

Abstract
In this work, the reduction of iron oxide γ-Fe2O3 nanoparticles by hydrogen at high pressures is studied. Increasing the hydrogen pressure enables reduction of γ-Fe2O3 to α-Fe at significantly lower temperatures. At low pressures, a temperature of 390 °C is necessary whereas at 530 bar complete reduction can be realized at temperatures as low as 210 °C. This leads to significant improvement in the final particle morphology, maintaining high surface-to-volume ratio of the nanoparticles with an average size of 47 ± 5 nm which is close to that of the precursor γ-Fe2O3. Neck formation, coalescence and growth during reduction can be significantly suppressed. Investigations of magnetic properties show that saturation magnetization of the reduced α-Fe nanoparticles decreases with particle size from 209 A m2 kg−1 at 390 °C reduction temperature to 204 A m2 kg−1 at 210 °C. Coercivity for the fine iron particles reaches 0.076 T which exceeds the theoretical anisotropy field. This is attributed to nano-scale surface effects.
In this work, the reduction of iron oxide γ-Fe2O3 nanoparticles by hydrogen at high pressures is studied.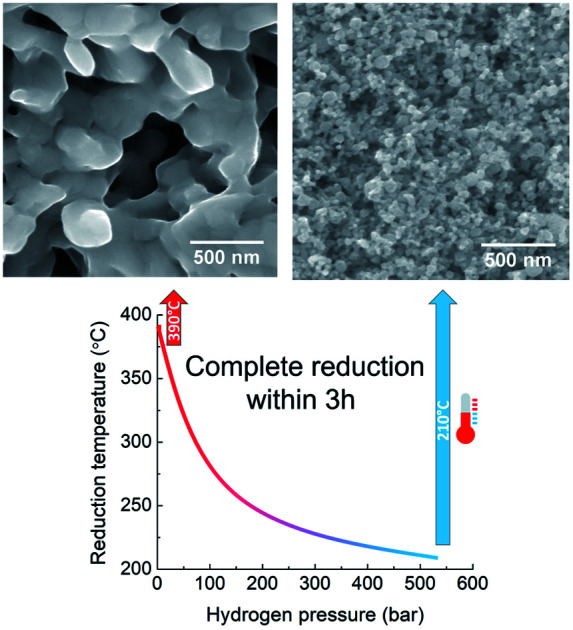
1. Introduction
Iron nanoparticles have important applications due to their unique chemical and magnetic properties.1 For example, it has been shown that compared to micrometer-sized particles, the catalytic activity of nanostructured iron for the Fischer–Tropsch process is much higher due to the larger surface area.2 Similar catalytic activity to platinum-based materials for chemical hydrogen storage applications has been reported for amorphous Fe nanoparticles.3 It has also been demonstrated by Dirba et al. that fine Fe nanoparticles are crucial for formation of the α′′-Fe16N2 phase using low-temperature nitriding with ammonia.4 Furthermore, since iron has the highest saturation magnetization among all ferromagnetic elements, yet is a cheap, abundant and environmentally friendly material, numerous applications of Fe nanoparticles in magnetism are promising. Exchange-coupled nanocomposite magnets for remanence and maximum energy product enhancement, consisting of nanoscale α-Fe as the magnetically soft phase and SmCo5, Nd2Fe14B or Sm2Fe17N3 as the hard phases, have been investigated.5–8 Furthermore, the potential in biomedical applications, such as magnetic resonance imaging9 or hyperthermia mediated drug release for cancer therapy10 has also been discussed if a proper surface coating (e.g. Au) is provided.11
Synthesis of nanoparticles with controlled particle size, morphology and surface modifications can be achieved by solution chemistry techniques.12–14 This approach is well suited for biomedical applications where minimizing reactivity and agglomeration is important. However, for magnetic nanocomposites and catalysts, the presence of an organic shell around the nanoparticles is not favorable, because it competes with the exchange length (<5 nm for typical hard magnetic materials; e.g. 1.9 nm in for Nd2Fe14B15) in the former and hinders active surface sites for the latter. Therefore, in this work we demonstrate how to produce fine α-Fe nanoparticles using a gas–solid high-pressure hydrogen reduction process.
