

Abstract
The synthesis of self-assembled peptide nanoparticles using a facile one-pot synthesis approach is gaining increasing attention, allowing therapy in combination with diagnosis. Their drawback is limited diagnostic potential, which can be improved after necessary modifications and efficacious functionalization. Herein, a cyclic heptapeptide having the Arg-Gly-Asp-Lys-Leu-Ala-Lys sequence was modified by conjugation of the ε-amino group of the terminal lysine residue with diethylenetriamine pentaacetic acid (DTPA) as a bifunctional chelating agent (BFC) for radiolabeling with a γ-emitting radionuclide (99mTc, half-life 6.01 h; energy 140 keV). Further, the free amino group of the middle lysine residue was successfully conjugated with near-infrared fluorescence (NIRF) dye Cyanine5.5 N-succinimidyl ester (Ex/Em = 670/701 nm) by a co-assembly method to form newly designed novel NIRF dye conjugated self-assembled peptide–DTPA (Cy5.5@SAPD) nanoparticles. The fluorescent nanoparticle formation was confirmed by using a fluorescence spectrophotometer (Ex/Em = 650/701 nm), and the transmission electron microscope (TEM) images showed a size of ∼ 40 nm with a lattice fringe distance of 0.294 nm. Cytotoxicity and confocal laser scanning microscopy (CLSM) studies showed that these nanoparticles possess a high affinity for the αvβ3-integrin receptor overexpressed on brain tumor glioblastoma with an EC50 = 20 μM. Moreover, these nanoparticles were observed to have potential to internalize into U87MG cells more prominently than HEK-293 cancer cells and induce apoptosis. The apoptosis assay showed 79.5% apoptotic cells after 24 h treatment of Cy5.5@SAPD nanoparticles. Additionally, these nanoparticles were also radiolabeled with 99mTc for the single photon emission computed tomography (SPECT) imaging study in tumor-bearing female Balb/c mice. The excellent imaging feature of Cy5.5@SAPD-99mTc nanoparticles as a multimodal (SPECT/NIRF) diagnostic probe, as well as noteworthy therapeutic potential was observed. The results suggested that our newly designed novel dual-targeting dual-imaging nanoparticles may serve as an admirable theranostic probe to treat brain tumor glioblastoma multiforme.
This study describes the development of self-assembled peptide nanoparticles with improved functionality via modifications by using multimodal imaging tools and enhanced the therapeutic efficacy against brain tumor glioblastoma multiforme.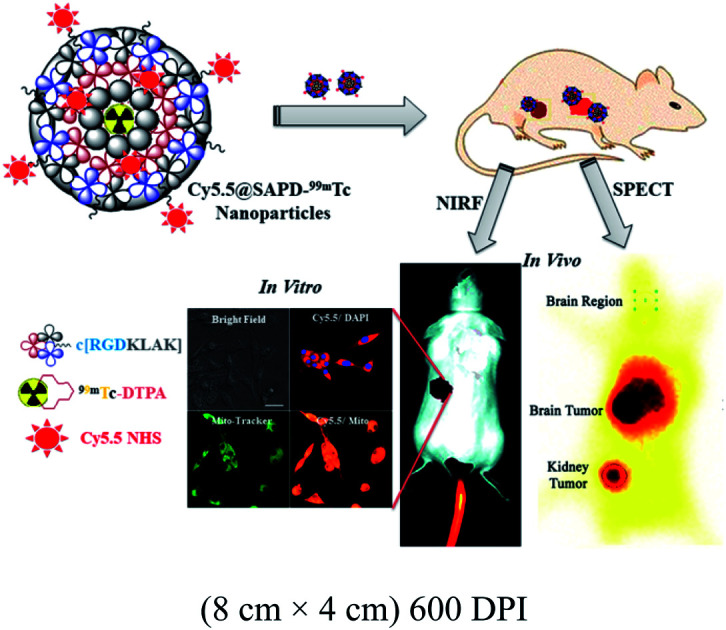
1. Introduction
The development of malignant cells in the circulatory system is progressively assisted by the growth of new blood vessels by utilizing oxygen and nutrients, ultimately increasing tumor angiogenesis. Self-regulation of angiogenesis causes the progression of diseases such as the proliferation of cancer cells, glioblastoma multiforme (GBM), myocardial infarction, and atherosclerosis.1 For the treatment of cancer, anti-angiogenic therapy in combination with anticancer therapy has emerged as a novel strategy to combat the growth of tumor cells by discontinuing their nutrient oxygen supply. Multidomain-targeting drug delivery serves as a guided missile to effectively and efficiently target the cancer cell vasculature. These targeted drugs have been developed using peptides/proteins, integrin-receptor ligands, antibodies, and aptamers possessing recognition domains and effector domains.2 There are many cognate receptors and pro-angiogenic factors involved to promote vessel formation in tumors such as the fibroblast growth factor-2 (FGF-2), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF).3 These growth factors up-regulate the expression of integrins including α1β1, α2β1, α4β1, α5β1, α9β1, and αvβ3-integrins on blood and lymphatic vessels. Several investigations implicate integrins as key regulators of tumor angiogenesis which also regulate endothelial cell survival and migration and mediate cell–cell adhesion and cell–extracellular matrix (ECM) events.4,5 Basic clinical studies reveal that angiogenesis can be blocked by inhibiting the angiogenic signaling pathways, ultimately resulting in tumor dormancy and metastasis.6,7
Integrin-mediated signaling pathways play an important role in tissue development and homeostasis, while its deregulation causes multiple brain diseases.8 Interestingly, integrins are crucial glycoproteins essential for many physiological processes such as proliferation, cell migration, wound healing, hemostasis, bone remodeling, and oncogenic transformation.9,10 They are composed of nineteen α- and eight β-subunits,11,12 and among them, αvβ3-, αvβ5-, αvβ8-, α5β1-, and αIIbβ3-integrins have been extensively studied for their active role as an excellent candidate for cancer theranostics.13 More specifically, the αvβ3-integrin serves as a receptor for ECM proteins such as fibronectin, vitronectin, fibrinogen, collagen, laminin, and osteopontin with an exposed arginine-glycine-aspartic acid (RGD) sequence.14 It is expressed on epithelial and mature epithelial cells at low levels, while highly expressed on the surface of many tumors including carcinomas of breast and lungs, melanomas, osteosarcomas, and glioblastoma.15–17 Hence, the αvβ3-integrin is considered as a molecular target of interest for the early diagnosis of cancer and selective attachment and internalization of RGD-containing peptides and peptidomimetics for cancer therapy.18 The cyclic monomeric (RGDfV) and multimeric (Galacto-RGD) peptides have emerged in phase-III clinical trials for the diagnosis of glioblastoma and in phase-II trials for many other tumors.19 It is the first anti-angiogenic small molecule drug that specifically targets the αvβ3-, αvβ5-, and α5β1-integrins and is the first potent and superactive αvβ3-integrin receptor antagonist.
