

Abstract
Immunogenicity has imposed a challenge to efficacy and safety evaluation of adeno-associated virus (AAV) vector-based gene therapies. Mild to severe adverse events observed in clinical development have been implicated with host immune responses against AAV gene therapies, resulting in comprehensive evaluation of immunogenicity during nonclinical and clinical studies mandated by health authorities. Immunogenicity of AAV gene therapies is complex due to the number of risk factors associated with product components and pre-existing immunity in human subjects. Different clinical mitigation strategies have been employed to alleviate treatment-induced or -boosted immunogenicity in order to achieve desired efficacy, reduce toxicity, or treat more patients who are seropositive to AAV vectors. In this review, the immunogenicity risk assessment, manifestation of immunogenicity and its impact in nonclinical and clinical studies, and various clinical mitigation strategies are summarized. Last, we present bioanalytical strategies, methodologies, and assay validation applied to appropriately monitor immunogenicity in AAV gene therapy-treated subjects.
Keywords: adeno-associated virus, gene therapy, immunogenicity, risk assessment, clinical mitigation, bioanalytical methodologies and validation strategies, nonclinical and clinical outcomes
Graphical abstract
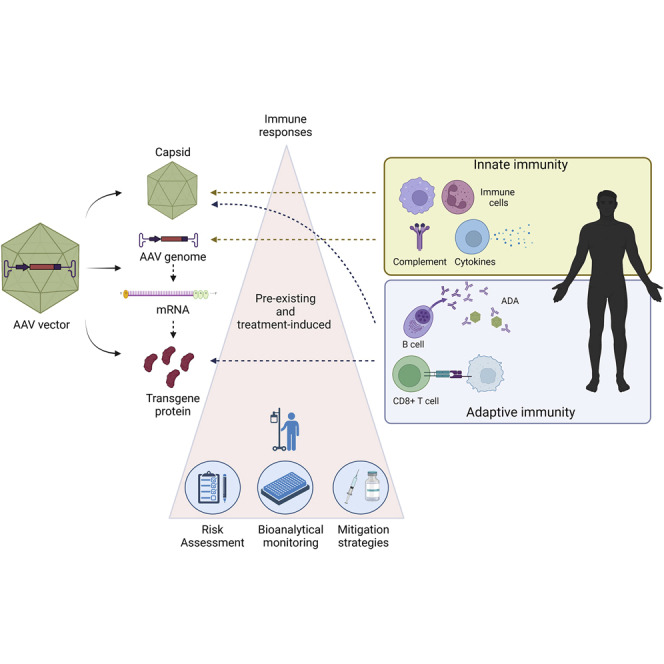
This paper summarizes current knowledge and understanding related to immunogenicity during discovering and developing AAV-vector-based gene therapies. There are three main sections: (1) overview of immunogenicity risks, and their impact on nonclinical and clinical studies; (2) bioanalytical methodologies for monitoring pre-existing and treatment-emergent immunogenicity; and (3) clinical mitigation strategies.
Introduction
Innovative medicines, such as cell and gene therapies (GTs), have expanded the landscape of medicinal products beyond small molecules and therapeutic proteins and opened new avenues for treating debilitating diseases. While these novel modalities carry a lot of promise, their development is riddled with unique challenges. To help accelerate discovery and clinical development of cell therapies and GTs and their availability to patients, the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ) launched a joint working group between the Clinical Pharmacology Leadership Group (CPLG) and the Translational and ADME Sciences Leadership Group (TALG) with several sub-teams focusing on different modalities (e.g., adeno-associated virus vector [AAV]-based GTs, chimeric antigen receptor [CAR]-T cell therapy) and aspects related to their development. This white paper summarizes the immunogenicity potential and risks associated with recombinant AAV (rAAV)-based GTs and provides a framework for bioanalytical approaches related to the immunogenicity assessment of rAAV-based GTs.
Although safety setbacks1 and ethical considerations2 presented significant challenges for GT development in the past, the recent approvals of Luxturna (voretigene neparvovec) and Zolgensma (onasemnogene abeparvovec), both using rAAV vectors, generated renewed excitement and boosted confidence in the field. Luxturna is an AAV serotype 2 vector encoding retinal pigment epithelium-specific 65 kDa (RPE65) protein for the treatment of Leber’s congenital amaurosis by subretinal injection. Zolgensma uses an AAV serotype 9 vector encoding survival motor neuron 1 (SMN1) protein for the treatment of spinal muscular atrophy by intravenous administration. The clinical successes of these therapies, using different serotypes and routes of administration, demonstrated that rAAV vector-based GTs have the potential and versatility to deliver breakthrough treatments for a range of diseases. A search of the US National Library of Medicine Database (ClinicalTrials.gov) using AAV as a keyword yielded 146 active clinical trials as of June 28, 2022, for a variety of indications.
AAVs are small, non-enveloped, non-replicative viruses that depend on other viruses like adenovirus or herpes virus for their replication. While AAVs can infect humans, they only induce a mild immune response and are not known to cause any disease. AAVs can transduce both dividing and non-dividing cells within a variety of tissues (tropism) and persist as concatemers in an extrachromosomal state with limited integration potential into the genome of the host cell.3,4 These features make AAVs attractive candidates for the safe and efficient delivery of GTs.5 However, the broad application of AAV GTs might be limited by the presence of natural immunity (anti-AAV capsid antibodies or cytotoxic T cells) induced by wild type (wt)AAV infections in humans.6 Although prevalence data always need careful interpretation as they are strongly dependent on the assay used for assessment, the published data demonstrate that prevalence of pre-existing immunity varies widely across age, geographical location, and serotype and may be as high as 80% for some serotypes.7, 8, 9, 10 An overview of wtAAV serotypes, tropism, and corresponding neutralizing antibody (NAb) prevalence can be found in Table 1.
Table 1.
wtAAV serotypes, tropism, and NAb prevalence
| AAV serotype | Preferential tissue tropism | NAb seroprevalence (%)a,4,8,11 |
|---|---|---|
| AAV1 | skeletal muscle, lung, CNS, retina, pancreas | 27–50.5 |
| AAV2 | smooth muscle, skeletal muscle, CNS, liver, kidney | 47–74 |
| AAV3 | hepatocarcinoma, skeletal muscle, inner ear | 35 |
| AAV4 | CNS, retina | NA |
| AAV5 | skeletal muscle, CNS, lung, retina, liver | 20–59 |
| AAV6 | skeletal muscle, heart, lung, bone marrow | 37 |
| AAV7 | skeletal muscle, retina, CNS | NA |
| AAV8 | liver, skeletal muscle, CNS, retina, pancreas, heart | 32–63 |
| AAV9 | liver, heart, brain, skeletal muscle, lungs, pancreas, kidney | 33.5 |
| AAV10 | liver | 21 |
Important: geographical differences (not captured in table); higher seroprevalence in racial minorities in US.8
CNS, central nervous system; NA, not available.
Neutralizing factors in healthy subjects; percentage of subjects with neutralizing factors.
The de novo host immune response (innate and adaptive) against viral vector components as well as the pre-existing host immunity (humoral or cellular) to rAAV vectors can be immunological barriers to safe and effective treatment with rAAV-based GTs.6,12 In addition, the transgene proteins, whether secreted, present on the cell surface, or intracellular, can also potentially induce immune responses in the host. It has been observed in the clinic that pre-existing immunity or treatment-induced immune responses against viral capsid and transgene proteins can diminish efficacy and may contribute to severe adverse events (SAEs).13, 14, 15 In this paper, we provide a comprehensive overview of immunogenicity risks associated with rAAV GTs, as well as describing the methodologies for monitoring pre-existing and treatment-boosted/or -induced immune responses. In addition, we discuss current clinical mitigation strategies deployed to reduce immunogenicity upon administration of rAAV-based GT.5,6
Intrinsic properties of AAVs implicated in immunogenicity
In rAAV-based GT constructs, the inverted terminal repeats (ITRs) that are necessary for packaging viral genome into the AAV capsid are utilized, but the genome of the wtAAV, which encodes the viral functional and structural genes, is completely replaced by the genetic construct of the therapeutic. The therapeutic construct, thus embedded between the AAV ITRs, is usually composed of the therapeutic transgene and a promoter regulating the transgene expression. The promoter can be tissue-specific, inducible, or ubiquitously active and its strength can also vary widely.16 The transgenic protein expressed by the transduced cells may be engineered to remain intracellular, be expressed on the cell surface, or for secretion for systemic distribution and action at a remote site. Some transgenes code for RNA constructs (e.g., microRNA) with pharmacological function that do not require translation. The recombinant capsid used for packaging the genetic construct can derive from a wtAAV serotype or a modified form that was engineered to overcome pre-existing immunity and/or improve transduction efficiency and/or tropism.17, 18, 19 All the components mentioned above (the viral capsid, the genetic construct, and the transgene product) are potential contributors to the overall immunogenicity risk of an AAV GT product.6,12,20
Innate immunity induced by AAV capsid and viral genome
The host innate immunity can be activated by either AAV capsids or the viral genome. As the first line of host defense, the innate immune system recognizes pathogen-associated molecular patterns (PAMPs) in the AAV components via Toll-like receptor (TLR) 2 and TLR9, resulting in activation of factors that induce expression of pro-inflammatory cytokines or type I interferons (IFNs). Type I IFNs may then activate CD8+ T cells. It has been demonstrated that viral capsids can bind to cell-surface TLR220,21 and complement proteins,22,23 potentially inducing an innate immune response.
The complement system, another component of the innate immune system, is composed of more than 30 proteins that circulate in serum and interstitial fluid and plays an important role in recognition and elimination of pathogens.24 Activation of complement proteins by immune complexes formed with pre-existing anti-capsid immunoglobulin (Ig) M or IgG antibodies with the AAV after administration of high vector doses may cause complement-mediated cell damage.25 Furthermore, complement component C3b bound to AAV particles may induce production of pro-inflammatory cytokines following uptake of the vector in human monocytes, amplifying capsid immune responses.22
Once the viral particles are uncoated within the host cells following endocytosis, single-stranded vector DNA can also be recognized by TLR9, located in the late endosomal and lysosomal compartments.26,27 Unmethylated CpG motifs, usually present at high frequency in viral and bacterial DNA, can trigger TLR9-mediated innate immune activation characterized by the production of pro-inflammatory cytokines like tumor necrosis factor alpha (TNF-α) and IL-6.28,29 Notably, unmethylated CpG motifs are so highly immunogenic that they are widely used as adjuvants in vaccine development.28,29 Unmethylated CpG motifs are found not only in the AAV ITRs and in the regulatory elements of the genomic construct but also in the transgene sequence. Although CpG motifs are rare and highly methylated in the mammalian genome, minimization or elimination of CpG content during codon optimization during rAAV GT lead candidate selection can reduce overall host immune responses against rAAV vectors.30 Double-stranded viral RNAs of rAAV GT trigger host innate immune responses via the TLR pathway as well,31,32 which potentiate risks with viral DNAs.
Innate immune responses caused primarily by activation of local macrophages23 that are triggered against an rAAV GT product may be transient (e.g., lasting only as short as about 6 h after exposure), but could lead to enhanced adaptive immune responses.26,27 This type of immunologic response, however, is highly dependent on the components (e.g., AAV capsid serotype, AAV ITR serotype, composition and sequence of the transgene expression construct) of the rAAV GT product. For example, it was demonstrated in a mouse model that self-complementary (sc)AAV vectors pseudotyped with capsids of serotypes 2, 7, or 8 induced more potent transgene product-specific CD8+ T cell and antibody responses than the corresponding single-stranded (ss)AAV vectors.33 Upregulation of innate immunity that resulted in neurotoxicity in nonhuman primates (NHPs) had been attributed to the promoter activities of 3′ ITR sequences.34 The interplay of host innate and adaptive immune responses after treatment with rAAV GTs is illustrated in Figure 1. The intensity of the responses and potential clinical consequences are determined by amalgamation of product- and host-related, as discussed in later sections.
Figure 1.

Interplay of innate and adaptive immune responses for AAV-based GTs
Humoral immune response against AAV capsid
Due to natural exposure, most humans develop antibodies against various wtAAV serotypes during their lives. The same AAV serotypes are commonly used as vectors in GT, and pre-existing antibodies are known to cross-react with not only the wtAAV but also with some engineered AAV capsids.11 The seroprevalence of pre-existing antibodies to wtAAV has been observed to vary by capsid serotype, patient’s age, and geographic location.10,11 Pre-existing antibodies can directly bind to the capsid, potentially leading to the formation of large immune complexes. Complement proteins, like those forming the C1 complement complex, may bind to these immune complexes to recruit macrophages, resulting in phagocytosis and rapid clearance of the viral particles.35 Pre-existing antibodies can also prevent the binding and internalization of the transgene-carrying viral vector to receptors on the target cells, thus inhibiting transduction and neutralizing the rAAV GT therapeutic activity.36 Other antibody-mediated mechanisms of viral neutralization and/or clearance that may result from the presence of pre-existing anti-AAV antibodies include antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and opsonization.37 Post treatment with rAAV GT, it has been observed that most patients with no detectable pre-existing anti-wtAAV antibodies at baseline will develop treatment-induced antibodies to rAAV. These treatment-induced antibodies may limit or prevent re-dosing of patients previously treated with an rAAV GT.
It may be possible to mitigate the effects of pre-existing anti-AAV antibodies by engineering vectors that are specifically depleted of immunogenic motifs,25 while still retaining sequences that are important for cellular transduction, trafficking, and capsid uncoating. Two general approaches by which capsids can be engineered to reduce antibody epitopes are site-directed (i.e., rationale design)38 or by random mutagenesis (i.e., directed evolution).39 While engineered AAV capsids need further evaluation in nonclinical species and humans, they should reduce the impact of pre-existing antibodies on safety and efficacy. Furthermore, it is unlikely that engineered capsids would pose additional safety risk compared with more commonly used rAAV capsids, and risks may be reduced by enabling lower dosages and more tissue-specific transduction. Using directed mutagenesis, Wang et al. recently reported an engineered capsid with specific tropism for the liver that did show transduction of other organs (e.g., heart, skeletal, muscle, kidney, lung, brain, and spleen) in mice. Additionally, the seroreactivity of this engineered capsid variant to NAbs in serum from hemophilia B patient serum was similar to AAV5, the parental capsid serotype.40
Cellular immune response against AAV capsid
Depending on serotype and the method (e.g., intracellular cytokine staining, enzyme-linked immunosorbent spot [ELISpot]) used for determination, up to ∼50% of healthy human adults had low but detectable anti-AAV capsid T cell responses, which can be reactivated after administration of rAAV GT.41 These T cell responses are mediated by major histocompatibility complex (MHC), class I and/or class II presentation of viral capsid peptides. MHC class I molecules are expressed by all nucleated cells, whereas MHC class II molecules are only expressed on specialized cells (i.e., antigen-presenting cells [APCs])).
