

Abstract
A mesoporous silica-based drug delivery device potentially useful for bone-specific drug delivery has been designed, developed and characterized starting from MSU-type mesoporous silica. The proposed system consists of a mesoporous silica nanoparticles (MSN) based vehicle, presenting alendronate as a targeting functionality for bone tissue while ibuprofen is used as a model molecule for the drugs to be delivered. The particles are functionalized on the external surface using a propionitrile derivative that is successively hydrolyzed to a carboxylic group. Alendronate, one of the most used member of the diphosphonate drug class, is electrostatically bonded to the external carboxyl functionalities of mesoporous silica. The obtained material has been characterized by powder X-ray diffraction, N2 adsorption–desorption porosimetry, UV-vis spectrophotometry, FT-IR spectrometry and MAS-NMR 13C and 29Si. Hydroxyapatite, which simulates the bone matrix, has been synthesized with the aim of testing the targeting activity of the obtained device. In a separate test, the MSNs have been loaded with ibuprofen, a non-steroidal anti-inflammatory drug (NSAID), and its release has been determined under neutral conditions by HPLC. Moreover, biological tests were carried out. The tested devices did not show any toxicity towards normal cells, confirming their high biocompatibility and the lack of off-target effects.
A mesoporous silica-based drug delivery device potentially useful for bone-specific drug delivery has been designed, developed and characterized starting from MSU-type mesoporous silica.
Introduction
Bisphosphonates (BPs) are a major class of drugs used in the treatment of bone diseases or metastasis because they inhibit re-absorption of bone tissues. Bisphosphonates can bind hydroxyapatite very strongly, one of the main components of bones, preventing crystal growth and dissolution. BPs are chemically stable analogues of pyrophosphate.1 However, instead of the P–O–P structure present in pyrophosphate, geminal BPs contain a P–C–P backbone. The P–C–P structure is responsible for the strong affinity for bone minerals and allows for several variations in the structure based on substitution in the R1 and R2 positions on the geminal carbon (Scheme 1).2
Scheme 1. Representation of the bisphosphonate structure.
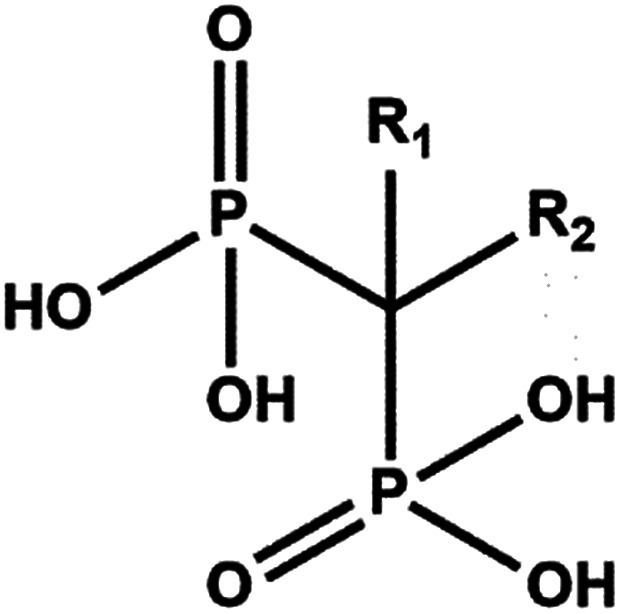
The presence of a hydroxyl group at the R1 position increases the affinity for calcium and thus bone minerals, enhancing chemisorption to minerals, presumably via tridentate binding to calcium; R2 substituents improve antiresorptive potency.3 Alendronate is a bisphosphonate widely used in the clinical treatment of systemic metabolic bone diseases such as Paget's disease, osteoporosis, and hypercalcemia of malignancy as well as inflammation-related bone loss. Alendronate induces in vitro functional and morphological modification of osteoclasts that lose their ability to acidify the surrounding media, becoming unable to dissolve hydroxyapatite crystals.4
The molecular mechanism of BPs varies with their particular structure. At the cellular level, BPs cause apoptosis of bone resorbing cells, osteoclasts: BPs bind to bone minerals in the body and expose them to osteoclasts at the resorption site, then the BPs are taken up intracellularly by osteoclasts during bone resorption and finally the absorbed BP causes inhibition of osteoclast function and apoptosis. In addition to their pharmacological effects, BPs are used as carriers for therapeutic agents to bone tissue. Bisphosphonates can be employed in the design and development of bone-specific drug delivery of therapeutics such as radio drugs, anti-inflammatory drugs, and small organic molecules such as oestrogen and macromolecules such as proteins.