In literature, reduction of iron oxides to α-Fe with hydrogen is typically conducted at temperatures of at least 390 °C–500 °C.16–18 This choice of temperature is supported by hygrometry measurements, where the maximum rate of H2O production was from 389 °C to 522 °C19,20 depending on the heating rate. Heat treatment at such elevated temperatures leads to particle coalescence and growth of their effective size, followed by a detrimental reduction of their surface area due to temperature-enhanced diffusion which increases exponentially with temperature. A sintering prevention layer, such as Al2O3,21 hydroxyapatite22 or carbon23 can be helpful, which, however, covers the produced nanoparticles and, therefore is also not suitable for catalyst or exchange-coupled nanocomposite applications due to the additional shell.
Assuming a single-step reaction, hydrogen reduction of Fe2O3 can be written as
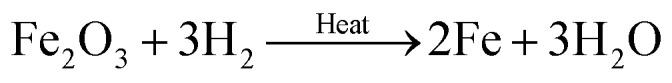 |
1 |
With a reaction constant Kr = pH2O3/pH23, where pH2O and pH2 are the partial pressures of water vapor and hydrogen respectively. By using the thermodynamic data from ref. 24, the calculated reaction Gibbs free energy of ΔG0 = 55 kJ mol−1 and enthalpy of ΔH0 = 98 kJ mol−1 are obtained. Thus, the reaction (1) is endergonic and endothermic, and does not occur spontaneously at ambient conditions. Taking the expression for Kr into account, a shift of the chemical equilibria to the right (towards products) and complete reduction of the iron oxide to Fe at lower temperatures is achievable at higher hydrogen partial pressure pH2. Therefore, in this work, we attempt to achieve complete reduction of Fe2O3 at significantly lower temperatures by using high hydrogen pressures. This approach should result in improved morphology of the produced nanoparticles since the detrimental coarsening during the reduction step is suppressed.
2. Experimental
Commercial maghemite γ-Fe2O3 nanoparticles from Alfa Aesar with an average particle size of 20–40 nm according to the manufacturer's datasheet, were used as a precursor. Reduction experiments were done in a hydrogen (99.999% purity, Linde) atmosphere in a custom-made autoclave system equipped with high pressure/high temperature vessel (series 4740, Parr Instrument Company) and an external vertical tube heater assembly (Model 4921, Parr Instrument Company). The working volume of the vessel was 75 cm3. 0.050 g of iron oxide nanoparticles were filled in a steel crucible and mounted in the pressure vessel. After twice evacuating and purging with Ar (99.999% purity, Air Liquide), the reactor was heated to target temperature under continuous pumping and then pressurized with hydrogen. The reduced samples were handled in an Ar filled glovebox (p(O2) < 0.1 ppm, MBraun) in order to avoid oxidation.
Magnetic properties of the nanoparticles were measured using vibrating sample magnetometer (VSM) (7400 Series, Lake Shore). Microstructural investigations were carried out using a scanning electron microscope (SEM, Philips XL30 FEG, 15 kV acceleration voltage). For scanning transmission electron microscopy (STEM) a JEOL JEM 2100F was used. The microscope was operated at 200 kV. The sample was prepared on a lacey carbon Cu grid in a glovebox and transferred to the TEM in a Gatan 648 double tilt vacuum transfer holder under nitrogen atmosphere to prevent oxidation. In addition to high-resolution (HR) TEM imaging and energy dispersive X-ray spectroscopy (EDS) in scanning transmission electron microscopy (STEM), nanobeam diffraction was performed to acquire electron diffraction data from different regions of the particle. The full width half maximum of the electron beam was 2 nm. Crystal structure and phase purity of the nanoparticles were investigated by X-ray diffraction (XRD) using a powder diffractometer (STOE, Stadi P) with Mo Kα1-radiation (λ = 0.70930 Å) in Debye–Scherrer geometry with powders sealed in quartz capillaries to avoid oxidation. X-ray photoelectron spectroscopy (XPS) measurements were performed on compacted nanoparticle pellets using a PHI Versaprobe 5000 spectrometer with monochromatic Al Kα radiation.
3. Results and discussion
The initial γ-Fe2O3 is a typical brown rust-like powder. The average particle size is expected to be around 20–40 nm according to manufacturer's data. SEM and TEM images of the powder are presented in Fig. 1. Particles show a broad size distribution from fine 20 nm nanoparticles to several large particles in the range of hundreds of nanometers. The average particle size estimated from the SEM images is 52 ± 31 nm (152 particles counted). The average crystallite size obtained from XRD peak broadening is 55 ± 5 nm. The TEM image shows that the fine nanoparticles mostly have well-defined hexagonal shapes.
