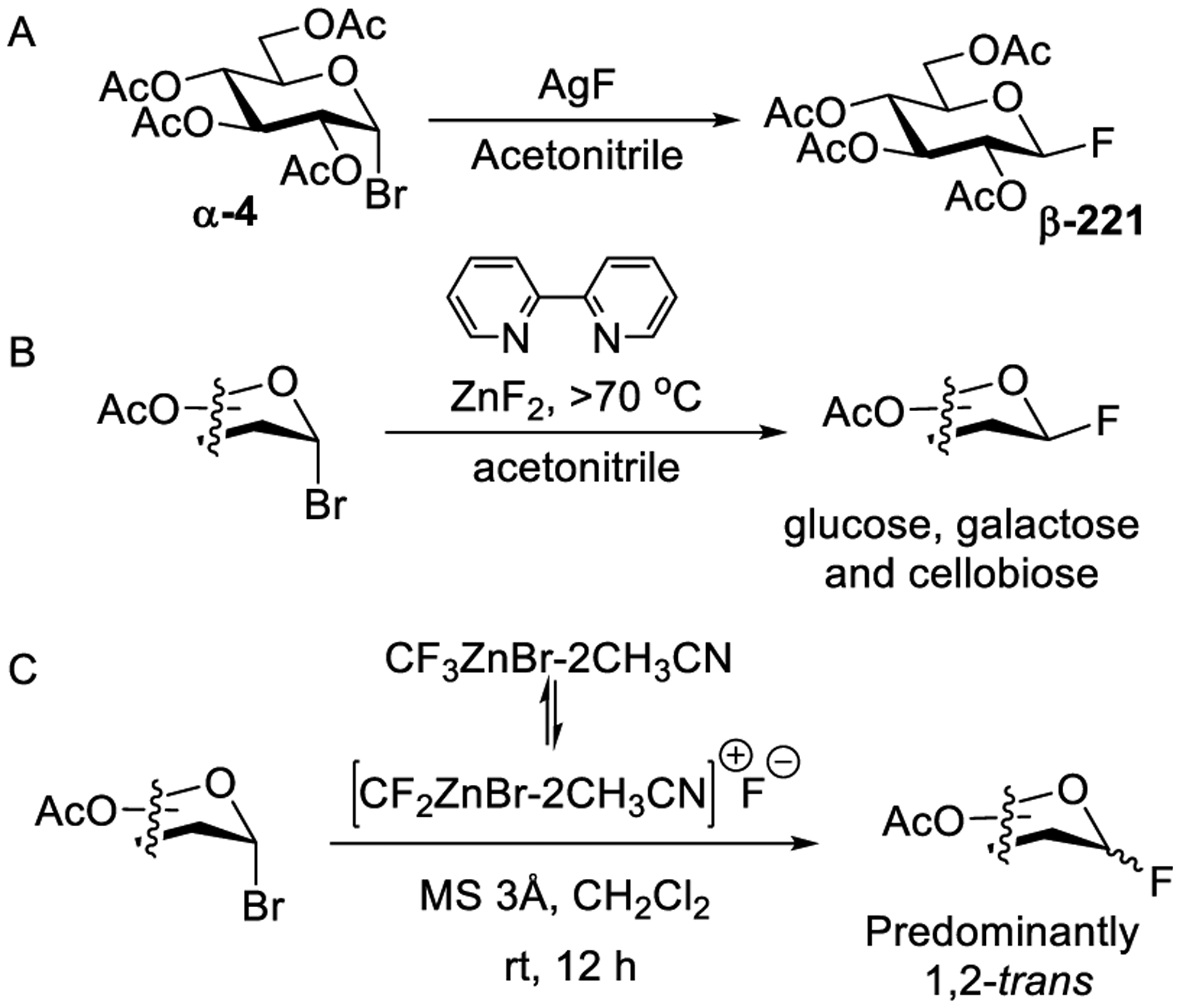
Synthesis of Glycosyl Fluorides from Other Halides
Walinsky and co-workers also investigated a halogen exchange procedure for the synthesis of glycosyl fluorides.412 For that, they chose zinc fluoride and the conversion of glycosyl bromides to the corresponding fluorides was successfully conducted in the presence of ZnF2 alone or in combination with 2,2′-bipyridine (Scheme 53B). Because of the poor solubility of zinc fluoride, these reactions worked best at higher temperatures in acetonitrile. Reactions in the presence of 2,2′-bipyridine were slower, but these conditions led to increased yields of glucosyl fluoride products.
Trifluoromethylzinc bromide (CF3ZnBr-2CH3CN) was known as a reagent for difluoromethylation reactions.413–415 Naumann et al. suggested that it can also act as a potential nucleophilic fluorinating reagent.416 Miethchen and co-workers successfully fluorinated α-bromides of per-acetylated glucose, galactose, mannose, lyxose, and rhamnose in DCM with CF3ZnBr-2CH3CN in DCM (Scheme 53C).417 Predominantly, 1,2-trans fluorides were obtained in these reactions. This protocol also worked with hemiacetals (vide infra), and the yield could be enhanced with TiF4 additive. Miethchen and co-workers also achieved bromine–fluorine exchange in the presence of two-phase system Et3N-3HF/CCl4.418,419 This protocol also worked with hemiacetals as starting materials.
6.1.3. Preparation from Hemiacetals.
Mukaiyama and co-workers reported the synthesis of glycosyl fluorides from the corresponding hemiacetals.420 Synthesis of 2,3,5-tri-O-benzyl-α/β-D-ribofuranosyl fluoride 224 was achieved by treating the corresponding hemiacetal 222 with 2-fluoro-l-methylpyridinium tosylate 223 in the presence of triethylamine as shown in Scheme 54A. The anomeric fluorides were separated by column chromatography, and the α-anomer was shown to equilibrate to β-anomer in the presence of boron trifluoride etherate. Kunz and Sager applied a modified Mitsunobu reaction421,422 to the synthesis of glycosyl fluorides equipped with acid-sensitive protecting groups.423 For example, isopropylidene protected mannofuranose 225 was treated with triphenylphosphine (Ph3P), diethyl azodicarboxylate (DEAD), and triethyloxonium tetrafluoroborate to afford fluoride 226 in 54% yield (Scheme 54B).
Scheme 54.
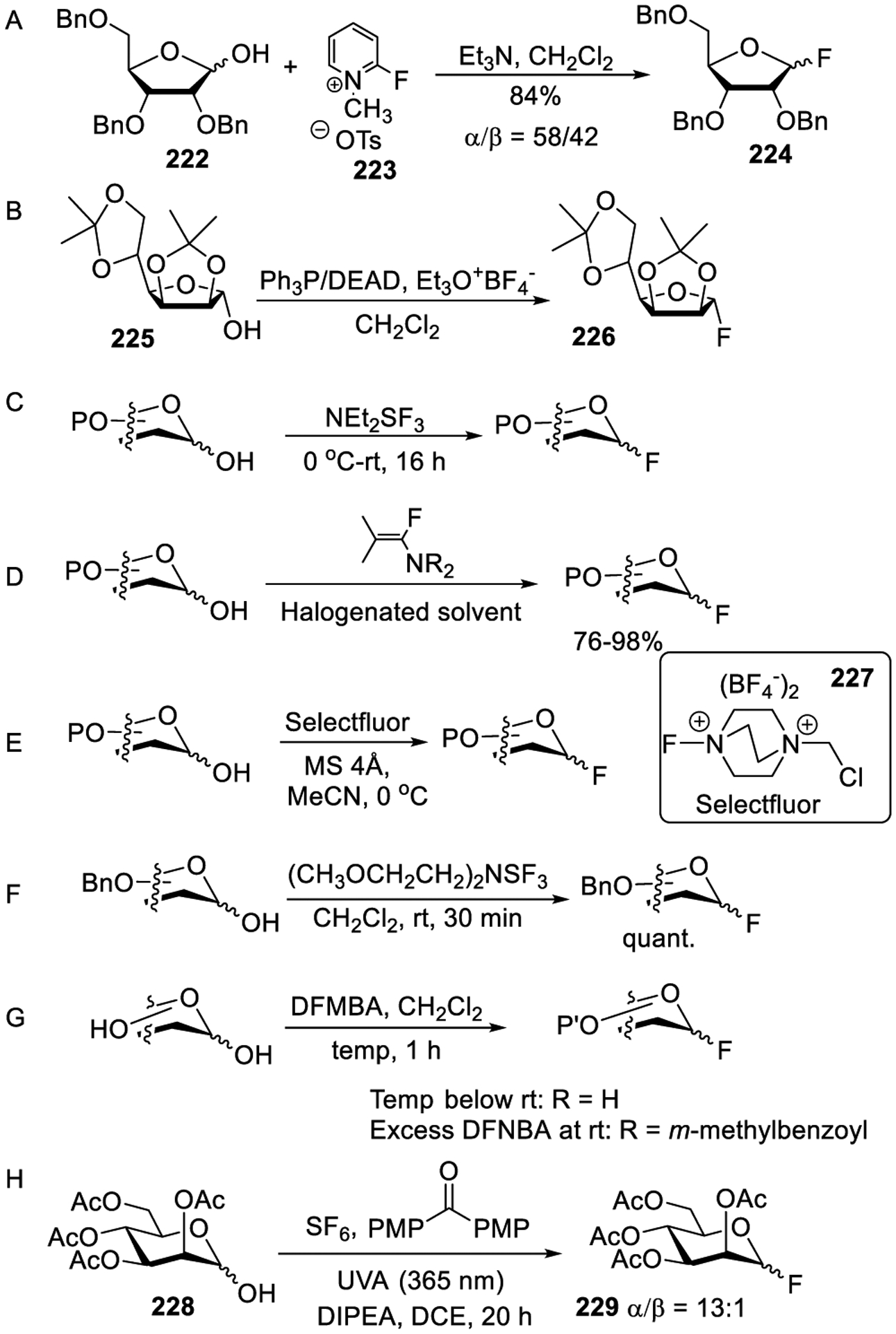
Synthesis of Glycosyl Fluorides from Hemiacetals
Rosenbrook and co-workers successfully applied diethylaminosulfur trifluoride (DAST), a well-known reagent for the direct conversion of alcohols to fluorides424 to sugar hemiacetals. Thus, when hemiacetals were treated with neat DAST at 0 °C followed by warming to rt, the corresponding anomeric fluorides were produced in good yields (60–91%, Scheme 54C).425 Concomitantly, Posner and Haines reported DAST-mediated fluorination reactions that were surprisingly fast (20 min) when conducted in THF as the reaction solvent.426
Ernst and Winkler extended the fluorinating property of α-haloenamines, known reagents for the synthesis of acyl427 and alkyl halides,428,429 to the synthesis of glycosyl fluorides.355 Thus, when hemiacetals of pyranose of furanose sugars were treated with 1-fluoro-N,N,2-trimethylprop-1-en-1-amine or a similar reagent, glycosyl fluorides where produced in 76–98% yield (Scheme 54D). The mild nature of α-haloenamines along with the neutral reaction conditions were found to be compatible with various protecting groups including acetyl, benzoyl, isopropylidene, benzyl, and silyl. 1-Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) 227,430 commonly known as Selectfluor, was found to be an efficient reagent for converting hemiacetals to the corresponding fluorides (Scheme 54E).431 This electrophilic fluorinating reagent was also found to be effective for converting glycals into 2-deoxyglycosyl fluorides. In the presence of SMe2 additive, Selectfluor also reacted with thioglycosides to produce glycosyl fluorides (vide infra).
Thermal instability of DAST hampered its application in large-scale syntheses or reactions at high temperatures.432 Lal and co-workers developed the bis(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) reagent, as a thermally stable analogue of DAST.433 Deoxo-Fluor is also a more efficient fluorination reagent compared to DAST as evidenced by a quantitative conversion of hemiacetal derivatives to the corresponding anomeric fluorides (Scheme 54F). Hara and co-workers reported the synthesis of glycosyl fluorides by treating hemiacetal derivatives with N,N-diethyl-α,α-difluoro-(m-methylbenzyl)amine (DFMBA) as shown in Scheme 54G.434,435 It was noticed that hydroxyl groups at nonanomeric positions were not affected at low temperatures. However, the reactions performed in the presence of excess DFMBA would lead to m-methylbenzoylation along with the anomeric fluorination.
Nagorny and co-workers developed photochemical reaction conditions for the synthesis of glycosyl fluorides from the corresponding hemiacetals.436 For that, the authors employed sulfur(VI) hexafluoride, which is an inexpensive and mild fluorinating reagent, along with a photocatalyst. As shown in Scheme 54H, fluorination of 2,3,4,6-tetra-O-acetyl-d-mannose 228 with SF6 (gas, 1 atm) in the presence of photocatalyst 4,4′- dimethoxybenzophenone (30 mol %) using a UV-A LED source (λmax = 365 nm) produced the corresponding mannosyl fluoride 229 in 70% yield (α/β = 13:1). Different sugar series with a variety of protecting groups were investigated, and glycosyl fluorides were obtained in yields of 43–97%. Further, the authors successfully showed gram scale formation of glycosyl fluorides in continuous flow systems and using electrochemical synthesis.437
6.1.4. Preparation from Thio- and Selenoglycosides.
Nicolaou and co-workers reported the direct conversion of phenylthio glycosides to glycosyl fluorides.333 Phenylthio glycosides were first treated with DAST followed by N-bromosuccinimide (NBS) to afford the corresponding glycosyl fluorides (Scheme 55A). This method is very effective, and several common protecting groups and O-glycosidic linkages were tolerated effortlessly. This method has created a basis for developing a two-stage activation and orthogonal strategies for glycan assembly (vide infra). Because of the similarity between thioglycosides and other chalcogen glycosides, Horne and Mackei converted phenylseleno and phenyltelluro glycosides to the corresponding glycosyl fluorides in the presence of DAST and NBS/NIS.438 The stereochemistry of obtained glycosyl fluorides was found to depend on various factors such as substituents at C-2, the nature of solvents, and the stereochemistry of starting materials.
Scheme 55.
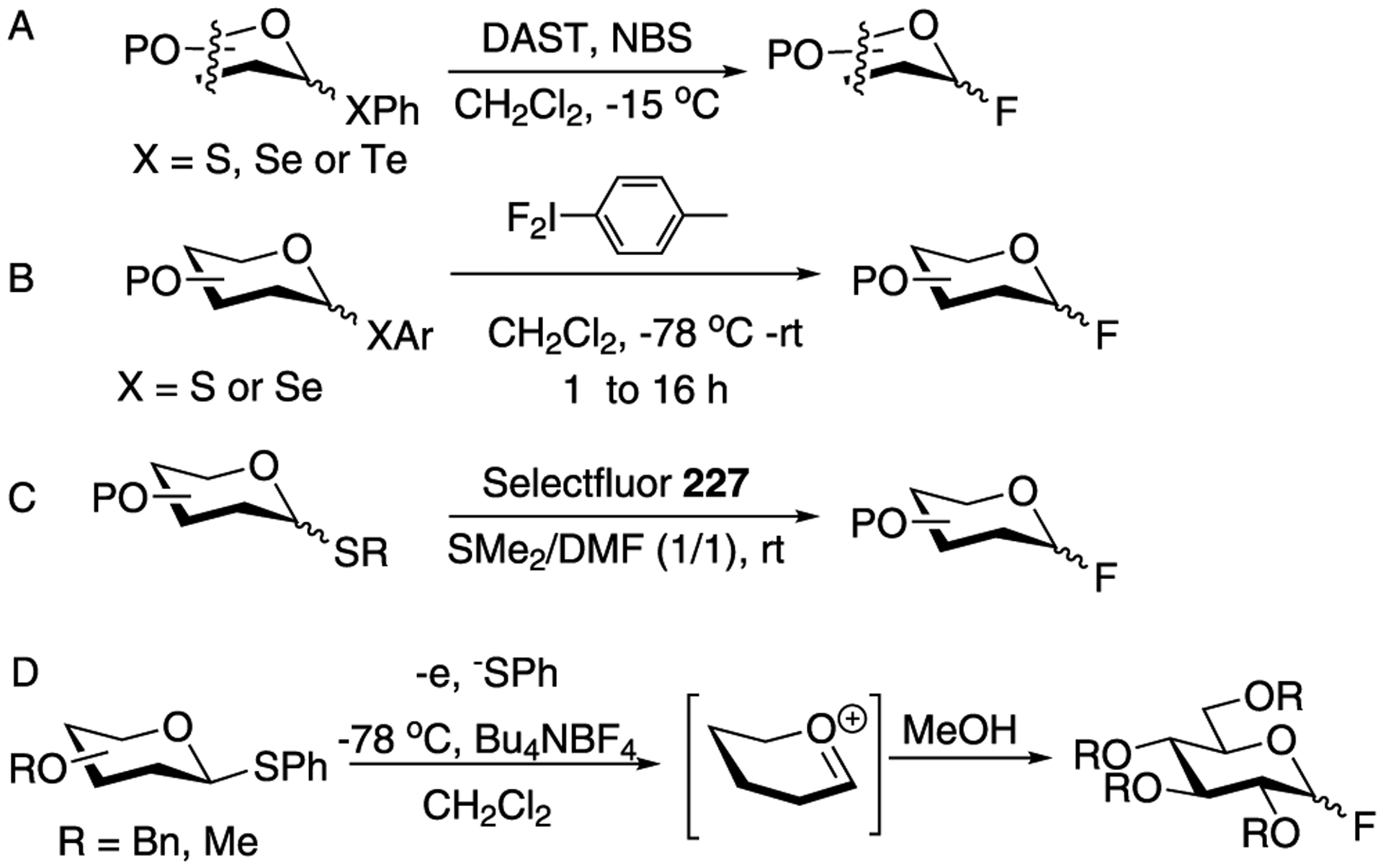
Synthesis of Glycosyl Fluorides from Chalcone Glycosides
Synthesis of several glucosyl fluorides was also achieved by treating arylthio glycosides with iodoarene difluoride reagents. Originally developed for noncarbohydrate substrates,439 Motherwell and co-workers then extended their studies to the synthesis of glycosyl fluorides from thioglycosides440 and selenoglycosides (Scheme 55B).441 As aforementioned, Wong’s approach to synthesizing fluorides with Selectfluor could be applied to hemiacetals, glycals, and thioglycosides as substrates.431 The latter required the use of dimethyl sulfide along with Selectfluor (Scheme 55C). Yoshida and co-workers attempted to isolate glycosyl cation intermediate by the “cation pool” method.442 Although this attempt was unsuccessful, the formation of glycosyl fluorides was achieved with the right combination of electrolytes.443 A low-temperature electrolysis reaction was conducted in the presence of Bu4NBF4 in CH2Cl2. Subsequent addition of methanol did not produce methyl glycosides, but rather afforded corresponding glycosyl fluorides in good yields (Scheme 55D). Subsequently, the authors figured out the one-pot glycosylation method by replacing Bu4NBF4 with other suitable electrolytes.
6.1.5. Preparation from Other Substrates.
Danishefsky and Gordon reported the synthesis of 3,4,6-tri-O-benzyl-β-d-glucopyranosyl fluoride 231 by treating 1,2-anhydro-3,4,6-tri-O-benzyl-α-d-glucopyranose 230 with tetrabutylammonium fluoride (TBAF, Scheme 56A).444 Though the yield was moderate (53%), the method is uniquely suited to produce the β-anomer only. Miethchen and co-workers prepared a fluorinating reagent by mixing anhydrous HF with acylating reagents to perform glycosyl fluoride synthesis and protection in one pot.445 Thus, a homogeneous mixture of HF/MeNO2/Ac2O (2/5/1.5, Method A) or HF/MeNO2/Piv2O (2/5/1.5, Method B) was found to transform 1,2:3,4-di-O-isopropylidene-6-O-methyl-α-d-galactopyranose 232 and other similar derivatives to the corresponding tri-O-acylated fluorides such as 233 and 234 as depicted in Scheme 56B. Protecting groups that are compatible with these conditions are alkyl, acyl, alkyl sulfone, etc.
Scheme 56.
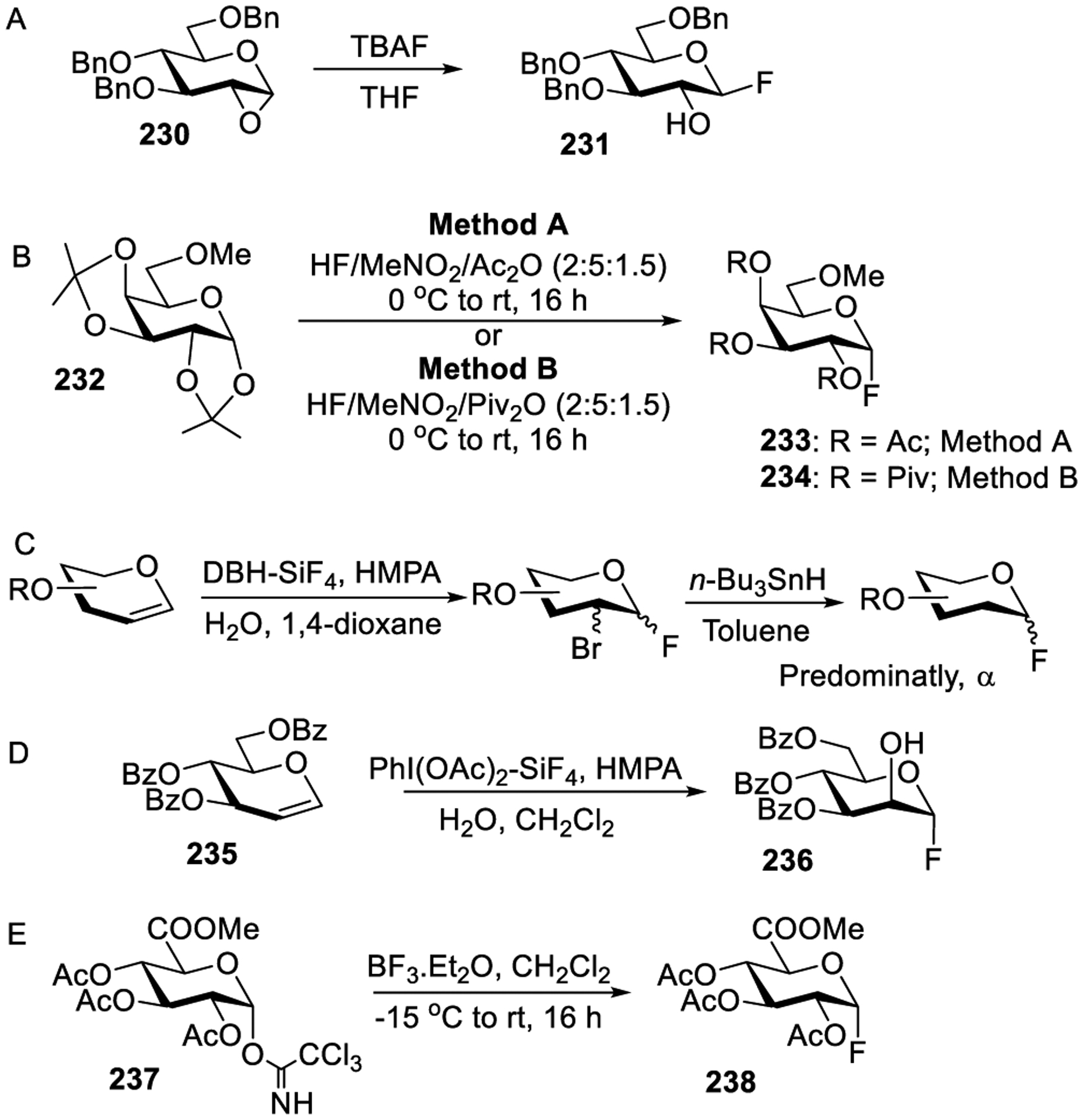
Synthesis of Glycosyl Fluorides from Other Starting Materials
As aforementioned, Selectfluor is another efficient method for converting glycals to the corresponding 2-deoxy-2-fluoro derivatives.431 Shimizu and co-workers developed a protocol for bromofluorination of olefins with silicon tetrafluoride and 1,3-dibromo-5,5-dimethylhydantoin (DBH),446 which was later applied to the synthesis of 2-deoxyglycosyl fluorides.447 This was accomplished via sequential bromofluorination of glycals followed by debromination as shown in Scheme 56C. First, bromofluorination of glycals derived from glucose, galactose, rhamnose, and fucose was conducted with SiF4, DBH, hexamethylphosphoric triamide (HMPA) in water/1,4-dioxane to produce the corresponding 2-bromoglycosyl fluoride derivatives. The latter were then subjected to debromination with tributyltin hydride (n-Bu3SnH) to afford the corresponding 2-deoxyglycosyl fluorides. The same authors also described hydroxyfluorination of glycals in the presence of PhI(OAc)2, SiF4, and HMPA. As shown in Scheme 56D, under these reaction conditions, α-mannosyl fluoride 236 was obtained from glucal precursor 235 in 73% yield.
Von Itzstein and co-workers observed the formation of glycosyl fluorides from O-imidates.448 As shown in Scheme 56E, the treatment of glycosyl imidate 237 with BF3-Et2O (1.0 equiv) resulted in the formation of α-fluoride 238 in 24%. This reaction was accompanied by the formation of a hemiacetal derivative because of competing hydrolysis. Jones and co-workers reported the synthesis of 1-fluorocellobiosyl fluoride by treating the corresponding diazirine derivative with xenon difluoride (XeF2).449
6.2. Glycosidation of Glycosyl Fluorides
Many different reagents and cooperative systems for the activation of glycosyl fluorides have been developed. To streamline the discussion, we chose to divide the activating reagents into different categories based on their fluorophilic nature (Table 2). The discussion begins from tin salts that were very instrumental for understanding the metal salt involvement in splitting the anomeric C–F bond and investigating the effects of different additives and the counteranions. We will then focus the discussion on how those studies enabled scientists to develop improved methods, and how the improved methods enhanced our synthetic capabilities. Recent studies dedicated to conformational analysis of glycosyl cations generated from glycosyl fluorides significantly enhanced our understanding of processes behind typical glycosylation reactions.450
Table 2.
Activation of Glycosyl Fluoride Donors
| promoter | additive | |
|---|---|---|
| tin- and silicon-based reagents | ||
| SnCl28,420 | AgClO4,8,451 TrClO4,420 AgOTf,451,452 AgB(C6F5)4451,453,454 | |
| SnCl4455 | AgB(C6F5)4453 | |
| SnF4456 | ||
| R3SnCl or R2SnCl2457 | AgClO4457 | |
| Sn(OTf)2458 | BF3-Et2O, TiCl4, La(OTf)3, Yb(OTf)3 or La(ClO4)3-nH2O459 | |
| SiF4404 | ||
| CF3SO3SiMe3404,456,460 | ||
| SiCl4 or Ph3SiCl453 | AgB(C6F5)4453 | |
| boron-, aluminum-, and gallium-based reagents | ||
| BF3-Et2O423,455,461 | ||
| TrB (C6F5)4462,463 | ||
| HB(C6F5)4464,465 | ||
| B(C6F5)3466 | ||
| AlMe3455 | ||
| Me2GaCl, Me2GaOTf or MeGa(OTf)2467 | ||
| main group metal salts | ||
| MgBr2-Et2O455 | ||
| LiClO4468,469 | CsF468,469 | |
| CaBr2 or Ca(NO3)2470 | NMe3470 | |
| Ca(OTf)2470 | NMe3470,471 | |
| Ca(OH)2471 | ||
| transition metal salts | ||
| TiF4456,460 | AgClO4,472 AgBF4473 | |
| Cp2TiCl2474 | AgClO4474 AgB(C6F5)4453 | |
| TiCl2453 | AgB(C6F5)4453 | |
| Cp2ZrCl2474 | AgClO4474,475 AgOTf, AgBF4, AgPF6 or AgSbF6,475 AgB(C6F5)4453 | |
| SO4/ZrO2476 | ||
| Cp2HfCl2474 | AgClO4,474,477,478 AgB(C6F5)4453 | |
| Hf(OTf)4479 | ||
| Cu(OTf)2480 | ||
| lanthanide metal salts | ||
| Yb(OTf)3481 | K2CO3,481,482 CaCO3, K2CO3/ ZnCl2 or K2CO3/Ba(ClO4)2481 | |
| YbCl3481 | CaCO3481 | |
| Yb-Amberlyst-15483 | ||
| Yb[N(O2SC4F9)2]3484 | ||
| La(ClO4)3-7H2O481,482 | K2CO3481,482 | |
| La(ClO4)3-nH2O, Pr(ClO4)3-nH2O, or Eu(ClO4)3-nH2O482 | K2CO3482 | |
| Ce(ClO4)3-nH2O482,485 | K2CO3482,485 | |
| anhydrides, protic acids, and other reagents | ||
| Tf2O460 | ||
| TfOH465,486–488 | ||
| HClO4, HOSO2C4F9, HNTf2 or HSbF6464–465 | ||
| RN+B(C6F5)4, RN+OTf, RN+SbF6, RN+BF4 or RN+ClO4489 | ||
| solvolysis490 | ||
6.2.1. Activation with Group 14-Based Reagents (Tin and Silicon).
Mukaiyama and co-workers discovered that glycosyl fluorides can be activated by a cooperative effect of tin(II) chloride (SnCl2, stannous chloride) and silver perchlorate (AgClO4) to afford glycosides in excellent yields.8 The glycosylation under these reaction conditions often proceeds in a stereoselective manner affording 1,2-cis α-glycosides predominantly. As shown in Scheme 57A, glycosidation of 2,3,4,6-tetra-O-benzyl-β-d-glucopyranosyl fluoride 239 with various aliphatic and sugar acceptors provided the respective products in good yields of 76–96% and respectable stereoselectivity. A similar approach was extended to the synthesis of 1,2-cis glycofuranosides.420 However, these reactions were predominantly 1,2-trans stereoselective. Interestingly, replacing silver perchlorate with trityl perchlorate (TrClO4) additive allowed to obtain the respective products with predominant 1,2-cis stereoselectivity (Scheme 57B).
Scheme 57.

Glycosidation of Glycosyl Fluoride in the Presence of Tin(II)-Based Reagents
Subsequently, Mukaiyama and co-workers extended their study to SnCl2 in combination with AgB(C6F5)4 as a stable and useful reagent for generating the active catalyst.451 This study also revealed that MS 5 Å additive (3 g/mmol) is necessary to achieve efficient activations. The cooperative catalytic activation was achieved by the combined use of SnCl2 and AgB(C6F5)4, 20 mol % each, in toluene. It was suggested that SnB(C6F5)4Cl is an active catalyst for the activation of glycosyl fluoride 240 (Scheme 57C). An important aspect of this cooperative catalyst is that it does not work well in polar solvents, which was attributed to deactivation of Lewis acid by coordination to stannous cation. This cooperative system led to a successful installation of different linked disaccharides. For example, Yamada and co-workers activated the 3,6-O-bridged glucosyl fluoride donor 241 under these reaction conditions to obtain β-linked products, for example 242, with complete stereoselectivity (Scheme 57D).454 The key to this exclusive β-stereoselectivity was due to the following two factors. First, the ability of the bridging positions C-3 and C-6 to force the sugar substituents to lock into the axial positions. Second, isomerization propensity to produce thermodynamically stable anomer. Upon glycosylation, α/β-glycosides, such as 242, are produced along with HB(C6F5)4, and the latter can catalyze the anomerization cycle of the α- to β-glycosides and, along with SnClF, induce the regeneration of SnB(C6F5)4Cl. Mechanistic investigation confirmed that SnB(C6F5)4Cl is the active catalyst for glycosylation.
Beyond initial studies by Mukaiyama and co-workers wherein the activation was achieved with SnCl2,420 several other tin salts are investigated such as SnF2, SnBr2, Sn(OAc)2. However, none of the salts provided satisfactory results without a copromoter.8,420 Similar observations were made by Nicolaou for SnCl4455 and by Shibasaki for Sn-(OSO2CF3)2.458 Thiem and co-workers reported the activation of glycosyl fluorides with tin(IV) fluoride (stannic fluoride, SnF4) without any additive.456 Glycosidation of acylated glycosyl fluoride β-221 having 2-participating group with glycosyl acceptor 55 or its 6-O-TMS-protected counterpart 243 in the presence of SnF4 produced 1,2-trans glycoside 244 (Scheme 58A).
Scheme 58.
