

Summary
Understanding lung immunity requires an unbiased profiling of tissue-resident T cells at their precise anatomical locations within the lung, but such information has not been characterized in the immunized mouse model. In this pilot study, using 10x Genomics Chromium and Visium platform, we performed an integrative analysis of spatial transcriptome with single-cell RNA-seq and single-cell ATAC-seq on lung cells from mice after immunization using a well-established Klebsiella pneumoniae infection model. We built an optimized deconvolution pipeline to accurately decipher specific cell-type compositions by anatomic location. We discovered that combining scATAC-seq and scRNA-seq data may provide more robust cell-type identification, especially for lineage-specific T helper cells. Combining all three modalities, we observed a dynamic change in the location of T helper cells as well as their corresponding chemokines. In summary, our proof-of-principle study demonstrated the power and potential of single-cell multi-omics analysis to uncover spatial- and cell-type-dependent mechanisms of lung immunity.
Subject areas: Biological sciences, Immunology, Omics, Transcriptomics
Graphical abstract
Highlights
-
•
Deconvolution workflow was verified to study lung immunity using ST
-
•
15 lung cell types were identified by integrating scRNA-seq and scATAC-seq data
-
•
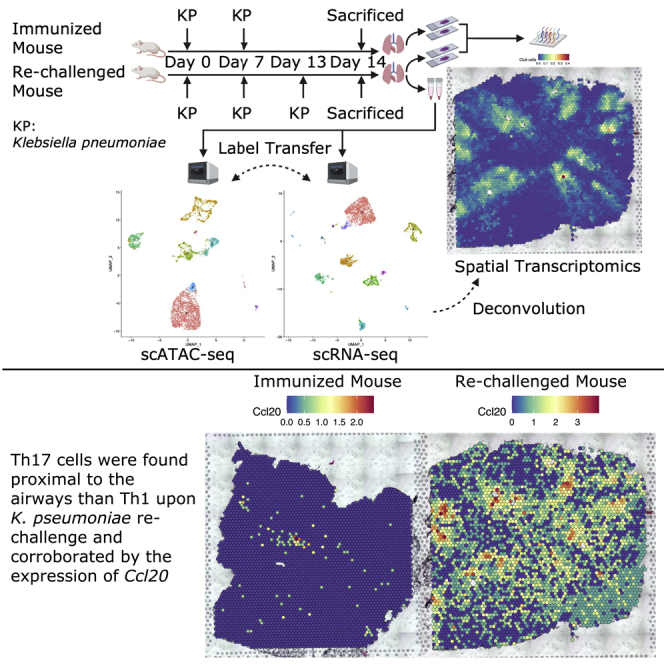
Th17 cells were found proximal to airways than Th1 upon Klebsiella pneumoniae re-challenge
-
•
Massive immune responses were activated in airways upon K. pneumoniae re-challenge
Biological sciences; Immunology; Omics; Transcriptomics
Introduction
Immunological memory, consisting of B cell and T cell memory, is a key characteristic of adaptive immunity upon encountering pathogen invasion. Tissue-resident memory T cells have more recently been defined as a new subset, predominantly residing in mucosal tissues, barrier surfaces, and other non-lymphoid organs but are also present in lymphoid sites (Turner and Farber, 2014). Tissue localization of these T cells has been investigated thoroughly in the past, and comparisons between mouse and human have been characterized extensively using the well-established flow cytometric and transcriptomic approaches (Szabo et al., 2019). However, an unbiased, high throughput gene expression profiling of tissue-resident immune cells residing in various anatomical locations within the lung, such as airway versus parenchyma, has not been possible using conventional flow cytometry, transcriptomic approaches, and even the most recently developed single-cell sequencing technology. These methodologies are limited as anatomic location-specific information is lost after the single-cell suspension is acquired. As a consequence, this leads to missing key information on location-specific downstream signaling events from structural cells targeted by cytokines and chemokines produced by immune cells. Furthermore, depending on the enzymes used to digest the target tissues, fragile cells like epithelial cells can be lost to a certain extent during single-cell suspension preparation. To overcome this issue, researchers have to optimize their protocols to preserve specific cell types and/or enrich them with cell-type specific markers by flow cytometry or magnetic beads, therefore missing a global assessment on cell-cell interaction among all cell types.
Single-cell RNA-seq (scRNA-seq) has been gradually applied to study immune cells and immune responses in mouse lungs (Ballesteros et al., 2020; Cohen et al., 2018; Kurihara et al., 2021; Wang et al., 2021). Few recent studies utilize single-cell ATAC-seq (scATAC-seq) to measure the chromatin architecture of immune cells in mouse lungs (Delacher et al., 2021). Although these technologies provide rich information in understanding cell heterogeneity and biological information of mouse lungs, spatial information of a single cell is lost in the process. Spatial transcriptomics (ST) is a recently developed technology and has the ability to map transcriptional signatures to distinct anatomical regions. To date, it has rarely been used in understanding lung tissues, and often time naive lungs without immunological perturbation such as vaccination and re-challenge. In this first-in-class study, we employed a commercially available ST platform to investigate the spatial topography of gene expression of mouse lungs after immunization and re-challenge. To overcome the resolution limitation of current ST technology, we also applied state-of-art single-cell transcriptomics and single-cell epigenomics to jointly study the spatial localization, transcriptome, and epigenome of T cells induced by this immunization/re-challenge. The integrative analysis of three types of omics data provides an unprecedented and comprehensive way to examine the dynamics of lung immunization on a genomic scale, which will be valuable for hypothesis generation and the design of hypothesis-testing experiments.
Results
Cataloging 12 lung cell types using scRNA-seq data
In this study, we inoculated mice with heat-killed Klebsiella pneumoniae and assigned them into two groups, the immunized and the re-challenged groups, based on our previous finding that heat-killed K. pneumoniae is sufficient to induce antigen-specific Th17 responses in mouse lungs (Chen et al., 2011). Because few Th17 cells exist in naive unimmunized mice, we did not include a blank control group in this study. The immunized group was inoculated twice on day 0 and day 7, whereas the re-challenged group was additionally inoculated on day 13. On day 14, both groups of mice were sacrificed for tissue harvesting. Slices A1 and A2 were sectioned at different regions from the fresh frozen lung of the immunized mouse, whereas slices A3 and A4 were from the re-challenged mouse. In a separate cohort, lung tissue from the re-challenged mouse was harvested, and two tubes of single-cell suspension were obtained after enzymatic digestion. The four lung slices were used for spatial transcriptomics, and the two tubes of single-cell suspension from the re-challenged mouse were subjected to scRNA-seq and scATAC-seq analyses. scRNA-seq data were integrated with scATAC-seq data via label transfer (Stuart et al., 2019) and were used as a reference to deconvolute spatial transcriptomics data (Figure 1A).
Figure 1.

Generation of multi-omics datasets of mice lungs after immunization, see also Figure S1
(A) Overview of study design. scRNA-seq data work as a bridge to link scATAC-seq and spatial transcriptomics data. UMAP plots were shown as examples, not for interpretation.
(B) UMAP plot of 12 lung cell types identified in scRNA-seq data, with manual annotation according to canonical markers.
(C) Dot plot showing selected canonical markers for each celltype.
(D) Heatmap showing top three (by log2-Fold Change) markers for each celltype.
We generated scRNA-seq data from 3,337 cells collected from the re-challenged mouse. Graph-based clustering identified ten clusters (Figures S1A and S1C). Cluster 1, which has an extremely high percentage of mitochondrial genes (Figure S1B), was excluded from downstream analyses. Cluster 6 consisted of several sporadic sub-clusters and was re-clustered into four clusters (Figures S1D and S1E). 2,834 cells with good quality were retained for the downstream analyses.
We carried out fine cluster annotation according to canonical markers and identified 12 lung cell types (Figure 1B), including alveolar epithelial cells, club cells, fibroblasts, endothelial cells, monocytes, macrophages, dendritic cells, neutrophils, B cells, T cells, NK cells, and erythrocytes. Selected canonical markers for each cell type were shown in the dot plot (Figure 1C, e.g., Sftpb for alveolar epithelial cells, Scgb1a1 for club cells, and Cd3d for T cells). The top three (by log2-Fold Change) markers for each cell type were visualized using a heatmap (Figure 1D).
Robust cell type decomposition (RCTD) of spatial transcriptome using scRNA-seq data
Although spatial transcriptomics provides additional spatial information, its resolution has not reached single-cell level. Therefore, the expression profile of each spot in spatial transcriptomics is from a mixture of a few cells, typically one to ten. To better interpret spatial transcriptome data, it is vital to determine the proportions of different cell types within each spot. Using our finely annotated scRNA-seq data as a reference, we carried out deconvolution for each spot in the four slices using the robust cell-type decomposition (RCTD) method (Cable et al., 2022), one of the top-performing methods for cell-type deconvolution (Li et al., 2022). The deconvolution results for slice A3 were shown as proportions of 12 cell types across slice A3 (Figure S2A).