Over the last two decades, both linear and cyclic RGD peptide analogs have been discovered, radiolabeled (99mTc, 111In, 68Ga, and 18F), and evaluated as radiotracers for tumor diagnosis using single-photon emission computed tomography (SPECT) or positron emission tomography (PET) imaging.20 Linear RGD peptides showed a lack of αvβ3-integrin specificity, low binding affinity, and high enzymatic degradation of the aspartic acid residue due to free rotation around a single bond. In contrast, cyclic RGD peptides were observed to be highly stable towards proteases and had increased affinity to the αvβ3-integrin receptor with reduced structural flexibility.21 The literature provides ample knowledge on the development of hybridized RGD conjugated anticancer peptide analogs including RGD-111In-DTPA-octreotate,22,2318F-bombesin-RGD,2499mTc-RGD-bombesin,2564Cu-RGD2-PG12-bombesin heterodimers,26 and 5FU-loaded SF-cRGDfK-Ce6 (ref. 27) for targeted drug delivery and improved diagnostic as well as therapeutic efficacy.
In the light of the above knowledge, we hypothesized that designing a short head-to-tail cyclic heptapeptide sequence using the RGD (αvβ3-integrin receptor binding) motif along with the KLAK (mitochondria targeting) motif without the addition of unnecessary linking amino acids can improve integrin targeting and mitochondrial damaging potential for therapeutic applications. Secondly, the modification of this cyclic heptapeptide by incorporating a NIRF dye for optical imaging as well as a BFC for radiolabeling with β/γ-emitting radionuclides for the SPECT imaging study resulted in self-assembled peptide nanoparticles that could be effective as a novel dual-imaging agent for SPECT/NIRF diagnosis.
To the best of our knowledge, we are the first who have designed this novel head-to-tail cyclic RGD-KLAK heptapeptide sequence.2 To evaluate the validation of our hypothesis, first we modified the ε-amino group of the terminal lysine residue with diethylenetriamine pentaacetic acid (DTPA; as the BFC). Secondly, this peptide–DTPA complex was intrinsically converted to self-assembled peptide nanoparticles via co-assembly with Cy5.5 (as the NIRF-probe), which covalently conjugated with the free amino group of the intermediate lysine residue and introduced optical imaging features. Further aim was to investigate the effectiveness and efficacy of this newly developed novel NIRF-dye conjugated self-assembled peptide nanoparticles radiolabeled with 99mTc (Cy5.5@SAPD-99mTc) for in vitro and in vivo diagnosis of glioblastoma multiforme (GBM) as well as therapeutic potential as a multimodal-imaging (SPECT/NIRF) and dual-targeting probe.
2. Materials and methods
2.1. Chemistry
2.1.1. Synthesis of the cyclic peptide–DTPA complex
A simple condensation reaction of DTPA dianhydride for coupling with the cyclic peptide was carried out using the protocol reported by Shi et al. 2011 after fewer modifications.28 Briefly, DTPA dianhydride (cDTPA; 1.5 mg, 0.0042 mmol) was dissolved in dimethyl formamide (DMF; 50 μL) and dimethyl sulfoxide (DMSO; 150 μL). The deprotected free carboxylic group of cDTPA was converted to an activated ester by treatment with hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU; 1.59 mg, 0.0042 mmol) dissolved in DMF (50 μL) and stirred for 30 min at room temperature. Then, the white crystals of the cyclic peptide c[RGD(Boc)KLAK] (3.65 mg, 0.0042 mmol) were dissolved in Milli-Q water (200 μL) and were added dropwise over 30 min into a cDTPA mixture and stirred for another 30 min. Later on, the coupling agent N,N-diisopropylethylamine (DIPEA; 20 μL, 0.115 mmol) dissolved in DMSO (50 μL) was added to the reaction mixture to obtain pH 8.5–9.0, followed by stirring at 900 rpm for an additional 6 h at room temperature under N2-purging. Upon completion of the reaction, the Boc protecting group was cleaved by treatment with 95% trifluoroacetic acid in water followed by stirring for 2 h at room temperature. Finally, the mixture was diluted with ultrapure deionized water and was adjusted to pH 6.0 using neat trifluoroacetic acid (TFA). The final product (c[RGDKLAK]-DTPA; cPD) was lyophilized to get white crystalline powder (3.68 mg, ∼78% yield) and stored at −20 °C until further use. The complex was characterized by qualitative analysis by using a high pressure liquid chromatography system equipped with a C-18 column and UV detector (λ = 220 nm). An isocratic mobile phase system having solvent A (0.1% TFA in acetonitrile; 87.5%) and solvent B (0.1% TFA in water; 12.5%) was used with a flow rate of 1 mL min−1. The formation of the required product was confirmed by measuring the molar mass using liquid chromatography-mass spectrometry (LC-MS) analysis, while the presence of additional functional groups was confirmed by Fourier-transform infrared-attenuated total reflection (FTIR-ATR) analysis of the cPD complex.