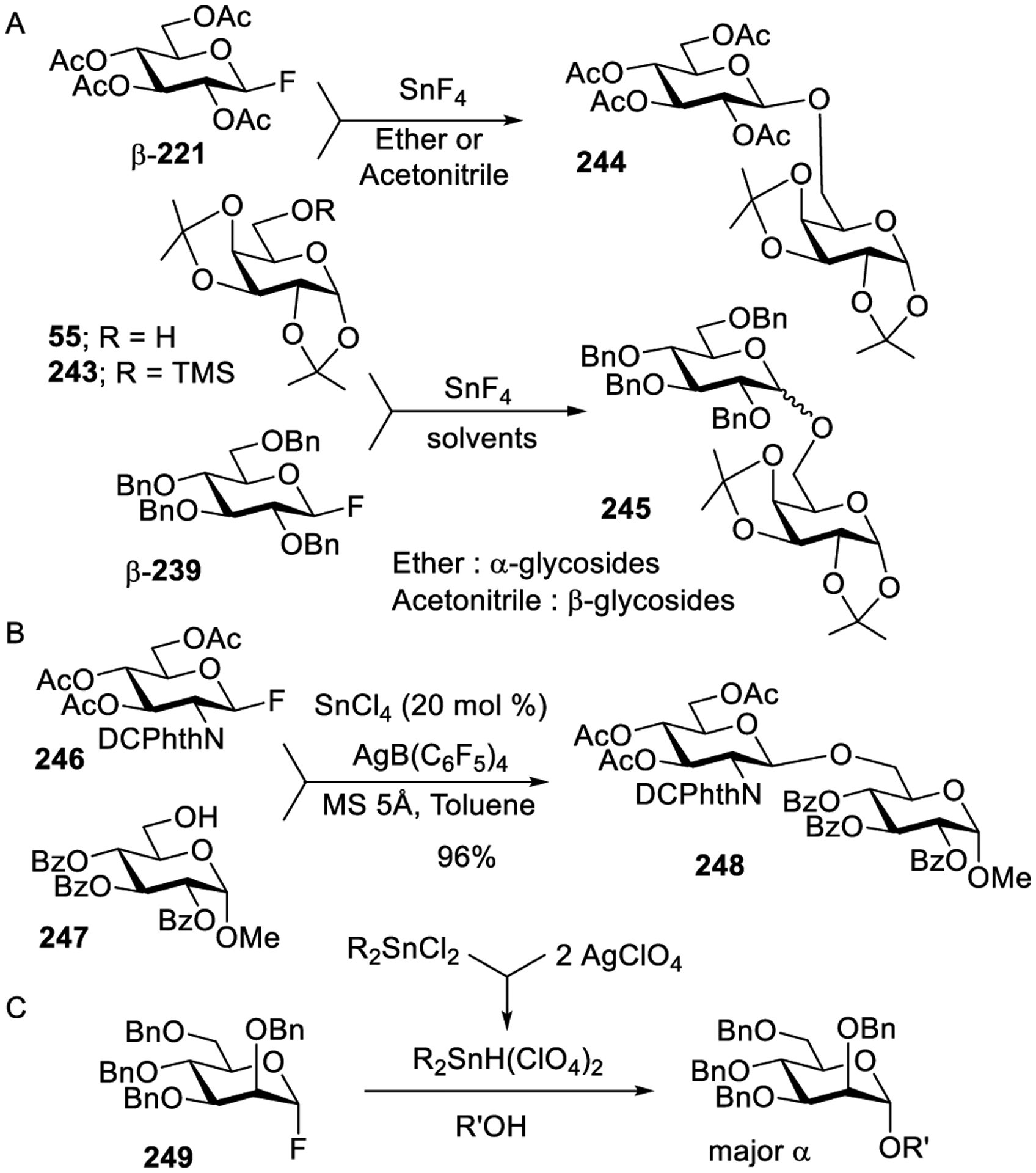
Glycosidation of Glycosyl Fluoride in the Presence of Other Tin-Based Reagents
However, the yields of glycosides were below the preparative value (42–48%). Conversely, glycosidation of the benzylated glycosyl fluoride β-239 was more efficient and produced glycoside 245 in up to 93% yield, and the distribution of the products was found to be solvent-dependent, α-linked in ether or β-linked disaccharides in acetonitrile. It was then discovered that SnCl4 is able to efficiently activate glycosyl fluorides in the presence of AgB(C6F5)4.453 Surprisingly, SnCl2 in combination with AgB(C6F5)4 (20 mol % each), only produced the expected disaccharide 248 in trace amounts when glycosyl donor 246 was glycosidated with acceptor 247. In comparison, SnCl4 in combination with AgB(C6F5)4 (20 mol % each) produced disaccharide 248 in an excellent yield (Scheme 58B). Therefore, SnCl4 was employed for further optimization of the amount of AgB(C6F5)4 that helped to establish 20 mol % of SnCl4 and 40 mol % of AgB(C6F5)4 as the most effective catalytic system for activating glycosyl donor 246 in toluene at −20 °C. Finally, the optimized reaction conditions were extended to several disarmed glycosyl donors as well as acceptors to synthesize the desired glycosides in excellent yields.
A metallocene-Ag salt-catalyzed approach was extended to organotin(IV) compounds to generate new activators for glycosyl fluoride donors.457 Two types of organotin compounds, R2SnCl2 and R3SnCl (R = aryl or alkyl), were used in the presence of AgClO4. Whereas glycosidation of fluoride donor 249 in the presence of R2SnCl2 and AgClO4 was almost instantaneous, reactions in the presence of R3SnCl and AgClO4 proceed slower. R2Sn(ClO4)2 was proposed as the active catalyst for these reactions (Scheme 58C), and the stereoselectivity was found to depend on the nature of the alkyl/aryl ligands of tin.
Noyori co-workers presented the synthesis of glycosides by coupling glycosyl fluorides and trimethylsilyl ether-protected glycosyl acceptors in the presence of tetrafluorosilane (SiF4) or trimethylsilyl triflate (TMSOTf).404 Glycosidation of α- and β-glycosyl fluoride 239 was successfully achieved and the selectivity of glycoside products was found to depend on the type of solvents employed (Scheme 59A). Expectedly, acetonitrile predominantly produced 1,2-trans glycosides whereas ether favored 1,2-cis glycosides irrespective of anomeric conformation of glycosyl fluorides.
Scheme 59.
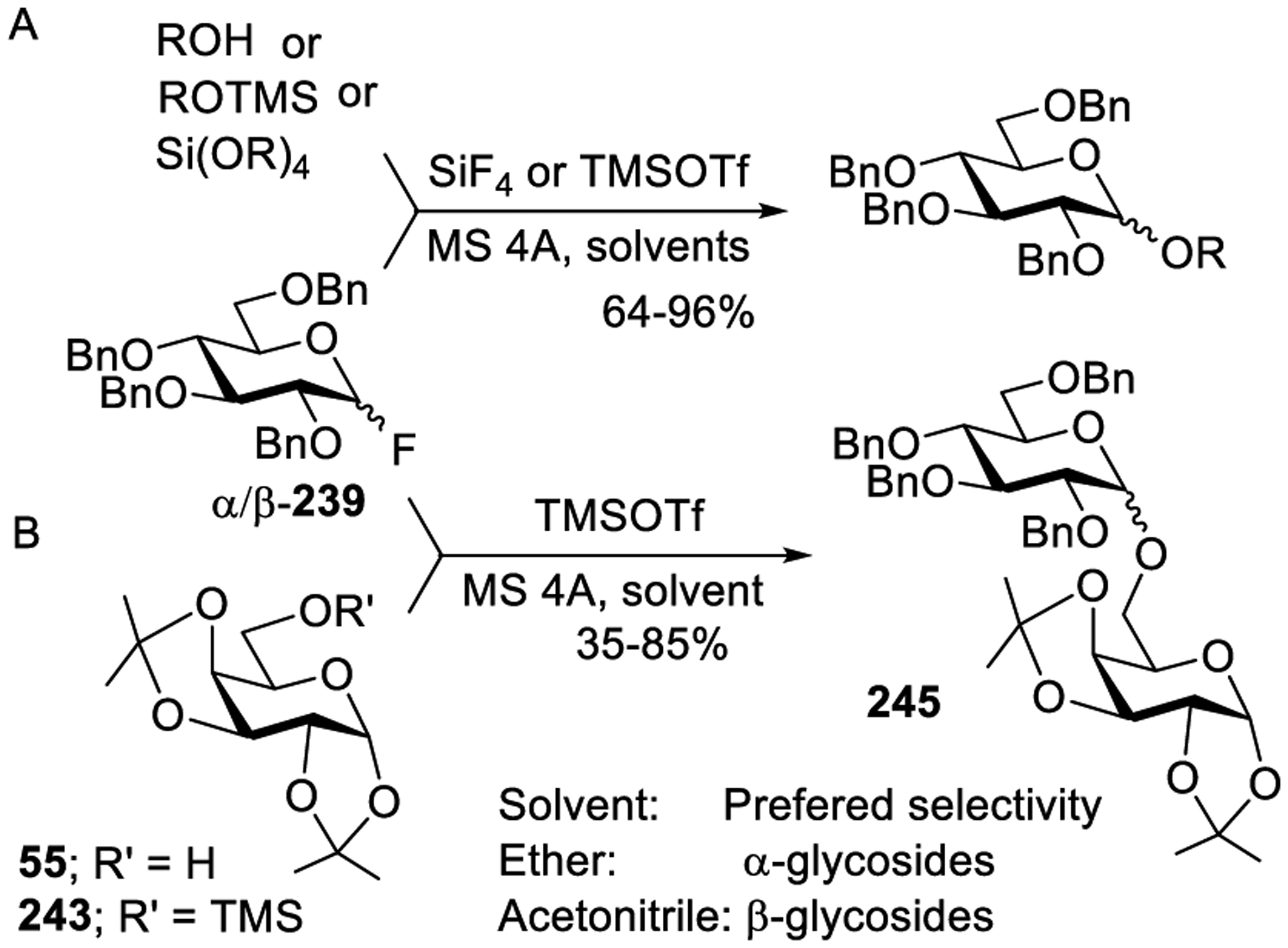
Glycosidation of Glycosyl Fluoride in the Presence of Silicon-Based Reagents
Further, it was confirmed that glycosides do not undergo anomerization over the course of the reaction. The efficiency of glycosylation reaction with glycosyl fluorides depends on the affinity of silicon atom to fluorine. Similarly, Thiem and co-workers also reported the activation of per-benzylated glycosyl fluorides in the presence of TMSOTf.456 For example, glycosidation of donor 239 with glycosyl acceptors 55 or 243 produced disaccharide 245. The yield of 245 could fall below preparative standards and the stereoselectivity of glycosides was also solvent dependent, as shown in Scheme 59B. An efficient activation was observed when glycosylation was conducted in the presence of SiCl4 in combination with AgB(C6F5)4 (20 mol % each). However, only moderate efficiency was achieved when the same glycosylation was conducted in the presence of Ph3SiCl.453
6.2.2. Activation with Group 13-Based Reagents (Boron, Aluminum, and Gallium).
Nicolaou and co-workers described the synthesis of O-, S-, and N-glycosides employing glycosyl fluoride donors in the presence of BF3-Et2O (0.3 equiv).455 This activator was found to be effective in most cases, however, there were specific cases where other metal promoters were found to be more effective (vide infra). Following essentially the same principle, Kunz and Waldman synthesized a variety of pyranosides and furanosides. Among these, coupling β-2-fluoro-neuraminic acid donor 250 with diacetone galactose acceptor 55 in the presence of BF3-Et2O afforded disaccharide 251 in 44% yield (α/β = 1:5, Scheme 60A).423,461 Recently, BF3-Et2O-mediated glycosylation was revisited by Fukase and co-workers wherein it was discovered that an efficient activation of glycosyl fluorides can be achieved in the presence of only 1 mol % of BF3-Et2O in a nitrogen-filled glovebox.491 An interesting feature of this reaction is that it does not require any dehydrating reagent compared to most glycosylation reactions. Armed per-benzylated glycosyl fluoride 239 and disarmed per-benzoylated glycosyl fluoride 240 both were successfully glycosidated with glycosyl acceptor 36 in the presence of 1 mol % of BF3-Et2O in nitrogen-filled glovebox and to produce disaccharides 165 in 91% yield (α/β = 1:5/1) and 102 in 85% yield (β only), respectively (Scheme 60B). When the reaction was conducted in a polytetrafluoroethylene vessel, the yield has drastically declined suggesting the involvement of a glass vessel in glycosylation reaction. Earlier, a similar observation of glass vessel’s involvement was made by Pedersen and co-workers wherein the formation of SiF4 was suggested to occur from HF and glass vessel.492 On the basis of the observations, the authors proposed a tentative mechanism according to which the anomeric activation is initiated by BF3-Et2O that produces HF. The latter then reacts with glass and produces SiF4, which acts as the activator of the anomeric fluoride.
Scheme 60.
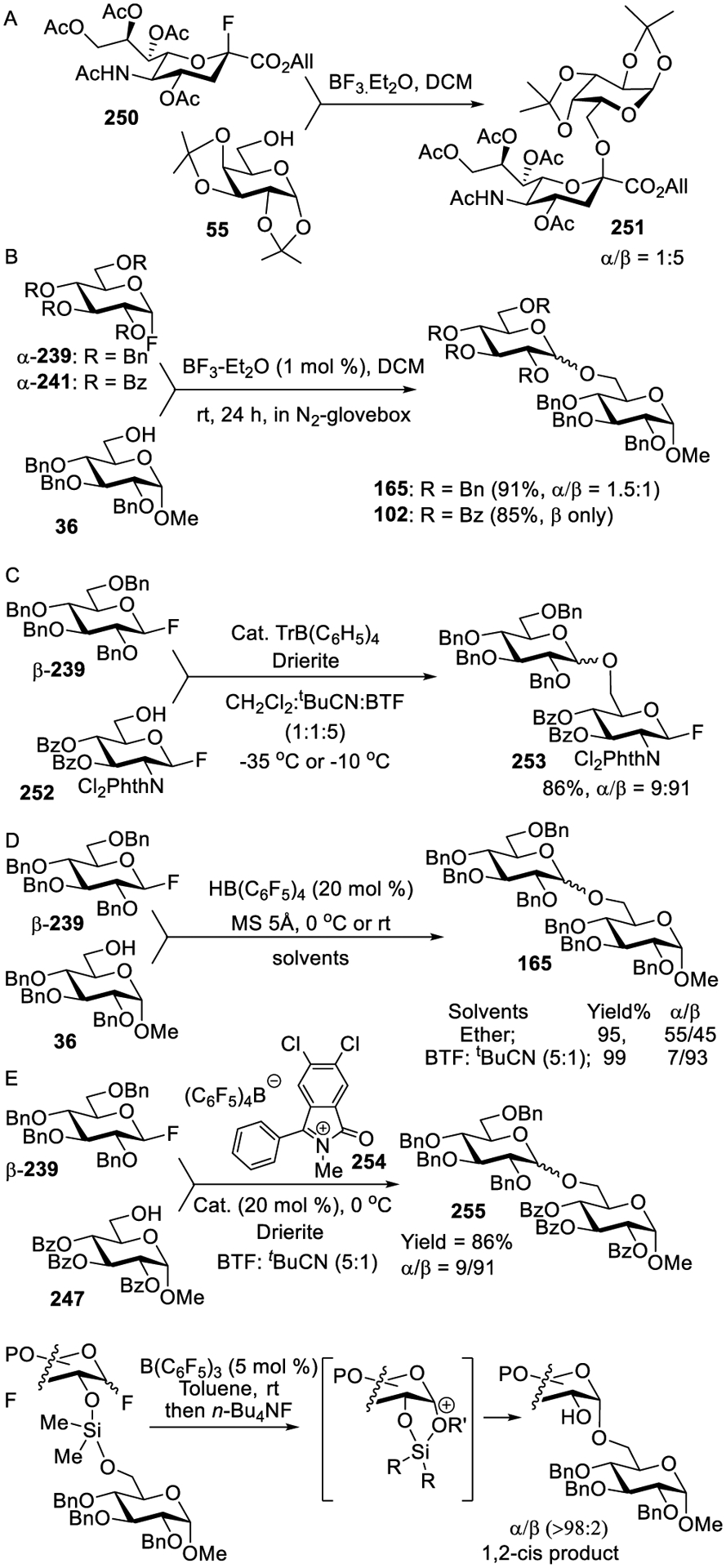
Glycosidation of Glycosyl Fluoride in the Presence of Boron-Based Reagents
Mukaiyama and Takeuchi reported a stereocontrolled catalytic activation of glycosyl fluorides in the presence trityl tetrakis(pentafluorophenyl)borate.462 Optimization of the reaction conditions such as catalyst loading, solvents, and temperature led to the establishment of a reaction conditions, which favored 1,2-trans β-glycosides. Differently linked disaccharides were synthesized in presence of 10–20 mol % of trityl tetrakis(pentafluorophenyl) borate and Drierite in pivalonitrile-benzotrifluride (1:5) at −10 to 0 °C. Similarly, β-stereoselective glycosylation was observed with tetrabenzylated galactosyl fluoride and various glycosyl acceptors.486 Further, TrB(C6F5)4-catalyzed glycosylation was successfully applied to an armed-disarmed glycosylation concept, wherein armed glycosyl fluoride donor β-239 was coupled with disarmed glycosyl fluoride acceptor 252 to afford the corresponding disaccharide 253 in a good yield (Scheme 60C).486 Several other disarmed fluoride acceptors were successfully glycosylated with armed glycosyl donor β-239.
Tetrakis(pentafluorophenyl)boric acid-catalyzed activation was also investigated. As shown in Scheme 60D, reaction of glycosyl fluoride donor β-239 with acceptor 36 produced disaccharide 165 with practically no stereoselectivity.464 Solvent investigation studies showed that HB(C6F5)4-catalyzed glycosylation in a mixture of solvent, i.e. benzotrifluoride/tert-butyl nitrile (5:1), produced disaccharides with excellent β-selectivity. The above observations suggested that the counteranion together with solvents play a crucial role in directing the stereoselectivity of the reaction. β-Stereoselective glycosidation of glycosyl fluoride β-239 was extended with a variety of glycosyl acceptors. In all cases disaccharides were obtained in high yields and with excellent β-selectivity (up to α/β = 2/98). HB(C6F5)4-catalyzed α-stereoselective glycosylation was also achieved when a glycosyl fluoride having diethylthiocarbamoyl group at C-6 was used as the glycosyl donor and diethyl ether was used as the reaction solvent.493
Mukaiyama and co-workers investigated the activation of glycosyl fluorides in the presence of catalytic carbocationic species paired with counterion such as tetrakis (pentafluorophenyl) borate or other anions.489 Glycosidation of glycosyl fluoride β-239 with glycosyl acceptor 247 in tBuCN in the presence of a cationic catalyst 254 produced disaccharide 255 in 86% yield with excellent β-selectivity (Scheme 60E). Boron-based catalytic activation of glycosyl fluorides was extended by Montgomery and co-workers for silyl ethers as glycosyl acceptors.466 The Frustrated Lewis Pair (FLP) characteristics of B(C6F5)3494,495 and B(C6F5)3-catalyzed trifluoromethylation via fluoride-rebound mechanism496,497 inspired the idea for the coupling glycosyl fluoride with silyl ethers in the presence of B(C6F5)3.
Efficient coupling of numerous glycosyl fluorides equipped with a 2-O-acyl group with silyl ether acceptors in the presence of 5 mol % of B(C6F5)3 rapidly provided the corresponding 1,2-trans-linked glycosides in good to excellent yields at rt. Exploitation of silicon-tethered intramolecular aglycone delivery (IAD) method was employed to construct 1,2-cis linkages. As shown in Scheme 60F, the glycosidation of the tethered donor–acceptor pair led to glycosides with excellent stereoselectivity. It was assumed that the reaction proceeds via the key five-membered intermediate that collapsed to produce 1,2-cis-linked products with concomitant removal of the silicon tether.
Nicolaou and co-workers described the synthesis of O-, S-, and N-glycosides employing glycosyl fluoride donors in the presence of AlMe3.455 Kobayashi and co-workers utilized different methylgallium chlorides and triflates that were found to activate glycosyl fluorides.467 Simple aliphatic alcohols produced glycosides in excellent yields (77% to quantitative) with moderate stereoselectivity; however, switching to hindered or sugar alcohols led to decrease in yields.
6.2.3. Activation with Main Group Metal Salts (Magnesium, Lithium, and Calcium).
Nicolaou and co-workers investigated the synthesis of O-, S-, and N-glycosides employing glycosyl fluoride donors in the presence of MgBr2-Et2O.455 Waldmann and Bohm discovered neutral reaction conditions for the activation of glycosyl fluorides.468 As shown in Scheme 61A, fluoride donor 256 was reacted with glycosyl acceptor 257 in a 1 M solution of LiClO4 in the presence of CsF as the proton scavenger to afford disaccharide 258 in 98% yield with exclusive α-stereoselectivity. Different types of glycosyl acceptors produced both glycosides with predominant α-stereoselectivity. This methodology was later applied to glycosidation of benzylated glucosyl fluorides.498 The glucosides were obtained in moderate yields, but increasing the concentration of LiClO4 promoted the formation of the respective 1,6-anhydro derivative. Further, acylated glucosyl fluorides were tested; while pivaloyl-protected fluoride produced 1,2-trans-linked products, acetylated fluoride was unreactive.469
Scheme 61.
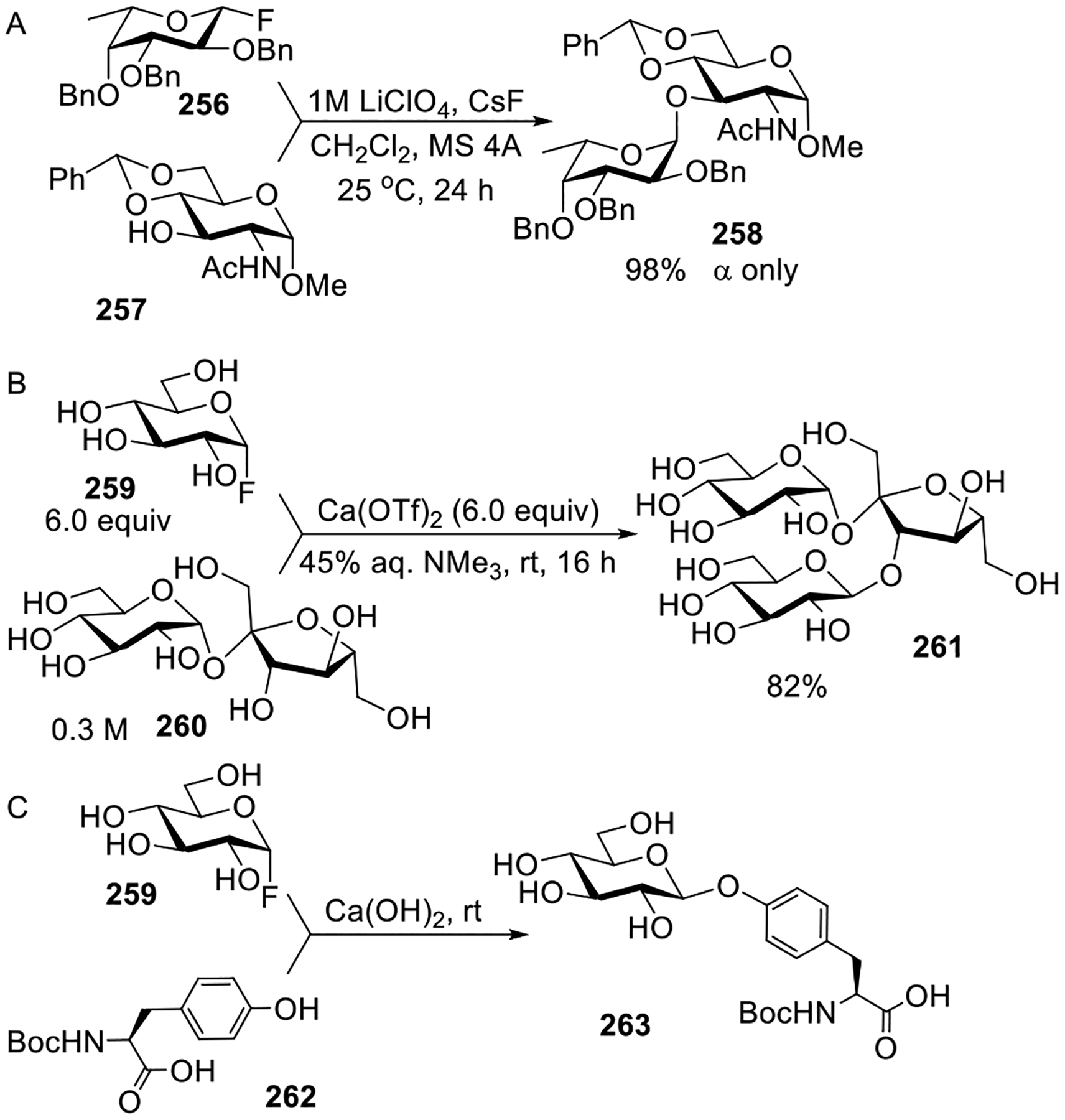
Glycosyl Fluoride Activation with Main Group Metal Salts
Glycosylations with unprotected sugars in aqueous solutions are challenging.499–501 Encouraged by the seminal work of Jencks502–504 and Bennett,490,505 Miller and co-workers described the activation of glycosyl fluorides in the aqueous phase to produce glycosides.470 After investigation of a number of metal salts and careful optimization of the reaction conditions, glycosidation of glycosyl fluoride 259 (6.0 equiv) with sucrose 260 (1.0 equiv, 0.3 M) was accomplished in the presence of Ca(OTf)2 (6.0 equiv) in aqueous trimethylamine to produce trisaccharide 261 in a good yield of 82% (Scheme 61B). The unique feature of this method is its β-stereoselectivity and regioselectivity for the 3′-position of sucrose. Extended glycosylation and NMR studies, similar to those described by Davies506–508 and by O’Leary,509–512 revealed that the unprecedented regioselectivity and reactivity of sucrose and its derivatives could be related to the hydrogen-bonding network. This network could have a significant impact on nucleophilicity of different hydroxyls of sugars and, hence, influence their interaction with Ca2+ during glycosylation.
In another report, Miller and co-workers extended the aqueous phase glycosidation of glycosyl fluorides to phenolic derivatives of biological relevance.471 Glycosylation of amino acids in aqueous phase remains an immense task,513–518 and calcium(II)-mediated aqueous phase glycosylation in the presence of Ca(OH)2 worked very well. As depicted in Scheme 61C, glycosylation between glycosyl fluoride 259 (3.0 equiv) and N-Boc-protected tyrosine derivative 262 (1.0 equiv) 1.0 M in H2O in the presence of Ca(OH)2 (3.0 equiv) produced glycoside 263 with complete β-stereoselectivity. The concentration dependence of the reaction rate offered strong evidence of the SN2-like mechanism.109,503,505,519 Subsequently, the standardized glycosylation conditions were applied to various phenolic derivatives wherein electron-rich (less acidic) phenolic derivatives turned out to perform better compared to the electron-deficient ones.
6.2.4. Activation with Transition Metal Salts (Titanium, Zirconium, Hafnium, and Copper).
Thiem and co-workers showed the activation of glycosyl fluorides with titanium fluoride (TiF4).456 Per-acetylated, per-benzylated, and 2-deoxyglycosyl fluorides were investigated. These studies evolved into investigation of the cooperative catalyst comprising titanium and silver salts.472 To enhance the stereoselectivity, the authors investigated brominated glycosyl fluoride donors depicted in Scheme 62A. Glycosidation of mannosyl fluoride in the presence of TiF4 and AgClO4 produced α-disaccharides irrespective of the solvents employed. When mannosyl fluoride 264 was reacted with glycosyl acceptor 265 in acetonitrile, diethyl ether, or hexane α-disaccharide 266 was obtained in a good yield and exclusive stereoselectivity. Montgomery and co-workers reported facile construction of 1,2-cis glycosidic linkages by means of silicon-directed intramolecular aglycone delivery (IAD) method (vide supra) in the presence of TiF4 and AgBF4.473 As depicted in Scheme 62B, silyl-tethered precursor 267 was subjected to intramolecular glycosylation to give the corresponding glycoside 268 in 78% yield. Functional groups such as ketones, hydroxy groups, silyl ethers, and esters were shown to remain unaffected during these transformations.
Scheme 62.
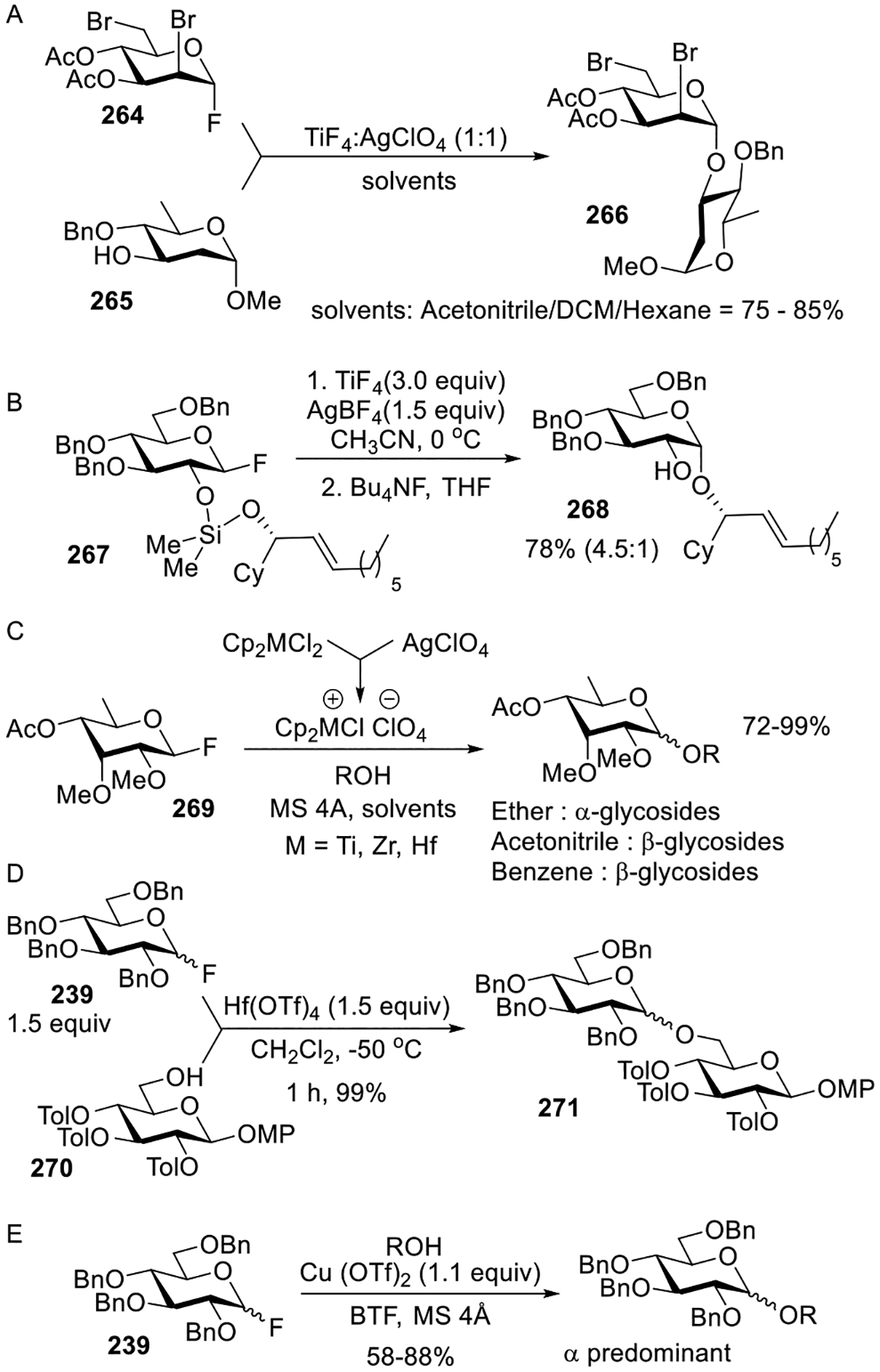
Glycosyl Fluoride Activation with Transition Metals Salts
Suzuki and co-workers discovered a new cooperative promoter system Cp2MCl2-AgClO4 (M = Zr, Hf, Ti) for glycosyl fluoride donors, which enabled very promising glycosylations (Scheme 62C).474 A controlled experiment showed that Cp2MCl2 alone is practically inert in glycosylations, and it was suggested that Cp2MCl2 and AgClO4 are required in molar equivalent to drive the reaction to completion. A rough order of reactivity of Zr ≥ Hf ≫ Ti was estimated by glycosylation between glycosyl fluoride 269 and cyclohexyl methanol. Later, the authors explored the activation of anomeric fluoride of amino sugars with Cp2MCl2-AgClO4 promoter systems.477 In this application, Cp2HfCl2-AgClO4 was superior to its Zr counterpart. Subsequently, the cooperative catalyst Cp2MCl2-AgClO4 (M = Zr, Hf) was utilized for the total synthesis of mycinamicin IV by the stereoselective glycosidation of D-mycinosyl and D-desosaminyl fluorides with mycinamicin VII.520
Further investigations revealed that Cp2MCl2-AgClO4 (M = Hf, Zr) in a ratio of 1:2 is more efficient for the activation of glycosyl fluorides.478,521 The authors suggested that the activation of glycosyl fluorides depends on the fluorophilicity of early transition metals and their ability to form the reactive promoter shown in Scheme 62C. The driving force for the glycosylation reaction is believed to be due to the formation of a strong M-F bond. To enhance the efficiency of glycosylation of hindered alcohols, several silver salts (AgBF4, AgOTf, AgPF6, and AgSbF6,) in combination with Cp2ZrCl2 were examined, and AgBF4 was found to be superior in this application, both in terms of reactivity and stereoselectivity.475
Matsumura and co-workers reported the stereocontrolled mannosylation in the presence of sulfated zirconia (SO4/ZrO2) and MS 5 Å.476 The advantage of solid acid-promoted glycosylation is that the acid can be recovered by filtration and then reused. Also, 2-deoxy-α-d-glucopyranosyl fluoride was investigated in the presence of sulfated zirconia.522
Ito and Manabe investigated hafnium(IV) reagents such as Hf(OTf)4, a well-known Lewis acid used for many synthetic transformations,523–526 for the activation of glycosyl fluorides.479 Optimization of the reaction conditions led to an effective glycosylation reaction between glycosyl donor 239 and acceptor 270 in CH2Cl2 to afford disaccharide 271 in 99% yield (α/β = 20:80, Scheme 62D). Participating solvents were found to influence the stereochemistry of glycosylated products,404,527,528 a mixture of dioxane/toluene provided mainly α-glycosides, and β-selectivity was observed in acetonitrile. Glycosyl fluorides of other sugar series with different protection patterns were glycosylated with primary and secondary glycosyl acceptors under the identical reaction conditions and the corresponding glycosides were obtained in good to excellent yields (58–94%). The method was then applied for solid-phase oligosaccharide synthesis (vide infra).