To assess the robustness of this deconvolution method, we inspected whether well-characterized cell types were colocalized with the expression of their canonical markers, as well as corresponding histological structures. In slice A3, spots with a high proportion of club cells were around the histological airways and colocalized with spots with increased expression of Scgb1a1 (Figures 2Ai–iii).
Figure 2.

Validation of the robustness of deconvolution method for spatial transcriptomics, see also Figure S2
(A) Proportion of club cells colocalizing with the expression of Scgb1a1 and the histological airways. (1) Histology of slice A3 showing the location of the airways. (2) Proportion of club cells across slice A3, deconvoluted using in-house scRNA-seq data. (3) Expression of Scgb1a1, a canonical marker for club cells, across slice A3.
(B) Proportions of T cells deconvoluted using the two independent scRNA-seq references were quite similar. (1) UMAP plot of 20 lung cell types identified in Cohen et al.’s public scRNA-seq data (GEO: GSE119228). (2) Proportion of T cells across slice A3, deconvoluted using in-house scRNA-seq data. (3) Proportion of T cells across slice A3, deconvoluted using Cohen et al.’s public scRNA-seq data.
(C) (i–iv) Correlation heatmap visualizing the proportions of cell types deconvoluted using in-house (in rows, 12 types) and public (in columns, 20 types) scRNA-seq data were highly correlated in slice A1-A4. Pearson’s r values were indicated by the color bars. Red boxes with solid lines highlighted selected adaptive immune cells, T cells and B cells. Red boxes with dashed lines highlighted selected innate immune cells, neutrophils and NK cells.
To examine the robustness of the method and the impact of different reference panel on deconvolution, another public scRNA-seq data (GEO: GSE119228) (Cohen et al., 2018), including 20 annotated mouse lung cell types (containing 20,931 cells) (Figure 2Bi), were also used as an independent reference to deconvolute spatial transcriptome for the four slices. The deconvolution results for slice A3, using public scRNA-seq reference, were shown (Figure S2B). In slice A3, proportions of T cells deconvoluted using in-house or public scRNA-seq references were quite similar (Figures 2Bii and 2Biii). Deconvolution results can also be represented using proportion matrices, whose rows indicate spots and columns indicate cell types. To quantify the similarity between the deconvolution results using the two independent references, Pearson correlation coefficients between the columns of the two proportion matrices were calculated and visualized using correlation heatmaps (Figure 2Ci–iv). Although we cannot perfectly match all the 12 cell types from our in-house data with the 20 cell types identified in this public dataset, we found the proportions of some adaptive immune cells (e.g., T cells and B cells) and innate immune cells (e.g., neutrophils and NK cells) were highly correlated with those deconvoluted using the other reference in all four slices. Other cell types were also highly correlated with their related cell types deconvoluted using the other reference (e.g., club cells in in-house data with ciliated cells in public data, alveolar epithelial cells in in-house data with AT2 in public data). These analyses validated the robustness of the robust cell-type decomposition (RCTD) method. They also supported the observation that the number of cells would not drastically impact the performance of deconvolution as long as a signature matrix can be built for each celltype in the reference panel.
Integrative analysis of scRNA-seq and scATAC-seq data enabling the identification of Th17 and Th1 cells
In parallel with scRNA-seq, we also generated scATAC-seq data from 4,908 cells collected from the re-challenged mouse. After excluding cells with low quality, 4,794 cells were retained for downstream analyses. Leveraging our finely annotated scRNA-seq data, we identified the 12 lung cell types in scATAC-seq data via label transfer (Figure 3A). Proportions of the 12 cell types in scRNA-seq and scATAC-seq data were quite similar (Figure 3B), showing a good biological agreement between the two types of data.
Figure 3.

Identification of subtypes of T cells by integrating scRNA-seq and scATAC-seq data, see also Figure S3
(A) UMAP plot of 12 lung cell types in scATAC-seq data identified via label transfer.
(B) Bar plot showing proportions of 12 cell types in scRNA-seq and scATAC-seq data were quite similar.
(C) UMAP plot of four subtypes of T cells in scATAC-seq data.
(D) Heatmap showing top 10 (by log2-Fold Change) markers (gene activity scores calculated using peaks) for each subtype of T cells in scATAC-seq data.
(E) (i–iv) Peaks in genomic regions around Il17a, Rorc, Ifng, and Tbx21, canonical markers for Th17 and Th1 cells.
(F) UMAP plot of four subtypes of T cells in scRNA-seq data identified via label transfer.
(G) Dot plot showing selected canonical markers for Th17 and Th1 cells in scRNA-seq data.
(H) UMAP plot of 15 lung cell types (including four subtypes of T cells) in scRNA-seq data.
Because we are interested in T cells in this study, we carried out re-clustering for T cells in scATAC-seq data and identified four subtypes of T cells (Figure 3C), including Th17 cells, Th1 cells, other T cells 1, and other T cells 2. The top 10 (by log2-Fold Change) markers (gene activity scores) for each subtype of T cells were visualized using a heatmap (Figure 3D). Canonical transcription factors for subtypes of T cells were shown on the heatmap (e.g., Rorc and Rora for Th17 cells, Tbx21 for Th1 cells). Cd8b1 was a marker for other T cells 1, suggesting they could be cytotoxic T cells.
To confirm the identity of Th17 and Th1 cells, further analyses were performed. For Th17 cells, there were much more peaks in genomic regions around Il17a and Rorc, compared with other subtypes of T cells (Figures 3Ei and 3Eii). In contrast, the peaks from Th1 cells dominated genomic regions around Ifng and Tbx21 (Figures 3Eiii and 3Eiv). Motif footprinting analysis further confirmed that RORC was dominated by Th17 cells, and TBX21 was dominated by Th1 cells (Figures S3Ai–ii and S3Bi–ii).
Although subtypes of lineage-specific T cells were not clearly distinguishable using scRNA-seq data alone (e.g., the public scRNA-seq data mentioned above), we managed to identify the four subtypes of T cells via label transfer from scATAC-seq data to scRNA-seq data (Figure 3F). To confirm the identity of Th17 and Th1 cells in scRNA-seq data, further analyses were performed. Selected canonical markers for Th17 and Th1 cells were shown in the dot plot (Figure 3G, e.g., Rora for Th17 cells, Tbx21 for Th1 cells). The top 10 (by log2-Fold Change) markers for each subtype of T cells were visualized using a heatmap (Figure S3C). Canonical markers for subtypes of T cells can be seen in the heatmap (e.g., Rora for Th17 cells, Ifng for Th1 cells, and Cd8b1 for other T cells 1). We also conducted gene regulatory network analysis using SCENIC (Aibar et al., 2017; Van deSande et al., 2020) and identified RORA as a cell-type specific regulator for Th17 cells in scRNA-seq data (Figure S3D). By integrative analysis of scRNA-seq and scATAC-seq data, 15 lung cell types (including four subtypes of T cells) were identified in scRNA-seq data (Figure 3H), which will facilitate the deconvolution analysis in spatial transcriptome.
Dynamic changes of cell locations upon K. pneumoniae Re-challenge
Using the updated scRNA-seq data with the final 15 cell types, we carried out deconvolution again for each spot in the four slices (Figure S4). The proportions of 15 cell types across four slices were summarized by a boxplot (Figure 4A). We performed t-tests (n = 14,244 spots) to compare the differences between the immunized (slice A1 and A2) and the re-challenged mouse (slice A3 and A4), with significant differences found for each celltype. Generally, there were more monocytes, macrophages, dendritic cells, neutrophils, Th1 cells, and NK cells in the re-challenged mouse. In contrast, the immunized mouse had more B cells, Th17 cells, and other T cells 1 (Figures 4A and S4).
Figure 4.

Spatial analyses of mice lungs after immunization, see also Figures S4 and S5
(A) Box plot showing proportions of 15 cell types across four slices were different. To compare the differences between the immunized (slice A1 and A2) and the re-challenged mouse (slice A3 and A4), t-tests (n = 14,244 spots) were performed for each cell type. ∗∗∗∗ p-value < 1x10−4.
(B) (i-ii) Correlation heatmap visualizing the same-spot co-occurrence of 15 cell types in the immunized and the re-challenged mouse. ST spots from the slices for the immunized (slice A1 and A2) or the re-challenged mouse (slice A3 and A4) were pooled together, respectively. Red boxes highlighted cell types tending to appear in the same spots, that is to say, possibly close to one another in vivo.
(C) Weighted distances to the airways for Th17 and Th1 cells in four slices, after excluding spots within the blood vessels and spots whose distances to the airways longer than 1,000 μm.