In 2001 several newly synthesized osteotropic diclofenac with bisphosphonic moieties (DIC-BP) were investigated towards site-specific and controlled delivery of diclofenac to the bone in rats.5
Geminal bisphosphonates have been studied as bone-targeting moieties for the treatment of bone-related diseases with the aim of using bone tropism to deliver therapeutics to the desired site of action, thus reducing unwanted side effects in other tissues.6 Prodrugs obtained from the combination of bisphosphonates and chemotherapeutic drugs have been proposed in order to target bone tissue and reduce drug side effects. The conjugation of camptothecin with bisphosphonates was proposed in order to improve the low water solubility of the drug and reduce severe toxic side effects during targeting.7 Chemical modifications with bisphosphonic acids can provide protein engineering to exhibit a strong bone affinity, allowing systemic administration that addresses the delivery of proteins selectively to the skeletal sites. The obtained molecules are potentially beneficial for the treatment of diseases such as osteoporosis and arthritis.8 Since 2003 we have studied the reactivity of silica surfaces in mesoporous silica and attempted the modification of external particles and internal pore surfaces using reactive organic molecules or organo-siloxane reagents for the purpose of creating specific surfaces which are able to host suitable molecules such as drugs or providing the particle with the desired functional groups or molecules.9,10 Silica-based mesoporous materials have been proposed for several applications such as drug targeting,11–14 sampling of peptide mixtures for mass spectrometry15 and enzyme immobilization.16 The regular nanostructure, good biocompatibility, large surface areas, large void volumes, high homogeneity in the pore diameter and high versatility of the procedures to modify the external and/or internal silica surface in mesoporous silica, make these materials a promising tool for biomedicine-applied nanotechnology. Silica-based mesoporous materials could serve for storing hydrophobic drugs inside the pores or in the delivery of covalently linked therapeutic agents.17 Furthermore, recently, silica nanoparticles have been approved by the Food and Drug Administration (FDA) for specific clinical applications.18 Two different kinds of amino-functionalized, alendronate-containing mesoporous silica (MCM-41 and SBA-15) have been presented by Vallet-Regi et al.19 for local treatments in bone tissue repairing and regeneration. The proposed application aims to overcome the low oral bioavailability of bisphosphonates, using mesoporous silica as a bone substitute material with local alendronate controlled delivery capability, by confining the drug in their pores.20 This work demonstrated that the amount and delivery rate of adsorbed bisphosphonates can be modulated through modification of the surface of the pore walls by means of organic molecules. At physiological pH (pH 7.4), the differences in polarity between the silica surface and the bisphosphonate, or between the PrNH2-covered surface and the bisphosphonate, induce weakening of the interaction between the silica surface and the adsorbed molecules, which are then slowly released into the media.8 In this paper we present the preparation and characterization of mesoporous silica-based carriers for bone hydroxyapatite targeting, aimed at the localized release of different kinds of drugs. The proposed MSN based system presents alendronate as a targeting functionality for bone tissue while ibuprofen is used as a model molecule for the drugs to be delivered. A preliminary investigation was performed in order to evaluate ibuprofen loading and release efficiency from silica carriers. Finally, the biocompatibility of the obtained materials for immortalized normal human foreskin fibroblast (BjhTERT) proliferation was tested.
Experimental
Materials
Triton X-100, neutral polyoxyethylene(10) octylphenyl ether (Aldrich cod. X100-1GA); tetraethylorthosilicate, TEOS (Aldrich cod. 86578, 99%); n-dodecane (Alfa Aesar, A14834, >99%); ultrapure water (MilliQ® water, Millipore); ethanol (VWR, cod. 20821.321, 99.95%); alendronate sodium trihydrate (Alfa Aesar, J61397); 4-(triethoxysilyl)butyronitrile-TSBN (Aldrich cod. 374156); HCl (VWR, cod. 20252, >36.4%); NaOH (Aldrich cod. S8045); ferric chloride hexahydrate, FeCl3·6H2O (Aldrich cod. 207926); perchloric acid, HClO4 2 M (Aldrich cod. 311421); calcium chloride, CaCl2 (Panreac, cod. 131232.1211); ammonium hydrogenphosphate, (NH4)2HPO4 (Aldrich cod. 428922); sodium dihydrogenphosphate, NaH2PO4 (Fluka, cod. 17844); disodium phosphate, Na2HPO4 (Aldrich cod. NIST2186II); and ibuprofen (Sigma Aldrich cod. PHR1004) were used.
Synthesis of MSNs
MSU-type mesoporous silica nanoparticles used in this study were obtained by a modified interfacial synthesis procedure carried out at room temperature and without mineralizing agents reported in a previous paper.21 Dodecane was introduced as the organic phase. Distilled H2O and Triton X-100 were completely mixed in a beaker, and then a solution of TEOS and dodecane was added causing the formation of a biphasic system. After 8 days of continuous stirring, the lower phase was filtered, and the obtained powder was washed with distilled H2O and then dried overnight.
Synthesis of MSN–COOH
Synthesized MSNs were modified by post-synthesis grafting introducing a carboxyl functional group, able to bind alendronate through electrostatic interactions. The preparation of MSN–COOH involves two steps: first MSN conjugation with cyanide groups (CN) and then their hydrolysis to carboxylic groups (COOH). An organic compound containing CN groups, TSBN, was first grafted onto the MSNs activated at 90 °C for 2 h (Scheme 2). The mixture was slowly stirred at 60 °C for 48 h and then filtered and washed twice with ethanol; the produced MSN–CN was dried at 80 °C for 24 h. Hydrolysis of the nitrile group to carboxylic acid was obtained by two treatments with 2.5 M HCl at 65 °C, under stirring for 30 h. Functionalization with carboxyl groups gives MSN a higher interaction capacity towards alendronate, acting as anchor sites for the BP. The mixture was filtered and washed with ultrapure H2O and ethanol. The sample was dried at 70 °C for 2 h to obtain a MSN–COOH powder.