2.1.2. Synthesis of fluorescent functionalized self-assembled peptide–DTPA nanoparticles
A facile self-assembling strategy was adopted for the synthesis of cyclic peptide nanoparticles as reported in the literature.29 Briefly, the cPD complex (1 mg, 0.87 mmol) was dissolved in 1.8 mL of Milli-Q water. Additionally, 0.2 mL of 0.1 M sodium borate solution (Na2B4O7, pH 8.3) containing an equivalent amount of Cy5.5-NHS ester (0.67 mg/20 μL DMSO) was added dropwise into the cPD solution. The pH of the reactions mixture was adjusted to pH 6 using neat TFA in a glass vial and the vial was capped air-tight. Further, the solution was stirred at 4 °C in the darkness under N2-purging for up to 48 h (high speed 24 h, medium 12 h, and low 12 h). On the next day, the reaction was quenched by adding acetic acid (150 μL; 5% in water), and the suspension was centrifuged at 13 000 rpm to isolate the NIRF-dye coupled self-assembled cyclic peptide–DTPA (Cy5.5@SAPD) nanoparticles, followed by washing with deionized (DI) water twice to remove the acidic water. Next, the mixture was sonicated for 2 h to obtain dispersed SAPD nanoparticles in DI water and they were characterized by fluorescence spectroscopy, dynamic light scattering (DLS), and transmission electron microscopy (TEM).
2.1.3. Radiosynthesis of Cy5.5@SAPD nanoparticles
The intrinsic radiolabeling of these NIRF-dye coupled self-assembled cyclic peptide–DTPA (Cy5.5@SAPD) nanoparticles with 99mTc (a γ-emitting radionuclide) was carried out by using sodium borohydride (NaBH4) as a reducing agent via an fac-[99mTc(CO)3(H2O)3]+ precursor, as reported previously.30 Briefly, the complex of the fac-[99mTc(CO)3(H2O)3]+ (99mTc-tricarbonyl) precursor was prepared by taking 4 mg of Na2CO3, 5.5 mg of NaBH4, and 10 mg of Na/K-tartrate in deionized water (0.5 mL), vortexing for 30 s, followed by purging with N2-gas for 5 min to remove bubbles in the solution. Then, ∼185 MBq of 99mTc was added to the solution vial and allowed to incubate for 30 min at 95 °C. After successful radiolabeling, the solution was cooled to room temperature and the pH of the solution was adjusted to pH 7 using 0.1 M HCl solution. The radiochemical purity of the resulting 99mTc-tricarbonyl complex was assessed by using thin-layer chromatography plates coated with silica gel (TLC-SG) as the stationary phase and by using a mixture of methanol : HCl (95 : 5% v/v) as the mobile phase (Rf for 99mTc-tricarbonyl = 0.85–0.95; Rf for Cy5.5@SAPD-99mTc(CO)3 = 0.55–0.75) as well as a radio-HPLC method. Approximately 98% pure 99mTc-tricarbonyl complex (∼185 MBq/0.5 mL saline) was filtered through a 0.22 μm Millipore membrane and added into Eppendorf tubes containing Cy5.5@SAPD nanoparticles (as the ligand; 20 μg/100 μL Na-PBS, pH 6.5). The reaction mixture was allowed to incubate for 20 min at 55 °C. After successful radiolabeling, the newly developed radiotracer (Cy5.5@SAPD-99mTc(CO)3 nanoparticles) was allowed to cool at room temperature, and the quality control analysis was performed using TLC-SG and ultra-centrifugation methods.
2.2. In vivo imaging, pharmacokinetics and therapeutic study
For the in vivo study, female Balb/c mice (5 weeks old, 16–18 g, 20 mice in total) were purchased from the National Institute of Health (NIH), Islamabad, Pakistan. All animal studies were carried out as per guidelines issued by the animal ethical committee of the National Institute of Health (NIH), Islamabad, Pakistan and National Regulation of China for Care and Use of Laboratory Animals (NRCCUL) of Lanzhou University, China. All the experiments performed in this study were approved by NIH and NRCCUL. Each mouse model bearing brain tumor glioblastoma and human embryonic kidney cancers was successfully established using U87MG and HEK-293 cells, respectively, using a concentration of 5 × 107 cells suspended in 100 μL PBS (pH 7.4). The cells were subcutaneously injected into the right flank area of each mouse and the mouse were kept at standard conditions till the tumor volume reached to 50–60 mm3. All mice were divided into two groups (n = 3 for each group) randomly to receive normal saline (control) and Cy5.5@SAPD-99mTc (treated). To observe the in vivo tumor accumulation and therapeutic potential, a single therapeutic dose of Cy5.5@SAPD in saline (30 mg per kg body weight; b.w) was injected intravenously via the tail vein of each group. The body weight and tumor size of each mouse were monitored after a couple of days. On the sixth day post-injection, mice were injected with 20 μg/200 μL of Cy5.5@SAPDN-99mTc(CO)3 and images were acquired using a SPECT camera. Later on, they were sacrificed after giving chloroform anesthesia; tumor mass, as well as other organs, were segregated and weighed and radioactivity was measured using a NaI(Tl) γ-scintillation counter. At the same time, the images were also acquired using an IN VIVO FX camera for optical imaging, and the segregated organs were also observed for the uptake of our newly designed nanoparticles by observing the fluorescence intensity in each organ.