Yamada and Hayashi reported activation of glycosyl fluorides in the presence of Cu(OTf)2.480 As depicted in Scheme 62E, glycosyl fluoride 239 was reacted with various alcohols in the presence of the stoichiometric amount of Cu(OTf)2 in benzotrifluoride at temperatures above 60 °C to afford the corresponding glycosides in good yields with predominant α-stereoselectivity.
6.2.5. Activation with Lanthanide Metal Salts (Lanthanum, Cerium, Ytterbium, etc.).
Relying on a high dissociation energy of the Ln–F bond,529,530 Shibasaki and co-workers employed rare earth metal salts for the activation of glycosyl fluorides.481 Ytterbium(III) triflate or chloride were found to be very effective for the activation of different glycosyl fluorides (Scheme 63A). The authors suggested that the reaction proceeds through the formation of the oxacarbenium ion intermediate since the stereoselectivity of glycosides was reaction solvent dependent. Predominant 1,2-trans stereoselective glycosylation was observed in the presence of Yb(OTf)3 and K2CO3 in acetonitrile, whereas 1,2-cis stereoselectivity was achieved in a similar reaction conducted in the presence of Yb(OTf)3 and CaCO3 in ether. The authors also showed that the addition of Lewis acid such as ZnCl2 or Ba(ClO4)2 to ytterbium(III)-promoted glycosylation enhances the rate and improves the yield of glycosides.481 Wang and co-workers immobilized lanthanide ions on ion exchange resins and utilized these catalysts in several chemical modifications.483 Glycosylation reaction between glycosyl fluorides and methanol was shown to be promoted by Yb-Amberlyst-15. Interestingly, complete inversion at the anomeric center was observed in these reactions. Inazu and co-workers showed the activation of glycosyl fluorides in the presence of catalytic amounts of ytterbium(III) tris[bis(perfluorobutylsulfonyl) amide] (Yb[N(O2SC4F9)2]3, 5 mol %).484
Scheme 63.
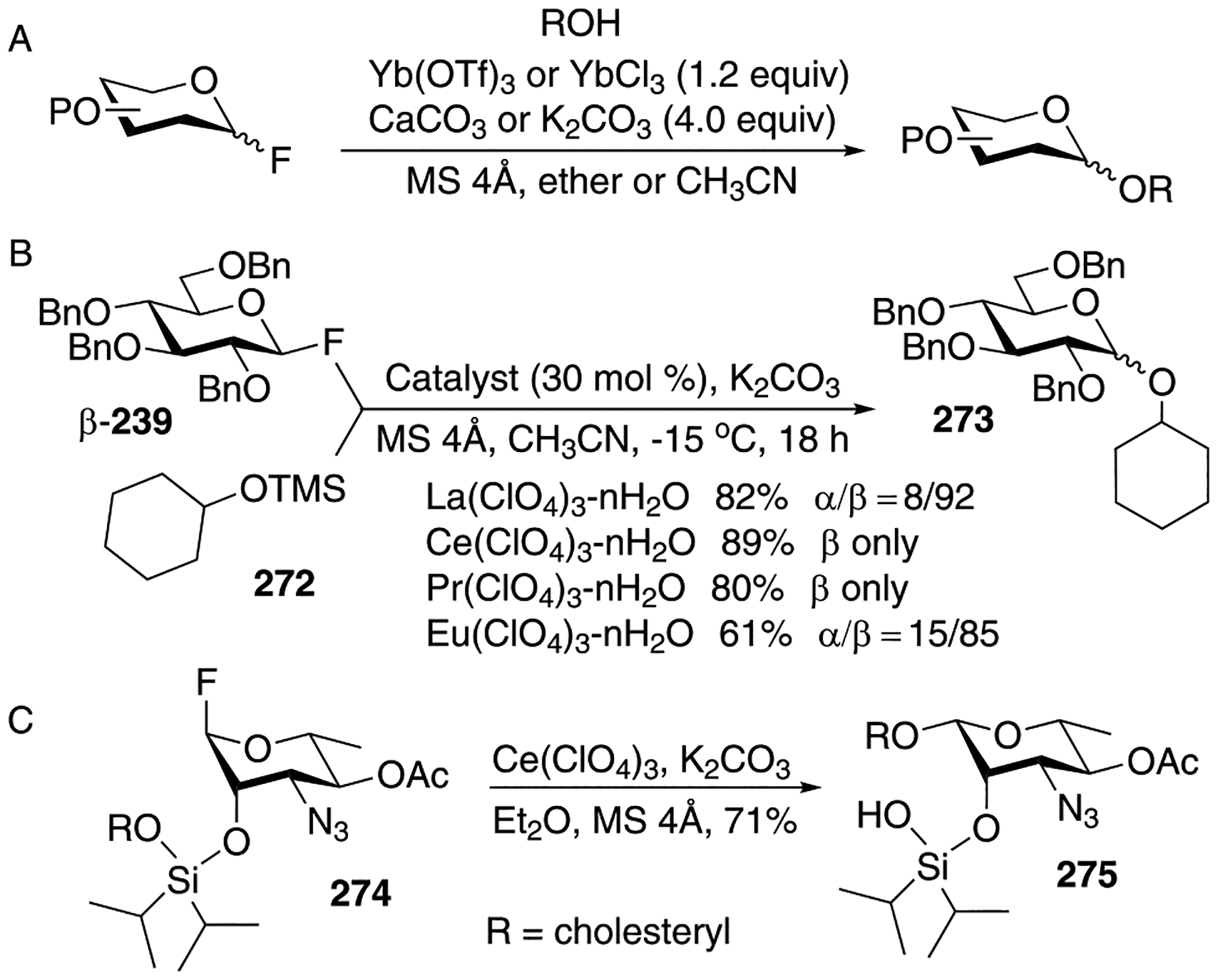
Glycosyl Fluoride Activation with Lanthanide Metals Salts
Other rare earth metals were found to promote the glycosidation of glycosyl fluorides.481,482 Detailed investigation led to the development of a catalytic activation of glycosyl fluorides with trimethylsilyl ether acceptors.482 Thus, coupling of glycosyl fluoride β-239 with trimethylsilyl hexyl ether 272 in the presence of rare earth metal perchlorate hydrates in acetonitrile produced product 273 in good yields and with high β-stereoselectivity (Scheme 63B). Catalytic activation of glycosyl fluorides with rare earth metal perchlorates was attributed to the increased fluorophilicity of metal cations.531–534 Further, 19F NMR experiments confirmed the formation of trimethylsilyl fluoride, which suggested the formation of oxacarbenium ion resulted from the interaction of naked metal cation (perchlorate) with anomeric fluoride which was then attacked by trimethylsilyl ether to produce glycosides, trimethyl fluoride and regenerates metal perchlorates to run the catalytic cycle. It was also suggested that glycosylation occurred with a certain range of ionic radii, it was supported by the failed glycosylation by metal perchlorate hydrates of gadolinium, holmium, ytterbium, and yttrium. Differently protected glycosyl fluorides were couples with several alcohols in the presence of La(ClO4)3-nH2O to obtain the respective glycosides in good to excellent yields.535 Packard and Rychnovsky investigated the activation of anomeric fluorides in the presence of Ce(ClO4)3,485 wherein β-stereoselective glycosidation of anomeric fluoride derivative of d-mycosamine 274 was conducted via the IAD.536,537 This reaction afforded cholesteryl β-glycoside 275 in 71% yield as shown in Scheme 63C.
6.2.6. Activation with Anhydrides, Protic Acids, and Other Reagents.
Wessel introduced triflic anhydride as a new activator for glycosyl fluorides.460 The author proposed that the activation of glycosyl fluorides occurred due to a higher affinity of CF3SO2δ+ to fluorine than oxygen. As depicted in Scheme 64A, glycosidation of fluoride β-239 with glycosyl acceptor 276 afforded α-glycoside 277 in 92% yield. Further, this promoter was applied for the synthesis of differently linked disaccharides.538 Interestingly, it was discovered that low and moderately reactive alcohols successfully delivered products, whereas reactive alcohols provided fair yields due to the competing sulfonation of the hydroxyl group. Mukaiyama and co-workers discovered the activation of glycosyl fluorides with cat. trifluoromethanesulfonic acid (TfOH).465,486,487 Glycosidation of glycosyl fluoride α- or β-239 with glycosyl acceptor 36 in dichloromethane in the presence of a catalytic amount of TfOH (5 mol %) produced disaccharide 165 within 2 h in a good yield (Scheme 64B).
Scheme 64.

Other Methods for the Activation of Glycosyl Fluorides
TfOH-catalyzed activation was extended to reactions of differently protected glycosyl fluoride donors with various glycosyl acceptors. Without a nonparticipating group at C-2, the stereochemical outcome was difficult to predict. To address the problem associated with stereoselectivity, a thorough investigation for optimal reaction conditions was conducted. It was established that TfOH-catalyzed glycosylations produce good to excellent 1,2-cis selectivity in ethereal solvents in the presence of MS 5 Å or Drierite.488 Further investigation revealed that efficient activation of glycosyl fluoride donors can also be achieved in the presence of other protic acids such as HClO4, HOSO2C4F9, HNTf2, HSbF6, and HB(C6F5)4.464 Some of the strong acids were generated in situ in a similar way to the procedures described by Kevill539 or Kato.540 The diastereomeric ratio of glycoside products was found to be a subject of reaction conditions such as the nature of solvents, counteranions, and protecting groups.493 As previously discussed, Mukaiyama and co-workers performed the activation of glycosyl fluorides in the presence of catalytic carbocationic species paired with counterion such as tetrakis(pentafluorophenyl)borate.489 Accordingly, trifluoromethanesulfonate and perchlorate were found to be effective in activating glycosyl fluoride donors.
Chan and Bennett described the solvolysis of glycosyl fluorides in weak nucleophilic solvents such as hexafluoro-2-propanol 278 (HFIP).490 The formation of the retained solvolysis product, 1,1,1,3,3,3-hexafluoropropan-2-yl α-d-glucopyranoside 279, and the inverted product, 1,6-anhydro-β-d-glucopyranose 280 from glucosyl fluoride α-259 in HFIP or HFIP-d was observed (Scheme 64C). To understand the product distribution and pathway of the formation, the solvolysis of fluoride α-259 in HFIP was conducted at different temperatures and the activation parameters were calculated from Eyring plot. The obtained results suggested that solvolysis occurs via a highly ordered transition state, which resulted from solvation of the fluoride ion by the HFIP solvent.541 The expansive experimental and computed KIE measurements suggest solvolysis of fluorides α-259 in HFIP solvent involves cleavage of the C–F bond as the rate-determining step, wherein proton transfer occurs from solvating HFIP molecule to the leaving group fluoride to afford the solvent-separated ion pair. Concurrently, the formation of solvolysis product 279 proceeds via the collapse of the solvent-separated ion pair in the SNi reaction, whereas the formation of inverted product 280 proceeds via the intramolecular capture of the solvent-equilibrated glycosyl cation generated from the dissociation of the solvent-separated ion pair. The authors also showed that an SN2-like nucleophilic displacement of anomeric fluoride of unprotected fluoride donor α-259 with an aqueous solution of sodium azide produces corresponding β-glycosyl azide,505,541 wherein the anomeric center displayed that the reaction proceeds through a loose (exploded) transition state.505
6.3. Glycosyl Fluorides in Glycan and Glycoconjugate Synthesis
6.3.1. Convergent Building Block Assembly.
Early examples of impressive convergent syntheses employing fluorides as glycosyl donors involve synthesis of α-cyclodextrin Ogawa and Takahashi wherein glycosylations were promoted with SnCl2+AgOTf.542 Nicolaou and co-workers employed the cooperative catalytic systems SnCl2-Ag salts and also Cp2MCl2-Ag salts for the total synthesis of the tumor-associated Lex glycosphingolipids and sialyl dimeric Lex.543,544 A representative example of such an approach is the synthesis of the high mannose-type N-glycan 286 illustrated in Scheme 65.545 Glycosyl acceptor diol 282 was bis-glycosylated with the bromide donor 281 in the presence of AgOTf to afford the pentasaccharide 283 in 72% yield. The latter was then converted into a glycosyl fluoride donor 284 by reaction with DAST in the presence of NBS. The presynthesized hexasaccharide acceptor 285 was then regioselectively glycosylated with fluoride donor 284 in the presence of a Cp2HfCl2/AgOTf promoter system to afford oligosaccharide 286 in an excellent yield of 87%.
Scheme 65.
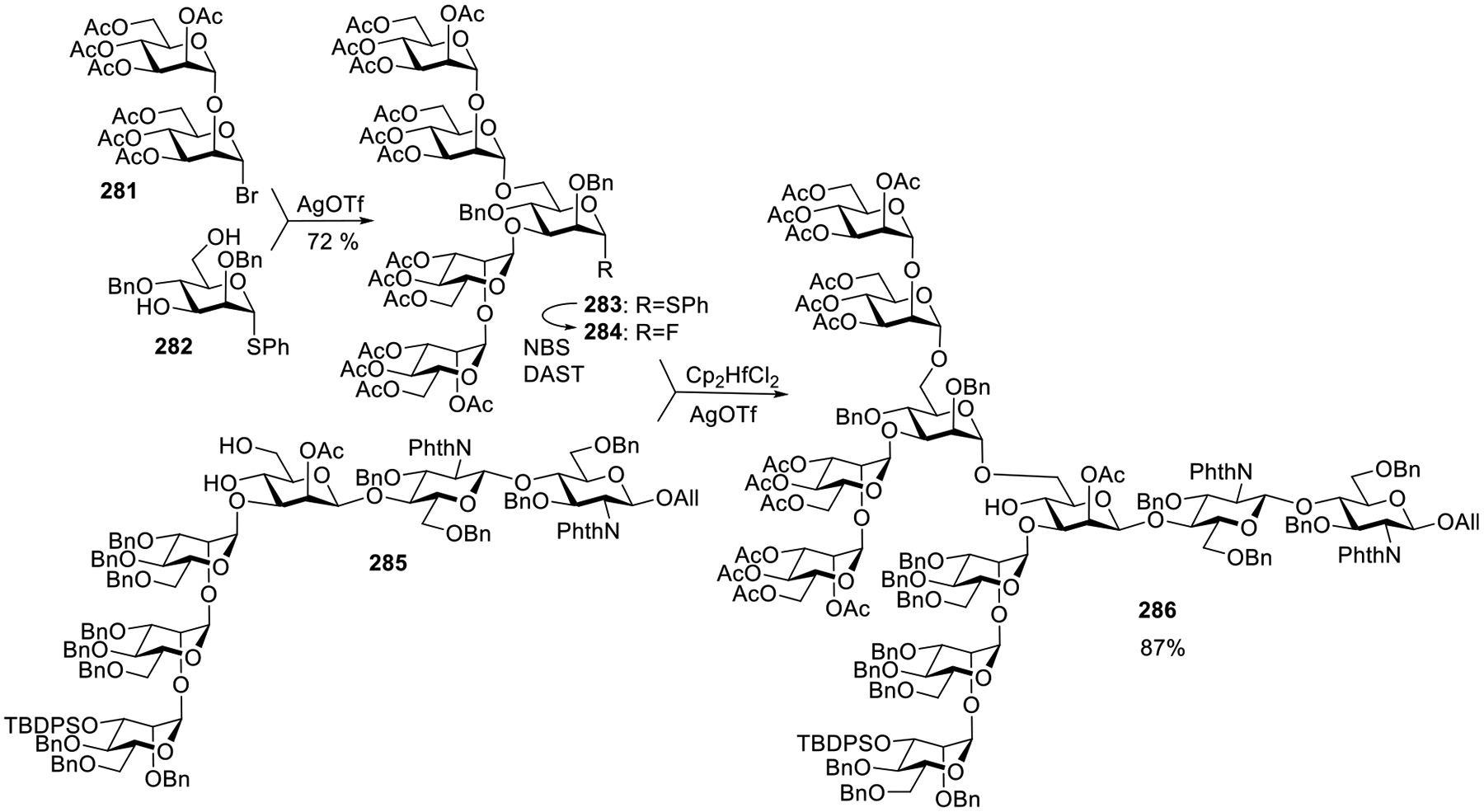
Convergent Assembly of High Mannose Type N-Glycan
A recent convergent assembly reported by Montgomery and co-workers comprises elements of one-pot synthesis.466 A three-component coupling was performed between monosaccharides 287–291 as shown in Scheme 66. The coupling of fluoride 287 with glycosyl acceptor 288 proceeded regioselectively at the C-4 position protected with TMS ether. Glycosyl fluoride donor 289 was then added to the reaction mixtures to produce the corresponding trisaccharide 292 in a good yield. In this reaction, TBS-protected C-6 position was glycosylated. Applying a similar approach for controlling the relative reactivity of silylated hydroxyl groups (TMS vs triethylsilyl vs triisopropylsilyl) trisaccharide 293 was synthesized. For this purpose, mannosyl fluoride donor 290 was coupled with glucosyl acceptor 291 to produce the corresponding disaccharide that was then reacted with mannosyl donor 287. Trisaccharide 293 was converted to glycosyl acceptor 294 by TIPS group removal with n-tetrabutylammonium fluoride (n-Bu4NF). Finally, n-pentenyl leaving group of trisaccharide donor 292 was activated for reaction with acceptor 294 in the presence of NIS/Et3SiOTf to afford hexasaccharide 295 in 61% yield.
Scheme 66.
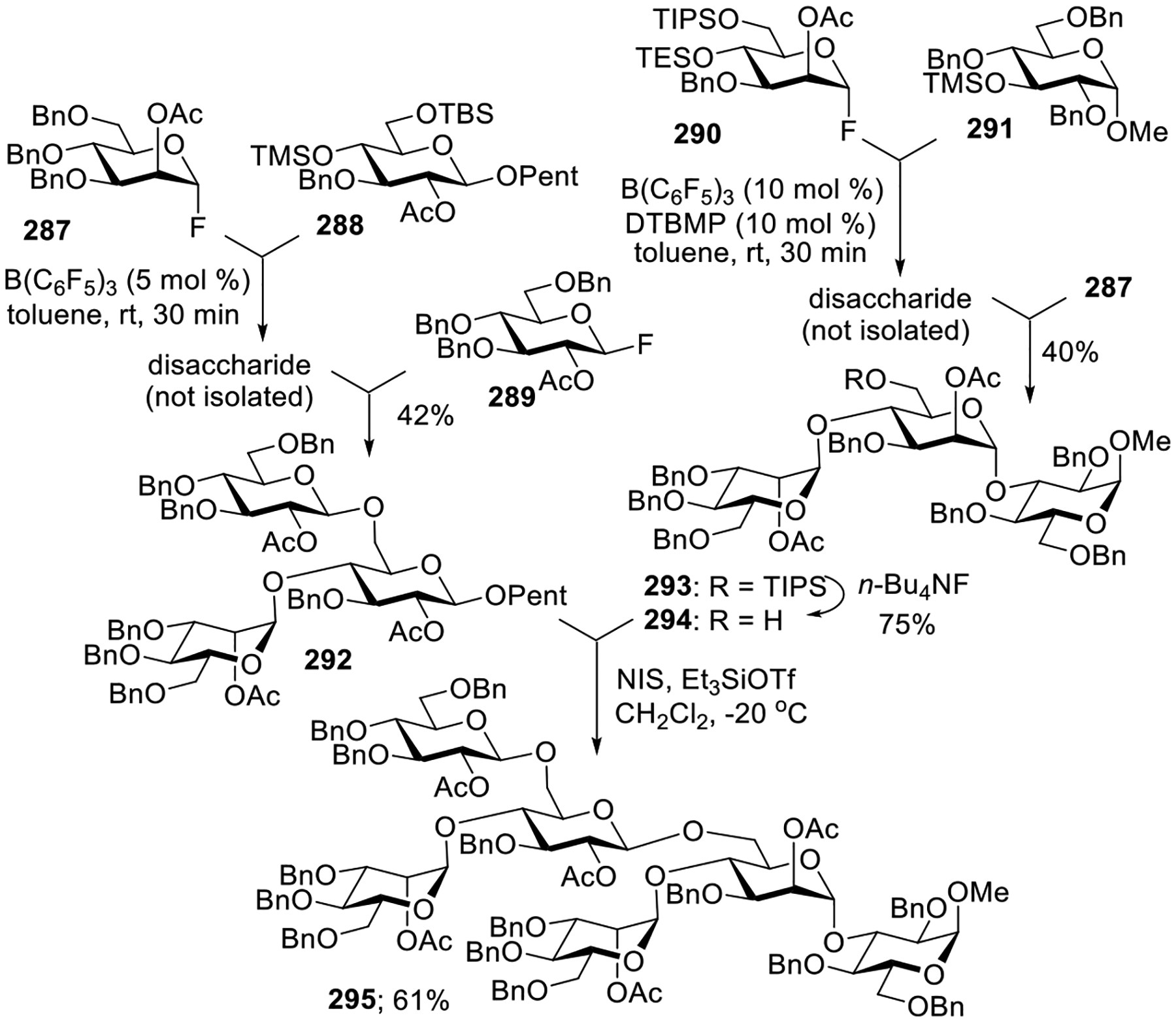
Borane-Catalyzed Convergent Oligosaccharides Synthesis
6.3.2. Two-Step Activation.
According to the two-stage activation approach, both glycosyl donor and glycosyl acceptor initially bear the same type of leaving group. However, to couple these two reactants, the LG of the potential glycosyl donor unit is first converted into a different (more reactive) LG, which is then selectively activated. This two-step activation sequence can be reiterated. This concept was discovered by Zen for S-ethyl and bromo groups329 and further expanded by Nicolaou for S-phenyl and fluoro groups.333,546 For a relevant example, see the synthesis of 302 depicted in Scheme 67.333 First thioglycoside 296 was converted into glycosyl fluoride 297 by the treatment with NBS/DAST. The latter was then coupled with a thioglycoside acceptor 298, which was generated from 296 by the treatment with TBAF, in the presence of SnCl2/AgClO4 to provide disaccharide 299 in 75% yield. This sequence is then repeated to furnish the desired fluoride donor 300 and thioglycoside acceptor 301, followed by glycosylation in a 2+2 fashion to provide tetrasaccharide 302. This approach combines advantages offered by the two-step activation and convergent strategies.
Scheme 67.

Two-Step Activation with Fluorides and Thioglycosides
6.3.3. Selective and Orthogonal Activation.
Another general concept to expedite oligosaccharide synthesis is to achieve selective activation of different leaving groups. In a typical application, a glycosyl fluoride donor is activated over a thioglycoside acceptor in the presence of a suitable promoter. The thio-leaving group of the resulting disaccharide is then activated directly to afford a trisaccharide. Examples of these applications include TfOH as the promoter for the first step, and NIS added for the second step.488 Another example involves HB(C6F5)4 and NIS as activators for the activation of fluoride and thioglycoside, respectively.465 Demchenko and co-workers synthesized a hexasaccharide in only five steps via sequential selective activation of five leaving groups, including fluorides.547 As a part of their strategy, S-benzoxazolyl (SBox) trisaccharide donor was coupled with fluoride acceptor in the presence of MeOTf, and the resulting tetrasaccharide was then activated in the presence of AgClO4/Cp2ZrCl2 to afford a pentasaccharide in 84% yield.
The combination of two chemically distinct glycosylation reactions, in which one of the leaving groups is activated while the other one remains intact, and vice versa, has led to the discovery of the orthogonal strategy for oligosaccharide synthesis.548 It requires the use of two orthogonal classes of glycosyl donors.549,550 As with the selective activation strategy, at the first step the glycosyl donor bearing LGa is activated over the glycosyl acceptor bearing LGb. Uniquely to the orthogonal strategy, the LGb is then activated over LGa of the new glycosyl acceptor (Scheme 68A). This activation sequence can then be reiterated to give straightforward access to larger oligosaccharides. Specifically, this unique strategy was discovered with glycosyl fluorides, and the classic variation of the orthogonal activation route also involves building blocks bearing the S-phenyl leaving group. Thus, Ogawa and co-workers illustrated that phenylthio glycoside 303 can be selectively activated over glycosyl fluoride 304 in the presence of NIS/AgOTf to afford disaccharide 305 (Scheme 68B).548 The fluoro leaving group of disaccharide donor 305 can then be activated over the SPh moiety of glycosyl acceptor 306 in the presence Cp2HfCl2 and AgClO4 to afford trisaccharide 307 in 72% yield. Phenylthio glycosyl donor 307 was then activated over fluoride acceptor 304 to provide tetrasaccharide 308 in 66% yield. These reactions have been performed both in solution548,551 and on the polymer support (vide infra).551,552
Scheme 68.

Orthogonal Activation Was Discovered with Fluorides and Thioglycosides
6.3.4. Polymer- and Tag-Supported Synthesis.
Among major breakthroughs that have emerged in the area of synthetic chemistry is the development of organic synthesis on the solid phase.553–556 As a consequence, the past two decades have witnessed dramatic improvements in the area of solid phase-supported oligosaccharide synthesis,557–561 particularly in the context of automation.3,562–566 Polymer-supported synthesis allows for rapid synthesis of oligosaccharide sequences without the necessity of purifying and characterizing the intermediates. Another advantage of oligosaccharide synthesis on solid-phase support is the ease of excess reagent removal (by filtration). Among multiple methods and approaches that have been developed to date, two main strategies for solid-phase saccharide synthesis that differ in the type of the attachment can be identified. In the most common glycosyl acceptor bound approach excess of the glycosyl donor and promoter are present in the solution phase. In the rarer glycosyl donor-bound approach, glycosyl acceptor and promoters are present in the solution phase. Application of the orthogonal strategy is a rare example of a donor-bound approach in polymer supported synthesis.551 As reported Kanie et al.,552,567 polymer-bound donor 309 was activated selectively over the solution-phase glycosyl fluoride acceptor 310 in the presence of dimethyl(methylthio)sulfonium trifluoromethanesulfonate (DMTST) shown in Scheme 69. The immobilized disaccharide fluoride was then activated over S-phenyl acceptor 215 in the presence of Cp2Hf(OTf)2. Finally, the immobilized S-phenyl trisaccharide was glycosidated with octanol 311 in the presence of DMTST. The resulting oligosaccharide 312 was cleaved off the polymer support.567 The Hf(OTf)4-promoted activation of solution phase mannosyl fluoride donor was applied to glycosylation of an acceptor immobilized on TentaGel.568
Scheme 69.
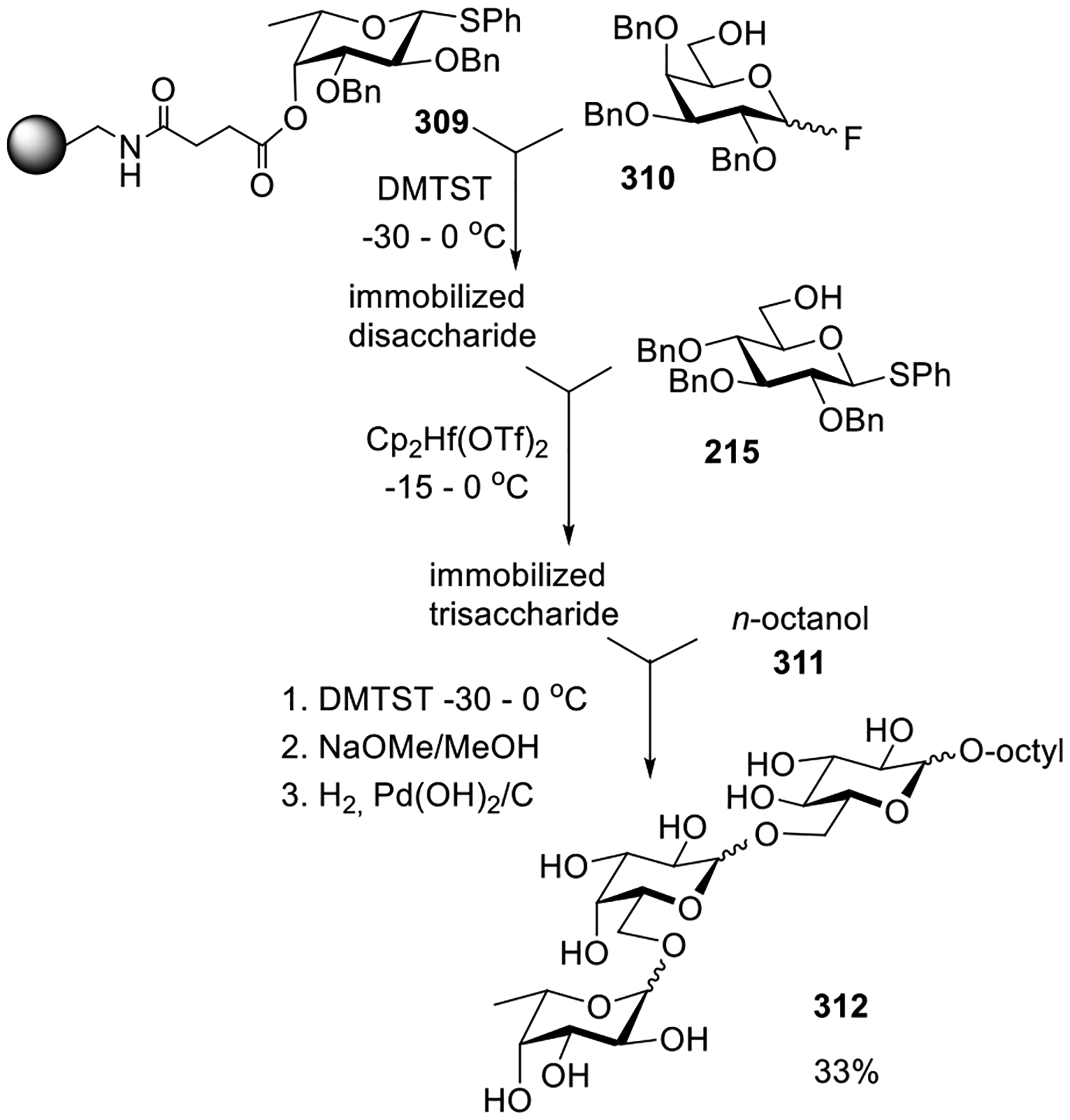
Orthogonal Synthesis on Solid Phase
Another promising technique for supported oligosaccharide synthesis makes use of an ionic-liquid tag.569,570 Ionic liquid-supported assembly also expedites oligosaccharide synthesis by eliminating the need for chromatographic purification of the intermediates. After the desired reaction of the tagged compound has been completed, the reaction mixture is concentrated. The excess of organic reagents is removed by extraction with low polarity solvents in which the tagged compounds are insoluble. This approach is illustrated by a synthesis that incorporates elements of an orthogonal strategy making use of alternating activations of STol and F leaving groups and the convergent approach.571,572 A (1-methylimda-zoliumhexafluorophospho) acetyl ionic liquid (IL) tag was introduced via the corresponding 6-chloroacetylated starting material by reaction with N-methylimidazole and potassium hexafluorophosphate. As depicted in Scheme 70, the tagged mannosyl fluoride donor 313 was glycosidated with thioglycoside acceptor 314 to afford the IL-tagged disaccharide. Meanwhile, disaccharide 315 was produced using tagged thioglycoside as the donor and a fluoride acceptor. Each disaccharide was split into portions, and the tag was removed from one portion. Coupling of the two disaccharides in the presence of NIS/TfOH produced a tagged tetrasaccharide. The latter was then coupled with tetrasaccharide 316, which was prepared in a similar fashion. The resulting glycan was then cleaved off from the IL support to afford mannan 317.
Scheme 70.
