

Abstract
The development of new active biocompatible materials and devices is a current need for their implementation in multiple fields, including the fabrication of implantable devices for biomedical applications and sustainable devices for bio-optics and bio-optoelectronics. This paper describes a simple strategy to use designed proteins to develop protein-based functional materials. Using simple proteins as self-assembling building blocks as a platform for the fabrication of new optically active materials takes previous work one step further towards the design of materials with defined structures and functions using naturally occurring protein materials, such as silk. The proposed fabrication strategy generates thin and flexible nanopatterned protein films by letting the engineered protein elements self-assemble over the surface of an elastomeric stamp with nanoscale features. These nanopatterned protein films are easily transferred onto 3D objects (flat and curved) by moisture-induced adhesion. Additionally, flexible nanopatterned protein films are prepared by incorporating a thin polymeric layer as a back support. Finally, taking advantage of the tunability of the selected protein scaffold, the flexible protein-based surfaces are endowed with optical functions, achieving efficient lasing features. As such, this work enables the simple and cost-effective production of flexible and nanostructured, protein-based, optically active biomaterials and devices over large areas toward emerging applications.
A simple approach for the fabrication of functional nanopatterned protein materials using protein engineering and soft-nanolithography and its implementation in optical devices based on distributed feedback (DFB) laser phenomena.
Introduction
Active biocompatible materials and devices are emerging as a new cornerstone in multiple sectors including healthcare, energy, lighting, information, computer technology, and environmental monitoring.1–3 A variety of bioinspired strategies have recently emerged to develop universal fabrication methodologies to create new functional biomaterials with improved properties and potential uses.4–7 Bioinspired materials are capable of recreating processes that occur in nature, where complex structures emerge from the combination of small components through self-assembly, and display a broad variety of functionalities. Although these bioinspired fabrication approaches have opened new routes toward the scalable production of biocompatible multifunctional systems in biooptics1 and lighting,1,8 the development of protein-based devices is still in its infancy. Significant progress has been made using proteins that encompass the building blocks of natural protein-based materials, mostly silk fibroin.9 Silk fibroin materials have been fabricated using various strategies, including spin coating, soft lithography, inkjet printing, and contact printing, of silk solutions.10–13 These pioneering studies illustrated the great potential of bioinspired building blocks for the fabrication of active functional materials. However, the use of natural materials usually requires several pre-processing steps, which are often complex and involve costly equipment, hampering their reproducibility in low-resource laboratories.14 Although there is an increasing understanding of the interactions and processes that govern the structuration of biomolecules,15,16 it remains insufficient, and the state-of-the-art research in the field of fabrication of hybrid biomaterials and devices relies on trial-error assays.
Protein engineering enables the creation—in addition to the naturally occurring building blocks—of protein-based synthetic building blocks, and significantly enhances reproducibility rates and facilitates the rational design and assembly of functional biomaterials.4,17 Modular building blocks with simple intermolecular interactions allow for better control of the assembly,18,19 synthesis of different nanomaterials,20 and even 3D structures21 by using simple building blocks with well-described intermolecular interactions, such as coiled-coils and amyloid peptides.22,23 Engineered building blocks enable the control over the organization from the simplest to more complicated structures. This is achieved using a bottom-up strategy based on highly specific biomolecular interactions, which facilitate the nanometer-scale arrangement of bio-components, tailoring their final features and functionalities.24–27 Moreover, engineered proteins display similar biocompatibility, biodegradability, and water-processability to their naturally occurring counterparts. Owing to their easily tunable structure, stability, self-assembly properties, and functional reactivities, engineered proteins constitute a promising biocompatible alternative for the fabrication of the next generation of bioinspired functional materials.
To study how small engineered proteins perform as building blocks for materials engineering, we chose a well-characterized system based on an engineered repeat protein domain, the consensus tetratricopeptide repeat (CTPR) proteins.28 CTPR proteins are formed by a variable number of identical repeated modules, each one comprising 34 amino acids that fold in a helix–turn–helix motif.29,30 Their modular architecture allows the elongation of the basic structural unit by varying the number of repeats. CTPR proteins adopt a superhelical structure with eight repeats per superhelical turn,29–31 whose stability is determined by the number of repeats.32,33 In previous studies, we demonstrated the self-assembly properties of CTPR proteins into nanofibers and thin films, which retained the architectural and functional features of the individual proteins under these conditions.34,35 In particular, CTPR-based solid films are mechanically robust and present hierarchical anisotropic mechanical properties that can be tuned by the constituent molecules.36,37 This adds an extra advantage over other broadly studied biomaterials like, for example, those based on silk fibroin. In addition, we have shown that CTPR-based proteins can encode diverse recognition activities,38–40 and can be further endowed with other functional elements,17,41,42 which can be exploited to introduce new functionalities in the final biomaterial.
In combination with bottom-up self-assembly approaches, it is interesting to explore simple top-down fabrication techniques that are usually applied to polymeric materials, such as replica molding, in the context of protein-based materials. Providing these biocompatible functional materials with nanoscale structures will add an extra level of complexity and open the door to applications that require nanostructuration, such as the development of optically active devices. However, many current techniques in nanofabrication are used in the semiconductor and electronics industry,43 and their adaptation to other fields of research such as biology,44,45 organic electronics,46 plasmonics,47 or catalysis,48,49 in which unconventional, soft, or degradable materials and substrates are used, represents a challenge.50
Unconventional nanofabrication techniques, such as soft lithography performed using elastomeric stamps, provide a simple, affordable, and versatile alternative to traditional lift-off methods to produce micro- and nano-scale patterns over large areas.43,51,52 Particularly, soft replica molding enables the simple transfer of nanostructures from a patterned polydimethylsiloxane (PDMS) stamp to a substrate by placing a molding agent—typically referred to as “ink”—between the stamp and sample. With the appropriate choice of stamp, the final resolution of soft replica molding at room temperature is only limited by van der Waals forces.43 A variety of soft lithographic techniques have been proposed to pattern biological materials, such as biomolecules, proteins, or nucleic acids, providing also control over the surface chemistry of the patterned substrate.53,54 Similar approaches have already been applied to control the structure of cellulose and silk-based materials,55,56 but have not been implemented in engineered building blocks.
Here, we explore the potential of engineered proteins to develop photoactive protein-based devices using a fabrication strategy that combines a bottom-up (functionalization of the protein-based unit cells) and a top-down (topographically guided molding) approach. This novel system combines the advantages of soft nanolithographic techniques and self-assembly of modular repetitive proteins as building blocks. In addition, we developed a mild crosslinking protocol that preserves the topographical features in an aqueous environment. The strategy is fully performed in an aqueous environment, preserving the structure and function of the constituent protein building blocks, and allowing the direct incorporation of different functional moieties. Thus, we were able to introduce a new functionality focusing on laser applications by incorporating a lasing dye into the functionalized protein films to provide the capability to absorb, emit, confine, and amplify light. The results of this work open up new perspectives for light-management in devices based on engineered recombinant proteins with a broader range of applications given the versatility of the methodology presented, and its potential implementation in other protein systems that may add different competitive advantages.
Overall, our technique to fabricate flexible and conformable nanopatterned protein layers over large areas has the potential to unveil new concepts at the interface of protein engineering, materials sciences, and biocompatible functional nanodevices. The possibility of generating fully biodegradable protein materials patterned on the nanoscale will pave the way towards the development of biosensors, bio-optics, lasers, nanoelectronics, plasmonics, implantable devices, tissue engineering, and fundamental studies in cell biology.