Scheme 2. Representation of the functionalization of MSNs with TSBN to give MSN-CN.

Synthesis of MSN–COOH–AL
Alendronate was loaded on hybrid silica through a double step procedure. 50 mg of alendronate were dissolved in 15 ml of ultrapure H2O, by monitoring the pH of the solution. The solution pH reached 4.5 when alendronate was completely dissolved and the solution was clear. Then 600 mg of MSN–COOH were added in six aliquots to the drug solution, and the final solution pH value was 1.9. The loading procedure was carried out at room temperature for 72 h. A second loading step was performed in order to maximise AL interaction with mesoporous silica: 30 mg of pure alendronate were dissolved in 15 ml of the filtered solution from the first step containing 4.5 mM alendronate (measured by UV-vis). Under continuous magnetic stirring, 400 mg of the powder derived from the first loading were added to drug solution, and the final pH value of the suspension was 2. The second loading step was carried out under the same operating conditions of the first one. After filtration and washing with ultrapure H2O, the sample was dried overnight at room temperature.
Alendronate quantification
UV-vis spectrophotometric analysis was performed to determine the amount of immobilized alendronate (AL) on the silica surface. AL is a non-chromophoric compound and so its determination by conventional spectrophotometric methods is not possible. So the UV-vis determination of AL is based on the formation of a chromophoric complex of the bisphosphonate with Fe3+ ions with an AL/Fe3+ molar ratio of 1 : 1, as reported by Kuljanin et al.22 An iron(iii) chloride standard solution was prepared by dissolving 5.5 mg of ferric chloride hexahydrate, FeCl3·6H2O, in 100 ml of perchloric acid, HClO4 2 M. For the calibration curve, five standard solutions at different concentrations of AL were prepared using a freshly prepared 5 mM AL/2 M HClO4 solution as the stock solution. In order to ensure the formation of the complex between AL and Fe3+, 394 µl of each AL-containing standard solution were stirred for 100 h with 40 ml of the iron-containing standard solution. Afterwards, absorbance of solutions was measured at 286 nm and a calibration curve (y = εx) was built with absorbance on the y axis and alendronate concentration on the x axis; slope ε = 0.0264, (R2 = 0.98).
Ibuprofen loading and release
Drug loading was achieved by soaking 0.2 g of the MSN–COOH–AL sample in 5 cm3 of ibuprofen solution (0.08 g cm−3) prepared in ethanol in tightly closed flasks. After 24 h the drug-loaded MSNs were filtered and underwent a second drug loading procedure with the same amount of ibuprofen. After 24 h the MSNs were filtered and stored in a fridge at 4 °C. To measure the drug loading, 1 cm3 of the filtrate was diluted to 10 cm3 and the absorbance of these samples was measured on a UV/vis spectrometer at 265 nm. Pure drug powder was used to prepare calibration standards of known drug concentration, which was set up with each assay. The encapsulation efficiency of ibuprofen onto the particles was about the 3%.
Ibuprofen release was quantified by HPLC analysis (Jasco HPLC system equipped with a Jasco Pu-2080 Plus Intelligent HPLC pump, Jasco DG-2080-53 3-Line Degasser, Jasco UV-2075 Plus Intelligent UV/vis detector, low pressure gradient unit 3-solvent LG-2080-02) using a Teknokroma® column (LICHROSORB RP-18 10 µm 25 cm × 0.46 cm TR-011499). In vitro drug release was performed in media simulating the normal blood/tissue environment (pH 7). The simulated body fluid (SBF pH 7) was prepared by dissolving NaH2PO4 (49.25 mg) and Na2HPO4 (20.3 mg) in 0.25 dm3 of ultrapure water.
To evaluate the kinetic drug release pattern, 4 mg of the MSNs loaded with IBU samples were dispersed in 15 cm3 of buffer solution under gentle magnetic stirring (30 rpm) for a few minutes. The release tests were carried out under static conditions at 37 °C, because prolonged mixing could negatively affect drug release. At predetermined time intervals (0.25, 0.5, 1.0, 3, 4, 5, and 6 h) 20 µl samples were collected and analysed at 220 nm to acquire the HPLC chromatograms. A 70/30% v/v of acetonitrile/water mobile phase was used for HPLC analyses with a flow rate of 1 ml min−1. The analyses were performed in triplicate.
Hydroxyapatite production and interaction with hybrid mesoporous silica particles
Hydroxyapatite (HA) was synthesized,23,24 characterized and pressed into pellets in order to use it as a substrate to test the targeting efficacy of alendronate present on the surface of mesoporous silica particles.
HA attracts great interest in catalysis, pharmaceutical products, and tissue engineering scaffolds, mostly because it is the main inorganic component of bone tissue. The synthesized HA has excellent mechanic properties as well as an ultra-fine structure similar to biological apatite.