Fig. 1. SEM image of the initial γ-Fe2O3 nanoparticles. The inset shows TEM image of a single nanoparticle.

Fig. 2 shows XRD patterns of the initial γ-Fe2O3 and the same nanoparticles reduced at a hydrogen pressure of 100 bar for 3 hours at different temperatures in the range from 230 °C to 390 °C. At 390 °C, in agreement to previous reports, reduction can be achieved even at low pressures (not shown here). 100 bar hydrogen pressure is sufficient for complete reduction at temperatures down to 270 °C within 3 h. Further lowering of the reduction temperature to 230 °C leads to partial reduction and remaining γ-Fe2O3 reflections are evident in the XRD data.
Fig. 2. XRD patterns of the γ-Fe2O3 nanoparticle powder before and after three-hour reduction in hydrogen at a pressure of 100 bar and temperatures between 230 °C and 390 °C.

In a next step, the reduction of γ-Fe2O3 nanoparticles at temperatures below 230 °C was investigated at hydrogen pressure above 100 bar. The corresponding XRD results are presented in Fig. 3. At 230 °C and 200 bar hydrogen pressure, a small signal from the highest intensity iron oxide diffraction peak can be identified at 16.1°.
Fig. 3. XRD patterns of the γ-Fe2O3 nanoparticle powders after three-hour reduction at temperatures of 210°C–230 °C and hydrogen pressures in the range between 100 bar and 530 bar.

Therefore, the pressure was increased further to 300 bar at which the γ-Fe2O3 nanoparticles are fully reduced; only reflections from α-Fe are visible in the corresponding diffractogram. At lower temperatures of 220 °C and 210 °C, a hydrogen pressure of 400 bar and 530 bar is necessary to complete the reduction in 3 hours. Thus, by increasing the hydrogen pressure to 530 bar, it is indeed possible to reduce the temperature necessary for complete reduction of γ-Fe2O3 nanoparticles to α-Fe from 390 °C to 210 °C. The pressure of 530 bar was the limit for the autoclave, a further decrease in the reduction temperature for higher pressures can be expected. On the other hand, it is possible that at a certain pressure the benefits from the lower temperature will be outweigh by a reduction of the diffusion barrier25 and gas-pressure sintering effects.26
In order to investigate the coalescence and growth of the nanoparticles at the proposed high-pressure, low-temperature hydrogen reduction, free-standing nanoparticles were examined using SEM. The reduction at 390 °C leads to severe bonding of the nanoparticles into sponge-like agglomerates with sizes in several tens of micrometers (Fig. 4a). A coalescence of the nanoparticles into large grains with much smaller surface area is observed in the high-resolution SEM image in Fig. 4b. Bridging between neighboring nanoparticles and their coalescence occur also at 310 °C (Fig. 4c) and at 270 °C (Fig. 4d), although, with much smaller increase in the size. However, after reduction at 230 °C (Fig. 4e) and especially at 210 °C (Fig. 4f), neck formation and coalescence are minimized, and individual nanoparticles can be distinguished. Thus, the coalescence of the nanoparticles during the reduction process has been suppressed. Clustering still takes place due to magnetostatic interactions, therefore monodisperse particles could not be achieved. The average size of the obtained phase-pure α-Fe nanoparticles is comparable with that of the γ-Fe2O3 precursor. A particle size analysis was conducted by the line interception method in which an average size was calculated for the grains along 4 lines (horizontal, vertical and both diagonals)27 in the respective SEM images. The obtained sizes of the nanoparticles are summarized in Table 1, including a comparison with the crystallite size, extracted from the XRD peak broadening. Usage of lower reduction temperatures is clearly beneficial and results in fine α-Fe nanoparticles with a size smaller than 50 nm. Clearly, lowering of the reduction step temperature results in much finer α-Fe particles. Error in the SEM data is caused mainly by a limited number and inaccurate size measurements of the analyzed nanoparticles in the SEM images, especially for large concrescent particles at high reduction temperatures.
Fig. 4. SEM images of the hydrogen reduced nanoparticles. (a) Low and (b) high magnification images of a sample reduced at 390 °C (100 bar). (c–f) High magnification SEM images of samples reduced at 310 °C (100 bar), 270 °C (100 bar), 230 °C (300 bar) and 210 °C (530 bar) respectively.

