

Graphical abstract
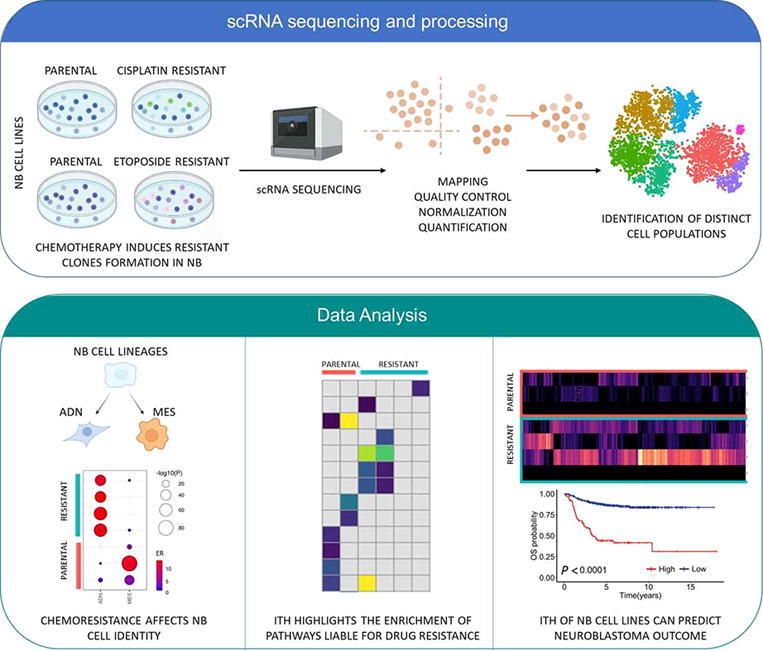
Keywords: Drug resistance, Intra-tumor heterogenity (ITH), Neuroblastoma (NB), Prognostic biomarkers, Single cell transcriptomics
Abstract
High-Risk neuroblastoma (NB) survival rate is still <50%, despite treatments being more and more aggressive. The biggest hurdle liable to cancer therapy failure is the drug resistance by tumor cells that is likely due to the intra-tumor heterogeneity (ITH). To investigate the link between ITH and therapy resistance in NB, we performed a single cell RNA sequencing (scRNAseq) of etoposide and cisplatin resistant NB and their parental cells. Our analysis showed a clear separation of resistant and parental cells for both conditions by identifying 8 distinct tumor clusters in etoposide-resistant/parental and 7 in cisplatin-resistant/parental cells. We discovered that drug resistance can affect NB cell identities; highlighting the bi-directional ability of adrenergic-to-mesenchymal transition of NB cells. The biological processes driving the identified resistant cell subpopulations revealed genes such as (BARD1, BRCA1, PARP1, HISTH1 axis, members of RPL family), suggesting a potential drug resistance due to the acquisition of DNA repair mechanisms and to the modification of the drug targets. Deconvolution analysis of bulk RNAseq data from 498 tumors with cell subpopulation signatures showed that the transcriptional heterogeneity of our cellular models reflected the ITH of NB tumors and allowed the identification of clusters associated with worse/better survival.
Our study demonstrates the distinct cell populations characterized by genes involved in different biological processes can have a role in NB drug treatment failure. These findings evidence the importance of ITH in NB drug resistance studies and the chance that scRNA-seq analysis offers in the identification of genes and pathways liable for drug resistance.
1. Introduction
Neuroblastoma (NB) is a clinically heterogeneous pediatric tumor of the sympathetic nervous system that accounts for 15% of total cancer-related childhood [1]. In the last years, the identification of diverse genomic markers has contributed to the risk stratification and improvement of survival rate of NB patients [2]. Indeed, several recurrent segmental chromosomal alterations have been demonstrated to discriminate between low-risk and high-risk patients [2], [3], [4]. Additionally, genome-wide association studies, high-throughput sequencing and microarray gene expression-based studies have identified multiple genetic changes that characterize NB both hereditable and somatically acquired [5], [6], [7], [8] and that are promising prognostic predictors and therapeutic targets. Genetic alterations occurring in non-coding DNA such as TERT rearrangements [9] and point mutations in regulatory elements of transcription factor binding sites [10], [11] also contribute to NB development. Moreover, we recently showed that common genetic variants of PARP1 gene have potential to predict the failure of therapy for high-risk NB patients [12]. Despite these advances in genomic research, treatment of NB is still unsuccessful in half of the patients diagnosed with the high-risk form. The standard therapeutic regimen includes 4 consecutive steps: induction chemotherapy (IC), local control, consolidation, and maintenance therapy; but there is no doubt that IC is the core of all therapeutic strategies for NBs. The Society of Pediatric Oncology Europe Neuroblastoma Group (SIOPEN) utilizes the COJEC regimen, a combination of vincristine, carboplatin, etoposide, cyclophosphamide, and cisplatin, for the cure of high-risk patients [13]. Unfortunately, the rate of patients that experience chemoresistance or relapse is still high and alternative therapeutic options in clinical practice are limited; therefore, there is an urgent need for developing additional treatment strategies.
Ongoing therapies treat cancer as a homogenous disease. However, it is now well known that tumors are not, genetically and phenotypically, a simple monolithic structure of cells, but they are more complex systems composed of different cellular entities. Indeed, within the same tumor, cells can adapt in response to the different selective environmental pressures with complex genetic and post-translational modifications, giving rise to the phenomenon of intra-tumor heterogeneity (ITH) [14], [15]. ITH currently represents a major obstacle to overcoming the problem of therapeutic resistance for the majority of human tumors [16].
During the last years, the understanding and characterization of ITH were made possible thanks to the analyses of gene expression and epigenomes’ profiles that can vary among malignant cells in the same tumor [17]. In NB, the analyses of transcriptional and epigenetic profiles of cell lines have demonstrated that the ITH has mainly denoted by adrenergic (ADN) and undifferentiated mesenchymal (MES) identities [18], [19]. Recently, the introduction of single-cell sequencing (sc-seq) technologies has given researchers a new set of tools to interrogate ITH and to understand the tumor origin and composition. Two relevant studies [20], [21], by performing a scRNA-seq analysis of normal developing human adrenal glands and a series of NB cases, have unraveled the NB founder cell. Both studies agree that NB cells match different stages along normal neuroblast differentiation trajectories and that neuroblastomas containing late, mature neuroblasts, have better prognoses than neuroblastomas with early neuroblasts. Another interesting work has applied scRNA-seq to demonstrate that cultured cell lines recapitulate the heterogeneity observed among malignant cells in tumors, suggesting thus their use for cancer research [22].
