

Abstract
The global COVID-19 pandemic continues due to emerging Severe Acute Respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern (VOC). Here, we performed comprehensive analysis of in-house sequenced SARS-CoV-2 genome mutations dynamics in the patients infected with the VOCs - Delta and Omicron, within Recovered and Mortality patients. Statistical analysis highlighted significant mutations - T4685A, N4992N, and G5063S in RdRp; T19R in NTD spike; K444N and N532H in RBD spike, associated with Delta mortality. Mutations, T19I in NTD spike, Q493R and N440K in the RBD spike were significantly associated with Omicron mortality. We performed molecular docking for possible effect of significant mutations on the binding of Remdesivir. We found that Remdesivir showed less binding efficacy with the mutant Spike protein of both Delta and Omicron mortality compared to recovered patients. This indicates that mortality associated mutations could have a modulatory effect on drug binding which could be associated with disease outcome.
Keywords: SARS-CoV-2, Genomic surveillance, Mutation analysis, Disease severity, Molecular docking, Drug efficacy
Graphical abstract
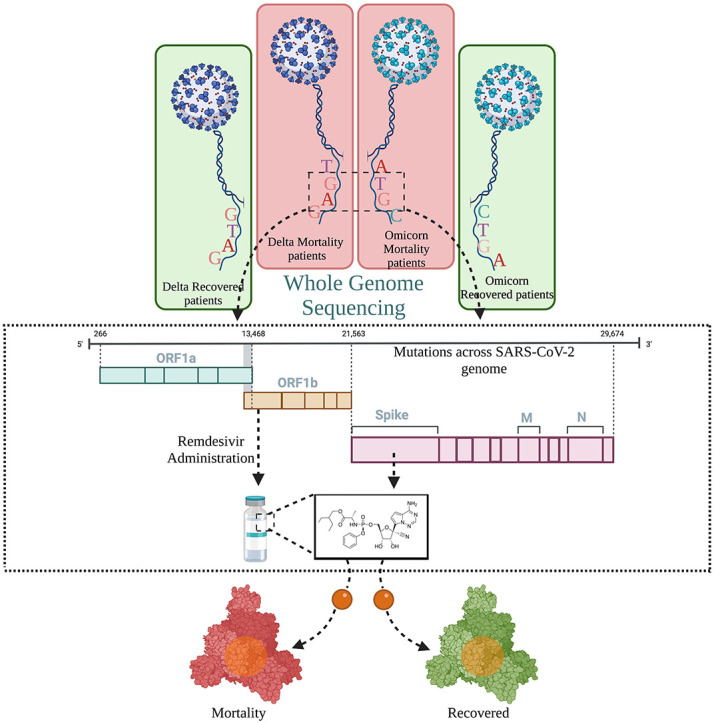
Fig. 1: Summary of the possible functional role of SARS-CoV-2 VOCs, mutations, and drug efficacy; with modulatory role in disease severity and clinical outcome. Distribution of SARS-CoV-2 infected individuals with VOCs - Delta and Omicron and segregation based on their disease outcome into Recovered and Mortality. Genome sequencing reveals the mutation spectrum of both VOCs across the different clinical outcome, and the modulatory potential of mutations vis-à-vis interaction to Remdesivir, a widely used drug during the different waves of COVID-19.
1. Introduction
The persistent upsurges of infectious cases engendered by the origin of SARS-CoV-2 and its novel variants of concern (VOC) resulting in >570 million cases worldwide have undoubtedly overburdened the public healthcare, medical infrastructure and posed economic challenges. The viral genome has been evolving since its advent, and surveilling viral genome diversity during SARS-CoV-2 evolution has therefore been a high priority to restrain viral prevalence [57]. Since the SARS-CoV-2 single-stranded RNA virus exhibits the ability to acquire rapid mutations as it proliferates within a geographical region, only a subset of mutations are naturally selected, favouring enhanced viral transmissibility, replication, and host immune evasion [34]. Consequently, several VOCs have emerged, challenging the containment measures of the pandemic in spite of practiced social measures and by reducing vaccines and available therapeutics efficacy [5].
Emergence of SARS-CoV-2 VOCs has led to natural selection of several mutations with distinct functional consequences that are being explored, elucidated and understood in greater detail through the usage of model systems, genomic means, and mechanism/s. Mutations, which are the drivers of potential evolutionary changes over time could have an effect on epidemiology, antigenicity, and immune escape mechanisms, thereby influencing the overall virus fitness [39,55]. The first widely studied substitution, D614G, became dominant and is present in most of the variants now with a role in enhancing viral replication [27]. Another substitution, N501Y, convergently evolved in the Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), and Omicron (B.1.1.529). The N501Y has been reported to enhance the spike protein interaction with the human host ACE2 receptor and has been shown to influence vaccine efficacy [12,49]. Numerous other studies have highlighted that Delta or B.1.617.2, had a higher proportion of infected patients who warranted ICU admission and unfortunate mortality due to the presence of nine mutations in the Spike protein of SARS-CoV-2 [31,56]. Omicron, harbouring the highest number of mutations in the Spike protein, compared to other existing VOCs, has been reported to cause a milder disease phenotype than the Delta variant [50,53]. Hence, VOCs have been attributed to exacerbate the disease severity sub-phenotypes and differential mortality rates reported worldwide (https://covid19.who.int).
Since the beginning of the pandemic, multiple drugs such as antivirals, antibiotics, and antimalarials have been predicted to be effective against SARS-CoV-2 [11]. Towards that, several clinical trial studies, and computational drug interaction studies have been done and published worldwide [1,3,43,46]. For instance, drugs such as chloroquine and hydroxychloroquine, antimalarial drugs, which were available in the markets, were widely utilized to treat/control COVID-19 symptoms in the beginning of the pandemic for quicker antiviral intervention. Later, it was realised that these drugs do not effectively inhibit SARS-CoV-2 replication and reduce the severity [20,48]. Notably, remdesivir, an adenosine analogue, which inhibits RdRp of SARS-CoV-2, was widely used in India during the Delta driven 2nd wave of COVID-19. However, a WHO trial which included 11,266 adults showed that the drug remdesivir had little or no impact towards improved survival of SARS-CoV-2 infected patients [6]. On the other hand, Gurung et al. found that the drugs, Paritaprevir and Emetine, could serve as an antiviral against SARS-CoV-2 RdRp, through molecular docking studies [18]. Nevertheless, it is important to note that mutations emerging in the drug interaction sites or in the target protein can potentially influence the drug binding efficiencies with a role in antiviral or vaccine development. Mohammed et.al showed that remdesivir binds more efficiently with RdRp mutant P323L than the wildtype by molecular simulation [35]. However, it was reported that nirmatrelvir, a drug targeting the main protease of SARS-CoV-2, showed similar binding potential with its variants as well as the wildtype [58].