(D) Formula defining weighted distance, allowing for each spot’s distance to the nearest airway and the proportions of Th17 and Th1 cells in each spot.
(E) (i-iv) Proportions of immune cells over distance to the airways showing the spatial distribution of immune cells in four slices, after excluding spots within the blood vessels and spots whose distances to the airways longer than 1,000 μm. The curves were obtained from natural spline (with three degrees of freedom) regression.
(F) Expression of Ccl20, a top distance-associated gene, across four slices.
(G) UMAP plot of 8 lung cell types identified in a scRNA-seq dataset of epithelial cells (CD45-CD31-Epcam+), harvested from K. pneumoniae-infected mouse lung. Mice were infected with 10ˆ3 CFU/mouse and sacrificed at 48 h after infection. Lungs were homogenized with collagenase and DNase. CD45-cells were enriched by magnetic beads, followed by CD45-CD31-Epcam+ cell sorting by FACS.
(H) UMAP plots showing the distribution of selected markers.
(I) Violin plots demonstrating Ccl20 was elevated in inflammatory type II cells.
After pooling spots from the slices for the immunized (slice A1 and A2) or the re-challenged mouse (slice A3 and A4), Pearson correlation coefficients between the columns of the proportion matrices were calculated. Significant same-spot co-occurrence of different cell types was found in the immunized and the re-challenged mouse (Figure 4B). For the immunized mouse, Th17 cells, monocytes, and endothelial cells tended to appear in the same spots. Dendritic cells, B cells, fibroblasts, and other T cells 1 were also close to one another. For the re-challenged mouse, other T cells 1, B cells, Th17 cells, Th1 cells, and dendritic cells often appeared in the same spots, whereas Th1 cells, NK cells, and myeloid cells were possibly close to one another in vivo. These observations remained unchanged with each slice analyzed separately (Figure S5C).
Localization and segmentation of airway and blood vessels are important in our analysis. We defined airways and blood vessels according to the proportion of club cells and the histological blood vessels (Figures S5A and S5B). We thought T cells within the spot overlapping with blood vessels could be circulating T cells, whereas T cells with no overlapping with blood vessels were more like tissue-resident T cells. To minimize the impact of circulating T cells, we excluded spots within the blood vessels from downstream analyses. After excluding spots within the blood vessels and spots whose distances to the airways are longer than 1,000 μm, we calculated weighted distances to the airways for Th17 and Th1 cells in four slices, according to the formula (Figure 4D). Th17 cells were found closer to the airways than Th1 cells in the re-challenged mouse, whereas Th1 cells were closer to the airways in the immunized mouse (Figure 4C). This conclusion was independent of the definition of airways and blood vessels, since it remained unchanged even if a set of cut-offs were used to define these structures (Figure S5D).
To find the spatial distribution patterns of immune cells, natural spline regression was performed to fit the non-linear relationship between the proportions of immune cells and the distances to the airways. Generally, B cells and other T cells 1 were proximal to the airways in the immunized mouse, whereas neutrophils were distal to the airways in the re-challenged mouse (Figures 4Ei–iv). The same analysis for all 15 cell types was also performed (Figures S5Ei–iv). We also performed natural spline regression between the expression of genes and the distances to the airways. 3,655 distance-associated genes (FDR-adjusted p-value < 0.05 in both slices A1 and A2) were identified in the immunized mouse (Table S1), whereas 3,407 (FDR-adjusted p-value < 0.05 in both slices A3 and A4) were identified in the re-challenged mouse (Table S2). Gene Ontology (GO) enrichment analysis was performed for these distance-associated genes (Ashburner et al., 2000; Mi et al., 2019; The Gene Ontology Consortium, 2021). 1,293 significantly (FDR-adjusted p-value < 0.05) over-represented biological processes were identified for the immunized mouse, and 1,294 for the re-challenged mouse. The top 20 (hierarchically sorted by fold-enrichment) over-represented biological processes for the re-challenged mouse included many immune responses (e.g., proliferation, differentiation, activation, aggregation, adhesion, and chemotaxis of immune cells). In contrast, few immune responses were over-represented in the immunized mouse (Table 1). Ccl20, a top distance-associated gene, was highly expressed around the airways in the re-challenged mouse, instead of the immunized mouse (Figure 4F). Because CCL20 is capable of binding to CCR6, a chemokine receptor expressed on Th17 cells, this finding possibly explains why Th17 cells were closer to the airways than Th1 cells in the re-challenged mouse.
Table 1.
Top 20 (hierarchically sorted by fold-enrichment) over-represented biological processes for both mice
| Immunized Mouse |
Re-challenged Mouse |
||||||
|---|---|---|---|---|---|---|---|
| GO Biological Process | Fold Enrichment | p-value | FDR-adjusted p-value | GO Biological Process | Fold Enrichment | p-value | FDR-adjusted p-value |
| protein localization to bicellular tight junction | 6.22 | 1.36E-03 | 2.04E-02 | protein localization to ciliary transition zone | 6.64 | 4.12E-04 | 7.33E-03 |
| inner dynein arm assembly | 6.22 | 2.60E-05 | 6.53E-04 | intraciliary retrograde transport | 6.2 | 4.72E-06 | 1.36E-04 |
| cranial ganglion development | 6.22 | 3.06E-03 | 3.94E-02 | inner dynein arm assembly | 6.09 | 5.62E-05 | 1.23E-03 |
| positive regulation of cell migration by vascular endothelial growth factor signaling pathway | 5.53 | 1.01E-03 | 1.60E-02 | hemoglobin biosynthetic process | 5.81 | 1.61E-03 | 2.33E-02 |
| peripheral nervous system myelin maintenance | 5.53 | 1.01E-03 | 1.60E-02 | protein localization to bicellular tight junction | 5.69 | 3.77E-03 | 4.65E-02 |
| intraciliary retrograde transport | 5.39 | 3.27E-05 | 8.07E-04 | negative regulation of skeletal muscle tissue development | 5.69 | 3.77E-03 | 4.65E-02 |
| outer dynein arm assembly | 5.29 | 2.42E-06 | 7.67E-05 | positive regulation of homotypic cell-celladhesion | 5.17 | 2.53E-03 | 3.37E-02 |
| cerebrospinal fluid circulation | 5.26 | 1.54E-04 | 3.18E-03 | epithelial cilium movement involved in determination of left/right asymmetry | 5.17 | 2.53E-03 | 3.37E-02 |
| establishment of blood-brain barrier | 5.18 | 3.35E-04 | 6.17E-03 | positive regulation of integrin-mediated signaling pathway | 5.17 | 2.53E-03 | 3.36E-02 |
| endothelial tube morphogenesis | 5.18 | 3.35E-04 | 6.17E-03 | negative regulation of CD4-positive, alpha-beta T cell proliferationa | 5.17 | 2.53E-03 | 3.36E-02 |
| glomerulus morphogenesis | 5.09 | 7.32E-04 | 1.21E-02 | negative regulation of actin nucleation | 5.17 | 2.53E-03 | 3.36E-02 |
| dichotomous subdivision of an epithelial terminal unit | 5.09 | 7.32E-04 | 1.21E-02 | antigen processing and presentation of exogenous peptide antigen via MHC class Ia | 5.17 | 2.53E-03 | 3.36E-02 |
| defense response to tumor cella | 4.98 | 1.60E-03 | 2.34E-02 | cerebrospinal fluid circulation | 5.11 | 3.27E-04 | 6.00E-03 |
| axoneme assembly | 4.92 | 6.34E-19 | 8.26E-17 | neutrophil activation involved in immune responsea | 4.83 | 1.68E-03 | 2.41E-02 |
| epithelial cilium movement involved in determination of left/right asymmetry | 4.84 | 3.52E-03 | 4.43E-02 | branching involved in labyrinthine layer morphogenesis | 4.74 | 4.95E-04 | 8.50E-03 |
| positive regulation of aspartic-type peptidase activity | 4.84 | 3.52E-03 | 4.43E-02 | defense response to tumor cella | 4.65 | 3.83E-03 | 4.70E-02 |
| surfactant homeostasis | 4.84 | 3.73E-05 | 9.05E-04 | outer dynein arm assembly | 4.65 | 4.46E-05 | 1.00E-03 |
| regulation of cilium beat frequency | 4.56 | 3.65E-04 | 6.65E-03 | positive regulation of superoxide anion generationa | 4.62 | 1.36E-05 | 3.51E-04 |
| negative regulation of endothelial cell differentiation | 4.52 | 2.43E-03 | 3.27E-02 | regulation of Fc receptor mediated stimulatory signaling pathwaya | 4.43 | 2.48E-03 | 3.34E-02 |
| regulation of aspartic-type endopeptidase activity involved in amyloid precursor protein catabolic process | 4.52 | 2.43E-03 | 3.27E-02 | collagen-activated tyrosine kinase receptor signaling pathway | 4.43 | 2.48E-03 | 3.33E-02 |
Biological processes related to immune responses.