Several recent studies have highlighted the impact of monocellular transcriptomics to understand the molecular mechanisms underlying ITH that promote cancer chemoresistance [23]. In triple negative breast cancer, it has been shown that transcriptional profiles were acquired by reprogramming in response to chemotherapy [14], and in cell mantle lymphoma (MCL) cell markers have been identified, including a set of genes associated with immune escape and drug resistance [24]. In addition, an increase in ITH has been also found in small cell lung tumors resistant to cytotoxic agents via scRNA-seq approach, suggesting that resistance to treatment is characterized by the appearance of coexistent subsets of cells with concurrent heterogeneous gene expression [15]. These studies suggest that the characterization of tumor heterogeneity through scRNA-seq is crucial to understanding the molecular mechanisms underlying the drug resistance.
So far, none has employed this advanced technology to study the link between ITH and therapy resistance in NB. The challenge is to unravel which are the molecular pathways and genes that drive a single cell to give rise to a chemoresistant clone and act accordingly on high-risk NB therapeutic failure. Here, we perform scRNA-seq analyses of two high-risk NB cell lines resistant to cisplatin and etoposide, respectively, in order to provide novel insights into drug resistance in NB.
2. Material and methods
2.1. Cell culture
The MYCN-amplified human stage-IV HTLA-230 NB cell line was obtained from (Gaslini Institute, Genoa, Italy). HTLA-230 etoposide resistant (ER) was selected by treating HTLA-230 cells, by incubating the cells with gradually increasing concentrations of etoposide (Calbiochem, Merck KGaA, Darmstadt, Germany) up to 1.25 μM (dose comparable to that clinically used) for 6 months. Cells were grown in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 2 mM l-glutamine, penicillin (100U/ml) and streptomycin (100 μg/ml) (Invitrogen), 1% sodium pyruvate (Sigma-Aldrich), and 1% of aminoacid solution (Sigma-Aldrich) at 37 °C, under 5% CO2 in a humidified atmosphere. HTLA-230 ER were grown in the same conditions of HTLA-230 with 1.25 μM Etoposide. The MYCN-amplified, human stage IV, 7q21 gain, UKF-NB-4 NB cell line, was established from the Goethe University in Frankfurt am Main, Germany. The UKF-NB-4 cisplatin resistant (CDDP) cell line was established from parental UKF-NB-4 cells by incubating t with gradually increasing concentrations of CDDP. The cells were grown at 37 °C and 5% CO2 in Iscove’s modified Dulbecco’s medium (IMDM) with 10% bovine serum. UKF-NB-4 CDDP cells were grown in the same conditions of UKF-NB-4 with CDDP (100 ng/mL).
2.2. Single-cell RNA sequencing (scRNAseq)
Single-cell mRNA sequencing was performed according to standard 10x Genomics 3′ v3.1 Single Cell Gene Expression dual index chemistry guidelines. NB cell lines at 75% confluence in P-100 plate were collected and counted to determine their viability. We assessed at least 90–95% of viability before cryopreserving, in a freezing medium 90% FBS+ 10% DMSO. We aliquoted 2,000,000 cells in 1 mL freezing media into cryovials, we put them in CoolCell (at room temperature) and then into a −80 °C freezer. The CoolCell ensures that the temperature decreases steadily by 1 °C/minute. After approximately 24 h, we removed the cryovials from the CoolCell and we transferred them into liquid nitrogen till the processing. Samples were thawed according to 10x genomics guidelines. Cells were washed, filtered, and counted. Cells were loaded in a concentration of 700–1200 cells/µl on a 10x Genomics chip, and the chip was run on a Chromium controller to initiate GEM formation. Library preparation was performed, following standard 10x Genomics guidelines to generate Gene Expression (GEX) libraries. The resulting DNA libraries were paired-end sequenced on an Illumina Novaseq S4 with an Illumina 2 × 150 bp kit, to obtain an average of 100 K reads per cell for the GEX library.
2.3. Processing of scRNA-seq data
FASTQ files were generated from BCL files with mkfastq from Cell Ranger v6 [25] (10× Genomics). Cell barcode filtering, alignment of reads and UMI counting were performed using Cell Ranger v6.0 (10× Genomics). UMI counts were imported into R with the Seurat package for quality control (QC) and analysis [26]. Cells were removed from the analysis if <2000 distinct genes, 20,000 counts or >30% of reads mapping to mitochondrial genes were detected. We further filtered out cells with >20% of reads mapping to the largest gene. A total of 4140 cells for UKF-NB-4 and UKF-NB-4 CDDP and 3618 cells for HTLA-230 and HTLA-230 ER passed the QC filters and were brought forward. We scored each cell based on the expression of G2/M and S phase markers using the Seurat CellCycleScoring function [27]. Data were normalized, scaled and log-transformed using the Seurat SCTransform function [28] and, to remove confounding sources of variation, percent of mitochondrial genes, read counts and cell cycle scores were regressed out during the normalization step. Data were clustered using the nearest neighbour algorithm and the FindClusters function with a resolution of 0.5. Uniform Manifold Approximation and Projection (UMAP) and t-distributed stochastic neighbor embedding (tSNE) dimensionality reduction were carried out with default methods implemented in Seurat and cells were colored according to the gene signatures on a dimensional reduction plot with the FeaturePlot function. Gene markers (differentially expressed genes) were determined by comparing each cluster with all the remaining clusters using a wilcoxon rank sum test. Gene markers for every cluster compared to all remaining cells in UKF-NB-4 and HTLA-230 parental and UKF-NB-4 CDDP and HTLA-230 ER resistant cells are reported in Table S1. Only genes detected in a minimum fraction (25%) of cells in either of the two conditions were tested and those with a positive fold change and false discovery rate (FDR) adjusted P < 0.01 were reported.