The global endeavour to track the evolutionary trajectory of SARS-CoV-2 throughout the pandemic and screen for efficacious drugs against newly emerging viral variants is crucial. In this context, we aimed to comprehensively analyse the mutational spectrum of in-house sequenced SARS-CoV-2 VOCs affected individuals with two distinct clinical outcomes of Recovered vs Mortality, to identify viral genomic variability/ies, possibly affecting the drug efficacy. To elucidate and understand this aspect, we investigated the molecular docking characteristics of different SARS-CoV-2 target proteins including Nsp12, Nsp13, and Spike protein. Findings indicate the functional role of mutations within the SARS-CoV-2 genome through sub-optimal drug binding compromising the efficacy.
2. Methods
2.1. Patient categorization and sample segregation
A total of 249 nasopharyngeal RNA samples were obtained from the hospital admitted SARS-CoV-2 infected patients for this study. All the patient samples were given anonymous identities at CSIR-IGIB. Further, the patients were classified into four groups based on the disease/clinical outcomes obtained from the clinical data (Recovered and Mortality for each of Delta and Omicron). This was further overlaid with the SARS-CoV-2 genome sequencing confirmed VOC, Delta and Omicron, leading to the infection (Supplementary file 1).
2.2. Whole genome sequencing
The whole genome sequencing of 249 SARS-CoV-2 genome sequences was performed utilising both Oxford Nanopore technology (ONT) and Illumina (Nextseq 2000 and MiSeq) platforms.
For Nanopore based sequencing: Out of 249 samples, 32 samples were sequenced through Rapid Barcoding kit protocol (SQK-RBK110.96) available for the ONT platform. In summary, 50 ng of the extracted viral RNA from the nasopharyngeal samples was used to reverse transcribe the RNA into complementary DNA using LunaScript RT SuperMix (New England Biolab. Cat No. E3010L). The single-stranded cDNA was further utilized to amplify the viral genome using rapid barcoding primers (IDT Product number: 10007184) and Q5 High-Fidelity 2× master mix (New England Biolabs, Cat. No. M0494S). The amplicons were then ligated to the unique barcode sequences and purified using SPRI beads. The prepared library was ligated to the adapter sequences and loaded on MinION Mk1C for sequencing ( Fig. 1A).
Fig. 1.

Methodology used in our study for SARS-CoV-2 whole genome sequencing and analysis. (A) Graphical representation of the sequencing workflow from nasopharyngeal RNA extraction to library preparation, followed by sequencing on different platforms– ONT and Illumina. (B) A workflow for sequencing analysis on both ONT and Illumina platform, highlighting the data quality control, read annotation and mutation calling.
Illumina sequencing: The remaining 217 samples were sequenced using Illumina COVIDSeq protocol (Cat. No.20043675 and reference guide: 1000000126053 v04). The cDNA was synthesised from the extracted nasopharyngeal RNA and was further used to amplify the SARS-CoV-2 genome through two separate multiplex PCR reactions. This was followed by tagmentation, a process to fragment and ligate amplicons with adapter sequences. The tagged amplicons then underwent a bead based purification where they were further amplified in a second round of PCR with the addition of index adapters and sequences essential for sequencing cluster generation. The indexed samples were pooled and purified using Illumina Tune beads. The final library was quantified using the Qubit dsDNA HS Assay kit (Cat. No. Q32854). The prepared library was denatured and diluted to prepare a loading concentration of 11pM and 1 nM, accordingly using the MiSeq and Nextseq 2000 System Denature and Dilute Libraries Guide respectively (Illumina, Document no. 15039740 v10 & Document # protocol 1,000,000,126,053 v04). The denatured library was then sequenced using the MiSeq reagent kit v3 or NextSeq 1000/2000 P2 flow cell.
2.3. Sequencing data analysis
The ONT minION raw fast5 files were analysed using the ARTIC end-to-end pipeline up to variant calling. These raw files were basecalled and demultiplexed through the Guppy basecaller that functions using the algorithms of ONT with a phred score cut-off >7 (Nanopore Community). Consequently, all the reads having a score below 7 were eliminated to filter out the low-quality reads which would compromise mutation calling confidence. The demultiplexed fast5 files were normalised according to the average size of the amplicon (1200 bp) and were aligned to the reference SARS-CoV-2 genome (MN908947.3) using Minimap2 v2.17 [32]. Following their alignment to the reference genome, the fast5 files were indexed through Nanopolish [33] for variant calling from the minimap output files. Bcftools v1.8 was used to create consensus fasta with normalised minimap output (Fig. 1B) [10].
For the fastq files generated in Illumina sequencing data, a FASTAQC was performed to examine the phred quality score with a cut-off >20 (Babraham Bioinformatics – FastQC, A Quality Control tool for High Throughput Sequence Data). The low quality reads were filtered out and the adapter sequences were trimmed through the Trim Galore tool (Babraham Bioinformatics - TrimGalore). In order to separate any human read contamination, the sequences were aligned using the HISAT2 algorithm on human data build hg38 [26]. The consensus fasta files were then generated using the BEDTools and variant calling was performed with high phred quality score reads [45]. The genomic coverage and depth for all the samples are available in Supplementary File 2.
2.4. Mutation analysis
The genomic diversity of the 249 SARS-CoV-2 samples was analysed by calculating the frequency of mutations vis-a-vis samples within the mortality and recovered patients across the VOCs – Delta and Omicron. Since two different sequencing platforms were utilized, in order to deduce the heterozygous calls, a haplotype based variant caller freebayes was used for Illumina sequencing data [16]. To further detect the genetic variation from the bam file, a filter of depth 20 was applied. Two public databases were used to obtain lineage defining mutations for Delta and Omicron variants (https://github.com/cov-lineages/constellations, https://covariants.org/shared-mutations).
2.5. Statistical analysis
The statistical analysis was performed using the R and Python programming languages. The Fisher exact test of independence for evaluating non-random association between the two categorical variables was performed to differentiate the mutation profile for all the four clinical groups by considering the Delta mortality and recovered as one group and the Omicron mortality and recovered as another group. The p-values were calculated from two-tailed tests, considering 0.05 as the significance level. A phi-correlation coefficient was calculated to quantify the strength of association between mutations and the clinical groups.
2.6. Phylogenetic analysis
The multiple sequence alignment of 249 SARS-CoV-2 genome sequences with the Wuhan reference strain (NC_045512.2) was performed through MAFFT (v7.475) [21]. The aligned sequences were manually trimmed and a phylogenetic tree was constructed through IQ-tree [37]. The lineage stratification was done using PANGOLIN (https://cov-lineages.org/resources/pangolin.html). The phylogenetic analysis was then visualised using the FIGTREE tool (http://tree.bio.ed.ac.uk/software/figtree/).