2.3. Statistical analysis
All experiments reported in this study were performed in triplicate and results are given as ±standard deviation (±SD) of ‘n’ independent measurements. Statistical significance was calculated using the Student's t-test. The significance level was assigned as p < 0.05.
3. Results and discussion
3.1. Chemistry
3.1.1. Synthesis of the cyclic peptide–DTPA complex
The Fmoc-synthesis approach was used to design a novel dual-targeting cyclic peptide–DTPA (cPD) complex with high chemical yield and efficacy. In this newly designed heptapeptide sequence, the receptor binding motif (RGD) was used as a vehicle to selectively deliver the mitochondria targeting peptide (KLAK) to the cancer cell surface.31 The free ε-amino group of the terminal lysine residue was coupled with the deprotected free β-carboxylic group of cDTPA via covalent linkage for labeling with 99mTc. The cyclic heptapeptide–DTPA complex was successfully synthesized with a satisfactory yield of ∼78% as presented in Step-I of Scheme 1. The purity of the cyclic peptide–DTPA (cPD) complex was confirmed by HPLC analysis, indicating a single peak with ≥98% purity at retention time Rt = 3.780 min (Fig. S1†), and HR-MS analysis showed molecular mass peaks calculated for C47H81N15O18 with m/z [M + 2H+] = 1144.01 a.m.u (Fig. S2†). Furthermore, FTIR-ATR analysis showed superimposed spectra of cDTPA (black line), the cyclic peptide (red line), and cPD complex (blue line) having peaks at 3265.1 cm−1 and 1640.2 cm−1 assigned to the stretching vibrations of the –NH2 and –NC O groups; respectively, and a peak at 2109.7 cm−1 assigned to the bending vibrations of the –COOH group (Fig. S3†).
Scheme 1. Schematic representation of the reaction mechanisms for the modification of cyclic peptide c[RGDKLAK] with cDTPA as the BFC to design the c[RGDKLAK]–DTPA complex (Step-I). Further, the complex was self-assembled to form nanoparticles in the presence of Cyanine5.5 NHS ester to form Cy5.5 coupled self-assembled cyclic peptide–DTPA (Cy5.5@SAPD) nanoparticles (Step-IIa), followed by radiolabeling with the 99mTc-Tricarbonyl precursor (Step-IIb).

3.1.2. Synthesis of Cy5.5@SAPD nanoparticles and radiolabeling with 99mTc
To rationalize the simplicity of the facile one-pot self-assembly strategy and functionalization of optical imaging modality, we choose Cy5.5 NHS ester as the NIRF dye. Next, the deprotected amino group of the central lysine residue of the cPD complex was covalently conjugated with Cy5.5 during the self-assembly process, as illustrated in StepII(a) of Scheme 1.32 The co-assembly of Cy5.5 with the cPD complex was successfully achieved to yield uniform size self-assembled peptide nanoparticles (Cy5.5@SAPD) with a well-defined spherical shape and “always ON” NIR-fluorescence properties by using the pH-sensitive method.29,33 This will facilitate in improving the diagnostic and therapeutic efficiencies of dual-targeting dual-imaging Cy5.5@SAPD nanoparticles, whilst maintaining the intrinsic biocompatibility and biodegradability.34 The effect of self-assembly on the change in fluorescence intensity was assessed by using a fluorescence spectrophotometer, and the spectrogram presented in Fig. 1a showed peaks at an excitation wavelength of 650 nm and emission wavelength of 702 nm in the aggregation state, which is nearly consistent with the parent NIRF dye (Cy5.5 NHS; Ex/Em = 650/700 nm) with a slight increase in the emission wavelength indicating successful co-assembly of Cy5.5 with the cPD complex.35 The freshly synthesized Cy5.5@SAPD nanoparticles were further radiolabeled with technetium-99m by using a fac-[99mTc-(CO)3(H2O)3]+ core complex for SPECT/CT imaging.
Fig. 1. Qualitative analysis of Cy5.5@SAPD and Cy5.5@SAPD-99mTc nanoparticles: (a) fluorescence intensity measurements for Cy5.5@SAPD nanoparticles, (b) percent radiochemical yield of Cy5.5@SAPD-99mTc nanoparticles via ultracentrifugation, and (c and d) TEM images acquired for dried Cy5.5@SAPD and Cy5.5@SAPD-99mTc nanoparticles, respectively. The inset figures show high-resolution TEM images of the respective nanoparticles.