2.4. Gene set enrichment analysis
Enrichment analysis of adrenergic (ADN) and undifferentiated mesenchymal (MES) signatures was performed with Fisher tests comparing the gene markers of each cluster (as defined above) with a list of genes from literature [18], [19]. This list of genes is the result of the merge of AND and MES genes from above cited Boeva and van Groningen works. The full list is reported in Table S2 representing the two cell lineages that typify NB tumors, using the whole set of gene markers as background (Fig. 2A, C). Preferential enrichment of ADN and MES signatures was tested within each cluster with logistic regression implemented in the logistf package and enrichment ratios represented in terms of log of absolute values (Fig. 2B, D). Balloon plots and heatmaps were produced with the ggplot package. [29] The lists of gene markers for every cluster (Table S1) these genes were used to query Gene Ontology databases. The functional enrichment analysis tool WebGestalt (WEBbased GEne SeT AnaLysis Toolkit) was used to detect significant enrichments for Gene Ontology filtered for non-redundant biological pathways and affinity propagation. The enrichment analysis was performed with hypergeometric test using FDR ≤ 0.01 and 10 as a minimum number of genes for a category. Then, to attenuate the redundancy of biological terms obtained in each subcellular cluster, we use REVIGO web tool [https://revigo.irb.hr/] Data generated during this analysis are included in Tables S4 and S5. The map of the GO processes was generated by GOView implemented in WebGestalt including the GO processes obtained by our enrichment analysis with FDR < 1 × 10−5.
Fig. 2.

Enrichment analysis of genes expressed in the cellular clusters. (A) Balloon plot showing the enrichment of ADN and MES signatures in each cluster identified in UKF-NB-4 CDDP and UKF-NB-4, and (C) in HTLA-230 ER and HTLA-230. Balloon size indicates the statistical significance in log scale, color indicates the Enrichment Ratio (ER) from the enrichment test. (B) Plot showing Log(ER) results of comparison analysis of MES and ADN signatures expressed in the cellular clusters of UKF-NB-4 CDDP and UKF-NB-4 and (D) HTLA-230 ER and HTLA-230.(E) Enrichment analysis for the top cluster markers in relation to GO terms in UKF-NB-4 CDDP and UKF-NB-4, and (F) in HTLA-230 ER and HTLA-230. Parental and resistant cell lines are delineated by pink and blue boxes respectively and clusters are delineated by colored dots. GO gene sets overlapping with at least 10 markers in one, were included. Data represents significant results from WebGestalt analysis (FDR < 0.01), and shown in color scale ranging from 0 to the 99th data quantile (to avoid highly significant pathways dominating the heatmap). Not significant enrichments (FDR > 0.01) are reported in gray. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.5. Deconvolution of bulk RNA-seq datasets and correlation analyses with clinical data
CIBERSORTx [30] was used to deconvolute bulk RNA-seq datasets with scRNA-seq-derived cell clusters. Bulk RNAseq data for 498 NB cases with available clinical data was downloaded from GEO (GSE62564). Top marker genes per single-cell cluster were selected using an adjusted P < 0.05 and a log2 fold change >0.8 as identified by Seurat. A reference matrix with the average expression of these top markers per cell cluster (511 genes for UKF-NB-4 CDDP and UKF-NB-4 and 219 genes for HTLA-230 ER and HTLA-230) was calculated using R base functions. This reference matrix was then used to deconvolute bulk RNA-seq data with CIBERSORTx.
Relative cluster abundances inferred from bulk RNAseq data were correlated with NB clinical data. The enrichment of MYCN amplification (amplified vs not amplified), age at diagnosis (≥18 months or <18 months), INSS stage (4 vs 1/2/3/4s) and risk groups (high-risk vs low/intermediate risk) was tested among clusters with Wilcoxon test. False discovery rate (FDR) adjustment was used to account for type I errors and significance threshold was set to FDR = 0.05. Boxplots and heatmaps were produced with ggplot and pheatmap R packages [29] respectively. Deconvolved cluster abundances were also tested in relation to overall survival data in order to identify cluster signatures associated with worse/better outcomes. Samples were split in two groups (high expression vs low expression) according to the median of CIBERSORTx absolute values and Kaplan-Meier curves were compared with log-rank test implemented in the ggsurv package https://rdocumentation.org/packages/survminer/versions/0.4.9.
2.6. Cell viability assay
NB cell lines UFK-NB-4 and UFK-NB-4 CDDP and HTLA-230 and HTLA230 ER were seeded as six replicates into 96-well plates at a density of 104 cells per well. Cell viability was determined after 24 h, 48 h and 72 h of treatment, by using the 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, according to the manufacture protocol (Promega, Milan, Italy). Plates were analyzed with EnVision multimode plate reader (Perkinelmer). The experiments have been repeated twice.
2.7. Colony formation assay in soft agar
NB cell lines UFK-NB-4 and UFK-NB-4 CDDP and HTLA-230 and HTLA230 ER cell lines were plated (2 × 105 cells) in 0.35% agar on a bottom layer of 1% agar in 35-mm dishes of 6-well plates (Corning, New York, NY, USA). The plates were incubated at 37 °C for 2 weeks and stained with 0.01% Crystal violet. Colonies with 20 cells or more were counted. The means and standard deviations were calculated from three independent experiments.
2.8. Real-time (RT)-PCR
The expression levels of genes were analyzed using real-time, quantitative PCR. Total RNA extraction using TRIzol LS Reagent (Invitrogen) and cDNA retrotranscription using the SensiFAST cDNA Synthesis KiT (Bioline) were performed according to the manufacturer protocol. The cDNA samples were diluted to 20 ng/μl. Gene-specific primers were designed by using PRIMEREXPRESS software (Applied Biosystems); a detailed list of primers is provided in Table S6. RT-PCR was performed using SYBR Green PCR Master Mix (AppliedBiosystems). All real-time PCR reactions were performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems). The experiments were carried out in triplicate for each data point. The housekeeping gene β -actin was used as the internal control. Relative gene expression was calculated using the 2−ΔCT method, where the ΔCT was calculated using the differences in the mean CT between the selected gene and the internal control (β -Actin). The mean fold change of 2−(average ΔΔCT) was determined using the mean difference in the ΔCT between the gene of interest and the relative internal control.
