

Abstract
Intermetallics are long-range-ordered alloys traditionally synthesized by annealing nanoparticles of a random alloy, which results in the sintering of the nanoparticles and leads to the formation of polydispersed samples. It thus remains a challenge to achieve a monodispersion of tiny intermetallics. In the current work, ultra-small monodisperse intermetallic NiZn nanoparticles were synthesized based on a low-temperature solution chemistry route involving the chemical conversion of metal nanoparticles into an ordered alloy using an organometallic zinc precursor. During the transformation of single metal nanoparticles into the corresponding alloy, the particles retained their morphology. The resulting ordered alloy made up of earth-abundant materials demonstrated high electrocatalytic performance for the oxygen evolution reaction (OER) with a low overpotential of 283 mV at a current density of 10 mA cm−2 and a small Tafel slope of 73 mV dec−1, along with excellent stability and durability. The prepared intermetallic NiZn exhibited outstanding OER efficacy, better than those of a Ni0.7Zn0.3 alloy, pure Ni nanoparticles and even state-of-the art RuO2. The atomic ordering as well as the modification of the electronic structure of Ni upon becoming alloyed with Zn, together with an atomic-scale synergistic effect produced from Ni and Zn, led to the enhanced intrinsic catalytic activity. The present findings point to a general route to produce nanoscale tiny alloys and also provide excellent electrocatalysts having exceptional energy conversion efficiency.
Ultra-small monodisperse intermetallic NiZn nanoparticles synthesized via a low-temperature solution chemistry route possess enhanced electrocatalytic activity during the oxygen evolution reaction.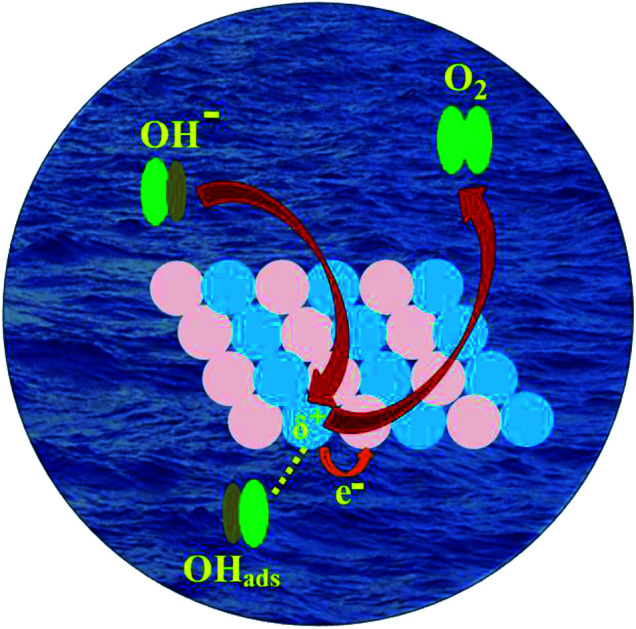
Introduction
The oxygen evolution reaction (OER) driven by renewable energy or electrochemical splitting of water represents an essentially perfectly green, sustainable and efficient approach for producing oxygen.1–5 This approach holds great promise to supply future oxygen demands due to the enormous availability of water, and is environmentally benign since no greenhouse gases are emitted. Moreover, this is believed to be a next-generation energy solution as an alternative to carbon-based energy sources. The most commonly used currently state-of-the-art catalysts for OERs are IrO2 or RuO2,6,7 but their widespread application may be impeded owing to their low abundance and high cost.8 Hence, there is a need to develop new efficient energy conversion systems. In electrochemical water splitting, the OER is the anodic reaction and its sluggish kinetics has led to extensive research in pursuing and designing proficient electrocatalysts to expedite the reaction by reducing the overpotential and hence subsequently boost the energy conversion efficacy.
Nanocrystalline alloys and intermetallics have emerged as a novel class of materials, which are being explored in the development of green sources of energy in fuel cells,9–14 Li-ion batteries,15,16 photocatalysts,17–19 thermoelectric power generation20,21 and other diverse applications22–25 owing to their robust stability and excellent activity. Since the discovery of intermetallic compounds, they demonstrated excellent prospects by playing a leading role for the fabrication of low-cost high performance potential catalysts.26–28 Conventionally, intermetallics are synthesized via metallurgical techniques that require high-temperature reductions followed by annealing for a long period of time29,30 or through the annealing of nanoparticles (NPs) of a random alloy,31,32 leading to the sintering of the particles and thus resulting in the formation of polydisperse particles. Unlike the disordered alloys, ordered intermetallics provide uniform active sites on the same surface plane owing to their compositional and positional order as well as possess precise structure accompanied by electronic effects. It is therefore really challenging to get desired monodisperse intermetallics with high surface area, as it is an important criterion interrelated to their physical properties that govern their catalytic activity. Interestingly, tuning their size, structure and composition at the nanometer scale is key to significantly impacting their catalytic activities, since their chemical and physical properties are associated with the electronic states of the constituent metals. A general objective of our research is to synthesize ultra-small intermetallic NPs of earth-abundant transition metals using a low-temperature solution chemistry approach since improving their physical properties by means of controlling the average size and composition of the NPs may lead to the enhanced activity. However, to date, the synthesis of tiny NPs of alloys with a tunable composition through a low-temperature route is still a challenging task. Nevertheless, developing cost-effective and high-performance electrocatalysts for oxygen production remains of immense importance.