While pre-existing anti-viral capsid antibodies, particularly NAbs, can affect the initial transduction efficiency of the rAAV vectors, host cellular immune responses have been implicated in the reduced long-term persistence of the therapy.42 Mechanistically, upon transduction of an AAV vector, the capsid proteins can be degraded by the proteasome with the resulting peptides presented on MHC class I molecules by a mechanism of cross presentation,19,43 where they could be recognized by CD8+ cytotoxic T lymphocytes (CTLs).18,43 Once a CTL binds to the peptide-MHC class I complex, the CTL becomes activated and secretes perforin and granzyme, resulting in the death of the transduced target cell. If the viral vectors were phagocytosed by APCs (e.g., plasmacytoid or conventional dendritic cells or macrophages), the capsid peptides from proteolytic degradation could be presented on MHC class II molecules on the cell surface of the APCs. The peptide-MHC class II complex may then be recognized by CD4+ T lymphocytes, which secrete interleukins to stimulate B lymphocytes, which thereafter could proliferate and produce AAV capsid-specific antibodies. AAV capsids may be engineered to reduce T cell responses. For example, AAV capsids with reduced surface exposed tyrosine residues showed reduced capsid-specific CTL responses in vitro and in nonclinical studies in mice, assuming a reduction in MHC (cross-) presentation.18,19
Additionally, the anti-wtAAV antibody status (e.g., seropositive or seronegative) of humans has been shown to result in two distinct cellular immune responses.44 In a recently published study, peripheral blood mononuclear cells (PBMCs) isolated from AAV-seropositive and -seronegative subjects were exposed to recombinant AAV. For the seropositive donor PBMCs, an activated effector CD8+ T cell response consistent with a memory response was noted against the capsid. Interestingly, the capsid-specific response of PBMCs from seronegative donors was an activation of a transient natural killer (NK) cell response. The role these activated NK cells may play in shaping the adaptive immune response to AAV capsids is not yet fully understood.
Humoral and cellular immune responses against transgene protein
Even though rAAV GTs encode human proteins, the protein can be potentially novel to the dosed subjects in cases where the patient has a null mutation and, therefore, does not express the protein related to their disease state such as in cross-reactive immunologic material (CRIM) negative patients with infantile Pompe disease.45 Cellular and humoral immune responses as described above for the viral capsid are also possible against the expressed transgene protein. This has been demonstrated in nonclinical studies by the generation of antibodies against transgene proteins,46,47 and in clinical studies with de novo CD8+ T cell responses.14,15 Even pre-existing immunity against the transgene protein is possible, especially in patients pre-treated with replacement therapies.48 The complexity of host immune responses against secreted transgene proteins has recently been reviewed.46 The consequences of immune responses against a transgene protein expressed on the cell surface may theoretically have greater impact compared with a secreted protein: immune effector functions mediated by anti-transgene antibodies (e.g., ADCC, ADCP, complement-dependent cytotoxicity [CDC]) could potentially result in direct destruction of tissues by immune cells (e.g., NK cells, macrophages, phagocytes) or direct activation of the complement cascade resulting in cell lysis (e.g, CDC). Despite the observed induction of innate and humoral responses, and possibly low cellular responses in some cases, a direct effect or correlation of these on efficacy or safety has not yet been clearly demonstrated and needs further evaluation.
Immunogenicity risks
Similar to immunogenicity risks of therapeutic proteins, the immunogenicity risk of rAAV GTs is a combination of product-, manufacturing process-, treatment-, and patient-related factors (Table 2).10 Risks to efficacy and safety to be considered are discussed in more detail below and include (1) the innate immune response, (2) pre-existing and treatment-induced or -boosted anti-AAV antibodies, (3) anti-transgene protein antibodies, and (4) cellular responses (mainly T cell responses) to AAV or transgene protein peptides displayed on the surface of transduced cells. Bioanalytical methods employed to assess immunogenicity are described the section “bioanalytical assays for immunogenicity assessment.”
Table 2.
Immunogenicity risk factors of AAV GTs
| AAV gene therapy immunogenicity risk factors | ||
|---|---|---|
| Product-related risk factors | Risk | Impact |
| Capsid-specific factors | ||
|
|
|
|
pre-existing antibodies (capsid-specific and/or cross-reactive antibodies) can potentially cause:50, 51, 52, 53 |
|
|
capsid protein sequences could trigger adaptive cellular and humoral immune responses via antigen processing and presentation pathways (i.e., MHC class I and II)54, 55, 56 | activation of the adaptive immune system may be associated with destruction of transduced cells/tissues; immunotoxicities (cellular responses) and/or loss of efficacy (cellular and humoral responses) humoral immune response: treatment-emergent anti-capsid antibodies may limit or prevent opportunity for re-dosing of patients |
|
tissues may have different immunogenicity risks depending on the tissue-specific immune environment. Risk factors include tissue accessibility to lymphocyte trafficking, tissue vascularization, tissue-resident immune cell populations4,10,57, 58, 59 | potential impact to safety and/or efficacy may be tissue dependent |
| Viral genome factors | ||
|
CpGs,30,60 scDNA,25,61,62 and viral dsRNA31,32 transcripts have increased risk to trigger innate immune responses (via TLR9), leading to production of pro-inflammatory cytokines and subsequent activation of the adaptive immune response. Single-stranded DNA genomes are likely lower risk for activating the TLR9 pathway33 | activation of the adaptive immune response may be associated with immunotoxicities (cellular response) and loss of efficacy (cellular and humoral response) |
|
potential immunogenicity risk is related to type of promoter enhancer utilized
|
potential impact on safety and/or efficacy |
|
several factors influence the immunogenicity risk of the transgene protein:10,14,64,65
|
activation of the adaptive immune response may be associated with immunotoxicities (cellular response) and loss of efficacy (cellular and humoral response) |
| Treatment-related factors | ||
|
higher dose levels may be associated with a higher immunogenicity risk, resulting in activation of innate and adaptive immune responses66,67 | activation of the adaptive immune system may be associated with immunotoxicities (cellular response) and loss of efficacy (cellular and humoral response) |
|
|
activation of the adaptive immune system may be associated with immunotoxicities (cellular response) and loss of efficacy (cellular and humoral response) |
| Manufacturing-related risk factors | ||
|
examples of process-related impurities that may increase immunogenicity risk include:70,71
|
potential to increase safety risk and reduce efficacy |
|
expression of immunogenic peptides and additional CpG motifs may trigger and/or boost immune responses via various mechanisms causing potential immunotoxicities | potential to increase safety risk and reduce efficacy |
| Patient-related factors | ||
|
|
disease-related factors having the potential to increase severity of (immuno)toxicities and/or reduce efficacy |
|
|
activation of an adaptive immune responses may be associated with immunotoxicities and loss of efficacy |
|
|
pre-existing immunity or inflammation may be associated with the potential of reduced transduction efficiency and/or increased severity of immunotoxicities74,75 |
|
|
pre-existing immunity or inflammation may be associated with the potential of reduced transduction efficiency and/or increased severity of immunotoxicities74,75 |
|
seroprevalence of pre-existing immunity or immunological memory to AAVs increases with age:
|
pre-existing immunity or inflammation may be associated with the potential of reduced transduction efficiency and/or increased severity of immunotoxicities74,75 |
Product-related immunogenicity risks associated with the rAAV vector construct include the immunogenic potential of the viral capsid, the DNA element of the transgene expression cassette, and the expressed transgene protein as discussed in the sections above. Manufacturing process-related immunogenicity risk factors are product- and process-related impurities (Table 2). Product-related impurities usually closely resemble the AAV viral particles, and reduction of these impurities is challenging, requiring upstream (cell culture) and downstream (purification) manufacturing process optimization. Such impurities can include oxidized, deamidated, degraded, and aggregated forms of the vector product as well as biosynthetic intermediates and particles of incorrect composition. The rAAV particles generated in cell culture may contain 50%–90% particles that have partial genome or no genetic content (empty capsids),76 yet these all have the potential to activate immune responses. CD8+ T cells directed against the AAV capsid were involved in the recognition and clearance of transduced hepatocytes in clinical trials using rAAV GT.13,54,77 Peptides derived from AAV empty capsids have been demonstrated to be presented on MHC class I,55 while deamidation within these peptides can certainly affect their binding to human leukocyte antigen (HLA) molecules, resulting in different T cell responses.78 Another example of product-related impurities is heterogeneous fragments of host cellular DNA unintentionally packaged in AAV capsid particles. AAV-encapsidated human genomic DNA from the host cells in the rAAV vector product may be associated with a greater risk of genotoxicity, due to the potential for homologous recombination with genomic sequences in transduced human cells. AAV-encapsidated insect cell genomic DNA may have a reduced genotoxic risk but a higher risk of immunotoxicity due to unintended expression of insect cell peptides or proteins in transduced tissues.70
Additional process-related impurities can derive from the manufacturing process of the raw materials, and their components and may not be structurally related to the rAAV product. Such impurities include host cell proteins, nuclease-sensitive nucleic acids, helper components, relevant viruses to production cell lines, and plasmid DNA. Such manufacturing process-related impurities usually can be monitored using analytical methods established for therapeutic proteins. Process optimization can help keep impurity levels low, thus reducing their contribution to host immune activation.
The risk of immunogenicity also depends on the route of administration and the administered dose (Table 2). Local administration to an immune-privileged site may be associated with a lower risk compared with systemic administration.57,68 It has been widely reported that induction of immune tolerance can be achieved via hepatic gene transfer of rAAV therapies both in animal and human studies.69,79, 80, 81 Hepatocyte-derived expression of a secreted transgene protein may induce tolerance against this transgene protein, as demonstrated in mice.69,82 This tolerance induction was mainly mediated by antigen-specific CD4+CD25+FoxP3+ regulatory T cells (Tregs).82,83 Interestingly the CD8+ T cell-mediated clearance and induction of tolerance is dictated by dose levels.66
A higher dose is thought to have higher immunogenicity risk, potentially leading to SAEs (Table 2). Dose-dependent adverse events (AEs) of varying severity following intravenous administration have been reported. Some recent examples include administration of high doses of rAAV9 (6 × 1013 to 2 × 1014 vector genomes per kilogram body weight [vg/kg]) in neonates with spinal muscle atrophy (SMA) leading to elevated liver enzymes84 and similar high doses in children with Duchenne Muscular Dystrophy (DMD) (5 × 1013 to 3 × 1014 vg/kg) leading to acute kidney injury, complement activation, reduced platelets and red blood cells (RBCs), and thrombocytopenia.85 A more profound impact of the high-dose systemic delivery was noted in the X-linked myotubular myopathy (XLMTM) where three young males in the high-dose (3 × 1015 vg/kg) cohort developed severe hepatobiliary disease (damage to the liver and bile ducts) resulting in two deaths from bacterial infections and sepsis.86
Immunogenicity assessment in nonclinical studies: in vitro and in vivo
Rare cases of SAEs were reported from nonclinical and clinical studies of a variety of rAAV-based GTs following both systemic and local (e.g., intrathecal) administration. These adverse effects (AEs)87 included thrombotic microangiopathy88 and hepatotoxicity84, 85, 86, 87, 88, 89 observed in clinical studies as well as loss of dorsal root ganglia observed in monkeys, piglets, and mice.90, 91, 92, 93, 94 In most cases, these AEs were observed after administration of very high doses (≥1 to 3 × 1014 vg/kg intravenous or >4 × 1014 vg total intrathecal) and may be associated with activation of the innate (including complement) and/or adaptive immune system.13,95,96 Transgene overexpression under a strong enhancer/promoter combination may also have contributed to AEs causing deleterious effects on normal cellular function and cellular stress.92,93 Reducing the dose by improving the rAAV vector transduction efficiency97 may have the potential to circumvent such high-dose AEs as well as associated immune reactions. Interestingly, the green fluorescence protein (GFP), a commonly used reporter protein, has been shown to cause dose-dependent targeted tissue toxicity when used in nonclinical biodistribution studies of rAAV vectors.98,99
Animal studies, primarily used to study general toxicology and pharmacology in correlation with biodistribution, failed to accurately predict AEs of rAAV GT products in humans.100 Additionally, animal models do not predict immunogenicity incidence or severity in humans, likely due to the differences between the immune systems of animals and human subjects.101 Nonetheless, findings from a combination of in silico tools, in vitro, and in vivo nonclinical studies can help to elucidate the potential of various immune mechanisms to trigger AEs in different species. A better understanding of the contribution of individual product components to the overall immunogenic potential of an rAAV GT product may help to improve vector design (e.g., CpG content, ssAAV or scAAV vector) and product quality (e.g., reduce impurities like empty capsid). It can also guide in the selection of the appropriate animal models, the design of nonclinical studies, and implementation of immunogenicity mitigation (pre-) treatments in clinical studies. Evaluating the immune response in nonclinical safety and efficacy studies may be important for interpretation of the study observations. Importantly, the same route of administration, administration device, and manufacturing process should be used in IND-enabling and clinical studies whenever possible. In summary, data generated during nonclinical development need to be carefully interpreted, and interspecies differences of the immune systems need to be considered when nonclinical data are translated to the clinics.
Nonclinical models for assessing innate immune pathway activation
AAV capsids can bind to cell-surface TLR220 and are recognized by complement factors like C3b,21 which can accentuate host adaptive immune responses. C3b binding can also lead to opsonization. TLR9-mediated innate immune response activation is triggered by unmethylated CpG motifs. Reduction or removal of such motifs from AAV vectors can reduce the subsequent host adaptive immune responses and prolong the persistence of transgene expression in animal models.30 As previously demonstrated in mice, increased numbers of unmethylated CpG contents in the genetic construct resulted in increased CD8+ CTL responses and decreased persistence of transgene expression in clinical hemophilia B trials with eight different rAAV constructs, even with immunosuppressant treatment, as reviewed in Wright 2020a.102
To optimize vector design and improve clinical efficacy and safety, in silico algorithms can be used to estimate the TLR9 activation potential of an AAV vector construct.103 In vitro animal or human cells22,23 and mouse models may also be used during construct selection, particularly for engineered capsids, due to high homology of the pattern recognition receptors (PRRs).104 Utilization of NHP models for assessment of innate immune responses has been summarized in recent reviews.100,101
Nonclinical models for assessing adaptive immunogenicity
Nonclinical studies in mice and monkeys demonstrated that pre-existing antibodies with a transduction neutralizing potential can reduce transduction efficiency and transgene expression.105,106 Therefore, patients with certain levels of pre-existing antibodies or pre-existing transduction inhibition (TI) activity were often excluded from clinical studies, especially when using systemic route of administration.77,107,108 Safety risks may be another reason to consider exclusion of patients with pre-existing immunity; for example, to avoid formation of large immune complexes activating the complement system and to avoid boosting immune reactions that may lead to more SAEs. To date, there are insufficient data to make firm predictions on how the observed pre-existing or treatment-related immunogenicity will affect safety or efficacy of GT in each individual, either in animals or in the clinic.
The translatability of animal studies/models to clinical AEs can be limited primarily due to the fundamental differences of the immune systems among species. Animal models including NHPs may not accurately predict immunogenicity incidence in humans due to differences in functions of immune cells. In NHPs, memory CD8+ T cells against AAV8 capsids acquired from natural infection were unable to eliminate rAAV8 transduced hepatocytes,109,110 in contrast to observations in clinical trials95 demonstrating differences in cellular responses between NHPs and humans. Immunological profiling showed that both CD4+ and CD8+ capsid-reactive T cells in NHPs displayed functional and phenotypic differences compared with human cells.111 However, characterizing adaptive immune responses in animal models has been informative,101 especially for the interpretation of the efficacy and safety outcome of nonclinical studies. While many preclinical studies are performed in immuno-incompetent or immunosuppressed animals, emphasis should be placed on developing CTL models in immunocompetent animals that are more reflective of the clinical scenario.112 Although there may be differences in the cellular responses observed in animals and humans, the correlation in animals (if any) between the response and measured persistence of transgene expression may be informative on the potential consequences of such a response in humans. The challenges of detailed characterization of immunogenicity to GTs, including the large blood volumes currently required for cellular immunogenicity assays and the relevance of peripheral blood sampling to immunogenicity in tissues, are described in more detail in the section “cellular immune response measurement against viral capsids and transgene product.”