3. Results
3.1. Cisplatin and etoposide treatment induces cell subpopulations formation characterized by distinct transcriptome profiles
We performed a single cell RNA-seq (scRNA-seq) of two pairs of NB cell lines: i) UKF-NB-4 parental and UKF-NB-4 cisplatin resistant (UKF-NB-4 CDDP) cell lines and ii) HTLA-230 parental and HTLA-230 etoposide resistant (HTLA-230 ER) cell lines. Cisplatin and etoposide drugs are included in COJEC treatment of NB patients [13]. We assessed the induction of Cisplatin and Etoposide chemoresistance in UFK-NB-4 and HTLA-230 cell lines with viability and clonogenic assays (Fig. S1a–d), and gene expression of noted drugs target genes [31], [32], [33], [34], [35], [36] (Fig. S1e-f). Drug resistance significantly increases the growth ability of NB cell lines and, moreover, we observed an increase of the number of cell colonies upon drug induction compared with parental cells. After data filtering and normalization, we obtained transcription profiles of 1514 UKF-NB-4 CDDP vs 2646 UKF-NB-4 cells and of 1674 HTLA-230 ER vs 1160 HTLA-230 cells. These cells exhibited distinct transcriptomic profiles, as shown in t-distributed stochastic neighbour embedding (tSNE) (Fig. 1A,B), and uniform manifold approximation and projection (UMAP) analyses (Fig. S2a,b). In both cell lines, drug resistant cells clustered apart from untreated cells (Fig. 1A,B), indicating that the drug treatment (with cisplatin or etoposide) significantly affects the transcriptomes of these cell lines.
Fig. 1.

Drug treatment induces cell subpopulations formation with distinct transcriptome profiles (A) tSNE plot of UKF-NB-4 cells in blue and UKF-NB-4CDDP NB cells in pink. (B) tSNE plot of HTLA-230 in blue and HTLA-230 ER NB cells in pink. (C) tSNE plot of cluster identification of UKF-NB-4 and UKF-NB-4 DDP cells (D) HTLA-230 and HTLA-230 ER cells (each color represents a single clusters of cells). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We then explored the heterogeneity of single-cell transcriptome profiles through high dimensional clustering and identified three clusters in UKF-NB-4, four clusters in UKF-NB-4 CDDP (Fig. 1C and Fig. S2c), four clusters in HTLA-230 and four clusters in HTLA-230ER (Fig. 1D and Fig. S2d). Gene markers for every cluster compared to all remaining cells in UKF-NB-4 and HTLA-230 parental and UKF-NB-4 CDDP and HTLA-230 ER resistant cells are reported in Table S1. These results provide evidence that NB cell lines are transcriptionally heterogeneous and that drug resistance can induce the development of cell subpopulations, thus emphasizing the phenomenon of ITH. A validation of the gene signature of each cluster was performed by RT-PCR (Fig. S3a–d).
To further strengthens the ITH between the two drug resistant NB cells, we analyzed the transcriptome at single cell level of the UFK-NB4-CDDP and HTLA-230 ER cells and their parental cells simultaneously. Cisplatin and etoposide resistant NB cell lines exhibited distinct transcriptomic profiles, clustering apart from each other, as shown in t-SNE and UMAP analyses in Fig. S4a-b. Moreover, this analysis identified two distinct clusters in UFK-NB-4 CDDP and three distinct clusters in HTLA-230 ER, and two and three distinct clusters in their parental cell lines respectively (Fig. S4c-d).
3.2. Chemoresistance affects NB cell identity
Two types of phenotypically divergent cells committed adrenergic (ADN) cells and undifferentiated mesenchymal (MES) cells are dominant in NB cell lines and tumors [37], [38]. Thus, to understand whether NB cell identity could be involved in chemoresistance, we assessed the enrichment of known ADN and MES signatures [37], [38] in the cell clusters (CL) obtained by the scRNA-seq analysis.
In the UKF-NB-4 CDDP vs UKF-NB-4 cells, the parental CL0, CL5 and CL6 clusters are significantly enriched in MES and ADN genes (Fig. 2A and y Table S2) but MES signature resulted preferentially represented when compared against ADN signature (Fig. 2B and Table S3). In contrast, all cisplatin resistant clusters are strongly enriched in ADN genes (Fig. 2A and B Table S2 and Table S3); thus, the cisplatin seems to induce higher expression of ADN genes.
In the parental HTLA-230 cells, we found all clusters (CL0, CL1, CL3 and CL7) enriched in ADN genes (Fig. 2C and Table S2) with a significant overrepresentation in CL3 and CL7 when compared to MES signature (Fig. 2D and Table S3). While in the HTLA-230-ER cells, we found all clusters equally enriched in MES and ADN genes (Fig. 2C, D and Table 2S and Table S3), these results may highlight a plasticity potential of etoposide resistant cells, so their ability to switch from MES or AND identity or vice versa.
3.3. ITH highlights the development of drug potential resistance
We have demonstrated that there is a substantial difference of subcellular clusters at the transcriptomic level between resistant and parental cell lines. To infer biological features relevant to each cluster, we used the markers of each cluster as defined by Seurat (FDR < 0.01 and logFC > 0, see Methods) to carry out over-representation analysis against the gene ontology (GO) database of biological processes. The results were then clustered for affinity propagation (see Methods). In the Fig. 2E,F are plotted the two heatmaps summarizing the results of the top 10 significant biological processes for each cluster ordered by P-value, the results of complete GO analysis are reported in the Table S4 and Table S5.
In the UKF-NB-4 cells, we observed two subcellular clusters characterized by significant biological processes (FDR < 0.01) (Fig. 2E): the CL0 was characterized by gene pathways involved in the regulation of DNA and RNA metabolic process and the CL5 by pathways related to tissues and organ development (with terms such as tissue migration, gastrulation, stem cell differentiation). In the UKF-NB-4 CDDP cells, all cisplatin resistant clusters showed significant biological processes (FDR < 0.01). Particularly, CL1 and CL2 are both characterized by pathways involved in cell cycle regulation, DNA repair, and protein post-trasdutional modification, while CL3 and CL4 share a pathway involved in protein maintenance to endoplasmic reticulum.