In the current work, we developed a general route to achieve tiny Ni and corresponding Ni0.7Zn0.3 and NiZn NPs from commercially available reagents via a solution-mediated reaction approach. After characterizing them using various physical methods, we studied their electrocatalytic activities towards OER in detail. In this studies, the intermetallic NiZn NPs displayed higher catalytic activity than that of Ni0.7Zn0.3, pure Ni NPs and even the commercial RuO2. Finally, we have compared the electrocatalytic efficacy of NiZn with those of reported Ni-based electrocatalysts.
Experimental
Synthesis of Ni, NiZn and Ni0.7Zn0.3 nanoparticles
The nanoscale metal and alloys were synthesized based on the thermal decomposition of subsequent metal salts or organometallic compounds in a hot organoamine solvent. All reactions were performed under a nitrogen atmosphere via standard air-free techniques.
First, Ni NPs were synthesized using the following method. Nickel(ii) acetylacetonate (0.5 mmol) was added in a mixture of oleylamine (9.25 mmol), octadecene (9.25 mmol) and trioctylphosphine (9.25 mmol) in a three-necked round-bottom flask. The flask was fitted with a condenser, rubber septum, thermocouple adaptor and sheath through which a thermocouple was inserted. The reaction mixture was degassed at 100 °C for 30 min and then heated to 220 °C using a heating mantle upon which the color of the solution gradually changed from blue to black, signifying the formation of Ni NPs. The black solution was then aged at 220 °C under a nitrogen atmosphere for 90 min. The synthesized particles were collected via centrifugation after addition of an acetone–hexane (3 : 1) mixture. Finally, the collected particles were redispersed in non-polar organic solvents, like hexane or toluene, and were stable for couple of months.
For the synthesis of NiZn NPs, Ni NPs were first synthesized by following the above-mentioned route. Then, the temperature of the reaction mixture containing Ni NPs was increased to 250 °C. A diethylzinc–oleylamine solution (0.5 mmol) was injected drop-wise into the reaction mixture once the reaction temperature reached the desired temperature and, following addition, the mixture was held at that temperature for 30 min. Then, an analogous procedure was followed to collect the NiZn NPs, which were redispersed in hexane or toluene. For the synthesis of the Ni0.7Zn0.3 alloy, the amount of diethylzinc solution was only changed to 0.18 mmol, keeping all other reaction conditions the same. After characterization, all of these as-synthesized NPs were explored as electrocatalysts for OER studies.
Electrochemical measurements
A CHI660E electrochemical workstation (CHI Instruments) was used to check the OER activities of the various catalysts. All of the electrochemical experiments were carried out at room temperature in a conventional three-electrode system of an electrochemical analyzer using glassy carbon (GC) as the working electrode, Hg/HgO (1.0 M KOH) as the reference electrode and Pt wire as the counter electrode. All potentials were referenced to the reversible hydrogen electrode (RHE) by using the equation E (vs. RHE) = E (vs. Hg/HgO) + 0.098 + 0.059 pH. The GC electrode was polished with 1.0, 0.3 and 0.05 μm alumina slurries successively and washed several times with distilled water before the catalyst was deposited on it. First, a mass of 4.0 mg (30 wt% sample in carbon black) of Ni NPs was dispersed in a mixture containing distilled water (250 μL), isopropanol (250 μL) and Nafion® solution (1%, 20 μL). Then, the mixture was sonicated for 60 min to make a homogeneous mixture. The working electrode was prepared by drop-casting 15 μL of the sample solution onto the GC electrode (diameter of 3 mm) and dried overnight. The final loading of each catalyst on the GC electrode was ∼0.51 mg cm−2. Linear sweep voltammetry (LSV) was performed with a scan rate of 5 mV s−1 in a 1.0 M KOH solution. Cyclic voltammetry (CV) and chronoamperometry (CA) analyses were done to measure, respectively, the electrochemically active surface area (ECSA) and stability of each catalyst. Electrochemical impedance spectroscopy (EIS) was performed in the frequency range 0.1 Hz to 100 kHz with an AC amplitude of 0.005 V. The pH of the electrolyte solution was estimated to be 14.0. For the preparations of the working electrodes using NiZn, Ni0.7Zn0.3 NPs and commercial RuO2 catalysts, respectively, similar procedures were followed. The overpotential (η) for each catalyst was calculated using the equation η = E (RHE) − 1.23 V.