In a validation cohort, we enriched and profiled epithelial cells from K. pneumoniae-infected mouse lungs (Figure 4G). The enrichment of epithelial cells complemented the in-house scRNA-seq data utilized above and helped us to trace back the source of Ccl20 around the airways—alveolar epithelial type II cells, specifically, inflammatory type II cells (Figures 4H and 4I).
Biological differences upon K. pneumoniae Re-challenge
Upon K. pneumoniae re-challenge, the spatial transcriptome was tremendously changed (Figure S6A), whereas little batch effects were found between the two slices from the same mouse. The spots we defined as airways according to the proportion of club cells were also shown in a Uniform Manifold Approximation and Projection (UMAP) plot (Figures S6B and S5A), with significant transcriptomic changes found between the immunized and the re-challenged mouse.
Differential expression (DE) analysis was performed to compare the airways in the re-challenged mouse versus the immunized mouse, with 2,071 significantly (FDR-adjusted p-value < 0.05) differentially expressed genes (DEGs) identified (Table S3). To overcome the limitations of the analysis based on different thresholds of selected DEGs, we performed Gene Set Enrichment Analysis (GSEA) using the unfiltered, ranked gene list (including 16,937 genes), and found 379 significantly (FDR-adjusted p-value < 0.05) enriched GO biological processes. The top 30 (by normalized enrichment score) up-regulated and down-regulated pathways were shown by a lollipop plot (Figure 5A). Compared with the immunized mouse, most up-regulated pathways in the airways of the re-challenged mouse were related to immune responses (e.g., migration and chemotaxis of immune cells, and response to stimuli), whereas most down-regulated pathways were related to catabolic and metabolic processes, as well as ciliary functions. AW112010, a top DE gene up-regulated in the airways upon re-challenge, was highly expressed in the airways of the re-challenged mouse (Figure 5B). AW112010 has also been reported capable of promoting the differentiation of inflammatory T cells (Yang et al., 2020). Cbr2, a top DE gene down-regulated in the airways upon re-challenge, was highly expressed in the airways of the immunized mouse (Figure 5C), and may function in the metabolism of endogenous carbonyl compounds (Nakanishi et al., 1995) and alveolar epithelial cell plasticity (Mutze et al., 2015).
Figure 5.

Differential expression analysis of spots annotated as airways in the immunized and the re-challenged mouse, see also Figure S6
(A) Gene Set Enrichment Analysis (GSEA) of all the 16,937 genes available in the DE analysis of the airways.
(B) Expression of AW112010, a top DE gene up-regulated in the airways upon re-challenge, across four slices.
(C) Expression of Cbr2, a top DE gene down-regulated in the airways upon re-challenge, across four slices.
Spatial transcriptomics showing the potential to analyze cell-cell communication
To perform cell-cell communication networks analysis, cell-type enriched spots were identified according to their ranks of proportions of cell types (Figure S6D), with little difference found between the two slices from the same mouse (Figure S6C). Fibroblasts, endothelial cells, and erythrocytes were not included in the analysis due to the difficulty of interpreting the interactions with these cells.
Cell-cell communication networks among the cell-type enriched spots were inferred in each slice using CellChat (Figure S6E) (Jin et al., 2021). To compare the differences in communication patterns between the re-challenged and the immunized mouse, differential interaction strength between cell-type enriched spots was shown by a heatmap (Figure 6A). The communication among myeloid cells, Th1 cells, and Th17 cells was increased in the re-challenged mouse, also revealed by our same-spot co-occurrence analysis. For the immunized mouse, dendritic cells could be the major sender of communication signals, whereas B cells and other T cells 1 were essential receivers of the signals.
Figure 6.

Cell-cell communication among cell-type enriched spots, see also Figure S6
(A) Heatmap showing differential interaction strength between cell-type enriched spots. Outgoing signals were shown in rows, while incoming signals were shown in columns. Increased (or decreased) signals in the re-challenged mouse compared to the immunized mouse were represented using red (or blue) in the color bar. The sum of values within the same column was summarized using the colored bar plot on the top. The sum of values within the same row was summarized using the colored bar plot on the right.
(B) Bar plots showing overall information flow of each signaling pathway. Relative information flow was shown in the stacked bar plot, while raw information flow was shown in the regular bar plot. Enriched signaling pathways were colored in red or cyan.
(C) Th17 enriched spots in the re-challenged mouse were the receivers of the IL6 signaling pathway. (i) Circle plot visualizing the inferred communication network of the IL6 signaling pathway in the re-challenged mouse. The network of the IL6 signaling pathway in the immunized mouse was not significant. Circle sizes represented the number of spots in each group. Edge colors were consistent with the senders of the signal (sources), and edge weights represented the interaction strength. (ii) Violin plot visualizing the expression of genes related to IL6 signaling pathway in cell-type enriched spots from the re-challenged mouse. Gene expression from slice A3 was colored in red, and that from slice A4 was colored in cyan. (iii-iv) Peaks in genomic regions around Il6 and Il6ra in scATAC-seq data.
(D) Th17 enriched spots in the re-challenged mouse were the receivers of the TGF-β signaling pathway. (i) Circle plot visualizing the inferred communication network of the TGF-β signaling pathway in the re-challenged mouse. The network of the TGF-β signaling pathway in the immunized mouse was also significant but not shown. Circle sizes represented the number of spots in each group. Edge colors were consistent with the senders of the signal (sources), and edge weights represented the interaction strength. (ii) Violin plot visualizing the expression of genes related to TGF-β signaling pathway in cell-type enriched spots from the re-challenged mouse. Gene expression from slice A3 was colored in red, and that from slice A4 was colored in cyan. (iii-v) Peaks in genomic regions around Tgfbr1, Tgfbr2, and Acvr1b in scATAC-seq data.
The conserved and context-specific signaling pathways were identified and visualized by bar plots (Figure 6B). TNF pathway was only turned on in the re-challenged mouse, whereas CXCL and CCL pathways were increased. We also confirmed some well-known signaling pathways in the cell-cell communication analysis of spatial transcriptome, outperforming the cell-cell communication analysis of scRNA-seq data, which could not identify these signaling pathways, possibly due to dropouts (Qiu, 2020). The performance of this analysis could be improved as the resolution of spatial transcriptomics evolves. We found Th17 enriched spots in the re-challenged mouse were the receivers of the IL6 signaling pathway (Figure 6Ci), given the receptor gene Il6ra was highly expressed in Th17 enriched spots from slice A3 and slice A4 (Figure 6Cii). Leveraging scATAC-seq data, we confirmed macrophages and dendritic cells were the sources of the IL6 signaling (Figure 6Ciii), and Th17 cells were the targets (Figure 6Civ). For the TGF-β signaling pathway, we also found Th17 enriched spots in the re-challenged mouse were its receivers (Figure 6Di), with the receptor genes Tgfbr1, Tgfbr2, and Acvr1b found highly expressed in Th17 enriched spots from slice A3 and slice A4 (Figure 6Dii). By checking peaks in genomic regions around these receptor genes, Th17 cells were confirmed to have the capacity to express the receptors for the TGF-β signaling (Figures 6Diii–v). These data suggested that spatial transcriptomics data can be extrapolated and utilized to perform cell-cell communication network analysis.
Discussion
With the development of single-cell sequencing technologies, scRNA-seq has been gradually now widely applied to study immune cells and immune responses in mouse lungs (Ballesteros et al., 2020; Cohen et al., 2018; Kurihara et al., 2021; Wang et al., 2021). In many cases, T helper cells and cytotoxic T cells can be identified in these scRNA-seq studies, but not their subtypes. Few studies additionally utilize scATAC-seq to measure the chromatin architecture of immune cells in mouse lungs (Delacher et al., 2021), which allows the identification of subtypes of T helper cells. With the birth of spatial transcriptomics, this cutting-edge technology is also adopted by this field to study mouse lungs infected by influenza (Boyd et al., 2020). The influenza study, containing four slices from infected mice, visualized genes of interest across the slices but did not try to decipher specific cell-type compositions across the slices. Our analytical pipeline could be applied to similar studies in the lung field and facilitate unraveling the spatial dynamics of lung immunization.