Results and discussion
To explore the potential of designed proteins to create nanopatterned biomaterials capable of acting as active optical devices, we use a designed protein system that exhibits large-scale self-assembling properties. Fig. 1 shows the topology-guided self-assembly strategy followed to create flexible nanopatterned films of engineered proteins. We cast a 0.4–3% water solution of CTPR protein onto the nanopatterned surface of the elastomeric stamp and allowed the water of the solution to slowly dry at room temperature, similarly to previously described protocols for generating CTPR protein films—see the Experimental section for more details.57 After 5 h, the solvent evaporates and generates a thin CTPR film (∼5 μm) that can be easily transferred to wet surfaces via moisture-induced adhesion. A thin layer of polymethylmethacrylate (PMMA) was deposited onto the unpatterned side of the film while this was still on the stamp to facilitate its handling (Fig. 1C and D). CTPR thin films can also be directly printed over any substrate by pressing the aqueous protein solution with the elastomeric stamp for 5 h over the target surface (Fig. 1E). After the protein film is dry, the PDMS stamp could be easily peeled off from the film exposing its patterned surface. We combined structural and optical methodologies to verify the structural integrity and high-resolution successful patterning of the protein films.
Fig. 1. Schematic strategy for obtaining nanostructured protein films. (A) PDMS stamp over a microscope slide covered with a plastic film in order to facilitate the stamp removal. (B) A solution of concentrated protein with PEG is deposited over the PDMS stamp and dried at room temperature. (C) Once the protein film is formed, a drop of 10% liquid PMMA is deposited over the film and dried at 70 °C. (D) When dried, the PMMA-protein film could be easily peeled off from the PDMS stamp. (E) As proteins display good adhesion properties, a glass support can be used instead of PMMA, and in this case the protein film is directly transferred to the surface of the glass. As an example, a protein film has been stamped on a glass bottle.

The optimum protein concentration required to create homogeneous nanopatterned films is a trade-off between the large protein packing degree to completely fill the nanofeatures on the stamp and the viscosity required to facilitate the appropriate flow of molecules inside the stamp channels (Fig. 1A and B). Bearing this in mind, a range of concentrations from 0.4 to 3% (w/v) of purified CTPR10 protein, a consensus CTPR protein with 10 identical repeats,32,58 were tested in the formation of nanopatterned films. The generation of continuous and homogeneous patterned protein films was first evaluated under an optical microscope, confirming the formation of patterned films at CTPR10 concentrations from 0.4% to 3%. Protein concentration below 0.4% leads to heterogeneous discontinuous films. A CTPR10 concentration of 1.5% was chosen for detailed characterization, as it led to the highest film homogeneity and reproducibility with the lowest film thickness (∼1 μm).
The nanopatterned CTPR films prepared by replica molding are highly transparent and flexible (Fig. 1D). Immediately after peeling the stamp from the nanopatterned CTPR film, characteristic iridescent reflections can be observed due to the diffraction of light by the protein nanofeatures on the film (Fig. 1D and E). We used this observation as the first evidence to verify that the molding process worked effectively. It is worth highlighting that adding a PMMA layer as a back support for the nanopatterned CTPR film is only required to facilitate the manipulation of the films, and that the free-standing protein films can also be fabricated and transferred to surfaces by simply omitting this back support. Also, as proteins display excellent adhesion properties, we can generate the films directly on substrates like quartz, polymers, glass, etc.Fig. 1E shows the lateral wall of a glass bottle coated with a nanopatterned CTPR film, demonstrating the good adhesion and conformability of CTPR films created on hydrophilic curved surfaces.
Importantly, the protein structure was altered neither during the film casting process, as demonstrated previously,57 nor during the nanopatterning process. We evaluated the protein structure in the patterned films by circular dichroism observing that the secondary structure of the CTPR protein remained helical, showing no evidence of structural alteration during the casting and drying processes (Fig. 2A). Furthermore, X-ray diffraction of patterned films showed the most intense broad signal at 2θ around 20° and a minor peak around 10° (Fig. 2B), and these values with a d-spacing of about 1 nm are similar to the values that have been previously reported for CTPR unpatterned films, which confirms the presence of the signature peaks ascribed to lamellar packing of CTPR proteins and indicates the directional order.41,57 These results are critical for further ordered functionalization of the protein films using the protein structure as a template to fabricate precisely organized functional elements.
Fig. 2. Structural characterization of the protein films. (A) Circular dichroism spectra of the nanostructured CTPR protein film deposited on a sandwich quartz cuvette. (B) X-ray diffraction spectra of the nanostructured CTPR protein film.

The nanopatterned protein films were first imaged after removal of the PDMS stamp under an optical transmission microscope, revealing in the samples patterned surfaces as large as 1 cm2. The surface morphology, topography, and roughness were characterized by scanning electron microscopy (SEM) and atomic force microscopy (AFM) techniques. SEM images revealed that the protein films accurately replicated the nanopatterns on the PDMS stamp (Fig. 3), and that the films are free-standing (Fig. 3B). Additionally, the nanopatterned film maintained the structural integrity for at least 6 months after fabrication, which provides additional value to the system (Fig. S1†). Using AFM, an area of 50 × 50 μm2 was imaged (Fig. 4). The profiles of four different areas of 10 × 10 μm2 scattered across a larger zone demonstrate the uniform topology of the protein nanopatterns throughout the film. The protein nanofeatures exhibit a height of ∼120–130 nm, while the nanoscale features on the PDMS stamp used as a template for the self-assembly of the protein have a height of ∼140 nm. This difference in height may be due to the shrinking caused by the evaporation of water from the protein structure of the film (Fig. 4A). The nanostructures of both the stamp and the molded protein film exhibited a pitch of 700 nm. We compared a section and the reconstruction of 2 × 2 μm2 of the PDMS stamp (Fig. 4B) and the patterned protein film (Fig. 4C). Using high-resolution AFM microscopy, we found that the root-mean-square roughness of the protein film on top of the pattern is below 10 nm.
Fig. 3. Scanning electron microscopy characterization of the nanostructured protein films. (A) SEM micrographs showing a comparison of the surface of the PDMS stamp and the surface of the protein patterned film, in which the nanostructure patterns are perfectly replicated. (B) From left to right, the optical image and SEM image of the film cross-section; the patterned protein films are self-standing. On the right a higher magnification SEM image of the protein film is shown.

Fig. 4. AFM characterization of the nanostructured protein films. For a PDMS stamp (A) and the protein replica (B), AFM images of 2 × 2 μm2 and a height profile are shown. The cross-section of the replica shows a perfect replication of the nanopatterns on the protein film, with a height of approximately 140 nm and a periodicity of 700 nm (C). The top panel shows a 50 × 50 μm2 image acquired by AFM, in which the film patterns can be observed. The black squares show the four areas of 10 × 10 μm2 that were selected to evaluate the topography of the surface. The bottom panels show the AFM profiles of the four areas selected, revealing a regular topography, with heights around 140 nm.

The low feature variation that soft replica molding provides over the relatively large area of the nanopatterned CTPR film enables the development of uniform bioactive elements for optical applications. UV-Vis characterization showed that the film presents high transmittance values (up to 95%) across the visible region (from 300 to 800 nm) (Fig. 5A). As expected for a protein film, absorption from the high content of aromatic amino acids was observed in the 200–300 nm range, in which the characteristic peak corresponding to the absorption of the proteins appeared at 280 nm (Fig. 5A). These parameters are similar in patterned and non-patterned protein films, presenting in both cases the characteristic high transmission of CTPR protein films. The real part of the refractive index of the material at different wavelengths shows a characteristic tail (Fig. 5B) with the refractive index values ranging from 1.518 to 1.552 across the visible spectrum. These optical parameters lay in a similar range to that of most transparent optical glasses,59 thus allowing the implementation of the nanopatterned CTPR films in conventional optical applications requiring highly transparent materials with a well-defined refractive index.