Particle and crystallite sizes extracted from the SEM and XRD measurements, respectively, of the α-Fe nanoparticles obtained by reduction at different temperatures and hydrogen pressures.
| Temperature (°C) | Particle size (nm) | Crystallite size (nm) | Hydrogen pressure (bar) |
|---|---|---|---|
| 390 | 215 ± 37 | 263 ± 46 | 100 |
| 310 | 96 ± 47 | 133 ± 12 | 100 |
| 270 | 67 ± 2 | 79 ± 1 | 100 |
| 210 | 47 ± 5 | 41 ± 1 | 530 |
The extracted value of the crystallite size is additionally influenced by the broadening of the XRD reflections due to microstrain. Fig. 5 summarizes the results of hydrogen reduction experiments. The data points represent temperature–pressure pairs where complete reduction of Fe2O3 to α-Fe was achieved according to the XRD data. It can be concluded that reduction of iron oxide nanoparticles at elevated hydrogen pressures and consequently lower temperatures can indeed be used for production of high-quality iron nanoparticles without the detrimental particle coarsening.
Fig. 5. Summary of the results from hydrogen reduction experiments. (a) Gradual increase in hydrogen pressure enables lowering of the reaction temperature. (b) At lower reduction temperatures particle growth is minimized.

Due to the broadening of the XRD reflections in nanoparticles and a possibility that some fraction of the material is amorphous, small amounts of residual iron oxide could remain unnoticed in the XRD data. For this reason, XPS measurements of the initial γ-Fe2O3 powder and the reduced α-Fe nanoparticles were conducted to analyze electronic structure of Fe (Fig. 6). Prior to the XPS measurements, the samples were sputtered for 10 minutes using Ar+ ions at 2 kV to remove the adventitious carbon contamination and oxidized layers on the sample surface due to contacts with atmospheric oxygen.
Fig. 6. XPS Fe2p spectra of the initial γ-Fe2O3 powder and α-Fe nanoparticles produced by reduction in hydrogen atmosphere at 530 bar and 210 °C.

The Fe2p spectra for the initial γ-Fe2O3 nanoparticles show emissions at 710.8 and 724.5 eV (a spin–orbit splitting of 13.7 eV) for the Fe2p1/2 and Fe2p3/2, respectively. The observed shake-up satellite peaks are shifted from the parent peaks by 8.5 eV. Within the measurement accuracy, the spectra correspond to those reported in literature for γ-Fe2O3.28,29 The Fe2p spectra of the obtained α-Fe nanoparticles reveal a textbook-like30 Fe2p1/2 and Fe2p3/2 emissions at 706.8 and 719.9 eV, respectively, with 13.1 eV spin–orbit splitting and absence of shake-up satellites. The XPS results indicate complete reduction of the γ-Fe2O3 powder at 210 °C and 530 bar hydrogen pressure. It has to be noted that the measured initial survey spectra show oxygen and carbon on the surface which can be removed after in situ Ar ion sputtering. This indicates surface oxidation of the α-Fe nanoparticles.
In the HRTEM image shown in Fig. 7a a spherical particle shape can be observed in accordance to the SEM imaging discussed above. A core–shell type oxidation layer is revealed. Since atomic resolution information is included in this image, it is possible to apply a bandpass filter for the lattice spacing of the Fe2O3 (210) lattice spacing (d = 2.519 Å). The filtered image shown in Fig. 7b clearly shows the occurrence of this spacing in the outer or shell regions of the particle. This coincides with increased oxygen concentration in the shell, which is evident in the STEM-EDS mapping, Fig. 7c. As a spatially resolved electron diffraction (ED) method, NBD allows for the collection of k-space data with a high real space resolution. A series of NBDED patterns was acquired along indicated line in Fig. 7a and the azimuthal integrated intensities are plotted in Fig. 7e. Characteristic Bragg intensities are marked for both phases, α-Fe and γ-Fe2O3, it can be observed that the core of the particle, the Fe intensities are prominent and with increasing distance from the center of the particle, the Fe2O3 phase intensities increase. In the experimental geometry, only a projection can be observed, which is causing a convolution of the probed volume in both the NBD patterns and the EDS intensities. This behavior is shown in Fig. 7f, where a homogeneous distribution of an element in a spherical particle is indicated. As discussed above, the core–shell structure of the particle becomes evident with the increased concentration of Fe in the center of the particle and with the increased O content for the shell, both deviating from the ideal distribution. These results are in agreement with the XPS survey measurements and show that surface oxidation during handling could not be fully avoided.