To our knowledge, this is the first study to integrate spatial transcriptome with single-cell transcriptome and single-cell epigenome in mouse lungs. A key advantage of our study lies in the capture of intact anatomical structures of mouse lungs, providing location-specific information and preserving information from cells prone to damage. Integrating with single-cell multi-omics profiled from matched tissue, sub-single-cell resolution was further achieved in our spatial analysis of cells residing in lung, especially TRM. In our study, we first identified 12 lung cell types in scRNA-seq data, covering major epithelial, mesenchymal, and immune cells. Using our finely annotated scRNA-seq data as a reference, we deconvoluted spatial transcriptome of the four slices and inferred proportions of cell types for each spot. We established an optimized deconvolution pipeline to accurately decipher specific cell-type compositions at sub-single-cell resolution, by checking the correlation of deconvolution results using two independent references and the colocalization of canonical markers and histological structures. Integrating scATAC-seq data from the same cells processed side by side with scRNA-seq, we found epigenomic profiles facilitate robust cell-type identification, especially for lineage-specific T helper cells, and in current study, we identified four subtypes of T cells. Combining all three data modalities, we mapped the 15 lung cell types to histological structures for the four slices at sub-single-cell resolution.
Our study as a proof of concept suggests that spatial transcriptomics study can provide further insights into dynamic changes of cell locations upon K. pneumoniae re-challenge. We found Th17 cells were closer to the airways than Th1 cells in the re-challenged mouse, whereas Th1 cells were closer to the airways in the immunized mouse without re-challenge. This could be explained by the increased expression of Ccl20 around the airways in the re-challenged mouse. Ccl20, a top distance-associated gene identified by our spatial analysis, has the ability to attract Ccr6 expressing cells. The CCL20/CCR6 axis has been shown to play crucial roles in recruiting Th17 cells in many organs as well as various disease settings (Turner et al., 2010; Wang et al., 2009). We discovered different spatial distribution patterns of immune cells in the lungs of the two mice, finding B cells were proximal to the airways in the immunized mouse, whereas neutrophils were distal to the airways in the re-challenged mouse. We also identified thousands of distance-associated genes for the two mice by natural spline regression, confirming immune responses related genes were over-represented in the re-challenged mouse. These spatial analyses were only possible when location-specific information was captured by spatial transcriptomics, highlighting the value of our study compared with conventional flow cytometric and transcriptomic approaches.
Comparing the biological differences in the airways upon K. pneumoniae re-challenge is also feasible using our established analytical pipeline, we found pathways related to migration and chemotaxis of immune cells, and response to stimuli were up-regulated in the re-challenged mouse, whereas pathways related to catabolic and metabolic processes, and ciliary functions were up-regulated in the immunized mouse, further warrant hypothesis-driven experiments. We also performed cell-cell communication analysis of spatial transcriptome, providing evidence of lineage-specific T helper cells receiving designated cytokine signaling. We acknowledged that cell-type enriched spots still contain several cell types at current resolution. We did not intend to interpret autocrine signaling for these cell-type enriched spots, since it could be real autocrine signaling or short-range communication between neighbor cell types. We thought cell-cell communications between different types of cell-type enriched spots could still be informative, which represents long-range communications between different cell types. A higher resolution of spatial transcriptomics could resolve our current challenges. Our study shows the potential to perform DE analysis between specific cells or regions across the slices and analyze cell-cell communication using spatial transcriptomics. The performance of these analyses could be improved as spatial transcriptomics advances toward single-cell resolution.
Limitations of the study
Several limitations were recognized in our study. First, we had a small sample size for our scRNA-seq and scATAC-seq data as the purpose of generating these data was to provide a reference for deconvolution, instead of carrying out a census of lung cell types. Still, we also observed that the cell number of scRNA-seq data would not drastically impact the performance of deconvolution as long as a signature matrix can be built for each celltype. Second, there is no ground truth to evaluate deconvolution results, although we optimized our deconvolution pipeline by assessing its robustness. This issue could eventually be resolved as the resolution of the spatial transcriptomics technology improves. Third, our method to define airways and blood vessels may not be fully accurate and can be further improved by both high-resolution histological images and enhanced resolution of spatial transcriptome. Finally, more biological replicates and experimental validations may be needed to make definitive biological conclusions. For example, the cellular source of CCL20 around the airways in the re-challenged mouse could be further determined by immunofluorescence. Follow-up studies like this are needed to sufficiently confirm the conclusion.
In summary, as a proof of concept, we presented a comprehensive single-cell multi-omics study on immunized mouse lungs and generated hypotheses for understanding underlying biological mechanisms. Recently, spatial analysis has been made compatible with Formalin-fixed paraffin-embedded (FFPE) tissue specimens. It is foreseeable that a massive amount of data will be generated from historically preserved samples. Our spatial transcriptomics data processing pipeline provides a timely solution to these analyses and contributes to advancing the field of lung biology and respiratory medicine.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Chromium Single Cell 3ʹ GEM, Library & Gel Bead Kit v3 | 10x Genomics | PN-1000075 |
| Chromium Chip B Single Cell Kit | 10x Genomics | PN-1000073 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| Chromium Single Cell ATAC Library & Gel Bead Kit | 10x Genomics | PN-1000110 |
| Chromium Chip E Single Cell ATAC Kit | 10x Genomics | PN-1000155 |
| Chromium i7 Multiplex Kit N, Set A | 10x Genomics | PN-1000084 |
| Visium Spatial Gene Expression Slide & Reagent Kit | 10x Genomics | PN-1000187 |
| Visium Accessory Kit | 10x Genomics | PN-1000194 |
| Dual index Kit TT set A | 10x Genomics | PN-1000215 |
| Deposited data | ||
| Raw and processed scRNA-seq data | This paper | GSE190225 |
| Raw and processed scATAC-seq data | This paper | GSE190225 |
| Raw and processed ST data | This paper; Mendeley Data | GSE190225; https://doi.org/10.17632/2yr6y6wnc5.1 |
| Cohen et al.’s raw and processed scRNA-seq data | Cohen et al., 2018 | GSE119228 |
| Experimental models: Organisms/strains | ||
| C57BL/6J, mus musculus | The Jackson Laboratory | IMSR_JAX:000664 |
| Software and algorithms | ||
| Cell Ranger | 10x Genomics | Version 3.1.0 |
| Cell Ranger ATAC | 10x Genomics | Version 1.1.0 |
| Space Ranger | 10x Genomics | Version 1.2.2 |
| Seurat | https://satijalab.org/seurat/index.html | Version 4.0.1 |
| RCTD | https://github.com/dmcable/RCTD | Version 1.2.0 |
| Signac | https://satijalab.org/signac/index.html | Version 1.1.1 |
| SCENIC | https://github.com/aertslab/SCENIC | Version 1.2.4 |
| AUCell | https://github.com/aertslab/AUCell | Version 1.12.0 |
| QuPath | https://qupath.github.io | Version 0.2.3 |
| limma | https://bioconductor.org/packages/release/bioc/html/limma.html | Version 3.46.0 |
| PANTHER Classification System | http://www.pantherdb.org | Version 16.0 |
| MAST | https://www.bioconductor.org/packages/release/bioc/html/MAST.html | Version 1.16.0 |
| clusterProfiler | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | Version 3.18.1 |
| CellChat | https://github.com/sqjin/CellChat | Version 1.1.0 |
| Other | ||
| Eclipse 90i Motorized Upright Microscope | Nikon | Eclipse 90i |
Resource availability
Lead contact
Additional information and requests for resources and reagents should be directed to and will be completed by the lead contact, Kong Chen (koc5@pitt.edu).
Materials availability
This study did not generate any unique reagents.
Experimental model and subject details
Mouse models
All mice used in this study were wild-type and purchased from Jackson Lab (Cat# 000664). Animals were maintained in pathogen-free conditions in the animal facility at the University of Pittsburgh Medical Center. All experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
In vivo inflammation induction
6–8 weeksold C57BL/6 mice were immunized with heat-killed K. Pneumoniae (ATCC-43816) as previously described (Chen et al., 2011). Briefly, mice were injected with heat-killed K. Pneumoniae twice (Day 0 and Day 7) or three times (Day 0, Day 7, and Day 13) intranasally and sacrificed on Day 14. Lungs were removed and digested by Collagenase/DNase to obtain a single-cell suspension. Mononuclear cells, after red blood cell lysis and filtration with a 40 μM cell strainer, were subjected to single-cell RNA-seq (scRNA-seq) and single-cell ATAC-seq (scATAC-seq) library prep following the protocols by 10x Genomics using the Chromium controller (10x Genomics). To yield sufficient IL-17A producing cells and reduce doublets formation, we targeted 3,000–5,000 cells/nuclei for recovery. Libraries were QC’ed on an Agilent TapeStation and sequenced on an Illumina Novaseq.
Method details
Spatial transcriptomics experiment
We conducted ST experiment using 10X Genomics Visium platform.
Tissue harvesting
Mouse lungs were harvested, and the left lobes were inflated with 1 mL mixture of 50% sterile PBS/50% Tissue-Tek OCT Compound (SAKURA FINETEK) followed by freezing in alcohol bath on dry ice. OCT blocks were stored at −80°C until further processing.