Recently, some acute immunotoxicities observed in clinical studies of rAAV GT products have been reproduced and evaluated in animal models. AAV-naive and AAV-primed C57Bl/6 mice have been used to evaluate the mechanism of developing uveitis after intravitreal administration of rAAV GT.113 Both innate and adaptive immune cell infiltrates were observed in the eye after intravitreal administration with intraocular elevation of CD45+ cells, particularly T cells, regardless of the prior immune status; however, the adaptive response was delayed in rAAV treatment-naive eyes.
As NHPs are thought to be the closest animal models for toxicology studies of rAAV GTs due to comparable physiology and anatomy, a clinical-grade rAAV8 encoding CNGA3 was administered to cynomolgus monkeys by intraocular surgery (either intravitreal at 1 × 1012 vg or subretinal administration at 1 × 1011 vg and 1 × 1012 vg) with concomitant systemic and local steroid treatment mimicking the clinical application.31 Despite the widespread notion that the eye is an immune-privileged organ, activation of immune responses within the eye was observed in the high-dose group during the first month after rAAV administration. Both innate and adaptive immune responses resolved by day 90 in peripheral blood samples. Genetic profiling revealed upregulation of IFN-γ-mediated cytokines of the pro-inflammatory Th1 pathway four weeks after subretinal injection. At the same time, immunohistochemistry showed evidence of microglial activation and the presence of adaptive immune cells (e.g., CD8+ T cells and CD20+ B cells) in the retina. This NHP study informed the subretinal delivery and accompanying cautious monitoring strategy in the subsequent clinical trial, including a 1-month delay between administration. Recent clinical observations of inflammatory responses to ocular rAAV GTs demonstrated translatability of this nonclinical phenomenon to humans. Additional nonclinical studies may help to further elucidate the mechanism of potential immune reactions and their impact on the safety and efficacy of ocular GTs.
Impact of pre-existing immunity and treatment-induced immune responses in nonclinical studies
Understanding the risk and impact of pre-existing immunogenicity to rAAV GTs in animal studies will facilitate the clinical study design and interpretation of study results.114 During the discovery stage of evaluating a dose-efficacy or -safety relationship, animal disease models are used and potentially screened for naive animals (not previously dosed with AAV, although they may have been exposed to natural AAV infections). Animals with pre-existing immunity against wtAAVs may be included in addition to naive animals in later nonclinical studies to evaluate the impact of pre-existing immunity on transduction efficiency115 and to determine the potential for immunotoxicities116 prior to clinical studies. Some regulatory guidance documents on selecting animal species for nonclinical studies are referenced in Table 3.
Table 3.
Regulatory guidance documents on immunogenicity considerations for rAAV GT study design
| Documents | Immunogenicity considerations |
|---|---|
| FDA Guidance for Human Somatic Cell Therapy and Gene Therapy, March 1998 | immunogenicity considerations in nonclinical study interpretation |
| FDA: Pre-clinical Assessment of Investigational Cellular and Gene Therapy Products, November 2013 | immunogenicity considerations to AAV capsids and transgene proteins |
| FDA: Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products, June 2015 | immunogenicity considerations in nonclinical and clinical studies |
| EMA: Guideline on the Quality, Non-Clinical and Clinical Aspects of Gene Therapy Medicinal Products, March 2018 | section 6.5 immunogenicity |
| FDA: Human Gene Therapy for Hemophilia, January 2020 | pre-existing antibody for patient selection; humoral and cellular immunogenicity against vector and transgene proteins |
| FDA: Human Gene Therapy for Retinal Disorders, January 2020 | nonclinical immunogenicity against vector and transgene protein; humoral and cellular immunogenicity against vector and transgene proteins |
| FDA: Human Gene Therapy for Rare Diseases, January 2020 | nonclinical immunogenicity against vector and transgene protein; pre-existing immunity; humoral and cellular immunogenicity against vector and transgene proteins |
| FDA: Human Gene Therapy for Neurodegenerative Diseases (draft) | pre-existing immunity considerations; humoral and cellular immunogenicity against vector and transgene proteins in clinical development |
| ICH S12 Nonclinical biodistribution considerations for gene therapy products (draft) | section 5.4 immunogenicity on species selection |
Immunogenicity assessment and the potential consequence of the immune response in clinical studies
After administration of rAAV GT products, an immune response is usually triggered regardless of the route of administration.117 This immune response is expected to include activation of the innate immune system including the complement system, as well as activation of the humoral and cell-based adaptive immune system.
Prior to the administration with rAAV GT products, patients may likely have pre-existing anti-AAV capsid antibodies that can be associated with reduced efficacy in clinical studies. These antibodies primarily result from previous naturally occurring infections with wtAAVs74 or are maternally derived in newborns. They may affect efficacy by potentially inhibiting viral transduction and/or increasing the clearance of the administered rAAV vector directly after administration. Additionally, pre-existing immunity may be boosted after administration of rAAV GT products and may be associated with a higher risk for certain SAEs. Reduction of pre-existing antibodies demonstrated improved efficacy in nonclinical studies.115 Therefore, many clinical studies with systemic administration excluded patients with pre-existing total antibody against AAV capsids or TI titers above a predefined threshold.96,118 Anti-AAV capsid antibodies are known to increase in titer after administration of an rAAV GT product and to persist up to several years,119 preventing re-dosing. The impact of this humoral immune response on safety and efficacy is not yet understood; however, its contribution to some antibody class-specific (immune) toxicities, observed in nonclinical and/or clinical studies, cannot be excluded.
The impact of pre-existing cellular immunity to the AAV capsid on efficacy or even on safety is not yet understood and will need further investigation. Cellular immune responses directed against the administered rAAV vectors are reported to affect efficacy and persistence after systemic13,54,77 or intramuscular administration.120,121 They may also trigger tissue damage that results in the release of tissue-specific biomarkers like creatine phosphokinase (a marker of muscle cell damage) and transaminases (suggesting liver cell damage) into the circulation.41,95 However, elevated safety or efficacy biomarker levels or detectable cellular immune responses against capsid and transgene are not always associated with clinical loss of transgene expression.119,122,123 The lack of correlation may be due to differences in the functionality of resident versus circulating CTLs (measured in clinical studies) or other underlying mechanisms of cellular immune responses against rAAV GT. The roles played by circulating or resident CTLs in the observed toxicities in transduced tissues,41 as well as the role of Tregs,120,124,125 are not yet completely understood and will need further investigation. In addition, PBMC sampling and assay sensitivity adopted for measuring cellular immune responses are not consistently performed (see section “cellular immune response measurement against viral capsids and transgene product.”). It is imperative to build a bioanalytical and biomarker strategy using well-characterized methodologies during nonclinical and clinical development to measure, monitor, and characterize potential host immune responses against rAAV GT products.
Mitigation strategies of immunogenicity
In general, immunogenicity mitigation strategies for rAAV GTs should start early during lead candidate design and selection stage, and should be made based on the individual risk-benefit assessment of each GT. In silico, in vitro, and in vivo nonclinical studies need to be conducted to estimate and understand the contribution of product components and critical quality attributes (CQAs)126 like product- and process-related impurities to immunogenicity of the rAAV GT.
Strategies to avoid immune reactions against the rAAV genome may include product optimization to block reverse-strand transcription, depletion of CpG motifs during early product development, and increasing methylation of CpG by improved production technologies. Incorporation of short non-coding DNA oligonucleotides into the vector genome to inhibit TLR9 activation may also reduce the immunogenic potential35,102,127 and therefore the risk of a CD8+ T cell response to the transgene product.30,61 Additionally, capsids may be optimized through protein engineering to reduce prevalence of (cross-reactive) pre-existing immunity against the rAAV GT or to be less immunogenic. Capsids may also be encapsulated in exosomes or coated with lipids to mask immunogenic epitopes.117,128
Effective immune mitigation strategies may also include reducing the impact of pre-existing anti-AAV capsid antibodies in nonclinical species and humans. Plasmapheresis has been used in nonclinical and clinical studies to remove immunoglobulins from the blood129,130 to overcome pre-existing anti-AAV capsid immunity. Imflidase/IdeS, a bacterial cysteine protease,131,132 can potentially be used to degrade circulating antibodies133 prior to treatment or re-administration of an rAAV GT.
Furthermore, immunomodulation has proved to be an effective mitigation strategy to reduce immune responses to rAAV GTs. An overview on immunosuppressive medications used to mitigate immune responses to AAV therapies in clinical or nonclinical studies is given in Table 4.35,41,134,135 Immunomodulatory drugs are often given in combination to achieve optimal reduction of the immune response.
Table 4.
Mitigation of immune responses to AAV therapies demonstrated in nonclinical or clinical studies
| Immunosuppressive medications | Mechanism of action | Examples |
|---|---|---|
| Rapamycin/ Sirolimus | general immunosuppressant. Inhibits activation of B and T cells. May lead to reduced anti-AAV antibody production but does not remove existing antibodies. Downstream effects include:136, 137, 138
|
rapamycin in combination with prednisolone prevents production of immunoglobulin G in the mouse (up to 93%) thereby reducing the pre-existing AAV capsid antibodies over time139 |
| Corticosteroids (methylprednisolone, prednisolone, prodrug prednisone) | inhibition of innate and adaptive immune cells and of T and B cell (lesser extent) production | treatment of transaminitis and CTL-induced injury associated with transgene loss in hemophilia B gene therapy77 used in approved GTs for inherited retinal dystrophy140 and SMA141 corticosteroid treatment alone might not inhibit AAV-mediated immune responses and formation of capsid-reactive T cells after administration of high doses142,143 combination of methylprednisolone with rituximab and rapamycin resulted in a lower increase of NAb and T cell response in a trial with intrathecal administration of 4.2 × 1014 vg of an AAVrh10-microRNA to adult patients144 |
| MMF | suppression of T and B cell proliferation inhibiting Inosine monophosphate dehydrogenase145 | immune suppression in a phase II/III AAV GT trial (NCT03612869) was achieved by administration of tacrolimus, MMF, and steroids146 MMF reduced AAV GT transduction efficiency, inhibiting second-strand synthesis of the vector genome in mice147 no difference in AAV transgene expression observed between immunosuppressed and non-immunosuppressed NHP following MMF administration110 |
| Calcineurin inhibitors: Ciclosporin, tacrolimus |
Glybera clinical studies incorporated treatment with ciclosporin, MMF, and methylprednisolone resulting in transient cellular responses without clinical manifestation150, 151, 152 | |
| Abatacept, belatacept | suppression of cytotoxic CD8+ T cell responses blocking CD28-mediated signals | abatacept suppressed anti-AAV T cell and neutralizing antibody response in a nonclinical mouse model153 |
| Rituximab | rituximab depletes CD20+ B cells to reduce levels of pre-existing anti-AAV capsid antibodies and post-treatment reduction of anti-AAV capsid and anti-transgene antibodies. Usually given in combination with other immunomodulatory drugs | rituximab in combination with methylprednisolone prior to dosing with AAV GT has led to reduced anti-capsid and anti-transgene antibody responses144,154 |
| Hydroxychloroquine | inhibition of TLRs and cyclic GMP-AMP synthase, reducing pro-inflammatory cytokine and type I IFN production increase of endosomal and lysosomal pH
|
subretinal injection of hydroxychloroquine resulted in improved photoreceptor transgene expression156 |
| Eculizumab | inhibition of activation of complement factor C5, thereby preventing membrane attack complex formation.157,158 approved for treatment of rare disorders involving complement hyperactivation159,160 or paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome157,158 |
eculizumab was used to treat a Duchenne muscular dystrophy patient that developed atypical hemolytic syndrome-like complement activation related to AAV GT in phase 1B study by Pfizer in 2020 |
| Proteasome inhibitors: Carfilzomib, bortezomib |
inhibition of AAV degradation after endosomal escape, thereby preventing presentation of capsid-derived peptides to CTL by MHC class I molecules.5,161 immunomodulatory role in dendritic cell suppression and T cell stimulation162 |
bortezomib and carfilzomib, both approved for treatment of multiple myeloma, enhanced transgene expression in mice163 bortezomib enhanced transgene expression in hemophilia A mice and hemophilia A dogs164 bortezomib in combination with prednisolone resulted in significant reduction in serum anti-AAV capsid antibodies139 |
MMF, mycophenolate mofetil.
In addition, drugs targeting members of the TLR signaling cascade like MyD88 or IRAK-4 may also be considered to inhibit the activation of naive T cells35 reducing immune responses to AAV GTs as well as drugs inhibiting inflammatory responses via TNF-α.41
As mentioned in the section “immunogenicity risks,” immune tolerance can be induced via hepatic transfer of rAAV GTs. Therefore, an AAV GT may be a good, or in some cases even a better, alternative to an enzyme replacement therapy (ERT).165 Furthermore, AAV GTs may also have the potential to treat autoimmune diseases.165
In cases in which immune responses against the transgene are observed, the immunomodulation strategies listed in Table 4 may be used for immunosuppression. Complement inhibitors like inhibitors of the C1 complex (sutimlimab)166 and C3 (APL-2)135 are under development or launched for treatment of rare or autoimmune diseases and may also be considered as additional immunosuppressive treatments in future clinical trials.
A better understanding of the immunogenic properties of, and the immune pathways activated by, an AAV GT may help to optimize the immunosuppressive treatment during clinical development.
Relevant regulatory guidance documents to consider for immunogenicity assessment during study design for rAAV GTs
Regulatory agencies have issued a series of guidance documents on study design for rAAV GTs. In these guidelines (examples listed in Table 3), immunogenicity considerations are central to study design, selection of subjects, impact on study outcome, and data interpretation. These guidance documents provide the foundation for designing the risk-based bioanalytical strategy for nonclinical and clinical studies of rAAV vector-based GTs.
Bioanalytical strategy and methodology for assessment of pre-existing and treatment-emergent immunogenicity
To address immunogenicity risk and align with regulatory expectations for nonclinical and clinical development, a program-tailored immunogenicity testing strategy needs to be implemented that potentially covers the detection of host innate as well as adaptive humoral and cellular immune responses. A suite of bioanalytical assays that align with the immunogenicity risk assessment should be developed and appropriately validated to enable nonclinical and clinical studies. These assays could include cytokine and complement analysis to cover innate responses, anti-AAV-capsid, and anti-transgene product antibody testing to cover pre-existing, treatment-boosted, or treatment-induced humoral responses, as well as testing for cellular immune responses against the capsid and transgene product (Table 5).
Table 5.