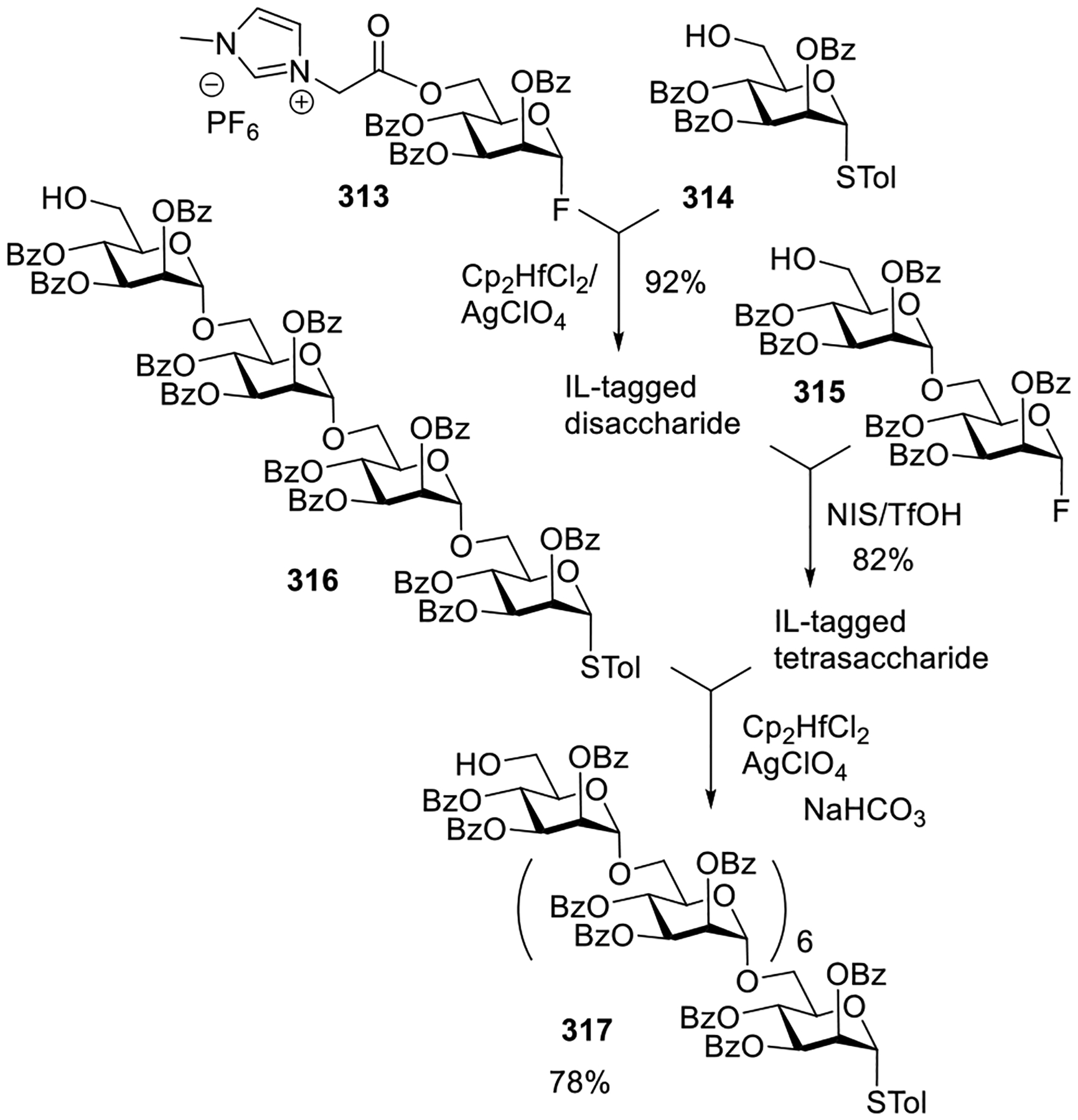
Ionic Liquid-Tagged Synthesis of Mannans
6.3.5. Oligosaccharide Synthesis with Hydrolases.
In the natural environment, these enzymes hydrolyze glycosidic bonds and therefore are responsible for degradation of oligosaccharides. However, the reverse hydrolytic activity of hydrolases can also be exploited for the glycosidic bond-formation process.573,574 Glycosyl hydrolases are much more readily available than glycosyltransferases. They also are typically less regioselective, and the transformation yields are lower. There are two main catalytic mechanisms for hydrolases: one leading to inversion of the anomeric configuration and the other leading to retention.575,576 The easiest way to employ this approach is to perform the glycosylation under thermodynamically controlled conditions, where the reverse hydrolysis is achieved at equilibrium, but the yields can be low. A significantly improved outcome can be achieved under kinetically controlled glycosylation conditions. In this case, activated glycosyl fluoride donors can be employed. Kinetically controlled glycosylations employing glycosidases (often called “transglycosylation” reactions) have been employed for the synthesis of a variety of oligosaccharides.574 As shown in Scheme 71, the application of glycosynthase, a synthetic enzyme derived from a retaining glycosidase, allowed irreversible glycosidation of glycosyl fluoride donor 318 with disaccharide acceptor 319.577 Application of the glycosynthase derived from the β-1,4-xylanase of Cellulomonas fimi resulted in a very efficient synthesis of a series of xylans. The (1→4)-linked xylooligosaccharides 320, ranging from tetra- to dodecasaccharides, have been obtained regio- and stereoselectively in over 60% combined yield.
Scheme 71.
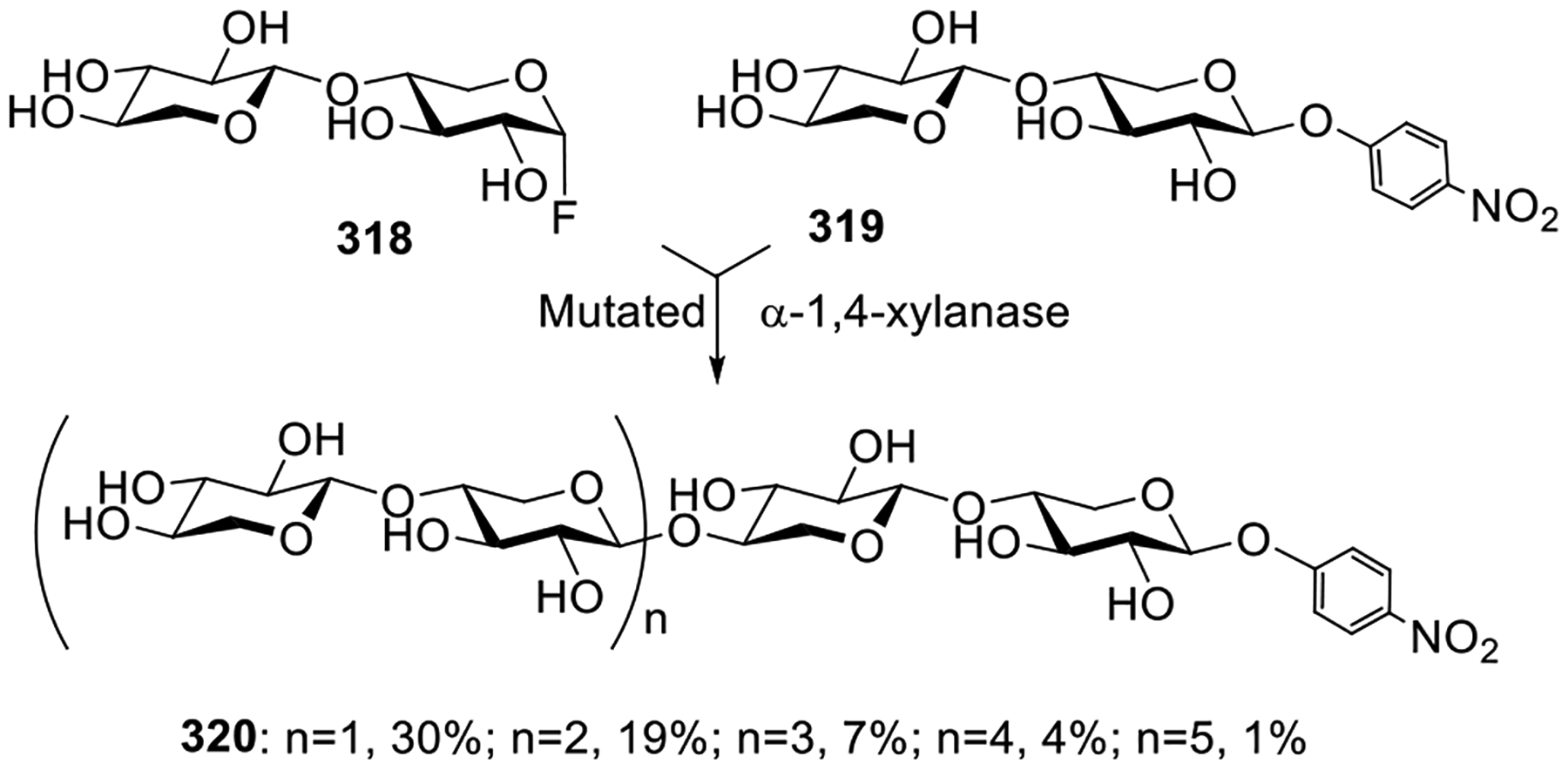
Synthesis of Xylans 320 Using an Engineered Xylanase
7. CONCLUSIONS AND OUTLOOK
To keep pace with the expanding areas of glycosciences, it is critical to make glycans more accessible to the general chemical, biomedical, and industrial audiences. The advancement of glycosylation methods and strategies and their broader adoption is crucial to meeting this need. While new developments are required both in the generalization of the strategies and in the optimization of methods for glycoside synthesis all existing methods of carbohydrate chemistry require further tuning of reactivity levels and reaction conditions. The goal of optimizing glycosylation has been pursued in many ways. This significantly enhanced our understanding and ability to refine the reaction conditions, suppress side reactions, understand stereoelectronics and conformation of the starting material and key reaction intermediates, and predict the outcomes of many glycosylations. Despite all these recent improvements, the challenge of glycosylation has remained, and many scientists have turned their attention to reinvestigating glycosyl halides. These simple donors are typically easily accessible from a variety of precursors, fairly stable, can be readily activated, and offer superior atom economy. Recent work has demonstrated that many glycosyl halides can be activated with less toxic promoters. These studies with glycosyl chlorides and bromides brought these glycosylation reactions to an entirely different level of flexibility and versatility. Conversely, glycosyl fluorides, once prominent glycosyl donors, have been used more and more rarely in the past decade. The authors of this review believe that the full potential of glycosyl fluorides is yet to be revealed, and the glycosynthetic audience will witness further improvements of this promising method already in the next decade.
With further improvement of our synthetic capabilities, new effective approaches to stereoselective chemical glycosylation that will be broadly applicable to a wide range of substrates and synthetic targets will emerge. We anticipate that exploring glycosyl halide donors in combination with other methods and approaches will contribute significantly to the general methodological field by enhancing our ability to monitor all steps of the glycosylation and study the key reaction intermediates. This, in turn, will open exciting opportunities for studying and understanding general aspects of glycosylation, reveal and solve problem spots, and develop reliable strategies for troubleshooting and sidetracking, all of which will improve our ability to obtain glycosides with high stereo-control. As a result, the methodological advances made in the synthetic field will boost innovations in the related fields of glycosciences. It is our belief that a dedicated study of the methods and mechanisms of the glycosylation process will strategically advance the field such that both one-step glycosylation and multistep syntheses of glycans will be considered standard.
Many current methods for chemical glycosylation remain highly sophisticated, operationally complex, and require significant user know-how. By contrast, automation of glycosylation reactions showcases a highly accessible method of synthesis because it offers an idea of operational simplicity and reproducibility. At this stage it is still unclear whether a dedicated glycosylation reaction needs to be developed or existing methods used in solution can simply be repurposed to fulfill the requirements of automated synthesis. Upon achieving a reliable and simple platform for completely automated glycosylation, anybody should be able to perform automated synthesis of glycans. Machine-assisted synthesis will help to eliminate variability, and to accurately reproduce experiments multiple times by different users.3,200,562–564,566,578–596 Synthesis of glycans using user-friendly automated platforms will accelerate discovery of new carbohydrate-based or carbohydrate-containing diagnostics,597–612 pharmaceuticals,597,613–624 and vaccines.625–635 This will lead to innovations in many scientific disciplines and can significantly impact technology, society, the economy, and public health.
ACKNOWLEDGMENTS
The authors are indebted to the National Science Foundation (CHE-1800350) and National Institute of General Medical Sciences (GM111835) for supporting their research program on developing new methods for glycosylation and glycan synthesis.
Biographies
Yashapal Singh graduated from Deen Dayal Upadhyaya Gorakhpur University, UP in India with a Master of Science (M.Sc.) degree in Chemistry (2008). In 2010, he joined Professor Narayanaswamy Jayaraman’s lab in the Department of Organic Chemistry at Indian Institute of Science Bangalore, India. In 2016, he received his Ph.D. degree for his work in the area of Macromolecular Chemistry including dendrimers and carbohydrates, directed particularly toward design and synthesis of organic materials for sensing applications. In February 2017, he joined the Demchenko lab at the University of Missouri–St. Louis (USA) as a postdoctoral research associate. His work entailed the identification of novel aglycones as organocatalysts for atom-efficient regenerative glycosylation reaction. Along with Professor Demchenko, he discovered the catalytic role of an acid in the Koenigs–Knorr glycosylation. He also worked on developing methods for the synthesis and activation of glycosyl nitrates and thioglycosides. Further, he was engaged in developing a robust large-scale method for the synthesis of human milk oligosaccharides, and HPLC-based automation for the synthesis of biologically relevant glycans. Currently, Dr. Singh is working as a postdoctoral research associate in Professor Philip S. Low’s lab at Purdue University (USA) where his research is directed toward cancer immunotherapy.
Scott Geringer graduated from the Southern Illinois University–Edwardsville (SIUe) with a Bachelor of Science (B.S.) in Chemistry in 2013. He continued his education at SIUe and graduated with a Master of Science (M.Sc.) in Chemistry in 2015. He worked with Dr. Cristina De Meo. He completed his thesis studying the effects of the picoloyl protecting group on sialic acid glycosylation as well as studying the conformational equilibrium of the oxacarbenium ion intermediate. In August of 2015, Scott joined Dr. Alexei Demchenko’s lab at University of Missouri–St. Louis (UMSL) as a Ph.D. student. He successfully graduated from UMSL in May of 2020 with a Doctor of Philosophy in Chemistry with an emphasis on Organic Chemistry. Here Scott worked on developing new catalytic reactions in carbohydrate chemistry. These methods include developing a method for catalytic glycosylation of glycosyl chlorides using iron(III) chloride, a new method for chemoselective picoloyl cleavage using catalytic iron(III) chloride, and activation of glycosyl chlorides using the cooperative silver(I) oxide-triflic acid catalyst system. Dr. Geringer currently works as a senior scientist at MilliporeSigma.
Alexei Demchenko graduated from the Mendeleev University of Chemical Technology of Russia with a Diploma (M.S.) in Chemical Engineering (1988) before joining the laboratory of the late Professor Kochetkov at the Zelinsky Institute of Organic Chemistry in Moscow. In 1993, he was awarded a Ph.D. in Organic Chemistry by the Russian Academy of Sciences for his work on the development of thiocyanate methodology for glycosylation. After two postdoctoral years under Kochetkov, he joined Professor Boons’ group at the University of Birmingham (UK) as a BBSRC postdoctoral research fellow. In 1998, he moved to the Complex Carbohydrate Research Center, University of Georgia (USA) as a research associate. In 2001, he joined the faculty at the University of Missouri–St. Louis (UMSL) as an assistant professor where he was promoted to the rank of associate professor with tenure (2007) and professor (2011). In 2014, Dr. Demchenko was appointed Curators’ Distinguished Professor of Chemistry and Biochemistry. In 2021, Demchenko joined the faculty at Saint Louis University as professor and department chair. His research interests are in the area of synthetic carbohydrate chemistry that include streamlined synthesis of carbohydrate building blocks, development of new glycosylation reactions, strategies for expeditious assembly of complex glycans and glycopharmaceuticals, and automated synthesis.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.2c00029
The authors declare no competing financial interest.
Contributor Information
Yashapal Singh, Department of Chemistry and Biochemistry, University of Missouri–St. Louis, St. Louis, Missouri 63121, United States.
Scott A. Geringer, Department of Chemistry and Biochemistry, University of Missouri–St. Louis, St. Louis, Missouri 63121, United States
Alexei V. Demchenko, Department of Chemistry and Biochemistry, University of Missouri–St. Louis, St. Louis, Missouri 63121, United States; Department of Chemistry, Saint Louis University, St. Louis, Missouri 63103, United States.
REFERENCES
- (1).Hehre EJ Glycosyl transfer: a history of the concept’s development and view of its major contributions to biochemistry. Carbohydr. Res 2001, 331, 347–368. [DOI] [PubMed] [Google Scholar]
- (2).Muthana S; Cao H; Chen X Recent progress in chemical and chemoenzymatic synthesis of carbohydrates. Curr. Opin. Chem. Biol 2009, 13, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Panza M; Pistorio SG; Stine KJ; Demchenko AV Automated chemical oligosaccharide synthesis: novel approach to traditional challenges. Chem. Rev 2018, 118, 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Michael A On the synthesis of helicin and phenolglucoside. Am. Chem. J 1879, 1, 305–312. [Google Scholar]
- (5).Fischer E Über die glucoside der alkohole. Ber. Dtsch. Chem. Ges 1893, 26, 2400–2412. [Google Scholar]
- (6).Fischer E; Armstrong EF Over isomers of the aceto-halogen derivative of grape sugar and the synthesis of the glucosides. Ber. Dtsch. Chem. Ges 1901, 34, 2885–2900. [Google Scholar]
- (7).Koenigs W; Knorr E Über einige derivate des traubenzuckers und der galactose. Ber. Dtsch. Chem. Ges 1901, 34, 957–981. [Google Scholar]
- (8).Mukaiyama T; Murai Y; Shoda S.-i. An efficient method for glucosylation of hydroxy compounds using glucopyranosyl fluoride. Chem. Lett 1981, 10, 431–432. [Google Scholar]
- (9).Gervay J; Hadd MJ Anionic additions to glycosyl iodides: highly stereoselective syntheses of β-C-, N-, and O-glycosides. J. Org. Chem 1997, 62, 6961–6967. [Google Scholar]
- (10).Helferich B; Schmitz-Hillebrecht E A new method for the synthesis of glucosides of the phenols. Ber. Dtsch. Chem. Ges 1933, 66B, 378–383. [Google Scholar]
- (11).Boursier M; Descotes G Activation of the anomeric carbon of sugars by the carbonate group and applications in glycosidic synthesis. C. R. Acad. Sci. Ser 2 1989, 308, 919–921. [Google Scholar]
- (12).Koide K; Ohno M; Kobayashi S A new glycosylation reaction based on a ″remote activation concept″: glycosyl 2-pyridinecarboxylate as a novel glycosyl donor. Tetrahedron Lett. 1991, 32, 7065–7068. [Google Scholar]
- (13).Kunz H; Zimmer J Glycoside synthesis via electrophile-induced activation of N-allyl carbamates. Tetrahedron Lett. 1993, 34, 2907–2910. [Google Scholar]
- (14).Ferrier RJ; Hay RW; Vethaviyasar N A potentially versatile synthesis of glycosides. Carbohydr. Res 1973, 27, 55–61. [Google Scholar]
- (15).Nicolaou KC; Seitz SP; Papahatjis DP A mild and general method for the synthesis of O-glycosides. J. Am. Chem. Soc 1983, 105, 2430–2434. [Google Scholar]
- (16).Garegg PJ; Henrichson C; Norberg T A reinvestigation of glycosidation reactions using 1-thioglycosides as glycosyl donors and thiophilic cations as promoters. Carbohydr. Res 1983, 116, 162–165. [Google Scholar]
- (17).Oscarson S General synthetic methods: Reactions at the anomeric center: S-glycosylation. In Glycoscience: Chemistry and Chemical Biology; Fraser-Reid B, Tatsuta K, Thiem J, Eds.; Springer: Berlin - Heidelberg - New York, 2001; Vol. 1; pp 643–671. [Google Scholar]
- (18).Bochkov AF; Kochetkov NK A new approach for the synthesis of oligosaccharides. Carbohydr. Res 1975, 39, 355–357. [Google Scholar]
- (19).Kochetkov NK; Backinowsky LV; Tsvetkov YE Sugar thio orthoesters as glycosylating agents. Tetrahedron Lett. 1977, 18, 3681–3684. [Google Scholar]
- (20).Pougny JR; Jacquinet JC; Nassr M; Duchet D; Milat ML; Sinay P A novel synthesis of 1,2-cis-disaccharides. J. Am. Chem. Soc 1977, 99, 6762–6763. [DOI] [PubMed] [Google Scholar]
- (21).Schmidt RR; Michel J Facile synthesis of α- and β-O-glycosyl imidates; Preparation of glycosides and disaccharides. Angew. Chem., Int. Ed. Engl 1980, 19, 731–732. [Google Scholar]
- (22).Adinolfi M; Barone G; Iadonisi A; Schiattarella M Iodine/triethylsilane as a convenient promoter system for the activation of disarmed glycosyl trichloro- and N-(phenyl)trifluoroacetimidates. Synlett 2002, 2002, 269–270. [Google Scholar]
- (23).Adinolfi M; Barone G; Iadonisi A; Schiattarella M Efficient activation of glycosyl N-(phenyl)trifluoroacetimidate donors with ytterbium(III) triflate in the glycosylation reaction. Tetrahedron Lett. 2002, 43, 5573–5577. [Google Scholar]
- (24).Mukaiyama T; Nakatsuka T; Shoda SI An efficient glucosylation of alcohol using 1-thioglucoside derivative. Chem. Lett 1979, 8, 487–490. [Google Scholar]
- (25).Hanessian S; Bacquet C; Lehong N Chemistry of the glycosidic linkage. Exceptionally fast and efficient formation of glycosides by remote activation. Carbohydr. Res 1980, 80, c17–c22. [Google Scholar]
- (26).Woodward RB; Logusch E; Nambiar KP; Sakan K; Ward DE; Au-Yeung BW; Balaram P; Browne LJ; Card PJ; Chen CH Asymmetric total synthesis of erythromycin. 3. Total synthesis of erythromycin. J. Am. Chem. Soc 1981, 103, 3215–3217. [Google Scholar]
- (27).Demchenko AV; Malysheva NN; De Meo C S-Benzoxazolyl (SBox) glycosides as novel, versatile glycosyl donors for stereoselective 1,2-cis glycosylation. Org. Lett 2003, 5, 455–458. [DOI] [PubMed] [Google Scholar]
- (28).Demchenko AV; Kamat MN; De Meo C S-Benzoxazolyl (SBox) glycosides in oligosaccharide synthesis: novel glycosylation approach to the synthesis of β-D-glucosides, β-D-galactosides, and α-D-mannosides. Synlett 2003, 2003, 1287–1290. [Google Scholar]
- (29).Demchenko AV; Pornsuriyasak P; De Meo C; Malysheva NN Potent, versatile, and stable: thiazolyl thioglycosides as glycosyl donors. Angew. Chem., Int. Ed 2004, 43, 3069–3072. [DOI] [PubMed] [Google Scholar]
- (30).Fraser-Reid B; Konradsson P; Mootoo DR; Udodong U Direct elaboration of pent-4-enyl glycosides into disaccharides. J. Chem. Soc., Chem. Commun 1988, 823–825. [Google Scholar]
- (31).Marra A; Esnault J; Veyrieres A; Sinay P Isopropenyl glycosides and congeners as novel classes of glycosyl donors: theme and variations. J. Am. Chem. Soc 1992, 114, 6354–6360. [Google Scholar]
- (32).Boons GJ; Isles S Vinyl glycosides in oligosaccharide synthesis. Part 1: A new latent-active glycosylation strategy. Tetrahedron Lett. 1994, 35, 3593–3596. [Google Scholar]
- (33).Brown DS; Ley SV; Vile S Preparation of cyclic ether acetals from 2-benzenesulphonyl derivatives: a new mild glycosidation procedure. Tetrahedron Lett. 1988, 29, 4873–4876. [Google Scholar]
- (34).Kochetkov NK; Klimov EM; Malysheva NN Novel highly stereospecific method of 1,2-cis-glycosylation. Synthesis of α-D-glucosyl-D-glucoses. Tetrahedron Lett. 1989, 30, 5459–5462. [Google Scholar]
- (35).Briner K; Vasella A Glycosylidene carbenes a new approach to glycoside synthesis. Part 1. Preparation of glycosylidene-derived diaziridines and diazirines. Helv. Chim. Acta 1989, 72, 1371–1382. [Google Scholar]
- (36).Friesen RW; Danishefsky SJ On the controlled oxidative coupling of glycals: A new strategy for the rapid assembly of oligosaccharides. J. Am. Chem. Soc 1989, 111, 6656–6660. [Google Scholar]
- (37).Halcomb RL; Danishefsky SJ On the direct epoxidation of glycals: application of a reiterative strategy for the synthesis of β-linked oligosaccharides. J. Am. Chem. Soc 1989, 111, 6661–6666. [Google Scholar]
- (38).Balmond EI; Coe DM; Galan MC; McGarrigle EM α-Selective organocatalytic synthesis of 2-deoxygalactosides. Angew. Chem., Int. Ed 2012, 51, 9152–9155. [DOI] [PubMed] [Google Scholar]
- (39).Palo-Nieto C; Sau A; Galan MC Gold(I)-catalyzed direct stereoselective synthesis of deoxyglycosides from glycals. J. Am. Chem. Soc 2017, 139, 14041–14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kinfe HH Versatility of glycals in synthetic organic chemistry: coupling reactions, diversity oriented synthesis and natural product synthesis. Org. Biomol. Chem 2019, 17, 4153–4182. [DOI] [PubMed] [Google Scholar]
- (41).Kahne D; Walker S; Cheng Y; van Engen D Glycosylation of unreactive substrates. J. Am. Chem. Soc 1989, 111, 6881–6882. [Google Scholar]
- (42).Marra A; Sinay P A novel stereoselective synthesis of N-acetyl-α-neuraminosyl-galactose disaccharide derivatives, using anomeric S-glycosyl xanthates. Carbohydr. Res 1990, 195, 303–308. [Google Scholar]
- (43).Mehta S; Pinto BM Phenylselenoglycosides as novel, versatile glycosyl donors. selective activation over thioglycosides. Tetrahedron Lett. 1991, 32, 4435–4438. [Google Scholar]
- (44).Kondo H; Ichikawa Y; Wong CH β-Sialyl phosphite and phosphoramidite: synthesis and application to the chemoenzymatic synthesis of CMP-sialic acid and sialyl oligosaccharides. J. Am. Chem. Soc 1992, 114, 8748–8750. [Google Scholar]
- (45).Martin TJ; Schmidt RR Efficient sialylation with phosphite as leaving group. Tetrahedron Lett. 1992, 33, 6123–6126. [Google Scholar]
- (46).Stick RV; Tilbrook DMG; Williams SJ The selective activation of telluro- over seleno- β-D-glucopyranosides as glycosyl donors: a reactivity scale for various telluro, seleno and thio sugars. Aust. J. Chem 1997, 50, 237–240. [Google Scholar]
- (47).Hinklin RJ; Kiessling LL Glycosyl sulfonylcarbamates: new glycosyl donor with tunable reactivity. J. Am. Chem. Soc 2001, 123, 3379–3380. [DOI] [PubMed] [Google Scholar]
- (48).Huchel U; Schmidt C; Schmidt RR Synthesis of hetaryl glycosides and their glycosyl donor properties. Eur. J. Org. Chem 1998, 1998, 1353–1360 and references therein. [Google Scholar]
- (49).Plante OJ; Andrade RB; Seeberger PH Synthesis and use of glycosyl phosphates as glycosyl donors. Org. Lett 1999, 1, 211–214. [DOI] [PubMed] [Google Scholar]
- (50).Davis BG; Ward SJ; Rendle PM Glycosyldisulfides: a new class of solution and solid phase glycosyl donors. Chem. Commun 2001, 189–190. [Google Scholar]
- (51).Kim KS; Kim JH; Lee YJ; Lee YJ; Park J 2-(Hydroxycarbonyl)benzyl glycosides: a novel type of glycosyl donors for highly efficient β-mannopyranosylation and oligosaccharide synthesis by latent-active glycosylation. J. Am. Chem. Soc 2001, 123, 8477–8481. [DOI] [PubMed] [Google Scholar]
- (52).Li Y; Yang Y; Yu B An efficient glycosylation protocol with glycosyl ortho-alkynylbenzoates as donors under the catalysis of Ph3PAuOTf. Tetrahedron Lett. 2008, 49, 3604–3608. [Google Scholar]
- (53).Mamidyala SK; Finn MG Glycosylation using unprotected alkynyl donors. J. Org. Chem 2009, 74, 8417–8420. [DOI] [PubMed] [Google Scholar]
- (54).Li Y; Yang X; Liu Y; Zhu C; Yang Y; Yu B Gold(I)-catalyzed glycosylation with glycosyl ortho-alkynylbenzoates as donors: general scope and application in the synthesis of a cyclic triterpene saponin. Chem.—Eur. J 2010, 16, 1871–1882. [DOI] [PubMed] [Google Scholar]
- (55).Yang W; Sun J; Lu W; Li Y; Shan L; Han W; Zhang W; Yu B Synthesis of kaempferol 3-O-(3″,6″-di-O-E-p-coumaroyl)-b-D-glucopyranoside, efficient glycosylation of flavonol 3-OH with glycosyl o-alkynylbenzoates as donors. J. Org. Chem 2010, 75, 6879–6888. [DOI] [PubMed] [Google Scholar]
- (56).Li Y; Sun J; Yu B Efficient synthesis of lupane-type saponins via gold(I)-catalyzed glycosylation with glycosyl ortho-alkynylbenzoates as donors. Org. Lett 2011, 13, 5508–5511. [DOI] [PubMed] [Google Scholar]
- (57).Ma Y; Li Z; Shi H; Zhang J; Yu B Assembly of Digitoxin by gold(I)-catalyzed glycosidation of glycosyl o-alkynylbenzoates. J. Org. Chem 2011, 76, 9748–9756. [DOI] [PubMed] [Google Scholar]
- (58).Zhu Y; Yu B Characterization of the Isochromen-4-yl-gold(I) intermediate in the gold(I)-catalyzed glycosidation of glycosyl ortho-alkynylbenzoates and enhacement of the catalytic efficiency thereof. Angew. Chem., Int. Ed 2011, 50, 8329–8332. [DOI] [PubMed] [Google Scholar]
- (59).Yang F; Wang Q; Yu B ortho-Alkynylphenyl thioglycosides as a new type of glycosylation donors under the catalysis of Au(I) complexes. Tetrahedron Lett. 2012, 53, 5231–5234. [Google Scholar]
- (60).Tang Y; Li J; Zhu Y; Li Y; Yu B Mechanistic insights into the gold(I)-catalyzed activation of glycosyl ortho-alkynylbenzoates for glycosidation. J. Am. Chem. Soc 2013, 135, 18396–18405. [DOI] [PubMed] [Google Scholar]
- (61).Mishra B; Neralkar M; Hotha S Stable alkynyl glycosyl carbonates: catalytic anomeric activation and synthesis of a tridecasaccharide reminiscent of Mycobacterium tuberculosis cell wall lipoarabinomannan. Angew. Chem., Int. Ed 2016, 128, 7917–7922. [DOI] [PubMed] [Google Scholar]
- (62).Nukada T; Berces A; Whitfield DM Can the stereochemical outcome of glycosylation reactions be controlled by the conformational preferences of the glycosyl donor? Carbohydr. Res 2002, 337, 765–774. [DOI] [PubMed] [Google Scholar]
- (63).Ranade SC; Demchenko AV Mechanism of chemical glycosylation: focus on the mode of activation and departure of anomeric leaving groups. J. Carbohydr. Chem 2013, 32, 1–43. [Google Scholar]
- (64).Kochetkov NK; Klimov EM; Malysheva NN; Demchenko AV A new stereospecific method for 1,2-cis-glycosylation. Carbohydr. Res 1991, 212, 77–91. [DOI] [PubMed] [Google Scholar]
- (65).Kochetkov NK; Klimov EM; Malysheva NN; Demchenko AV Stereospecific 1,2-cis-glycosylation: a modified thiocyanate method. Carbohydr. Res 1992, 232, C1–C5. [DOI] [PubMed] [Google Scholar]
- (66).Cato D; Buskas T; Boons G-J Highly efficient stereospecific preparation of Tn and TF building blocks using thioglycosyl donors and the Ph2SO/Tf2O promotor system. J. Carbohydr. Chem 2005, 24, 503–516. [Google Scholar]
- (67).Mydock LK; Kamat MN; Demchenko AV Direct synthesis of diastereomerically pure glycosyl sulfonium salts. Org. Lett 2011, 13, 2928–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hoang KLM; Liu X-W The intriguing dual-directing effect of 2-cyanobenzyl ether for a highly stereospecific glycosylation reaction. Nat. Commun 2014, 5, 5051. [DOI] [PubMed] [Google Scholar]
- (69).Issa JP; Bennett CS A reagent-controlled SN2-glycosylation for the direct synthesis of beta-linked 2-deoxy-sugars. J. Am. Chem. Soc 2014, 136, 5740–5744. [DOI] [PubMed] [Google Scholar]
- (70).Park Y; Harper KC; Kuhl N; Kwan EE; Liu RY; Jacobsen EN Macrocyclic bis-thioureas catalyze stereospecific glycosylation reactions. Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Adero PO; Amarasekara H; Wen P; Bohe L; Crich D The experimental evidence in support of glycosylation mechanisms at the SN1-SN2 Interface. Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Eby R; Schuerch C The use of 1-O-tosyl-D-glucopyranose derivatives in α-D-glucoside synthesis. Carbohydr. Res 1974, 34, 79–90 and references therein. [Google Scholar]
- (73).Marousek V; Lucas TJ; Wheat PE; Schuerch C The influence of reactant structure and solvent on galactoside synthesis from galactosyl sulfonates. Carbohydr. Res 1978, 60, 85–96. [Google Scholar]
- (74).Crich D; Sun S Are glycosyl triflates intermediates in the sulfoxide glycosylation method? a chemical and1H,13C, and19F NMR spectroscopic investigation. J. Am. Chem. Soc 1997, 119, 11217–11223. [Google Scholar]
- (75).Crich D; Sun S Direct chemical synthesis of β-mannopyranosides and other glycosides via glycosyl triflates. Tetrahedron 1998, 54, 8321–8348. [Google Scholar]
- (76).Crich D Chemistry of glycosyl triflates: synthesis of β-mannopyranosides (Reprinted from Glycochemistry: Principles, Synthesis, and Applications, pp 53–75, 2001). J. Carbohydr. Chem 2002, 21, 667–690. [Google Scholar]
- (77).Nokami T; Shibuya A; Tsuyama H; Suga S; Bowers AA; Crich D; Yoshida J Electrochemical generation of glycosyl triflate pools. J. Am. Chem. Soc 2007, 129, 10922–10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Garcia BA; Poole JL; Gin DY Direct glycosylations with 1-hydroxy glycosyl donors using trifluoromethanesulfonic anhydride and diphenyl sulfoxide. J. Am. Chem. Soc 1997, 119, 7597–7598. [Google Scholar]
- (79).Garcia BA; Gin DY Dehydrative glycosylation with activated diphenyl sulfonium reagents. Scope, mode of C(1)-hemiacetal activation, and detection of reactive glycosyl intermediates. J. Am. Chem. Soc 2000, 122, 4269–4279. [Google Scholar]
- (80).Nguyen HM; Chen YN; Duron SG; Gin DY Sulfide-mediated dehydrative glycosylation. J. Am. Chem. Soc 2001, 123, 8766–8772. [DOI] [PubMed] [Google Scholar]
- (81).Gin D Dehydrative glycosylation with 1-hydroxy donors (reprinted from Glycochemistry: Principles, Synthesis, and Applications, pg 33– 52, 2001). J. Carbohydr. Chem 2002, 21, 645–665. [Google Scholar]
- (82).Boebel TA; Gin DY Probing the mechanism of sulfoxide-catalyzed hemiacetal activation in dehydrative glycosylation. J. Org. Chem 2005, 70, 5818–5826. [DOI] [PubMed] [Google Scholar]
- (83).Romero JAC; Tabacco SA; Woerpel KA Stereo-chemical reversal of nuclephilic substitution reactions depending upon substituent: reactions of heteroatom-substituted six-membered-ring oxocarbenium ions through pseudoaxial conformers. J. Am. Chem. Soc 2000, 122, 168–169. [Google Scholar]
- (84).Ayala L; Lucero CG; Romero JAC; Tabacco SA; Woerpel KA Stereochemistry of nucleophilic substitution reactions depending upon substituent: evidence for electrostatic stabilization of pseudoaxial conformers of oxocarbenium ions by heteroatom substitution. J. Am. Chem. Soc 2003, 125, 15521–15528. [DOI] [PubMed] [Google Scholar]