Fig. 5. Optical properties of protein films. (A) Transmittance spectrum of a nanopatterned CTPR protein film of approximately 1 μm thickness over the UV-visible range showing values up to 95%. The inset shows the high transparency of the protein film. (B) Wavelength dependence of the refractive index of a spin-coated protein film. The plot is obtained from measurements of ψ and Δ at each wavelength on an ellipsometer.

Some interesting biomedical applications for these nanopatterned films involve their use in aqueous environments. In their current form, nanopatterned CTPR films quickly degrade when in contact with any aqueous solution due to the high solubility and weak non-covalent interactions of the integrating proteins. This property can be advantageous for the development of transient washable devices; however, for other applications there is a need for water-resistant films.
We infused glutaraldehyde (GA) through vapor diffusion over the nanopatterned CTPR film before its unmolding from the PDMS stamp to create a mild chemical crosslink across the proteins which made the nanostructured CTPR films water resistant—see the Experimental section for more details. We characterized the structural integrity of the nanostructures of the GA-crosslinked CTPR films using optical microscopy. The height and pitch of the nanopattern were unaltered and remained uniform throughout the entire film after GA exposition (Fig. 6B), when compared to untreated films (Fig. 6A). Likewise, optical features like transmittance and refractive index remained constant. We tested the efficiency of the chemical cross-linking process by immersing the films in a large amount of stirred warm water (∼37 °C) for 1 h. The structural integrity of the nanopatterned CTPR film remained unaltered after this solubility test (Fig. 6C and S2†). Moreover, it is worth mentioning that the lamellar packing of the film is also unaltered upon cross-linking and even under moisture conditions, i.e. upon water immersion, as shown by X-ray diffraction (Fig. S3†). Finally, the circular dichroism spectrum of the crosslinked film showed that mild GA crosslinking did not affect the protein structure (Fig. 6D). Therefore, this crosslinking procedure makes it possible to use these protein films in applications requiring the presence of aqueous media, further expanding the potential uses of nanostructured CTPR films.
Fig. 6. Crosslinking of protein films. Optical microscopy images of the nanostructured protein film (A), the film after the crosslinking reaction with glutaraldehyde (B), and the crosslinked film after immersion in water (C). (D) Circular dichroism spectrum of the crosslinked protein film showing the unaltered characteristic signal of the alpha-helical protein structure after wetting (in comparison with the non-crosslinked dry protein films, see Fig. 2A).

After demonstrating the possibility of controlling both the topology and the solubility of the CTPR films, we expanded the functionality of these systems from the mere biomolecular recognition surfaces. We transformed nanostructured CTPR films into active optical components using a simple yet effective bottom-up approach consisting of the fabrication of nanostructured protein films using CTPR proteins functionalized with rhodamine 6G (Rh6G), an efficient organic fluorescent dye commonly used in laser cavities.60
Note that our first top-down attempt to dope CTPR films with Rh6G resulted in a heterogeneous distribution of dye molecules within the protein films, significantly decreasing their efficiency as optical elements.61,62 We then used a bottom-up approach to achieve the covalent attachment of the dye to the protein following a procedure that proved to be successful for the functionalization of CTPR with porphyrins.41,63 The self-assembly properties of the CTPR–Rh6G conjugate proteins remained unaltered, allowing for the generation of nanopatterned protein films using the replica molding method described above. The resulting nanopatterned CTPR films exhibit structural features indistinguishable from those of the nanopatterned CTPR protein films. The emission features of Rh6G in the nanopatterned films were characterized to determine that their integration in the protein material did not hamper its function for lasing application—see the Experimental section for more details. Protein films with covalently linked Rh6G exhibited excitation and emission spectra peaking at 556 and 591 nm, respectively (Fig. S4†).
Both spectra resemble those of the corresponding CTPR–Rh6G conjugate solutions except for a slight dielectric red-shift and broadening in films, confirming minor dye aggregation in protein films (Fig. 7A). This is also confirmed by comparing other figures like the radiative rate constants and photoluminescence quantum yields (PLQYs). In short, the PLQY of Rh6G in solution is ca. 32%, whereas in thin films for lasing applications a significant reduction up to <1% is observed due to aggregation-induced emission quenching. In stark contrast, the CTPR–Rh6G conjugate showed a PLQY of 24.5% in solution and 17% in thin films. In addition, time-resolved fluorescence constants are similar in solution and films for the CTPR–Rh6G conjugates, concomitant to an increase in the non-radiative decay rates in films (6.2 × 108 s−1) with respect to solution (2.5 × 108 s−1) (Fig. S5 and Table S1†). Furthermore, the photostability of the films was studied under constant irradiation at 505 nm (6.6 mW cm−2) in air, showing a remarkable stability for ca. 100 h. The mechanical stability of these films was tested taking into account optical (emission band shape and PLQY values) and morphological (roughness and crack formation) changes under bending stress (single cantilever, 0.1% strain and 1 Hz, 10 000 cycles). As shown in Fig. S6,† the emission band of both the CTPR and Rh6G as well as the PLQY values remain constant upon bending tests. This is in line with the neglectable changes in the film morphology, lacking a critical change in roughness and/or crack formation (Fig. S7†). Finally, the PLQY of the CTPR–Rh6G film was not altered upon cross-linking and after water exposure demonstrating the optical stability and usability of the material (Fig. S8†).
Fig. 7. (A) Normalized photoluminescence excitation PLE (black) and photoluminescence (PL) (red) spectra of CTPR–Rh6G protein solutions (dashed) and films (solid). (B) Normalized PL (black) and ASE (blue) spectra of CTPR–Rh6G protein films and laser emission (red) from nanostructured CTPR–Rh6G protein films. The inset shows the picture of a CTPR–Rh6G film under UV illumination. (C) Vertical cut SEM image of nanostructured CTPR–Rh6G on top of IPS with 416 nm periodicity. (D) SEM image of an IPS template showing lines of 125 nm height and 416 nm period. (E) log–lin plot of the PL spectra as a function of excitation fluence. (F) log–log plot of the PL linewidth (circles, left Y-axis) and emission output normalized by the output at the highest fluence (squares, right Y-axis) as a function of excitation fluence (red and blue arrows indicate, respectively, their corresponding Y axis).