In the HTLA-230 cells, all clusters showed an enrichment of significant biological processes (Fig. 2F). The over expressed genes of subcellular cluster CL0 are involved in tissues and organ development (Fig. 2F and Table S5), those of the CL1 and CL3 in DNA and RNA metabolic and gene expression processes while the CL7 is fully characterized by biological processes involved in nervous system development and organization. In the HTLA-230 ER cells only the CL4 results significantly enriched in biological pathways; mainly involved in regulation of gene expression and protein post- translational modification. Together these results suggest that drug treatment induces the acquisition of DNA repair and post- translational modifications pathways as biological mechanisms liable for drug resistance in NB cells. Full results of GO analysis are reported in Table S4 and Table S5 while a map showing the connections among the most significant GO terms (FDR < 1x10-5) and the other known biological themes is in Fig. S5.
3.4. ITH of NB cell lines can predict neuroblastoma outcome
To analyze the composition and biological programs involved in drug resistance in a large cohort of NB samples, we used the expression signatures of each cluster derived from scRNAseq clustering analysis of UKF-NB-4 CDDP vs UFK-NB-4 and HTLA-ER vs HTLA-230 cells to decompose the transcriptomes of bulk NB tumors [39]. By this approach, we observed in NB samples (SEQC cohort) an ITH comparable to that observed in UKF-NB-4 and HTLA-230 cell line populations (Fig. 3A, B). Hierarchical clustering of bulk NB transcriptomes by their relative abundances of individual cell populations of each cluster indicated the presence of high ITH. Indeed, we observed that diverse clusters while belonging to the same cell population are associated with different, positive or negative, clinical factors. For instance (Fig. 3C) CL0 and CL5 (UKF NB-4 cells) are associated with high-risk (HR)-NBs (Padj = 9.2 × 10-9) and low-risk (LR)-NBs (Padj = 2.1 × 10-18), respectively while CL2 and CL3 (UKF NB4-CDDP cells) are associated with HR-NBs (Padj = 2 × 10-20) and LR-NBs (Padj = 6.1 × 10-9), respectively. Similarly (Fig. 3C), CL3 (belonging to HTLA-230 cells) is associated with HR-NBs (Padj = 1.6 × 10-12) while CL2 and CL5 (both belonging to HTLA-230 ER cells) are associated with LR-NBs (Padj = 7.8 × 10-33) and HR-NBs (Padj = 2 × 10-20), respectively. We also evaluated other NB clinical parameters with prognostic significance as MYCN amplification (Fig. 3D), stage (Fig. 3E) and age of tumor onset (Fig. 3F), and for each CL, we assessed the same clinical association observed in risk comparison (Fig. 3C).
Fig. 3.

Neuroblastoma cell type composition deduced from bulk transcriptomes. (A) Hierarchical clustering of SEQC cohort patients based on the relative abundance of UKF-NB-4 (CL0, CL5, CL6), UFK-NB-4 CDDP (CL1, CL2, CL3, CL4) and (B) HTLA-230 (CL0, CL1, CL3, CL7) and HTLA-230 ER (CL2, CL4, CL5, CL6) cell clusters. Clinical parameters associated with better and worse prognosis are reported in grey and black, respectively. Boxplots reporting correlations between UKF-NB-4, UFK-NB-4 CDDP, HTLA-230 and HTLA-230 ER clusters abundances in SEQC patients and risk classification (C), MYCN amplification (D), INSS stages (E) and age at the diagnosis (F). Concerning the latter two parameters, patients are divided in two groups (individuals classified as stage 4 against those classified as stage 1, 2, 3 and 4 s; individuals older than 18 months against those younger than 18 months at diagnosis). Grey boxes represent distributions of patients with clinical markers associated with worse prognosis. White boxes represent distributions of patients with clinical markers associated with a better prognosis. Boxes are ordered to show UKF-NB-4 (CL0, CL5 and CL6), UFK-NB-4 CDDP (CL1, CL2, CL3 and CL4), HTLA-230(CL0, CL1, CL3 and CL7) and HTLA-230 ER (CL2, CL4, CL5 and CL6) cluster abundances. Kaplan-Meier analysis of Overall Survival (OS) in neuroblastoma patients according to the absolute abundance of UFK-NB-4 CDDP CL2 (G). HTLA-230 ER CL5 (H) – values were calculated using log-rank test. Significant P-value are indicated by *(* <0.05 ** <0.01,*** <0.001,****< 1e-04) Ns indicates non significant P-value.
Notably, in the UKF-NB-4 CDDP cells, CL2 is associated with all worst prognostic factors evaluated (Fig. 3D,E,F): MYCN amplification (Padj = 2.7 × 10-12), stage 4 (Padj = 6.8 × 10-13), and age at diagnosis (Padj = 7.5 × 10-7) highlighting the presence of a more aggressive cell subclone (due to cisplatin treatment) and corroborating the biological significance observed in GO analysis (Fig. 2E). In the HTLA-230 ER cells CL5 (Fig. 3D,E,F) is relevantly associated with MYCN amplification (Padj = 3.6 × 10-19), stage 4 (Padj = 3.3 × 10-15), and age of onset of NB (Padj = 1.3 × 10-10), suggesting that this cell subclone might be a more aggressive clone (due to etoposide treatment); indeed, the GO analysis (Table S5) showed the enrichment of biosynthetic and metabolic biological processes.