ST library prep
OCT blocks were sectioned at 10 μm in thickness, 6.5 mm × 6.5 mm in size, attached to the Visium slides, then stained with hematoxylin and eosin following 10x Genomics Visium fresh frozen tissue processing protocol. H&E images were taken by a fluorescence and tile scanning microscope (Olympus FluoView 1000) then the slides underwent tissue removal and library generation per 10x Genomics demonstrated protocol.
Raw sequencing data processing
The sequenced scRNA-seq library was processed and aligned to mm10 mouse reference genome using Cell Ranger software (version 3.1.0) from 10x Genomics, with unique molecular identifier (UMI) counts summarized for each barcode. To distinguish cells from the background, cell calling was performed on the full raw UMI count matrix, with the filtered UMI count matrix generated (31,053 genes x 3,337 cells).
The sequenced scATAC-seq library was processed and aligned to mm10 mouse reference genome using Cell Ranger ATAC software (version 1.1.0) from 10x Genomics, with fragments and peak counts summarized for each barcode. To distinguish cells from the background, cell calling was performed on the full raw peak count matrix, with the filtered peak count matrix generated (84,317 peaks x 4,908 cells).
Each sequenced spatial transcriptomics library was processed and aligned to mm10 mouse reference genome using Space Ranger software (version 1.2.2) from 10x Genomics, with UMI counts summarized for each spot. To distinguish tissue overlaying spots from the background, tissue overlaying spots were detected according to the images. And only barcodes associated with these tissue overlaying spots were retained, with the filtered UMI count matrices generated. We also manually excluded spots not covered by tissue but not detected by Space Ranger and further filter the UMI count matrices (slice A1: 32,285 genes x 3,689 spots; slice A2: 32,285 genes x 2,840 spots; slice A3: 32,285 genes x 3,950 spots; slice A2: 32,285 genes x 3,765 spots).
Quantification and statistical analysis
scRNA-seq data analysis
After imported into R, the filtered UMI count matrix was analyzed using the R package Seurat (version 4.0.1) (Hao et al., 2021). The percentage of mitochondrial genes per cell was calculated for further check of the quality of cells. Regularized negative binomial regression (SCTransform) (Hafemeister and Satija, 2019) was used to normalize UMI count data, with the removal of confounding effects from mitochondrial mapping percentage. To improve the speed of the normalization, glmGamPoi (Ahlmann-Eltze and Huber, 2021) was invoked in the procedure. 3,000 highly variable genes were identified and used in principal component analysis to reduce dimensionality. We determined to use the first 50 principal components in clustering analysis according to the elbow plot. UMAP dimensionality reduction (McInnes et al., 2018) was performed with the first 50 principal components as input to visualize cells. Using the shared nearest-neighbor (SNN) graph as input, cells were then clustered using the original Louvain algorithm with resolution = 0.2.
Cluster 1 was marked as low-quality cells and excluded from downstream analysis (2,834 cells retained) because its median percentage of mitochondrial genes was 87.7%, whereas those of all other clusters were lower than 10.5%. Markers for each cluster were identified using a Wilcoxon rank sum test with only.pos = TRUE, min.pct = 0.25, and logfc.threshold = 0.25. According to the UMAP plot, cluster 6 was found to consist of several sporadic sub-clusters. We isolated cells within cluster 6 and repeated the procedures from normalization to dimensionality reduction. These cells were then clustered using the original Louvain algorithm with resolution = 0.6. Markers were also identified as described above.
Characterization of 12 lung cell types in scRNA-seq data
Fine cluster annotation was performed for the retained 12 clusters in scRNA-seq data according to canonical markers: alveolar epithelial cells (Sftpa1, Sftpb, Sftpc, and Sftpd), club cells (Scgb1a1, Muc5b, Scgb3a1, and Scgb3a2), fibroblasts (Col3a1, Col1a2, Col1a1, and Mfap4), endothelial cells (Cdh5, Mcam, Vcam1, and Pecam1), monocytes (Cd14 and Itgam), macrophages (Itgax, Cd68, Mrc1, and Marco), dendritic cells (Aif1, H2-DMb1, H2-Eb1, and H2-Aa), neutrophils (Gsr, Pglyrp1, S100a8, and Ly6g), B cells (Ms4a1, Cd79a, Igkc, and Cd19), T cells (Cd3d, Cd3e, and Cd3g), NK cells (Ncr1 and Nkg7), and erythrocytes (Hbb-bt and Hba-a2).
Spatial transcriptomics data analysis
After imported into R, the filtered UMI count matrix was analyzed using the R package Seurat (version 4.0.1) (Hao et al., 2021). Regularized negative binomial regression (SCTransform) (Hafemeister and Satija, 2019) was used to normalize UMI count matrices, and glmGamPoi (Ahlmann-Eltze and Huber, 2021) was invoked in the procedure to improve the speed of the normalization. Four matrices from the four slices were merged to analyze them together. 3,000 highly variable genes were identified in each matrix, and the union set of them was set as highly variable genes for the merged data and used in principal component analysis to reduce dimensionality. We determined to use the first 30 principal components in clustering analysis according to the elbow plot. UMAP dimensionality reduction (McInnes et al., 2018) was performed with the first 30 principal components as input to visualize spots.
Deconvolution of spatial transcriptome
Deconvolution for each spot in the four slices was performed using the robust cell-type decomposition (RCTD) method (Cable et al., 2022). Before running the R package RCTD (version 1.2.0), scRNA-seq data used as a reference were processed, with gene expression matrix, the annotation for each cell, and the total UMI count for each cell extracted and saved in the RDS object. Spatial transcriptome for the four slices was also processed, with spot location matrices, gene expression matrices, and the total UMI count for each spot extracted and saved in the RDS object.
RCTD objects were created for each slice from the processed RDS objects, with max_cores = 24, test_mode = F, and CELL_MIN_INSTANCE = 6. RCTD pipeline was run on the RCTD objects, with doublet_mode = full. The deconvolution results were matrices of celltype weights for each spot. The celltype weights were normalized to make the sum of celltype weights in each spot equal to 1. Proportion matrices, whose rows indicate spots and columns indicate cell types, were then created and stored in the analyzed spatial transcriptome for loading.
In total, three scRNA-seq references were used in deconvolution, which were in-house scRNA-seq data with 12 cell types, in-house scRNA-seq data with 15 cell types (including four subtypes of T cells), and public scRNA-seq data with 20 cell types.
Pearson correlation coefficients between the columns (indicating different cell types) of the two proportion matrices deconvoluted using in-house (12 cell types) and public (20 cell types) references were calculated in each slice. Correlation r matrices were hierarchically clustered and visualized in heatmaps.
scATAC-seq data analysis
After imported into R, the filtered peak count matrix was analyzed using the R package Signac (version 1.1.1) (Stuart et al., 2020). Gene annotations were extracted from Ensembl release 79 of the mm10 mouse reference genome. Nucleosome signal score and Transcriptional Start Site (TSS) enrichment score per cell were calculated for further check of the quality of cells. Cells whose fraction of fragments in peaks > 15, ratio of reads in genomic blacklist regions < 0.05, nucleosome signal score < 4, and TSS enrichment score > 2 were retained for downstream analysis. Latent Semantic Indexing (LSI) (Cusanovich et al., 2015), which is a combined step of Term Frequency–Inverse Document Frequency (TF-IDF) followed by Singular Value Decomposition (SVD), was used to normalize and reduce the dimensionality of peak count data, with all the peaks selected as variable features. We found the first LSI component was highly correlated with sequencing depth and determined to use the second to the fortieth LSI components in non-linear dimensionality reduction. UMAP dimensionality reduction (McInnes et al., 2018) was performed with the second to the fortieth LSI components as input to visualize cells.
Label transferring from scRNA-seq data to scATAC-seq data
A gene activity matrix was created in scATAC-seq data by counting the number of fragments mapping to promoter or gene body regions of all protein-coding genes for each cell. Regularized negative binomial regression (SCTransform) (Hafemeister and Satija, 2019) was used to normalize the gene activity matrix. Transfer anchors were identified by canonical correlation analysis between the normalized gene activity matrix in scATAC-seq data and the normalized gene expression matrix in scRNA-seq data (Stuart et al., 2019). Annotations were then transferred from scRNA-seq to scATAC-seq data with the second to the fortieth LSI components in scATAC-seq data used for weighting anchors. Canonical markers were checked in the gene activity matrix for each predicted celltype. The proportions of 12 cell types were also compared between scRNA-seq and scATAC-seq data.