The radiosynthon introduced by Alberto et al. has been widely used for preferential radiolabeling of peptides/proteins to achieve relatively high specificity, while retaining the biological properties of bioactive molecules.36 The radiosynthon was successfully prepared with a high radiochemical purity of ≥97% showing a single high-intensity peak at retention time Rt = 4.727 min, as indicated by radio-HPLC analysis (Fig. S4†). The inset figure shows the TLC-SG results which showed that fac-[99mTc(CO)3(H2O)3]+ moves with the solvent front at Rf = 0.90, while free 99mTc remain at the point of spotting (Rf = 0.00). The electron donor nitrogen and oxygen groups of DTPA provide an easy platform for radiolabeling with 99mTc in the presence of NaBH4 as the reducing agent and an optimum pH value, as shown in Step-II(b) of Scheme 1. Consequently, the radiolabelled nanoparticles were observed to remain stable at room temperature over a 4 h incubation period and no change in the radiolabeling efficiency was seen by increasing the concentration of Cy5.5@SAPD-99mTc nanoparticles with a percent radiochemical purity of >96% (Fig. 1b), obtained using the ultracentrifugation technique. The same results were obtained from the TLC-SG/methanol technique, and the inset images presented in Fig. S4† show ∼97% yield at Rf = 0.65, while <3% impurities were found in saline.30
Additionally, transmission electron microscope (TEM) images presented in Fig. 1c and d show a clear spherical morphology of Cy5.5@SAPD and Cy5.5@SAPD-99mTc nanoparticles, respectively, with uniform dispersion in an aqueous medium. The size of Cy5.5@SAPD nanoparticles was observed in between 30–40 nm, as indicated in the inset image Fig. 1c′ acquired by using a high resolution-TEM, and for Cy5.5@SAPD-99mTc nanoparticles, the size reduced to 20–25 nm, as shown in the inset image Fig. 1d′. The clear consecutive bight and dark lattice fringes with an interplanar lattice fringe distance of 0.294 nm, as presented in Fig. 1d′, indicates a tight interface among all three-ingredient (i.e. SAPD, NIRF-dye, and radionuclide 99mTc) which could facilitate in balancing the surface charge as well as demonstrates the successful self-assembly of the cPD complex with Cy5.5 having crystallinity in novel designed nanoparticles.37 The dynamic light scattering (DLS) measurements revealed that the hydrodynamic size of Cy5.5@SAPD nanoparticles with a diameter of less than 100 ± 28 nm, as depicted in Fig. S5,† enabled the capabilities of novel designed nanoparticles in vitro and in vivo.38
3.2. In vitro cell study
3.2.1. Cytotoxicity study
Upon successful modification of the dual-targeting cyclic peptide to design dual imaging self-assembled peptide nanoparticles, in vitro cytotoxicity studies were further carried out to evaluate the effectiveness, specificity, and efficacy. The dual-targeting capabilities of Cy5.5@SAPD nanoparticles were assessed by using the αvβ3-integrin positive cancer cell line (U87MG) and αvβ3-integrin negative cells (HEK-293). Both cancer cells were separately treated with Cy5.5@SAPD and Cy5.5@SAPD-99mTc nanoparticles to estimate the cytotoxic potential. The results presented in Fig. 2a showed that the cytotoxic effect against U87MG cells was significantly higher for Cy5.5@SAPD-99mTc nanoparticles as compared to Cy5.5@SAPD nanoparticles, with EC50 values of 20 nM and 25 nM, respectively (p < 0.05). In contrast, the MTT assay showed that both nanoprobes were weakly cytotoxic towards HEK-293 cancer cells as ≥80% viable cells were found in 96-well plates, as presented in the bar graph (Fig. 2b). The significantly higher cytotoxicity of Cy5.5@SAPD-99mTc nanoparticles might be due to the attachment of γ-emitting radionuclides compared with Cy5.5@SAPD nanoparticles.39 So, further cell studies were performed using Cy5.5@SAPD nanoparticles to avoid the effect of diagnostic radionuclides.
Fig. 2. In vitro cell study of novel peptide nanoparticles using HEK-293 and U87MG glioblastoma cancer cell lines. A cytotoxicity study was performed in (a) U87MG cancer cells, and (b) HEK-293 cells using Cy5.5@SAPD and Cy5.5@SAPD-99mTc nanoparticles and (c and d) show the CLSM images acquired with the co-localization of Cy5.5@SAPD nanoparticles along with DAPI staining dye. FACS analysis shows the apoptosis-inducing factor in U87MG cells (e) as the control experiment and (f) for the nanoparticle treated study, CLSM images acquired after co-localization of Mito-Tracker Green dye along with Cy5.5@SAPD nanoparticles (g) as well as caspase-3 staining dye (h). (i–k) Bio-TEM images acquired after treatment with Cy5.5@SAPD nanoparticles in U87MG cells, indicating successful mitochondrial damage.

3.2.2. Confocal laser scanning microscopy
The CLSM images acquired after treatment with Cy5.5@SAPD nanoparticles showed very less red fluorescence intensity in HEK-293 cells (Fig. 2c), and alternatively showed bright red fluorescence intensity in U87MG cells (Fig. 2d). The co-localization of DAPI staining in the nuclear region shows blue fluorescence, while the merged image shows localization of red fluorescence in the nuclear periphery region. These results highlighted the specificity and efficacy of the newly designed nanoparticles for αvβ3-integrin positive cancer cells due to the RGD tripeptide.11
3.2.3. Apoptosis assay
Further, we investigated the potential of Cy5.5@SAPD nanoparticles to induce apoptosis in U87MG cancer cells. The results presented in Fig. 2e showed 95.6% live cells as the control and Fig. 2f showed 17.2% live cells, 26.8% early apoptotic and 52.7% late apoptotic cells, and only 1.3% necrotic cells. This study gives validation of our hypothesis that the newly designed Cy5.5@SAPD nanoparticles have the potential to kill cancer cells by inducing apoptosis in glioblastoma cancer cells (U87MG) due to the presence of the KLAK motif.40 Moreover, to investigate the appropriate cell apoptosis-inducing pathway, we also performed the CLSM imaging study by treating the U87MG cancer cells with Cy5.5@SAPD nanoparticles and stained the cells with Mito-Tracker Green as well as caspase-3 dyes. The CLSM images acquired with a Cy5.5 (red) filter after 30 min incubation time showed the internalization of Cy5.5@SAPD nanoparticles into cancer cells via pinocytosis and enters into the mitochondria by disrupting the mitochondrial membrane, as indicated in Fig. 2g; and the merged image showed well-overlapped fluorescence to give yellowish-green color intensity, and a scale bar of 20 μm was set for all images. This ultimately produces reactive oxygen species (ROS) that causes mtDNA damage, whilst it releases cytochrome C to promote the formation of an apoptosome, causing the activation of the caspase-3 enzyme, as shown in Fig. 2h.41 The bright green fluorescence is due to activated caspase-3 and is well-overlapped with red fluorescence, as presented in the merged image, to give bright yellow fluorescence intensity in the nuclear periphery region.42,43 The merged CLSM images with blue colored holes show a stained nuclear region (Fig. 2c and d) while blank holes showed an unstained nuclear region (Fig. 2g and h), indicating improved specificity of our novel Cy5.5@SAPD nanoparticles to selectively target mitochondria and induce cell apoptosis.