Results and discussion
The properties of an alloy entirely originate from the composition and atomic ordering of the constituent metals. Keeping these aspects in mind, we synthesized Ni NPs and their corresponding NiZn and Ni0.7Zn0.3 alloys. Transmission electron microscopy (TEM) was used to characterize the morphologies and compositions of the NPs (Fig. 1). TEM images of the Ni NPs demonstrated formation of monodispersed spherical NPs having particle dimensions of ∼3 nm (Fig. 1A and B). The crystal structure of the Ni NPs (Fig. 1C) was examined using powder X-ray diffraction (XRD), which indicated the formation of face-centered cubic (fcc) Ni NPs (PDF no. 03-065-0380). An extremely broad peak corresponding to the (111) plane was observed and was due to the presence of smaller crystallites; two other peaks corresponding to (200) and (220) planes were suppressed, and this suppression was attributed to the ultra-small nature of the formed Ni NPs. It should be noted that there were no diffraction peaks corresponding to nickel oxides or other phases observed in the XRD pattern, indicating the formation of pure fcc Ni NPs.
Fig. 1. TEM micrographs of (A and B) ultra-small Ni NPs and corresponding (D–F) NiZn NPs at various magnifications. (C) XRD patterns of Ni NPs and the corresponding NiZn alloy. A simulated XRD pattern of tetragonal NiZn is also provided. (G) The EDS spectrum of NiZn illustrating the presence of both Ni and Zn in the alloy. (H) A STEM image of the NiZn alloy and elemental mapping of Ni (cyan) and Zn (yellow) in the NiZn alloy.

After the injection of equimolar diethylzinc (DEZ) to the reaction solution containing Ni NPs at 250 °C, NiZn NPs formed. TEM micrographs of the NiZn NPs shown in Fig. 1D–F at various magnifications demonstrated that they retained the spherical morphology of the Ni NPs. The estimated particle size of the NiZn NPs was ∼4.5 nm. The histograms of particle size for Ni and corresponding NiZn NPs are presented in the ESI section as Fig. S1.† The XRD pattern of NiZn NPs displayed the formation of a distinct tetragonal structure associated with the ordered alloyed NiZn (PDF no. 01-072-2668) without any crystallographic impurities, obtained with a 1 : 1 ratio of Ni and Zn (Fig. 1C). The observed broad peaks could be assigned to the characteristic (101) and (110) planes of NiZn respectively, whereas other peaks were suppressed because of their tiny sizes. We then performed a compositional analysis of the NiZn NPs using TEM energy-dispersive X-ray spectroscopy (TEM-EDS; Fig. 1G) and inductively coupled plasma atomic emission spectroscopy (ICP-OES). The overall Ni/Zn composition in the NiZn alloy was quantitatively determined to be 0.97/1.0 (49.2 and 50.8 atomic wt% values of Ni and Zn, respectively). To directly visualize the chemical composition and subsequent distribution of the constituent elements, scanning transmission electron microscopy-energy dispersive spectroscopy (STEM-EDS) elemental mapping was performed. The STEM image of the NiZn alloy is shown in Fig. 1H. The STEM-EDS elemental mapping indicated that the distribution of Ni and Zn was even throughout the NiZn, providing further evidence for the formation of an alloyed structure. Again, the composition of Zn was within the phase boundary of the β-phase, attributed to the formation of ordered NiZn alloys, i.e.; intermetallic NiZn.33,34 No other peaks were observed in the XRD pattern, indicating a high purity of the β-NiZn, which was produced due to the substitution of Ni atoms with Zn atoms.35
X-ray photoelectron spectroscopy (XPS) of the NiZn alloy was carried out to understand the elemental composition of the alloy and the electronic interaction between the Ni and Zn atoms in the alloy. The high-resolution spectrum in the Ni 2p region (Fig. 2A) displayed the characteristic spin–orbit peaks for Ni 2p3/2 and Ni 2p1/2 with their corresponding shake-up satellites.36,37 The observed binding energy (BE) of 852.8 eV for the Ni 2p3/2 peak and the BE of 870.2 eV for the Ni 2p1/2 peak were assigned to metallic Ni0. Similarly, the fine-scanned spectrum in the Zn 2p region (Fig. 2B) showed peaks at BE values of 1021.7 and 1044.7 eV, attributed to Zn 2p3/2 and Zn 2p1/2, that is with a BE difference of 23 eV, demonstrating the presence of Zn0 in the alloy.38,39 On the basis of their BEs, both Ni and Zn were established to be present in the metallic state in the NiZn alloy. However, a positive shift was noticed for the 2p3/2 and 2p1/2 peaks for Ni0 in the spectrum of NiZn compared to those in the spectrum of pure Ni NPs (Fig. S2†). This shift indicated the modification of the electronic structure of Ni in NiZn after becoming alloyed with Zn.40 All of the experimental analyses and corresponding collective results ascertained the successful synthesis of monodispersed NiZn alloys under the developed reaction route.