Characterization of four subtypes of T cells in scATAC-seq data
We isolated predicted T cells in scATAC-seq data and repeated the procedures from normalization to non-linear dimensionality reduction. Using the SNN graph leveraging the second to the fortieth LSI components as input, T cells were then clustered into four subtypes using the Smart Local Moving (SLM) algorithm with resolution = 0.2.
Markers for each subtype were identified in the gene activity matrix using a Wilcoxon rank sum test with only.pos = TRUE, min.pct = 0.25, and logfc.threshold = 0.25. Differential accessible analysis was performed between the possible Th17 and Th1 cells using logistic regression, with fraction of fragments in peaks set as latent variable and min.pct = 0.1. Peaks in genomic regions around (including all the differentially accessible regions or 10,000 bps apart from the gene bodies) canonical markers for Th17 and Th1 cells were visualized. Motif footprinting analysis was also performed to provide supportive evidence. Taken together, four subtypes of T cells were annotated according to canonical markers: Th17 cells (Il17a, Rorc, and Rora), Th1 cells (Ifng and Tbx21), other T cells 1, and other T cells 2.
Label transferring from scATAC-seq data to scRNA-seq data
We isolated annotated T cells in scRNA-seq data and repeated the procedures from normalization to non-linear dimensionality reduction. Transfer anchors were identified by canonical correlation analysis between the normalized gene activity matrix of T cells in scATAC-seq data and the normalized gene expression matrix of T cells in scRNA-seq data (Stuart et al., 2019). Annotations were then transferred from T cells in scATAC-seq to T cells in scRNA-seq data with the first 50 principal components in T cells from scRNA-seq data used for weighting anchors.
Characterization of four subtypes of T cells in scATAC-seq data
Canonical markers were checked for the predicted subtypes of T cells in scRNA-seq data (Rorc, Rora, Ccr6, and Ccr4 for Th17 cells; Ifng, Tbx21, Cxcr3, Ccr5, and Il12rb2 for Th1 cells). Markers for each predicted subtype of T cells were identified using a Wilcoxon rank sum test with only.pos = TRUE, min.pct = 0.25, and logfc.threshold = 0.25.
To detect active transcription factor (TF) modules, the R package SCENIC (version 1.2.4) (Aibar et al., 2017; Van deSande et al., 2020) was used to analyze the annotated scRNA-seq data, including 15 cell types. We downloaded the RcisTarget database containing transcription factor motif scores for gene promoter and around Transcription Start Site (TSS) for mm10 mouse reference genome from (https://resources.aertslab.org/cistarget/databases/mus_musculus/mm10/refseq_r80/mc9nr/gene_based/). The gene expression matrix was filtered according to default settings, and 9392 genes were retained and used to compute a gene-gene correlation matrix. Co-expression module detection was performed using the GENIE3 algorithm based on random forest. Transcription factor network analysis was performed to detect co-expression modules enriched for target genes of each candidate TF from the RcisTarget database, with regulons identified. The R package AUCell (version 1.12.0) was used to compute a score for each TF module in each cell.
Regulon Specificity Score (RSS) was calculated for regulons in each celltype according to area under the curve (AUC) of regulons. Cell-type specific regulators were then identified, and those for the four subtypes of T cells were visualized to provide supportive evidence (Rora_extended (15g) for Th17 cells).
Same-spot co-occurrence analysis
Pearson correlation coefficients between the columns (indicating different cell types) of the proportion matrices were calculated in each slice to broadly assess spatial celltype co-occurrence in the same spot. Spots from the slices for the immunized (slice A1 and A2) or the re-challenged mouse (slice A3 and A4) were pooled together, respectively, and Pearson correlation coefficients were also calculated for them. Correlation r matrices were hierarchically clustered and visualized in heatmaps.
Defining airways and blood vessels in four slices
Airways were defined according to the proportion of club cells in four slices. We manually set the thresholds in each slice to match the selected spots with the histological airways. Spots whose proportion of club cells is higher than the thresholds were defined as airways (20% for slice A1, 20% for slice A2, 10% for slice A3, and 10% for slice A4).
Blood vessels were defined according to the histological blood vessels. We created a training set using manual annotation of histological structures in the image of slice A1 and trained a random trees pixel classifier using QuPath (version 0.2.3) (Bankhead et al., 2017) with downsample = 16. The probability of blood vessels was predicted for each pixel in the image of four slices using the trained classifier. If the probability of blood vessels in the spot corresponding pixel was higher than 0.5, the spot would be defined as blood vessels.
Alternatively, the proportion of club cells and the expression of Mgp were used to define airways and blood vessels with different cut-offs, from 90th quantile to 95th quantile. Spots whose proportion or expression is higher than the selected quantile were defined as airways or blood vessels.
Spatial transcriptomics spot distance-based analyses
For all distance-based analyses, spots defined as blood vessels and spots whose distances to the airways are longer than 1,000 μm were excluded. Because cells within the blood vessels were different from those in the lung parenchyma, and the analyses including cells extremely distal to the airways were not stable, given the capture area of each slice was only 6.5 × 6.5 mm2. Only a few spots’ distances to the airways were longer than 1,000 μm (5.89% for slice A1, 5.75% for slice A2, 15.00% for slice A3, and 11.43% for slice A4).
Weighted distances to the airways for Th17 and Th1 cells were calculated in four slices according to the formula, allowing for each spot’s distance to the nearest airway and the proportions of Th17 and Th1 cells in each spot.
To find the spatial distribution patterns of cell types, natural spline regression (with three degrees of freedom) was performed to fit the non-linear relationship between the proportions of cell types and the distances to the airways.
To identify distance-associated genes, we constructed natural splines (with three degrees of freedom) for the distances to the airways in each slice and created design matrices. We then fitted linear models for each gene in normalized gene expression matrices, with the R package limma (version 3.46.0) (Ritchie et al., 2015) invoked to speed up the procedure. p-values for each spline in each slice were corrected for multiple testing using Benjamini-Hochberg correction. Genes with FDR-adjusted p-values < 0.05 for at least one spline were considered significant in that slice. And genes significant in both slices for the immunized (slice A1 and A2) or the re-challenged mouse (slice A3 and A4) were defined as distance-associated genes.
Gene ontology (GO) enrichment analysis
Gene ontology enrichment analysis was performed on the distance-associated genes identified in the immunized and re-challenged mouse using PANTHER Classification System (version 16.0) (Ashburner et al., 2000; Mi et al., 2019; The Gene Ontology Consortium, 2021). GO biological process complete was used as annotation dataset, and the analysis was performed using Fisher’s exact test, with FDR adjustment for multiple testing. Over-represented pathways were then hierarchically clustered.
Differential expression analysis
Differential expression analysis was performed to compare the airways in the re-challenged mouse versus the immunized mouse using the R package MAST (version 1.16.0) (Finak et al., 2015). MAST procedure was invoked in Seurat FindMarkers function (test.use = MAST) with logfc.threshold = 0, min.pct = 0, in order to obtain an unfiltered gene list. The gene list was then ranked by log2-fold change (L2FC). Genes whose L2FC is equal to 0 were excluded because their ranks were not available.
Gene Set Enrichment Analysis (GSEA)
To overcome the limitations of the analysis based on manually selected DEGs, Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) was performed on the unfiltered, ranked gene list (including 16,937 genes) using the R package clusterProfiler (version 3.18.1) (Yu et al., 2012). The parameters for the clusterProfiler gseGO function were set as ont = BP, keyType = SYMBOL, nPerm = 10,000, minGSSize = 3, maxGSSize = 800, pvalueCutoff = 0.05, OrgDb = org.Mm.eg.db, pAdjustMethod = fdr. The top 30 (by normalized enrichment score) up-regulated and down-regulated pathways were then visualized by a lollipop plot.
Defining cell-type enriched spot in spatial transcriptomics data
The expression profile of each spot in spatial transcriptomics is a mixture of a few cells, and it is irrational to annotate a spot with a cell type directly. To perform cell-cell communication analysis of spatial transcriptome, we annotated spots as cell-type enriched spots according to their proportions of cell types.
The mean proportions of cell types were available for each slice, as well as the number of spots for each slice. The mean proportions could be interpreted as the expected proportions of cell types in each spot. The product of the mean proportions and the number of spots could be interpreted as the average spots representing each cell type. The spots with the highest proportions were selected according to the product and defined as cell-type enriched spots. Spots defined as cell-type enriched spots for multiple cell types were then excluded.
For example, the mean proportion of club cells was 6.39% in slice A1, and the number of spots was 3,689 in slice A1. Thus, the top 236 spots with the highest proportion of club cells were defined as club cell enriched spots. A spot would be excluded if the spot was defined as a club cell enriched spot and a Th17 enriched spot simultaneously.