The emission features, photostability, and homogeneity of CTPR–Rh6G films give rise to the possibility of observing light amplification upon photoexciting the film with laser pulses. Upon pumping the protein films with a pulsed stripe (532 nm, 4 ns, 10 Hz, 3 mm × 700 μm size) above a certain fluence threshold (2.5 mJ cm−2), we observe emission linewidth narrowing due to the appearance of amplified spontaneous emission (ASE) at 621 nm arising from the edge of the film (Fig. 7B). The nanostructured protein films (Fig. 7C) obtained by depositing the protein on IPS grating templates of 416 nm periodicity (Fig. 7D) behaved as second order surface emitting distributed feedback (DFB) lasers leading to a sharp emission line (0.5 nm linewidth) centered at 625 nm (Fig. 7B). The onset for laser action measured upon changing the excitation density (Fig. 7E) indicated a linewidth collapse from 44 to 0.5 nm and a sudden change in the slope of the emission output (Fig. 7F) at fluences above a 55 μJ cm−2 lasing threshold (see the ESI† for a description on how the lasing threshold was obtained). This value is below those reported in DFB lasers based on chemically unbound Rh6G on different hosts (refer for instance to 1.7 mJ cm−2 found on Rh6G-doped silk fibroin or 140 μJ cm−2 observed in Rh6G-doped cellulose acetate).64,65 Our approach benefits from a combination of enhanced Rh6G emission in the solid state and efficiently supported feedback provided by the highly defined periodical patterning.66
Owing to its large fluorescence quantum yield (0.94)67 and outstanding optical gain properties, Rh6G has been the choice as the optical gain medium to develop optically-pumped lasers upon its dispersion on a myriad of organic matrices including for instance mesoporous silica,68,69 aerogels,70,71 latex,72 PMMA,73 methyl methacrylate copolymers,74,75 cellulose nanofibers76 or cellulose acetate,77,78 among many others. The solid-state biological lasing platform demonstrated in this work offers attractive assets for integration of light sources in biological media aiming for instance at in vivo imaging or diagnosis. Furthermore, our approach opens up possibilities to combine lasing and protein functionalities through protein engineering such as their unique recognition capabilities which can be exploited for constructing highly sensitive and selective bio-sensors.
Experimental
Protein production and purification
The gene encoding CTPR proteins were previously generated based on a consensus CTPR protein.24 The encoding gene was ligated into the His tag expression vector pPROEX-HTa vector, which produces a His-tag fused protein for affinity purification.79 The plasmid was transformed into BL21 (DE3) Escherichia coli cells. The cells were grown in LB medium with 0.1 mg ml−1 ampicillin under agitation to an O.D. between 0.6 and 0.8. Protein expression was induced with 0.6 mM IPTG for 5 hours, and after this, the cells were centrifuged at 4500 rpm and resuspended in 300 mM NaCl, 50 mM Tris pH = 8.0 lysis buffer with 1 mg ml−1 lysozyme, 5 mM β-mercaptoethanol, and 1 μl ml−1 protease inhibitor. The resulting lysate was sonicated for 2 minutes with 30 second intervals, and centrifuged at 10 000 rpm for 45 min. Protein purification was performed by affinity chromatography using high density cobalt ABT beads. The eluted protein was dialyzed overnight in PBS (150 mM NaCl, 50 mM phosphate buffer pH = 7.4 with 2.5 mM β-mercaptoethanol). Then, the protein was concentrated and purified by FPLC gel filtration chromatography over a Superdex 200 HiLoad column. Fractions containing the protein were analysed in 15% acrylamide gels to confirm the purity of the protein. Finally, the protein was concentrated until the desired concentration, from 50 to 300 μM, determined using the estimated molar extinction coefficient at 280 nm from the amino acid composition.80
Generation of nanopatterned protein films
In order to prepare the PDMS stamps, a section of a DVD was cut off with scissors. Carefully, the two polymer layers were peeled, keeping the polycarbonate layer as the master piece. Then, a 10 : 1 mix of Slygard prepolymer and curing agent was poured over the master piece and left curing at 70 °C for an hour. Finally, the PDMS stamp ready for use was removed with a razor blade. The approximate area of each stamp was 2.5 cm2 and the gratings can be defined based on the application. In this work we used gratings with periods of 700 nm and 416 nm and a height of 140 nm. The diffraction grating had a groove density of 1500 grooves per mm.
The PDMS stamps were washed with distilled water and 70% ethanol and dried at room temperature (ideally under a N2 flow, to avoid watermarks). Two approaches were used for the generation of nanopatterns on the protein films. First, for the generation of free-standing CTPR protein–PMMA films, a PDMS stamp was placed with the pattern facing-up over a plastic piece on a microscope slide. The plastic piece was useful to avoid the adhesion of the stamp to the slide. As shown in Fig. 1, protein solutions of 300 μM CTPR and 1% (w/v) PEG-400 were deposited over the PDMS stamp and left to dry for at least 4 hours at room temperature to ensure the formation of the film. PEG is used as a plasticizer as previously described.57 Once the film was formed, a solution of 10% PMMA 950 kDa in GBL (gamma butyrolactone) was deposited over the film and left until solidification at 70 °C. GBL was used as a solvent since it does not affect the protein film during its evaporation. The PMMA layer was added with two objectives: (i) to provide a back support that will facilitate handling of the films and (ii) to be used as an adhesive to transfer the patterns to other surfaces. The second approach was the generation of CTPR films directly on a variety of substrates, such as glass, quartz, or plastic. In this latter case, a protein solution with PEG-400 was deposited on the selected substrate and the PDMS stamp placed over the drop for drying the film at room temperature. Films were readily released from the PDMS stamp without requiring functionalization or surface treatment of the elastomer. The PDMS stamps were used several times to fabricate protein films without degradation in their performance.
Crosslinking of the nanostructured protein films
Glutaraldehyde at 0.5% was used as a crosslinking agent for a gentle vapor diffusion crosslinking.81 The reaction was carried out in wells of 1 ml for 24 h at room temperature. At the bottom of the well was added 500 μl glutaraldehyde solution while the protein film was fixed on a cover slip used to seal the well. After the reaction, the films were recovered and dipped into water solution to monitor the potential release of the protein into the solvent in order to evaluate the cross-linking efficiency.
Structural characterization of the nanostructured protein films
Scanning electron microscopy (SEM, Carl Zeiss AURIGA CrossBeam FIB/SEM) was used to image the surface patterning of the films. Films were mounted over a carbon tape and imaged under vacuum conditions, applying an electron high tension (EHT) of 3.00 kV, WD of 5.8 mm and an aperture size of 20 μm.
Atomic force microscopy (AFM), to determine the groove height, surface roughness and thickness of the patterned films, was performed using a JPK NanoWizard II, coupled to a Nikon Eclipse Ti inverted fluorescence optical microscope. An Olympus commercial silicon nitride cantilever tip (OMCL-RC800PSA) with a force constant of 0.76 N m−1 and a resonant frequency of 71 kHz was employed in dynamic mode. The surface morphology of the patterns was studied under ambient conditions in an air environment. To remove the large area background, 1st to 3rd order polynomial fitting was applied using JPK Data Processing software.
Film thickness was measured using a mechanical profilometer, “Dektak XT”, with a 2.5 mm radio stylus. The applied strength was the minimal (around 1 mg), and the length of analysis was 7500 μm.
Circular dichroism experiments to determine the secondary structure of the CTPR components within the films were performed using a Jasco J-815 spectropolarimeter. The solid films were deposited on a sandwich quartz cuvette (0.1 mm path length). The CD spectra were acquired at 1 nm increments and 10 second average time over a wavelength range of 190 to 260 nm.
X-ray diffraction was performed on a PANalytical X'Pert PRO diffractometer with a Cu tube (λ = 1.54187 Å) operated at 45 kV, 40 mA, a Ni beta filter, programmable divergence and anti-scatter slits working in fixed mode, and a fast linear detector (X'Celerator) working in scanning mode.
Optical characterization of the nanostructured protein films
Optical images of the patterned films were acquired on a Leica DMI 3000 B optical transmission microscope using a 63× objective lens, or using an Olympus optical microscope Model (BX51) with an objective of 100× (Nikon) and a numerical aperture of 0.70. Images were processed using ImageJ software for background subtraction.