Fig. 2. High-resolution XPS spectra of the NiZn alloy, showing the (A) Ni 2p and (B) Zn 2p regions. The peaks indicate the presence of metallic Ni0 and Zn0 in the alloy.

Here, ultra-small NiZn alloy particles were synthesized via a diffusion-based mechanism, which involved two steps. First, Ni NPs were formed from the thermolysis or reduction of the corresponding precursors in an organoamine solvent at high temperature, and then transformation of these Ni NPs into NiZn intermetallics occurred due to the reaction with diethylzinc, where organoamines acted as a reducing agent.41,42 Basically, Zn atoms diffused into the nanocrystalline Ni and produced the NiZn alloy. In our present system, the morphology of the NiZn alloy was templated by the morphology of the corresponding Ni NPs. Hence, the present synthetic approach provided the formation of uniform spherical NiZn intermetallics, which may have potential applications in energy storage and conversion owing to their alloyed structure.
OER activity
The electrocatalytic performances of the various electrocatalysts were investigated with respect to the OER using a 1.0 M KOH aqueous solution (4OH− → O2 + 2H2O + 4e−) in a conventional three-electrode configuration.43 The catalytic properties of the as-synthesized materials were measured after drop casting in each case a prepared ink—containing the desired sample and carbon black suspended in a mixture of isopropanol, Nafion and water—uniformly onto a GC electrode with a loading of ∼0.51 mg cm−2. To provide context for the activity of the as-synthesized catalyst, we also studied the electrocatalytic performance of a standard OER catalyst, namely RuO2, with a similar amount of loading under analogous experimental conditions.
The OER activities of the samples were first evaluated by performing linear sweep voltammetry (LSV) in aqueous solutions of 1.0 M KOH. Fig. 3A shows LSV polarization curves for all of the catalysts, where current density is plotted against potential. Interestingly, ultrasmall Ni NPs exhibited better electrocatalytic activity compare to the commercial RuO2. We then studied the OER activity of the NiZn alloy NPs and found them displaying much higher OER activity than RuO2 and pure Ni NPs. In an electrocatalytic reaction, current density is directly proportional to the OER activity of a catalyst. Thus, a higher current density at a same potential indicates a faster OER. In our system, the current densities at the same potential were always higher for the NiZn alloys. To reach a current density of 10 mA cm−2, the required overpotential (η10) values calculated from the LSV curves were 343, 332 and 283 mV for RuO2, Ni and NiZn, respectively. That is, the obtained η10 values of the catalysts were found to be in the order RuO2 > Ni > NiZn. To gain further insights into the OER kinetics, Tafel slopes were determined using the equation44η = b log(j/j0), where η indicates overpotential, b represents the Tafel slope, and j and j0 are the current density and exchange current density, respectively. The Tafel slope of NiZn was estimated to be 73 mV dec−1, lower than that of Ni (85 mV dec−1) and even RuO2 (92 mV dec−1), as shown in Fig. 3B. The obtained Tafel slope indicated a much faster OER when using the NiZn alloy, demonstrating prominent oxygen evolution activity of NiZn compared to those when using only Ni or RuO2.
Fig. 3. The OER activities of the NiZn alloy, Ni NPs and RuO2. (A) LSV curves of Ni, NiZn and RuO2, at a scan rate of 5 mV s−1. (B) Tafel plots of all the catalysts. (C) Nyquist plots of the OER using Ni and NiZn (with AC impedance data acquired at the η10 potential for each catalyst). The inset shows the corresponding equivalent circuit diagram. (D) Half of the capacitive current density differences (Δj/2) of Ni and NiZn plotted against the scan rate, where the double layer capacitance (Cdl) is equivalent to the linear slope. (E) The i–t curve of NiZn electrocatalyst for the OER up to 16 h at η10. (F) LSV curves of as-deposited fresh NiZn catalyst and that after continuous application to the OER for 16 h. Note the near-complete overlap of these curves.