In addition, we investigated the association of the expression signatures of each cluster with patient survival. Expectedly, CL1 and CL3 (UKF-NB-4 CDDP) (Fig. S6a,b) and CL2 (Fig. S7a) (HTLA-230 cells) that are directly associated with good clinical factors are also significantly associated with favorable prognosis. Interestingly, CL7 of HTLA-230 cells, as mentioned above, characterized by the expression of genes involved in nervous system development and organization process (Fig. 2F), is significantly associated with a high survival rate (Fig. S7c), reinforcing the hypothesis that this CL is composed of cells more differentiated and less aggressive. The clusters CL0 (UFK-NB4), CL2 (UFK-NB4 CDDP) (Fig. S6c and Fig. 3G), and CL3 (HTLA-230) and CL5 (HTLA-230 ER) (Fig. S7b and Fig. 3H) are associated with bad clinical factors have a significant worst prognosis. Notably, CL2 (cisplatin resistant cells) and CL5 (etoposide resistant cells) signatures might be of clinical relevance in non responders NB patients screening (Fig. 3G,H). These results emphasize the importance of ITH in drug resistance studies and the chance that scRNA-seq analysis offers in the identification of genes and pathways liable for drug resistance.
4. Discussion
The urgent medical need to overcome NB therapy resistance led us to use, for the first time, the single cell transcriptomic approach to examine the link between ITH and therapy resistance in NB. In this study, we investigate the change of transcriptomic profiles at single cell levels of NB cell lines parental and resistant to etoposide and cisplatin drugs that are currently used in the first line treatment of NB patients. The analysis described in this work has demonstrated the utility of single cell RNA data in: (1) identifying distinct tumor cell populations that determine the ITH of drug-resistant and their parental NB cells; (2) discovering the drug resistance effects on NB cell identity; (3) unraveling that ITH of NB cell lines reflects the cellular composition of NB tumors.
We have identified the concomitant presence of transcriptionally different subpopulations of cells acquired during cisplatin or etoposide treatment that clustered apart from parental NB cells, emphasizing the phenomenon of ITH and the link with chemoresistance. Moreover, we also demonstrated that the two drug-resistant NB cell lines clustered apart from each other, to further strengthens the role of ITH in NB drug resistance.
In line with our observation, recent scRNAseq studies, in colon and in non-small cell lung cancer, report that drug treatment induces distinct transcriptome phenotypes and unique cell populations compared to the untreated samples [37], [38].
Translating the mechanisms of differentiation between ADN to MES identity is a key step to better understand the NB biology and improve the therapeutic management of patients. The differences in chemoresistance and invasion/migration properties of these two identities may have important clinical relevance [18], [19]. Indeed, MES cells have been already reported as more resistant after in vitro treatment of NB cells to cisplatin, doxorubicin, and etoposide compared to ADN cells [18], [19], and also that post-chemotherapy or relapsed NB samples might be enriched in MES cells [40]. Unexpectedly, our analysis detected, in the parental UKF-NB4 cell lines, two cell clusters (CL0 and CL5) significantly enriched in MES genes while, in those that are cisplatin resistant (UKF-NB4 CDDP), all clusters were enriched in ADN genes. These results are in line with past observations indicating that tumors at relapse are not systematically enriched in MES cells [18], probably because cells switch from ADN to MES identity under chemotherapy, and switch back from MES to ADN after treatment, supporting the idea of plasticity understood as the reversion of cell identity [41]. Instead, in the parental HTLA-230 cells, we observed that two clusters are strongly enriched in ADN genes while etoposide treatment seems to emphasize NB cells plasticity potential, showing a significant increase in HTLA-230 ER clusters of both signatures (ADN and MES) and highlighting the bi-directional ability of NB cells to transdifferentiate [40].
We also performed a gene set enrichment analysis and found that the diverse subpopulations of cisplatin resistant cell lines are characterized by biological pathways related to the DNA damage response and protein localization to endoplasmic reticulum while in both cisplatin and etoposide resistant cells we found subcellular populations enriched in genes involved in post-translational modification processes. Of note, the alteration of endoplasmic reticulum functions and epiproteomic modifications have been recently proposed as the cause of cancer drug resistance [42], [43]. In cisplatin resistant cell clusters, we found BRCA1, BRCA2, BARD1, PARP1 genes involved in Double Strand Break Repair (DSBR) pathway that are known to play a relevant role in drug resistance and in NB therapy [12], [44], [45]. While surprisingly, we uncovered several genes belonging to Ribosomal Protein Large (RPL) and Ribosomal Protein Small (RPS) genes families. Beyond their essential roles in ribosome assembly and protein translation, ribosome-independent functions of ribosomal proteins have also been greatly appreciated, especially in the study achievements connecting cancer diagnosis and therapy [46]. Indeed, the alkylating and intercalating agents can induce ribosome biogenesis inhibition, contributing to their toxic action on cancer cells. This is in agreement with our results that show significant expression of ribosome protein genes in cisplatin resistant cluster cells [47], [48]. Furthermore, we also observed several genes belonging to Histone H1 axis, known to be a therapeutic target in cancer cell self-renewal [49]. Interestingly, in etoposide resistant clusters we found numerous genes belonging to PSMB, PSMC, and PSMD proteasome-subunit families, known to confer drug resistance in diverse cancers [50], [51], [52].
In contrast to the single-cell analysis, previous bulk analyses of the same cells that are used in this study, did not reveal [53], [54] the above-mentioned pathways or genes. This demonstrates the utility of scRNA-seq to identify tumor features that were previously unable to be found. Moreover, unlike what has been observed from past studies, our scRNA-seq strategy allowed us to highlight, in both parental cells, metabolic processes of nucleic acids and of pathways related to cardiac, muscle, respiratory, embryonic and neuronal tissues development, reflecting the dynamic cell origin of NB from the neural crest cells.
It is reasonable to believe that the analysis of gene expression signatures of drug resistance from scRNA-seq clustering integrated with the transcriptome of a large cohort of NB samples could advance translational science, corroborating the passage from in vitro to in vivo. In this regard by a deconvolution analysis of bulk transcriptomic data from 486 tumors using the expression signature of cell subpopulations, we observed that the transcriptional heterogeneity of our cellular models reflected the ITH of NB tumors. Moreover, the cell populations of CL2 in UKF-NB-4CDDP and of CL5 in HTLA-230 ER cell lines expressed gene signatures associated with worse NB patient survival and unfavorable clinical markers, suggesting their tumor aggressiveness features. These findings provide evidence that prediction based on single cell gene expression data may allow the identification of treatment strategies targeting cell population heterogeneity.