- (85).Shenoy SR; Woerpel KA Investigations into the role of ion pairing in reactions of heteroatom-substituted cyclic oxocarbenium ions. Org. Lett 2005, 7, 1157–1160. [DOI] [PubMed] [Google Scholar]
- (86).Billings SB; Woerpel KA Nucleophilic substitution reactions of sulfur-substituted cyclohexanone acetals: an analysis of the factors controlling stereoselectivity. J. Org. Chem 2006, 71, 5171–5178. [DOI] [PubMed] [Google Scholar]
- (87).Beaver MG; Billings SB; Woerpel KA C-Glycosylation reactions of sulfur-substituted glycosyl donors: Evidence against the role of neighboring-group participation. J. Am. Chem. Soc 2008, 130, 2082–2086. [DOI] [PubMed] [Google Scholar]
- (88).Chu A-HA; Nguyen SH; Sisel JA; Minciunescu A; Bennett CS Selective synthesis of 1,2-cis-α-glycosides without directing groups. Application to iterative oligosaccharide synthesis. Org. Lett 2013, 15, 2566–2569. [DOI] [PubMed] [Google Scholar]
- (89).Soliman SE; Bennett CS Reagent-controlled synthesis of the branched trisaccharide fragment of the antibiotic Saccharomicin B. Org. Lett 2018, 20, 3413–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Frihed TG; Bols M; Pedersen CM Mechanisms of glycosylation reactions studied by low-temperature nuclear magnetic resonance. Chem. Rev 2015, 115, 4963–5013. [DOI] [PubMed] [Google Scholar]
- (91).Whitfield DM Computational studies of the role of glycopyranosyl oxacarbenium ions in glycobiology and glycochemistry. Adv. Carbohydr. Chem. Biochem 2009, 62, 83–159. [DOI] [PubMed] [Google Scholar]
- (92).Paulsen H Selectivity and reactivity in oligosaccharide synthesis. In Selectivity - a Goal for Synthetic Efficiency; Bartmann W; Trost BM, Eds.; Verlag Chemie: Weinheim, 1984; pp 169–190. [Google Scholar]
- (93).Uriel C; Gomez AM; Lopez JC; Fraser-Reid B Reciprocal donor-acceptor selectivity: the influence of the donor O-2 substituent in the regioselective mannosylation of myo-inositol orthopentanoate. Eur. J. Org. Chem 2009, 2009, 403–411. [Google Scholar]
- (94).Jayaprakash KN; Lu J; Fraser-Reid B Synthesis of a lipomannan component of the cell-wall complex of Mycobacterium tuberculosis is based on Paulsen’s concept of donor/acceptor ″match″. Angew. Chem., Int. Ed 2005, 44, 5894–5898. [DOI] [PubMed] [Google Scholar]
- (95).Uriel C; Gomez AM; Lopez JC; Fraser-Reid B Further insight into ‘matching’ of donors and acceptors via reciprocal donor acceptor selectivity (RDAS) studies. Synlett 2003, 2003, 2203–2207. [Google Scholar]
- (96).Mach M; Schlueter U; Mathew F; Fraser-Reid B; Hazen KC Comparing n-pentenyl orthoesters and n-pentenyl glycosides as alternative glycosyl donors. Tetrahedron 2002, 58, 7345–7354. [Google Scholar]
- (97).Fraser-Reid B; Lopez JC; Radhakrishnan KV; Nandakumar MV; Gomez AM; Uriel C One pot/two donors/one diol give one differentiated trisaccharide: powerful evidence for reciprocal donor-acceptor selectivity (RDAS). Chem. Commun 2002, 2104–2105. [DOI] [PubMed] [Google Scholar]
- (98).Spijker NM; van Boeckel CAA Double stereodifferentiation in carbohydrate coupling reactions: the mismatched Interaction of donor and dcceptor as an unprecedented factor governing the α/β-ratio of glycoside formation. Angew. Chem., Int. Ed. Engl 1991, 30, 180–183. [Google Scholar]
- (99).Zhang Q; van Rijssel ER; Walvoort MTC; Overkleeft HS; van der Marel GA; Codée JDC Acceptor Reactivity in the Total Synthesis of Alginate Fragments Containing α-L-Guluronic Acid and β-D-Mannuronic Acid. Angew. Chem. Int. Ed 2015, 54, 7670–7673. [DOI] [PubMed] [Google Scholar]
- (100).van der Vorm S; Hansen T; Overkleeft HS; van der Marel GA; Codée JDC The influence of acceptor nucleophilicity on the glycosylation reaction mechanism. Chem. Sci 2017, 8, 1867–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).van der Vorm S; van Hengst JMA; Bakker M; Overkleeft HS; van der Marel GA; Codée JDC Mapping the relationship between gycosyl acceptor reactivity and glycosylation stereoselectivity. Angew. Chem., Int. Ed. Engl 2018, 57, 8240–8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).van der Vorm S; Hansen T; van Hengst JMA; Overkleeft HS; van der Marel GA; Codee JDC Acceptor reactivity in glycosylation reactions. Chem. Soc. Rev 2019, 48, 4688–4706. [DOI] [PubMed] [Google Scholar]
- (103).Crich D; Chandrasekera NS Mechanism of 4,6-O-benzylidene-directed β-mannosylation as determined by α-deuterium kinetic isotope effects. Angew. Chem., Int. Ed 2004, 43, 5386–5389 and references therein. [DOI] [PubMed] [Google Scholar]
- (104).El-Badri MH; Willenbring D; Tantillo DJ; Gervay-Hague J Mechanistic Studies on the Stereoselective Formation of beta-Mannosides from Mannosyl Iodides Using α-Deuterium Kinetic Isotope Effects. J. Org. Chem 2007, 72, 4663–4672. [DOI] [PubMed] [Google Scholar]
- (105).Huang M; Garrett GE; Birlirakis N; Bohe L; Pratt DA; Crich D Dissecting the mechanisms of a class of chemical glycosylation using primary13C kinetic isotope effects. Nat. Chem 2012, 4, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Adero PO; Furukawa T; Huang M; Mukherjee D; Retailleau P; Bohe L; Crich D Cation clock reactions for the determination of relative reaction kinetics in glycosylation reactions: Applications to gluco- and mannopyranosyl sulfoxide and trichloroacetimidate type donors. J. Am. Chem. Soc 2015, 137, 10336–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Crich D Mechanism of a chemical glycosylation reaction. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- (108).Huang M; Retailleau P; Bohe L; Crich D Cation clock permits distinction between the mechanisms of α- and β-O- and β-C-glycosylation in the mannopyranose series: evidence for the existence of a mannopyranosyl oxocarbenium ion. J. Am. Chem. Soc 2012, 134, 14746–14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Bohe L; Crich D A propos of glycosyl cations and the mechanism of chemical glycosylation; the current state of the art. Carbohydr. Res 2015, 403, 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Andreana PR; Crich D Guidelines for O-glycoside formation from first principles. ACS Cent. Sci 2021, 7, 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Crich D En route to the transformation of glycoscience: A chemist’s perspective on internal and external crossroads in glycochemistry. J. Am. Chem. Soc 2021, 143, 17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Crich D Methodology development and physical organic chemistry: a powerful combination for the advancement of glycochemistry. J. Org. Chem 2011, 76, 9193–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Demchenko AV General aspects of the glycosidic bond formation. In Handbook of Chemical Glycosylation; Demchenko AV, Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp 1–27. [Google Scholar]
- (114).Christensen HM; Oscarson S; Jensen HH Common side reactions of the glycosyl donor in chemical glycosylation. Carbohydr. Res 2015, 408, 51–95. [DOI] [PubMed] [Google Scholar]
- (115).Baghdasarian G; Woerpel KA Electrostatic effects on the reactions of cyclohexanone oxocarbenium ions. J. Org. Chem 2006, 71, 6851–6858. [DOI] [PubMed] [Google Scholar]
- (116).Yang MT; Woerpel KA The effect of electrostatic interactions on conformational equlibria of multiply substituted tetrahydropyran oxocarbenium ions. J. Org. Chem 2009, 74, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Bérces A; Enright G; Nukada T; Whitfield DM The conformational origin of the barrier to the formation of neighboring group assistance in glycosylation reactions: a dynamical density functional theory study. J. Am. Chem. Soc 2001, 123, 5460–5464. [DOI] [PubMed] [Google Scholar]
- (118).Nukada T; Berces A; Wang L; Zgierski MZ; Whitfield DM The two-conformer hypothesis: 2,3,4,6-tetra-O-methyl-mannopyranosyl and -glucopyranosyl oxacarbenium ions. Carbohydr. Res 2005, 340, 841–852. [DOI] [PubMed] [Google Scholar]
- (119).Whitfield DM Plausible transition states for glycosylation reactions. Carbohydr. Res 2012, 356, 180–190. [DOI] [PubMed] [Google Scholar]
- (120).Jensen HH; Lyngbye L; Bols M A free-energy relationship between the rate of acidic hydrolysis of glycosides and the pKa of isofagomines. Angew. Chem., Int. Ed 2001, 113, 3555–3557. [DOI] [PubMed] [Google Scholar]
- (121).Jensen HH; Bols M Steric effects are not the cause of the rate difference in hydrolysis of stereoisomeric glycosides. Org. Lett 2003, 5, 3419–3421. [DOI] [PubMed] [Google Scholar]
- (122).Jensen HH; Nordstrom LU; Bols M The disarming effect of the 4,6-acetal group on glycoside reactivity: torsional or electronic? J. Am. Chem. Soc 2004, 126, 9205–9213. [DOI] [PubMed] [Google Scholar]
- (123).Jensen HH; Bols M Stereoelectronic substituent effects. Acc. Chem. Res 2006, 39, 259–265. [DOI] [PubMed] [Google Scholar]
- (124).Jensen HH; Pedersen CM; Bols M Going to extremes: “super” armed glycosyl donors in glycosylation chemistry. Chem.—Eur. J 2007, 13, 7576–7582. [DOI] [PubMed] [Google Scholar]
- (125).Pedersen CM; Nordstrom LU; Bols M Super armed” glycosyl donors: conformational arming of thioglycosides by silylation. J. Am. Chem. Soc 2007, 129, 9222–9235. [DOI] [PubMed] [Google Scholar]
- (126).Pedersen CM; Marinescu LG; Bols M Conformationally armed glycosyl donors: reactivity quantification, new donors and one pot reactions. Chem. Commun 2008, 2465–2467. [DOI] [PubMed] [Google Scholar]
- (127).Heuckendorff M; Pedersen CM; Bols M Quantifying electronic effects of common carbohydrate protecting groups in a piperidine model system. Chem.—Eur. J 2010, 16, 13982–13994. [DOI] [PubMed] [Google Scholar]
- (128).Beaver MG; Woerpel KA Erosion of stereochemical control with increasing nucleophilicity: O-glycosylation at the diffusion limit. J. Org. Chem 2010, 75, 1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Garcia A; Sanzone JR; Woerpel KA Participation of alkoxy groups in reactions of acetals: violation of the reactivity/selectivity principle in a Curtin- Hammett kinetic scenario. Angew. Chem., Int. Ed. Engl 2015, 54, 12087–12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Mootoo DR; Konradsson P; Udodong U; Fraser-Reid B ″Armed″ and ″disarmed″ n-pentenyl glycosides in saccharide couplings leading to oligosaccharides. J. Am. Chem. Soc 1988, 110, 5583–5584. [Google Scholar]
- (131).Fraser-Reid B; Udodong UE; Wu ZF; Ottosson H; Merritt JR; Rao CS; Roberts C; Madsen R n-Pentenyl glycosides in organic chemistry: a contemporary example of serendipity. Synlett 1992, 1992, 927–942 and references therein. [Google Scholar]
- (132).Fraser-Reid B; Wu Z; Andrews CW; Skowronski E; Bowen JP Torsional effects in glycoside reactivity: saccharide couplings mediated by acetal protecting groups. J. Am. Chem. Soc 1991, 113, 1434–1435. [Google Scholar]
- (133).Wilson BG; Fraser-Reid B n-Pentenyl glycoside based methodology for determining the relative reactivities of variously protected pairs of glycosides. J. Org. Chem 1995, 60, 317–320. [Google Scholar]
- (134).Douglas NL; Ley SV; Lucking U; Warriner SL Tuning glycoside reactivity: new tool for efficient oligosaccharides synthesis. J. Chem. Soc., Perkin Trans 1 1998, 51–65. [Google Scholar]
- (135).Zhang Z; Ollmann IR; Ye XS; Wischnat R; Baasov T; Wong CH Programmable one-pot oligosaccharide synthesis. J. Am. Chem. Soc 1999, 121, 734–753. [Google Scholar]
- (136).Fridman M; Solomon D; Yogev S; Baasov T One-pot synthesis of glucosamine oligosaccharides. Org. Lett 2002, 4, 281–283. [DOI] [PubMed] [Google Scholar]
- (137).Bandara MD; Yasomanee JP; Demchenko AV Application of armed, disarmed, superarmed and superdisarmed building blocks in stereocontrolled glycosylation and expeditious oligosaccharide synthesis. In Selective Glycosylations: Synthetic Methods and Catalysts; Bennett CS, Ed.; Wiley, 2017; pp 29–58. [Google Scholar]
- (138).Zhu T; Boons GJ Thioglycosides protected as trans-2,3-cyclic carbonates in chemoselective glycosylations. Org. Lett 2001, 3, 4201–4203. [DOI] [PubMed] [Google Scholar]
- (139).Kamat MN; Demchenko AV Revisiting the armed-disarmed concept rationale: chemoselective activation of the S-benzoxazolyl glycosides in oligosaccharide synthesis. Org. Lett 2005, 7, 3215–3218. [DOI] [PubMed] [Google Scholar]
- (140).Mydock LK; Demchenko AV Superarming the S-benzoxazolyl glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. Org. Lett 2008, 10, 2103–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Mydock LK; Demchenko AV Application of the superarmed glycosyl donor to chemoselective oligosaccharide synthesis. Org. Lett 2008, 10, 2107–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Premathilake HD; Mydock LK; Demchenko AV Superarming common glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. J. Org. Chem 2010, 75, 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Panza M; Civera M; Yasomanee JP; Belvisi L; Demchenko AV Bromine-promoted glycosidation of conformationally superarmed thioglycosides. Chem.—Eur. J 2019, 25, 11831–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Nogueira JM; Issa JP; Chu A-HA; Sisel JA; Schum RS; Bennett CS Halide effects on cyclopropenium cation promoted glycosylation with deoxy sugars: highly α-selective glycosylations using a 3,3-dibromo-1,2-diphenylcyclopropene promoter. Eur. J. Org. Chem 2012, 2012, 4927–4930. [Google Scholar]
- (145).Sun L; Wu X; Xiong DC; Ye XS Stereoselective Koenigs-Knorr glycosylation catalyzed by urea. Angew. Chem., Int. Ed 2016, 55, 8041–8044. [DOI] [PubMed] [Google Scholar]
- (146).Singh Y; Demchenko AV Koenigs-Knorr glycosylation reaction catalyzed by trimethylsilyl trifluoromethanesulfonate. Chem.—Eur. J 2019, 25, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Yu F; Dickson JL; Loka RS; Xu H; Schaugaard RN; Schlegel HB; Luo L; Nguyen HM Diastereoselective sp3 CO bond formation via visible light-Induced, copper-catalyzed cross couplings of glycosyl bromides with aliphatic alcohols. ACS Catal. 2020, 10, 5990–6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Fischer E; Fischer H Über einige Derivate des Milchzuckers und der Maltose und über zwei neue Glucoside. Ber. Dtsch. Chem. Ges 1910, 43, 2521–2536. [Google Scholar]
- (149).Fischer E Acetohalogen Glucoses and Maltose and Melibiose p-Bromophenylosazones. Ber. Dtsch. Chem. Ges 1911, 44, 1898–1904. [Google Scholar]
- (150).Fischer E; von Mechel L Synthesis of phenyl glucosides. Ber. Dtsch. Chem. Ges 1916, 49, 2813–2820. [Google Scholar]
- (151).Fischer E; Helferich B Uber neue synthetische glucoside. Ann. Chem 1911, 383, 68–91. [Google Scholar]
- (152).Helferich B; Klein W Disaccharides. IV. Two tetraacetyl-b-D-glucoses. Liebigs Ann. 1926, 450, 219–229. [Google Scholar]
- (153).Helferich B; Bohn E; Winkler S Unsaturated derivatives of gentiobiose and cellobiose. Ber. Dtsch. Chem. Ges 1930, 63B, 989–998. [Google Scholar]
- (154).Reynolds DD; Evans WL The preparation of α-and β-gentiobiose octaacetates. J. Am. Chem. Soc 1938, 60, 2559–2561. [Google Scholar]
- (155).Kaeothip S; Demchenko AV Expeditious oligosaccharide synthesis via selective and orthogonal activation. Carbohydr. Res 2011, 346, 1371–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Helferich B; Bredereck H Zuckersynthesen. VIII. Liebigs Ann. Chem 1928, 465, 166–184. [Google Scholar]
- (157).Brigl P; Keppler H Carbohydrates. IV. Synthesis of α-glucosides. Ber. Dtsch. Chem. Ges 1926, 59, 1588–1591. [Google Scholar]
- (158).Schlubach HH; Schroter GA Die Gewinnung von Glucosiden der α-Reihe: Krystallisiertes α-Methyl-fructosid. Ber. 1928, 61, 1216–1222. [Google Scholar]
- (159).Hickinbottom WJ Glucosides. Part II. The preparation of α-glucosides from β-glucosyl chlorides. J. Chem. Soc 1929, 0, 1676–1687. [Google Scholar]
- (160).Zemplén G Einwirkung von Aluminiummetall und Quecksilbersalzen auf Aceto-halogen-zucker, I.: Synthese von α-Biosiden. Ber. Dtsch. Chem. Ges 1929, 62, 990–993. [Google Scholar]
- (161).Zemplén G; Nagy ZS Einwirkung von Quecksilbersalzen auf Aceto-halogenzucker. II. Mitteil.: Bildungs-Bedingungen des α-Phenyl-cellobiosids. Ber. Dtsch. Chem. Ges 1930, 63, 368–368. [Google Scholar]
- (162).Zemplen G; Gerecs A Action of mercury salts on acetohalogenosugars. IV. Direct preparation of alkyl biosides of the α-series. Ber. Dtsch. Chem. Ges 1930, 63B, 2720–2729. [Google Scholar]
- (163).Zemplén G Einwirkung von Quecksilbersalzen auf Acetohalogenzucker, III. Mitteil.: Synthese der achtfach methylierten Cellobiose. Ber. Dtsch. Chem. Ges 1930, 63, 1820–1823. [Google Scholar]
- (164).Fischer E; Bergmann M; Rabe A Acetobromorhanose and its use for the synthesis of rhamnosides. Ber. Dtsch. Chem. Ges. B 1920, 53B, 2362–2388. [Google Scholar]
- (165).Dale JK Three isomeric crystalline tetra-acetyl-methyl-D-mannosides. J. Am. Chem. Soc 1924, 46, 1046–1051. [Google Scholar]
- (166).Levene PA; Wolfrom ML Acetylmonoses. V. The rates of hydrolysis of tetraacetylmethylmannosides and of triacetylmethyllyxosides. J. Biol. Chem 1928, 79, 471–474. [Google Scholar]
- (167).Haworth WN; Hirst EL; Miller EJ Development of a novel form of stereoisomerism in the sugar series. I. The third variety of triacetylmethylrhamnoside. J. Chem. Soc 1929, 0, 2469–2479. [Google Scholar]
- (168).Hudson C Relations between rotatory power and structure in the sugar group. XXV. The ring structures of various monosaccharides. J. Am. Chem. Soc 1930, 52, 1680–1700. [DOI] [PubMed] [Google Scholar]
- (169).Isbell H The structures of the acetylmethylmannosides. J. Am. Chem. Soc 1930, 52, 5298–5298. [Google Scholar]
- (170).Haworth WN; Hirst EL; Streight HRL; Thomas HA; Webb JI CCCXLIX.—The structure of carbohydrates and their optical rotatory power. Part II. 4-Glucosido-α-mannose and its derivatives. J. Chem. Soc 1930, 0, 2636–2644. [Google Scholar]
- (171).Freudenberg K; Braun E A new isomerism in the sugar group. Naturwissenschaften 1930, 18, 393. [Google Scholar]
- (172).Freudenberg K; Scholz H Acetonesugars and other compounds of the carbohydrates. XXII. Cyclic acetates in the sugar group. Ber. Dtsch. Chem. Ges. B 1930, 63B, 1969–1972. [Google Scholar]
- (173).Bott HG; Haworth WN; Hirst EL Development of a novel form of isomerism in the sugar series. II. The third variety of tetraacetylmethylmannoside. J. Chem. Soc 1930, 0, 1395–1405. [Google Scholar]
- (174).Haworth WN; Hirst EL; Stacey M Walden inversion in the α-glucoheptose series. Preparation of new derivatives and the determination of the structure of methyl α-glucoheptoside. J. Chem. Soc 1931, 0, 2864–2872. [Google Scholar]
- (175).Isbell HS Derivatives of 4-glucosidomannose. Bur. Stand. J. Res 1931, 7, 1115–1131. [Google Scholar]
- (176).Isbell HS Chemistry of the Carbohydrates and Glycosides. Annu. Rev. Biochem 1940, 9, 65–92. [DOI] [PubMed] [Google Scholar]
- (177).Frush HL; Isbell HS Sugar acetates, acetylglycosyl halides and orthoacetates in relation to the Walden inversion. J. Res. Natl. Bur. Stand. (U. S.) 1941, 27, 413–428. [Google Scholar]
- (178).Frush HL; Isbell H Acetyl derivatives of certain heptoses, of gulose, and of lactulose. J. Res. Natl. Bur. Stds 1945, 34, 111–116. [Google Scholar]
- (179).Isbell HS; Frush HL Mechanisms for the formation of acetylglycosides and orthoesters from acetylglycosyl halides. J. Res. Natl. Bur. Stand 1949, 43, 161–171. [Google Scholar]
- (180).Helferich B; Doppstadt A; Gottschlich A Synthesen von Glykosiden im homogenen Medium. Naturwissenschaften 1953, 40, 441–442. [Google Scholar]
- (181).Helferich B; Weis K Zur Synthese von Glucosiden und von nicht-reduzierenden Disacchariden. Chem. Ber 1956, 89, 314–321. [Google Scholar]
- (182).Lemieux R; Morgan A The mechanism for the formation of 1, 2-cis-pyridine nucleosides from 1, 2-cis-acetohalogenosugars. A novel rearrangement. J. Am. Chem. Soc 1963, 85, 1889–1890. [Google Scholar]
- (183).Lemieux GA; Morgan AR The preparation and configurations fo tri-O-acetyl-α-D-glucopyranose 1,2-(orthoesters). Can. J. Chem 1965, 43, 2198–2204. [Google Scholar]
- (184).Kochetkov NK; Khorlin AY; Bochkov AF A new method of glycosylation. Tetrahedron 1967, 23, 693–707. [Google Scholar]
- (185).Schulz M; Steinmaus H 1.2-Orthoacetate aus 1.2-cis-Acetohalogenosen. Zeitschrift für Naturforschung B 1964, 19, 263–263. [Google Scholar]
- (186).Wulff G; Krüger W Untersuchungen Glykosidsynthese: III. Teil. Eine neue darstellungsmethode für 1, 2-D-glucose-orthoester. Carbohydr. Res 1971, 19, 139–142. [Google Scholar]
- (187).Hanessian S; Banoub J Chemistry of the glycosidic linkage. A rapid and efficient synthesis of carbohydrate 1, 2-orthoesters. Carbohydr. Res 1975, 44, C14–C17. [Google Scholar]
- (188).Wulff G; Schröder U; Wilchelhaus J On the stereochemical control of glycosylation reactions by the addition of tetrahydrofuran. Carbohydr. Res 1979, 72, 280–284. [Google Scholar]
- (189).Wang W; Kong F New synthetic methodology for regio-and stereoselective synthesis of oligosaccharides via sugar ortho ester intermediates. J. Org. Chem 1998, 63, 5744–5745. [DOI] [PubMed] [Google Scholar]
- (190).Kong F Recent studies on reaction pathways and applications of sugar orthoesters in synthesis of oligosaccharides. Carbohydr. Res 2007, 342, 345–373. [DOI] [PubMed] [Google Scholar]
- (191).Talley EA; Reynolds DD; Evans WL The synthesis of certain disaccharide acetates in the mannose series. J. Am. Chem. Soc 1943, 65, 575–582. [Google Scholar]
- (192).Perlin A Formation of diastereomeric orthoacetates of D-mannose: N.M.R. spectral evidence. Can. J. Chem 1963, 41, 399–406. [Google Scholar]
- (193).Ditmar R Über Methylglucoside und andere Derivate des Milchzuckers. Monatsh. Chem 1902, 23, 865–876. [Google Scholar]
- (194).Koto S; Yoshida T; Takenaka K; Zen S A Through-process for the preparation of methyl per-O-acetyl 1-thio-glycosides from aldoses. Bull. Chem. Soc. Jpn 1982, 55, 3667–3668. [Google Scholar]
- (195).Koto S; Morishima N; Shichi S; Haigoh H; Hirooka M; Okamoto M; Higuchi T; Shimizu K; Hashimoto Y; Irisawa T; Kawasaki H; Takahashi Y; Yamazaki M; Mori Y; Kudo K; Ikegaki T; Suzuki S; Zen S Dehydrative glycosylation using heptabenzyl derivatives of glucobioses and lactose. Bull. Chem. Soc. Jpn 1992, 65, 3257–3274. [Google Scholar]
- (196).Kartha KPR; Jennings HJ A simplified, one-pot preparation of acetobromosugars from reducing sugars. J. Carbohydr. Chem 1990, 9, 777–781. [Google Scholar]
- (197).Hunsen M; Long DA; D’Ardenne CR; Smith AL Mild one-pot preparation of glycosyl bromides. Carbohydr. Res 2005, 340, 2670–2674. [DOI] [PubMed] [Google Scholar]
- (198).Zhao J.-z.; Zhang X.-q.; Wu X; Xing Z.-b.; Yue A.-q.; Shao H.-w. Rapid synthesis of glycosyl bromides by ultrasound irradiation. Chem. Res. Chin. Univ 2013, 29, 71–75. [Google Scholar]
- (199).Huang LC; Liang PH; Liu CY; Lin CC Large-scale synthesis of per-O-acetylated saccharides and their sequential transformation to glycosyl bromides and thioglycosides. J. Carbohydr. Chem 2006, 25, 303–313. [Google Scholar]
- (200).Yalamanchili S; Nguyen TA; Zsikla A; Stamper G; DeYong AE; Florek J; Vasquez O; Pohl NLB; Bennett CS Automated, Multistep Continuous-Flow Synthesis of 2,6-Dideoxy and 3-Amino-2,3,6-trideoxy Monosaccharide Building Blocks. Angew. Chem., Int. Ed. Engl 2021, 60, 23171–23175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Bredereck H; Hambsch E About alkali metal compounds of 2.3.4.6-tetramethyl- d -glucopyranose and 1.3.4.6-tetramethyl- d -fructofuranose and their use in glycoside and disaccharide syntheses. Chem. Ber 1954, 87, 38–45. [Google Scholar]
- (202).Zemplén G; Gerecs Á Einwirkung von Quecksilbersalzen auf Aceto-halogen-zucker, IX. Mitteil.: Synthese von Derivaten der β-1-l-Rhamnosido-6-d-glykose. Ber. Dtsch. Chem. Ges 1934, 67, 2049–2051. [Google Scholar]
- (203).Kochetkov NK; Klimov EM; Malysheva NN; Demchenko AV Stereospecific synthesis of 1,2-cis-glycosides of 2-amino sugars. Carbohydr. Res 1993, 242, C7–C10. [DOI] [PubMed] [Google Scholar]
- (204).Barker R; Fletcher HG Jr 2,3,5-Tri-O-benzyl-D-ribosyl and -L-arabinosyl Bromides. J. Org. Chem 1961, 26, 4605–4609. [Google Scholar]
- (205).Lemieux RU; Kondo T Benzyl trifluoromethanesulfonate. Preparation of tri-O-acetyl-2-O-benzyl-α-D-galactopyranosyl bromide from 1,3,4,6-tetra-O-acetyl-α-D-galactopyranose. Carbohydr. Res 1974, 35, C4–C6. [Google Scholar]
- (206).Excoffier G; Gagnaire DY; Vignon MR Synthesis of oligosaccharides on a polymeric support. VII. Stereospecificity of glycosylation reactions: synthesis of isomaltose. Carbohydr. Res 1976, 46, 215–226. [Google Scholar]
- (207).Thiem J; Meyer B Synthesen mit Iod- und Bromtrimethylsilan in der Saccharidchemie. Chem. Ber 1980, 113, 3075–3085. [Google Scholar]
- (208).Gillard JW; Israel M Trimethylsilyl bromide as a mild, stereoselective anomeric brominating agent. Tetrahedron Lett. 1981, 22, 513–516. [Google Scholar]
- (209).Montero J-L; Winum J-Y; Leydet A; Kamal M; Pavia AA; Roque J-P A convenient synthesis of peracetylated glycosyl halides using bismuth (III) halides as catalysts. Carbohydr. Res 1997, 297, 175–180. [Google Scholar]
- (210).Tojino M; Hirose Y; Mizuno M Convenient synthesis of glycosyl bromide from 1-O-acetyl sugars by photo-irradiative phase-vanishing reaction of molecular bromine. Tetrahedron Lett. 2013, 54, 7124–7126. [Google Scholar]
- (211).Fletcher HG Jr; Hudson, C. 1, 5-Anhydro-xylitol. J. Am. Chem. Soc 1947, 69, 921–924. [DOI] [PubMed] [Google Scholar]
- (212).Ness RK; Fletcher HG Jr; Hudson C The reaction of 2, 3, 4, 6-tetrabenzoyl-α-D-glucopyranosyl bromide and 2, 3, 4, 6-tetrabenzoyl-α-D-mannopyranosyl bromide with methanol. Certain benzoylated derivatives of D-Glucose and D-Mannose. J. Am. Chem. Soc 1950, 72, 2200–2205. [Google Scholar]
- (213).Wen P; Simmons CJ; Ma Z.-x.; Blaszczyk SA; Balzer PG; Ye W; Duan X; Wang H-Y; Yin D; Stevens CM; Tang W Synthesis of glycosyl chlorides and bromides by chelation assisted activation of picolinic esters under mild neutral conditions. Org. Lett 2020, 22, 1495–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Weygand F; Ziemann H; Bestmann HJ Eine neue methode zur darstellung von acetobromzuckern. Chem. Ber 1958, 91, 2534–2537. [Google Scholar]
- (215).Weygand F; Ziemann H Glycosyl bromides from ethyl 1-thioglycosides. II. Ann. 1962, 657, 179–198. [Google Scholar]
- (216).Fugedi P; Garegg PJ; Lonn H; Norberg T Thioglycosides as glycosylating agents in oligosaccharide synthesis. Glycoconjugate J. 1987, 4, 97–108. [Google Scholar]
- (217).Singh Y; Wang T; Geringer SA; Stine KJ; Demchenko AV Regenerative glycosylation. J. Org. Chem 2018, 83, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Shingu Y; Nishida Y; Dohi H; Matsuda K; Kobayashi K Convenient access to halide ion-catalyzed α-glycosylation free from noxious fumes at the donor synthesis. J. Carbohydr. Chem 2002, 21, 605–611. [Google Scholar]
- (219).Polat T; Linhardt RJ Zinc triflate-benzoyl bromide: a versatile reagent for the conversion of ether into benzoate protecting groups and ether glycosides into glycosyl bromides. Carbohydr. Res 2003, 338, 447–449. [DOI] [PubMed] [Google Scholar]
- (220).Helferich B; Gootz R Synthesis of a tetrasaccharide acetate. Ber. Dtsch. Chem. Ges 1931, 64B, 109–114. [Google Scholar]
- (221).Singh Y; Demchenko AV Defining the scope of the acid-catalyzed glycosidation of glycosyl bromides. Chem.—Eur. J 2020, 26, 1042–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Knöchel A; Rudolph G Anionenaktivierung I: Anionenaktivierung durch makrocyklische Polyaether und ihr Einfluß auf nucleophile Substitutionsreaktionen. Tetrahedron Lett. 1974, 15, 3739–3740. [Google Scholar]
- (223).Knöchel A; Rudolph G; Thiem J Homogene koenigsknorr-reaktion mit hilfe zyklischer polyäther. Tetrahedron Lett. 1974, 15, 551–552. [Google Scholar]
- (224).Gouliaras C; Lee D; Chan L; Taylor MS Regioselective activation of glycosyl acceptors by a diarylborinic acid-derived catalyst. J. Am. Chem. Soc 2011, 133, 13926–13929. [DOI] [PubMed] [Google Scholar]