Calcium chloride and ammonium hydrogen phosphate were first dissolved in ultrapure H2O to form 0.5 M and 0.3 M aqueous solutions respectively. The two solutions were separately heated at 70 °C and then mixed under vigorous stirring, and the mixture pH was adjusted to 10 by adding ammonium hydroxide. After 24 h of ripening time, the precipitates were recovered by three centrifugation cycles at 1000 rpm. A washing step with water was carried out to remove any by-product trace, ammonium chloride. The centrifuged fraction was dried at 80 °C for 24 h. Calcination of the synthesized hydroxyapatite was carried out by first drying the sample at 70 °C for 24 h and then calcination at 650 °C with a ramp rate of 5 °C min−1. Hydroxyapatite crystals were pelletized using a hydraulic press, by exerting a pressure of 2000 ton per m2 for about 2 minutes on the sample. A compact disk was formed and afterwards the disk was subjected to thermal treatment at 150 °C for 24 h and drying at 100 °C for 6 h. The hydroxyapatite used was selected using a 710–900 µm mesh size sieve. This size allows us to visually distinguish pellets from the mesoporous silica system during separation steps involved in the study and at the same time neglect mass transport resistances on pellets.
The capability of the produced hybrid MSN–COOH–AL system to bind hydroxyapatite has been evaluated at pH 7.4 in phosphate buffer: this environment well simulates the physiological one. The buffer solution was prepared by dissolving 9.85 mg of sodium dihydrogenphosphate (NaH2PO4) and 4.06 mg of disodium phosphate (Na2HPO4) in 50 ml of ultrapure H2O. 50 mg of MSN–COOH–AL were resuspended in 15 ml of the buffer. This volume was chosen to avoid the saturation of the delivery media during the interaction tests and ensure resuspension of silica. Two steps of filtration were subsequently performed to separate the pellets first and then the mesoporous silica particles from the suspension.
Chemical/physical characterization
Fourier transform infrared (FTIR) spectra were recorded with an infrared spectrometer (FT/IR-4600 FT-IR, Jasco; Germany).
The ordered mesoporous framework of the synthesized materials was studied by small angle powder X-ray diffraction (XRD) on a MiniFlex 600 Rigaku diffractometer operating at 40 kV and 15 mA, employing Ni b-filtered Cu Kα radiation in the 2θ range of 0.3–10° with a scan speed of 0.3 deg per min.
Scanning electron microscopy was performed with a Philips Quanta 200 F microscopy. The specific surface area of the functionalized materials was evaluated by physical adsorption measurements of nitrogen, using a Micromeritics ASAP 2020 gas adsorption device. The isotherms were elaborated for the assessment of the surface area and porosity. Alendronate concentration, loaded on the MSN system, was determined using a 300 UV-Vis Evolution, Thermo Fisher Scientific.
Cell culture conditions and viability test
HTERT-immortalized normal human foreskin fibroblasts BJ-hTERT were purchased from ATCC. The cells were maintained in Dulbecco's modified Eagle medium (DMEM) plus 10% fetal bovine serum, 100 IU ml−1 penicillin, 100 mg ml−1 streptomycin, and 0.2 mM l-glutamine (all reagents were from Invitrogen, Italy), and were maintained as a monolayer culture in a humidified incubator at 5% CO2 and 37 °C. The effect of MSN–COOH–AL on BjhTERT cell proliferation was assessed by trypan blue exclusion assay. The cells were seeded in 12-well plates in growing medium in triplicate for each condition and synchronized overnight in serum free medium (SFM). MSN–COO–AL particles were used in two forms: as-synthesized (surfactant containing) or extracted (ext), those after being processed for complete surfactant removal from the mesoporous structure. Both MSN–COO–AL particle forms were resuspended in serum free medium and added to the cells at a concentration of 10 µg ml−1 equal to 10 µg per well, containing 7.7% of the alendronate drug (77 ng). An untreated cell triplicate was used as the negative control. After 1 h, the cells were shifted to 1% FBS containing medium and incubated up to 3 days. At selected time points (1, 2 and 3 days), the cells were harvested by trypsinization and incubated in a 0.5% trypan blue solution at room temperature. Cell viability was determined by counting trypan blue negative cells by means of a Countess® automated cell counter (Thermo Fisher Scientific Inc.).
Results and discussion
Morphological analysis of mesoporous silica particles
Fig. 1 illustrates the hypothesized bone-specific drug delivery of our mesoporous silica-based hybrid material. The synthesized MS particles have an average diameter between 100 and 200 nm as shown in the SEM image (Fig. 2a), while the TEM micrograph (Fig. 2b) shows a porous texture in accordance with the mesoporous materials of the MSN family.
Fig. 1. Representation of bone-specific drug delivery of the developed mesoporous silica-based hybrid materials.

Fig. 2. (a) Electron scanning microscopy (SEM) image of the MSN sample and (b) TEM micrograph of the same sample.

Chemical–physical characterization of materials
AL interacts both with carboxylic groups on silica on one side and with the HA pellet surface on the other side, providing the mesoporous silica particles with the required targeting specificity towards HA to obtain bone-specific drug delivery.
Fig. 3a presents the X-ray diffraction pattern of the sample before and after –CN functionalization. All samples show a highly ordered structure, indicating that the functionalization procedures did not affect the order inducing pore collapse. The obtained MSN–COOH material has been characterized by nitrogen adsorption–desorption porosimetry, from which a type IV reversible isotherm is obtained (Fig. 3b). BJH analysis indicates an average pore diameter of 70 Å. The sample exhibits a broad pore size distribution due to incomplete surfactant removal during the modification and hydrolysis reaction. Table 1 summarizes these results.
