

Abstract
We report a genome-wide association study (GWAS) of coronary artery disease (CAD) incorporating nearly a quarter million cases, in which existing studies are integrated with data from cohorts of White, Black, and Hispanic individuals from the Million Veteran Program. We document near equivalent heritability of CAD across multiple ancestral groups, identify 95 novel loci, including the first nine to be identified on the X-chromosome, detect the first eight genome-wide significant loci among Blacks and Hispanics, and demonstrate that two common haplotypes at the 9p21 locus are responsible for risk stratification in all populations except those of African origin, where these haplotypes are virtually absent. Moreover, in the largest GWAS for angiographically derived coronary atherosclerosis performed to date, we find 15 genome-wide significant loci that robustly overlap with established loci for clinical CAD. Phenome-wide association analyses of novel loci and polygenic risk scores (PRS) augment signals related to insulin resistance, extend pleiotropic associations of these loci to include smoking and family history, and precisely document the markedly reduced transferability of existing PRS to Black individuals. Downstream integrative analyses reinforce the critical roles of vascular endothelial, fibroblast, and smooth muscle cells in CAD susceptibility, but also point to a shared biology between atherosclerosis and oncogenesis. Our study highlights the value of diverse populations in further characterizing the genetic architecture of CAD.
Editor summary:
To overcome limitations of previous genome-wide association studies of coronary artery disease, this study incorporates a cohort of individuals containing large fractions of Black and Hispanic individuals, providing a wider perspective of the genetic landscape of this disease.
Introduction
Remarkable progress in the prevention and treatment of coronary artery disease (CAD) has been made over the last half century. Yet, the rate of decrease in the age-adjusted prevalence of CAD has slowed substantially in the last decade, and CAD remains the leading cause of death worldwide1. Sizeable differences in the age-adjusted fatality rates of CAD persist between men and women and among the major populations in the US with non-Hispanic Black men persistently demonstrating the highest risk of fatal CAD2. These disparities, largely driven by structural racism3, may be amplified in the era of precision medicine due to little or no inclusion of Blacks and Hispanics in large-scale genetic studies of cardiovascular disease to date4–6. Thus, a persistent need exists to further understand both the between-population and the population-specific genetic causes of CAD as an avenue towards improved risk prediction and the development of novel therapies.
Large-scale population genetic studies provide an opportunity to improve our understanding of the inherited basis of complex traits. Twin studies report a heritability of 40–60% for fatal CAD7,8 and genome-wide association studies (GWAS) to date have identified 208 susceptibility loci9,10. These loci explain a modest fraction (~15%) of this heritability, have largely been identified in European populations, and are exclusively autosomal9,10. Approximately one half of established loci appear to confer risk through effects on traditional risk factors such as lipids and blood pressure with fewer links to other risk factors9,11. Several loci discovered in Europeans have also reached genome-wide significance in South and East Asian populations suggesting an overlap in the genetic architecture of CAD across these three populations10,12,13. Yet, 14 years after the discovery of the first susceptibility locus at 9p21, no region has convincingly reached genome-wide significance in Black or Hispanic populations, which represent a sizable and growing proportion of the US population14,15.
New DNA biobanks with enrollment of diverse populations are poised to fulfill this knowledge gap. Here we describe results from analyses of the Million Veteran Program (MVP), a nationwide cohort drawn from an integrated health care system serving a diverse population including many Blacks and Hispanics. By meta-analyzing these new large-scale, multi-population GWAS data with extant GWAS of CAD from public resources, we extend discovery of CAD loci within and across populations for both the autosomes and the X-chromosome (X-chr). In addition, we incorporate data from a national registry of cardiac catheterization procedures in the discovery of novel CAD loci and in the interpretation of the mechanism of action of established loci and polygenic risk scores.
Results
Population diversity in the MVP population
Fig. 1a summarizes new and existing cohorts included in our analyses stratified by population and the analytic approach for the clinical CAD phenotype. A majority (90.8%) of veteran participants are male with 95,151 cases and 197,287 controls being classified as non-Hispanic White, hereinafter referred to as White, (73.1%), 17,202 cases and 59,507 controls as non-Hispanic Blacks, hereinafter referred to as Black, (19.2%), and 6,378 and 24,270 as Hispanic (7.7%) (Extended Data Table 1). Most cases (85.6%) showed evidence of CAD at the time of enrollment in the MVP (i.e., “prevalent”). The mean age at first evidence of CAD in the electronic health record (EHR) was 63 years with a mean combined EHR follow-up either prior to and/or after enrollment of 10 years.