Fig. 7. (a) HRTEM image of a representative nanoparticle, the NBD scan direction is indicated originating at the center of the particle. (b) Bandpass/Fourier filtered image with the Fe2O3 (210) distance selected, note that this lattice spacing is only observed for the shell of the particle. (c) Color-coded EDS map of the particle (red: Fe–La, green O–K). (d) Exemplary NBD electron diffraction pattern with the radius in k-space indicated. (e) Intensity profile of rotational averaged NBD patterns recorded along the scan line indicated in (a). Note that for the center of the particle, the intensities of the Fe lattice spacings are prominent and for the edge/shell of the particle the Fe2O3 lattice spacings are stronger. (f) Line profiles of the characteristic line intensities plotted in (c). An ideal distribution for a homogeneous and spherical particle is also plotted.

Magnetic properties of the precursor Fe2O3 and hydrogen reduced Fe nanoparticles are summarized in Fig. 8. Room temperature M(H) loops for pure samples reduced at 390 °C, 310 °C, 270 °C and 210 °C are shown in Fig. 8a with the corresponding Ms and Hc values as a function of the size of the reduced particles, plotted in Fig. 8b. The initial γ-Fe2O3 powder shows Ms = 70 A m2 kg−1 which is slightly lower than the bulk value of 74 A m2 kg−1.31 There is a decrease in Ms with particle size for the α-Fe samples from 209 A m2 kg−1 (390 °C reduction temperature) to 204 A m2 kg−1 (210 °C reduction temperature). Similar findings have been reported by Hsu et al.,32 where the lower saturation magnetization was explained by the semi-infinite nature of fine particles. The atoms on the free surface of nanoparticles possess a lower magnetization than the bulk material due to the lack of translational symmetry and broken magnetic exchange bonds on the surface. For this reason, finer particles lead to reduced Ms due to the large surface area. Another contributing factor to the reduced magnetization as a function of the reciprocal particle size is also the slight surface oxidation during handling and measurement as evident from the TEM and XPS data.
Fig. 8. (a) Hysteresis loops for the initial γ-Fe2O3 and α-Fe particles produced by hydrogen reduction at 390 °C, 310 °C, 270 °C and 210 °C. (b) Saturation magnetization Ms and coercivity Hc at room temperature as a function of the particle size for the hydrogen reduced α-Fe nanoparticles.

Coercivity Hc scales inversely with the particle size and increases more than 10-fold from 0.0069 T (390 °C reduction temperature) to 0.076 T (390 °C reduction temperature). The increased coercivity for smaller particles (lower reduction temperatures) is consistent with the expectation that the nucleation field is maximum for single-domain particles.33 For a spherical iron particle, the single-domain diameter is about 10 nm,15 which is smaller than the experimental particle size. Thus, a further coercivity enhancement for even finer particles can be expected and indeed Hc values of 0.106 T for the mean particle size of approximately 30 nm have been reported.32 According to the Stoner–Wohlfarth model,34 the maximum coercivity is defined by the anisotropy field Ha = 2K1/μ0Ms, where K1 is the principal anisotropy constant. For iron, K1 ≈ 50 kJ m−3, μ0Ms ≈ 2.15 T and, therefore Ha ≈ 0.06 T. However, typically the measured coercivities reach only small fraction, approximately 25% of the theoretical Ha which is known as the Brown's paradox.35,36 Interestingly, as demonstrated in the discussion above, coercivity of fine Fe particles exceeds the calculated theoretical upper limit – the anisotropy field Ha. Therefore, most likely additional surface-related phenomena take place and account for the discrepancy. For example, an increase in the anisotropy constant with the reciprocal particle diameter up to 300 kJm−3 for 2 nm α-Fe nanoparticles has been reported37 and is attributed presumably to influence of surface effects. Also, the observed surface oxidation may play a role due to exchange coupling between the core/shell layers.
4. Summary
In this work, we have demonstrated that by increasing the hydrogen pressure up to 530 bar, it is possible to lower the temperature necessary for complete reduction of γ-Fe2O3 nanoparticles to α-Fe from 390 °C down to 210 °C. This significant improvement in reduction temperature was shown to be beneficial for the final particle morphology. Coalescence and sintering of the particles accompanied by surface area loss which occurs at elevated temperatures (e.g. 390 °C), can be suppressed when reduction is performed at 210 °C. The saturation magnetization of the reduced α-Fe nanoparticles decreases with particle size from 209 A m2 kg−1 at 390 °C reduction temperature to 204 A m2 kg−1 at 210 °C. Coercivity shows opposite behavior and even exceeds the theoretical anisotropy field for the fine 47 nm particles. This is attributed to surface effects. TEM investigations reveal that the Fe nanoparticles are passivated with a Fe2O3 layer resulting in a core–shell structure.