Bioanalytical methods for monitoring host immune responses to rAAV GTs
| Immune response | Endpoints | Assays (examples) |
|---|---|---|
| Capsid immunogenicity assays | ||
| Innate | cytokine and chemokine secretion/expression | |
| complement factors and activation169 |
|
|
| Humoral | anti-capsid binding antibodies (i.e., total antibodies, TAb) | immunoassay (e.g., bridging and sandwich assay formats)170 |
| anti-capsid TI (i.e., neutralizing antibodies) | cell-based neutralization assay171, 172, 173, 174 | |
| Cellular | T cell response to capsid by measuring secreted factors (e.g., IFN-γ) | |
| T cell phenotypes responding to capsid antigen | ||
| Transgene product immunogenicity assays | ||
| Humoral | anti-transgene product binding antibodies | immunoassay (e.g., bridging and sandwich assay formats)177 |
| NAbs | ||
| Cellular | T cell response to transgene antigen by secreted factors (e.g., IFN-γ) | |
| T cell phenotypes responding to transgene product antigen | ||
It is important to consider the right sampling time points, blood volumes, and sample matrices (pre- and post-dosing) to evaluate the different types of host immune responses (see also section “bioanalytical strategies”). For example, early collection for cytokine and complement factor assessment is advisable to assess innate immune responses (within 24 h), while pre-dose, 2–4 weeks post-treatment, as well as late (months) sample collection is beneficial to assess antibody responses (humoral) as well as T cell-mediated immune responses (cellular). It is also important to consider sampling alignment with other types of readouts, e.g., biodistribution and biomarkers, when possible, to enable correlation with clinical findings. For complement measurement, plasma collected by EDTA tube is the appropriate matrix for testing.
For locally delivered GTs, it may be informative to assess pre-existing immunity and post-treatment immune reactions from the systemic circulation.182 However, antibody titers determined in plasma/serum are unlikely to be the same as in the local compartment (e.g., cerebrospinal fluid for intrathecal administration or vitreous humor for ocular administration), so it is important to correlate data obtained in these surrogate matrices with efficacy and safety outcomes. This practice has been successfully employed in ocular programs with therapeutic proteins.182 Regarding monitoring cellular responses, it is also noted that memory T cells reside in lymphoid organs or even in the tissues in which the immune response occurred and, if at all, only a small percentage may be accessible in the systemic circulation. Therefore, data obtained from blood should be interpreted with caution.183
Bioanalytical strategies for nonclinical and clinical studies
Data on humoral immune responses against the capsid and transgene in combination with biodistribution data are used for interpretation of nonclinical toxicity studies. Infiltration of immune cells into transduced tissues is generally assessed during histopathology examination. Further characterization of the infiltrated immune cells might be helpful for data interpretation as well as for implementation of safety measures and mitigation strategies in clinical trials. To study dose-response relationships, pre-existing antibody-negative animals are typically used for nonclinical toxicity studies. However, this practice is evolving and the understanding of the impact of pre-existing antibody titers on dose and exposure may change. Importantly, if all nonclinical studies were performed with animals selected based on low or no anti-AAV capsid antibody titers, this selection criterion will likely also be required in the clinical phase and after launch. When using an animal species with a higher likelihood for pre-existing immunity, like NHPs, pre-screening is often necessary to identify the status of pre-existing immunity in animals for data interpretation. Animals with pre-existing immunity may help to evaluate the impact of pre-existing immunity on transduction and transgene expression and to assess the potential for additional AEs associated with pre-existing immunity.
For clinical development, monitoring of both humoral and cellular immune responses against the AAV capsid and the transgene protein are requested by authorities depending on the route of administration and the target organ. In cases where one or the other assessment is deemed not necessary, a justification may be required. Measurements of the immune responses should be implemented, starting in phase I since only a small number of subjects may be available to provide sufficient data for an overall meaningful immunogenicity evaluation. In addition, monitoring immunogenicity can retrospectively guide clinical mitigation strategy and justify clinical intervention if SAEs are observed that correlate with immunogenicity. Excluding patients from clinical trials based on their pre-existing immunity may require development of a companion diagnostic test (CDx).
Cytokine and complement measurements should be included as standard clinical pathology assessments during nonclinical and clinical GT studies. It is recommended that cytokines relevant to innate immune responses to AAV, which include IL-1β, TNF-α, IL-10, and IL-6, are monitored. These cytokines may be assessed in serum or plasma samples using available protein detection assays, including multiplexed immunoassay platforms. Since cytokines typically appear and resolve quickly, earlier time points, usually within 24 h after dosing, will need to be collected. Complement activation has been observed clinically after administration of high-dose rAAV GT product35 and can be monitored using complement activity assays or by detection of selected complement factors like C1, C3, C4, and C5.
Regulatory guidance on bioanalytical assay development and validation for rAAV GTs
The appropriate regulatory aspects of assays to be used for immunogenicity evaluation of rAAV GTs depend upon the stages of the development and the use of the data (context of use). The 2017 European Medicines Agency (EMA)/Committee for Medicinal Products for Human Use (CHMP) guidance (“Guideline on Immunogenicity Assessment of Biotechnology-derived Therapeutic Proteins”) and the 2019 US Food and Drug Administration (FDA) guidance (“Immunogenicity Testing of Therapeutic Protein Products–Developing and Validating Assays for Anti-Drug Antibody Detection”) do not apply to cell and GT products. Nevertheless, the principles of assay design, development, and validation found in these documents can be useful to sponsors developing rAAV GT products, especially regarding anti-capsid and anti-transgene protein antibody assays in a regulated good-practice (GxP) environment.
The methods used for nonclinical safety, clinical trial, and post-marketing use need to be conducted in the appropriate GxP environment. Assays for detection of pre-existing antibodies to AAV capsids may be used to exclude patients from GT treatment, especially when the route of administration is systemic. In those cases, the sponsor(s) should consider developing a CDx at least during pivotal studies for its availability upon market approval. A CDx is defined as an in vitro diagnostic test providing information that is essential for the safe and effective use of a corresponding drug or biological product. Such tests are regulated (in the US) by the Center for Devices and Radiological Health (CDRH) and should be performed in a Clinical Laboratory Improvement Amendments (CLIA)-compliant laboratory. Sponsors can refer to FDA guidance “In Vitro Companion Diagnostic Devices (2014)” and “Principles for Co-development of an In Vitro Companion Diagnostic Device with a Therapeutic Product (2016)” and local/regional regulations as appropriate (e.g., State of New York). In such cases, co-approval of the CDx together with the Biologics License Application (BLA) is anticipated; thus, it is advisable to start the co-development of the CDx latest in pivotal studies. Methods used for pre-existing immunity assessment without affecting patient selection or immunogenicity evaluations after treatment are usually validated and performed in accordance with GxP requirements. In the European Union, diagnostic tests are regulated in Regulation (EU) 2017/746 (L 117/192), and diagnostic laboratories are typically accredited according to ISO 15189-2012(E) by country-specific agencies, and other country/local/regional requirements like Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen (Rili-BAEK).
Other types of assays, such as ELISpot, complement assessment, and cytokine assays, have no formal guidance document at present. However, several publications exist for related types of assays that might be helpful to consider for the sponsor.175,184 In addition, the FDA has published a draft guidance document, “Biomarker Qualification: Evidentiary Framework,” that proposes general approaches for fit-for-purpose validation within their context of use.
Bioanalytical assays for immunogenicity assessment
Anti-viral capsid antibody assays
Humoral immune responses against the capsid are addressed using ligand binding assays or functional cell-based TI assays. Although the ligand binding assays have the potential to determine the complete humoral immune response (total binding antibodies against capsids [TAbs]), the cell-based TI assays are expected to only detect the transduction neutralizing capacity (neutralizing antibodies [NAbs] or transduction-inhibiting antibodies, terms that have been used interchangeably in the literature).
A major challenge for development and validation of immunogenicity assays in the rAAV GT space is the high prevalence of pre-existing antibodies.11,50 This positivity makes it difficult to select the true-negative samples required to evaluate the natural variance of the antibody-negative population and to set an appropriate cut point. For some serotypes or in specific matrices, the standard cut point approach may be applicable, and positive individual samples can be removed via outlier test as applied commonly to therapeutic proteins. For serotypes with high prevalence of positive samples, however, an outlier test is probably not useful and alternative criteria for elimination of such samples from cut point determination may be required. The use of immunodepleted samples might be an option by either removal of all immunoglobulins or selective depletion of AAV serotype-specific antibodies. The former approach needs careful evaluation as nonspecific removal of all immunoglobulins might have a huge impact on the variance of the samples and thus on cut point determination. The latter, depletion of AAV serotype-specific antibodies, might not alter the overall sample background and variance.
Ligand binding assays to assess the TAb response (TAb assay)
Serotype-specific direct, indirect, or bridging ligand binding assays are used to determine the total antibody response against the AAV capsid. Depending on the assay design and the detection reagents used, such assays will detect all immunoglobulin subtypes (such as a conventional bridging assay format) or specific to IgG or IgM detection (i.e., direct or indirect assay). An electrochemiluminescence (ECLIA) or absorbance (ELISA) readouts are commonly used. In these TAb assay formats, the assay signal is directly proportional to the amount of antibodies in the sample.
Many sponsors follow the process typical for therapeutic proteins when developing and validating methods for assessing humoral immune responses to rAAV GT products, including screening, confirmatory, and titration steps.
For study sample bioanalysis, the screening or even the confirmatory step may be omitted depending on the sampling time point (pre-dose versus post-dose) and on the use of the generated data (screening for patient inclusion/exclusion versus immune response after treatment). In some cases, a comparison of the titer between pre- and post-treatment samples can be sufficient to report the treatment-boosted positive samples.
Assay parameters that usually are validated include a screening and confirmatory cut point, and, if necessary, a titer cut point could also be determined. The sensitivity, selectivity, and precision of a method are explored using a positive control antibody. Specificity and reproducibility should also be evaluated.170
Cell-based assays to assess vector TI
TI assays are in vitro cell-based assays detecting all sample components that affect viral transduction independent of the nature of the component.171 Small molecules or other components present in the sample matrix (e.g., heparin) that inhibit or enhance viral transduction185,186 will influence the assay outcome and could lead to false-positive or false-negative results. Common serum components, like C-reactive protein, or galectin 3 binding protein, are also known to inhibit rAAV transduction efficiency.187,188 Such matrix interferences should be minimized during assay development and, therefore, a careful characterization of the TI assay is important to ensure meaningful data interpretation.
In principle, the same assay development and validation approaches as for the TAb assays described above can be followed for the cell-based TI assays. In many cases, a screening cut point is determined, but a confirmatory step is rarely performed due to the large amount of viral capsid required. Sometimes, a general immunodepleting step is used to confirm that the positive assay result was caused by antibodies and not by a matrix effect.
As already described above, the high prevalence of pre-existing antibodies is a major challenge for development and validation of TI assays. Alternative approaches to select negative samples for cut point determination might be used. One way to remove most of the true-positive samples is to exclude samples based on their consistent generation of inhibition above a predefined threshold (e.g., 30%–50% inhibition) in the confirmatory step in multiple repeated runs. After that, individual samples with screening assay responses higher than three times standard deviation (SD) and the median response of the rest of individual panels in multiple runs may be removed. To be more conservative, even 20% inhibition and two times SD can be applied. Alternatives to a statistical cut point, like the half maximal inhibitory concentration (IC50) titer approach, where a 50% reduction relative to the same dilution of control serum is used,105 may also be considered.
When designing a TI assay, the first step is the selection of a permissive cell line. Cell lines like HEK293, HuH-7, and HeLa are usually used. Although HEK293 cells have been recommended,171 in some cases, and depending on the AAV serotype, the use of other cell lines might provide the most sensitive method. In case the drug substance is used to transduce the cells in the in vitro assay, cell transduction and its inhibition are evaluated by analysis of the intracellular vector DNA using a qPCR readout.189 In most cases, an AAV reporter construct is used as a surrogate for the drug substance. These surrogate constructs usually use the same capsid as the drug substance but contain a reporter gene like luciferase, instead of the therapeutic transgene, under a cytomegalovirus (CMV) promoter. It has been reported that using secreted luciferase, NanoLuc, as the reporter in TI assays can greatly improve TI assay sensitivity and reproducibility, which may help develop NAb assay as a CDx assay during/or after BLA approval.172
During method development, the following parameters should be considered and optimized
-
•
Cell number seeded per well
-
•
Rate of confluency of the cell culture before cell seeding
-
•
Number of capsids per cell (multiplicity of infection [MOI]), usually in the range of 102–105
-
•
Optional: use of helper adenovirus or chemicals to enhance AAV transduction and reporter gene expression if a permissive cell line is not readily available
-
•
Optional: heat inactivation of negative control serum when complement proteins interfere with transduction
-
•
Time point to start transduction after cell seeding
-
•
Duration of incubation with the substrate solution at readout
In cases where sponsors exclude patients based on pre-existing anti-AAV antibodies from clinical trials, a robust CDx demonstrating stable assay performance over years will probably be required post-regulatory approval for physicians to make treatment decisions. Establishing an assay fulfilling these requirements may be technically challenging when a cell-based TI assay is used for patient selection. Therefore, increasingly more sponsors use TAb assays for patient selection, which will detect all antibodies, including NAbs.96,118 Even more important, TAbs are potentially associated with AEs observed in clinical studies.13,20,51
Anti-transgene product antibody measurement
Due to the fact that patients may not express the protein (i.e., double-null mutation, CRIM negative) that is homologous to the expressed transgene product or express a truncated or mutated version, the transgene protein that is produced after treatment with an rAAV GT product may be regarded as foreign protein by a patient’s immune system. Consequently, the patient may mount an immune response to the transgene protein. Patients receiving a protein replacement therapy before being treated with GT might have pre-existing antibodies directed against the transgene protein.
The humoral immune response, including TAbs and NAbs, can be measured using methods very analogous to standard immunogenicity assays used for therapeutic proteins. Anti-transgene product antibody (i.e., TAb or NAb) assays may be developed and validated following the industry white papers and respective guidelines (FDA, EMA). Direct, indirect, or bridging ligand binding assays with enzymatic or ECLIA readout can be commonly used for such purposes. The development of additional assays for determination of neutralizing activity might be necessary depending on the nature of the transgene product and its mechanism of action. For example, NAbs against transgene proteins with enzymatic activity like factor IXa or enzymatic co-factors like factor VIIIa can be detected using enzymatic assays or functional activity assays (e.g., clinically approved coagulation tests). When the transgene product is a lysosomal enzyme, it may be necessary to consider characterizing NAbs that inhibit cellular uptake of lysosomal enzyme via the cell-surface cation-independent mannose-6 phosphate receptor, which delivers the enzyme to the lysosome.190 This could be important, as the class of NAbs could prevent cross-correction of other cell types that are not directly targeted by the AAV GT. Additionally, neutralization of their enzymatic activities may also be relevant to evaluate. For structural proteins like dystrophin, development of a functional NAb assay like a cell-based internalization assay may not be feasible. In such cases, a competitive ligand binding assay may be used. In this format, the labeled drug molecule competes with the NAbs for binding to its target molecule, which is coated to the plate. For an intracellular protein, such as retinitis pigmentosa GPTase regulator protein or RPE65, development of a NAb assay may not be scientifically justified.