Fig. 3. Different characterization techniques for MSN samples functionalized with different functional groups: (a) X-ray diffraction patterns of MSN-CN; (b) nitrogen adsorption–desorption isotherm of the MSN–COOH material; comparison between FT-IR spectra: (c) MSN and MSN-CN; (d) magnification of (c) between 3000 and 1800 cm−1; (e) FT-IR spectra of MSN-CN and MSN–COOH; (f) magnification of (e) between 2750 and 1500 cm−1.

Nitrogen adsorption–desorption data for the MSN–COOH sample.
| Pore diameter | [nm] | 7 |
| Pore volume@0.95 P/P0 | [cm3 g−1] | 0.59 |
| BET surface | [m2 g−1] | 501 |
The FT-IR spectra displayed in Fig. 3c and e show new vibration bands due to changes of the functional group population on the surface of MSNs after functionalization with TSBN and hydrolysis with HCl.
The FT-IR spectrum of the synthesized mesoporous silica (Fig. 3c upper and lower curves, details in Fig. 3d) shows a broad band centered at around 3300 cm−1 assigned to the stretching vibration of OH groups of Si–OH species. Intense bands at around 2900 cm−1 are due to the C–H stretching of the alkyl chains of the surfactant. The peak at 1622 cm−1 represents the bending vibration of H2O, while the strong band observed at 1056 cm−1 is assigned to the asymmetric Si–O–Si stretching vibration; finally, the symmetric Si–O–Si stretching vibration (Fig. 3c) is observed in the region between 950 and 800 cm−1. A peak centered at around 2256 cm−1 assigned to C N stretching vibration of 3-cyanopropyl groups (Fig. 3c lower curve, details in Fig. 3d) confirms the grafting of CN groups on the MSN silica surface.
The intensity of the band at 2256 cm−1 is greatly reduced after hydrolysis with HCl (Fig. 3e and f), thus confirming that most –CN groups have been hydrolyzed. The peaks related to the carboxyl groups in MSN–COOH spectrum are clearly shown in Fig. 3e and f, and the C O stretching band is observed at 1640 cm−1. Solid-state 29Si and 13C CP-MAS NMR spectra are presented in the ESI.† Fig. S1 and S2† show the 29Si NMR spectra of MSN and MSN–CN materials respectively. The line at −109.70 ppm stems from the Si(OSi)4 structural units, while the lines at −100.91 and −91.09 ppm are due to Si(OSi)3OH and Si(OSi)2(OH)2 units, respectively (Fig S1†). 29Si NMR of the cyano-propyl derivatized material shows an adjunctive series of low-intensity peaks between −66 and −53 ppm assigned to functionalized Si atoms but indicates also a low functionalization degree (Fig. S2†).
The 13C CP-MAS NMR spectrum of the MSN–CN sample confirmed the successful functionalization of MSNs (Fig. S3†).
The resonances between 10 and 20 ppm are assigned to the methylene carbon atoms of the propyl chain while the signal at 118.96 ppm is attributed to the carbon atom of the cyano group.
The spectrum also exhibited additional resonances corresponding to carbon atoms of the surfactant (Triton X-100), among which the peaks at around 60 and 70 ppm are the most intense (asterisks mark the positions of residual surfactant carbons present in the spectrum).
Hydrolysis of nitrile groups and the relative presence of carboxylic functionalities on the mesoporous silica surface create an intriguing material potentially useful as a drug delivery system, suitable to bind bisphosphonates. The presence of alendronate was assessed by UV-vis spectrophotometric analysis after the formation of a chromophoric complex of bisphosphonates with Fe3+ ions.
The FT-IR spectrum of alendronate is shown in the ESI (Fig. S4a–d†), and the peaks are consistent with those reported in the literature for the pure molecule.25 A broad O–H stretching band is observed at 3500–3100 cm−1, corresponding to the symmetric and asymmetric stretching vibrations of the NH3+ group and C–H stretching bands are located between 3000 and 2800 cm−1 (2959 cm−1 and 2797 cm−1) (ESI Fig. S4b†). The N–H bending vibration of the primary ammonium ion, observed in the 1650–1580 cm−1 region, is shown in Fig. S4c† (1643 cm−1, 1606 cm−1, and 1544 cm−1). Finally, the peaks between 1200 and 900 cm−1 are due to P O, P–O–H, P–O–C stretching and bending vibrations (ESI Fig. S4d†).
FT-IR investigation was carried out to verify the success of the interaction between the mesoporous silica surface and alendronate. Fig. 4 shows the comparison between the spectra of MSN–COOH and MSN–COOH–AL functionalized silica, respectively, before and after the AL loading step. After the second loading step, N–H bending vibrations of the primary ammonium ion among the characteristic peaks of alendronate can be observed.
Fig. 4. FT-IR spectra of MSN–COOH and MSN–COOH–AL functionalized silica before and after the AL loading step in the range of 1650–1450 cm−1.