These findings are also relevant for applications such as catalysis and exchange-coupled nanocomposites, where fine iron nanoparticles with high surface area are required. The presented method can be extended to other metal-oxide systems.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the German Federal Ministry of Education and Research (BMBF) within the project 03X3582. This work was also supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Project ID No. 405553726, TRR 270.
References
- Huber D. L. Synthesis, properties, and applications of iron nanoparticles. Small. 2005;1:482–501. doi: 10.1002/smll.200500006. doi: 10.1002/smll.200500006. [DOI] [PubMed] [Google Scholar]
- Suslick K. S. Hyeon T. Fang M. Nanostructured materials generated by high-intensity ultrasound: sonochemical synthesis and catalytic studies. Chem. Mater. 1996;8:2172–2179. doi: 10.1021/cm960056l. doi: 10.1021/cm960056l. [DOI] [Google Scholar]
- Yan J. M. Zhang X. B. Han S. Shioyama H. Xu Q. Iron-nanoparticle-catalyzed hydrolytic dehydrogenation of ammonia borane for chemical hydrogen storage. Angew. Chem., Int. Ed. 2008;47:2287–2289. doi: 10.1002/anie.200704943. doi: 10.1002/anie.200704943. [DOI] [PubMed] [Google Scholar]
- Dirba I. Schwöbel C. A. Diop L. V. B. Duerrschnabel M. Molina-Luna L. Hofmann K. Komissinskiy P. Kleebe H. J. Gutfleisch O. Synthesis, morphology, thermal stability and magnetic properties of α′′-Fe16N2 nanoparticles obtained by hydrogen reduction of γ-Fe2O3 and subsequent nitrogenation. Acta Mater. 2017;123:214–222. doi: 10.1016/j.actamat.2016.10.061. doi: 10.1016/j.actamat.2016.10.061. [DOI] [Google Scholar]
- Zeng Q. Zhang Y. Bonder M. J. Hadjipanayis G. C. Radhakrishnan R. Bulk SmCo5/α-Fe Composite by Plasma Pressure Consolidation. IEEE Trans. Magn. 2003;39:2974–2976. doi: 10.1109/TMAG.2003.816711. doi: 10.1109/TMAG.2003.816711. [DOI] [Google Scholar]
- Hu J. Hu B. P. Wang Y. Wang K. Wang Z. Hard magnetic behaviours of composite materials of the iron nanoparticles and HDDR NdDy-FeCo-B powders. J. Phys.: Condens. Matter. 1995;7:L271–L273. doi: 10.1088/0953-8984/7/20/001. doi: 10.1088/0953-8984/7/20/001. [DOI] [Google Scholar]
- Hermann R. Schultz L. O'Donnell K. Kuhrt C. Givord D. Magnetic reversal in single-phase nanocrystalline sm-fe-n alloys and a two-phase remanence enhanced (Sm2fe17n3 + α-fe) alloy. J. Phys. D: Appl. Phys. 1995;28:2411–2417. doi: 10.1088/0022-3727/28/12/004. doi: 10.1088/0022-3727/28/12/004. [DOI] [Google Scholar]
- Sepehri-Amin H. Dirba I. Tang X. Ohkubo T. Schrefl T. Gutfleisch O. Hono K. Development of high coercivity anisotropic Nd-Fe-B/Fe nanocomposite powder using hydrogenation disproportionation desorption recombination process. Acta Mater. 2019;175:276–285. doi: 10.1016/j.actamat.2019.06.017. doi: 10.1016/j.actamat.2019.06.017. [DOI] [Google Scholar]
- Bautista M. C. Bomati-Miguel O. Zhao X. Morales M. P. González-Carreño T. Pérez de Alejo R. Ruiz-Cabello J. Veintemillas-Verdaguer S. Comparative study of ferrofluids based on dextran-coated iron oxide and metal nanoparticles for contrast agents in magnetic resonance imaging. Nanotechnology. 2004;15(4):S154–S159. doi: 10.1088/0957-4484/15/4/008. doi: 10.1088/0957-4484/15/4/008. [DOI] [Google Scholar]