Electrochemical impedance spectroscopy (EIS) was performed at a potential of 10 mA cm−2 (η10) for each of the catalysts to understand the nature of the charge transfer between the tested catalyst and electrolyte as well as to characterize the OER kinetics of the catalyst. The semicircle of a Nyquist plot (Fig. 3C) is associated with charge-transfer resistance (Rct). The corresponding equivalent circuit diagram is presented in the inset of Fig. 3C, where Rs is the solution resistance, Rct is the charge-transfer resistance and CPE represents a constant-phase element. It is important to note that lowering Rct is crucial for a catalyst from the both scientific and practical viewpoints. The Rct of NiZn was determined from the Nyquist plot to be 22 Ω, much lower than the 51.5 Ω value for Ni NPs, and revealing faster electron transfer occurring in NiZn and thus facilitated the OER process. The faster electron transfer was attributed to the synergistic effect of Ni and Zn in the NiZn alloy, resulting in the excellent electrocatalytic activity of NiZn. The electrocatalytic active surface area (ECSA) is another important factor, as it regulates the intrinsic activity of an electrocatalyst. First, double-layer capacitance (Cdl) was measured in order to obtain an estimate of the ECSA of the catalyst using scan-rate-dependent CVs in a non-faradaic region,45,46 as they are proportional to each other (Fig. S3 in the ESI section†). Half capacitive current density differences (Δj/2) of Ni and NiZn obtained from CVs were each plotted against scan rate. The double layer capacitance (Cdl) was determined from the slope of the linear plot, as shown in Fig. 3D. The Cdl value of NiZn was determined to be 1.84 mF cm−2 and that of Ni was 0.98 mF cm−2. The greater Cdl value of NiZn was attributed to the higher ECSA of NiZn, which contributed to the enhanced electrocatalytic activity of the NiZn alloy. Stability and long-standing durability are important requirements for the practical application of any designed electrocatalyst. To demonstrate the stability of NiZn, potentiostatic analysis at an overpotential of η10 was carried out using NiZn for a prolonged period of time; the result indicated almost no change in the current density even after 16 h (Fig. 3E). Furthermore, NiZn subjected to continuous OER at η10 for a long period of time showed almost the same onset potential in its LSV curve as did the as-deposited fresh catalyst (Fig. 3F). Both analyses pointed to the excellent stability as well as durability of NiZn even after repeated usage.
In addition to the electrochemical analysis of the stability, TEM analysis of the NiZn alloy was carried out after the OER study. This TEM analysis revealed its intact morphology, even after a longer chronoamperometry study (Fig. S4†). Then, an XPS analysis was performed on NiZn after the OER process to demonstrate whether any chemical change occurred (Fig. S5†). From the XPS spectra of NiZn alloy, it was noticed that along with the metallic Ni0 (852.8 eV for Ni 2p3/2) and Zn0 (1021.7 eV for Zn 2p3/2) states, their oxides were also present. Deconvolution of the signal in the Ni 2p region of the high-resolution spectrum demonstrated the presence of new peaks, at 855.7 eV for Ni 2p3/2 and at 873.5 eV for Ni 2p1/2, suggesting the formation of Ni2+ along with the metallic Ni0.47 Similarly, analysis of the Zn 2p region of the high-resolution spectrum indicated the presence of Zn2+ along with the metallic Zn0, as new peaks were observed at 1022.3 eV and 1045.1 eV for Zn 2p3/2 and Zn 2p1/2, respectively.48 Although the TEM analysis demonstrated an unaltered morphology, the XPS analysis suggested a slight oxidation of the NiZn alloy after the prolonged electrochemical study.
It is then interesting to us to study the electrochemical properties of the alloy upon tuning the composition of its constituents. For this purpose, we prepared nanoscale fcc Ni0.7Zn0.3 alloy (PDF no. 01-072-2672) with particle dimensions similar to those of the original NiZn alloy by changing the reaction conditions (Fig. S6†). The overall percentages of Ni and Zn in the prepared Ni0.7Zn0.3 alloy were quantitatively assessed using ICP-OES analysis to be 74.0 and 26.0 atomic wt%. Ni0.7Zn0.3 showed an overpotential (η10) of 320 mV, which is bit lower than that of pure Ni NPs but much higher compare to intermetallic NiZn (shown in Fig. S7†). The estimated overpotentials and Tafel slopes for all of the catalysts are summarized in Table 1. All these results indicated that the OER activity was sensitive to the atomic ordering and composition of the alloy, leading to the superior activity of order NiZn intermetallics than that of Ni0.7Zn0.3 alloy.
A summary of the overpotentials and Tafel slopes for all the catalysts.