Cellular immune response measurement against viral capsids and transgene product
Currently the standard methodology for measuring cellular immune responses against either viral capsid or transgene protein is ELISpot assay due to assay reproducibility.191 ELISpot is a cell-based assay with cytokine-specific antibodies immobilized on a multi-well plate to detect secreted cytokine from cells in the presence of stimulating agents. This method offers higher sensitivity and functionality detection at the single-cell level, (e.g., number of responding cells), with a readout of spot-forming units/well (SFU/well) or SFU/million cells. A common ELISpot assay is the detection of IFN-γ secreted by activated CD4+ and CD8+ T cells. Antigen-specific cellular immune responses to an AAV capsid or transgene encoded protein can be detected using cytokine secretion as functional measure in an ELISpot assay.119
Two factors are very critical to generate reliable ELISpot assay results. The first factor is the prompt PBMC sample isolation and appropriate processing after whole-blood collection as done during vaccine development.192 In order to maintain required cell viability (>70%) and functionality, PBMCs should be isolated within 24 h after whole-blood collection, carefully cryopreserved, and require a conscientious selection of thawing and medium conditions.192 Another key factor is the generation and qualification of synthetic peptide pools derived from the primary amino acid sequences of either the capsid or transgene protein. These peptide pools are made of individual peptides generally of ∼15 amino acids in length overlapping by 10–11 amino acids to measure T cell responses after incubation with PBMCs. Biochemical and biophysical characterization of these peptide pools, such as purity (>90% without aggregation), is critical to reduce false-positive responses due to impurities, sub-optimal length of peptides, and so forth. Several positive control systems can be adopted during the development of an ELISpot assay. Well-characterized peptides, such as CEF pool (CMV, Epstein-Barr virus [EBV], influenza),175 can trigger strong T cell responses from normal PBMCs after stimulation. In addition, sponsors can also screen for positive PBMCs against the same serotype of viral capsid from subjects with pre-existing antibodies. Threshold for positivity may be an arbitrary or empirical cut point (e.g., >50 SFU/million cells) or relative to a negative control signal (e.g., >NC+3SD), or, alternatively, a statistical cut point.
The general validation strategy will primarily be based on experience from ELISpot assay validation used to support vaccine development as well as published white papers,175,184 since there is no specific regulatory guidance available yet. Another consideration is to assess the total number of PBMCs used versus assay precision based on other bioanalytical practice193 when pediatric/juvenile patients are included in the study, resulting in very limited volume of whole blood collected.
Besides ELISpot assay, sponsors are evaluating other bioanalytical platforms, such as FluoroSpot, flow cytometry,119,194 and other cellular markers,195 for multiplexing and improved sensitivity. The FluoroSpot assay allows detection of antigen-specific responses, combining the sensitivity of ELISpot and simultaneously detecting several analytes (antibodies, cytokines), enabling studies of cell populations with different functional profiles.44 Flow cytometry is a cell-based assay using target-specific antibodies to stain and detect simultaneously cell-surface expression of co-stimulatory molecules, intracellular stress markers, transcription factors, and cytokines from activated cells. This method offers a multiplexed detection of cytokines and other immune-related proteins like TNF-α, IL-2, IL-6, or granzyme B, and provides information on various immune cell populations or phenotype of the immune cells, with a readout of cell frequency and/or absolute cell numbers. Flow cytometry can be used to study AAV capsid epitope antigen presentation and CD8+ T cell activation potential by MHC class I tetramers specific to the AAV serotype.72
With the availability of these bioanalytical methods, CD8+ T cell responses against viral capsids were identified in patients receiving rAAV GT.41 A study identified the specific immunodominant peptides from AAV capsids and mapped them to common HLA types by using spleens isolated from subjects undergoing splenectomy for non-malignant indications. The spleens provided a source of large numbers of lymphocytes that were then restimulated in vitro with single AAV capsid peptides. Further experiments confirmed that these epitopes are naturally processed and functionally relevant. The design of more effective and less immunogenic rAAV vectors, as well as a precise immune response monitoring in terms of sampling time points of vector-dosed subjects, are facilitated by these findings.72
Determination of cellular immune responses against either viral capsid or transgene protein could provide insights into mechanisms that trigger safety-related events such as elevation of liver transaminase activities.42 Such data may be used to support the immunomodulatory treatment strategy to reduce the cellular immune response in patients (see section “mitigation strategies of immunogenicity” for further details). As mentioned previously, the cellular immune response data might also help to elucidate mechanisms involved in efficacy loss.6,25
Conclusion
Recombinant AAV-vector-based GT has demonstrated promising therapeutic efficacy in human studies. Nevertheless, the host immune response is one of the biggest barriers for broad application of rAAV vector-based GTs. The initial hurdle to overcome for successful systemic delivery is pre-existing immunity from natural exposure to wtAAV serotypes, which can also potentially be reactivated after administration of rAAV GT products.117 Pre-existing immunity can lead to lack of transduction efficiency and may have the potential to accelerate immune reactions after treatment, which may be associated with AEs in nonclinical and clinical studies. The most significant outcome of pre-existing reactivity to rAAV GT product and its components is the exclusion of patients from enrollment into clinical studies due to the risks of either lack of transduction or safety concerns derived from immune-mediated AEs. While nonclinical studies in animal models can provide insights into the AEs, the extent of translatability may be limited due to the fundamental differences of immune systems among species. Development of more relevant in vitro predictive assays using primary cells from target tissues would be needed to help anticipate and mitigate the risks in the clinic and could guide decisions on dose selection for clinical development.
There are multiple immunogenicity risk factors associated with development of rAAV GT-based therapeutics that can be mitigated at each stage. These include an amalgamation of intrinsic and extrinsic factors related to the GT product as well as factors related to the patient, disease, and route of administration. Currently, manufacturing of rAAV GT products is not fully optimized,97 and the key CQAs’ impact on immunogenicity risks needs to be better understood. A thorough assessment of the boosted immune reactions and evaluation of the underlying immunogenic trigger (e.g., total AAV capsid dose, CpG content, scDNA, residual host cell DNA and proteins, formulation) may help to set the optimal ranges for CQAs. The outcome of better-defined CQAs could lead to safer rAAV GT products with improved efficacy. In theory, this could include dosing with fewer total viral particles, resulting in lower exposure in humans. It is a current hypothesis that greater numbers of total viral particles administered can lead to increased immunotoxicities:196 dosing more optimized material with less empty and thus total amount of viral particles might lead to safer product.
Clinical implications of immune responses to safety and efficacy have been observed both in nonclinical and clinical studies regardless of route of administration. Thus, improved understanding of how immune responses affect rAAV GT, and the development of robust immunogenicity assays and mitigation strategies for achieving optimal clinical outcomes are required.
The most common measure of immune response in the clinic is humoral immunogenicity or anti-capsid and anti-transgene antibody responses. Although current clinical data may show an association of humoral responses with AEs, a clear correlation of humoral responses with the observed immunotoxicities could not be demonstrated. This suggests that the interplay between host innate and adaptive immunity, including both humoral and cellular immunogenicity, determines the immunogenicity to the rAAV GTs. Unraveling the complexity of host immune responses may help address the inconsistencies in AEs observed in subjects and the range of responses observed to immune conditioning treatments. In order to interpret the impact of immunogenicity on nonclinical study results and the clinical relevance, an arsenal of bioanalytical assays is needed to accurately measure and monitor host innate and adaptive immune responses after dosing. There is a great opportunity to standardize some of these methodologies across the industry, such as critical reagents (e.g., positive controls), assay formats, sensitivities, and PBMC processing (e.g., isolation, storage). Standardizing these methodologies will make the overall immunogenicity assessment more comparable between studies and sponsors. Since the number of subjects in each study from each sponsor is typically much smaller (<100) compared with trials for more traditional therapeutic modalities (e.g., small-molecule drugs, therapeutic proteins), a comparison of immunogenicity assay and study results across different clinical trials and sponsors may help to elucidate the contribution of immune responses to AEs. The combined immunogenicity results will potentially allow assessment of clinical relevance and development of effective mitigation strategies. Another benefit of universal/transferrable bioanalytical assays across laboratories would be to enable informing clinical development strategies based on global prevalence of pre-existing immunity in large patient populations.197 More robust and sensitive methodologies will help decipher the underlying biological mechanisms related to immunogenicity and potentially provide better correlation between immunogenicity and efficacy/safety. These results will further guide optimal design and selection of lead capsids, lead to better defined CQAs, optimize the route of administration and repeat dosing strategies, and contribute to precision medicine development (e.g., immunosuppression regimens).
The severity of the immune responses and potential clinical consequences are determined by the immunogenicity risk factors of rAAV GTs. A better understanding of the contribution of immune reactions to SAEs and the nature of these immune reactions may aid in “reverse translation” to design/optimize clinical candidate(s) with reduced immunogenic potential. Additionally, it may also lead to implementation of more effective clinical mitigation strategies, such as immunosuppressive regimens. For example, it is not well understood why some SAEs are observed in a small subset of patients, whereas these severe events can be prevented via immunosuppressive treatments in others. The wide clinical application of immunosuppressants before and during treatment with rAAV GTs further complicates building correlations of efficacy/safety with detectable immunogenicity.119
In summary, a better understanding of the components and mechanism(s) triggering immune reactions will provide tremendous value in optimizing and accelerating the discovery and development of rAAV-based GTs. This starts as early as optimal vector design and includes, but is not limited to, control of the manufacturing process, well-characterized patient populations, repeat dosing evaluation and feasibility, and clinical risk mitigation. Each of these factors is expected to lead to more successful GT development by reducing AEs and achieving optimal efficacy for patients with unmet medical needs.
Acknowledgments
The authors would like to thank IQ TALG for support of this work. Julia Fakhiri (Roche) contributed the graphical abstract. This work received no external funding. The content of this article represents the authors’ opinions and does not necessarily represent the views of their employers.
Author contributions
T.Y.Y. and M.B. drafted and finalized the manuscript. All authors have contributed to the contents, review, and references of the article.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Tong-yuan Yang, Email: tyang9@its.jnj.com.
Manuela Braun, Email: manuela.braun@bayer.com.
References
- 1.Weber G.F. Gene therapy--why can it fail? Med. Hypotheses. 2013;80:613–616. doi: 10.1016/j.mehy.2013.01.037. [DOI] [PubMed] [Google Scholar]
- 2.Hunt S.Y. Controversies in treatment approaches: gene therapy, IVF, stem cells, and pharmacogenomics. Nat. Educ. 2008;1:222. [Google Scholar]
- 3.Dalwadi D.A., Calabria A., Tiyaboonchai A., Posey J., Naugler W.E., Montini E., Grompe M. AAV integration in human hepatocytes. Mol. Ther. 2021;29:2898–2909. doi: 10.1016/j.ymthe.2021.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balakrishnan B., Jayandharan G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene. Ther. 2014;14:86–100. doi: 10.2174/1566523214666140302193709. [DOI] [PubMed] [Google Scholar]
- 5.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronzitti G., Gross D.A., Mingozzi F. Human immune responses to adeno-associated virus (AAV) vectors. Front. Immunol. 2020;11:670. doi: 10.3389/fimmu.2020.00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hüser D., Khalid D., Lutter T., Hammer E.M., Weger S., Heßler M., Kalus U., Tauchmann Y., Hensel-Wiegel K., Lassner D., Heilbronn R. High prevalence of infectious adeno-associated virus (AAV) in human peripheral blood mononuclear cells indicative of T lymphocytes as sites of AAV persistence. J. Virol. 2017;91:e02137-16. doi: 10.1128/JVI.02137-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruzik A., Fetahagic D., Hartlieb B., Dorn S., Koppensteiner H., Horling F.M., Scheiflinger F., Reipert B.M., de la Rosa M. Prevalence of anti-adeno-associated virus immune responses in international cohorts of healthy donors. Mol. Ther. Methods Clin. Dev. 2019;14:126–133. doi: 10.1016/j.omtm.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D., Zhong L., Li M., Li J., Tran K., Ren L., He R., Xie J., Moser R.P., Fraser C., et al. Adeno-associated virus neutralizing antibodies in large animals and their impact on brain intraparenchymal gene transfer. Mol. Ther. Methods Clin. Dev. 2018;11:65–72. doi: 10.1016/j.omtm.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verdera H.C., Kuranda K., Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boutin S., Monteilhet V., Veron P., Leborgne C., Benveniste O., Montus M.F., Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene. Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 12.Shirley J.L., de Jong Y.P., Terhorst C., Herzog R.W. Immune responses to viral gene therapy vectors. Mol. Ther. 2020;28:709–722. doi: 10.1016/j.ymthe.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Rasko J., Ozelo M.C., Hoots K., Blatt P., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 14.Mendell J.R., Campbell K., Rodino-Klapac L., Sahenk Z., Shilling C., Lewis S., Bowles D., Gray S., Li C., Galloway G., et al. Dystrophin immunity in Duchenne's muscular dystrophy. N. Engl. J. Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calcedo R., Somanathan S., Qin Q., Betts M.R., Rech A.J., Vonderheide R.H., Mueller C., Flotte T.R., Wilson J.M. Class I-restricted T-cell responses to a polymorphic peptide in a gene therapy clinical trial for alpha-1-antitrypsin deficiency. Proc. Natl. Acad. Sci. USA. 2017;114:1655–1659. doi: 10.1073/pnas.1617726114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domenger C., Grimm D. Next-generation AAV vectors-do not judge a virus (only) by its cover. Hum. Mol. Genet. 2019;28:R3–R14. doi: 10.1093/hmg/ddz148. [DOI] [PubMed] [Google Scholar]