- Li Y. Dhawan U. Wang H. Y. Liu X. Ku H. H. Tsai M. T. Yen H. W. Chung R. J. Theranostic Iron@Gold Core–Shell Nanoparticles for Simultaneous Hyperthermia-Chemotherapy upon Photo-Stimulation. Part. Part. Syst. Charact. 2019;36:1–14. doi: 10.1002/ppsc.201800419. [DOI] [Google Scholar]
- Cho S. J. Jarrett B. R. Louie A. Y. Kauzlarich S. M. Gold-coated iron nanoparticles: a novel magnetic resonance agent for T 1 and T2 weighted imaging. Nanotechnology. 2006;17:640–644. doi: 10.1088/0957-4484/17/3/004. doi: 10.1088/0957-4484/17/3/004. [DOI] [Google Scholar]
- Willard M. A. Kurihara L. K. Carpenter E. E. Calvin S. Harris V. G. Chemically prepared magnetic nanoparticles. Int. Mater. Rev. 2004;49:125–170. doi: 10.1179/095066004225021882. doi: 10.1179/095066004225021882. [DOI] [Google Scholar]
- Watt J. Bleier G. C. Austin M. J. Ivanov S. A. Huber D. L. Non-volatile iron carbonyls as versatile precursors for the synthesis of iron-containing nanoparticles. Nanoscale. 2017;9:6632–6637. doi: 10.1039/c7nr01028a. doi: 10.1039/c7nr01028a. [DOI] [PubMed] [Google Scholar]
- Cheong S. Ferguson P. Feindel K. W. Hermans I. F. Callaghan P. T. Meyer C. Slocombe A. Su C. H. Cheng F. Y. Yeh C. S. Ingham B. Toney M. F. Tilley R. D. Simple synthesis and functionalization of iron nanoparticles for magnetic resonance imaging. Angew. Chem., Int. Ed. 2011;50:4206–4209. doi: 10.1002/anie.201100562. doi: 10.1002/anie.201100562. [DOI] [PubMed] [Google Scholar]
- Coey J. M. D., Magnetism and Magnetic Materials, Cambridge University Press, Cambridge, 2010, 10.1017/CBO9780511845000 [DOI] [Google Scholar]
- Yamashita S. Masubuchi Y. Nakazawa Y. Okayama T. Tsuchiya M. Kikkawa S. Crystal structure and magnetic properties of α′′-Fe 16N 2 containing residual α-Fe prepared by low-temperature ammonia nitridation. J. Solid State Chem. 2012;194:76–79. doi: 10.1016/j.jssc.2012.07.025. doi: 10.1016/j.jssc.2012.07.025. [DOI] [Google Scholar]
- Widenmeyer M. Hansen T. C. Niewa R. Formation and decomposition of metastable α′′-Fe 16N2 from in situ powder neutron diffraction and thermal analysis. Z. Anorg. Allg. Chem. 2013;639:2851–2859. doi: 10.1002/zaac.201300379. doi: 10.1002/zaac.201300379. [DOI] [Google Scholar]
- Kikkawa S. Kubota K. Takeda T. Particle size dependence in low temperature nitridation reaction for Fe16N2. J. Alloys Compd. 2008;449:7–10. doi: 10.1016/j.jallcom.2005.12.134. doi: 10.1016/j.jallcom.2005.12.134. [DOI] [Google Scholar]
- Tiernan M. J. Barnes P. A. Parkes G. M. B. Reduction of iron oxide catalysts: the investigation of kinetic parameters using rate perturbation and linear heating thermoanalytical techniques. J. Phys. Chem. B. 2001;105:220–228. doi: 10.1021/jp003189+. doi: 10.1021/jp003189+. [DOI] [Google Scholar]
- Jung S. S. Lee J. S. In-situ kinetic study of hydrogen reduction of Fe2O3 for the production of Fe nanopowder. Mater. Trans. 2009;50:2270–2276. doi: 10.2320/matertrans.MRA2008472. doi: 10.2320/matertrans.MRA2008472. [DOI] [Google Scholar]
- Ogi T. Dani Nandiyanto A. B. Kisakibaru Y. Iwaki T. Nakamura K. Okuyama K. Facile synthesis of single-phase spherical α′′-Fe16N 2/Al2O3 core-shell nanoparticles via a gas-phase method. J. Appl. Phys. 2013;113(16):164301. doi: 10.1063/1.4798959. [DOI] [Google Scholar]