2.7. Molecular docking
Fasta sequence of proteins are taken from Uniprot [Id: P0DTD1 (ORF1ab), P0DTC2 (Spike glycoprotein)], and modelled using SWISS-MODEL [60]. The modelled protein was verified using PROCHECK, wherein the Ramachandran plot revealed 70% of the amino acid residues to be present in the most favoured region, 23.4% in the additional allowed region, 5% in generously allowed regions and only 1.5% in the disallowed region. Also, the RMSD value of modelled protein structure with respect to the protein crystal structure available in Uniport was observed to be 1.4 (Supplementary file 3).
The structure for remdesivir were downloaded from NCBI-PUBCHEM database (cid:121304016) in SDF format. SDF format of ligand was converted into PDB format using pymol (ref: The PyMOL Molecular Graphics System, Version 2.5.2 Schrödinger, LLC). Docking has been done through Autodock vina 4.2 [36]. PDBQT formats of protein and ligand were then prepared using Autodock Tools v1.5.7 [36]. Subsequently, free ions, water, and ligand were removed, kollman charges and polar hydrogens were introduced to the protein structure prior to docking. Visualizations of docked structures were done using PyMOL v2.5.2 and ChimeraX [42]. Further interaction studies have been done using discovery studio [41].
Remdesivir, a prodrug with broad spectrum antiviral activity has been widely utilized against SARS-CoV-2 infections [25]. An in-vitro study by Wang et al. have demonstrated its antiviral mechanism as a nucleotide analogue in VERO-E6 cell lines [59]. Moreover, clinical trials have shown the improvement of clinical outcomes in patients presenting moderate to severe COVID-19 upon Remdesivir administration [17]. These findings prompted us to consider and evaluate the binding strength of remdesivir to different viral mutant proteins in variable clinical outcomes.
2.8. Molecular dynamics simulation
MD simulation was performed to verify the docking results. CHARMM36 force-field [52] was used where ligands have been parameterized using CGENEFF server. The protein-ligand complexes of all the subgroups (Delta Recovered, Delta Mortality, Omicron Recovered, and Omicron Mortality) along with the wildtype prepared from Autodock-vina 4.2 were subjected to 100 ns MD simulations on the Nvidia-AVX2_256 GPU using the Gromacs-2020.2 CUDA module [30]. The protein has been prepared using the TIP3 water model with a cubic box of 1 Å of buffer distance. Energy minimization was done using the steepest descent method, for 50,000 steps to remove the bad contacts and clashes. All the complexes then went through two steps of equilibrium after energy minimization, the first being 100 ps of NVT [number of particles (N), system volume (V) and temperature (T)] equilibration, and the second one at 100 ps of NPT [number of particles (N), system volume (V) and pressure (P)] equilibration. A Berendsen thermostat was used to maintain the system's temperature at 300 °C [7] [40]. Moreover, the LINCS approach was used to address the system's long-range interaction [19]. Further downstream analysis of simulation results was performed using xmgrace 5.1.2 (Turner, P. J. “XMGRACE, Version 5.1. 19.” Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR 2 (2005)).
3. Results
3.1. Mutation profile of SARS-CoV-2 VOCs within distinct clinical outcomes
In alignment with our study theme, segregation based on clinical outcome and SARS-CoV-2 whole genome sequencing data within our cohort of 249 patients, was categorised into four groups - Delta recovered (67), Delta mortality (69), Omicron recovered (96) and Omicron mortality (17), respectively. The rationale was to evaluate the association of mutations with different clinical outcomes presented by the COVID-19 patients and their possible impact on the drug efficacy. Mutation spectrum analysis across our samples yielded a total of 697 mutations across the cohort, of which 418 mutations were observed in the Delta subclinical groups (recovered and mortality) and 279 mutations within the Omicron subgroups (recovered and mortality), respectively (Supplementary file 4). This evinces towards a higher number of mutations exhibited by the Delta variant (3.07/sample) relative to that of the Omicron (2.46/sample). Fig. 2B represents the cohort frequency of mutations and their corresponding genomic orientation across the viral genome for all the four subgroups. Delving further, we performed SARS-CoV-2 gene normalisation to observe the relative abundance of mutations with respect to the gene length across the viral genome (Fig. 2D). This revealed an overrepresentation of mutations in the ORF8 region in the Delta and within the 3’UTR region in the Omicron. However, the least number of normalised mutations were observed in the Membrane gene within the Delta and the ORF1ab gene in the Omicron.
Fig. 2.

Distribution of mutation frequency across the viral genome in our cohort of 249 COVID-19 patients. (A) Sample distribution for patients infected with Delta and Omicron variant, categorised based on their clinical outcomes - recovered and mortality. (B) Mutational landscape for all the four subgroups, depicting the cohort frequency of mutations according to its genome coordinates. (C) Intersection of mutations between the subgroups – Delta mortality, Omicron mortality, Omicron recovered and Delta recovered. (D) Visual representation of gene normalisation for both the VOCs, wherein the number of mutations were normalised with respect to the length of the gene.
3.2. Phylogenetic analysis
Comparative genomic analysis revealed distribution of the samples across the SARS-CoV-2 lineages of B.1.617.2 (43.37%) and BA.2 (36.14%). Several B.1.617.2 (Delta) sub-lineages including, AY.112, AY.120, AY.122, AY.127, AY.46.5, AY.98 were together categorised as AY.* lineage (11.24%). Moreover, sub-lineages of BA.2 (Omicron), as evident in Fig. 3 , were BA.2.10, BA.2.10.1, BA.2.2, BA.2.12, BA.2.23, and BA.2.3.4 (6.42%). We further observed the presence of an Omicron lineage B.1.1.529 (1.2%) followed by the sub-lineages XQ (0.8%), XT (0.4%) and BA.1.1.18 (0.4%). Subsequently, stratification of lineages based on the clinical outcome revealed the overrepresentation of B.1.617.2 across Delta recovered (58%) and mortality patients (100%) and BA.2 dominance for the Omicron recovered (79.16%) and mortality (82.35%) patients. Importantly, all the Delta mortality patients in this study belonged to the lineage B.1.617.2.
Fig. 3.