3.2.4. Bio-TEM imaging
Additionally, we also performed a bio-TEM imaging study using Cy5.5@SAPD nanoparticle treated U87MG cancer cells, and the TEM images showed internalization of nanoparticles through pinocytosis, as shown in Fig. 2i. The magnified image showed the accumulation of nanoparticles in the inner-layers of the cell membrane (Fig. 2j) which targets the mitochondria to induce apoptosis by damaging the mitochondrial membrane, as illustrated by the high-resolution TEM image presented in Fig. 2k. Bio-TEM images are in agreement with the CLSM study evidenced the induction of cancer cell apoptosis by damaging the mitochondria with high specificity and improved efficacy.38,44,45
3.3. Pharmacokinetics, in vivo imaging, and therapeutic study
Additionally, we also investigated the dual-imaging potential of Cy5.5@SAPD-99mTc nanoparticles in brain tumor glioblastoma (U87MG cells) and human embryonic kidney (HEK-293) tumor-induced (5 × 107 cells per mice subcutaneously) female Balb/c mouse models. Firstly, the nanoparticles with a concentration of 20 μg/200 μL (74 MBq) saline were injected via the tail vein, and images were acquired using a dynamic SPECT/CT camera (NM/CT 670 Pro Discovery) as well as a fluorescence imaging camera (IN VIVO FX Pro Carestream). The images presented in Fig. 3a–c showed CT, SPECT, and SPECT/CT images acquired after 30 min post-injection (p.i) with coronal, sagittal, and transaxial directions, indicating the occurrence of brain tumor glioblastoma and the kidney tumor (as indicated in CT images) with excellent accumulation of novel Cy5.5@SAPD-99mTc nanoparticles (SPECT images), as shown in the tumor-to-background contrast at the left side of Balb/c mice (presented in SPECT/CT images).46 The results presented in Fig. 3d–g show planar SPCET images acquired at 30 min p.i (Fig. 3d left) and 2 h p.i (right) before therapeutic dose treatment, showing high internalization of the radiotracer in brain tumor glioblastoma with 4.7 ± 0.8% ID g−1 as compared to the kidney tumor (2.1 ± 0.9% ID g−1) with a target-to-nontarget ratio (T/NT) of 2.24 ± 0.6 at 30 min p.i. The results of the planar SPECT imaging study are comparable with those of the biodistribution study, as presented in the bar graph in Fig. S6 (see the ESI†).
Fig. 3. In vivo SPECT/CT and fluorescence imaging studies in tumor-bearing animal models. (a–c) Images show the SPECT/CT study of Cy5.5@SAPD-99mTc nanoparticles in the brain tumor and kidney tumor-induced mouse models, (d) shows in vivo static SPECT images after 30 min and 2 h post-injection (p.i) before therapeutic dose treatment, (e) shows ex vivo images indicating the pharmacokinetic study, (f) shows SPECT images after 30 min and 2 h p.i after treatment, and (g) shows the ex vivo image. (h) shows in vivo and (i) 30 min and (j) 2 h ex vivo fluorescence images in the U87MG glioblastoma tumor-bearing mouse model.

After therapeutic dose (30 mg per kg b.w) treatment of Cy5.5@SAPD within a very short period of time (say nearly one week), the planar SPECT imaging study was performed again. Fig. 3f clearly showed much less or negligible accumulation of the radiotracer at the site of a brain tumor while prominent uptake can be seen at the kidney tumor site; this is because of the binding potential of the RGD motif to the αvβ1-integrin overexpressed on HEK-293 cells.9 A sharp decrease in the brain tumor size indicates the excellent therapeutic potential of Cy5.5@SAPD nanoparticles for GBM. Moreover, Fig. 3e shows an ex vivo image presenting the pharmacokinetics of Cy5.5@SAPD-99mTc nanoparticles before and (Fig. 3g) after therapeutic dose treatment. The main accumulation of the radiotracer was found in the brain, liver, lungs, and kidneys and nonspecific uptake was observed in the heart, stomach, and spleen. Furthermore, the brain tumor-bearing female Balb/c mice models were subjected to fluorescence camera imaging, and the results presented in Fig. 3h showed in vivo and those in Fig. 3i and j showed ex vivo images after 30 min and 2 h p.i; respectively. The in vivo live imaging study showed the accumulation of Cy5.5@SAPD nanoparticles at the site of brain glioma tumors. The tumor uptake was observed to be highly prominent than normal brain tissues, and other body organs pointed out the efficacy, specificity, and effectiveness of our newly designed novel Cy5.5@SAPD nanoparticles as compared to those of previously reported nanoparticles.47–50
4. Conclusion
In the light of the above-mentioned results, we presented the successful development of a dual-targeting peptide sequence consisting of the RGD motif for targeting the αvβ3-integrin and KLAK pro-apoptotic motif for targeting the mitochondria, which ultimately resulted in inducing cancer cell apoptosis by the activation of the caspase-3 enzyme. This dual-targeting peptide probe was further modified with DTPA. Further, the cyclic peptide–DTPA complex was self-assembled into peptide nanoparticles via co-assembly with NIRF-dye Cy5.5 to form uniform spherical shaped nanoparticles, followed by radiolabeling with 99mTc for molecular imaging studies as a novel dual-imaging probe. Consequently, these novel dual-imaging and dual-targeting self-assembled cyclic peptide nanoparticles were successfully designed to possess improved diagnostic and therapeutic capabilities with enhanced specificity and efficiency for GBM. The in vitro cytotoxicity assay, apoptosis assay, and CLSM imaging studies illustrated that these newly synthesized Cy5.5@SAPD nanoparticles have potential to internalize specifically and efficiently into U87MG brain tumor cells as compared to HEK-293 kidney tumor cells for early diagnosis of GBM. SPECT/NIRF diagnostic studies in tumor-bearing female Balb/c mouse models showed the excellent potential of our novel designed Cy5.5@SAPD-99mTc nanoparticles to diagnose brain tumor more prominently. The therapeutic dose treatment showed effective depletion in tumor tissues, indicating the remarkable therapeutic effectiveness within one-week treatment. These outcomes suggested that our novel theranostic nanoparticles (Cy5.5@SAPD-99mTc) may serve efficiently, and specifically as a potential SPECT/NIRF nanoprobe for future pre-clinical and clinical studies on brain tumor glioblastoma multiforme.