A slight shift can be observed in the N–H bending vibration and C–H stretching vibration of the alkyl chain bands compared to the pure AL spectrum after loading because of the formation of chemical bonds between AL and the silica surface. After the second loading step, AL will still occupy the available sites. The electrostatic bond is labile at acidic pH9,26 but it ensures high stability at neutral pH and under the conditions of the intended use. The measurement of the zeta potential (ZP) showed a value increase from −21 mV for unmodified MSNs to −7 mV after functionalization of the MSN external surface. After interaction with AL, a new decrease of the zeta potential to −11 mV was observed, confirming successful conjugation. The amount of AL immobilized on the silica surface was calculated by measuring the absorbance of the filtered solution after each loading step. The starting amount of the drug used is known, and AL concentration in the filtered solution is measured by UV-vis spectrophotometric analysis; so the AL amount immobilized on the surface of the carrier is derivable. The data calculated by UV-vis analysis are shown in Table S1 in the ESI.† After the first loading step, the filtered solution has a volume of 26 ml and an AL concentration of 4.5 mM. The AL amount for the first step was 50 mg, and accordingly 20.87 mg of AL are on the mesoporous silica surface and the remaining 29.13 mg are in solution. The AL/silica ratio is 0.034. The total amount of AL used for the 2nd loading step is 32 mg (22 mg in the filtered solution after the 1st step, measured with UV-vis, and 10 mg of pure AL). During the 2nd loading step, 17.5 ml of the filtered solution are collected with an AL concentration of 5 mM. Hence, 22 mg is the amount of AL in the filtered solution and the remaining 10 mg of AL are on the silica surface. Overall, 30.87 mg of AL have been loaded on mesoporous silica (400 mg). The AL/silica ratio is 0.077.
Hydroxyapatite characterization
The morphology of HA crystals is shown through SEM analysis in Fig. 5a and b. The smaller crystals measure 9 × 2.6 µm and the larger ones are 21 × 5 µm.
Fig. 5. Hydroxyapatite characterization: (aand b) SEM image of the HA sample; (c) XRD spectra of HA before and after calcination; (d) FT-IR spectrum of the HA sample.

The peak of calcium hydroxide at 39° visible on HA crystals (Fig. 5a) disappears after calcination. Calcined HA powders show sharper diffraction peaks due to the loss of water (dehydration) during the calcination process (Fig. 5c). Fig. 5d shows the FT-IR spectrum of the HA sample, and the characteristic bands for HA are exhibited in the spectrum in the region of 1200–900 cm−1 for phosphate stretching and 1530–1450 cm−1 for carbonate groups, while in the region of 3700–2500 cm−1 the reflection of combined water in HA crystals is observed.
Interactions between the MSN–COOH–AL (MSN–AL) carrier and HA
Interactions between the MSN–COOH–AL carrier and HA pellets are studied in phosphate buffer with pH 7.4. A schematic representation of the hypothesized mechanism of interaction between MSN–COOH–AL and HA is shown Scheme 3. The mechanism of interaction between BP molecules and apatite nanocrystals of bone is not well understood. Nevertheless it is widely recognized that BPs present strong affinity for the apatite surface.27 The expected molecular configuration of the system proposed in this study implies the interaction between MSN–COOH and the amine group of alendronate. For this reason, MSN–COOH–AL would expose the P–C–P backbone of alendronate to the external medium, which is the portion of BP molecules with high affinity for HA. HA pellets are analysed by XRD after interaction with MSN–AL. The peaks at 2θ 25.78, 26.68, 29.19, and 30.64, typical of hydroxyapatite, show reduced intensities in the diffraction pattern of HA after interaction with MSN–COOH–AL. This result suggests that the interaction of alendronate on the mesoporous silica surface is successful and, because of bone-specificity, AL interacts both with carboxylic groups on silica on one side and with the HA pellet surface on the other side. The buffer solution does not induce changes during interaction tests, as shown in the ESI Fig. S5b.†
Scheme 3. Representation of AL–HA interactions.

Ibuprofen loading on the mesoporous carrier MSN–COOH–AL–IBU
Successful loading of ibuprofen on mesoporous silica nanoparticles was investigated with Fourier transform infrared (FTIR) spectroscopy, comparing the free ibuprofen spectrum with the one from carriers before and after drug loading. As Fig. 6 shows, loading of ibuprofen on nanoparticles is evidenced in the region between 2000 and 750 cm−1. The FTIR spectrum shows the presence of a free acid carbonyl peak around 1720 cm−1 with high intensity, CH2 scissoring vibrations (1462 cm−1) and CH–CO deformation (1420 cm−1) in accordance with the literature.28
Fig. 6. (a) FT-IR spectra of samples MSN–COO–Al, MSN–COO–Al–IBU and ibuprofen (IBU); (b) magnification of (a) in the range between 2000 and 750 cm−1.