Phylogenetic analysis of 249 SARS-CoV-2 genome sequences in comparison to the wild type strain. The distribution of the lineages and sub-lineages across in-house sequenced Delta and Omicron VOCs is representative of the community prevalence in India during their respective waves. Different SARS-CoV-2 sub-lineages are depicted by different colours with recovered (green) and mortality (red). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Statistical & correlation analysis reveals mutations with potential significance
To further comprehend the significance of mutations associated with clinical outcomes across the VOCs, Fisher's exact test was performed, considering a p-value of <0.05. We observed 50 statistically significant mutations in the Delta clinical subgroups and 11 significant mutations in the Omicron (available as supplementary file 4). Phi-coefficient correlation analysis of these significant mutations revealed 20 mutations to be associated with the Delta recovered patients and 30 mutations were found to be associated with the Delta mortality patients. The same analysis highlighted three mutations for their association with Omicron recovered and eight mutations with the Omicron mortality patients. The distribution of significant mutations in different regions of the SARS-CoV-2 genome, including ORF1ab, Spike, ORF7a, ORF8 and Nucleocapsid protein has been elucidated in Fig. 4 .
Fig. 4.

Statistically significant mutations within the recovered and mortality patients of Delta and Omicron variants infections. The pan genomes distribution of the significant mutations across VOCs observed for the recovered (green) and mortality patients (red) during the waves of higher SARS-CoV-2 infectivity driven by the VOCs. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Intriguingly, most of the significant mutations (n = 28) in the Delta patients were seen in the ORF1ab region, and notably, three mutations T4685A (p = 4.61E-23), N4992N (p = 0.0006) and G5063S (p = 0.0005) were observed in the Nsp12/RdRp region of the Delta mortality patients, whereas no mutations in the RdRp were seen in the Delta Recovered. This could possibly emphasise the critical role of RdRp in viral replication, transcription and its subsequent contribution in elevating viral load, thereby influencing the progression of disease severity. Contrarily, two mutations, P5401L and A5811A in the Nsp13/Helicase region, were found to be associated (p-value = 4.30E-06 and 0.0012) within the Delta recovered patients.
Furthermore, among the significant mutations observed in the spike region for the Delta infected patients, six mutations were associated with mortality and five mutations with the recovered, respectively. It is important to mention that one of the mutations observed in the NTD region of the Spike protein, T19R in the Delta mortality patients (p-value = 9.93E-13) has been previously predicted to reduce the inhibitory effect of monoclonal antibodies. Interestingly, two significant mutations, K444N and N532H (p-value = 0.0006, 0.0006) in the mortality patients were observed to be present in the RBD region of spike protein, which could possibly impact the Spike-RBD interaction with the ACE2 receptor.
Upon examining the mutations present in the Omicron subgroups, we observed two mutations, Q493R and N440K in the RBD region of Spike protein to be strongly associated (p-value = 1.55E-12 and 0.0029) with the mortality. Spike mutations, particularly in the RBD region, have been attributed to conferring immune escape properties to the virus and thereby assisting in the progression of the disease severity. At the same time, T19I in the NTD region of Spike protein was found to be associated (p-value = 0.0045) with the recovered patients. Also, other significant mutations, such as G18C and R413S in the Nucleocapsid gene, were found to be associated with mortality (p-value = 0.0214) and recovered (p-value = 0.0214) patients, respectively. A comprehensive literature search for all the significant mutations was performed to elucidate the functional consequences of these mutations in altering the viral genome characteristics (supplementary file 5).
3.4. Interactions of Remdesivir with SARS-CoV-2 Nsp12, Nsp13 protein & Spike protein
To explore and understand the modulatory potential of the observed significant mutations towards available therapeutics, molecular docking was performed to assess the interaction strength and affinity of drugs with the mutated SARS-CoV-2 proteins. We investigated the interaction of the drug Remdesivir with the viral proteins, Nsp12, Nsp13, and Spike. The significant mutations observed in the Delta and Omicron patients were considered for the 3D homology modelling of the respective proteins. Table 1 enumerates the binding affinity for each ligand-protein complex, mutations in the active sites, interacting residues present at the active site of protein and the number of hydrogen bonds involved in the interaction of remdesivir with the predicted region of protein in each group.
Table 1.
Docking results of SARS-CoV-2 Nsp12, Nsp13 and Spike mutant proteins against Remdesivir.
| Drug | Protein | Patient Group | Mutations | Mutation in Active site | Binding Affinity | Interacting Residues | Number of H-Bonds |
|---|---|---|---|---|---|---|---|
| Remdesivir | Nsp12 Wildtype |
Wildtype | −8.1 | Lys47, Tyr129, Ala130, Asp135, Ser709, Lys780, Asn781 |
6 | ||
| Nsp12 mutation |
Delta Mortality | T4685A, N4992N, G5063S | None | −8.2 | Leu142, His133, Ala130, Thr141, Tyr32, Asp126, Ala34, Tyr129, Lys47 |
6 | |
| Nsp13 Wildtype |
Wildtype | −8.7 | Pro406, Pro408, Leu412, Phe422, Tyr515, Asn516, Thr532, Ala553, His554, Arg560 |
8 | |||
| Nsp13 Mutation |
Delta Recovered | P5401L, A5811A | None | −8.5 | Arg409, His554, Ala553, Leu412, Leu417 | 3 | |
| Remdesivir | S protein Wildtype | Wildtype | −6.3 | Asp40, Lys206, Tyr38, Ala222 |
5 | ||
| S protein Mutation |
Delta Recovered | G142D, A222V, Q677L, H1101Q, 1147S | None | −7.3 | Val367, Ser371, Asn343, Leu441, Phe374, Trp436 |
10 | |
| S protein Mutation |
Delta Mortality | T19R, K444N, N532H, D950N, 1061 V | K444N, N532H | −5.8 | Asn331, Gln56, Pro521, Pro330 |
5 | |
| S protein Mutation |
Omicron Recovered | T19I | None | −7.1 | Phe565, Asp428, Thr430, Phe515, Phe464, Glu516, Leu517, Pro426 | 6 | |
| S protein Mutation |
Omicron Mortality | N440K, Q493R | N440K, Q493R | −6.4 | Arg567, Thr573, Leu546, Val382, Ala522, Leu517, Gly545, Phe565 |
3 | |
The interaction study by docking revealed that the remdesivir drug binds to Nsp12 wildtype and Nsp12 mutant in the Delta mortality patients with nearly the same binding efficacy. Consequently, we observed six hydrogen bonds in the active site of Nsp12 wildtype and mutant proteins, four hydrophobic bonds in the active site of the wildtype and three hydrophobic bonds in the active site of mutant Nsp12 protein interacting with the remdesivir, respectively. Notably, hydrophobic interactions in the wildtype Nsp12 were formed through three amino acids - Ala130, Tyr129 and Lys780 and the mutant Nsp12 hydrophobic interactions involved the amino acids - Tyr129, Ala130, and Lys47 (Table 1). The appearance of a salt bridge during docking can indicate Nsp12 as a preferential binding site for Remdesivir. On the other hand, docking of remdesivir with Nsp13 wildtype and Nsp13 mutant in the Delta recovered patients revealed that the efficiency of binding is greater in the wildtype than in the mutant protein. We also observed more hydrogen and hydrophobic bonds in the drug interaction with the wildtype as compared with the mutant Nsp13 protein (supplementary file 6). Though remdesivir is widely studied to interact with Nsp12, Nsp14, and Nsp5/3CL Protease, we observed a considerable binding affinity of remdesivir with the Spike protein.