- 17.Büning H., Srivastava A. Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol. Ther. Methods Clin. Dev. 2019;12:248–265. doi: 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martino A.T., Basner-Tschakarjan E., Markusic D.M., Finn J.D., Hinderer C., Zhou S., Ostrov D.A., Srivastava A., Ertl H.C.J., Terhorst C., et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood. 2013;121:2224–2233. doi: 10.1182/blood-2012-10-460733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong L., Li B., Mah C.S., Govindasamy L., Agbandje-McKenna M., Cooper M., Herzog R.W., Zolotukhin I., Warrington K.H., Jr., Weigel-Van Aken K.A., et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA. 2008;105:7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nidetz N.F., McGee M.C., Tse L.V., Li C., Cong L., Li Y., Huang W. Adeno-associated viral vector-mediated immune responses: understanding barriers to gene delivery. Pharmacol. Ther. 2020;207:107453. doi: 10.1016/j.pharmthera.2019.107453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabinowitz J., Chan Y.K., Samulski R.J. Adeno-associated virus (AAV) versus immune response. Viruses. 2019;11 doi: 10.3390/v11020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaiss A.K., Cotter M.J., White L.R., Clark S.A., Wong N.C.W., Holers V.M., Bartlett J.S., Muruve D.A. Complement is an essential component of the immune response to adeno-associated virus vectors. J. Virol. 2008;82:2727–2740. doi: 10.1128/JVI.01990-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaiss A.K., Liu Q., Bowen G.P., Wong N.C.W., Bartlett J.S., Muruve D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002;76:4580–4590. doi: 10.1128/jvi.76.9.4580-4590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holers V.M. Complement and its receptors: new insights into human disease. Annu. Rev. Immunol. 2014;32:433–459. doi: 10.1146/annurev-immunol-032713-120154. [DOI] [PubMed] [Google Scholar]
- 25.Hamilton B.A., Wright J.F. Challenges posed by immune responses to AAV vectors: addressing root causes. Front. Immunol. 2021;12:675897. doi: 10.3389/fimmu.2021.675897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashley S.N., Somanathan S., Giles A.R., Wilson J.M. TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell. Immunol. 2019;346:103997. doi: 10.1016/j.cellimm.2019.103997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J., Huang X., Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Invest. 2009;119:2388–2398. doi: 10.1172/JCI37607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bode C., Zhao G., Steinhagen F., Kinjo T., Klinman D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines. 2011;10:499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheiermann J., Klinman D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine. 2014;32:6377–6389. doi: 10.1016/j.vaccine.2014.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faust S.M., Bell P., Cutler B.J., Ashley S.N., Zhu Y., Rabinowitz J.E., Wilson J.M. CpG-depleted adeno-associated virus vectors evade immune detection. J. Clin. Invest. 2013;123:2994–3001. doi: 10.1172/JCI68205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reichel F.F., Dauletbekov D.L., Klein R., Peters T., Ochakovski G.A., Seitz I.P., Wilhelm B., Ueffing M., Biel M., Wissinger B., et al. RD-CURE Consortium AAV8 can induce innate and adaptive immune response in the primate eye. Mol. Ther. 2017;25:2648–2660. doi: 10.1016/j.ymthe.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao W., Earley L.F., Chai Z., Chen X., Sun J., He T., Deng M., Hirsch M.L., Ting J., Samulski R.J., Li C. Double-stranded RNA innate immune response activation from long-term adeno-associated virus vector transduction. JCI Insight. 2018;3:120474. doi: 10.1172/jci.insight.120474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu T., Töpfer K., Lin S.W., Li H., Bian A., Zhou X.Y., High K.A., Ertl H.C.J. Self-complementary AAVs induce more potent transgene product-specific immune responses compared to a single-stranded genome. Mol. Ther. 2012;20:572–579. doi: 10.1038/mt.2011.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keiser M.S., Ranum P.T., Yrigollen C.M., Carrell E.M., Smith G.R., Muehlmatt A.L., Chen Y.H., Stein J.M., Wolf R.L., Radaelli E., et al. Toxicity after AAV delivery of RNAi expression constructs into nonhuman primate brain. Nat. Med. 2021;27:1982–1989. doi: 10.1038/s41591-021-01522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muhuri M., Maeda Y., Ma H., Ram S., Fitzgerald K.A., Tai P.W., Gao G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Invest. 2021;131:143780. doi: 10.1172/JCI143780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fitzpatrick Z., Leborgne C., Barbon E., Masat E., Ronzitti G., van Wittenberghe L., Vignaud A., Collaud F., Charles S., Simon Sola M., et al. Influence of pre-existing anti-capsid neutralizing and binding antibodies on AAV vector transduction. Mol. Ther. Methods Clin. Dev. 2018;9:119–129. doi: 10.1016/j.omtm.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsic D., Govindasamy L., Currlin S., Markusic D.M., Tseng Y.S., Herzog R.W., Agbandje-McKenna M., Zolotukhin S. Vector design Tour de Force: integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol. Ther. 2014;22:1900–1909. doi: 10.1038/mt.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lochrie M.A., Tatsuno G.P., Christie B., McDonnell J.W., Zhou S., Surosky R., Pierce G.F., Colosi P. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J. Virol. 2006;80:821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perabo L., Endell J., King S., Lux K., Goldnau D., Hallek M., Büning H. Combinatorial engineering of a gene therapy vector: directed evolution of adeno-associated virus. J. Gene. Med. 2006;8:155–162. doi: 10.1002/jgm.849. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y., Yang C., Hu H., Chen C., Yan M., Ling F., Wang K.C., Wang X., Deng Z., Zhou X., et al. Directed evolution of adeno-associated virus 5 capsid enables specific liver tropism. Mol. Ther. Nucleic Acids. 2022;28:293–306. doi: 10.1016/j.omtn.2022.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ertl H.C.J. T cell-mediated immune responses to AAV and AAV vectors. Front. Immunol. 2021;12:666666. doi: 10.3389/fimmu.2021.666666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J., et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C., He Y., Nicolson S., Hirsch M., Weinberg M.S., Zhang P., Kafri T., Samulski R.J. Adeno-associated virus capsid antigen presentation is dependent on endosomal escape. J. Clin. Invest. 2013;123:1390–1401. doi: 10.1172/JCI66611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuranda K., Jean-Alphonse P., Leborgne C., Hardet R., Collaud F., Marmier S., Costa Verdera H., Ronzitti G., Veron P., Mingozzi F. Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J. Clin. Invest. 2018;128:5267–5279. doi: 10.1172/JCI122372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berrier K.L., Kazi Z.B., Prater S.N., Bali D.S., Goldstein J., Stefanescu M.C., Rehder C.W., Botha E.G., Ellaway C., Bhattacharya K., et al. CRIM-negative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genet. Med. 2015;17:912–918. doi: 10.1038/gim.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herzog R.W. Complexity of immune responses to AAV transgene products - example of factor IX. Cell. Immunol. 2019;342:103658. doi: 10.1016/j.cellimm.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herzog R.W., Yang E.Y., Couto L.B., Hagstrom J.N., Elwell D., Fields P.A., Burton M., Bellinger D.A., Read M.S., Brinkhous K.M., et al. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat. Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- 48.Gorovits B., Clements-Egan A., Birchler M., Liang M., Myler H., Peng K., Purushothama S., Rajadhyaksha M., Salazar-Fontana L., Sung C., Xue L. Pre-existing antibody: biotherapeutic modality-based review. AAPS J. 2016;18:311–320. doi: 10.1208/s12248-016-9878-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hösel M., Broxtermann M., Janicki H., Esser K., Arzberger S., Hartmann P., Gillen S., Kleeff J., Stabenow D., Odenthal M., et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology. 2012;55:287–297. doi: 10.1002/hep.24625. [DOI] [PubMed] [Google Scholar]
- 50.Calcedo R., Vandenberghe L.H., Gao G., Lin J., Wilson J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Louis Jeune V., Joergensen J.A., Hajjar R.J., Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods. 2013;24:59–67. doi: 10.1089/hgtb.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kotterman M.A., Schaffer D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erles K., Sebökovà P., Schlehofer J.R. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV) J. Med. Virol. 1999;59:406–411. doi: 10.1002/(sici)1096-9071(199911)59:3<406::aid-jmv22>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 54.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E.J., Ragni M.V., Manno C.S., Sommer J., Jiang H., et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 55.Pien G.C., Basner-Tschakarjan E., Hui D.J., Mentlik A.N., Finn J.D., Hasbrouck N.C., Zhou S., Murphy S.L., Maus M.V., Mingozzi F., et al. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J. Clin. Invest. 2009;119:1688–1695. doi: 10.1172/JCI36891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shirley J.L., Keeler G.D., Sherman A., Zolotukhin I., Markusic D.M., Hoffman B.E., Morel L.M., Wallet M.A., Terhorst C., Herzog R.W. Type I IFN sensing by cDCs and CD4(+) T cell help are both requisite for cross-priming of AAV capsid-specific CD8(+) T cells. Mol. Ther. 2020;28:758–770. doi: 10.1016/j.ymthe.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boisgerault F., Mingozzi F. The skeletal muscle environment and its role in immunity and tolerance to AAV vector-mediated gene transfer. Curr. Gene. Ther. 2015;15:381–394. doi: 10.2174/1566523215666150630121750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao O., Dobrzynski E., Wang L., Nayak S., Mingle B., Terhorst C., Herzog R.W. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reichel F.F., Peters T., Wilhelm B., Biel M., Ueffing M., Wissinger B., Bartz-Schmidt K.U., Klein R., Michalakis S., Fischer M.D., RD-CURE Consortium Humoral immune response after intravitreal but not after subretinal AAV8 in primates and patients. Invest. Ophthalmol. Vis. Sci. 2018;59:1910–1915. doi: 10.1167/iovs.17-22494. [DOI] [PubMed] [Google Scholar]
- 60.Konkle B.A., Walsh C.E., Escobar M.A., Josephson N.C., Young G., von Drygalski A., McPhee S.W.J., Samulski R.J., Bilic I., de la Rosa M., et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. 2021;137:763–774. doi: 10.1182/blood.2019004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rogers G.L., Suzuki M., Zolotukhin I., Markusic D.M., Morel L.M., Lee B., Ertl H.C.J., Herzog R.W. Unique roles of TLR9- and MyD88-dependent and -independent pathways in adaptive immune responses to AAV-mediated gene transfer. J. Innate Immun. 2015;7:302–314. doi: 10.1159/000369273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martino A.T., Suzuki M., Markusic D.M., Zolotukhin I., Ryals R.C., Moghimi B., Ertl H.C.J., Muruve D.A., Lee B., Herzog R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood. 2011;117:6459–6468. doi: 10.1182/blood-2010-10-314518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun B., Zhang H., Franco L.M., Brown T., Bird A., Schneider A., Koeberl D.D. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol. Ther. 2005;11:889–898. doi: 10.1016/j.ymthe.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 64.Herzog R.W., Cooper M., Perrin G.Q., Biswas M., Martino A.T., Morel L., Terhorst C., Hoffman B.E. Regulatory T cells and TLR9 activation shape antibody formation to a secreted transgene product in AAV muscle gene transfer. Cell. Immunol. 2019;342:103682. doi: 10.1016/j.cellimm.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L., Cao O., Swalm B., Dobrzynski E., Mingozzi F., Herzog R.W. Major role of local immune responses in antibody formation to factor IX in AAV gene transfer. Gene. Ther. 2005;12:1453–1464. doi: 10.1038/sj.gt.3302539. [DOI] [PubMed] [Google Scholar]
- 66.Kumar S.R.P., Hoffman B.E., Terhorst C., de Jong Y.P., Herzog R.W. The balance between CD8(+) T cell-mediated clearance of AAV-encoded antigen in the liver and tolerance is dependent on the vector dose. Mol. Ther. 2017;25:880–891. doi: 10.1016/j.ymthe.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Herzog R.W., Fields P.A., Arruda V.R., Brubaker J.O., Armstrong E., McClintock D., Bellinger D.A., Couto L.B., Nichols T.C., High K.A. Influence of vector dose on factor IX-specific T and B cell responses in muscle-directed gene therapy. Hum. Gene. Ther. 2002;13:1281–1291. doi: 10.1089/104303402760128513. [DOI] [PubMed] [Google Scholar]
- 68.Yiu G., Chung S.H., Mollhoff I.N., Nguyen U.T., Thomasy S.M., Yoo J., Taraborelli D., Noronha G. Suprachoroidal and subretinal injections of AAV using transscleral microneedles for retinal gene delivery in nonhuman primates. Mol. Ther. Methods Clin. Dev. 2020;16:179–191. doi: 10.1016/j.omtm.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mingozzi F., Liu Y.L., Dobrzynski E., Kaufhold A., Liu J.H., Wang Y., Arruda V.R., High K.A., Herzog R.W. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J. Clin. Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wright J.F. Product-related impurities in clinical-grade recombinant AAV vectors: characterization and risk assessment. Biomedicines. 2014;2:80–97. doi: 10.3390/biomedicines2010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kishimoto T.K., Samulski R.J. Addressing high dose AAV toxicity - 'one and done' or 'slower and lower'? Expert Opin. Biol. Ther. 2022:1–5. doi: 10.1080/14712598.2022.2060737. [DOI] [PubMed] [Google Scholar]
- 72.Hui D.J., Edmonson S.C., Podsakoff G.M., Pien G.C., Ivanciu L., Camire R.M., Ertl H., Mingozzi F., High K.A., Basner-Tschakarjan E. AAV capsid CD8+ T-cell epitopes are highly conserved across AAV serotypes. Mol. Ther. Methods Clin. Dev. 2015;2:15029. doi: 10.1038/mtm.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka T., Narazaki M., Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014;6:a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li C., Narkbunnam N., Samulski R.J., Asokan A., Hu G., Jacobson L.J., Manco-Johnson M.J., Monahan P.E., Joint Outcome Study Investigators Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene. Ther. 2012;19:288–294. doi: 10.1038/gt.2011.90. [DOI] [PubMed] [Google Scholar]
- 75.Calcedo R., Morizono H., Wang L., McCarter R., He J., Jones D., Batshaw M.L., Wilson J.M. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin. Vaccine Immunol. 2011;18:1586–1588. doi: 10.1128/CVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cole L., Fernandes D., Hussain M.T., Kaszuba M., Stenson J., Markova N. Characterization of recombinant adeno-associated viruses (rAAVs) for gene therapy using orthogonal techniques. Pharmaceutics. 2021;13:586. doi: 10.3390/pharmaceutics13040586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nathwani A.C., Tuddenham E.G.D., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C., et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bing S.J., Justesen S., Wu W.W., Sajib A.M., Warrington S., Baer A., Thorgrimsen S., Shen R.-F., Mazor R. Differential T cell immune responses to deamidated adeno-associated virus vector. Mol. Ther. Methods Clin. Dev. 2022;24:255–267. doi: 10.1016/j.omtm.2022.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dobrzynski E., Mingozzi F., Liu Y.L., Bendo E., Cao O., Wang L., Herzog R.W. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood. 2004;104:969–977. doi: 10.1182/blood-2004-03-0847. [DOI] [PubMed] [Google Scholar]