Funding
None of the author(s) received funding from any institution for the execution of this research work and/or publication of the article.
Author contributions
S. F. A. Rizvi: conceptualization, methodology, validation, investigation, visualization, writing – original draft, writing – review & editing. A. Ali: formal analysis, visualization. M. Ahmad: resources, software, data curation. S. Mu: validation, formal analysis. H. Zhang: validation, writing – review & editing, supervision, project administration.
Conflicts of interest
All authors declare no potential conflict of interest in any way.
Supplementary Material
Acknowledgments
The authors are cordially thankful to the Director of the Institute of Nuclear Medicine and Oncology, Lahore, Pakistan for providing hot-lab facilities and the necessary radioisotopes and SPECT/CT camera facility for animal study.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00597a
References
- Shi H. Kwok R. T. K. Liu J. Xing B. Tang B. Z. Liu B. J. Am. Chem. Soc. 2012;134:17972. doi: 10.1021/ja3064588. [DOI] [PubMed] [Google Scholar]
- Rizvi S. F. A. Mu S. Wang Y. Li S. Zhang H. Biomed. Pharmacother. 2020;127:110179. doi: 10.1016/j.biopha.2020.110179. [DOI] [PubMed] [Google Scholar]
- Lugano R. Ramachandran M. Dimberg A. Cell. Mol. Life Sci. 2020;77:1745. doi: 10.1007/s00018-019-03351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraamides C. J. Garmy-Susini B. Varner J. A. Nat. Rev. Canc. 2008;8:604. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S. Kodama S. Kunstfeld R. Kajiya K. Brown L. F. Detmar M. J. Exp. Med. 2005;201:1089. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin E. Y. Pollard J. W. Cancer Res. 2007;67:5064. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Kerbel R. S. Nature. 2005;438:967. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- Ellert-Miklaszewska A. Poleszak K. Pasierbinska M. Kaminska B. Int. J. Mol. Sci. 2020;21:888. doi: 10.3390/ijms21030888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittert J. Bansal R. Storm G. Prakash J. Adv. Drug Delivery Rev. 2018;129:37. doi: 10.1016/j.addr.2018.01.020. [DOI] [PubMed] [Google Scholar]
- Ganguly K. K. Pal S. Moulik S. Chatterjee A. Cell Adhes. Migrat. 2013;7:251. doi: 10.4161/cam.23840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danhier F. Le Breton A. Préat V. Mol. Pharm. 2012;9:2961. doi: 10.1021/mp3002733. [DOI] [PubMed] [Google Scholar]
- Zheng Y. Ji S. Czerwinski A. Valenzuela F. Pennington M. Liu S. Bioconjugate Chem. 2014;25:1925. doi: 10.1021/bc500452y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrake H. M. Patterson L. H. J. Med. Chem. 2014;57:6301. doi: 10.1021/jm5000547. [DOI] [PubMed] [Google Scholar]
- Park E. J. Myint P. K. Ito A. Appiah M. G. Darkwah S. Kawamoto E. Shimaoka M. Front. Cell Dev. Biol. 2020;8:588066. doi: 10.3389/fcell.2020.588066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung K. Y. Dickinson A. Donoghue J. F. Polekhina G. White S. J. Grammatopoulos D. K. McKenzie M. Johns T. G. St John J. C. Acta Neuropathol. Commun. 2014;2:1. doi: 10.1186/2051-5960-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malric L. Monferran S. Gilhodes J. Boyrie S. Dahan P. Skuli N. Sesen J. Filleron T. Kowalski-Chauvel A. Cohen-Jonathan Moyal E. Toulas C. Lemarié A. Oncotarget. 2017;8:86947. doi: 10.18632/oncotarget.20372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J. Guo A. A. Chowdhury I. Guo S. Hibbert J. Wang G. Liu M. Front. Oncol. 2019;9:1413. doi: 10.3389/fonc.2019.01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas-Moruno C. Rechenmacher F. Kessler H. Anticancer Agents Med. Chem. 2010;10:753. doi: 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamous M. Haberkorn U. Mier W. Molecules. 2013;18:3379. doi: 10.3390/molecules18033379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S. Czerwinski A. Zhou Y. Shao G. Valenzuela F. Sowiński P. Chauhan S. Pennington M. Liu S. Mol. Pharm. 2013;10:3304. doi: 10.1021/mp400085d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornesello A. L. Buonaguro L. Tornesello M. L. Buonaguro F. M. Molecules. 2017;22:1282. doi: 10.3390/molecules22081282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capello A. Krenning E. P. Bernard B. F. Breeman W. A. van Hagen M. P. de Jong M. J. Nucl. Med. 2004;45:1716. [PubMed] [Google Scholar]