| Catalyst | Overpotential (mV) η10 | Tafel slope (mV dec−1) |
|---|---|---|
| RuO2 | 343 | 92 |
| Ni | 332 | 85 |
| Ni0.7Zn0.3 | 320 | 82 |
| NiZn | 283 | 73 |
In the present system, the diffusion of Zn atoms into nanocrystalline Ni was considered to be key to enhancing the catalytic performances of the NiZn alloy. The higher OER activity of the NiZn alloy was apparently associated with the increase in the ECSA together with their intrinsic catalytic activity as well as the possible synergistic effect between the Ni and Zn metals. The 2p and valence electrons of Ni in case of NiZn alloy shifted toward Zn,40,49,50 resulting in the charge transfer from Ni to Zn and subsequently making the Ni centre slightly positive. In our experiments, Ni served as the active center, whereas Zn acted as a promoter in the alloy during the OER process. Consequently, the OER proceeded very rapidly via Ni centres in the NiZn alloy, as schematically described in Fig. 4. Basically, upon alloying of Zn into Ni, a modification of the electronic structure of Ni tailored by Zn took place in the NiZn alloy. Furthermore, the ordered alloys provided uniform active sites on the same surface plane owing to their compositional and positional order, in addition to the precise control over the structure accompanied by electronic effects. Finally, we compared the electrocatalytic activities of the NiZn alloy with the previously reported Ni-based OER catalysts, as shown in Table S1 in the ESI section.† The as-synthesized NiZn displayed better electrocatalytic activity toward the OER than did the reported catalysts. In a nutshell, based on the electrocatalytic activity as well as comparison with the expensive conventional electrocatalysts and even several other reported electrocatalysts, our study overall demonstrated that intermetallic NiZn NPs can be considered as an effective electrocatalyst for oxygen production.
Fig. 4. A schematic representation of oxygen evolution electrocatalysis on the surface of the NiZn alloy. Ni and Zn centers in NiZn are shown as blue and violet spheres, respectively.

Conclusions
In conclusion, the efficient synthesis of ultra-small NiZn intermetallics was demonstrated via the thermolysis of the corresponding metal precursors in a mixture of hot organic solvents. The present synthetic strategy comprised the chemical conversion of preformed metal NPs into alloys at low temperature via reaction with another metal precursor. The current finding also demonstrated that the Ni NPs acted as a precursor to prepare NiZn alloy and retained their morphology during the transformation to the NiZn alloy, attributing to a general synthesis pathway aimed at making ultra-small M-Zn alloys. The intermetallic NiZn NPs exhibited enhanced electrocatalytic activity and excellent stability during the OER, better than those of a Ni0.7Zn0.3 alloy, pure fcc Ni and RuO2. The present study demonstrated that activity of the alloy improved as a result of the composition of constituents, which govern their OER performances. The superior electrocatalytic efficiency of NiZn was ascribed to the modification of the electronic structure of Ni upon the incorporation of Zn, together with an atomic scale synergistic effect produced from Ni and Zn in the NiZn alloy. Thus, the novelty of our work was based on the controlled fabrication of monodisperse tiny NiZn alloy particles having excellent OER activity, better than those of expensive conventional catalysts as well as several other reported electrocatalysts. Our findings also suggested that catalysts with superior activity can be developed by engineering the composition of the constituent metals during the fabrication of desired alloyed nanoparticles.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was funded by a Nano Mission Research Grant (SR/NM/NS-18/2014) and SERB Women Excellence Award Grant (SB/WEA/08/2016) from the Department of Science and Technology (DST), Government of India. The authors also acknowledge the S. N. Bose National Centre for Basic Sciences, Kolkata, India.