- 80.Franco L.M., Sun B., Yang X., Bird A., Zhang H., Schneider A., Brown T., Young S.P., Clay T.M., Amalfitano A., et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol. Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 81.Ziegler R.J., Lonning S.M., Armentano D., Li C., Souza D.W., Cherry M., Ford C., Barbon C.M., Desnick R.J., Gao G., et al. AAV2 vector harboring a liver-restricted promoter facilitates sustained expression of therapeutic levels of alpha-galactosidase A and the induction of immune tolerance in Fabry mice. Mol. Ther. 2004;9:231–240. doi: 10.1016/j.ymthe.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 82.Martino A.T., Nayak S., Hoffman B.E., Cooper M., Liao G., Markusic D.M., Byrne B.J., Terhorst C., Herzog R.W. Tolerance induction to cytoplasmic beta-galactosidase by hepatic AAV gene transfer: implications for antigen presentation and immunotoxicity. PLoS One. 2009;4:e6376. doi: 10.1371/journal.pone.0006376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chand D., Mohr F., McMillan H., Tukov F.F., Montgomery K., Kleyn A., Sun R., Tauscher-Wisniewski S., Kaufmann P., Kullak-Ublick G. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J. Hepatol. 2021;74:560–566. doi: 10.1016/j.jhep.2020.11.001. [DOI] [PubMed] [Google Scholar]
- 85.Chand D.H. 2021. Clinical Findings of Thrombotic Microangiopathy (TMA) [Google Scholar]
- 86.Paulk N.K. 2020. Gene Therapy: It's Time to Talk about High-Dose AAV. [Google Scholar]
- 87.Administration U.S.F.D. 2021. Toxicity Risks of Adeno-Associated Virus (AAV) Vectors for Gene Therapy (GT) pp. 1–47. [Google Scholar]
- 88.Chand D.H., Zaidman C., Arya K., Millner R., Farrar M.A., Mackie F.E., Goedeker N.L., Dharnidharka V.R., Dandamudi R., Reyna S.P. Thrombotic microangiopathy following onasemnogene abeparvovec for spinal muscular atrophy: a case series. J. Pediatr. 2021;231:265–268. doi: 10.1016/j.jpeds.2020.11.054. [DOI] [PubMed] [Google Scholar]
- 89.High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020;38:910. doi: 10.1038/s41587-020-0642-9. [DOI] [PubMed] [Google Scholar]
- 90.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene. Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bolt M.W., Brady J.T., Whiteley L.O., Khan K.N. Development challenges associated with rAAV-based gene therapies. J. Toxicol. Sci. 2021;46:57–68. doi: 10.2131/jts.46.57. [DOI] [PubMed] [Google Scholar]
- 92.Hordeaux J., Buza E.L., Dyer C., Goode T., Mitchell T.W., Richman L., Denton N., Hinderer C., Katz N., Schmid R., et al. Adeno-associated virus-induced dorsal root ganglion pathology. Hum. Gene. Ther. 2020;31:808–818. doi: 10.1089/hum.2020.167. [DOI] [PubMed] [Google Scholar]
- 93.Hordeaux J., Buza E.L., Jeffrey B., Song C., Jahan T., Yuan Y., Zhu Y., Bell P., Li M., Chichester J.A., et al. MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates. Sci. Transl. Med. 2020;12:eaba9188. doi: 10.1126/scitranslmed.aba9188. [DOI] [PubMed] [Google Scholar]
- 94.Hordeaux J., Hinderer C., Buza E.L., Louboutin J.P., Jahan T., Bell P., Chichester J.A., Tarantal A.F., Wilson J.M. Safe and sustained expression of human iduronidase after intrathecal administration of adeno-associated virus serotype 9 in infant rhesus monkeys. Hum. Gene. Ther. 2019;30:957–966. doi: 10.1089/hum.2019.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ertl H.C.J., High K.A. Impact of AAV capsid-specific T-cell responses on design and outcome of clinical gene transfer trials with recombinant adeno-associated viral vectors: an evolving controversy. Hum. Gene. Ther. 2017;28:328–337. doi: 10.1089/hum.2016.172. [DOI] [PubMed] [Google Scholar]
- 96.Mingozzi F., High K.A. Overcoming the host immune response to adeno-associated virus gene delivery vectors: the race between clearance, tolerance, neutralization, and escape. Annu. Rev. Virol. 2017;4:511–534. doi: 10.1146/annurev-virology-101416-041936. [DOI] [PubMed] [Google Scholar]
- 97.Tabebordbar M., Lagerborg K.A., Stanton A., King E.M., Ye S., Tellez L., Krunnfusz A., Tavakoli S., Widrick J.J., Messemer K.A., et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell. 2021;184:4919–4938.e22. doi: 10.1016/j.cell.2021.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khabou H., Cordeau C., Pacot L., Fisson S., Dalkara D. Dosage thresholds and influence of transgene cassette in adeno-associated virus-related toxicity. Hum. Gene. Ther. 2018;29:1235–1241. doi: 10.1089/hum.2018.144. [DOI] [PubMed] [Google Scholar]
- 99.Klein R.L., Dayton R.D., Leidenheimer N.J., Jansen K., Golde T.E., Zweig R.M. Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol. Ther. 2006;13:517–527. doi: 10.1016/j.ymthe.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ertl H.C.J. Preclinical models to assess the immunogenicity of AAV vectors. Cell. Immunol. 2019;342:103722. doi: 10.1016/j.cellimm.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 101.Martino A.T., Markusic D.M. Immune response mechanisms against AAV vectors in animal models. Mol. Ther. Methods Clin. Dev. 2020;17:198–208. doi: 10.1016/j.omtm.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wright J.F. Codon modification and PAMPs in clinical AAV vectors: the tortoise or the hare? Mol. Ther. 2020;28:701–703. doi: 10.1016/j.ymthe.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wright J.F. Quantification of CpG motifs in rAAV genomes: avoiding the Toll. Mol. Ther. 2020;28:1756–1758. doi: 10.1016/j.ymthe.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Armant M.A., Fenton M.J. Toll-like receptors: a family of pattern-recognition receptors in mammals. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-8-reviews3011. REVIEWS3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang L., Calcedo R., Bell P., Lin J., Grant R.L., Siegel D.L., Wilson J.M. Impact of pre-existing immunity on gene transfer to nonhuman primate liver with adeno-associated virus 8 vectors. Hum. Gene Ther. 2011;22:1389–1401. doi: 10.1089/hum.2011.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Davidoff A.M., Gray J.T., Ng C.Y.C., Zhang Y., Zhou J., Spence Y., Bakar Y., Nathwani A.C. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol. Ther. 2005;11:875–888. doi: 10.1016/j.ymthe.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 107.Long B.R., Sandza K., Holcomb J., Crockett L., Hayes G.M., Arens J., Fonck C., Tsuruda L.S., Schweighardt B., O'Neill C.A., et al. The impact of pre-existing immunity on the non-clinical pharmacodynamics of AAV5-based gene therapy. Mol. Ther. Methods Clin. Dev. 2019;13:440–452. doi: 10.1016/j.omtm.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nathwani A.C., Rosales C., McIntosh J., Rastegarlari G., Nathwani D., Raj D., Nawathe S., Waddington S.N., Bronson R., Jackson S., et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol. Ther. 2011;19:876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li H., Murphy S.L., Giles-Davis W., Edmonson S., Xiang Z., Li Y., Lasaro M.O., High K.A., Ertl H.C. Pre-existing AAV capsid-specific CD8+ T cells are unable to eliminate AAV-transduced hepatocytes. Mol. Ther. 2007;15:792–800. doi: 10.1038/sj.mt.6300090. [DOI] [PubMed] [Google Scholar]
- 110.Jiang H., Couto L.B., Patarroyo-White S., Liu T., Nagy D., Vargas J.A., Zhou S., Scallan C.D., Sommer J., Vijay S., et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108:3321–3328. doi: 10.1182/blood-2006-04-017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li H., Lasaro M.O., Jia B., Lin S.W., Haut L.H., High K.A., Ertl H.C.J. Capsid-specific T-cell responses to natural infections with adeno-associated viruses in humans differ from those of nonhuman primates. Mol. Ther. 2011;19:2021–2030. doi: 10.1038/mt.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Basner-Tschakarjan E., Bijjiga E., Martino A.T. Pre-clinical assessment of immune responses to adeno-associated virus (AAV) vectors. Front. Immunol. 2014;5:28. doi: 10.3389/fimmu.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tummala G., Crain A., Rowlan J., Pepple K.L. Characterization of gene therapy associated uveitis following intravitreal adeno-associated virus injection in mice. Invest. Ophthalmol. Vis. Sci. 2021;62:41. doi: 10.1167/iovs.62.2.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Calcedo R., Franco J., Qin Q., Richardson D.W., Mason J.B., Boyd S., Wilson J.M. Preexisting neutralizing antibodies to adeno-associated virus capsids in large animals other than monkeys may confound in vivo gene therapy studies. Hum. Gene Ther. Methods. 2015;26:103–105. doi: 10.1089/hgtb.2015.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xiao W., Gao G., Ling C., Herzog R.W., Xiao X., Samulski R.J. Impact of neutralizing antibodies against AAV is a key consideration in gene transfer to nonhuman primates. Nat. Med. 2018;24:699. doi: 10.1038/s41591-018-0062-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Assaf B.T., Whiteley L.O. Considerations for preclinical safety assessment of adeno-associated virus gene therapy products. Toxicol. Pathol. 2018;46:1020–1027. doi: 10.1177/0192623318803867. [DOI] [PubMed] [Google Scholar]
- 117.Vandamme C., Adjali O., Mingozzi F. Unraveling the complex story of immune responses to AAV vectors trial after trial. Hum. Gene Ther. 2017;28:1061–1074. doi: 10.1089/hum.2017.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weber T. Anti-AAV antibodies in AAV gene therapy: current challenges and possible solutions. Front. Immunol. 2021;12:658399. doi: 10.3389/fimmu.2021.658399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Long B.R., Veron P., Kuranda K., Hardet R., Mitchell N., Hayes G.M., Wong W.Y., Lau K., Li M., Hock M.B., et al. Early phase clinical immunogenicity of valoctocogene roxaparvovec, an AAV5-mediated gene therapy for hemophilia A. Mol. Ther. 2021;29:597–610. doi: 10.1016/j.ymthe.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Flotte T.R., Trapnell B.C., Humphries M., Carey B., Calcedo R., Rouhani F., Campbell-Thompson M., Yachnis A.T., Sandhaus R.A., McElvaney N.G., et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: interim results. Hum. Gene Ther. 2011;22:1239–1247. doi: 10.1089/hum.2011.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mingozzi F., Meulenberg J.J., Hui D.J., Basner-Tschakarjan E., Hasbrouck N.C., Edmonson S.A., Hutnick N.A., Betts M.R., Kastelein J.J., Stroes E.S., High K.A. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114:2077–2086. doi: 10.1182/blood-2008-07-167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Doshi B.S., Arruda V.R. Gene therapy for hemophilia: what does the future hold? Ther. Adv. Hematol. 2018;9:273–293. doi: 10.1177/2040620718791933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwäble J., Bonig H., et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gernoux G., Gruntman A.M., Blackwood M., Zieger M., Flotte T.R., Mueller C. Muscle-directed delivery of an AAV1 vector leads to capsid-specific T cell exhaustion in nonhuman primates and humans. Mol. Ther. 2020;28:747–757. doi: 10.1016/j.ymthe.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mueller C., Chulay J.D., Trapnell B.C., Humphries M., Carey B., Sandhaus R.A., McElvaney N.G., Messina L., Tang Q., Rouhani F.N., et al. Human Treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J. Clin. Invest. 2013;123:5310–5318. doi: 10.1172/JCI70314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wright J.F. Quality control testing, characterization and critical quality attributes of adeno-associated virus vectors used for human gene therapy. Biotechnol. J. 2021;16:e2000022. doi: 10.1002/biot.202000022. [DOI] [PubMed] [Google Scholar]
- 127.Chan Y.K., Wang S.K., Chu C.J., Copland D.A., Letizia A.J., Costa Verdera H., Chiang J.J., Sethi M., Wang M.K., Neidermyer W.J., Jr., et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021;13:eabd3438. doi: 10.1126/scitranslmed.abd3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Meliani A., Boisgerault F., Fitzpatrick Z., Marmier S., Leborgne C., Collaud F., Simon Sola M., Charles S., Ronzitti G., Vignaud A., et al. Enhanced liver gene transfer and evasion of preexisting humoral immunity with exosome-enveloped AAV vectors. Blood Adv. 2017;1:2019–2031. doi: 10.1182/bloodadvances.2017010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chicoine L.G., Montgomery C.L., Bremer W.G., Shontz K.M., Griffin D.A., Heller K.N., Lewis S., Malik V., Grose W.E., Shilling C.J., et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol. Ther. 2014;22:338–347. doi: 10.1038/mt.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Monteilhet V., Saheb S., Boutin S., Leborgne C., Veron P., Montus M.F., Moullier P., Benveniste O., Masurier C. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol. Ther. 2011;19:2084–2091. doi: 10.1038/mt.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Elmore Z.C., Oh D.K., Simon K.E., Fanous M.M., Asokan A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight. 2020;5:139881. doi: 10.1172/jci.insight.139881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leborgne C., Barbon E., Alexander J.M., Hanby H., Delignat S., Cohen D.M., Collaud F., Muraleetharan S., Lupo D., Silverberg J., et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020;26:1096–1101. doi: 10.1038/s41591-020-0911-7. [DOI] [PubMed] [Google Scholar]
- 133.Lonze B.E., Tatapudi V.S., Weldon E.P., Min E.S., Ali N.M., Deterville C.L., Gelb B.E., Benstein J.A., Dagher N.N., Wu M., Montgomery R.A. IdeS (imlifidase): a novel agent that cleaves human IgG and permits successful kidney transplantation across high-strength donor-specific antibody. Ann. Surg. 2018;268:488–496. doi: 10.1097/SLA.0000000000002924. [DOI] [PubMed] [Google Scholar]
- 134.Chu W.S., Ng J. Immunomodulation in administration of rAAV: preclinical and clinical adjuvant pharmacotherapies. Front. Immunol. 2021;12:658038. doi: 10.3389/fimmu.2021.658038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zelek W.M., Xie L., Morgan B.P., Harris C.L. Compendium of current complement therapeutics. Mol. Immunol. 2019;114:341–352. doi: 10.1016/j.molimm.2019.07.030. [DOI] [PubMed] [Google Scholar]
- 136.Limon J.J., So L., Jellbauer S., Chiu H., Corado J., Sykes S.M., Raffatellu M., Fruman D.A. mTOR kinase inhibitors promote antibody class switching via mTORC2 inhibition. Proc. Natl. Acad. Sci. USA. 2014;111:E5076–E5085. doi: 10.1073/pnas.1407104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Delgoffe G.M., Kole T.P., Zheng Y., Zarek P.E., Matthews K.L., Xiao B., Worley P.F., Kozma S.C., Powell J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zeiser R., Leveson-Gower D.B., Zambricki E.A., Kambham N., Beilhack A., Loh J., Hou J.Z., Negrin R.S. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111:453–462. doi: 10.1182/blood-2007-06-094482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Velazquez V.M., Meadows A.S., Pineda R.J., Camboni M., McCarty D.M., Fu H. Effective depletion of pre-existing anti-AAV antibodies requires broad immune targeting. Mol. Ther. Methods Clin. Dev. 2017;4:159–168. doi: 10.1016/j.omtm.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K., et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 142.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shieh P.B., Bönnemann C.G., Müller-Felber W., Blaschek A., Dowling J.J., Kuntz N.L., Seferian A.M. Re: "moving forward after two deaths in a gene therapy trial of myotubular myopathy" by Wilson and flotte. Hum. Gene Ther. 2020;31:787. doi: 10.1089/hum.2020.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mueller C., Berry J.D., McKenna-Yasek D.M., Gernoux G., Owegi M.A., Pothier L.M., Douthwright C.L., Gelevski D., Luppino S.D., Blackwood M., et al. SOD1 suppression with adeno-associated virus and MicroRNA in familial ALS. N. Engl. J. Med. 2020;383:151–158. doi: 10.1056/NEJMoa2005056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Allison A.C. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14(Suppl 1) doi: 10.1191/0961203305lu2109oa. s2-8. [DOI] [PubMed] [Google Scholar]