Thermogravimetric analysis of the sample before and after ibuprofen loading shows an increment of the organic compound mass on the sample by 60%. Fig. S6† in the ESI shows the release curve of the sample MSN–COO–AL–IBU as a function of time at pH 7. The release starts at ca. 30 min reaching a concentration of 26 ppm which is about 70% of the total amount of ibuprofen loaded on the nanoparticles, and the drug release is complete after 120 min of gentle mixing. The abundant release of the drug from the vehicle is due to the large pore diameter of MSNs (7 nm) compared to the size of ibuprofen molecules (1.0 × 0.6 nm). The free space among the MSN pore walls allows the free entrance of the buffer solution and causes the transport of ibuprofen from the pores to the solution.29 Controlled release of ibuprofen from the silica nanoparticles is beyond the aim of this study; in fact, the technique used to load the drug was expected to produce only drug adsorption on the particle surface.30 In particular ibuprofen–MSN interactions are expected to rely on the potential interactions between free silanol and the organic functional groups of the guest molecule.29 Prolonged release might be considered in the future with customized chemical bonds between ibuprofen and the nanoparticle surface, as already proposed in the literature.19,31,32 Fig. S7† in the ESI presents a characteristic HPLC chromatogram of the sample collected at 30 min of release process time. IBU has a retention time of 6.1 min with a sharp and well-defined peak.
In vitro experiments
To assess MSN–AL biocompatibility, the conditions necessary for drug targeting purposes, MSN–COO–AL and MSN–COO–AL (ext) (ext = particles after surfactant removal from the mesoporous structure) were tested on normal human fibroblast BJ-hTERT cells. Interestingly, both samples did not significantly affect cell proliferation, compared to the untreated control (Fig. 7). Starting from MSN, a hybrid system in which alendronate electrostatically interacts with carboxylic group functionalization on the silica surface has been developed and characterized. The established interaction between alendronate, an efficient bone targeting drug, and silica opportunely functionalized on the surface, creates a promising drug delivery system for bone diseases. The synthesized system would target the bone thanks to alendronate specificity and can be used as a delivery system for several kinds of drugs. Combining the bone specificity of alendronate and the well-known potentiality of drug delivery of silica, the synthesized system shows great potential for bone targeting applications.
Fig. 7. Toxicity test of MSN–COO–AL on normal human fibroblasts. BJ-hTERT cells were treated with MSN–COO–AL (ext) (gray line with triangles) or MSN–COO–AL (orange line with square symbols), or left untreated (C – blue line). Cell viability was determined as described in the Experimental after 1, 2 and 3 days of treatment. The values represent the mean of three independent experiments and are reported as the number of viable cells.

Conclusions
We have developed hybrid mesoporous silica-based particles functionalized on the external surface using alendronate, a drug of the bisphosphonate series, potentially useful for bone-specific drug delivery. The tendency of the material to interact with hydroxyapatite has been investigated as well as the possibility to load it with a pharmacologically active molecule, specific for bone-targeted therapy. In conclusion, mesoporous silica particles represent a versatile starting architecture for the development of bi/multifunctional nanostructured devices. We do not know how many solutions or approaches, among the proposed ones in open or patented literature, will really contribute to the improvement of the state of the art of the concerned therapies. Nevertheless, we are sure that the ferment determining the design, synthesis and optimization of a new material with biological activity is a very positive stimulus for the rapid progress of material science.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors acknowledge the research fund from ex MIUR 60% 2014.
Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00249a
Notes and references
- Fleisch H. Russell R. G. Francis M. D. Science. 1969;165:1262–1264. doi: 10.1126/science.165.3899.1262. [DOI] [PubMed] [Google Scholar]
- Boanini E. Torricelli P. Gazzano M. Giardino R. Bigi A. Biomaterials. 2008;29:790–796. doi: 10.1016/j.biomaterials.2007.10.040. [DOI] [PubMed] [Google Scholar]
- Nancollas G. H. Tang R. Phipps R. J. Henneman Z. Gulde S. Wu W. Mangood A. Russell R. G. Ebetino F. H. Bone. 2006;38:617–627. doi: 10.1016/j.bone.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Fleisch H., in Bisphosphonates in Bone Disease, ed. H. Fleisch, Academic Press, San Diego, 4th edn, 2000, pp. 1–26, 10.1016/B978-012260371-6.50001-X [DOI] [Google Scholar]