Intriguingly, both the mortality group patients demonstrated a decrease in binding affinity and number of H-bonds in comparison to that of the recovered patients for the Spike protein. Notably, the presence of two mutations in the RBD region of spike protein can possibly influence the binding characteristics, thereby resulting in a weaker interaction between Remdesivir and spike protein across both the mortality group patients. An increase in binding affinity and number of H-bonds ( Fig. 5, Fig. 6 ) revealed by the recovered patients, when compared to the wildtype and mortality patients, is indicative of the possibility of these mutations promoting a stronger interaction between Remdesivir and the SARS-CoV-2 Spike protein.
Fig. 5.

3D and 2D view of interactions observed between Remdesivir and viral mutant Spike proteins of the Delta variant recovered and mortality patients. (A), (B) and (C) are showing 3D & 2D view of interactions between Remdesivir and RBD- Spike region in the Delta Recovered patients. (D), (E) and (F) highlights 3D & 2D view of interactions between Remdesivir and RBD-Spike region in the Delta Mortality patients.
Fig. 6.

3D and 2D view of interactions observed between Remdesivir and viral mutant proteins of the Omicron variant recovered and mortality patients. (A), (B) and (C) highlights 3D & 2D view of interactions between Remdesivir and Spike-RBD mutant in the Omicron Recovered. (D), (E) and (F) emphasizes 3D & 2D view of interactions between Remdesivir and Spike-RBD mutant in Omicron Mortality patients.
3.5. Molecular dynamics simulation of Remdesivir and SARS-CoV-2 Spike proteins
Based on the findings of molecular docking, MD simulation was performed to verify the difference in the stability of the predicted Remdesivir-Spike complex across all the four sub-groups: Delta recovered, Delta mortality, Omicron recovered and Omicron mortality. The structural dynamics of both the ligand and ligand-protein complex were evaluated by measuring their respective RMSD and RMSF values. Delving further, we observed the average RMSD values for Remdesivir-spike complex to be 1.54 nm and convergence of the RMSD curve after 85 ns for the Delta mortality group. Although the average RMSD value of the same complex was found to be 1.3 nm for the Delta recovered group along with the curve stabilization after 85 ns of MD simulation (Fig. 7A). The variability in the average RMSD values between the clinically distinct groups suggests a more stabilised and enclosed accommodation of Remdesivir within the predicted binding pockets of the RBD-spike region in the Delta recovered group. This was further substantiated by the ligand RMSD trajectories observed in Fig. 7B, wherein Remdesivir confers a ligand shift around 30 ns and fluctuates randomly from the active binding pocket until 100 ns in the Delta mortality group. On the other hand, the ligand RMSD curve of the Delta recovered group revealed a stable accommodation of remdesivir within the active pockets of the protein up to 45 ns. Moreover, we observed greater residual fluctuations in the Delta mortality group as compared to the Delta recovered group (Fig. 7C).
Fig. 7.

Structural Dynamics of Remdesivir and SARS-CoV-2 Spike protein across the recovered and mortality patients of Delta and Omicron. (A) RMSD plot of Remdesivir-Spike complex as a function of time for the Delta subgroups, (B) RMSD plot of Remdesivir for Delta subgroups, (C) Root Mean Square Fluctuation vs residue for the Delta subgroups, (D) RMSD plot of Remdesivir-Spike complex as a function of time for the Omicron subgroups, (E) RMSD plot of Remdesivir for Omicron subgroups, and (F) Root Mean Square Fluctuation vs residue for the Omicron subgroups.
Further investigation of the structural dynamics for the Omicron mortality group revealed a constant fluctuation in the RMSD values of the Remdesivir-spike complex until the completion of MD simulation ( Fig. 7D). The average RMSD for the complex was found to be 1.74 nm. However, we observed the RMSD trajectory of the Remdesivir-Spike complex to be stable around 50 ns to 75 ns for the Omicron recovered group, contributing to an average RMSD of 1.18 nm. Interestingly, the ligand RMSD curve of the Omicron mortality group exhibited a similar pattern of ligand shift from the binding site of the protein around 30 ns as that of the Delta mortality group and endured random fluctuations up to 100 ns of the simulation ( Fig. 7E). We further observed the stabilization of the ligand RMSD curve from 5 ns to 50 ns for the Omicron recovered group. A comparatively stable interaction of Remdesivir within the binding pockets of the Spike protein was observed for the Omicron recovered group than the mortality group, although the Root Mean Square Fluctuations per residue were observed to be higher in the recovered group ( Fig. 7F).
4. Discussion
Several vaccines and drugs are being used globally towards pandemic management with differential efficacy with hierarchical layers of modulation. This is in combination with the vaccine development for the below 18 age groups, with specific success towards the same as well. Hence, global efforts are directed towards developing, exploring, and repurposing several antiviral drugs to subside the clinical severity, especially the continuously evolving and emerging SARS-CoV-2 variants of concern. Towards that, repurposing of available drugs, such as remdesivir and dexamethasone, has been shown to be efficacious in managing the disease severity symptoms [22]. At the same time, we have observed differential efficacy of the drug in a subset of the SARS-CoV-2 infected patients. What could be the reason? Is it due to the different VOCs or mutations or genomic region-specific mutations? However, our understanding of the association of mutations with different clinical outcomes and the potency of these drugs is restricted [54]. To evaluate and elucidate this aspect, we stratified SARS-CoV-2 infected patients based on the clinical outcome and assessed the binding interactions of Remdesivir with different mutant viral proteins. Integrative studies, computational and experimental, involving the screening of drugs with patients presenting variable clinical symptoms, are important to optimise treatment and identify other available treatment options as well.
In the present study, through genomic analysis of 249 SARS-CoV-2 genome sequences, we sought to investigate the differences in the association of mutations manifested by SARS-CoV-2 VOCs infected individuals with the disease severity, leading to differential clinical outcomes of recovery or mortality. All the mutations observed within our cohort were analysed for their functional relevance and their respective viral genome coordinates. This revealed the distribution of significant mutations in the ORF1ab (28), Spike (11), ORF8 (3), Intergenic (3), Orf7a (2), Nucleocapsid (2), and only one mutation in the 3’UTR region across the viral genomes in the Delta infected patients. Contrarily, we observed a greater percentage of significant mutations in the Spike (5) followed by mutations in the Nucleocapsid (2), 3’UTR (2), 5’UTR (1), and the ORF1ab (1) genes in the Omicron patients. Furthermore, we found the co-presence of five mutations (P309L in NSP2, P1640L in NSP3, A3209V in NSP4, V3718A in NSP6, and R385K in the N gene) in the Delta mortality patients, which has been recently reported to be more prevalent in the symptomatic patients than the asymptomatic ones [4]. On the other hand, three co-occurring mutations, G142D in the Spike, P2287S in the Nsp3 and T3255I in the Nsp4 region of the ORF1ab were observed in the Delta recovered patients, which has been previously reported for their correlation with milder infectious cases of SARS-CoV-2 [38].