- 146.de Castro M.J., Del Toro M., Giugliani R., Couce M.L. Gene therapy for neuronopathic mucopolysaccharidoses: state of the art. Int. J. Mol. Sci. 2021;22:9200. doi: 10.3390/ijms22179200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Montenegro-Miranda P.S., ten Bloemendaal L., Kunne C., de Waart D.R., Bosma P.J. Mycophenolate mofetil impairs transduction of single-stranded adeno-associated viral vectors. Hum. Gene Ther. 2011;22:605–612. doi: 10.1089/hum.2010.222. [DOI] [PubMed] [Google Scholar]
- 148.Miroux C., Morales O., Ghazal K., Othman S.B., de Launoit Y., Pancré V., Conti F., Delhem N. In vitro effects of cyclosporine A and tacrolimus on regulatory T-cell proliferation and function. Transplantation. 2012;94:123–131. doi: 10.1097/TP.0b013e3182590d8f. [DOI] [PubMed] [Google Scholar]
- 149.Akimova T., Kamath B.M., Goebel J.W., Meyers K.E.C., Rand E.B., Hawkins A., Levine M.H., Bucuvalas J.C., Hancock W.W. Differing effects of rapamycin or calcineurin inhibitor on T-regulatory cells in pediatric liver and kidney transplant recipients. Am. J. Transplant. 2012;12:3449–3461. doi: 10.1111/j.1600-6143.2012.04269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gaudet D., Stroes E.S., Méthot J., Brisson D., Tremblay K., Bernelot Moens S.J., Iotti G., Rastelletti I., Ardigo D., Corzo D., et al. Long-term retrospective analysis of gene therapy with alipogene tiparvovec and its effect on lipoprotein lipase deficiency-induced pancreatitis. Hum. Gene Ther. 2016;27:916–925. doi: 10.1089/hum.2015.158. [DOI] [PubMed] [Google Scholar]
- 151.Gaudet D., Méthot J., Déry S., Brisson D., Essiembre C., Tremblay G., Tremblay K., de Wal J., Twisk J., van den Bulk N., et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–369. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ferreira V., Petry H., Salmon F. Immune responses to AAV-vectors, the glybera example from bench to bedside. Front. Immunol. 2014;5:82. doi: 10.3389/fimmu.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Frentsch M., Japp A.S., Dingeldey M., Matzmohr N., Thiel A., Scheiflinger F., Reipert B.M., de la Rosa M. Blockade of the costimulatory CD28-B7 family signal axis enables repeated application of AAV8 gene vectors. J. Thromb. Haemost. 2020;18:1075–1080. doi: 10.1111/jth.14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Corti M., Liberati C., Smith B.K., Lawson L.A., Tuna I.S., Conlon T.J., Coleman K.E., Islam S., Herzog R.W., Fuller D.D., et al. Safety of intradiaphragmatic delivery of adeno-associated virus-mediated alpha-glucosidase (rAAV1-CMV-hGAA) gene therapy in children affected by Pompe disease. Hum. Gene Ther. Clin. Dev. 2017;28:208–218. doi: 10.1089/humc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schrezenmeier E., Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol. 2020;16:155–166. doi: 10.1038/s41584-020-0372-x. [DOI] [PubMed] [Google Scholar]
- 156.Chandler L.C., Barnard A.R., Caddy S.L., Patrício M.I., McClements M.E., Fu H., Rada C., MacLaren R.E., Xue K. Enhancement of adeno-associated virus-mediated gene therapy using hydroxychloroquine in murine and human tissues. Mol. Ther. Methods Clin. Dev. 2019;14:77–89. doi: 10.1016/j.omtm.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee J.W., Sicre de Fontbrune F., Wong Lee Lee L., Pessoa V., Gualandro S., Füreder W., Ptushkin V., Rottinghaus S.T., Volles L., Shafner L., et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133:530–539. doi: 10.1182/blood-2018-09-876136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McKeage K. Ravulizumab: first global approval. Drugs. 2019;79:347–352. doi: 10.1007/s40265-019-01068-2. [DOI] [PubMed] [Google Scholar]
- 159.Legendre C.M., Licht C., Muus P., Greenbaum L.A., Babu S., Bedrosian C., Bingham C., Cohen D.J., Delmas Y., Douglas K., et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 160.Łoboda A., Dulak J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future. Pharmacol. Rep. 2020;72:1227–1263. doi: 10.1007/s43440-020-00134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Finn J.D., Hui D., Downey H.D., Dunn D., Pien G.C., Mingozzi F., Zhou S., High K.A. Proteasome inhibitors decrease AAV2 capsid derived peptide epitope presentation on MHC class I following transduction. Mol. Ther. 2010;18:135–142. doi: 10.1038/mt.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Naujokat C., Berges C., Höh A., Wieczorek H., Fuchs D., Ovens J., Miltz M., Sadeghi M., Opelz G., Daniel V. Proteasomal chymotrypsin-like peptidase activity is required for essential functions of human monocyte-derived dendritic cells. Immunology. 2007;120:120–132. doi: 10.1111/j.1365-2567.2006.02487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mitchell A.M., Samulski R.J. Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. J. Virol. 2013;87:13035–13041. doi: 10.1128/JVI.01826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Monahan P.E., Lothrop C.D., Sun J., Hirsch M.L., Kafri T., Kantor B., Sarkar R., Tillson D.M., Elia J.R., Samulski R.J. Proteasome inhibitors enhance gene delivery by AAV virus vectors expressing large genomes in hemophilia mouse and dog models: a strategy for broad clinical application. Mol. Ther. 2010;18:1907–1916. doi: 10.1038/mt.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Keeler G.D., Kumar S., Palaschak B., Silverberg E.L., Markusic D.M., Jones N.T., Hoffman B.E. Gene therapy-induced antigen-specific Tregs inhibit neuro-inflammation and reverse disease in a mouse model of multiple sclerosis. Mol. Ther. 2018;26:173–183. doi: 10.1016/j.ymthe.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Röth A., Barcellini W., D'Sa S., Miyakawa Y., Broome C.M., Michel M., Kuter D.J., Jilma B., Tvedt T.H.A., Fruebis J., et al. Sutimlimab in cold agglutinin disease. N. Engl. J. Med. 2021;384:1323–1334. doi: 10.1056/NEJMoa2027760. [DOI] [PubMed] [Google Scholar]
- 167.Qiu J.G., Mei X.L., Chen Z.S., Shi Z. Cytokine detection by flow cytometry. Methods Mol. Biol. 2014;1172:235–242. doi: 10.1007/978-1-4939-0928-5_21. [DOI] [PubMed] [Google Scholar]
- 168.Forlenza M., Kaiser T., Savelkoul H.F.J., Wiegertjes G.F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol. Biol. 2012;820:7–23. doi: 10.1007/978-1-61779-439-1_2. [DOI] [PubMed] [Google Scholar]
- 169.Kirschfink M., Mollnes T.E. Modern complement analysis. Clin. Diagn. Lab. Immunol. 2003;10:982–989. doi: 10.1128/cdli.10.6.982-989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gorovits B., Azadeh M., Buchlis G., Harrison T., Havert M., Jawa V., Long B., McNally J., Milton M., Nelson R., et al. Evaluation of the humoral response to adeno-associated virus-based gene therapy modalities using total antibody assays. AAPS J. 2021;23:108. doi: 10.1208/s12248-021-00628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gorovits B., Fiscella M., Havert M., Koren E., Long B., Milton M., Purushothama S. Recommendations for the development of cell-based anti-viral vector neutralizing antibody assays. AAPS J. 2020;22:24. doi: 10.1208/s12248-019-0403-1. [DOI] [PubMed] [Google Scholar]
- 172.Baatartsogt N., Kashiwakura Y., Hayakawa M., Kamoshita N., Hiramoto T., Mizukami H., Ohmori T. A sensitive and reproducible cell-based assay via secNanoLuc to detect neutralizing antibody against adeno-associated virus vector capsid. Mol. Ther. Methods Clin. Dev. 2021;22:162–171. doi: 10.1016/j.omtm.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kasprzyk T., Triffault S., Long B.R., Zoog S.J., Vettermann C. Confirmatory detection of neutralizing antibodies to AAV gene therapy using a cell-based transduction inhibition assay. Mol. Ther. Methods Clin. Dev. 2022;24:222–229. doi: 10.1016/j.omtm.2022.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Meliani A., Leborgne C., Triffault S., Jeanson-Leh L., Veron P., Mingozzi F. Determination of anti-adeno-associated virus vector neutralizing antibody titer with an in vitro reporter system. Hum. Gene Ther. Methods. 2015;26:45–53. doi: 10.1089/hgtb.2015.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yang F., Patton K., Kasprzyk T., Long B., Gupta S., Zoog S.J., Tracy K., Vettermann C. Validation of an IFN-gamma ELISpot assay to measure cellular immune responses against viral antigens in non-human primates. Gene. Ther. 2022;29:41–54. doi: 10.1038/s41434-020-00214-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vandamme C., Xicluna R., Hesnard L., Devaux M., Jaulin N., Guilbaud M., Le Duff J., Couzinié C., Moullier P., Saulquin X., Adjali O. Tetramer-based enrichment of preexisting anti-AAV8 CD8(+) T cells in human donors allows the detection of a TEMRA subpopulation. Front. Immunol. 2019;10:3110. doi: 10.3389/fimmu.2019.03110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Partridge M.A., Purushothama S., Elango C., Lu Y. Emerging technologies and generic assays for the detection of anti-drug antibodies. J. Immunol. Res. 2016;2016:6262383. doi: 10.1155/2016/6262383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu B., Chung S., Jiang X.R., McNally J., Pedras-Vasconcelos J., Pillutla R., White J.T., Xu Y., Gupta S. Strategies to determine assay format for the assessment of neutralizing antibody responses to biotherapeutics. AAPS J. 2016;18:1335–1350. doi: 10.1208/s12248-016-9954-6. [DOI] [PubMed] [Google Scholar]
- 179.Gupta S., Indelicato S.R., Jethwa V., Kawabata T., Kelley M., Mire-Sluis A.R., Richards S.M., Rup B., Shores E., Swanson S.J., Wakshull E. Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J. Immunol. Methods. 2007;321:1–18. doi: 10.1016/j.jim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 180.Séllos-Moura M., Barzegar S., Pan L., Shi P., Oommen S., Durant J., Ruiz J.A. Development of a panel of highly sensitive, equivalent assays for detection of antibody responses to velaglucerase alfa or imiglucerase enzyme replacement therapy in patients with Gaucher disease. J. Immunol. Methods. 2011;373:45–53. doi: 10.1016/j.jim.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 181.White J.T., Argento Martell L., Prince W.S., Boyer R., Crockett L., Cox C., Van Tuyl A., Aguilera A., Foehr E. Comparison of neutralizing antibody assays for receptor binding and enzyme activity of the enzyme replacement therapeutic Naglazyme (galsulfase) AAPS J. 2008;10:439–449. doi: 10.1208/s12248-008-9048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wessels U., Zadak M., Reiser A., Brockhaus J., Ritter M., Abdolzade-Bavil A., Heinrich J., Stubenrauch K. Immunogenicity testing of therapeutic antibodies in ocular fluids after intravitreal injection. Bioanalysis. 2018;10:803–814. doi: 10.4155/bio-2018-0047. [DOI] [PubMed] [Google Scholar]
- 183.Farber D.L., Yudanin N.A., Restifo N.P. Human memory T cells: generation, compartmentalization and homeostasis. Nat. Rev. Immunol. 2014;14:24–35. doi: 10.1038/nri3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Patton K.S., Harrison M.T., Long B.R., Lau K., Holcomb J., Owen R., Kasprzyk T., Janetzki S., Zoog S.J., Vettermann C. Monitoring cell-mediated immune responses in AAV gene therapy clinical trials using a validated IFN-gamma ELISpot method. Mol. Ther. Methods Clin. Dev. 2021;22:183–195. doi: 10.1016/j.omtm.2021.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Arnett A.L.H., Beutler L.R., Quintana A., Allen J., Finn E., Palmiter R.D., Chamberlain J.S. Heparin-binding correlates with increased efficiency of AAV1- and AAV6-mediated transduction of striated muscle, but negatively impacts CNS transduction. Gene. Ther. 2013;20:497–503. doi: 10.1038/gt.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cabanes-Creus M., Westhaus A., Navarro R.G., Baltazar G., Zhu E., Amaya A.K., Liao S.H.Y., Scott S., Sallard E., Dilworth K.L., et al. Attenuation of heparan sulfate proteoglycan binding enhances in vivo transduction of human primary hepatocytes with AAV2. Mol. Ther. Methods Clin. Dev. 2020;17:1139–1154. doi: 10.1016/j.omtm.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Denard J., Beley C., Kotin R., Lai-Kuen R., Blot S., Leh H., Asokan A., Samulski R.J., Moullier P., Voit T., et al. Human galectin 3 binding protein interacts with recombinant adeno-associated virus type 6. J. Virol. 2012;86:6620–6631. doi: 10.1128/JVI.00297-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Denard J., Marolleau B., Jenny C., Rao T.N., Fehling H.J., Voit T., Svinartchouk F. C-reactive protein (CRP) is essential for efficient systemic transduction of recombinant adeno-associated virus vector 1 (rAAV-1) and rAAV-6 in mice. J. Virol. 2013;87:10784–10791. doi: 10.1128/JVI.01813-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Guo P., Zhang J., Chrzanowski M., Huang J., Chew H., Firrman J.A., Sang N., Diao Y., Xiao W. Rapid AAV-neutralizing antibody determination with a cell-binding assay. Mol. Ther. Methods Clin. Dev. 2019;13:40–46. doi: 10.1016/j.omtm.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cheung R., deHart G.W., Jesaitis L., Zoog S.J., Melton A.C. Detection of antibodies that neutralize the cellular uptake of enzyme replacement therapies with a cell-based assay. J. Vis. Exp. 2018:e57777. doi: 10.3791/57777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang W., Caspell R., Karulin A.Y., Ahmad M., Haicheur N., Abdelsalam A., Johannesen K., Vignard V., Dudzik P., Georgakopoulou K., et al. ELISPOT assays provide reproducible results among different laboratories for T-cell immune monitoring--even in hands of ELISPOT-inexperienced investigators. J. Immunotoxicol. 2009;6:227–234. doi: 10.3109/15476910903317546. [DOI] [PubMed] [Google Scholar]
- 192.Slota M., Lim J.B., Dang Y., Disis M.L. ELISpot for measuring human immune responses to vaccines. Expert Rev. Vaccines. 2011;10:299–306. doi: 10.1586/erv.10.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Allan A.L., Keeney M. Circulating tumor cell analysis: technical and statistical considerations for application to the clinic. J. Oncol. 2010;2010:426218. doi: 10.1155/2010/426218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gazagne A., Claret E., Wijdenes J., Yssel H., Bousquet F., Levy E., Vielh P., Scotte F., Goupil T.L., Fridman W.H., Tartour E. A fluorospot assay to detect single T lymphocytes simultaneously producing multiple cytokines. J. Immunol. Methods. 2003;283:91–98. doi: 10.1016/j.jim.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 195.Malyguine A.M., Strobl S., Dunham K., Shurin M.R., Sayers T.J. ELISPOT assay for monitoring cytotoxic T lymphocytes (CTL) activity in cancer vaccine clinical trials. Cells. 2012;1:111–126. doi: 10.3390/cells1020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wright J.F. AAV empty capsids: for better or for worse? Mol. Ther. 2014;22:1–2. doi: 10.1038/mt.2013.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Falese L., Sandza K., Yates B., Triffault S., Gangar S., Long B., Tsuruda L., Carter B., Vettermann C., Zoog S.J., Fong S. Strategy to detect pre-existing immunity to AAV gene therapy. Gene. Ther. 2017;24:768–778. doi: 10.1038/gt.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]