- (225).Nigudkar SS; Stine KJ; Demchenko AV Regenerative glycosylation under nucleophilic catalysis. J. Am. Chem. Soc 2014, 136, 921–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Shadrick M; Singh Y; Demchenko AV Stereocontrolled α-galactosylation under cooperative catalysis. J. Org. Chem 2020, 85, 15936–15944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Bredereck H; Wagner A; Faber G; Ott H; Rauther J Eine vereinfachte Oligosaccharid-Synthese. Chem. Ber 1959, 92, 1135–1139. [Google Scholar]
- (228).Bredereck H; Wagner A; Kuhn H; Ott H Oligosacchar-idsynthesen, II. Synthesen von Di-und Trisacchariden des Gentio-biosetyps. Chem. Ber 1960, 93, 1201–1206. [Google Scholar]
- (229).Khorlin AY; Nesmeyanov VA; Zurabyan SE Glycosylation of sugar 2, 3-diphenyl-2-cyclopropen-1-yl ethers. A new route to oligosaccharides. Carbohydr. Res 1975, 43, 69–77. [Google Scholar]
- (230).Kronzer FJ; Schuerch C The methanolysis of some derivatives of 2,3,4-tri-O-benzyl-α-D-glucopyranosyl bromide in the presence and absence of silver salts. Carbohydr. Res 1973, 27, 379–390. [Google Scholar]
- (231).Kihlberg JO; Leigh DA; Bundle DR The in situ activation of thioglycosides with bromine: an improved glycosylation method. J. Org. Chem 1990, 55, 2860–2863. [Google Scholar]
- (232).Wulff G; Röhle G; Krüger W New methods for preparation of glycosides. Angew. Chem., Int. Ed 1970, 9, 455–456. [Google Scholar]
- (233).Wulff G; Roehle G; Krueger W Glycoside synthesis. IV. Novel silver salts in glycoside synthesis. Chem. Ber 1972, 105, 1097–1110. [DOI] [PubMed] [Google Scholar]
- (234).Wulff G; Rohle G Results and problems of O-glycoside synthesis. Angew. Chem., Int. Ed. Engl 1974, 13, 157–170. [DOI] [PubMed] [Google Scholar]
- (235).Paulsen H; Lockhoff O Building units for oligosaccharides. XXX. New effective β-glycoside synthesis of mannose glycosides. Syntheses of mannose containing oligosaccharides. Chem. Ber 1981, 114, 3102–3114. [Google Scholar]
- (236).Garegg PJ; Ossowski P Silver zeolite as promoter in glycoside synthesis. The synthesis of β-D-mannopyranosides. Acta Chem. Scand., Ser. B 1983, 37b, 249–250. [Google Scholar]
- (237).Van Boeckel C; Beetz T; Kock-van Dalen A; Van Bekkum H A note on the use of porous silver silicates as promoter in carbohydrate coupling reactions. Recl. Trav. Chim. Pays-Bas 1987, 106, 596–598. [Google Scholar]
- (238).Garegg PJ; Johansson R; Samuelsson B Silver imidazolate-assisted glycosidations. Part 6. Synthesis of 1,2-trans-linked disaccharides. Acta Chem. Scand., Ser. B 1982, 36b, 249–250. [Google Scholar]
- (239).Helferich B; Wedemeyer KF Preparation of glucosides from acetobromoglucose. Ann. 1949, 563, 139–145. [Google Scholar]
- (240).Schroeder LR; Green JW Koenigs-Knorr syntheses with mercuric salts. J. Chem. Soc. C: Organic 1966, 0, 530–531. [Google Scholar]
- (241).Sinay P Recent advances in glycosylation reactions. Pure Appl. Chem 1978, 50, 1437–1452. [Google Scholar]
- (242).Paulsen H; Kolář Č Bausteine von Oligosacchariden, XX. Synthese der Oligosaccharid-Determinanten der Blutgruppensubstanzen der Type 2 des ABH-Systems. Diskussion der α-Glycosid-Synthese. Chem. Ber 1981, 114, 306–321. [Google Scholar]
- (243).Helferich B; Wedemeyer KF Zur Darstellung von Glykosiden aus Acetobromhalogenosen. Chem. Ber 1950, 83, 538–540. [Google Scholar]
- (244).Bock K; Meldal M Mercury iodide as a catalyst in oligosaccharide synthesis. Acta Chem. Scand 1983, 37b, 775–783. [DOI] [PubMed] [Google Scholar]
- (245).Higashi K; Nakayama K; Soga T; Shioya E; Uoto K; Kusama T Novel stereoselective glycosidation by the combined use of trityl halide and Lewis acid. Chem. Pharm. Bull 1990, 38, 3280–3282. [Google Scholar]
- (246).Murakami T; Sato Y; Shibakami M Stereoselective glycosylations using benzoylated glucosyl halides with inexpensive promoters. Carbohydr. Res 2008, 343, 1297–1308. [DOI] [PubMed] [Google Scholar]
- (247).Higashi K; Nakayama K; Uoto K; Shioya E; Kusama T A novel glycoside anomerization catalyzed by trimethylsilyl bromide and zinc bromide in combination. Chem. Pharm. Bull 1991, 39, 590–592. [Google Scholar]
- (248).Higashi K; Nakayama K; Shioya E; Kusama T Direct transformation of O-glycosides into other O-glycosides using trimethylsilyl bromide and zinc bromide. Chem. Pharm. Bull 1992, 40, 1042–1043. [Google Scholar]
- (249).Bernstein S; Conrow R Steroid conjugates. VI. Improved Koenigs-Knorr synthesis of aryl glucuronides using cadmium carbonate, a new and effective catalyst. J. Org. Chem 1971, 36, 863–870. [DOI] [PubMed] [Google Scholar]
- (250).Wimmer Z; Pechová L; Saman D Koenigs-Knorr synthesis of cycloalkyl glycosides. Molecules 2004, 9, 902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Dick WE Jr Efficiency of cadmium carbonate as an aryl glycosidation catalyst: effects of lot variations on product compositions. Carbohydr. Res 1979, 70, 313–318. [Google Scholar]
- (252).Mukherjee D; Kumar Ray P; Sankar Chowdhury U Synthesis of glycosides via indium (III) chloride mediated activation of glycosyl halide in neutral condition. Tetrahedron 2001, 57, 7701–7704. [Google Scholar]
- (253).Chandra S; Yadav RN; Paniagua A; Banik BK Indium salts-catalyzed O and S-glycosylation of bromo sugar with benzyl glycolate: an unprecedented hydrogenolysis. Tetrahedron Lett. 2016, 57, 1425–1429. [Google Scholar]
- (254).Li C; Liang H; Zhang Z.-x.; Wang Z; Yu L; Liu H; An F; Wang S; Ma L; Xue W A catalytic Koenigs-Knorr glycosylation based on acceptor activation with In(NTf2)3. Tetrahedron 2018, 74, 3963–3970. [Google Scholar]
- (255).Ogawa T; Matsui M An approach to synthesis of glycosides: enhancement of nucleophilicity of hydroxyl groups by trialkylstannylation. Carbohydr. Res 1976, 51, C13–C18. [Google Scholar]
- (256).Lubineau A; Malleron A Stannous triflate mediated glycosidations. A stereoselective synthesis of β-D-glucosides. Tetrahedron Lett. 1985, 26, 1713–1716. [Google Scholar]
- (257).Yu F; Li J; DeMent P; Tu Y; Schlegel H; Nguyen H Phenanthroline-catalyzed stereoretentive glycosylations. Angew. Chem., Int. Ed 2019, 58, 6957–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Lemieux RU; Hendriks KB; Stick RV; James K Halide ion catalyzed glycosylation reactions. Syntheses of α-linked disaccharides. J. Am. Chem. Soc 1975, 97, 4056–4062 and references therein. [Google Scholar]
- (259).Lemieux RU; Driguez H Chemical synthesis of 2-acetamido-2-deoxy-4-O-(. alpha.-L-fucopyranosyl)-3-O-(. beta.-D-galactopyranosyl)-D-glucose. Lewis a blood-group antigenic determinant. J. Am. Chem. Soc 1975, 97, 4063–4069. [DOI] [PubMed] [Google Scholar]
- (260).Kartha KPR; Aloui M; Field RA Iodine: a versatile reagent in carbohydrate chemistry III. Efficient activation of glycosyl halides in combination with DDQ. Tetrahedron Lett. 1996, 37, 8807–8810. [Google Scholar]
- (261).Lanz G; Madsen R Glycosylation with disarmed gycosyl bromides promoted by iodonium ions. Eur. J. Org. Chem 2016, 2016, 3119–3125. [Google Scholar]
- (262).Kartha KR; Field RA Glycosylation chemistry promoted by iodine monobromide: Efficient synthesis of glycosyl bromides from thioglycosides, and O-glycosides from ‘disarmed’thioglycosides and glycosyl bromides. Tetrahedron Lett. 1997, 38, 8233–8236. [Google Scholar]
- (263).Stachulski AV Glucuronidation of alcohols using the bromosugar-iodonium reagent method. Tetrahedron Lett. 2001, 42, 6611–6613. [Google Scholar]
- (264).Micheel F; Micheel H Configuration of the α- and β-forms in the sugar series. Ber. Dtsch. Chem. Ges. B 1930, 63B, 386–393. [Google Scholar]
- (265).West AC; Schuerch C The reverse anomeric effect and the synthesis of α-glycosides. J. Am. Chem. Soc 1973, 95, 1333–1335. [Google Scholar]
- (266).Kronzer FJ; Schuerch C Use of positively charged, leaving groups in the synthesis of α-D-linked galactosides. Attempted synthesis of 3-O-α-(D-(galactopyranosyl)-D-galactose. Carbohydr. Res 1974, 33, 273–280. [Google Scholar]
- (267).Ishikawa T; Fletcher HG The synthesis and solvolysis of some D-glucopyranosyl bromides having a benzyl group at C-2. J. Org. Chem 1969, 34, 563–571. [DOI] [PubMed] [Google Scholar]
- (268).Frechet JM; Schuerch C Solid-phase synthesis of oligosaccharides. II. Steric control by C-6 substituents in glucoside syntheses. J. Am. Chem. Soc 1972, 94, 604–609. [Google Scholar]
- (269).Wulff G; Röhle G Untersuchungen zur Glykosidsynthese, VI. Kinetische Untersuchungen zum Mechanismus der Koenigs-Knorr-Reaktion. Chem. Ber 1972, 105, 1122–1132. [DOI] [PubMed] [Google Scholar]
- (270).Wulff G; Röhle G; Schmidt U Untersuchungen zur Glykosidsynthese, V. Reaktionsprodukte und Stereospezifität der Glucosylierung in Gegenwart unlöslicher Silbersalze in Diäthylather. Chem. Ber 1972, 105, 1111–1121. [DOI] [PubMed] [Google Scholar]
- (271).Kaneko M; Herzon SB Scope and limitations of 2-deoxy- and 2,6-dideoxyglycosyl bromides as donors for the synthesis of beta-2-deoxy- and beta-2,6-dideoxyglycosides. Org. Lett 2014, 16, 2776–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Lee D; Williamson CL; Chan L; Taylor MS Regioselective, borinic acid-catalyzed monoacylation, sulfonylation and alkylation of diols and carbohydrates: expansion of substrate scope and mechanistic studies. J. Am. Chem. Soc 2012, 134, 8260–8267. [DOI] [PubMed] [Google Scholar]
- (273).Lee D; Taylor MS Borinic acid-catalyzed regioselective acylation of carbohydrate derivatives. J. Am. Chem. Soc 2011, 133, 3724–3727. [DOI] [PubMed] [Google Scholar]
- (274).Chan L; Taylor MS Regioselective alkylation of carbohydrate derivatives catalyzed by a diarylborinic acid derivative. Org. Lett 2011, 13, 3090–3093. [DOI] [PubMed] [Google Scholar]
- (275).Yang W; Mortier WJ The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc 1986, 108, 5708–5711. [DOI] [PubMed] [Google Scholar]
- (276).Nigudkar SS; Wang T; Pistorio SG; Yasomanee JP; Stine KJ; Demchenko AV OFox imidates as versatile glycosyl donors for chemical glycosylation. Org. Biomol. Chem 2017, 15, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Escopy S; Singh Y; Stine KJ; Demchenko AV A Streamlined regenerative glycosylation reaction: direct, acid-free activation of thioglycosides. Chem.—Eur. J 2021, 27, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Fraser-Reid B; Wu Z; Udodong UE; Ottosson H Armed/disarmed effects in glycosyl donors: rationalization and sidetracking. J. Org. Chem 1990, 55, 6068–6070. [Google Scholar]
- (279).Demchenko AV; Rousson E; Boons GJ Stereoselective 1,2-cis-galactosylation assisted by remote neighboring group participation and solvent effects. Tetrahedron Lett. 1999, 40, 6523–6526. [Google Scholar]
- (280).Creutz SE; Lotito KJ; Fu GC; Peters JC Photoinduced Ullmann C-N coupling: demonstrating the viability of a radical pathway. Science 2012, 338, 647–651. [DOI] [PubMed] [Google Scholar]
- (281).Ahn JM; Ratani TS; Hannoun KI; Fu GC; Peters JC Photoinduced, copper-catalyzed alkylation of amines: a mechanistic study of the cross-coupling of carbazole with alkyl bromides. J. Am. Chem. Soc 2017, 139, 12716–12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Kochi J; Subramanian R Kinetics of Electron-Transfer Oxidation of Alkyl Radicals by Copper (II) Complexes. J. Am. Chem. Soc 1965, 87, 4855–4866. [Google Scholar]
- (283).Kochi JK Mechanisms of organic oxidation and reduction by metal complexes. Science 1967, 155, 415–424. [DOI] [PubMed] [Google Scholar]
- (284).Kochi J; Bemis A; Jenkins C Mechanism of electron transfer oxidation of alkyl radicals by copper (II) complexes. J. Am. Chem. Soc 1968, 90, 4616–4625. [Google Scholar]
- (285).De La Mare H; Kochi J; Rust F The oxidation of free radicals by metal ions. J. Am. Chem. Soc 1961, 83, 2013–2015. [Google Scholar]
- (286).Wayner DDM; McPhee DJ; Griller D Oxidation and reduction potentials of transient free radicals. J. Am. Chem. Soc 1988, 110, 132–137. [Google Scholar]
- (287).Zhang Z; Wang F; Mu X; Chen P; Liu G Copper-catalyzed regioselective fluorination of allylic halides. Angew. Chem., Int. Ed 2013, 52, 7549–7553. [DOI] [PubMed] [Google Scholar]
- (288).Andrews RS; Becker JJ; Gagne MR Intermolecular addition of glycosyl halides to alkenes mediated by visible light. Angew. Chem., Int. Ed. Engl 2010, 49, 7274–7276. [DOI] [PubMed] [Google Scholar]
- (289).Zemplen G; Gerecs A; Flesch H Action of mercury salts on acetohalosugars. XI. Synthesis of some derivatives of β-1-l-rhamnosido-6-d-galactose. Ber. Dtsch. Chem. Ges. B 1938, 71B, 774–776. [Google Scholar]
- (290).Zemplen G; Bognar R Action of mercury salts on acetohalo sugars. XIII. Synthesis of isoprimeverose (6-α-d-xylosido-d-glucose), the α-isomer of primeverose, and its derivatives. Ber. Dtsch. Chem. Ges. B 1939, 72B, 1160–1167. [Google Scholar]
- (291).Helferich B; Berger A Über die Synthese von Glucuroniden. Chem. Ber 1957, 90, 2492–2498. [Google Scholar]
- (292).Helferich B; Zirner J Synthesis of tetra-O-acetyhexoses with a free 2-hydroxyl group. Synthesis of disaccharides. Chem. Ber 1962, 95, 2604–2611. [Google Scholar]
- (293).Lemieux RU Some implications in carbohydrate chemistry of theories relating to the mechanisms of replacement reactions. Adv. Carbohydr. Chem. Biochem 1954, 9, 1–57 and references therein. [DOI] [PubMed] [Google Scholar]
- (294).Lemieux R; Brice C A comparison of the properties of pentaacetates and methyl 1, 2-orthoacetates of glucose and mannose. Can. J. Chem 1955, 33, 109–119. [Google Scholar]
- (295).Guthrie RD; McCarthy JF Acetolysis. Adv. Carbohydr. Chem 1967, 22, 11–23. [DOI] [PubMed] [Google Scholar]
- (296).Conchie J; Levvy G; Marsh C Methyl and phenyl glycosides of the common sugars. Adv. Carbohydr. Chem 1957, 12, 157–187. [DOI] [PubMed] [Google Scholar]
- (297).Bishop C; Cooper F Glycosidation of sugars: I. Formation of methyl-D-xylosides. Can. J. Chem 1962, 40, 224–232. [Google Scholar]
- (298).Bishop C; Cooper F Glycosidation of sugars: II. Methanolysis of d-xylose, d-arabinose, d-lyxose, and d-ribose. Can. J. Chem 1963, 41, 2743–2758. [Google Scholar]
- (299).Schmidt RR New methods for the synthesis of glycosides and oligosaccharides - are there alternatives to the Koenigs-Knorr method. Angew. Chem., Int. Ed. Engl 1986, 25, 212–235. [Google Scholar]
- (300).Nakayama K; Higashi K; Soga T; Uoto K; Kusama T Zinc triflate-promoted glycosidation: synthesis of lipid A disaccharide intermediates. Chem. Pharm. Bull 1992, 40, 2909–2912. [DOI] [PubMed] [Google Scholar]
- (301).Kusama T; Soga T; Ono Y; Kumazawa E; Shioya E; Nakayama K; Uoto K; Osada Y Synthesis and biological activities of lipid A analogs: modification of a glycosidically bound group with chemically stable polar acidic groups and lipophilic groups on the disaccharide backbone with tetradecanoyl or N-dodecanoylglycyl groups. Chem. Pharm. Bull 1991, 39, 3244–3253. [DOI] [PubMed] [Google Scholar]
- (302).Bernstein S; Joseph JP; Dusza JP Steroid conjugates. VII. Preparation of N-acetylglucosaminides of 17α-and 17β-estradiol. Biochemistry 1971, 10, 2941–2947. [DOI] [PubMed] [Google Scholar]
- (303).Iida T; Tazawa S; Ohshima Y; Niwa T; Goto J; Nambara T Analysis of conjugated bile acids in human biological fluids. Synthesis of hyodeoxycholic acid 3-and 6-glycosides and related compounds. Chem. Pharm. Bull 1994, 42, 1479–1484. [DOI] [PubMed] [Google Scholar]
- (304).Mukherjee D; Sarkar SK; Chowdhury US; Taneja SC A rapid stereoselective C-glycosidation of indoles and pyrrole via indium trichloride promoted reactions of glycosyl halides. Tetrahedron Lett. 2007, 48, 663–667. [Google Scholar]
- (305).Lemieux R; Hayami J -i. The mechanism of the anomerization of the tetra-O-acetyl-D-glucopyranosyl chlorides. Can. J. Chem 1965, 43, 2162–2173. [Google Scholar]
- (306).Lubineau A; Le Gallic J; Malleron A Stannous triflate mediated glycosidations. A stereoselective synthesis of 2-amino-2-deoxy-β-D-glucopyranosides directly with the natural N-acetyl protecting group. Tetrahedron Lett. 1987, 28, 5041–5044. [Google Scholar]
- (307).Renaudet O; Dumy P Synthesis of glycosylated-β (1–4)-amino (methoxy) and-oxyamino carbohydrate analogues. Tetrahedron 2002, 58, 2127–2135. [Google Scholar]
- (308).Steber HB; Singh Y; Demchenko AV Bismuth(III) triflate as a novel and efficient activator for glycosyl halides. Org. Biomol. Chem 2021, 19, 3220–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Lemieux RU; Morgan AR The abnormal conformations of pyridinium α-glycopyranosides. Can. J. Chem 1965, 43, 2205–2213. [Google Scholar]
- (310).Lemieux RU; Morgan AR Mechanisms for the reactions of the anomeric tetra-O-acetyl-D-glucopyranosyl bromides with pyridine and 2-pyridone. Can. J. Chem 1965, 43, 2214–2221. [Google Scholar]
- (311).Micheel F; Micheel H Configuration of the α- and β-forms in the sugar series. II. Configuration of glucosamine. Ber. Dtsch. Chem. Ges. B 1932, 65B, 253–258. [Google Scholar]
- (312).Hess K; Heumann KE The question of the determination of configuration at the glucosidic carbon atom in sugars by ammonium salt formation. Ber. Dtsch. Chem. Ges. B 1939, 72B, 1495–1499. [Google Scholar]
- (313).Eby R; Schuerch C The use of positively charged leaving-groups in the synthesis of α-D-linked glucosides. Synthesis of methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-α-D-glucopyranoside. Carbohydr. Res 1975, 39, 33–38. [Google Scholar]
- (314).Igarashi K The Koenigs-Knorr reaction. Adv. Carbohydr. Chem. Biochem 1977, 34, 243–283. [Google Scholar]
- (315).Kronzer FJ; Schuerch C The use of 2,3,4,6-tetra-O-benzyl-α-D-glycopyranosyl iodides in α-glycoside synthesis. Carbohydr. Res 1974, 34, 71–78 and references therein. [Google Scholar]
- (316).Lucas TJ; Schuerch C Methanolysis as a model reaction for oligosaccharide synthesis of some 6-substituted 2,3,4-tri-O-benzyl-D-galactopyranosyl derivatives. Carbohydr. Res 1975, 39, 39–45. [Google Scholar]
- (317).Srivastava VK; Schuerch C Synthesis of β-D-mannopyranosides and β-L-rhamnopyranosides by glycosidation at C-1. J. Org. Chem 1981, 46, 1121–1126. [Google Scholar]
- (318).Eby R; Schuerch C The synthesis of α-and β-(1->3)-linked glucopyranose disaccharides and their protein conjugates. Carbohydr. Res 1982, 102, 131–138 and references therein. [DOI] [PubMed] [Google Scholar]
- (319).Lemieux RU; Driguez H The chemical synthesis of 2-O-(α-L-fucopyranosyl)-3-O-(α-D-galactopyranosyl)-D-galactose. The terminal structure of blood-group B antigenic determinant. J. Am. Chem. Soc 1975, 97, 4069–4075. [DOI] [PubMed] [Google Scholar]
- (320).Kartha KR; Ballell L; Bilke J; McNeil M; Field RA Iodine and its interhalogen compounds: versatile reagents in carbohydrate chemistry. XIV. Glycosylated amino acid synthesis. J. Chem. Soc. Perkin Trans 1 2001, 770–772. [Google Scholar]
- (321).Kaeothip S; Yasomanee JP; Demchenko AV Glycosidation of thioglycosides in the presence of bromine: mechanism, reactivity, and stereoselectivity. J. Org. Chem 2012, 77, 291–299. [DOI] [PubMed] [Google Scholar]
- (322).Mannino MP; Demchenko AV Hydrogen-bond-mediated Aglycone Delivery (HAD) and related methods in carbohydrate chemistry. In Carbohydrate Chemistry: Chemical and Biological Approaches (Specialist Periodical Reports); Rauter AP, Lindhorst TK, Queneau Y, Eds.; RSC, 2021; Vol. 44, pp 93–116. [Google Scholar]
- (323).Yasomanee JP; Demchenko AV Hydrogen bond-mediated aglycone delivery (HAD): a highly stereoselective synthesis of 1,2-cis α-D-glucosides from common glycosyl donors in the presence of bromine. Chem.—Eur. J 2015, 21, 6572–6581. [DOI] [PubMed] [Google Scholar]
- (324).Frechet JM; Schuerch C Solid-phase synthesis of oligosaccharides. I. Preparation of the solid support. Poly[p-(1-propen-3-ol-1-yl)styrene]. J. Am. Chem. Soc 1971, 93, 492–496. [Google Scholar]
- (325).Frechet JM; Schuerch C Solid-phase synthesis of oligosacchapides: III. Preparation of some derivatives of di-and trisaccharides via a simple alcoholysis reaction. Carbohydr. Res 1972, 22, 399–412. [Google Scholar]
- (326).Serbian I; Prell E; Al-Harrasi A; Csuk R Stereoselective synthesis of alkyl pyranosides on a laboratory scale. Mediterranean J. Chem 2020, 10, 269–276. [Google Scholar]
- (327).Cardona A; Boutureira O; Castillón S; Díaz Y; Matheu MI Metal-free and VOC-free O-glycosylation in supercritical CO2. Green Chem. 2017, 19, 2687–2694. [Google Scholar]
- (328).Takiura K; Honda S; Endo T; Kakehi K Studies of oligosaccharides. IX. Synthesis of gentiooligosaccharides by bock condensation. Chem. Pharm. Bull 1972, 20, 438–442. [Google Scholar]
- (329).Koto S; Uchida T; Zen S Syntheses of isomaltose, isomaltotetraose, and isomaltooctaose. Bull. Chem. Soc. Jpn 1973, 46, 2520–2523. [Google Scholar]
- (330).Paulsen H Advances in selective chemical syntheses of complex oligosaccharides. Angew. Chem., Int. Ed. Engl 1982, 21, 155–173. [Google Scholar]
- (331).Ogawa T; Yamamoto H; Nukada T; Kitajima T; Sugimoto M Synthetic approach to glycan chains of a glycoprotein and proteoglycan. Pure Appl. Chem 1984, 56, 779–795. [Google Scholar]
- (332).Depre D; Duffels A; Green LG; Lenz R; Ley SV; Wong CH Synthesis of glycans from the glycodelins: two undeca-, two deca-, three nona-, an octa- and a heptasaccharide. Chem.—Eur. J 1999, 5, 3326–3340. [Google Scholar]
- (333).Nicolaou KC; Dolle RE; Papahatjis DP; Randall JL Practical synthesis of oligosaccharides. Partial synthesis of avermectin B1a. J. Am. Chem. Soc 1984, 106, 4189–4192. [Google Scholar]
- (334).Randall JL; Nicolaou KC Preparation and reactions of glycosyl fluorides. ACS Symp. Ser 1988, 374, 13–28. [Google Scholar]
- (335).Lonn H Synthesis of a tri- and a heptasaccharide which contain α-L-fucopyranosyl groups and are part of the complex type of carbohydrate moiety of glycoproteins. Carbohydr. Res 1985, 139, 105–113. [DOI] [PubMed] [Google Scholar]
- (336).Lonn H Synthesis of a tetra- and a nona-saccharide which contain α-L-fucopyranosyl groups and are part of the complex type of carbohydrate moiety of glycoproteins. Carbohydr. Res 1985, 139, 115–121. [DOI] [PubMed] [Google Scholar]
- (337).Garegg PJ; Oscarson S A synthesis of 8-methoxycarbonyloct-1-yl O-α-D-galactopyranosyl-(1–3)-O-β-D-galactopyranosyl-(1–4)-2-acetamido-2-deoxy-β-D-glucopyranoside. Carbohydr. Res 1985, 136, 207–213. [Google Scholar]
- (338).Leontein K; Nilsson M; Norberg T Synthesis of the methyl and 1-octyl glycosides of the P-antigen tetrasaccharide (globotetraose). Carbohydr. Res 1985, 144, 231–240. [DOI] [PubMed] [Google Scholar]
- (339).Paulsen H; Heume M; Nurnberger H Synthese der verzwiegten Nonasaccharid-Sequenz der ″bisected″ Structur von N-Glycoproteinen. Carbohydr. Res 1990, 200, 127–166. [DOI] [PubMed] [Google Scholar]
- (340).Mehta S; Pinto BM Novel glycosidation methodology. The use of phenylselenoglycosides as glycosyl donors and acceptors in oligosaccharide synthesis. J. Org. Chem 1993, 58, 3269–3276. [Google Scholar]
- (341).Baeschlin DK; Chaperon AR; Charbonneau V; Green LG; Ley SV; Lucking U; Walther E Rapid assembly of oligosaccharides: Total synthesis of a glycosylphosphatidylinositol anchor of trypanosoma brucei. Angew. Chem., Int. Ed 1998, 37, 3423–3428. [DOI] [PubMed] [Google Scholar]
- (342).Colley MA Action des haloides libres et de quelques chlorures sur la glucose. Ann. Chem. Phys 1870, 21, 363–377. [Google Scholar]
- (343).Kononov LO; Magnusson G Synthesis of methyl and allyl α-glycosides of N-acetylneuraminic acid in the absence of added promoter. Acta Chem. Scand 1998, 52, 141–144. [Google Scholar]
- (344).Shpirt AM; Kononov LO; Torgov VI; Shibaev VN Conversion of N-acetylneuraminic acid glycosyl chloride into dibenzyl glycosyl phosphate: O-glycosylation in the absence of a promoter. Russ. Chem. Bull 2004, 53, 717–719. [Google Scholar]
- (345).Bonomi P; Attieh MD; Gonzato C; Haupt K A New Versatile Water-Soluble Iniferter Platform for the Preparation of Molecularly Imprinted Nanoparticles by Photopolymerisation in Aqueous Media. Chem.—Eur. J 2016, 22, 10150–10154. [DOI] [PubMed] [Google Scholar]
- (346).Singhamahapatra A; Sahoo L; Paul KJV; Varghese B; Loganathan D Improved synthesis of per-O-acetylated C1 hydroxyglycopyranose and structural study as non-covalent organic framework. Tetrahedron Lett. 2013, 54, 6121–6124. [Google Scholar]
- (347).Ibatullin FM; Selivanov SI Reaction of 1,2-trans-glycosyl acetates with phosphorus pentachloride: new efficient approach to 1,2-trans-glycosyl chlorides. Tetrahedron Lett. 2002, 43, 9577–9580. [Google Scholar]
- (348).Bednarczyk D; Walczewska A; Grzywacz D; Sikorski A; Liberek B; Myszka H Differently N-protected 3,4,6-tri-O-acetyl-2-amino-2-deoxy-d-glucopyranosyl chlorides and their application in the synthesis of diosgenyl 2-amino-2-deoxy-β-d-glucopyranoside. Carbohydr. Res 2013, 367, 10–17. [DOI] [PubMed] [Google Scholar]
- (349).Ziegler T; Seidl U Preparation of some Amygdalin-Derived Gentiobiosyl Donors and Acceptors for Oligosaccharide Syntheses. J. Carbohydr. Chem 1991, 10, 813–831. [Google Scholar]
- (350).Watt GM; Boons G-J A convergent strategy for the preparation of N-glycan core di-, tri-, and pentasaccharide thioaldoses for the site-specific glycosylation of peptides and proteins bearing free cysteines. Carbohydr. Res 2004, 339, 181–193. [DOI] [PubMed] [Google Scholar]
- (351).Iversen T; Bundle DR Antigenic determinants of Salmonella serogroups A and D1. Synthesis of trisaccharide glycosides for use as artificial antigens. Carbohydr. Res 1982, 103, 29–40. [DOI] [PubMed] [Google Scholar]
- (352).Encinas L; Chiara JL Polymer-assisted solution-phase synthesis of glycosyl chlorides and bromides using a supported dialkylformamide as catalyst. J. Comb. Chem 2008, 10, 361–363. [DOI] [PubMed] [Google Scholar]
- (353).Gómez AM; Pedregosa A; Casillas M; Uriel C; López JC Synthesis of C-1 alkyl and aryl glycals from pyranosyl or furanosyl chlorides by treatment with organolithium reagents. Eur. J. Org. Chem 2009, 2009, 3579–3588. [Google Scholar]
- (354).Hung SC; Wong CH Synthesis of glycosyl chlorides with acid-labile protecting groups. Tetrahedron Lett. 1996, 37, 4903–4906. [Google Scholar]
- (355).Ernst B; Winkler T Preparation of glycosyl halides under neutral conditions. Tetrahedron Lett. 1989, 30, 3081–3084. [Google Scholar]
- (356).Cicchillo RM; Norris P A convenient synthesis of glycosyl chlorides from sugar hemiacetals using triphosgene as the chlorine source. Carbohydr. Res 2000, 328, 431–434. [DOI] [PubMed] [Google Scholar]
- (357).Traboni S; Liccardo F; Bedini E; Giordano M; Iadonisi A Solvent-free synthesis of glycosyl chlorides based on the triphenyl phosphine/hexachloroacetone system. Tetrahedron Lett. 2017, 58, 1762–1764. [Google Scholar]
- (358).Huy PH; Filbrich I A General Catalytic Method for Highly Cost- and Atom-Efficient Nucleophilic Substitutions. Chem.—Eur. J 2018, 24, 7410–7416. [DOI] [PubMed] [Google Scholar]
- (359).Tatina MB; Khong DT; Judeh ZMA Efficient synthesis of α-glycosyl chlorides using 2-chloro-1,3-dimethylimidazolinium chloride: A convenient protocol for quick one-pot glycosylation. Eur. J. Org. Chem 2018, 2018, 2208–2213. [Google Scholar]
- (360).Pongener I; Nikitin K; McGarrigle EM Synthesis of glycosyl chlorides using catalytic Appel conditions. Org. Biomol. Chem 2019, 17, 7531–7535. [DOI] [PubMed] [Google Scholar]
- (361).Sugiyama S; Diakur JM A convenient preparation of glycosyl chlorides from aryl/alkyl thioglycosides. Org. Lett 2000, 2, 2713–2715. [DOI] [PubMed] [Google Scholar]
- (362).Verma VP; Wang C-C Highly Stereoselective Glycosyl-Chloride-Mediated Synthesis of 2-Deoxyglucosides. Chem.—Eur. J 2013, 19, 846–851. [DOI] [PubMed] [Google Scholar]
- (363).Chang CW; Wu CH; Lin MH; Liao PH; Chang CC; Chuang HH; Lin SC; Lam S; Verma VP; Hsu CP; Wang CC Establishment of guidelines for the control of glycosylation reactions and intermediates by quantitative assessment of reactivity. Angew. Chem., Int. Ed. Engl 2019, 58, 16775–16779. [DOI] [PubMed] [Google Scholar]
- (364).Helferich B; Wedemeyer KF Zur Darstellung von Glucosiden aus Acetobromglucose. Justus Liebigs Ann. Chem 1949, 563, 139–145. [Google Scholar]