Ellipsometry measurements were performed on a spectroscopic rotating compensator ellipsometer (M2000V, Woollam, NE, USA) at room temperature (21 ± 1 °C), operating in the wavelength range from 380 to 1000 nm, using an angle of incidence of 70°. The protein sample was deposited on silicon wafers (SiO2) using a spin coating method (Laurell Technologies Corporation, Model WS-400B-6NPP/LITE) at 1000 rpm for 10 min. First, the silicon surface was cleaned in pure acetone to remove the coated surface, and rinsed thoroughly with MilliQ water. Second, it was cleaned with 96% v/v ethanol and rinsed with water. Finally, the surface was dipped in 2% w/v solution of SDS for 30 min and then rinsed with water. In order to ensure the complete surface cleaning, i.e. the surface is free from organic molecules, UV/ozone treatment was carried out using a UV/ozone ProCleaner™ (Bioforce Nanosciences). To calculate the refractive index the data were processed using the software CompleteEASE (Woollam, NE, USA) and the Cauchy model was used to fit the results.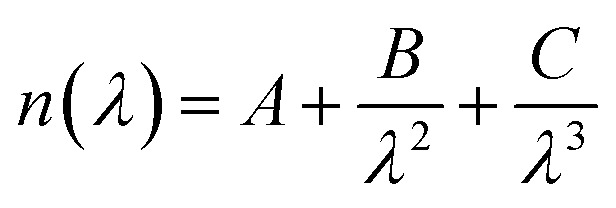
The steady-state transmittance and emission measurements were performed in both solution and thin films using Varian Cary 50 UV-VIS (spot size of 1 mm2 in the 200–700 nm spectral region) and FS5 (Edinburgh) spectrophotometers (spot size of 1 mm2 and excitation at 500 nm) coupled with an integrating sphere for PLQY measurements. The photostability of both CTPR–Rh6G films was analyzed using a home-made system that consists of a high power excitation station – LED WINGER® WEPGN3-S1 Power LED Star green (520 nm) 3 W – 120 lm (505 nm and 6.6 mW cm−2) – coupled to an IR camera (FLIR FS5), and a detection station consisting of a spectrophotometer (Avaspec-ULS2048L-USB2). Both temperature and emission changes of the films were monitored over time.
Bending assays were performed with a DMA Q800 from TA Instruments in a multi-frequency-strain module with a single-cantilever clamp. The experiment was carried out with 0.1% strain and a frequency of 1 Hz at room temperature on a sample size of 17.5 mm × 5.2 mm × 0.2 mm. Optical images were acquired using a 5× objective lens. Atomic force microscopy was performed to measure the film roughness in an area of 15 × 15 μm2.
Time-resolved photoluminescence emission measurements were performed using a pulsed laser diode (405 nm, 80 ps, LDH Picoquant) for photoexcitation. The emission intensity of the sample (in 10−2 mbar vacuum) was collected in free space, subsequently dispersed by the grating of a 1/2 m monochromator and detected with a PMT assembly (PMA – Hybrid from Hamamatsu Photonics) coupled to a Picoquant Hydraharp time-correlated single photon counting system.
Laser action in distributed feedback (DFB) lasers based on CTPR–Rh6G proteins was performed by exciting with a fraction of the 532 nm output provided by a narrow-band OPO (Continuum Sunlite, bandwidth 0.075 cm−1, pulse width 3 ns) pumped by a seeded frequency-tripled Nd:YAG laser (Continuum Powerlite PL8000) and equipped with a frequency doubler delivering 300 ps pulses at 532 nm with a 70 kHz repetition rate (Continuum FX-1). The pump beam was focused under oblique incidence on the DFB surface (placed in a vacuum) using a 200 mm focal length spherical lens, with the sample placed slightly out of focus (9.4 × 10−2 cm2). Laser action emitted perpendicularly from the DFB surface was detected with a spectrometer (SP2500, Acton Research) equipped with a liquid nitrogen cooled back-illuminated deep depletion CCD (Spec-10:400BR, Princeton Instruments). A 533 nm notch filter was placed in front of the spectrometer slit to selectively remove pump laser scattering. Intensity dependent measurements were performed upon pump beam attenuation with a set of neutral density filters. Amplified spontaneous emission (ASE) measurements on CTPR–Rh6G protein films were performed with the same photoexcitation as lasing measurements but using a 200 mm focal length cylindrical lens and a 3 mm slit width to define a photoexcited rectangular area of 700 μm × 3 mm on the sample, and the photoluminescence (PL) was collected from the edge of the sample (90° with respect to incidence) and sent onto the spectrometer. The pump thresholds for laser action and ASE were obtained as the incident fluences at which the FWHM linewidth falls to half of the difference between the linewidths of the PL and laser and ASE spectra, respectively.
Distributed feedback (DFB) lasers from nanostructured CTPR–Rh6G films
In order to obtain a DFB protein based system, nanostructured protein films doped with a laser dye were fabricated. For this purpose two approximations were tested: (1) mixing of the laser dye with the protein solution and (2) covalent coupling of the laser dye to the protein unit. In the first approach, to obtain nanostructured protein films doped with Rh6G, 15 μl of 300 μM CTPR10 were mixed with PEG 1% (w/v) and Rh6G 1 mg ml−1. The solution was mixed gently for 10 min and left in the dark for the formation of the nanostructured film following the procedure described above. In the second approach, a CTPR10 protein with a single cysteine at the C-terminal end was used to generate a CTPR–Rh6G covalent conjugate for a better organization of the Rh dye in the protein films. Tetramethylrhodamine-5-maleimide (Sigma-Aldrich) was attached to the CTPR10-cys protein following previously described protocols.61,82 Nanopatterned films for DFB lasers were produced using a solution of the CTPR–Rh6G conjugate at 300 μM and PDMS stamps with a period of 416 nm as described above.
For efficient lasing an optimized nanoimprinting process was required. A grating with a period of 416 nm was fabricated by thermal nanoimprinting lithography (T-NIL) on a soft material, frequently used as an intermediate polymer stamp (IPS®, Obducat) (Fig. 7D). The grating period was chosen considering the Bragg’s condition given by eqn (1) in order to obtain a constructive interference of light:
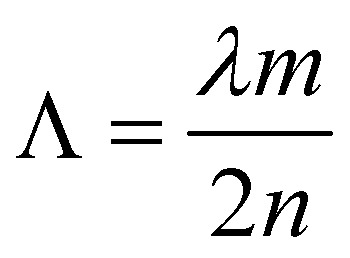 |
1 |
where the Λ is the grating period, m is the order of Bragg diffraction induced by the grating, λ is the target wavelength, and n is the refractive index. The pattern was replicated from a Si master mold (NIL Technology) by using an EITRE Nano Imprint Lithography system (Obducat) as schematically depicted in Fig. S9.† The imprinting parameters were adjusted according to the manufacturer specifications. The T-NIL was accomplished in a sequential process. Temperature and pressure profiles during the process are described in Fig. S5.† Initially, the temperature was raised up to 155 °C, then the pressure was increased sequentially to 20 and 40 bar and kept constant during the rest of the process. After 3 minutes at 155 °C the temperature was decreased in two steps down to 100 °C and 70 °C before releasing the pressure. Finally, the sample was cooled down to room temperature before unmolding. Subsequently, CTPR–Rh6G protein films were deposited on top by spincoating protein solutions (CTPR–Rh6G at 500 μM) in PBS on top of IPS templates. The SEM image of the vertical cut of CTPR–Rh6G on nanostructured IPS (Fig. 7C) depicts a fibrillar protein network which replicates the ripples underneath.