We are aware that our analysis has limitations imposed by the use of cell lines to investigate the ITH, but so far, cultured cell lines represent the core of cancer research because are simple and controlled systems where to fine-tune the experimental approach. Indeed, a recent scRNA-seq work, that describes the landscape of heterogeneity across 198 cell lines from 22 cancer types, demonstrated that they surprisingly recapitulate the heterogeneity observed among malignant cells in tumors [22]. In this perspective, our scRNAseq approach combined with deconvolution analysis of bulk transcriptomic data from NB tumors provides interesting preliminary results for the understanding of drug resistance in NB. Whereas further analyses are required, our next intent is to move from the analysis of effects derived by the in cellulo modelling of the drug treatment to a more patient-relevant system, using NB tissues and patient-derived tumor xenograft models.
5. Conclusions
In conclusion, our study demonstrates the distinct cell populations characterized by expressed genes involved in different biological processes can have a role in drug resistance in NB treatment and further support the evidence that single-cell sequencing allows for a better understanding of the genomic principles of tumor heterogeneity and might represent the basis for more successful tumor treatments.
Author contribution
MA performed the research and wrote the paper, FB performed the research and analysed the data VA analysed the data, SC, TM, AL performed the research, CD, BM, HZ, AV contributed essential material reagents and tools, AI designed the research study, MC designed the research study and wrote the paper.
Funding
This study was supported by grants from Associazione Italiana per la Ricerca sul Cancro (Grant No. 25796 to M.C. and Grant No. 20757 to A.I.); Fondazione Italiana per la Lotta al Neuroblastoma (to M.C.); Associazione Oncologia Pediatrica e Neuroblastoma (to M.C.); Regione Campania “SATIN” grant 2018–2020 (to M.C.).
Availability of data and material
The data generated and analyzed during the current study are available from the corresponding author on reasonable request. Public data and data repositories are referenced within the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.08.031.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Maris J.M. Recent advances in neuroblastoma. N Engl J Med. 2010;362(23):2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capasso M., Diskin S.J. Genetics and genomics of neuroblastoma. Cancer Treat Res. 2010;155:65–84. doi: 10.1007/978-1-4419-6033-7_4. [DOI] [PubMed] [Google Scholar]
- 3.Depuydt P., Boeva V., Hocking T.D., et al. Genomic amplifications and distal 6q loss: novel markers for poor survival in high-risk neuroblastoma patients. J Natl Cancer Inst. 2018;110(10):1084–1093. doi: 10.1093/jnci/djy022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lasorsa V.A., Cimmino F., Ognibene M., et al. 19p loss is significantly enriched in older age neuroblastoma patients and correlates with poor prognosis. NPJ Genom Med. 2020;5:18. doi: 10.1038/s41525-020-0125-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esposito M.R., Binatti A., Pantile M., et al. Somatic mutations in specific and connected subpathways are associated with short neuroblastoma patients' survival and indicate proteins targetable at onset of disease. Int J Cancer. 2018;143(10):2525–2536. doi: 10.1002/ijc.31748. [DOI] [PubMed] [Google Scholar]
- 6.Formicola D., Petrosino G., Lasorsa V.A., et al. An 18 gene expression-based score classifier predicts the clinical outcome in stage 4 neuroblastoma. J Transl Med. 2016;14(1):142. doi: 10.1186/s12967-016-0896-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lasorsa V.A., Formicola D., Pignataro P., et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget. 2016;7(16):21840–21852. doi: 10.18632/oncotarget.8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tonini G.P., Capasso M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Rev. 2020;39(1):275–285. doi: 10.1007/s10555-020-09843-4. [DOI] [PubMed] [Google Scholar]
- 9.Peifer M., Hertwig F., Roels F., et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015;526(7575):700–704. doi: 10.1038/nature14980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capasso M., Lasorsa V.A., Cimmino F., et al. Transcription factors involved in tumorigenesis are over-represented in mutated Active DNA-binding sites in neuroblastoma. Cancer Res. 2020;80(3):382–393. doi: 10.1158/0008-5472.CAN-19-2883. [DOI] [PubMed] [Google Scholar]
- 11.Lasorsa V.A., Montella A., Cantalupo S., et al. Somatic mutations enriched in cis-regulatory elements affect genes involved in embryonic development and immune system response in neuroblastoma. Cancer Res. 2022 doi: 10.1158/0008-5472.CAN-20-3788. [DOI] [PubMed] [Google Scholar]
- 12.Avitabile M., Lasorsa V.A., Cantalupo S., et al. Association of PARP1 polymorphisms with response to chemotherapy in patients with high-risk neuroblastoma. J Cell Mol Med. 2020;24(7):4072–4081. doi: 10.1111/jcmm.15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith V., Foster J. High-risk neuroblastoma treatment review. Children (Basel) 2018;5(9) doi: 10.3390/children5090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim C., Gao R., Sei E., et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173(4):879–893 e813. doi: 10.1016/j.cell.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart C.A., Gay C.M., Xi Y., et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat Cancer. 2020;1:423–436. doi: 10.1038/s43018-019-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Sayes N., Vito A., Mossman K. Tumor heterogeneity: a great barrier in the age of cancer immunotherapy. Cancers (Basel) 2021;13(4) doi: 10.3390/cancers13040806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suva M.L., Tirosh I. Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol Cell. 2019;75(1):7–12. doi: 10.1016/j.molcel.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Boeva V., Louis-Brennetot C., Peltier A., et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet. 2017;49(9):1408–1413. doi: 10.1038/ng.3921. [DOI] [PubMed] [Google Scholar]
- 19.van Groningen T., Koster J., Valentijn L.J., et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. 2017;49(8):1261–1266. doi: 10.1038/ng.3899. [DOI] [PubMed] [Google Scholar]
- 20.Jansky S., Sharma A.K., Korber V., et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat Genet. 2021;53(5):683–693. doi: 10.1038/s41588-021-00806-1. [DOI] [PubMed] [Google Scholar]