- (365).Chauvin AL; Nepogodiev SA; Field RA Synthesis of an apiose-containing disaccharide fragment of rhamnogalacturonan-II and some analogues. Carbohydr. Res 2004, 339, 21–27. [DOI] [PubMed] [Google Scholar]
- (366).Ito Y; Ogawa T Highly stereoselective glycosylation of sialic acid aided by stereocontrolling auxiliaries. Tetrahedron 1990, 46, 89–102. [Google Scholar]
- (367).Kondo T; Tomoo T; Abe H; Isobe M; Goto T Simple construction of Neu5Ac(α2–8)Neu5Ac and total synthesis of ganglioside GD3. J. Carbohydr. Chem 1996, 15, 857–878. [DOI] [PubMed] [Google Scholar]
- (368).Chernyak A; Oscarson S; Turek D Synthesis of the Lewis b hexasaccharide and squarate acid-HSA conjugates thereof with various saccharide loadings. Carbohydr. Res 2000, 329, 309–316. [DOI] [PubMed] [Google Scholar]
- (369).Haberman JM; Gin DY A new C(1)-auxiliary for anomeric stereocontrol in the synthesis of α-sialyl glycosides. Org. Lett 2001, 3, 1665–1668. [DOI] [PubMed] [Google Scholar]
- (370).Beale TM; Moon PJ; Taylor MS Organoboron-catalyzed regio- and stereoselective formation of beta-2-deoxyglycosidic linkages. Org. Lett 2014, 16, 3604–3607. [DOI] [PubMed] [Google Scholar]
- (371).Geringer SA; Singh Y; Hoard DJ; Demchenko AV A highly efficient glycosidation of glycosyl chlorides using cooperative silver(I) oxide - triflic acid catalysis. Chem.—Eur. J 2020, 26, 8053–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Castelli R; Schindler S; Walter SM; Kniep F; Overkleeft HS; Van der Marel GA; Huber SM; Codee JDC Activation of glycosyl halides by halogen bonding. Chem.—Asian J 2014, 9, 2095–2098. [DOI] [PubMed] [Google Scholar]
- (373).Walter SM; Kniep F; Herdtweck E; Huber SM Halogen-bond-induced activation of a carbon-heteroatom bond. Angew. Chem., Int. Ed 2011, 50, 7187–7191. [DOI] [PubMed] [Google Scholar]
- (374).Geringer SA; Demchenko AV Iron(III) chloride-catalyzed activation of glycosyl chlorides. Org. Biomol. Chem 2018, 16, 9133–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).An S; Wang Q; Zhu W; Sun Q; He G; Chen G Palladium-catalyzed O- and N-glycosylation with glycosyl chlorides. CCS Chemistry 2021, 3, 1821–1829. [Google Scholar]
- (376).Wang Q; An S; Deng Z; Zhu W; Huang Z; He G; Chen G Palladium-catalysed C-H glycosylation for synthesis of C-aryl glycosides. Nat. Catal 2019, 2, 793–800. [Google Scholar]
- (377).Kondo T; Tomoo T; Abe H; Isobe M; Goto T Total synthesis of GD3, a ganglioside. Chem. Lett 1996, 25, 337–338. [DOI] [PubMed] [Google Scholar]
- (378).Gervay J; Nguyen TN; Hadd MJ Mechanistic studies on the stereoselective formation of glycosyl iodides: first characterization of glycosyl iodides. Carbohydr. Res 1997, 300, 119–125. [Google Scholar]
- (379).Bhat AS; Gervay-Hague J Efficient Syntheses of β-Cyanosugars Using Glycosyl Iodides Derived from Per-O-silylated Mono- and Disaccharides. Org. Lett 2001, 3, 2081–2084. [DOI] [PubMed] [Google Scholar]
- (380).Kulkarni SS; Gervay-Hague J Efficient Synthesis of a C-Analogue of the Immunogenic Bacterial Glycolipid BbGL2. Org. Lett 2006, 8, 5765–5768. [DOI] [PubMed] [Google Scholar]
- (381).Lam SN; Gervay-Hague J Efficient Route to 2-Deoxy β-O-Aryl-d-Glycosides via Direct Displacement of Glycosyl Iodides. Org. Lett 2003, 5, 4219–4222. [DOI] [PubMed] [Google Scholar]
- (382).Lam SN; Gervay-Hague J Efficient Synthesis of Man2, Man3, and Man5 Oligosaccharides, Using Mannosyl Iodide Donors1. J. Org. Chem 2005, 70, 8772–8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Salvadó M; Amgarten B; Castillón S; Bernardes GJL; Boutureira O Synthesis of Fluorosugar Reagents for the Construction of Well-Defined Fluoroglycoproteins. Org. Lett 2015, 17, 2836–2839. [DOI] [PubMed] [Google Scholar]
- (384).Zhang W; Luo X; Wang Z; Zhang J One-pot synthesis of β-2,6-dideoxyglycosides via glycosyl iodide intermediates. J. Carbohydr. Chem 2016, 35, 315–325. [Google Scholar]
- (385).Baldoni L; Marino C Synthetic tools for the characterization of galactofuranosyl transferases: glycosylations via acylated glycosyl iodides. Carbohydr. Res 2013, 374, 75–81. [DOI] [PubMed] [Google Scholar]
- (386).Wałejko P; Baj A The synthesis of vitamin E sugar 1,2-orthoesters. Monatsh. Chem 2019, 150, 275–282. [Google Scholar]
- (387).Du W; Kulkarni SS; Gervay-Hague J Efficient, one-pot syntheses of biologically active α-linked glycolipids. Chem. Commun 2007, 2336–2338. [DOI] [PubMed] [Google Scholar]
- (388).Schombs M; Park FE; Du W; Kulkarni SS; Gervay-Hague J One-Pot Syntheses of Immunostimulatory Glycolipids. J. Org. Chem 2010, 75, 4891–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Jervis PJ; Veerapen N; Bricard G; Cox LR; Porcelli SA; Besra GS Synthesis and biological activity of alpha-glucosyl C24:0 and C20:2 ceramides. Bioorg. Med. Chem. Lett 2010, 20, 3475–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Veerapen N; Reddington F; Bricard G; Porcelli SA; Besra GS Synthesis and biological activity of α-l-fucosyl ceramides, analogues of the potent agonist, α-d-galactosyl ceramide KRN7000. Bioorg. Med. Chem. Lett 2010, 20, 3223–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Gu X; Chen L; Wang X; Liu X; You Q; Xi W; Gao L; Chen G; Chen Y-L; Xiong B; Shen J Direct Glycosylation of Bioactive Small Molecules with Glycosyl Iodide and Strained Olefin as Acid Scavenger. J. Org. Chem 2014, 79, 1100–1110. [DOI] [PubMed] [Google Scholar]
- (392).Kulkarni SS; Gervay-Hague J Glycosyl chlorides, bromides and iodides. In Handbook of Chemical Glycosylation; Demchenko AV, Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp 59–93. [Google Scholar]
- (393).Meloncelli PJ; Martin AD; Lowary TL Glycosyl iodides. History and recent advances. Carbohydr. Res 2009, 344, 1110–1122. [DOI] [PubMed] [Google Scholar]
- (394).Gervay-Hague J Taming the reactivity of glycosyl iodides to achieve stereoselective glycosidation. Acc. Chem. Res 2016, 49, 35–47. [DOI] [PubMed] [Google Scholar]
- (395).Wang H; Cui Y; Zou R; Cheng Z; Yao W; Mao Y; Zhang Y Synthesis of oligosaccharides using per-O-trimethylsilyl-glycosyl iodides as glycosyl donor. Carbohydr. Res 2016, 427, 1–5. [DOI] [PubMed] [Google Scholar]
- (396).Oka N; Kajino R; Takeuchi K; Nagakawa H; Ando K α-Selective Ribofuranosylation of Alcohols with Ribofuranosyl Iodides and Triphenylphosphine Oxide. J. Org. Chem 2014, 79, 7656–7664. [DOI] [PubMed] [Google Scholar]
- (397).Houston TA; Koreeda M Iodine-promoted ribosylation leads to a facile acetonide-forming reaction. Carbohydr. Res 2009, 344, 2240–2244. [DOI] [PubMed] [Google Scholar]
- (398).Park SS; Gervay-Hague J Step-economy synthesis of β-steryl sialosides using a sialyl iodide donor. J. Antibiot 2019, 72, 449–460. [DOI] [PubMed] [Google Scholar]
- (399).Vannam R; Pote AR; Peczuh MW Formation and selective rupture of 1,4-anhydroseptanoses. Tetrahedron 2017, 73, 418–425. [Google Scholar]
- (400).Nogueira JM; Nguyen SH; Bennett CS Cyclopropenium cation promoted dehydrative glycosylations using 2-deoxy- and 2,6-dideoxy-sugar donors. Org. Lett 2011, 13, 2814–2817. [DOI] [PubMed] [Google Scholar]
- (401).Brauns D Fluoro-acetyl derivatives of sugars. I. J. Am. Chem. Soc 1923, 45, 833–835. [Google Scholar]
- (402).Olah GA; Nojima M; Kerekes I Synthetic methods and reactions II. Hydrofluorination of alkenes, cyclopropane and alkynes with poly-hydrogen fluoride/pyridine (trialkylamine) reagents. Synthesis 1973, 1973, 779–780. [Google Scholar]
- (403).Sharma MN; Eby R Synthesis and conformational studies of 2-β-chloro, 2-α-fluoro, and 2-b-fluoro derivatives of 2-deoxy-N-acetylneuraminic acid. Carbohydr. Res 1984, 127, 201–210. [DOI] [PubMed] [Google Scholar]
- (404).Hashimoto S; Hayashi M; Noyori R Glycosylation using glucopyranosyl fluorides and silicon-based catalysts. Solvent dependency of the stereoselection. Tetrahedron Lett. 1984, 25, 1379–1382. [Google Scholar]
- (405).Hayashi M; Hashimoto S.-i.; Noyori R Simple synthesis of glycosyl fluorides. Chem. Lett 1984, 13, 1747–1750. [Google Scholar]
- (406).Szarek WA; Grynkiewicz G; Doboszewski B; Hay GW The synthesis of glycosyl fluorides using pyridinium poly (hydrogen fluoride). Chem. Lett 1984, 13, 1751–1754. [Google Scholar]
- (407).Helferich B; Gootz R Über Fluor-Derivate von Kohlehydraten. Ber. Dtsch. Chem. Ges 1929, 62, 2505–2507. [Google Scholar]
- (408).Micheel F; Klemer A Über den Reaktionsmechanismus der Glykosidbildung aus α-und β-1-Fluor-Derivaten der d-Glucose und d-Mannose. Chem. Ber 1958, 91, 663–667. [Google Scholar]
- (409).Micheel F; Klemer A Glycosyl fluorides and azides. Adv. Carbohydr. Chem 1962, 16, 85–103. [DOI] [PubMed] [Google Scholar]
- (410).Hall L; Manville J; Bhacca N Studies of specifically fluorinated carbohydrates. Part I. Nuclear magnetic resonance studies of hexopyranosyl fluoride derivatives. Can. J. Chem 1969, 47, 1–17. [Google Scholar]
- (411).Igarashi K; Honma T; Irisawa J Reaction of glycosyl chlorides with silver tetrafluoroborate. Carbohydr. Res 1969, 11, 577–578. [Google Scholar]
- (412).Goggin KD; Lambert JF; Walinsky SW Synthesis of peraceto-β-D-glycosyl fluorides using zinc fluoride. Synlett 1994, 1994, 162–164. [Google Scholar]
- (413).Naumann D; Möckel R; Tyrra W Difluoromethyl compounds of chalcogens: A NMR study of the reactions of Cd (CF3) 2· 2MeCN and ZnBr (CF3)· 2MeCN with dialkyl chalcogenides and boron trifluoride. Angew. Chem., Int. Ed 1994, 33, 323–325. [Google Scholar]
- (414).Miethchen R; Hein M; Naumann D; Tyrra W Organofluorine compounds and fluorinating agents, 15. O-difluoromethylations of monosaccharides by trifluoromethylzinc bromide. Liebigs Ann. 1995, 1995, 1717–1719. [Google Scholar]
- (415).Tyrra W; Naumann D; Pasenok SV; Yagupolskii YL Carbenoid reactions of trifluoromethylelement compounds. Part 4. Reactions of trifluoromethylzinc bromide with enamines and methylene bases. J. Fluorine Chem 1995, 70, 181–185. [Google Scholar]
- (416).Tyrra W; Naumann D Trifluormethylzinkbromid - ein bequem handhabbares Trifluor- und Difluormethylierungsmittel. Journal für Praktische Chemie/Chemiker-Zeitung 1996, 338, 283–286. [Google Scholar]
- (417).Miethchen R; Hager C; Hein M Organofluorine compounds and fluorinating agents; 18: trifluoromethylzinc bromide as a reagent for the preparation of glycosyl fluorides. Synthesis 1997, 1997, 159–161. [Google Scholar]
- (418).Miethchen R; Kolp G; Peters D; Holz J Reactions with and in water-free hydrogen fluoride. Part 3. Selective syntheses of gycosyl fluorides. Z. Chem 1990, 2 (30), 56. [Google Scholar]
- (419).Miethchen R; Kolp G Reactions with and in anhydrous hydrogen fluoride systems. Part 8. Triethylamine trishydrofluoride—a convenient reagent for the stereoselective synthesis of glycosyl fluorides. J. Fluorine Chem 1993, 60, 49–55. [Google Scholar]
- (420).Mukaiyama T; Hashimoto Y; Shoda S Stereoselective synthesis of 1,2-cis-glycofuranosides using glycofuranosyl fluorides. Chem. Lett 1983, 12, 935–938. [Google Scholar]
- (421).Mitsunobu O The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1981, 1–28. [Google Scholar]
- (422).Loibner H; Zbiral E Reaktionen mit phosphororganischen Verbindungen. XLI [1]. Neuartige synthetische Aspekte des Systems Triphenylphosphin-Azodicarbonsäureester-Hydroxyverbindung. Helv. Chim. Acta 1976, 59, 2100–2113. [Google Scholar]
- (423).Kunz H; Sager W Stereoselective glycosylation of alcohols and silyl ethers using glycosyl fluorides and boron trifluoride etherate. Helv. Chim. Acta 1985, 68, 283–287. [Google Scholar]
- (424).Penglis AAE Fluorinated carbohydrtaes. Adv. Carbohydr. Chem. Biochem 1981, 38, 195–285. [Google Scholar]
- (425).Rosenbrook W Jr.; Riley DA; Lartey PA A new method for the synthesis of glycosyl fluorides. Tetrahedron Lett. 1985, 26, 3–4. [Google Scholar]
- (426).Posner GH; Haines SR A convenient, one-step, high-yield replacement of an anomeric hydroxyl group by a fluorine atom using DAST. Preparation of glycosyl fluorides. Tetrahedron Lett. 1985, 26, 5–8. [Google Scholar]
- (427).Devos A; Remion J; Frisque-Hesbain A-M; Colens A; Ghosez L Synthesis of acyl halides under very mild conditions. J. Chem. Soc., Chem. Commun 1979, 1180–1181. [Google Scholar]
- (428).Schmidt U; Lieberknecht A; Griesser H; Utz R; Beuttler T; Bartkowiak F Amino acids and peptides. 55. Synthesis of biologically active cyclopeptides. 7. Total synthesis of chlamydocin. Synthesis 1986, 1986, 361–366. [Google Scholar]
- (429).Ernst B; Wagner B Preliminary communication. Helv. Chim. Acta 1989, 72, 165–171. [Google Scholar]
- (430).Abdul-Ghani M; Banks ER; Besheesh MK; Sharif I; Syvret RG N-Halogeno compounds. Part 14.”Transfer fluorination” of quinuclidine using F-TEDA-BFc (SelectfluorTM reagent): laboratory synthesis of N-fluoroquinuclidinium salts not requiring the use of elemental fluorine. J. Fluorine Chem 1995, 73, 255–257. [Google Scholar]
- (431).Burkart MD; Zhang Z; Hung S-C; Wong C-H A new method for the synthesis of fluoro-carbohydrates and glycosides using selectfluor. J. Am. Chem. Soc 1997, 119, 11743–11746. [Google Scholar]
- (432).Middleton WJ Explosive hazards with DAST (diethylaminosulfur trifluoride). Chem. Eng. News 1979, 57, 43. [Google Scholar]
- (433).Lal GS; Pez GP; Pesaresi RJ; Prozonic FM; Cheng H Bis (2-methoxyethyl) aminosulfur trifluoride: a new broad-spectrum deoxofluorinating agent with enhanced thermal stability. J. Org. Chem 1999, 64, 7048–7054. [Google Scholar]
- (434).Kobayashi S; Yoneda A; Fukuhara T; Hara S Selective synthesis of fluorinated carbohydrates using N, N-diethyl-α, α-difluoro-(m-methylbenzyl) amine. Tetrahedron Lett. 2004, 45, 1287–1289. [Google Scholar]
- (435).Kobayashi S; Yoneda A; Fukuhara T; Hara S Deoxyfluorination of alcohols using N, N-diethyl-α, α-difluoro-(m-methylbenzyl) amine. Tetrahedron 2004, 60, 6923–6930. [Google Scholar]
- (436).Kim S; Khomutnyk Y; Bannykh A; Nagorny P Synthesis of glycosyl fluorides by photochemical fluorination with sulfur(VI) hexafluoride. Org. Lett 2021, 23, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (437).Kim S; Nagorny P Electrochemical synthesis of glycosyl fluorides using sulfur(VI) hexafluoride as the fluorinating agent. Org. Lett 2022, 24, 2294–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Horne G; Mackie W Preparation of glycosyl fluorides from phenyl 1-seleno-and phenyl 1-telluroglycosides. Tetrahedron Lett. 1999, 40, 8697–8700. [Google Scholar]
- (439).Edmunds JJ; Motherwell WB Observations on the reaction of some electron rich dienes with hypervalent iodoarene difluorides: a novel mechanism for fluorination. J. Chem. Soc., Chem. Commun 1989, 881–883. [Google Scholar]
- (440).Caddick S; Motherwell WB; Wilkinson JA A concise method for the preparation of glycosyl fluorides via displacement reactions of 1-arylthioglycosides with 4-methyl (difluoroiodo) benzene. J. Chem. Soc., Chem. Commun 1991, 674–675. [Google Scholar]
- (441).Caddick S; Gazzard L; Motherwell W; Wilkinson J Preparation of 1-fluoroglycosides from 1-arylthio and 1-arylseleno-glycosides using 4-methyl (difluoroiodo) benzene. Tetrahedron 1996, 52, 149–156. [Google Scholar]
- (442).Yoshida J. i.; Suga S Basic concepts of “cation pool” and “cation flow” methods and their applications in conventional and combinatorial organic synthesis. Chem.—Eur. J 2002, 8, 2650–2658. [DOI] [PubMed] [Google Scholar]
- (443).Suzuki S; Matsumoto K; Kawamura K; Suga S; Yoshida J -i. Generation of alkoxycarbenium ion pools from thioacetals and applications to glycosylation chemistry. Org. Lett 2004, 6, 3755–3758. [DOI] [PubMed] [Google Scholar]
- (444).Gordon DM; Danishefsky SJ Displacement reactions of a 1, 2-anhydro-α-d-hexopyranose: installation of useful functionality at the anomeric carbon. Carbohydr. Res 1990, 206, 361–366. [DOI] [PubMed] [Google Scholar]
- (445).Miethchen R; Gabriel T; Kolp G Reaktionen mit und in wasserfreiem Fluorwasserstoft 7.1 Von Isopropylidenzuckern zu acylierten Pyranosyl-oder Furanosylfluoriden in einem Schritt. Synthesis 1991, 1991, 885–888. [Google Scholar]
- (446).Shimizu M; Nakahara Y; Yoshioka H Selective bromofluorination of alkenes with 1,3-dibromo-5,5-dimethylhydantoin and silicon tetrafluoride. J. Chem. Soc., Chem. Commun 1989, 1881–1882. [Google Scholar]
- (447).Shimizu M; Nakahara Y; Yoshioka H Stereocontrolled halofluorination of glycals with silicon tetrafluoride, leading to a facile synthesis of glycosyl fluorides. J. Fluorine Chem 1999, 97, 57–60. [Google Scholar]
- (448).Pearson AG; Kiefel MJ; Ferro V; von Itzstein M Towards the synthesis of aryl glucuronides as potential heparanase probes. An interesting outcome in the glycosidation of glucuronic acid with 4-hydroxycinnamic acid. Carbohydr. Res 2005, 340, 2077–2085. [DOI] [PubMed] [Google Scholar]
- (449).Koivula A; Ruohonen L; Wohlfahrt G; Reinikainen T; Teeri TT; Piens K; Claeyssens M; Weber M; Vasella A; Becker D; Sinnott ML; Zou J.-y.; Kleywegt GJ; Szardenings M; Ståhlberg J; Jones TA The active site of cellobiohydrolase cel6A from trichoderma reesei: The roles of aspartic acids D221 and D175. J. Am. Chem. Soc 2002, 124, 10015–10024. [DOI] [PubMed] [Google Scholar]
- (450).Martin A; Arda A; Désiré J; Martin-Mingot A; Probst N; Sinaÿ P; Jiménez-Barbero J; Thibaudeau S; Blériot Y Catching elusive glycosyl cations in a condensed phase with HF/SbF 5 superacid. Nat. Chem 2016, 8, 186. [DOI] [PubMed] [Google Scholar]
- (451).Mukaiyama T; Maeshima H; Jona H Catalytic and stereoselective glycosylation with disarmed glycosyl fluoride by using a combination of stannous (II) chloride (SnCl2) and silver tetrakis (pentafluorophenyl) borate [AgB (C6F5) 4] as a catalyst. Chem. Lett 2001, 30, 388–389. [Google Scholar]
- (452).Ogawa T; Sugimoto M Synthesis of α-Neu5Acp-(2—>3)-D-Gal and α-Neu5Acp-(2—>3)-β-D-Galp-(1—>4)-D-Glc. Carbohydr. Res 1985, 135, c5–c9. [Google Scholar]
- (453).Jona H; Maeshima H; Mukaiyama T Catalytic and stereoselective glycosylation with disarmed glycosyl fluoride having phthaloyl or dichlorophthaloyl protected amino function using a 1:2 combination of stannic chloride and silver tetrakis (pentafluorophenyl) borate. Chem. Lett 2001, 30, 726–727. [Google Scholar]
- (454).Okada Y; Asakura N; Bando M; Ashikaga Y; Yamada H Completely β-selective glycosylation using 3,6-O-(o-xylylene)-bridged axial-rich glucosyl fluoride. J. Am. Chem. Soc 2012, 134, 6940–6943. [DOI] [PubMed] [Google Scholar]
- (455).Nicolaou K; Chucholowski A; Dolle RE; Randall JL Reactions of glycosyl fluorides. Synthesis of O-, S-, and N-glycosides. J. Chem. Soc., Chem. Commun 1984, 1155–1156. [Google Scholar]
- (456).Kreuzer M; Thiem J Synthesis of oligosaccharides with glycosyl fluorides under Lewis acid catalysis. Carbohydr. Res 1986, 149, 347–361. [Google Scholar]
- (457).Maeta H; Matsumoto T; Suzuki K ″Dibutyltin diperchlorate″ for activation of glycosyl fluoride. Carbohydr. Res 1993, 249, 49–56. [Google Scholar]
- (458).Kim W-S; Sasai H; Shibasaki M β-Selective glycosylation with α-mannosyl fluorides using tin (II) triflate and lanthanum perchlorate. Tetrahedron Lett. 1996, 37, 7797–7800. [Google Scholar]
- (459).Kandasamy J; Hurevich M; Seeberger PH Automated solid phase synthesis of oligoarabinofuranosides. Chem. Commun 2013, 49, 4453–4455. [DOI] [PubMed] [Google Scholar]
- (460).Wessel HP Comparison of catalysts in α-glucosylation reactions and identification of triflic anhydride as a new reactive promoter. Tetrahedron Lett. 1990, 31, 6863–6866. [Google Scholar]
- (461).Kunz H; Waldmann H Directed stereoselective synthesis of α- and β-N-acetyl neuraminic acid-galactose disaccharides using 2-chloro and 2-fluoro derivatives of neuraminic acid allyl ester. J. Chem. Soc., Chem. Comm 1985, 638–640. [Google Scholar]
- (462).Takeuchi K; Mukaiyama T Trityl tetrakis (pentafluorophenyl) borate catalyzed stereoselective glycosylation using glycopyranosyl fluoride as a glycosyl donor. Chem. Lett 1998, 27, 555–556. [Google Scholar]
- (463).Mukaiyama T; Yanagisawa M; Iida D; Hachiya I A novel carbocationic species paired with tetrakis (pentafluorophenyl) borate anion in catalytic Aldol reaction. Chem. Lett 2000, 29, 606–607. [DOI] [PubMed] [Google Scholar]
- (464).Jona H; Mandai H; Mukaiyama T A catalytic and stereoselective glycosylation with glucopyranosyl fluoride by using various protic acids. Chem. Lett 2001, 30, 426–427. [Google Scholar]
- (465).Jona H; Mandai H; Chavasiri W; Takeuchi K; Mukaiyama T Protic acid catalyzed stereoselective glycosylation using glycosyl fluorides. Bull. Chem. Soc. Jpn 2002, 75, 291–309 and references therein. [Google Scholar]
- (466).Sati G; Martin J; Xu Y; Malakar T; Zimmerman PM; Montgomery J Fluoride migration catalysis enables simple, stereoselective, and iterative glycosylation. J. Am. Chem. Soc 2020, 142, 7235–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Kobayashi S; Koide K; Ohno M Gallium reagents in organic synthesis: Dimethylgallium chloride and triflate as activators in glycosidation using glycopyranosyl fluorides. Tetrahedron Lett. 1990, 31, 2435–2438. [Google Scholar]
- (468).Bohm G; Waldmann H Synthesis of glycosides of fucose under neutral conditions in solutions of LiClO4 in organic solvents. Tetrahedron Lett. 1995, 36, 3843–3846. [Google Scholar]
- (469).Böhm G; Waldmann H O-Glycoside Synthesis under Neutral Conditions in Concentrated Solutions of LiClO4 in Organic Solvents Employing O-Acyl-Protected Glycosyl Donors. Liebigs Ann. 1996, 1996, 621–625. [Google Scholar]
- (470).Pelletier G; Zwicker A; Allen CL; Schepartz A; Miller SJ Aqueous glycosylation of unprotected sucrose employing glycosyl fluorides in the presence of calcium ion and trimethylamine. J. Am. Chem. Soc 2016, 138, 3175–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Wadzinski TJ; Steinauer A; Hie L; Pelletier G; Schepartz A; Miller SJ Rapid phenolic O-glycosylation of small molecules and complex unprotected peptides in aqueous solvent. Nat. Chem 2018, 10, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Junnemann J; Lundt I; Thiem J 2-Deoxyglycosyl fluorides in oligosaccharide synthesis. Liebigs Ann. Chem 1991, 1991, 759–764. [Google Scholar]
- (473).Partridge KM; Bader SJ; Buchan ZA; Taylor CE; Montgomery J A streamlined strategy for aglycone assembly and glycosylation. Angew. Chem., Int. Ed 2013, 52, 13647–13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (474).Matsumoto T; Maeta H; Suzuki K; Tsuchihashi GI New glycosidation reaction 1. Combination use of Cp2ZrCl2-AgClO4 for activation of glycosyl fluorides and application to highly β-selective glycosidation of D-mycinose. Tetrahedron Lett. 1988, 29, 3567–3570. [Google Scholar]
- (475).Suzuki K; Maeta H; Suzuki T; Matsumoto T Cp2ZrCl2-AgBF4 in benzene: a new reagent system for rapid and highly selective α-mannoside synthesis from tetra-O-benzyl-D-mannosyl fluoride. Tetrahedron Lett. 1989, 30, 6879–6882. [Google Scholar]
- (476).Toshima K; Kasumi K; Matsumura S Novel stereocontrolled glycosidations using a solid acid, SO4/ZrO2, for direct syntheses of α- and β-mannopyranosides. Synlett 1998, 1998, 643–645. [Google Scholar]
- (477).Suzuki K; Maeta H; Matsumoto T; Tsuchihashi GI New glycosidation reaction 2. Preparation of 1-fluoro-D-desosamine derivative and its efficient glycosidation by the use of Cp2HfCl2-AgClO4 as the activator. Tetrahedron Lett. 1988, 29, 3571–3574. [Google Scholar]
- (478).Suzuki K; Maeta H; Matsumoto T An improved procedure for metallocene-promoted glycosidation. Enhanced reactivity by employing 1:2-ratio of Cp2HfCl2-AgClO4. Tetrahedron Lett. 1989, 30, 4853–4856. [Google Scholar]
- (479).Manabe S; Ito Y Hafnium (IV) tetratriflate as a glycosyl fluoride activation reagent. J. Org. Chem 2013, 78, 4568–4572. [DOI] [PubMed] [Google Scholar]
- (480).Yamada H; Hayashi T A substrate-unspecified glycosylation reaction promoted by copper (II) trifluoromethanesulfonate in benzotrifluoride. Carbohydr. Res 2002, 337, 581–585. [DOI] [PubMed] [Google Scholar]
- (481).Hosono S; Kim WS; Sasai H; Shibasaki M A new glycosylation procedure utilizing rare earth salts and glycosyl fluorides, with or without the requirement of Lewis acid. J. Org. Chem 1995, 60, 4–5. [Google Scholar]
- (482).Kim WS; Hosono S; Sasai H; Shibasaki M Rare earth perchlorate catalyzed glycosidation of glycosyl fluorides with trimethylsilyl ethers. Tetrahedron Lett. 1995, 36, 4443–4446. [Google Scholar]
- (483).Yu L; Chen D; Li J; Wang PG Preparation, characterization, and synthetic uses of lanthanide (III) catalysts supported on ion exchange resins. J. Org. Chem 1997, 62, 3575–3581. [Google Scholar]
- (484).Yamanoi T; Nagayama S; Ishida H.-k.; Nishikido J; Inazu T Ytterbium (III) tris {bis (perfluoro-butylsulfonyl) amide] as a Lewis acid for glycosylations. Synth. Commun 2001, 31, 899–903. [Google Scholar]
- (485).Packard GK; Rychnovsky SD β-Selective glycosylations with masked D-mycosamine precursors. Org. Lett 2001, 3, 3393–3396. [DOI] [PubMed] [Google Scholar]
- (486).Mukaiyama T; Takeuchi K; Jona H; Maeshima H; Saitoh T A catalytic and stereoselective glycosylation with β-glycosyl fluorides. Helv. Chim. Acta 2000, 83, 1901–1918. [Google Scholar]
- (487).Mukaiyama T; Jona H; Takeuchi K Trifluoromethanesulfonic acid (TfOH)-catalyzed stereoselective glycosylation using glycosyl fluoride. Chem. Lett 2000, 29, 696–697. [Google Scholar]
- (488).Jona H; Takeuchi K; Mukaiyama T 1,2-Cis selective glycosylation with glycosyl fluoride by using a catalytic amount of trifluoromethanesulfonic acid(TfOH) in the coexistence of molecular sieve 5A(MS5A). Chem. Lett 2000, 29, 1278–1279. [Google Scholar]
- (489).Yanagisawa M; Mukaiyama T Catalytic and stereoselective glycosylation with glycosyl fluoride using active carbocationic species paired with tetrakis(pentafluorophenyl)borate or trifluoromethanesulfonate. Chem. Lett 2001, 30, 224–225. [Google Scholar]
- (490).Chan J; Tang A; Bennet AJ A stepwise solvent-promoted SNi reaction of α-D-glucopyranosyl fluoride: Mechanistic implications for retaining glycosyltransferases. J. Am. Chem. Soc 2012, 134, 1212–1220. [DOI] [PubMed] [Google Scholar]
- (491).Manabe Y; Matsumoto T; Ikinaga Y; Tsutsui Y; Sasaya S; Kadonaga Y; Konishi A; Yasuda M; Uto T; Dai C; Yano K; Shimoyama A; Matsuda A; Fukase K Revisiting glycosylations using glycosyl fluoride by BF3-Et2O: activation of disarmed glycosyl fluorides with high catalytic turnover. Org. Lett 2022, 24, 6–10. [DOI] [PubMed] [Google Scholar]
- (492).Nielsen MM; Qiao Y; Wang Y; Pedersen CM Vessel effect in C-F bond activation prompts revised mechanism and reveals an autocatalytic glycosylation. Eur. J. Org. Chem 2020, 2020, 140–144. [Google Scholar]
- (493).Mukaiyama T; Suenaga M; Chiba H; Jona H Highly α-selective glycosylation with glycopyranosyl fluorides having diethylthiocarbamoyl group. Chem. Lett 2002, 31, 56–57. [Google Scholar]
- (494).Stephan DW The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, aaf7229. [DOI] [PubMed] [Google Scholar]
- (495).Caputo CB; Stephan DW Activation of alkyl C-F bonds by B (C6F5) 3: stoichiometric and catalytic transformations. Organometallics 2012, 31, 27–30. [Google Scholar]
- (496).Levin MD; Chen TQ; Neubig ME; Hong CM; Theulier CA; Kobylianskii IJ; Janabi M; O’Neil JP; Toste FD A catalytic fluoride-rebound mechanism for C (sp3)-CF3 bond formation. Science 2017, 356, 1272–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Chitnis SS; LaFortune JH; Cummings H; Liu LL; Andrews R; Stephan DW Phosphorus coordination chemistry in catalysis: air stable P (III)-dications as lewis acid catalysts for the allylation of C-F bonds. Organometallics 2018, 37, 4540–4544. [Google Scholar]
- (498).Böhm G; Waldmann H O-Glycoside Synthesis under Neutral Conditions in Concentrated Solutions of LiClO4 in Organic Solvents Employing Benzyl-Protected Glycosyl Donors. Liebigs Ann. 1996, 1996, 613–619. [Google Scholar]
- (499).Delacroix S; Bonnet J-P; Courty M; Postel D; Van Nhien AN Glycosylation mediated—BAIL in aqueous solution. Carbohydr. Res 2013, 381, 12–18. [DOI] [PubMed] [Google Scholar]