Conclusions
Bioinspired approaches to produce biocompatible protein-based devices are highly promising; however, protein-based devices are not a reality yet. The use of bottom-up approaches to produce biocompatible protein-based surfaces has proven to be useful to fabricate a variety of biomaterials.4,83 Unfortunately, the applicability of these protein-based materials as active devices has been limited by the difficulty to tune their microstructure using top-down approaches without altering the self-assembly of its constituents. Here, we explored the potential of engineered proteins to develop protein-based optically active materials using a fabrication strategy that combines a bottom-up (functionalization of unit cells) and a top-down (topologically guided self-assembly) approach. In addition, compared to previous reports, inspired by naturally existing protein-based materials such as silk, we provide a significant step further, using engineered recombinant proteins as platforms for the generation of active materials.
We reported the generation of nanopatterned protein films using simple designed modular proteins. We achieved macroscale materials composed of structured nanoscale elements ordered across different size scales. In addition, these materials were mouldable using a cost-effective top-down soft-nanolithography technique. The protein material replicated with high fidelity the pattern of the stamp, resulting in bio-compatible nanopatterned films. These films are flexible, easy to handle, and highly transparent. We improved the stability of the protein materials for their use in aqueous environments using mild cross-linking without affecting the optical or structural properties of the material. Finally, we exploited the unique modularity and designability of the protein scaffold of choice to introduce an optical functional element, i.e. an active laser dye, in the material. This resulted in a nanopatterned DFB laser based on protein thin films with good lasing efficiency.
Hence, we demonstrate a biocompatible and cost efficient technology to fabricate functional materials by a combination of bottom-up and top-down approaches. Active elements are integrated in the basic structural unit of this material, and in addition to bottom up approaches that result in an ordered protein material, top-down soft nanolithography approaches can be implemented for the next level of organization. We present a cost-efficient and easy-to-follow technology for patterning nanostructured thin films using PDMS stamps that can embed different functional elements of industrial interest maintaining their functionality. This work represents a starting point to implement this technique in the future to produce protein-based materials with a set of different topologies and patterns, by using molds with different shapes ranging from a few nanometres to the limits of microfabrication. Furthermore, the properties of these scaffold proteins can be easily tuned to embrace different functionalities and protein variants can easily be expressed recombinantly, making the described process versatile, affordable and scalable.
This work is a step toward the development of bioinspired multifunctional devices. Overall this work has deep implications in the design of new bioinspired devices and functional materials based on a combination of bottom-up and top-down approaches using simple protein based scaffolds. Further research is needed in order to establish the real potential of these approaches in materials sciences. The application of this strategy could drive the design of next generation biomaterials following a combined bottom-up/top-down strategy and choosing between a variety of engineered proteins, shapes, topologies, and functionalities, to be applied in very diverse fields including regenerative tissue medicine, nanobiocatalysis, photonics, bioelectronics, biosupporting or biosensing.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was partially supported by the European Research Council ERC-CoG-648071-ProNANO (ALC), the Spanish Ministry of Economy and Competitiveness (BIO2016-77367-R) (ALC), the Basque Government (Elkartek KK-2017/00008) and the IMDEA Nanociencia. DR thanks the IMDEA-Nanociencia for financial support through an “Ayuda de Iniciación a la Investigación” fellowship. This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720 (CIC BIOMAGUNE). A. E. and R. D. C. acknowledge the program “Ayudas para la atracción de talento investigador – Modalidad 1 of the Consejería de Educación, Juventud y Deporte – Comunidad de Madrid with reference number 2016-T1/IND-1463” and ERC2018-092825 (Convocatoria 2018 de acciones de dinamización “Europa Excelencia” – Ministerio de Ciencia, Innovación y Universidades). R. D. C. acknowledges the Ramón y Cajal program from the Spanish MINECO (RYC-2016-20891) and the 2018 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation. J. C.-G. acknowledges financial support from the Spanish Ministry of Science, Innovation and Universities (RTI2018-097508-B-I00), and from the Regional Government of Madrid (S2018/NMT4511). IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant No. SEV-2016-0686). We thank Judith Langer from the optical spectroscopy platform at CIC biomaGUNE for help with the acquisition of optical images.
Electronic supplementary information (ESI) available: Figures and tables. See DOI: 10.1039/c9na00289h
Notes and references
- Fresta E. Fernandez-Luna V. Coto P. B. Costa R. D. Adv. Funct. Mater. 2018;28:1707011. doi: 10.1002/adfm.201707011. [DOI] [Google Scholar]
- Kuehne A. J. Gather M. C. Chem. Rev. 2016;116:12823–12864. doi: 10.1021/acs.chemrev.6b00172. [DOI] [PubMed] [Google Scholar]
- Meglinski I., Biophotonics for Medical Applications, Woodhead Publishing, Cambridge, UK, 2015 [Google Scholar]
- Abascal N. C. Regan L. Open Biol. 2018;8:180113. doi: 10.1098/rsob.180113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. Gou Z. Nanoscale Horiz. 2019;4:52–76. doi: 10.1039/C8NH00223A. [DOI] [PubMed] [Google Scholar]
- Liu Z. Zhang Z. Ritchie R. O. Adv. Mater. 2018;30:e1705220. doi: 10.1002/adma.201705220. [DOI] [PubMed] [Google Scholar]