Electronic supplementary information (ESI) available: Materials and characterization techniques; particle size distribution data; XPS spectra of Ni 2p in Ni NPs and the corresponding NiZn alloy; ECSA calculations for the catalysts; TEM images and XPS analysis after potentiostatic measurements; TEM and XRD analyses of Ni0.7Zn0.3 and its OER activity; a comparison of the OER activity of the NiZn alloy with the reported Ni-based electrocatalysts. See DOI: 10.1039/c9na00611g
Notes and references
- Turner J. A. Science. 2004;305:972–974. doi: 10.1126/science.1103197. [DOI] [PubMed] [Google Scholar]
- Walter M. G. Warren E. L. McKone J. R. Boettcher S. W. Mi Q. Santori E. A. Lewis N. S. Chem. Rev. 2010;110:6446–6473. doi: 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- Tang C. Cheng N. Pu Z. Xing W. Sun X. Angew. Chem., Int. Ed. 2015;54:9351–9355. doi: 10.1002/anie.201503407. [DOI] [PubMed] [Google Scholar]
- Samanta A. Das S. Jana S. ACS Sustainable Chem. Eng. 2019;7:12117–12124. [Google Scholar]
- Zheng S. Guo X. Xue H. Pan K. Liu C. Pang H. Chem. Commun. 2019;55:10904–10907. doi: 10.1039/C9CC06113D. [DOI] [PubMed] [Google Scholar]
- Wang H. Lu Z. Xu S. Kong D. Cha J. J. Zheng G. Hsu P. C. Yan K. Bradshaw D. Prinz F. B. Cui Y. Proc. Natl. Acad. Sci. U. S. A. 2013;110:19701–19706. doi: 10.1073/pnas.1316792110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. Suntivich J. May K. J. Perry E. E. Shao-Horn Y. J. Phys. Chem. Lett. 2012;3:399–404. doi: 10.1021/jz2016507. [DOI] [PubMed] [Google Scholar]
- Concepcion J. J. Jurss J. W. Brennaman M. K. Hoertz P. G. Patrocinio A. O. T. Iha N. Y. M. Templeton J. L. Meyer T. J. Acc. Chem. Res. 2009;42:1954–1965. doi: 10.1021/ar9001526. [DOI] [PubMed] [Google Scholar]
- Wang Y.-J. Zhao N. Fang B. Li H. Bi X. T. Wang H. Chem. Rev. 2015;115:3433–3467. doi: 10.1021/cr500519c. [DOI] [PubMed] [Google Scholar]
- Stamenkovic V. R. Mun B. S. Arenz M. Mayrhofer K. J. J. Lucas C. A. Wang G. Ross P. N. Markovic N. M. Nat. Mater. 2007;6:241–247. doi: 10.1038/nmat1840. [DOI] [PubMed] [Google Scholar]
- Shao M. H. Sasaki K. Adzic R. R. J. Am. Chem. Soc. 2006;128:3526–3527. doi: 10.1021/ja060167d. [DOI] [PubMed] [Google Scholar]
- Lim B. Jiang M. Camargo P. H. C. Cho E. C. Tao J. Lu X. Zhu Y. Xia Y. N. Science. 2009;324:1302–1305. doi: 10.1126/science.1170377. [DOI] [PubMed] [Google Scholar]
- Bu L. Guo S. Zhang X. Shen X. Su D. Lu G. Zhu X. Yao J. Guo J. Huang X. Nat. Commun. 2016;7:11850–11859. doi: 10.1038/ncomms11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y. Zhou Y. Wang D. Wu X. Li J. Xi J. Adv. Energy Mater. 2018;8:1701759–1701765. doi: 10.1002/aenm.201701759. [DOI] [Google Scholar]
- Yi Z. Wang Z. Cheng Y. Wang L. Energy Environ. Mater. 2018;1:132–147. doi: 10.1002/eem2.12016. [DOI] [Google Scholar]
- Obrovac M. N. Chevrier V. L. Chem. Rev. 2014;114:11444–11502. doi: 10.1021/cr500207g. [DOI] [PubMed] [Google Scholar]
- Maeda K. Teramura K. Masuda H. Takata T. Saito N. Inoue Y. Domen K. J. Phys. Chem. B. 2006;110:13107–13112. doi: 10.1021/jp0616563. [DOI] [PubMed] [Google Scholar]
- Jiao Z. Zhang Y. Ouyang S. Yu H. Lu G. Ye J. Bi Y. ACS Appl. Mater. Interfaces. 2014;6:19488–19493. doi: 10.1021/am506030p. [DOI] [PubMed] [Google Scholar]
- Cai X. Wang A. Wang J. Wang R. Zhong S. Zhao Y. Wu L. Chen J. Bai S. J. Mater. Chem. A. 2018;6:17444–17456. doi: 10.1039/C8TA06204H. [DOI] [Google Scholar]
- Pei Y. LaLonde A. D. Heinz N. A. Snyder G. J. Adv. Energy Mater. 2012;2:670–675. doi: 10.1002/aenm.201100770. [DOI] [Google Scholar]
- Takagiwa Y. Utada S. Kanazawa I. Kimura K. J. Mater. Chem. C. 2015;3:10422–10429. doi: 10.1039/C5TC01608H. [DOI] [Google Scholar]
- Taub A. I. Fleischer R. L. Science. 1989;243:616–621. doi: 10.1126/science.243.4891.616. [DOI] [PubMed] [Google Scholar]
- Krenke T. Duman E. Acet M. Wassermann E. F. Moya X. Manosa L. Planes A. Nat. Mater. 2005;4:450–454. doi: 10.1038/nmat1395. [DOI] [PubMed] [Google Scholar]