- (500).Yoshida N; Noguchi M; Tanaka T; Matsumoto T; Aida N; Ishihara M; Kobayashi A; Shoda S. i. Direct dehydrative pyridylthio-glycosidation of unprotected sugars in aqueous media using 2-chloro-1, 3-dimethylimidazolinium chloride as a condensing agent. Chem.—Asian J 2011, 6, 1876–1885. [DOI] [PubMed] [Google Scholar]
- (501).Sato S; Naito Y; Aoki K Scandium cation-exchanged montmorillonite catalyzed direct C-glycosylation of a 1, 3-diketone, dimedone, with unprotected sugars in aqueous solution. Carbohydr. Res 2007, 342, 913–918. [DOI] [PubMed] [Google Scholar]
- (502).Banait NS; Jencks WP Reactions of anionic nucleophiles with. alpha.-D-glucopyranosyl fluoride in aqueous solution through a concerted, ANDN (SN2) mechanism. J. Am. Chem. Soc 1991, 113, 7951–7958. [Google Scholar]
- (503).Banait NS; Jencks WP General-acid and general-base catalysis of the cleavage of. alpha.-D-glucopyranosyl fluoride. J. Am. Chem. Soc 1991, 113, 7958–7963. [Google Scholar]
- (504).Sinnott ML; Jencks WP Solvolysis of D-glucopyranosyl derivatives in mixtures of ethanol and 2, 2, 2-trifluoroethanol. J. Am. Chem. Soc 1980, 102, 2026–2032. [Google Scholar]
- (505).Chan J; Sannikova N; Tang A; Bennet AJ Transition-state structure for the quintessential SN2 reaction of a carbohydrate: Reaction of α-glucopyranosyl fluoride with azide ion in water. J. Am. Chem. Soc 2014, 136, 12225–12228. [DOI] [PubMed] [Google Scholar]
- (506).Christofides JC; Davies DB; Martin JA; Rathbone EB Intramolecular hydrogen bonding in 1’-sucrose derivatives determined by SIMPLE proton NMR spectroscopy. J. Am. Chem. Soc 1986, 108, 5738–5743. [DOI] [PubMed] [Google Scholar]
- (507).Christofides JC; Davies DB Co-operative and competitive hydrogen bonding in sucrose determined by SIMPLE 1 H nmr spectroscopy. J. Chem. Soc., Chem. Commun 1985, 1533–1534. [DOI] [PubMed] [Google Scholar]
- (508).Christofides JC; Davies DB Secondary isotope multiplet NMR spectroscopy of partially labeled entities. Carbon-13 SIMPLE NMR of carbohydrates. J. Am. Chem. Soc 1983, 105, 5099–5105. [Google Scholar]
- (509).Anderson CE; Pickrell AJ; Sperry SL; Vasquez TE Jr; Custer TG; Fierman MB; Lazar DC; Brown ZW; Iskenderian WS; Hickstein DD NMR detection of intramolecular OH/OH hydrogen bond networks: an approach using isotopic perturbation and hydrogen bond mediated OH… OH J-coupling. Heterocycles 2007, 72, 469–495. [Google Scholar]
- (510).O’Leary DJ; Hickstein DD; Hansen BK; Hansen PE Theoretical and NMR studies of deuterium isotopic perturbation of hydrogen bonding in symmetrical dihydroxy compounds. J. Org. Chem 2010, 75, 1331–1342. [DOI] [PubMed] [Google Scholar]
- (511).Craig BN; Janssen MU; Wickersham BM; Rabb DM; Chang PS; O’Leary DJ Isotopic perturbation of intramolecular hydrogen bonds in rigid 1, 3-diols: NMR studies reveal unusually large equilibrium isotope effects. J. Org. Chem 1996, 61, 9610–9613. [Google Scholar]
- (512).Vasquez TE; Bergset JM; Fierman MB; Nelson A; Roth J; Khan SI; O’Leary DJ Using equilibrium isotope effects to detect intramolecular OH/OH hydrogen bonds: Structural and solvent effects. J. Am. Chem. Soc 2002, 124, 2931–2938. [DOI] [PubMed] [Google Scholar]
- (513).Boutureira O; Bernardes GJ; D’Hooge F; Davis BG Direct radiolabelling of proteins at cysteine using [18 F]-fluorosugars. Chem. Commun 2011, 47, 10010–10012. [DOI] [PubMed] [Google Scholar]
- (514).Adamo R; Nilo A; Castagner B; Boutureira O; Berti F; Bernardes GJ Synthetically defined glycoprotein vaccines: current status and future directions. Chem. Sci 2013, 4, 2995–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Boutureira O; Bernardes GJL; Fernández-González M; Anthony DC; Davis BG Selenenylsulfide-Linked Homogeneous Glycopeptides and Glycoproteins: Synthesis of Human “Hepatic Se Metabolite A. Angew. Chem., Int. Ed 2012, 51, 1432–1436. [DOI] [PubMed] [Google Scholar]
- (516).Ribeiro Morais G; Falconer RA; Santos I Carbohydrate-based molecules for molecular imaging in nuclear medicine. Eur. J. Org. Chem 2013, 2013, 1401–1414. [Google Scholar]
- (517).Grayson EJ; Bernardes GJ; Chalker JM; Boutureira O; Koeppe JR; Davis BG A coordinated synthesis and conjugation strategy for the preparation of homogeneous glycoconjugate vaccine candidates. Angew. Chem., Int. Ed 2011, 50, 4127–4132. [DOI] [PubMed] [Google Scholar]
- (518).Gamblin DP; Scanlan EM; Davis BG Glycoprotein synthesis: an update. Chem. Rev 2009, 109, 131–163. [DOI] [PubMed] [Google Scholar]
- (519).Stubbs JM; Marx D Glycosidic bond formation in aqueous solution: on the oxocarbenium intermediate. J. Am. Chem. Soc 2003, 125, 10960–10962. [DOI] [PubMed] [Google Scholar]
- (520).Matsumoto T; Maeta H; Suzuki K First total synthesis of mycinamicin IV and VII.: Successful application of new glycosidation reaction. Tetrahedron Lett. 1988, 29, 3575–3578. [Google Scholar]
- (521).Suzuki K; Matsumoto T New methods in gycoside synthesis by using early transition metal-derived reagent. J. Synth. Org. Chem. Jpn 1993, 51, 718–732. [Google Scholar]
- (522).Toshima K; Kasumi K.-i.; Matsumura S Novel stereocontrolled glycosidations of 2-deoxyglucopyranosyl fluoride using a heterogeneous solid acid, sulfated zirconia (SO4/ZrO2). Synlett 1999, 1999, 813–815. [Google Scholar]
- (523).Hachiya I; Moriwaki M; Kobayashi S Catalytic Friedel-Crafts acylation reactions using hafnium triflate as a catalyst in lithium perchlorate-nitromethane. Tetrahedron Lett. 1995, 36, 409–412. [Google Scholar]
- (524).Waller FJ; Barrett AG; Braddock DC; Ramprasad D Hafnium (IV) and zirconium (IV) triflates as superior recyclable catalysts for the atom economic nitration of o-nitrotoluene. Tetrahedron lett. 1998, 39, 1641–1642. [Google Scholar]
- (525).Noji M; Konno Y; Ishii K Metal triflate-catalyzed cationic benzylation and allylation of 1, 3-dicarbonyl compounds. J. Org. Chem 2007, 72, 5161–5167. [DOI] [PubMed] [Google Scholar]
- (526).Wu Y-C; Zhu J Hafnium trifluoromethanesulfonate (hafnium triflate) as a highly efficient catalyst for chemoselective thioacetalization and transthioacetalization of carbonyl compounds. J. Org. Chem 2008, 73, 9522–9524. [DOI] [PubMed] [Google Scholar]
- (527).Braccini I; Derouet C; Esnault J; de Penhoat C; Mallet J-M; Michon V; Sinaÿ P Conformational analysis of nitrilium intermediates in glycosylation reactions. Carbohydr. Res 1993, 246, 23–41. [Google Scholar]
- (528).Satoh H; Hansen HS; Manabe S; van Gunsteren WF; Hunenberger PH Theoretical investigation of solvent effects on glycosylation reactions: stereoselectivity controlled by preferential conformations of the intermediate oxacarbenium-counterion complex. J. Chem. Theory Comput 2010, 6, 1783–1797. [DOI] [PubMed] [Google Scholar]
- (529).Kleinschmidt P; Lau K; Hildenbrand D Thermochemistry of the gaseous fluorides of samarium, europium, and thulium. J. Chem. Phys 1981, 74, 653–660. [Google Scholar]
- (530).Zmbov KF; Margrave JL Mass spectrometric studies at high temperatures. XII. Stabilities of dysprosium, holmium, and erbium subfluorides. J. Phys. Chem 1966, 70, 3379–3382. [Google Scholar]
- (531).Di Bernardo P; Choppin GR; Portanova R; Zanonato PL Lanthanide (III) trifluoromethanesulfonate complexes in anhydrous acetonitrile. Inorg. Chim. Acta 1993, 207, 85–91. [Google Scholar]
- (532).Favier F; Pascal J-L Synthesis and structural analysis of a homogeneous series of anhydrous rare-earth-metal perchlorates. J. Chem. Soc., Dalton Trans 1992, 1997–2002. [Google Scholar]
- (533).Bünzli J-CG; Kasparek V FT-IR and fluorometric investigation of rare-earth and metal ion solvation. Part 10. Inner-sphere interaction between perchlorate and lanthanide ions in anhydrous acetonitrile. Inorg. Chim. Acta 1991, 182, 101–107. [Google Scholar]
- (534).Buenzli JCG; Yersin JR; Mabillard C FT IR and fluorometric investigation of rare earth and metallic ion solvation. 1. Europium perchlorate in anhydrous acetonitrile. Inorg. Chem 1982, 21, 1471–1476. [Google Scholar]
- (535).Qiu D; Schmidt RR Glycosyl imidates, 52. Synthesis of globotriaosylceramide (Gb3) and isoglobotriaosylceramide (isoGb3). Liebigs Ann. Chem 1992, 1992, 217–224. [Google Scholar]
- (536).Stork G; La Clair JJ Stereoselective synthesis of β-mannopyranosides via the temporary silicon connection method. J. Am. Chem. Soc 1996, 118, 247–248. [Google Scholar]
- (537).Bols M Stereocontrolled synthesis of α-glucosides by intramolecular glycosidation. J. Chem. Soc., Chem. Commun 1992, 913–914. [Google Scholar]
- (538).Wessel HP; Ruiz N α-Glucosylation reactions with 2,3,4,6-tetra-O-benzyl-b-D-glucopyranosyl fluoride and triflic anhydride as promoter. J. Carbohydr. Chem 1991, 10, 901–910. [Google Scholar]
- (539).Kevill DN; Sutthoff RF Kinetics of the reactions of t-butyl chloride and [2 H 9] t-butyl chloride with silver salts in acetonitrile. J. Chem. Soc., Perkin Trans 2 1977, 201–206. [Google Scholar]
- (540).Kato T; Tanaka M; Hoshikawa M; Yagi M An efficient construction of trinervitane and kempane skeletons from the common intermediate. Tetrahedron Lett. 1998, 39, 7553–7556. [Google Scholar]
- (541).Zhang Y; Bommuswamy J; Sinnott ML Kinetic isotope effect study of transition states for the hydrolyses of a- and b-glucopyranosyl fluorides. J. Am. Chem. Soc 1994, 116, 7557–7563. [Google Scholar]
- (542).Ogawa T; Takahashi Y Total synthesis of α-cyclodextrin. Carbohydr. Res 1985, 138, c5–c9. [Google Scholar]
- (543).Nicolaou K; Caulfield T; Kataoka H; Stylianides N Total synthesis of the tumor-associated Lex family of glycosphingolipids. J. Am. Chem. Soc 1990, 112, 3693–3695. [Google Scholar]
- (544).Nicolaou KC; Hummel CW; Iwabuchi Y Total synthesis of sialyl dimeric Lex. J. Am. Chem. Soc 1992, 114, 3126–3128. [Google Scholar]
- (545).Matsuo I; Totani K; Tatami A; Ito Y Comprehensive synthesis of ER related high-mannose-type sugar chains by convergent strategy. Tetrahedron 2006, 62, 8262–8277. [Google Scholar]
- (546).Nicolaou KC; Caulfield T; Kataoka H; Kumazawa T A practical and enantioselective synthesis of glycosphingolipids and related compounds. Total synthesis of globotriaosylceramide (Gb3). J. Am. Chem. Soc 1988, 110, 7910–7912. [Google Scholar]
- (547).Kaeothip S; Demchenko AV On orthogonal and selective activation of glycosyl thioimidates and thioglycosides: application to oligosaccharide assembly. J. Org. Chem 2011, 76, 7388–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Kanie O; Ito Y; Ogawa T Orthogonal glycosylation strategy in oligosaccharide synthesis. J. Am. Chem. Soc 1994, 116, 12073–12074. [Google Scholar]
- (549).Paulsen H Progress in oligosaccharide synthesis through a new orthogonal glycosylation strategy. Angew. Chem., Int. Ed. Engl 1995, 34, 1432–1434. [Google Scholar]
- (550).Kanie O Orthogonal strategy in oligosaccharide synthesis. In Carbohydrates in Chemistry and Biology; Ernst B, Hart GW, Sinay P, Eds.; Wiley-VCH: Weinheim, Germany, 2000; Vol. 1, pp 407–426. [Google Scholar]
- (551).Ito Y; Kanie O; Ogawa T Orthogonal glycosylation strategy for rapid assembly of oligosaccharides on a polymer support. Angew. Chem., Int. Ed 1996, 35, 2510–2512. [Google Scholar]
- (552).Kanie O; Ohtsuka I; Ako T; Daikoku S; Kanie Y; Kato R Orthogonal glycosylation reactions on solid phase and synthesis of a library consisting of a complete set of fucosyl galactose isomers. Angew. Chem., Int. Ed 2006, 45, 3851–3854. [DOI] [PubMed] [Google Scholar]
- (553).Winter M Supports for solid-phase organic synthesis. In Combinatorial peptide and nonpeptide libraries: a handbook; Jung G, Ed.; Wiley-VCH: Wienheim, Germany, 1996; pp 465–510. [Google Scholar]
- (554).Fruchtel JS; Jung G Organic chemistry on solid supports. Angew. Chem., Int. Ed. Engl 1996, 35, 17–42. [Google Scholar]
- (555).Hermkens PHH; Ottenheijm HCJ; Rees D Solid-phase organic reactions: a review of the recent literature. Tetrahedron 1996, 52, 4527–4554. [Google Scholar]
- (556).Brown RCD Recent developments in solid-phase organic synthesis. J. Chem. Soc., Perkin Trans 1 1998, 3293–3320. [Google Scholar]
- (557).Osborn HMI; Khan TH Recent developments in polymer supported syntheses of oligosaccharides and glycopeptides. Tetrahedron 1999, 55, 1807–1850. [Google Scholar]
- (558).Krepinsky JJ; Douglas SP Polymer-supported synthesis of oligosaccharides. In Carbohydrates in Chemistry and Biology; Ernst B, Hart GW, Sinay P, Eds.; Wiley-VCH: Weinheim, Germany, 2000; Vol. 1, pp 239–265. [Google Scholar]
- (559).Seeberger PH; Haase WC Solid-phase oligosaccharide synthesis and combinatorial carbohydrate libraries. Chem. Rev 2000, 100, 4349–4393. [DOI] [PubMed] [Google Scholar]
- (560).Fukase K Oligosaccharides: Synthesis: Combinatorial and solid phase methods in oligosaccharide synthesis. In Glycoscience: Chemistry and Chemical Biology; Fraser-Reid B, Tatsuta K, Thiem J, Eds.; Springer: Berlin - Heidelberg - New York, 2001; Vol. 2, pp 1621–1660. [Google Scholar]
- (561).Seeberger PH Solid phase oligosaccharide synthesis (reprinted from Glycochemistry: principles, synthesis, and applications, pg 1– 32, 2001). J. Carbohydr. Chem 2002, 21, 613–643. [Google Scholar]
- (562).Seeberger PH The logic of automated glycan assembly. Acc. Chem. Res 2015, 48, 1450–1463. [DOI] [PubMed] [Google Scholar]
- (563).Hahm HS; Schlegel MK; Hurevich M; Eller S; Schuhmacher F; Hofmann J; Pagel K; Seeberger PH Automated glycan assembly using the Glyconeer 2.1 synthesizer. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E3385–E3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (564).Wen L; Edmunds G; Gibbons C; Zhang J; Gadi MR; Zhu H; Fang J; Liu X; Kong Y; Wang PG Toward automated enzymatic synthesis of oligosaccharides. Chem. Rev 2018, 118, 8151–8187. [DOI] [PubMed] [Google Scholar]
- (565).Guberman M; Seeberger PH Automated Glycan Assembly: A Perspective. J. Am. Chem. Soc 2019, 141, 5581–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (566).Shadrick M; Panza M; Vijaya Ganesh N; Pistorio SG; Stine KJ; Demchenko AV HPLC-Based automated oligosaccharide synthesis. Comprehensive Glycoscience 2021, 623–636. [Google Scholar]
- (567).Ako T; Daikoku S; Ohtsuka I; Kato R; Kanie O A method of orthogonal oligosaccharide synthesis leading to a combinatorial library based on stationary solid-phase reaction. Chem.—Asian J 2006, 1, 798–813. [DOI] [PubMed] [Google Scholar]
- (568).Manabe S; Ishii K; Ito Y N-Benzyl-2,3-trans-carbamate-bearing glycosyl donors for 1,2-cis-selective glycosylation reactions. Eur. J. Org. Chem 2011, 2011, 497–516. [Google Scholar]
- (569).Huo C; Chan TH A novel liquid-phase strategy for organic synthesis using organic ions as soluble supports. Chem. Soc. Rev 2010, 39, 2977–3006. [DOI] [PubMed] [Google Scholar]
- (570).Galan MC; Corfield AP Ionic liquids in oligosaccharide synthesis: Towards mucin-type glycan probes. Biochem. Soc. Trans 2010, 38, 1368–1373. [DOI] [PubMed] [Google Scholar]
- (571).Pathak AK; Yerneni CK; Young Z; Pathak V Oligomannan synthesis using ionic liquid supported glycosylation. Org. Lett 2008, 10, 145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (572).Yerneni CK; Pathak V; Pathak AK Imidazolium cation supported solution-phase assembly of homolinear α(1→6)-linked octamannoside: an efficient alternate approach for oligosaccharide synthesis. J. Org. Chem 2009, 74, 6307–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (573).Scigelova M; Singh S; Crout DHG Glycosidases - a great synthetic tool. J. Mol. Catal. B-Enzym 1999, 6, 483–494. [Google Scholar]
- (574).Vocadlo DJ; Withers SG Glycosidase-catalyzed oligosaccharide synthesis. In Carbohydrates in Chemistry and Biology; Ernst B, Hart GW, Sinay P, Eds.; Wiley-VCH: Weinheim, Germany, 2000; Vol. 1, pp 723–844. [Google Scholar]
- (575).Kren V; Thiem J Glycosylation employing bio-systems: from enzymes to whole cells. Chem. Soc. Rev 1997, 26, 463–473. [Google Scholar]
- (576).Hehre EJ A fresh understanding of the stereochemical behavior of glycosylases: structural distinction of ″inverting″ (2-MCO-type) versus ″retaining″ (1-MCO-type) enzymes. Adv. Carbohydr. Chem. Biochem 2000, 55, 265–310. [DOI] [PubMed] [Google Scholar]
- (577).Kim YW; Fox DT; Hekmat O; Kantner T; McIntosh LP; Warren RAJ; Withers SG Glycosynthase-based synthesis of xylo-oligosaccharides using an engineered retaining xylanase from Cellulomonas fimi. Org. Biomol. Chem 2006, 4, 2025–2032. [DOI] [PubMed] [Google Scholar]
- (578).Plante OJ; Palmacci ER; Seeberger PH Automated solid-phase synthesis of oligosaccharides. Science 2001, 291, 1523–1527. [DOI] [PubMed] [Google Scholar]
- (579).Sears P; Wong CH Toward automated synthesis of oligosaccharides and glycoproteins. Science 2001, 291, 2344–2350. [DOI] [PubMed] [Google Scholar]
- (580).Seeberger PH; Werz DB Automated synthesis of oligosacchraides as a basis for drug discovery. Nat. Rev. Drug Discovery 2005, 4, 751–763. [DOI] [PubMed] [Google Scholar]
- (581).Tanaka H; Matoba N; Tsukamoto H; Takimoto H; Yamada H; Takahashi T Automated parallel synthesis of a protected oligosaccharide library based upon the structure of dimeric Lewis X by one-pot sequential glycosylation. Synlett 2005, 2005, 824–828. [Google Scholar]
- (582).Codée JDC; Kröck L; Castagner B; Seeberger PH Automated solid-phase synthesis of protected oligosaccharides containing β-mannosidic linkages. Chem.—Eur. J 2008, 14, 3987–3994. [DOI] [PubMed] [Google Scholar]
- (583).Seeberger PH Automated oligosaccharide synthesis. Chem. Soc. Rev 2008, 37, 19–28. [DOI] [PubMed] [Google Scholar]
- (584).Machida K; Hirose Y; Fuse S; Sugawara T; Takahashi T Development and application of a solution-phase automated synthesizer, ‘ChemKonzert’. Chem. Pharm. Bull 2010, 58, 87–93. [DOI] [PubMed] [Google Scholar]
- (585).Hsu CH; Hung SC; Wu CY; Wong CH Toward automated oligosaccharide synthesis. Angew. Chem., Int. Ed 2011, 50, 11872–11923. [DOI] [PubMed] [Google Scholar]
- (586).Krock L; Esposito D; Castagner B; Wang C-C; Bindschadler P; Seeberger PH Streamlined access to conjugation-ready glycans by automated synthesis. Chem. Sci 2012, 3, 1617–1622. [Google Scholar]
- (587).Vijaya Ganesh N; Fujikawa K; Tan YH; Stine KJ; Demchenko AV HPLC-assisted automated oligosaccharide synthesis. Org. Lett 2012, 14, 3036–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (588).Nokami T; Isoda Y; Sasaki N; Takaiso A; Hayase S; Itoh T; Hayashi R; Shimizu A; Yoshida J -i. Automated electrochemical assembly of the protected potential TMG-chitotriomycin precursor based on rational optimization of the carbohydrate building block. Org. Lett 2015, 17, 1525–1528. [DOI] [PubMed] [Google Scholar]
- (589).Tang S-L; Pohl NLB Automated solution-phase synthesis of β-1,4-mannuronate and β-1,4-mannan. Org. Lett 2015, 17, 2642–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (590).Pistorio SG; Nigudkar SS; Stine KJ; Demchenko AV HPLC-assisted automated oligosaccharide synthesis: the implementation of the autosampler as a mode of the reagent delivery. J. Org. Chem 2016, 81, 8796–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (591).Trobe M; Burke MD The molecular industrial revolution: automated synthesis of small molecules. Angew. Chem., Int. Ed 2018, 57, 4192–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Li T; Liu L; Wei N; Yang J-Y; Chapla DG; Moremen KW; Boons G-J An automated platform for the enzyme-mediated assembly of complex oligosaccharides. Nat. Chem 2019, 11, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (593).Mattes DS; Jung N; Weber LK; Brase S; Breitling F Miniaturized and Automated Synthesis of Biomolecules-Overview and Perspectives. Adv. Mater 2019, 31, e1806656. [DOI] [PubMed] [Google Scholar]
- (594).Chatterjee S; Guidi M; Seeberger PH; Gilmore K Automated radial synthesis of organic molecules. Nature 2020, 579, 379–384. [DOI] [PubMed] [Google Scholar]
- (595).Kern MK; Pohl NLB Automated Solution-Phase Synthesis of S-Glycosides for the Production of Oligomannopyranoside Derivatives. Org. Lett 2020, 22, 4156–4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Gromski PS; Granda JM; Cronin L Universal chemical synthesis and discovery with ‘The Chemputer’. Trends Chem. 2020, 2, 4–12. [Google Scholar]
- (597).Dube DH; Bertozzi CR Glycans in cancer and inflammation-potential for therapeutics and diagnostics. Nat. Rev. Drug Discovery 2005, 4, 477–488. [DOI] [PubMed] [Google Scholar]
- (598).Lebrilla CB; An HJ The prospects of glycan biomarkers for the diagnosis of diseases. Mol. Biosystems 2009, 5, 17–20. [DOI] [PubMed] [Google Scholar]
- (599).Cheng Y; Li M; Wang S; Peng H; Reid S; Ni N; Fang H; Xu W; Wang B Carbohydrate biomarkers for future disease detection and treatment. Sci. China Chem 2010, 53, 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Chaubard JL; Krishnamurthy C; Yi W; Smith DF; Hsieh-Wilson LC Chemoenzymatic probes for detecting and imaging fucose-alpha(1–2)-galactose glycan biomarkers. J. Am. Chem. Soc 2012, 134, 4489–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (601).Konforte D; Diamandis EP Is early detection of cancer with circulating biomarkers feasible? Clin. Chem 2013, 59, 35. [DOI] [PubMed] [Google Scholar]
- (602).Mahon E; Mouline Z; Silion M; Gilles A; Pinteala M; Barboiu M Multilayer lectin-glyconanoparticles architectures for QCM enhanced detection of sugar-protein interaction. Chem. Commun 2013, 49, 3004–3006. [DOI] [PubMed] [Google Scholar]
- (603).Ruhaak LR; Miyamoto S; Lebrilla CB Developments in the identification of glycan biomarkers for the detection of cancer. Mol. Cell. Proteomics 2013, 12, 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (604).Hu X-L; Jin H-Y; He X-P; James TD; Chen G-R; Long Y-T Colorimetric and Plasmonic Detection of Lectins Using Core-Shell Gold Glyconanoparticles Prepared by Copper-Free Click Chemistry. ACS Appl. Mater. Interfaces 2015, 7, 1874–1878. [DOI] [PubMed] [Google Scholar]
- (605).Oyelaran O; Gildersleeve JC Glycan arrays: recent advances and future challenges. Curr. Opin. Chem. Biol 2009, 13, 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (606).Rillahan CD; Paulson JC Glycan microarrays for decoding the glycome. Annu. Rev. Biochem 2011, 80, 797–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (607).Laurent N; Haddoub R; Voglmeir J; Flitsch SL MALDI-ToF MS analysis of gycosyltransferase activities on gold surface arrays. Carbohydrate Microarrays, in Methods in Molecular Biology 2012, 808, 269–284. [DOI] [PubMed] [Google Scholar]
- (608).Song X; Heimburg-Molinaro J; Smith DF; Cummings RD Glycan microarrays. Methods Mol. Biol 2012, 800, 163–171. [DOI] [PubMed] [Google Scholar]
- (609).Park S; Gildersleeve JC; Blixt O; Shin I Carbohydrate microarrays. Chem. Soc. Rev 2013, 42, 4310–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (610).Smith DF; Cummings RD Investigating virus-glycan interactions using glycan microarrays. Curr. Opin. Virol 2014, 7, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (611).Wang L; Cummings RD; Smith DF; Huflejt M; Campbell CT; Gildersleeve JC; Gerlach JQ; Kilcoyne M; Joshi L; Serna S; Reichardt NC; Parera Pera N; Pieters RJ; Eng W; Mahal LK Cross-platform comparison of glycan microarray formats. Glycobiology 2014, 24, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (612).Li Z; Feizi T The neoglycolipid (NGL) technology-based microarrays and future prospects. FEBS letters 2018, 592, 3976–3991. [DOI] [PubMed] [Google Scholar]
- (613).Wennekes T; van den Berg RJBHN; Boot RG; Van der Marel GA; Overkleeft HS; Aerts JMFG Glycosphingolipids - nature, function, and pharmacological modulation. Angew. Chem., Int. Ed 2009, 48, 8848–8869. [DOI] [PubMed] [Google Scholar]
- (614).Carbohydrate-based vaccines and immunotherapies; Guo Z, Boons GJ, Eds.; Wiley: Hoboken, 2009. [Google Scholar]
- (615).Cipolla L; Arajo AC; Bini D; Gabrielli L; Russo L; Shaikh N Discovery and design of carbohydrate-based therapeutics. Expert Opin. Drug Discovery 2010, 5, 721–737. [DOI] [PubMed] [Google Scholar]
- (616).Kaeothip S; Paranjape G; Terrill SE; Bongat AFG; Udan MLD; Kamkhachorn T; Johnson HL; Nichols MR; Demchenko AV Development of LPS antagonistic therapeutics: synthesis and evaluation of glucopyranoside-spacer-amino acid motifs. RSC Adv. 2011, 1, 83–92. [Google Scholar]
- (617).Liu L; Zha J; DiGiandomenico A; McAllister D; Stover CK; Wang Q; Boons GJ Synthetic Enterobacterial Common Antigen (ECA) for the Development of a Universal Immunotherapy for Drug-Resistant Enterobacteriaceae. Angew. Chem., Int. Ed. Engl 2015, 54, 10953–10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (618).Carbohydrate-based drug discovery; Wong CH, Ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- (619).Carbohydrate drug design; Klyosov AA, Witczak ZJ, Platt D, Eds.; American Chemical Society: Washington, DC, 2006; Vol. 932. [Google Scholar]
- (620).Ernst B; Magnani JL From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discovery 2009, 8, 661–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (621).Galan MC; Benito-Alifonso D; Watt GM Carbohydrate chemistry in drug discovery. Org. Biomol. Chem 2011, 9, 3598–3610. [DOI] [PubMed] [Google Scholar]
- (622).Klyosov AA Carbohydrates and Drug Design. In Glycobiology and Drug Design; ACS Symposium Series 1102; American Chemical Society: Washington, DC, 2012; Vol. 1102, pp 3–22. [Google Scholar]
- (623).Hsieh-Wilson LC; Griffin ME Improving Biologic Drugs via Total Chemical Synthesis. Science 2013, 342, 1332–1333. [DOI] [PubMed] [Google Scholar]
- (624).Wong CF Flexible receptor docking for drug discovery. Expert Opin. Drug Discovery 2015, 10, 1189–1200. [DOI] [PubMed] [Google Scholar]
- (625).Guo Z; Wang Q Recent development in carbohydrate-based cancer vaccines. Curr. Opinion in Chem. Biology 2009, 13, 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (626).Daum RS; Spellberg B Progress Toward a Staphylococcus aureus Vaccine. Clin. Infect. Dis 2012, 54, 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (627).Yin Z; Huang X Recent Development in Carbohydrate Based Anticancer Vaccines. J. Carbohydr. Chem 2012, 31, 143–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (628).Huang YL; Hung JT; Cheung SK; Lee HY; Chu KC; Li ST; Lin YC; Ren CT; Cheng TJ; Hsu TL; Yu AL; Wu CY; Wong CH Carbohydrate-based vaccines with a glycolipid adjuvant for breast cancer. Proc. Nat. Acad. Sci 2013, 110, 2517–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (629).Peri F Clustered Carbohydrates in Synthetic Vaccines. Chem. Soc. Rev 2013, 42, 4543–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (630).Khatun F; Stephenson RJ; Toth I An Overview of Structural Features of Antibacterial Glycoconjugate Vaccines That Influence Their Immunogenicity. Chem.—Eur. J 2017, 23, 4233–4254. [DOI] [PubMed] [Google Scholar]
- (631).Gouttefangeas C; Rammensee HG Personalized cancer vaccines: adjuvants are important, too. Cancer Immunol. Immunother 2018, 67, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (632).Li Q; Guo Z Recent advances in Toll like receptor-targeting glycoconjugate vaccines. Molecules 2018, 23, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (633).Behera A; Rai D; Kulkarni SS Total Syntheses of Conjugation-Ready Trisaccharide Repeating Units of Pseudomonas aeruginosa O11 and Staphylococcus aureus Type 5 Capsular Polysaccharide for Vaccine Development. J. Am. Chem. Soc 2020, 142, 456–467. [DOI] [PubMed] [Google Scholar]
- (634).Wang P; Huo CX; Lang S; Caution K; Nick ST; Dubey P; Deora R; Huang X Chemical Synthesis and Immunological Evaluation of a Pentasaccharide Bearing Multiple Rare Sugars as a Potential Anti-pertussis Vaccine. Angew. Chem., Int. Ed. Engl 2020, 59, 6451–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (635).Seeberger PH Discovery of Semi- and Fully-Synthetic Carbohydrate Vaccines Against Bacterial Infections Using a Medicinal Chemistry Approach. Chem. Rev 2021, 121, 3598–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]