Mutations in the Spike RBD region are crucial for increased infectivity and antibody resistance for SARS-CoV-2 emerging variants, and thereby promoting viral pathogenesis [5]. Keeping a constant track of these RBD mutations along with non-RBD mutations is imperative for the development of potential vaccines and therapies as well as monitoring the efficacy of the ones currently in use [2]. Celik et al. evaluated SARS-CoV-2 variants, including Iota, Mu, Delta plus, Kappa, Lambda, and C.1.2 for their changes in the interacting residues between spike-RBD region and hACE2 receptor, and highlighted the increase in the stability of the interacting complex as compared to the wild-type, leading to enhanced virulence [9]. Furthermore, Shafiq et al. demonstrated that one of the SARS-CoV-2 variants in Tanzania, the A.30 strain, manifesting the mutations R346K, T478K, and E484K in the Spike-RBD region, aids the virus with immune escape properties [51]. The Omicron variant, harbours a large number of mutations in the RBD region of the spike protein, including K417N, G446S, Q493R, and Q498R, leading to more stable interactions with the hACE2 protein [23]. Hence, these hotspot mutations in the Omicron have been considered as a potential target for drug design and repurposing to combat the viral spread [24].
Interestingly, one of the significant mutations, K444N in the RBD region of the spike protein, observed in the Delta mortality patients was identified by Weisblum et al. for conferring resistance to neutralising antibodies against the spike protein [61]. Other significant mutations in the Spike NTD region, such as T19R and D950N, observed in the Delta mortality have been attributed as a potential target for anti-NTD neutralising antibodies [44]. Furthermore, the Omicron mortality patients also manifested mutations in the RBD region of the Spike protein, including Q943R and N440K, which have been reported to enhance the binding affinity towards the hACE2 receptor and escape from the antibody neutralization, consequently increasing chances of reinfection [15,28].
To evaluate the propensity of these associated mutations towards functional modulation during SARS-CoV-2 pathogenesis, we computationally evaluated the docking parameters across both the Delta and Omicron sub-groups of patients for different viral proteins. Since remdesivir has been previously shown to have high binding affinity to RdRp, we observed that RdRp mutants (T4685A, N4992N and G5063S) in the Delta mortality exhibited nearly the same binding affinity for Remdesivir as that of the wildtype [8,25]. This possibly suggest that the RdRp mutants identified in the study does not affect remdesivir binding and does not modulate efficacy between recovered and the mortality clinical outcome. Nsp13 mutants (P5401L and A5811A) conferred decreased binding affinity towards remdesivir, indicating a weaker interaction between the protein and the ligand. Notably, the mutations K444N and N532H in the Delta mortality, and Q493R and N440K in the Omicron mortality within the RBD region of Spike protein resulted in a lower binding affinity for remdesivir than their respective recovered patients (Table 1). Consequently, these mutations observed across both VOCs hold the potential to ameliorate the disease severity by reducing the binding efficacy of remdesivir.
Considering molecular dynamics simulation as an effective technique to validate the stability profiles of the protein-ligand complexes presented in our study, we performed 100 ns simulation for all the clinical groups. Intriguingly, the Remdesivir-Spike protein complex of the Delta and Omicron mortality groups exhibited a higher average RMSD as compared to their respective recovered groups. This can thereby signify the relative instability of the Remdesivir-Spike protein complex of the Delta and Omicron mortality groups. The dynamic behaviour of Remdesivir observed as ligand shift from the binding pockets of the Spike protein in both the mortality groups further evinces towards inadequate accommodation of Remdesivir within the Spike protein. However, further in-vitro validation studies are required to support the role of these mutant proteins in disease transmission and pathogenicity.
At present, the availability of several antiviral drugs, immune modulators, and monoclonal antibody treatments has comparatively reduced the current disease burden [13,29]. The global vaccination data highlights a substantial number of individuals to be fully vaccinated worldwide, in addition to booster doses, further aiding in the reduction of morbidity and mortality [47]. Nevertheless, robust genomic surveillance can highlight emerging SARS-CoV-2 variants and help identify mutations manifesting characteristics affecting public health and clinical interventions.
5. Conclusion
The present study was initiated to, i) comprehensively investigate the viral genome heterogeneity represented by SARS-CoV-2 infected individuals with different VOCs, ii) identify significant mutations potentially modulating the different clinical outcomes, and iii) explore the functional aspect of such mutations for their propensity to impact the efficacy of available and future therapeutics. Our findings reveal significant mutations, T4685A, N4992N, and G5063S in the RdRp; T19R in the NTD spike; and K444N and N532H in the RBD spike region to be associated with the Delta mortality patients. At the same time, we observed the mutations, T19I in the NTD Spike, Q493R and N440K in the RBD spike, G18C, and R413S in the Nucleocapsid gene to be significantly associated with the Omicron mortality patients. Subsequently, docking studies of Remdesivir with mutant viral proteins including Nsp12, Nsp13, and Spike RBD region demonstrated a reduction in the binding strength of Remdesivir to mutant Spike proteins for both the mortality groups. The docking results were further verified through MD simulation. In combination, these findings suggest the possible role of these mutations in reduced drug efficacy and a potential role in the progression of disease severity.
Data availability
The cumulated and analysed clinical data as a part of this study is available in Supplementary file 1. Other supplementary files as mentioned in the text are available collectively. The SARS-CoV-2 genomic data has been uploaded to GISAID (IDs available in supplementary file 2).
Author contributions
VR and SpS performed analysis; SS, VR and AS wrote the manuscript; NSC did sequencing of the samples; RP designed, conceptualized, implemented and coordinated the study. All authors contributed to the article, have read and approved the final submission.
Funding support
This research was funded by the Bill and Melinda Gates Foundation (BMGF), project code - CLP0036, Foundation for Innovative New Diagnostics (FIND), project code – GAP0249, AIDS Healthcare Foundation (AHF), project code CLP0043.
Authors' statement
-
•
Varsha Ravi and Sparsh Sharma performed analysis;
-
•
Sheeba Saifi, Varsha Ravi and Aparna Swaminathan wrote the manuscript;
-
•
Nar Singh Chauhan did sequencing of the samples;
-
•
Rajesh Pandey designed, conceptualized, implemented and coordinated the study
# All authors contributed to the article, have read and approved the final submission.
Declaration of Competing Interest
Authors wish to declare no conflict of interest and funders did not have a role in planning and execution of the study.