- Hirabayashi H. Takahashi T. Fujisaki J. Masunaga T. Sato S. Hiroi J. Tokunaga Y. Kimura S. Hata T. J. Controlled Release. 2001;70:183–191. doi: 10.1016/S0168-3659(00)00355-2. [DOI] [PubMed] [Google Scholar]
- Bulman Page P. C. Moore J. P. G. Mansfield I. McKenzie M. J. Bowler W. B. Gallagher J. A. Tetrahedron. 2001;57:1837–1847. doi: 10.1016/S0040-4020(00)01164-9. [DOI] [Google Scholar]
- Erez R. Ebner S. Attali B. Shabat D. Bioorg. Med. Chem. Lett. 2008;18:816–820. doi: 10.1016/j.bmcl.2007.11.029. [DOI] [PubMed] [Google Scholar]
- Bansal G. Wright J. E. I. Kucharski C. Uludaǧ H. Angew. Chem., Int. Ed. 2005;44:3710–3714. doi: 10.1002/anie.200500350. [DOI] [PubMed] [Google Scholar]
- Pasqua L. Testa F. Aiello R. Cundari S. Nagy J. B. Microporous Mesoporous Mater. 2007;103:166–173. doi: 10.1016/j.micromeso.2007.01.045. [DOI] [Google Scholar]
- Pasqua L. Testa F. Aiello R. Madeo G. Nagy J. B. Phys. Chem. Chem. Phys. 2003;5:640–645. doi: 10.1039/B209444D. [DOI] [Google Scholar]
- Ceresa C. Nicolini G. Rigolio R. Bossi M. Pasqua L. Cavaletti G. Curr. Med. Chem. 2013;20:2589–2600. doi: 10.2174/0929867311320200007. [DOI] [PubMed] [Google Scholar]
- Morelli C. Maris P. Sisci D. Perrotta E. Brunelli E. Perrotta I. Panno M. L. Tagarelli A. Versace C. Casula M. F. Testa F. Ando S. Nagy J. B. Pasqua L. Nanoscale. 2011;3:3198–3207. doi: 10.1039/C1NR10253B. [DOI] [PubMed] [Google Scholar]
- Pasqua L. Leggio A. Sisci D. Ando S. Morelli C. Mini reviews in medicinal chemistry. 2016;16:743–753. doi: 10.2174/1389557516666160321113620. [DOI] [PubMed] [Google Scholar]
- Nigro A. Pellegrino M. Greco M. Comandè A. Sisci D. Pasqua L. Leggio A. Morelli C. Pharmaceutics. 2018;10:250. doi: 10.3390/pharmaceutics10040250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano R. Casadonte F. Pasqua L. Candeloro P. Di Fabrizio E. Urbani A. Savino R. Talanta. 2010;80:1532–1538. doi: 10.1016/j.talanta.2009.03.060. [DOI] [PubMed] [Google Scholar]
- Casadonte F. Pasqua L. Savino R. Terracciano R. Chem.–Eur. J. 2010;16:8998–9001. doi: 10.1002/chem.201000120. [DOI] [PubMed] [Google Scholar]
- Botella P. Rivero-Buceta E. J. Controlled Release. 2017;247:28–54. doi: 10.1016/j.jconrel.2016.12.023. [DOI] [PubMed] [Google Scholar]
- Bobo D. Robinson K. J. Islam J. Thurecht K. J. Corrie S. R. Pharm. Res. 2016;33:2373–2387. doi: 10.1007/s11095-016-1958-5. [DOI] [PubMed] [Google Scholar]
- Balas F. Manzano M. Horcajada P. Vallet-Regi M. J. Am. Chem. Soc. 2006;128:8116–8117. doi: 10.1021/ja062286z. [DOI] [PubMed] [Google Scholar]
- Colilla M. Izquierdo-Barba I. Vallet-Regí M. Microporous Mesoporous Mater. 2010;135:51–59. doi: 10.1016/j.micromeso.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqua L., Testa F. and Aiello R., in Studies in Surface Science and Catalysis, ed. J. Čejka, N. Žilková and P. Nachtigall, Elsevier, 2005, vol. 158, pp. 557–564 [Google Scholar]
- Kuljanin J. Janković I. Nedeljković J. Prstojević D. Marinković V. J. Pharm. Biomed. Anal. 2002;28:1215–1220. doi: 10.1016/S0731-7085(02)00021-3. [DOI] [PubMed] [Google Scholar]
- Pang Y. X. Bao X. J. Eur. Ceram. Soc. 2003;23:1697–1704. doi: 10.1016/S0955-2219(02)00413-2. [DOI] [Google Scholar]
- Sarig S. Kahana F. J. Cryst. Growth. 2002;237–239:55–59. doi: 10.1016/S0022-0248(01)01850-4. [DOI] [Google Scholar]
- Thoke S. P Sharma Y. Rawat s. r. Satish N. J. Drug Delivery Ther. 2013;3:65–74. [Google Scholar]
- Pasqua L. Procopio A. Oliverio M. Paonessa R. Prete R. Nardi M. Casula M. F. Testa F. Nagy J. B. J. Porous Mater. 2013;20:865–873. doi: 10.1007/s10934-012-9663-1. [DOI] [Google Scholar]
- Pascaud P. Bareille R. Bourget C. Amédée J. Rey C. Sarda S. Biomed. Mater. 2012;7:054108. doi: 10.1088/1748-6041/7/5/054108. [DOI] [PubMed] [Google Scholar]
- Acharya M. Mishra S. Sahoo R. N. Mallick S. Acta Chim. Slov. 2017;64:45–54. doi: 10.17344/acsi.2016.2772. [DOI] [PubMed] [Google Scholar]
- Vallet-Regi M. Rámila A. del Real R. P. Pérez-Pariente J. Chem. Mater. 2001;13:308–311. doi: 10.1021/cm0011559. [DOI] [Google Scholar]
- Cavallaro G. Pierro P. Palumbo F. S. Testa F. Pasqua L. Aiello R. Drug Delivery. 2004;11:41–46. doi: 10.1080/10717540490265252. [DOI] [PubMed] [Google Scholar]
- Rehman F. Ahmed K. Airoldi C. Gaisford S. Buanz A. Rahim A. Muhammad N. Volpe P. L. O. Mater. Sci. Eng., C. 2017;72:34–41. doi: 10.1016/j.msec.2016.11.040. [DOI] [PubMed] [Google Scholar]
- Vall M. Zhang P. Gao A. Frykstrand S. Cheung O. Stromme M. Int. J. Pharm. 2017;524:141–147. doi: 10.1016/j.ijpharm.2017.03.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.