- Zhang C. McAdams II D. A. Grunlan J. C. Adv. Mater. 2016;28:8566. doi: 10.1002/adma.201604494. [DOI] [PubMed] [Google Scholar]
- Fernandez-Luna V. Coto P. B. Costa R. D. Angew. Chem. 2018;57:8826–8836. doi: 10.1002/anie.201711433. [DOI] [PubMed] [Google Scholar]
- Huang W. Ling S. Li C. Omenetto F. G. Kaplan D. L. Chem. Soc. Rev. 2018;47:6486–6504. doi: 10.1039/C8CS00187A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H. Kaplan D. L. Omenetto F. G. Adv. Mater. 2012;24:2824–2837. doi: 10.1002/adma.201104477. [DOI] [PubMed] [Google Scholar]
- Wang Y. Aurelio D. Li W. Tseng P. Zheng Z. Li M. Kaplan D. L. Liscidini M. Omenetto F. G. Adv. Mater. 2017;29:1702769. doi: 10.1002/adma.201702769. [DOI] [PubMed] [Google Scholar]
- Zheng Z. Wu J. Liu M. Wang H. Li C. Rodriguez M. J. Li G. Wang X. Kaplan D. L. Adv. Healthcare Mater. 2018;7:1701026. doi: 10.1002/adhm.201701026. [DOI] [PubMed] [Google Scholar]
- Lawrence B. D. Cronin-Golomb M. Georgakoudi I. Kaplan D. L. Omenetto F. G. Biomacromolecules. 2008;9:1214–1220. doi: 10.1021/bm701235f. [DOI] [PubMed] [Google Scholar]
- Holland C. Numata K. Rnjak-Kovacina J. Seib F. P. Adv. Healthcare Mater. 2018:e1800465. doi: 10.1002/adhm.201800465. [DOI] [PubMed] [Google Scholar]
- Gazit E. Nat. Chem. 2010;2:1010–1011. doi: 10.1038/nchem.912. [DOI] [PubMed] [Google Scholar]
- Gazit E. Nat. Nanotechnol. 2008;3:8–9. doi: 10.1038/nnano.2007.435. [DOI] [PubMed] [Google Scholar]
- Mejias S. H. Aires A. Couleaud P. Cortajarena A. L. Adv. Exp. Med. Biol. 2016;940:61–81. doi: 10.1007/978-3-319-39196-0_4. [DOI] [PubMed] [Google Scholar]
- Bozic S. Doles T. Gradisar H. Jerala R. Curr. Opin. Chem. Biol. 2013;17:940–945. doi: 10.1016/j.cbpa.2013.10.014. [DOI] [PubMed] [Google Scholar]
- Main E. R. Phillips J. J. Millership C. Biochem. Soc. Trans. 2013;41:1152–1158. doi: 10.1042/BST20130102. [DOI] [PubMed] [Google Scholar]
- Banwell E. F. Abelardo E. S. Adams D. J. Birchall M. A. Corrigan A. Donald A. M. Kirkland M. Serpell L. C. Butler M. F. Woolfson D. N. Nat. Mater. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. S. Lockyer N. P. Vaidyanathan S. Vickerman J. C. Anal. Chem. 2007;79:2199–2206. doi: 10.1021/ac061370u. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. Oppenheim T. W. Buell A. K. Chirgadze D. Y. Welland M. E. Nat. Nanotechnol. 2010;5:204–207. doi: 10.1038/nnano.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J. M. Harniman R. L. Barnes F. R. Boyle A. L. Collins A. Mantell J. Sharp T. H. Antognozzi M. Booth P. J. Linden N. Miles M. J. Sessions R. B. Verkade P. Woolfson D. N. Science. 2013;340:595–599. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romera D. C. Couleaud P. Mejias S. H. Aires A. Cortajarena A. L. Biochem. Soc. Trans. 2015;43:825–831. doi: 10.1042/BST20150077. [DOI] [PubMed] [Google Scholar]
- Woolfson D. N. Mahmoud Z. N. Chem. Soc. Rev. 2010;39:3464–3479. doi: 10.1039/C0CS00032A. [DOI] [PubMed] [Google Scholar]
- Grove T. Z. Regan L. Curr. Opin. Struct. Biol. 2012;22:451–456. doi: 10.1016/j.sbi.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Ulijn R. V. Smith A. M. Chem. Soc. Rev. 2008;37:664–675. doi: 10.1039/B609047H. [DOI] [PubMed] [Google Scholar]
- D'Andrea L. D. Regan L. Trends Biochem. Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Kajander T. Cortajarena A. L. Main E. R. Mochrie S. G. Regan L. J. Am. Chem. Soc. 2005;127:10188–10190. doi: 10.1021/ja0524494. [DOI] [PubMed] [Google Scholar]
- Kajander T. Cortajarena A. L. Mochrie S. Regan L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007;63:800–811. doi: 10.1107/S0907444907024353. [DOI] [PubMed] [Google Scholar]
- Cortajarena A. L. Wang J. Regan L. FEBS J. 2010;277:1058–1066. doi: 10.1111/j.1742-4658.2009.07549.x. [DOI] [PubMed] [Google Scholar]
- Kajander T. Cortajarena A. L. Main E. R. Mochrie S. G. Regan L. J. Am. Chem. Soc. 2005;127:10188–10190. doi: 10.1021/ja0524494. [DOI] [PubMed] [Google Scholar]
- Cortajarena A. L. Mochrie S. G. Regan L. Protein Sci. 2011;20:1042–1047. doi: 10.1002/pro.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove T. Z. Regan L. Cortajarena A. L. J. R. Soc., Interface. 2013;10:20130051. doi: 10.1098/rsif.2013.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejias S. H. Sot B. Guantes R. Cortajarena A. L. Nanoscale. 2014;6:10982–10988. doi: 10.1039/C4NR01210K. [DOI] [PubMed] [Google Scholar]
- Carter N. A. Grove T. Z. Biomacromolecules. 2015;16:706–714. doi: 10.1021/bm501578j. [DOI] [PubMed] [Google Scholar]
- Carter N. A. Grove T. Z. J. Am. Chem. Soc. 2018;140:7144–7151. doi: 10.1021/jacs.8b02663. [DOI] [PubMed] [Google Scholar]
- Jackrel M. E. Cortajarena A. L. Liu T. Y. Regan L. ACS Chem. Biol. 2010;5:553–562. doi: 10.1021/cb900272j. [DOI] [PubMed] [Google Scholar]
- Cortajarena A. L. Yi F. Regan L. ACS Chem. Biol. 2008;3:161–166. doi: 10.1021/cb700260z. [DOI] [PubMed] [Google Scholar]
- Cortajarena A. L. Kajander T. Pan W. Cocco M. J. Regan L. Protein Eng., Des. Sel. 2004;17:399–409. doi: 10.1093/protein/gzh047. [DOI] [PubMed] [Google Scholar]
- Mejías S. H. López-Andarias J. Sakurai T. Yoneda S. Erazo K. P. Seki S. Atienza C. Martín N. Cortajarena A. L. Chem. Sci. 2016;7:4842–4847. doi: 10.1039/C6SC01306F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couleaud P. Adan-Bermudez S. Aires A. Mejías S. Sot B. Somoza A. Cortajarena A. Biomacromolecules. 2015;16:3836–3844. doi: 10.1021/acs.biomac.5b01147. [DOI] [PubMed] [Google Scholar]
- Lipomi D. J. Martinez R. V. Cademartiri L. Whitesides G. M. Polym. Sci. Compr. Ref. 2012;7:211–231. [Google Scholar]
- Ramanathan K. Bangar M. A. Yun M. Chen W. Myung N. V. Mulchandani A. J. Am. Chem. Soc. 2005;127:496–497. doi: 10.1021/ja044486l. [DOI] [PubMed] [Google Scholar]
- Hong J. Edel J. B. deMello A. J. Drug Discovery Today. 2009;14:134–146. doi: 10.1016/j.drudis.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Menard E. Meitl M. A. Sun Y. Park J. U. Shir D. J. Nam Y. S. Jeon S. Rogers J. A. Chem. Rev. 2007;107:1117–1160. doi: 10.1021/cr050139y. [DOI] [PubMed] [Google Scholar]