Cell-cell communication analysis of spatial transcriptome
Cell-cell communication analysis was performed using CellChat (Jin et al., 2021). Before running the R package CellChat (version 1.1.0), spatial transcriptome for the four slices was processed, with normalized gene expression matrices, the annotations for cell-type enriched spots extracted and saved in the CellChat object. The processed data from both slices for the immunized (slice A1 and A2) or the re-challenged mouse were also pooled together and saved in the CellChat objects to compare the differences between the two mice.
A manually curated database of literature-supported ligand-receptor interactions in mouse was loaded for the analysis. And all ligand-receptor interactions, including paracrine/autocrine signaling interactions, extracellular matrix (ECM)-receptor interactions, and cell-cell contact interactions, were included in the analysis.
Over-expressed ligands or receptors were first identified for each type of cell-type enriched spots in each CellChat object. Over-expressed ligand-receptor interactions were also identified if either ligand or receptor was over-expressed. Then, we computed the communication probability and inferred the cellular communication network according to default settings. The communication probability at signaling pathway level was computed by summarizing the communication probabilities of all ligand-receptor interactions associated with each signaling pathway. The aggregated cell-cell communication network was calculated by counting the number of links or summarizing the communication probability and visualized in four slices using heatmaps.
To figure out in which type of cell-type enriched spots interactions significantly changed in the re-challenged mouse versus the immunized mouse, differential interaction strength was identified and visualized using a heatmap. The conserved and context-specific signaling pathways were identified by comparing the information flow for each signaling pathway, which was defined by the total weights in the network. Selected signaling pathways were visualized by circle plots using CellChat netVisual_aggregate function.
Acknowledgments
This project is supported by HL137709 and P01AI106684 from National Institute of Health. This research is also supported in part by the University of Pittsburgh Center for Research Computing through the resources provided.
Author contributions
Z.X., W.C., and K.C. conceived the project and designed the experiments. L.F., F.W., B.L., and G.T. performed scRNA-seq, scATAC-seq, and ST experiments. Z.X. and X.W. performed scRNA-seq, scATAC-seq, and ST data analysis. J.W. provided guidance for the deconvolution of ST data. Z.X., W.C., and K.C. wrote the manuscript with input from all authors. W.C. and K.C. supervised the work.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects. One or more authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: September 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104900.
Contributor Information
Wei Chen, Email: wec47@pitt.edu.
Kong Chen, Email: koc5@pitt.edu.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. Raw and processed data of scRNA-seq, scATAC-seq, and ST are deposited in Gene Expression Omnibus (GEO) (GSE190225). Processed ST data with deconvolution results are deposited in Mendeley Data: https://doi.org/10.17632/2yr6y6wnc5.2 (Xu et al., 2022). Code and scripts necessary to repeat analyses in this paper are available at GitHub (https://github.com/xzlandy/Spatial_immunized_mouse_lungs). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ahlmann-Eltze C., Huber W. glmGamPoi: fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics. 2021;36:5701–5702. doi: 10.1093/bioinformatics/btaa1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros I., Rubio-Ponce A., Genua M., Lusito E., Kwok I., Fernández-Calvo G., Khoyratty T.E., van Grinsven E., González-Hernández S., Nicolás-Ávila J.Á., et al. Co-Option of neutrophil fates by tissue environments. Cell. 2020;183:1282–1297.e18. doi: 10.1016/j.cell.2020.10.003. [DOI] [PubMed] [Google Scholar]
- Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd D.F., Allen E.K., Randolph A.G., Guo X.-Z.J., Weng Y., Sanders C.J., Bajracharya R., Lee N.K., Guy C.S., Vogel P., et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature. 2020;587:466–471. doi: 10.1038/s41586-020-2877-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cable D.M., Murray E., Zou L.S., Goeva A., Macosko E.Z., Chen F., Irizarry R.A. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 2022;40:517–526. doi: 10.1038/s41587-021-00830-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., McAleer J.P., Lin Y., Paterson D.L., Zheng M., Alcorn J.F., Weaver C.T., Kolls J.K. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity. 2011;35:997–1009. doi: 10.1016/j.immuni.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M., Giladi A., Gorki A.-D., Solodkin D.G., Zada M., Hladik A., Miklosi A., Salame T.-M., Halpern K.B., David E., et al. Lung single-cell signaling interaction map reveals basophil role in macrophage imprinting. Cell. 2018;175:1031–1044.e18. doi: 10.1016/j.cell.2018.09.009. [DOI] [PubMed] [Google Scholar]
- Cusanovich D.A., Daza R., Adey A., Pliner H.A., Christiansen L., Gunderson K.L., Steemers F.J., Trapnell C., Shendure J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910–914. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M., Simon M., Sanderink L., Hotz-Wagenblatt A., Wuttke M., Schambeck K., Schmidleithner L., Bittner S., Pant A., Ritter U., et al. Single-cell chromatin accessibility landscape identifies tissue repair program in human regulatory T cells. Immunity. 2021;54:702–720.e17. doi: 10.1016/j.immuni.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.-H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara C., Lecuona E., Wu Q., Yang W., Núñez-Santana F.L., Akbarpour M., Liu X., Ren Z., Li W., Querrey M., et al. Crosstalk between nonclassical monocytes and alveolar macrophages mediates transplant ischemia-reperfusion injury through classical monocyte recruitment. JCI Insight. 2021;6:e147282. doi: 10.1172/jci.insight.147282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Zhang W., Guo C., Xu H., Li L., Fang M., Hu Y., Zhang X., Yao X., Tang M., et al. Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nat. Methods. 2022;19:662–670. doi: 10.1038/s41592-022-01480-9. [DOI] [PubMed] [Google Scholar]
- McInnes L., Healy J., Melville J. UMAP: Uniform Manifold approximation and projection for dimension reduction. arXiv. 2018 doi: 10.48550/arXiv.1802.03426. Preprint at. [DOI] [Google Scholar]
- Mi H., Muruganujan A., Ebert D., Huang X., Thomas P.D. Panther version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47:D419–D426. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutze K., Vierkotten S., Milosevic J., Eickelberg O., Königshoff M. Enolase 1 (ENO1) and protein disulfide-isomerase associated 3 (PDIA3) regulate Wnt/β-catenin-driven trans-differentiation of murine alveolar epithelial cells. Dis. Model. Mech. 2015;8:877–890. doi: 10.1242/dmm.019117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M., Deyashiki Y., Ohshima K., Hara A. Cloning, expression and tissue distribution of mouse tetrameric carbonyl reductase. Identity with an adipocyte 27-kDa protein. Eur. J. Biochem. 1995;228:381–387. [PubMed] [Google Scholar]
- Qiu P. Embracing the dropouts in single-cell RNA-seq analysis. Nat. Commun. 2020;11:1169. doi: 10.1038/s41467-020-14976-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Srivastava A., Lareau C., Satija R. Multimodal single-cell chromatin analysis with Signac. bioRxiv. 2020 doi: 10.1101/2020.11.09.373613. Preprint at. [DOI] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P.A., Miron M., Farber D.L. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 2019;4:eaas9673. doi: 10.1126/sciimmunol.aas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D.L., Farber D.L. Mucosal resident memory CD4 T cells in protection and immunopathology. Front. Immunol. 2014;5:331. doi: 10.3389/fimmu.2014.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J.-E., Paust H.-J., Steinmetz O.M., Peters A., Riedel J.-H., Erhardt A., Wegscheid C., Velden J., Fehr S., Mittrücker H.W., et al. CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis. J. Am. Soc. Nephrol. 2010;21:974–985. doi: 10.1681/ASN.2009070741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q., et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- Wang C., Kang S.G., Lee J., Sun Z., Kim C.H. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol. 2009;2:173–183. doi: 10.1038/mi.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Netto K.G., Zhou L., Liu X., Wang M., Zhang G., Foster P.S., Li F., Yang M. Single-cell transcriptomic analysis reveals the immune landscape of lung in steroid-resistant asthma exacerbation. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2005590118. e2005590118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Chen W., Chen K. Integrative analysis of spatial transcriptome with single-cell transcriptome and single-cell epigenome in mouse lungs after immunization. bioRxiv. 2022 doi: 10.1101/2021.09.17.460865. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Bam M., Becker W., Nagarkatti P.S., Nagarkatti M. Long noncoding RNA AW112010 promotes the differentiation of inflammatory T cells by suppressing IL-10 expression through histone demethylation. J. Immunol. 2020;205:987–993. doi: 10.4049/jimmunol.2000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. Raw and processed data of scRNA-seq, scATAC-seq, and ST are deposited in Gene Expression Omnibus (GEO) (GSE190225). Processed ST data with deconvolution results are deposited in Mendeley Data: https://doi.org/10.17632/2yr6y6wnc5.2 (Xu et al., 2022). Code and scripts necessary to repeat analyses in this paper are available at GitHub (https://github.com/xzlandy/Spatial_immunized_mouse_lungs). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.