- Nagai D. Kinemuchi Y. Suzuki K. Towata A. Yasuoka M. Highly dispersive α′′-Fe16N2 particle synthesis using hydroxyapatite coating. J. Solid State Chem. 2015;225:455–458. doi: 10.1016/j.jssc.2015.01.024. doi: 10.1016/j.jssc.2015.01.024. [DOI] [Google Scholar]
- Bridges C. A. Rios O. Allard L. F. Meyer H. M. Huq A. Jiang Y. Wang J. P. Brady M. P. The impact of carbon coating on the synthesis and properties of α′′-Fe16N2 powders. Phys. Chem. Chem. Phys. 2016;18:13010–13017. doi: 10.1039/c6cp00737f. doi: 10.1039/c6cp00737f. [DOI] [PubMed] [Google Scholar]
- Atkins P. and Paula J., Atkins' physical chemistry, Oxford University press, 2008, http://www.worldcat.org/isbn/9780195685220 [Google Scholar]
- Horch S. Lorensen H. T. Helveg S. Lægsgaard E. Stensgaard I. Jacobsen K. W. Nørskov J. K. Besenbacher F. Enhancement of surface self-disfusion of platinum atoms by adsorbed hydrogen. Nature. 1999;398:134–136. doi: 10.1038/18185. doi: 10.1038/18185. [DOI] [Google Scholar]
- Atkinson H. V. Davies S. Fundamental aspects of hot isostatic pressing: an overview. Metall. Mater. Trans. A. 2000;31:2981–3000. doi: 10.1007/s11661-000-0078-2. doi: 10.1007/s11661-000-0078-2. [DOI] [Google Scholar]
- ASTM, ASTM E112-13, Standard test methods for determining average grain size, ASTM Int., 2013, pp. 1–28, 10.1520/E0112-13.1.4 [DOI] [Google Scholar]
- McIntyre N. S. Zetaruk D. G. X-ray Photoelectron Spectroscopic Studies of Iron Oxides. Anal. Chem. 1977;49:1521–1529. doi: 10.1021/ac50019a016. doi: 10.1021/ac50019a016. [DOI] [Google Scholar]
- Grosvenor A. P. Kobe B. A. Biesinger M. C. McIntyre N. S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004;36:1564–1574. doi: 10.1002/sia.1984. doi: 10.1002/sia.1984. [DOI] [Google Scholar]
- Moulder J. F., Stickle W. F., Sobol P. E. and Bomben K. D., Handbook of XPS.pdf, 1995, p. 255 [Google Scholar]
- Speliotis D. E. Magnetic recording materials. J. Appl. Phys. 1967;38:1207–1214. doi: 10.1063/1.1709541. doi: 10.1063/1.1709541. [DOI] [Google Scholar]
- Hsu C. M. Lin H. M. Tsai K. R. Lee P. Y. High resolution transmission electron microscopy and magnetic properties of nanocrystalline iron particles with oxidized and nitrided surfaces. J. Appl. Phys. 1994;76:4793–4799. doi: 10.1063/1.358491. doi: 10.1063/1.358491. [DOI] [Google Scholar]
- Kronmüller H. and Fähnle M., Micromagnetism and the Microstructure of Ferromagnetic Solids, Cambridge Univ. Press, 2003, http://www.cambridge.org/asia/catalogue/catalogue.asp?isbn=9780521331357&ss=exc, accessed May 25, 2018 [Google Scholar]
- Stoner E. C. Wohlfarth E. P. A mechanism of magnetic hysteresis in heteresis in heterogeneous alloys. Philos. Trans. R. Soc., A. 1948;240:599–642. doi: 10.1098/rsta.1948.0007. doi: 10.1098/rsta.1948.0007. [DOI] [Google Scholar]
- Brown W. F. Criterion for Uniform Micromagnetization. Phys. Rev. 1957;105:1479–1482. doi: 10.1103/PhysRev.105.1479. doi: 10.1103/PhysRev.105.1479. [DOI] [Google Scholar]
- Skokov K. P. Gutfleisch O. Heavy rare earth free, free rare earth and rare earth free magnets - Vision and reality. Scr. Mater. 2018:1–6. doi: 10.1016/j.scriptamat.2018.01.032. [DOI] [Google Scholar]
- Bødker F. Mørup S. Linderoth S. Surface effects in metallic iron nanoparticles. Phys. Rev. Lett. 1994;72:282–285. doi: 10.1103/PhysRevLett.72.282. doi: 10.1103/PhysRevLett.72.282. [DOI] [PubMed] [Google Scholar]