- 21.Kameneva P., Artemov A.V., Kastriti M.E., et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat Genet. 2021;53(5):694–706. doi: 10.1038/s41588-021-00818-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinker G.S., Greenwald A.C., Tal R., et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet. 2020;52(11):1208–1218. doi: 10.1038/s41588-020-00726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Wang D., Peng M., et al. Single-cell RNA sequencing in cancer research. J Exp Clin Cancer Res. 2021;40(1):81. doi: 10.1186/s13046-021-01874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L., Mo S., Li X., He Y., Yang J. Single-cell RNA-seq reveals the immune escape and drug resistance mechanisms of mantle cell lymphoma. Cancer Biol Med. 2020;17(3):726–739. doi: 10.20892/j.issn.2095-3941.2020.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng G.X., Terry J.M., Belgrader P., et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuart T., Butler A., Hoffman P., et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–1902 e1821. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tirosh I., Venteicher A.S., Hebert C., et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539(7628):309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wickham H. ggplot2. Springers Link. 2016 [Google Scholar]
- 30.Newman A.M., Steen C.B., Liu C.L., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodrigo M.A.M., Michalkova H., Strmiska V., et al. Metallothionein-3 promotes cisplatin chemoresistance remodelling in neuroblastoma. Sci Rep. 2021;11(1):5496. doi: 10.1038/s41598-021-84185-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hientz K., Mohr A., Bhakta-Guha D., Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8(5):8921–8946. doi: 10.18632/oncotarget.13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paul I., Chacko A.D., Stasik I., et al. Acquired differential regulation of caspase-8 in cisplatin-resistant non-small-cell lung cancer. Cell Death Dis. 2012;3:e449. doi: 10.1038/cddis.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yasui K., Mihara S., Zhao C., et al. Alteration in copy numbers of genes as a mechanism for acquired drug resistance. Cancer Res. 2004;64(4):1403–1410. doi: 10.1158/0008-5472.can-3263-2. [DOI] [PubMed] [Google Scholar]
- 35.Wu C., Yang T., Liu Y., et al. ARNT/HIF-1beta links high-risk 1q21 gain and microenvironmental hypoxia to drug resistance and poor prognosis in multiple myeloma. Cancer Med. 2018;7(8):3899–3911. doi: 10.1002/cam4.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao H., Zheng Y., Ma L., Tian L., Sun Q. Clinically-relevant ABC transporter for anti-cancer drug resistance. Front Pharmacol. 2021;12:648407. doi: 10.3389/fphar.2021.648407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aissa A.F., Islam A., Ariss M.M., et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat Commun. 2021;12(1):1628. doi: 10.1038/s41467-021-21884-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park S.R., Namkoong S., Friesen L., et al. Single-cell transcriptome analysis of colon cancer cell response to 5-fluorouracil-induced DNA damage. Cell Rep. 2020;32(8):108077. doi: 10.1016/j.celrep.2020.108077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W., Yu Y., Hertwig F., et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015;16:133. doi: 10.1186/s13059-015-0694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thirant C, Peltier A, Durand S, et al. Interplay between intrinsic reprogramming potential and microenvironment controls neuroblastoma cell plasticity and identity. 2021:2021.2001.2007.425710.
- 41.Gautier M., Thirant C., Delattre O., Janoueix-Lerosey I. Plasticity in neuroblastoma cell identity defines a noradrenergic-to-mesenchymal transition (NMT) Cancers (Basel) 2021;13(12) doi: 10.3390/cancers13122904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bahar E., Kim J.Y., Yoon H. Chemotherapy resistance explained through endoplasmic reticulum stress-dependent signaling. Cancers (Basel) 2019;11(3) doi: 10.3390/cancers11030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song H., Liu D., Dong S., et al. Epitranscriptomics and epiproteomics in cancer drug resistance: therapeutic implications. Signal Transduct Target Ther. 2020;5(1):193. doi: 10.1038/s41392-020-00300-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cimmino F., Avitabile M., Lasorsa V.A., et al. Functional characterization of full-length BARD1 strengthens its role as a tumor suppressor in neuroblastoma. J Cancer. 2020;11(6):1495–1504. doi: 10.7150/jca.36164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhillon K.K., Swisher E.M., Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011;102(4):663–669. doi: 10.1111/j.1349-7006.2010.01840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Penzo M., Montanaro L., Trere D., Derenzini M. The ribosome biogenesis-cancer connection. Cells. 2019;8(1) doi: 10.3390/cells8010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruno P.M., Liu Y., Park G.Y., et al. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med. 2017;23(4):461–471. doi: 10.1038/nm.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burger K., Muhl B., Harasim T., et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010;285(16):12416–12425. doi: 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Izzo A., Kamieniarz K., Schneider R. The histone H1 family: specific members, specific functions? Biol Chem. 2008;389(4):333–343. doi: 10.1515/BC.2008.037. [DOI] [PubMed] [Google Scholar]
- 50.Shi C.X., Kortum K.M., Zhu Y.X., et al. CRISPR genome-wide screening identifies dependence on the proteasome subunit PSMC6 for bortezomib sensitivity in multiple myeloma. Mol Cancer Ther. 2017;16(12):2862–2870. doi: 10.1158/1535-7163.MCT-17-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi C.X., Zhu Y.X., Bruins L.A., et al. Proteasome subunits differentially control myeloma cell viability and proteasome inhibitor sensitivity. Mol Cancer Res. 2020;18(10):1453–1464. doi: 10.1158/1541-7786.MCR-19-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xuan D.T.M., Wu C.C., Kao T.J., et al. Prognostic and immune infiltration signatures of proteasome 26S subunit, non-ATPase (PSMD) family genes in breast cancer patients. Aging (Albany NY) 2021;13(22):24882–24913. doi: 10.18632/aging.203722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marengo B., Monti P., Miele M., et al. Etoposide-resistance in a neuroblastoma model cell line is associated with 13q14.3 mono-allelic deletion and miRNA-15a/16-1 down-regulation. Sci Rep. 2018;8(1):13762. doi: 10.1038/s41598-018-32195-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodrigo M.A.M., Buchtelova H., Jimenez A.M.J., et al. Transcriptomic landscape of cisplatin-resistant neuroblastoma cells. Cells. 2019;8(3) doi: 10.3390/cells8030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated and analyzed during the current study are available from the corresponding author on reasonable request. Public data and data repositories are referenced within the manuscript.