Acknowledgement
We acknowledge the role of Dr. Aradhita Baral as Research Manager and coordination with the funders. The authors also acknowledge the support of Anil Kumar and Nisha Rawat towards SARS-CoV-2 sample transport and sample management. We also would further like to acknowledge the role of Dr. Tavpritesh Sethi, for providing us access to the IIITD server during revision of the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygeno.2022.110466.
Appendix A. Supplementary data
Supplementary material 1
Supplementary material 2
Supplementary material 3
Supplementary material 4
Supplementary material 5
Supplementary material 6
Supplementary material 7
Supplementary material 8
Data availability
All the SARS-CoV-2 sequences used in the study, have been uploaded on GISAID.
References
- 1.Ahmed S., Mahtarin R., Ahmed S.S., Akter S., Islam M.S., Mamun A.A., et al. Investigating the binding affinity, interaction, and structure-activity-relationship of 76 prescription antiviral drugs targeting RdRp and Mpro of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021;39:6290–6305. doi: 10.1080/07391102.2020.1796804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almehdi A.M., Khoder G., Alchakee A.S., Alsayyid A.T., Sarg N.H., Soliman S.S.M. SARS-CoV-2 spike protein: pathogenesis, vaccines, and potential therapies. Infection. 2021;49:855–876. doi: 10.1007/s15010-021-01677-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aouidate A., Ghaleb A., Chtita S., Aarjane M., Ousaa A., Maghat H., et al. Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2021;39:4522–4535. doi: 10.1080/07391102.2020.1779130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee A., Mazumder A., Roy J., Majumdar A., Chatterjee A., Biswas N.K., et al. Evolution of Delta variant by non-Spike signature co-appearing mutations: trailblazer of COVID-19 disease outcome. BioRxiv. 2022 doi: 10.1101/2022.04.05.487103. [DOI] [Google Scholar]
- 5.Barton M.I., MacGowan S.A., Kutuzov M.A., Dushek O., Barton G.J., van der Merwe P.A. Effects of common mutations in the SARS-CoV-2 spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. ELife. 2021;10 doi: 10.7554/eLife.70658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., et al. Remdesivir for the treatment of Covid-19 - final report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 8.Byléhn F., Menéndez C.A., Perez-Lemus G.R., Alvarado W., de Pablo J.J. Modeling the binding mechanism of Remdesivir, Favilavir, and Ribavirin to SARS-CoV-2 RNA-dependent RNA polymerase. ACS Cent. Sci. 2021;7:164–174. doi: 10.1021/acscentsci.0c01242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celik I., Khan A., Dwivany F.M., Fatimawali Wei D.-Q., Tallei T.E. Computational prediction of the effect of mutations in the receptor-binding domain on the interaction between SARS-CoV-2 and human ACE2. Mol. Divers. 2022 doi: 10.1007/s11030-022-10392-x. [DOI] [PubMed] [Google Scholar]
- 10.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drożdżal S., Rosik J., Lechowicz K., Machaj F., Szostak B., Przybyciński J., et al. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist. Updat. 2021;59 doi: 10.1016/j.drup.2021.100794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escalera A., Gonzalez-Reiche A.S., Aslam S., Mena I., Laporte M., Pearl R.L., et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe. 2022;30:373–387.e7. doi: 10.1016/j.chom.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favalli E.G., Biggioggero M., Maioli G., Caporali R. Baricitinib for COVID-19: a suitable treatment? Lancet Infect. Dis. 2020;20:1012–1013. doi: 10.1016/S1473-3099(20)30262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Focosi D., Novazzi F., Genoni A., Dentali F., Gasperina D.D., Baj A., et al. Emergence of SARS-COV-2 Spike protein escape mutation Q493R after treatment for COVID-19. Emerg. Infect. Dis. 2021;27:2728–2731. doi: 10.3201/eid2710.211538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. ArXiv. 2012 doi: 10.48550/arxiv.1207.3907. [DOI] [Google Scholar]
- 17.Gottlieb R.L., Vaca C.E., Paredes R., Mera J., Webb B.J., Perez G., et al. Early Remdesivir to prevent progression to severe Covid-19 in outpatients. N. Engl. J. Med. 2022;386:305–315. doi: 10.1056/NEJMoa2116846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurung A.B., Ali M.A., Lee J., Farah M.A., Al-Anazi K.M. The potential of Paritaprevir and emetine as inhibitors of SARS-CoV-2 RdRp. Saudi J. Biol. Sci. 2021;28:1426–1432. doi: 10.1016/j.sjbs.2020.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 20.Hoffmann M., Mösbauer K., Hofmann-Winkler H., Kaul A., Kleine-Weber H., Krüger N., et al. Chloroquine does not inhibit infection of human lung cells with SARS-CoV-2. Nature. 2020;585:588–590. doi: 10.1038/s41586-020-2575-3. [DOI] [PubMed] [Google Scholar]
- 21.Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelleni M.T. Tocilizumab, Remdesivir, Favipiravir, and Dexamethasone repurposed for COVID-19: a comprehensive clinical and Pharmacovigilant reassessment. SN Compr. Clin. Med. 2021;3:919–923. doi: 10.1007/s42399-021-00824-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khan A., Randhawa A.W., Balouch A.R., Mukhtar N., Sayaf A.M., Suleman M., et al. Blocking key mutated hotspot residues in the RBD of the omicron variant (B.1.1.529) with medicinal compounds to disrupt the RBD-hACE2 complex using molecular screening and simulation approaches. RSC Adv. 2022;12:7318–7327. doi: 10.1039/d2ra00277a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan A., Waris H., Rafique M., Suleman M., Mohammad A., Ali S.S., et al. The Omicron (B.1.1.529) variant of SARS-CoV-2 binds to the hACE2 receptor more strongly and escapes the antibody response: insights from structural and simulation data. Int. J. Biol. Macromol. 2022;200:438–448. doi: 10.1016/j.ijbiomac.2022.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan F.I., Kang T., Ali H., Lai D. Remdesivir strongly binds to RNA-dependent RNA polymerase, membrane protein, and main protease of SARS-CoV-2: indication from molecular modeling and simulations. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.710778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kullappan M., Mary U., Ambrose J.M., Veeraraghavan V.P., Surapaneni K.M. Elucidating the role of N440K mutation in SARS-CoV-2 spike - ACE-2 binding affinity and COVID-19 severity by virtual screening, molecular docking and dynamics approach. J. Biomol. Struct. Dyn. 2021:1–18. doi: 10.1080/07391102.2021.2014973. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S., Chandele A., Sharma A. Current status of therapeutic monoclonal antibodies against SARS-CoV-2. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindahl Hess, Spoel V.D. 2020. GROMACS 2020.2 Manual. Zenodo. [DOI] [Google Scholar]