- Körner W. Krugel G. Elsässer C. Sci. Rep. 2016;6:24686–24694. doi: 10.1038/srep24686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N. Li Q. Yuan M. Guo X. Zheng S. Pang H. Chem.–Asian J. 2019;14:2259–2265. doi: 10.1002/asia.201900361. [DOI] [PubMed] [Google Scholar]
- Ding J. Bu L. Guo S. Zhao Z. Zhu E. Huang Y. Huang X. Nano Lett. 2016;16:2762–2767. doi: 10.1021/acs.nanolett.6b00471. [DOI] [PubMed] [Google Scholar]
- Liu Y. Liu X. Feng Q. He D. Zhang L. Lian C. Shen R. Zhao G. Ji Y. Wang D. Zhou G. Li Y. Adv. Mater. 2016;28:4747–4754. doi: 10.1002/adma.201600603. [DOI] [PubMed] [Google Scholar]
- Furukawa S. Komatsu T. ACS Catal. 2017;7:735–765. doi: 10.1021/acscatal.6b02603. [DOI] [Google Scholar]
- Kuttiyiel K. A. Sasaki K. Su D. Wu L. Zhu Y. Adzic R. R. Nat. Commun. 2014;5:5185–5192. doi: 10.1038/ncomms6185. [DOI] [PubMed] [Google Scholar]
- Yan Y. Du J. S. Gilroy K. D. Yang D. Xia Y. Zhang H. Adv. Mater. 2017;29:1605997–1606025. doi: 10.1002/adma.201605997. [DOI] [PubMed] [Google Scholar]
- Wang D. Xin H. L. Hovden R. Wang H. Yu Y. Muller D. A. DiSalvo F. J. Abruña H. D. Nat. Mater. 2013;12:81–87. doi: 10.1038/nmat3458. [DOI] [PubMed] [Google Scholar]
- Casado-Rivera E. Volpe D. J. Alden L. Lind C. Downie C. Vazquez-Alvarez T. Angelo A. C. DiSalvo F. J. Abruna H. D. J. Am. Chem. Soc. 2004;126:4043–4049. doi: 10.1021/ja038497a. [DOI] [PubMed] [Google Scholar]
- Vassilev G. Gomez-Acebo T. Tedenac J.-C. J. Phase Equilib. 2000;21:287–301. doi: 10.1361/105497100770340075. [DOI] [Google Scholar]
- Schmetterer C. Rajamohan D. Ipser H. Flandorfer H. Intermetallics. 2011;19:1489–1501. doi: 10.1016/j.intermet.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana S. Dalton Trans. 2015;44:18692–18717. doi: 10.1039/C5DT03699B. [DOI] [PubMed] [Google Scholar]
- Wu L. S. Dai H. B. Wen X. P. Wang P. ChemElectroChem. 2017;4:1944–1949. doi: 10.1002/celc.201700234. [DOI] [Google Scholar]
- Biesinger M. C. Payne B. P. Grosvenor A. P. Lau L. W. Gerson A. R. Smart R. S. C. Appl. Surf. Sci. 2011;257:2717–2730. doi: 10.1016/j.apsusc.2010.10.051. [DOI] [Google Scholar]
- Yin G. Abe H. Kodiyath R. Ueda S. Srinivasan N. Yamaguchi A. Miyauchi M. J. Mater. Chem. A. 2017;5:12113–12119. doi: 10.1039/C7TA00353F. [DOI] [Google Scholar]
- Sivagami I. N. Prasanna K. Santhoshkumar P. Jo Y. N. Seo G. Y. Lee C. W. J. Alloys Compd. 2017;697:450–460. doi: 10.1016/j.jallcom.2016.08.177. [DOI] [Google Scholar]
- Rodriguez J. A. Kuhn M. J. Phys. Chem. 1996;100:381–389. doi: 10.1021/jp952249m. [DOI] [Google Scholar]
- Jana S. Chang J. W. Rioux R. M. Nano Lett. 2013;13:3618–3625. doi: 10.1021/nl401467r. [DOI] [PubMed] [Google Scholar]
- Puntes V. F. Zanchet D. Erdonmez C. K. Alivisatos A. P. J. Am. Chem. Soc. 2002;124:12874–12880. doi: 10.1021/ja027262g. [DOI] [PubMed] [Google Scholar]
- Jia Y. Zhang L. Gao G. Chen H. Wang B. Zhou J. Soo M. T. Hong M. Yan X. Qian G. Zou J. Adv. Mater. 2017;29:1700017–1700024. doi: 10.1002/adma.201700017. [DOI] [PubMed] [Google Scholar]
- Xuan C. Wang J. Xia W. Zhu J. Peng Z. Xia K. Xiao W. Xin H. L. Wang D. J. Mater. Chem. A. 2018;6:7062–7069. doi: 10.1039/C8TA00410B. [DOI] [Google Scholar]
- Yu M. Moon G. Bill E. Tüysüz H. ACS Appl. Energy Mater. 2019;2:1199–1209. doi: 10.1021/acsaem.8b01769. [DOI] [Google Scholar]
- McCrory C. C. Jung S. Peters J. C. Jaramillo T. F. J. Am. Chem. Soc. 2013;135:16977–16987. doi: 10.1021/ja407115p. [DOI] [PubMed] [Google Scholar]
- Naghash A. R. Etsell T. H. Xu S. Chem. Mater. 2006;18:2480–2488. doi: 10.1021/cm051910o. [DOI] [Google Scholar]
- Guillory II R. J. Sikora-Jasinska M. Drelich J. W. Goldman J. ACS Appl. Mater. Interfaces. 2019;11:19884–19893. doi: 10.1021/acsami.9b05370. [DOI] [PubMed] [Google Scholar]
- Rodriguez J. A. Surf. Sci. Rep. 1996;24:223–287. doi: 10.1016/0167-5729(96)00004-0. [DOI] [Google Scholar]
- Pan Z. Wang R. Chen J. Appl. Catal., B. 2018;224:88–100. doi: 10.1016/j.apcatb.2017.10.040. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.