- Hofland L. J. Capello A. Krenning E. P. de Jong M. van Hagen M. P. J. Nucl. Med. 2005;46:191. [PubMed] [Google Scholar]
- Yan Y. Chen K. Yang M. Sun X. Liu S. Chen X. Amino Acids. 2011;41:439. doi: 10.1007/s00726-010-0762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Huang J. Dong C. Cui L. Jin X. Jia B. Zhu Z. Li F. Wang F. Mol. Pharm. 2012;9:1409. doi: 10.1021/mp200661t. [DOI] [PubMed] [Google Scholar]
- Lucente E. Liu H. Liu Y. Hu X. Lacivita E. Leopoldo M. Cheng Z. Bioconjugate Chem. 2018;29:1595. doi: 10.1021/acs.bioconjchem.8b00113. [DOI] [PubMed] [Google Scholar]
- Mao B. Liu C. Zheng W. Li X. Ge R. Shen H. Guo X. Lian Q. Shen X. Li C. Biomaterials. 2018;161:306. doi: 10.1016/j.biomaterials.2018.01.045. [DOI] [PubMed] [Google Scholar]
- Shi J. Kim Y. S. Chakraborty S. Zhou Y. Wang F. Liu S. Amino Acids. 2011;41:1059. doi: 10.1007/s00726-009-0439-0. [DOI] [PubMed] [Google Scholar]
- Katyal P. Meleties M. Montclare J. K. ACS Biomater. Sci. Eng. 2019;5:4132. doi: 10.1021/acsbiomaterials.9b00408. [DOI] [PubMed] [Google Scholar]
- Gaonkar R. H. Baishya R. Paul B. Dewanjee S. Ganguly S. Debnath M. C. Ganguly S. MedChemComm. 2018;9:812. doi: 10.1039/C7MD00638A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Niu G. Wang F. Chen X. Eur. J. Nucl. Med. Mol. Imag. 2009;36:1483. doi: 10.1007/s00259-009-1123-z. [DOI] [PubMed] [Google Scholar]
- Nie Z. Luo N. Liu J. Zeng X. Zhang Y. Su D. Nanoscale Res. Lett. 2020;15:81. doi: 10.1186/s11671-020-03309-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. Fan Z. Wang Y. Huang Y. Schmidt M. Zhang M. Soft Matter. 2015;11:3822. doi: 10.1039/C5SM00533G. [DOI] [PubMed] [Google Scholar]
- Chen Q. Wang X. Wang C. Feng L. Li Y. Liu Z. ACS Nano. 2015;9:5223. doi: 10.1021/acsnano.5b00640. [DOI] [PubMed] [Google Scholar]
- Cheng Z. Wu Y. Xiong Z. Gambhir S. S. Chen X. Bioconjugate Chem. 2005;16:1433. doi: 10.1021/bc0501698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberto R. Schibli R. Egli A. Schubiger A. P. Abram U. Kaden T. A. J. Am. Chem. Soc. 1998;120:7987. doi: 10.1021/ja980745t. [DOI] [PubMed] [Google Scholar]
- Song C. Wang Y. Rosi N. L. Angew. Chem., Int. Ed. 2013;52:3993. doi: 10.1002/anie.201209910. [DOI] [PubMed] [Google Scholar]
- Yang P.-P. Zhang K. He P.-P. Fan Y. Gao X. J. Gao X. Chen Z.-M. Hou D.-Y. Li Y. Yi Y. Cheng D.-B. Zhang J.-P. Shi L. Zhang X.-Z. Wang L. Wang H. Sci. Adv. 2020;6:4107. doi: 10.1126/sciadv.aaz4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askari Rizvi S. F. Zhang H. Eur. J. Med. Chem. 2021;221:113538. doi: 10.1016/j.ejmech.2021.113538. [DOI] [PubMed] [Google Scholar]
- Dufort S. Sancey L. Hurbin A. Foillard S. Boturyn D. Dumy P. Coll J.-L. J. Drug Target. 2011;19:582. doi: 10.3109/1061186X.2010.542245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J. Tait S. W. G. Br. J. Cancer. 2015;112:957. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P. Pan W. Li N. Tang B. Chem. Sci. 2019;10:6035. doi: 10.1039/C9SC01652J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Sun Q. Huang Z. Huang L. Xiao Y. J. Mater. Chem. B. 2019;7:2749. doi: 10.1039/C9TB00043G. [DOI] [PubMed] [Google Scholar]
- Tang W. Fan W. Lau J. Deng L. Shen Z. Chen X. Chem. Soc. Rev. 2019;48:2967. doi: 10.1039/C8CS00805A. [DOI] [PubMed] [Google Scholar]
- Ayo A. Laakkonen P. Pharmaceutics. 2021;13:481. doi: 10.3390/pharmaceutics13040481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.-Q. Yang Y. Fang W. Liu S. Nucl. Med. Biol. 2016;43:661. doi: 10.1016/j.nucmedbio.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel S. England C. G. Chen F. Cai W. Adv. Drug Deliv. Rev. 2017;113:157. doi: 10.1016/j.addr.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key J. Leary J. F. Int. J. Nanomedicine. 2014;9:711. doi: 10.2147/IJN.S53717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. Yang S. Shi J. Zhao H. Zhong L. Liu Z. Jia B. Wang F. Sci. Rep. 2016;6:18905. doi: 10.1038/srep18905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangger C. Helbok A. Sosabowski J. Kremser C. Koehler G. Prassl R. Andreae F. Virgolini I. J. von Guggenberg E. Decristoforo C. Int. J. Nanomedicine. 2013;8:4659. doi: 10.2147/IJN.S51927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.