- 31.Lin L., Liu Y., Tang X., He D. The disease severity and clinical outcomes of the SARS-CoV-2 variants of concern. Front. Public Health. 2021;9 doi: 10.3389/fpubh.2021.775224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loman N.J., Quick J., Simpson J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods. 2015;12:733–735. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 34.McCormick K.D., Jacobs J.L., Mellors J.W. The emerging plasticity of SARS-CoV-2. Science. 2021;371:1306–1308. doi: 10.1126/science.abg4493. [DOI] [PubMed] [Google Scholar]
- 35.Mohammad A., Al-Mulla F., Wei D.-Q., Abubaker J. Remdesivir MD simulations suggest a more favourable binding to SARS-CoV-2 RNA dependent RNA polymerase mutant P323L than wild-type. Biomolecules. 2021;11 doi: 10.3390/biom11070919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nilgiriwala K., Kadam P., Patel G., Shaikh A., Mestry T., Vaswani S., et al. Genomics of post-vaccination SARS-CoV-2 infections during the Delta dominated second wave of COVID-19 pandemic, from Mumbai metropolitan region (MMR), India. J Med Virol. 2022 doi: 10.1002/jmv.27861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ong S.W.X., Chiew C.J., Ang L.W., Mak T.-M., Cui L., Toh M.P.H.S., et al. Clinical and virological features of SARS-CoV-2 variants of concern: a retrospective cohort study comparing B.1.1.7 (Alpha), B.1.315 (Beta), and B.1.617.2 (Delta) Clin. Infect. Dis. 2021 doi: 10.1093/cid/ciab721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parrinello M. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 41.Pawar S.S., Rohane S.H. Review on discovery studio: an important tool for molecular docking. Asia J. Rese Chem. 2021;14:1–3. doi: 10.5958/0974-4150.2021.00014.6. [DOI] [Google Scholar]
- 42.Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pirzada R.H., Haseeb M., Batool M., Kim M., Choi S. Remdesivir and Ledipasvir among the FDA-approved antiviral drugs have potential to inhibit SARS-CoV-2 replication. Cells. 2021;10 doi: 10.3390/cells10051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Planas D., Veyer D., Baidaliuk A., Staropoli I., Guivel-Benhassine F., Rajah M.M., et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021;596:276–280. doi: 10.1038/s41586-021-03777-9. [DOI] [PubMed] [Google Scholar]
- 45.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribaudo G., Ongaro A., Oselladore E., Zagotto G., Memo M., Gianoncelli A. A computational approach to drug repurposing against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) J. Biomol. Struct. Dyn. 2022;40:1101–1108. doi: 10.1080/07391102.2020.1822209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ritchie H., Mathieu E., Rodés-Guirao L., Appel C., Giattino C., Ortiz-Ospina E., et al. 2020. Coronavirus (COVID-19) Vaccinations - Our World in Data. Our World in Data. [Google Scholar]
- 48.Saghir S.A.M., AlGabri N.A., Alagawany M.M., Attia Y.A., Alyileili S.R., Elnesr S.S., et al. Chloroquine and hydroxychloroquine for the prevention and treatment of COVID-19: a fiction, hope or hype? An updated review. Ther. Clin. Risk Manag. 2021;17:371–387. doi: 10.2147/TCRM.S301817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salehi-Vaziri M., Fazlalipour M., Seyed Khorrami S.M., Azadmanesh K., Pouriayevali M.H., Jalali T., et al. The ins and outs of SARS-CoV-2 variants of concern (VOCs) Arch. Virol. 2022;167:327–344. doi: 10.1007/s00705-022-05365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saxena S.K., Kumar S., Ansari S., Paweska J.T., Maurya V.K., Tripathi A.K., et al. Characterization of the novel SARS-CoV-2 Omicron (B.1.1.529) variant of concern and its global perspective. J. Med. Virol. 2022;94:1738–1744. doi: 10.1002/jmv.27524. [DOI] [PubMed] [Google Scholar]
- 51.Shafiq A., Zubair F., Ambreen A., Suleman M., Yousafi Q., Rasul Niazi Z., et al. Investigation of the binding and dynamic features of A.30 variant revealed higher binding of RBD for hACE2 and escapes the neutralizing antibody: a molecular simulation approach. Comput. Biol. Med. 2022;146:105574. doi: 10.1016/j.compbiomed.2022.105574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.da Silva T.U., de Pougy K.C., Albuquerque M.G., da Silva Lima C.H., de Machado S.P. Development of parameters compatible with the CHARMM36 force field for [Fe4S4]2+ clusters and molecular dynamics simulations of adenosine-5′-phosphosulfate reductase in GROMACS 2019. J. Biomol. Struct. Dyn. 2022;40:3481–3491. doi: 10.1080/07391102.2020.1847687. [DOI] [PubMed] [Google Scholar]
- 53.Singhal T. The emergence of omicron: challenging times are here again! Indian J. Pediatr. 2022;89:490–496. doi: 10.1007/s12098-022-04077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stevens L.J., Pruijssers A.J., Lee H.W., Gordon C.J., Tchesnokov E.P., Gribble J., et al. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022:eabo0718. doi: 10.1126/scitranslmed.abo0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tao K., Tzou P.L., Nouhin J., Gupta R.K., de Oliveira T., Kosakovsky Pond S.L., et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021;22:757–773. doi: 10.1038/s41576-021-00408-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tian D., Sun Y., Zhou J., Ye Q. The global epidemic of the SARS-CoV-2 delta variant, key spike mutations and immune escape. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.751778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Troyano-Hernáez P, Reinosa R, Holguín Á. Evolution of SARS-CoV-2 envelope, membrane, nucleocapsid, and Spike structural proteins from the beginning of the pandemic to September 2020: A global and regional approach by epidemiological week. Viruses 2021;13. doi: 10.3390/v13020243. [DOI] [PMC free article] [PubMed]
- 58.Ullrich S., Ekanayake K.B., Otting G., Nitsche C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022;62 doi: 10.1016/j.bmcl.2022.128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J.C., et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. ELife. 2020;9 doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1
Supplementary material 2
Supplementary material 3
Supplementary material 4
Supplementary material 5
Supplementary material 6
Supplementary material 7
Supplementary material 8
Data Availability Statement
The cumulated and analysed clinical data as a part of this study is available in Supplementary file 1. Other supplementary files as mentioned in the text are available collectively. The SARS-CoV-2 genomic data has been uploaded to GISAID (IDs available in supplementary file 2).
All the SARS-CoV-2 sequences used in the study, have been uploaded on GISAID.