- Arakawa H. Aresta M. Armor J. N. Barteau M. A. Beckman E. J. Bell A. T. Bercaw J. E. Creutz C. Dinjus E. Dixon D. A. Domen K. DuBois D. L. Eckert J. Fujita E. Gibson D. H. Goddard W. A. Goodman D. W. Keller J. Kubas G. J. Kung H. H. Lyons J. E. Manzer L. E. Marks T. J. Morokuma K. Nicholas K. M. Periana R. Que L. Rostrup-Nielson J. Sachtler W. M. Schmidt L. D. Sen A. Somorjai G. A. Stair P. C. Stults B. R. Tumas W. Chem. Rev. 2001;101:953–996. doi: 10.1021/cr000018s. [DOI] [PubMed] [Google Scholar]
- Stewart M. E. Anderton C. R. Thompson L. B. Maria J. Gray S. K. Rogers J. A. Nuzzo R. G. Chem. Rev. 2008;108:494–521. doi: 10.1021/cr068126n. [DOI] [PubMed] [Google Scholar]
- Wiley B. J. Lipomi D. J. Bao J. Capasso F. Whitesides G. M. Nano Lett. 2008;8:3023–3028. doi: 10.1021/nl802252r. [DOI] [PubMed] [Google Scholar]
- Kim D. H. Ahn J. H. Choi W. M. Kim H. S. Kim T. H. Song J. Huang Y. Y. Liu Z. Lu C. Rogers J. A. Science. 2008;320:507–511. doi: 10.1126/science.1154367. [DOI] [PubMed] [Google Scholar]
- Gates B. D. Xu Q. Stewart M. Ryan D. Willson C. G. Whitesides G. M. Chem. Rev. 2005;105:1171–1196. doi: 10.1021/cr030076o. [DOI] [PubMed] [Google Scholar]
- Xia Y. Rogers J. A. Paul K. E. Whitesides G. M. Chem. Rev. 1999;99:1823–1848. doi: 10.1021/cr980002q. [DOI] [PubMed] [Google Scholar]
- Mujahid A. Iqbal N. Afzal A. Biotechnol. Adv. 2013;31:1435–1447. doi: 10.1016/j.biotechadv.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Brenckle M. A. Tao H. Kim S. Paquette M. Kaplan D. L. Omenetto F. G. Adv. Mater. 2013;25:2409–2414. doi: 10.1002/adma.201204678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z. Shi Z. Cai X. Zhang S. Corder S. G. Li X. Zhang Y. Zhang G. Chen L. Liu M. Kaplan D. L. Omenetto F. G. Mao Y. Tao Z. Tao T. H. Adv. Mater. 2017;29:1605471. doi: 10.1002/adma.201605471. [DOI] [PubMed] [Google Scholar]
- Espinha A. Dore C. Matricardi C. Alonso M. I. Goni A. R. Mihi A. Nat. Photonics. 2018;12:343–348. doi: 10.1038/s41566-018-0152-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove T. Z. Regan L. Cortajarena A. L. J. R. Soc., Interface. 2013;10:20130051. doi: 10.1098/rsif.2013.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortajarena A. L. Regan L. Protein Sci. 2011;20:341–352. doi: 10.1002/pro.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G. Appl. Opt. 1997;36:1540–1546. doi: 10.1364/AO.36.001540. [DOI] [PubMed] [Google Scholar]
- Vasileva E. Li Y. Sychugov I. Mensi M. Berglund L. Popov S. Adv. Opt. Mater. 2017;5:1700057. doi: 10.1002/adom.201700057. [DOI] [Google Scholar]
- de Queiroz T. B. Botelho M. B. S. De Boni L. Eckert H. de Camargo A. S. S. J. Appl. Phys. 2013;113:113508. doi: 10.1063/1.4795506. [DOI] [Google Scholar]
- Forget S. and Chenais S., Organic solid state lasers, Organic Materials for Solid-State Lasers, Springer, 2013 [Google Scholar]
- López-Andarias J. Mejías S. H. Sakurai T. Matsuda W. Seki S. Feixas F. Osuna S. Atienza C. Martín N. Cortajarena A. L. Adv. Funct. Mater. 2018;28:1704031. doi: 10.1002/adfm.201704031. [DOI] [Google Scholar]
- da Silva R. R. Dominguez C. T. dos Santos M. V. Barbosa-Silva R. Cavicchioli M. Christovan L. M. de Melo L. S. A. Gomes A. S. L. de Araújo C. B. Ribeiro S. J. L. J. Mater. Chem. C. 2013;1:7181–7190. doi: 10.1039/C3TC30903G. [DOI] [Google Scholar]
- Tsutsumi N. Shinobu M. Appl. Phys. B. 2011;105:839–845. doi: 10.1007/s00340-011-4688-8. [DOI] [Google Scholar]
- Xu Y. Hai G. Xu H. Zhang H. Zuo Z. Zhang Q. Xia R. Sun C. Castro-Smirnov J. Sousaraei A. Casado S. Osorio M. R. Granados D. Rodriguez I. Cabanillas-Gonzalez J. Adv. Opt. Mater. 2018;6:1800263. doi: 10.1002/adom.201800263. [DOI] [Google Scholar]
- Fischer M. Georges J. Chem. Phys. Lett. 1996;260:115–118. doi: 10.1016/0009-2614(96)00838-X. [DOI] [Google Scholar]
- Yang P. Wirnsberger G. Huang H. C. Cordero S. R. McGehee M. D. Scott B. Deng T. Whitesides G. M. Chmelka B. F. Buratto S. K. Stucky G. D. Science. 2000;287:465–468. doi: 10.1126/science.287.5452.465. [DOI] [PubMed] [Google Scholar]
- Marlow F. McGehee M. D. Zhao D. Chmelka B. F. Stucky G. D. Adv. Mater. 1999;11:632–636. doi: 10.1002/(SICI)1521-4095(199906)11:8<632::AID-ADMA632>3.0.CO;2-Q. [DOI] [Google Scholar]
- Altman J. C. Stone R. E. Dunn B. Nishida F. IEEE Photonics Technol. Lett. 1991;3:189–190. [Google Scholar]
- Wetter N. U. Ramos de Miranda A. Pecoraro E. Lima Ribeiri S. J. Jimenez-Villar E. RSC Adv. 2018;8:29678–29685. doi: 10.1039/C8RA04561E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdán L. Enciso E. Martin V. Bañuelos Prieto J. López-Arbeloa I. Costela A. Garcia-Moreno I. Nat. Photonics. 2012;6:621–626. doi: 10.1038/nphoton.2012.201. [DOI] [Google Scholar]
- Kuriki K. Kobayashi T. Imai N. Tamura T. Nishihara S. Nishizawa Y. Tagaya A. Koike Y. Okamoto Y. Appl. Phys. Lett. 2000;77:331. doi: 10.1063/1.126967. [DOI] [Google Scholar]
- Costela A. Florido F. Garcia-Moreno I. Duchowicz R. Amat-Guerri F. Figuera J. M. Sastre R. Appl. Phys. B. 1995;60:383–389. doi: 10.1007/BF01082275. [DOI] [Google Scholar]
- Costela A. Garcia-Moreno I. Figuera J. M. Arnat-Guerri F. Mallavia R. Santa-Maria M. D. Sastre R. J. Appl. Phys. 1996;80:3167. doi: 10.1063/1.363255. [DOI] [Google Scholar]
- Vasileva E. Li Y. Sychugov I. Mensi M. Berglund L. Popov S. Adv. Opt. Mater. 2017;5:1700057. doi: 10.1002/adom.201700057. [DOI] [Google Scholar]
- Tsutsumi N. Hirano Y. Kinashi K. Sakai W. Langmuir. 2018;34:7527–7535. doi: 10.1021/acs.langmuir.8b01107. [DOI] [PubMed] [Google Scholar]
- Tsutsumi N. Nagi S. Kinashi K. Sakai W. Sci. Rep. 2016;6:34741. doi: 10.1038/srep34741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajander T. Cortajarena A. L. Regan L. Methods Mol. Biol. 2006;340:151. doi: 10.1385/1-59745-116-9:151. [DOI] [PubMed] [Google Scholar]
- Pace C. N. Vajdos F. Fee L. Grimsley G. Gray T. Protein Sci. 1995;4:2411. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-deAlcazar D. Velasco-Lozano S. Zeballos N. Lopez-Gallego F. Cortajarena A. L. ChemBioChem. 2019;20:1977–1985. doi: 10.1002/cbic.201900047. [DOI] [PubMed] [Google Scholar]
- Cortajarena A. L. Lois G. Sherman E. O'Hern C. S. Regan L. Haran G. J. Mol. Biol. 2008;382:203. doi: 10.1016/j.jmb.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. Sun Y. Li Z. Wu A. Wei G. Materials. 2016;9:E53. doi: 10.3390/ma9010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.