Fig. 1: Design of multi-population genome wide association study (GWAS) of coronary artery disease (CAD) and estimates of heritability (h2) of CAD using GREML-LDMS-I for four populations.

a, Study design. GWAS was first performed stratified by population group. GWAS for Whites was then meta-analyzed with 2 existing GWAS for initial discovery among Whites. The GWAS for MVP Hispanics and MVP Blacks as well as the Biobank Japan GWAS of CAD was further incorporated into a single multi-population meta-analysis. Two-stage joint meta-analysis of the most promising SNPs was performed for the Hispanics and Blacks with multiple external cohorts for population-specific discovery. b-d, Heritability (h2) analyses for CAD in four major racial groups using GREML-LDMS-I. b. Principal component analysis of MVP participants combined with 1000 genomes was first performed to identify a random subset of 19,395 Hispanics with the highest proportion of Indigenous American ancestry (pink). A random subset of the 19,392 least admixed Whites (dark green) and the 19,392 least admixed Blacks (dark blue), respectively, were then matched 1:1 on case-control status, age of first EHR evidence of CAD, type of CAD presentation, and age of controls to the Hispanics. Similar matching was performed for 18,747 participants from the Biobank Japan study. c, Observed narrow-sense h2 within each cohort defined in b using a multi-component model, GREML-LDMS-I, implemented in GCTA, with age, sex, and a genetic relatedness matrix as covariates. h2 estimate and respective standard error (SE) of that estimate is shown for each of 24 bins of imputed SNPs defined by linkage disequilibrium score quartiles and six minor allele frequency thresholds (top panel) with the corresponding absolute number of SNPs contributing to this h2 shown on the bottom panel. Total h2 is calculated by summing 24 estimates with SE for this estimate calculated by delta method. d, h2 on the liability scale for each population in c as a function of a range of presumed population prevalence of CAD. Error bars denote +/− one SE around each point estimate.
Estimation of CAD heritability across multiple ancestries
We first estimated the SNP-based heritability using GREML-LDMS-I in equally sized subsets of MVP Whites, Blacks with the least European admixture, and Hispanics with the least African admixture, as well as Japanese participants from Biobank Japan after matching on the age of onset and severity of disease of cases and the age of controls observed among the MVP Hispanics (Methods, Fig. 1b, Extended Data Table 2). Assuming a prevalence of CAD of 8.2%, 6.5%, 4.9%, and 6.0% in the same populations16,17, we derived roughly equivalent heritability on the liability scale of 36.3% (±7.0%), 30.0% (±8.1%), 32.6% (±3.9%), and 36.0% (±5.4%), respectively (Fig. 1c–d).
GWAS in MVP and meta-analysis with existing studies
We conducted a GWAS of autosomes and X-chr stratified by population of White, Black, and Hispanic MVP participants. The genomic control inflation (λ) for these GWAS was 1.360 (Whites), 0.988 (Blacks), and 0.986 (Hispanics). The LD score regression intercept for Whites was 1.077 (±0.014), indicating most of the inflation was polygenic in nature. We found a high rate of replication of established loci as of 20199 among Whites with 100% of 163 known lead SNPs being directionally concordant, 67.5% reaching Bonferroni significance (P<3.1×10−4), and 36 (22.1%) reaching genome-wide significance (GWS). Effect sizes were also highly correlated (Pearson rho=0.94) (Supplementary Table 1).
The GWAS of MVP Whites was followed by a meta-analysis with existing predominantly European-ancestry GWAS from CARDIoGRAMplusC4D18 and the UK Biobank9 yielding 33 novel loci at GWS (lead SNP P<5×10−8), including five on the X-chr (Methods, Fig. 2, Supplementary Table 2). Our multi-population meta-analysis further incorporated the GWAS data from MVP Blacks and MVP Hispanics and Biobank Japan19, yielding an additional 62 novel autosomal loci including four more loci on the X-chr (Fig. 2, Supplementary Table 3). All lead SNPs showed no significant heterogeneity across studies (lowest p=0.0017), both within the meta-analysis of Whites as well as the multi-population meta-analysis using either METAL or MR-MEGA. We annotate lead SNPs from these 95 loci by providing hyperlinks to five comprehensive variant-based portals (Supplementary Table 4).
Fig. 2: Population-specific GWAS and multi-population meta-analysis.
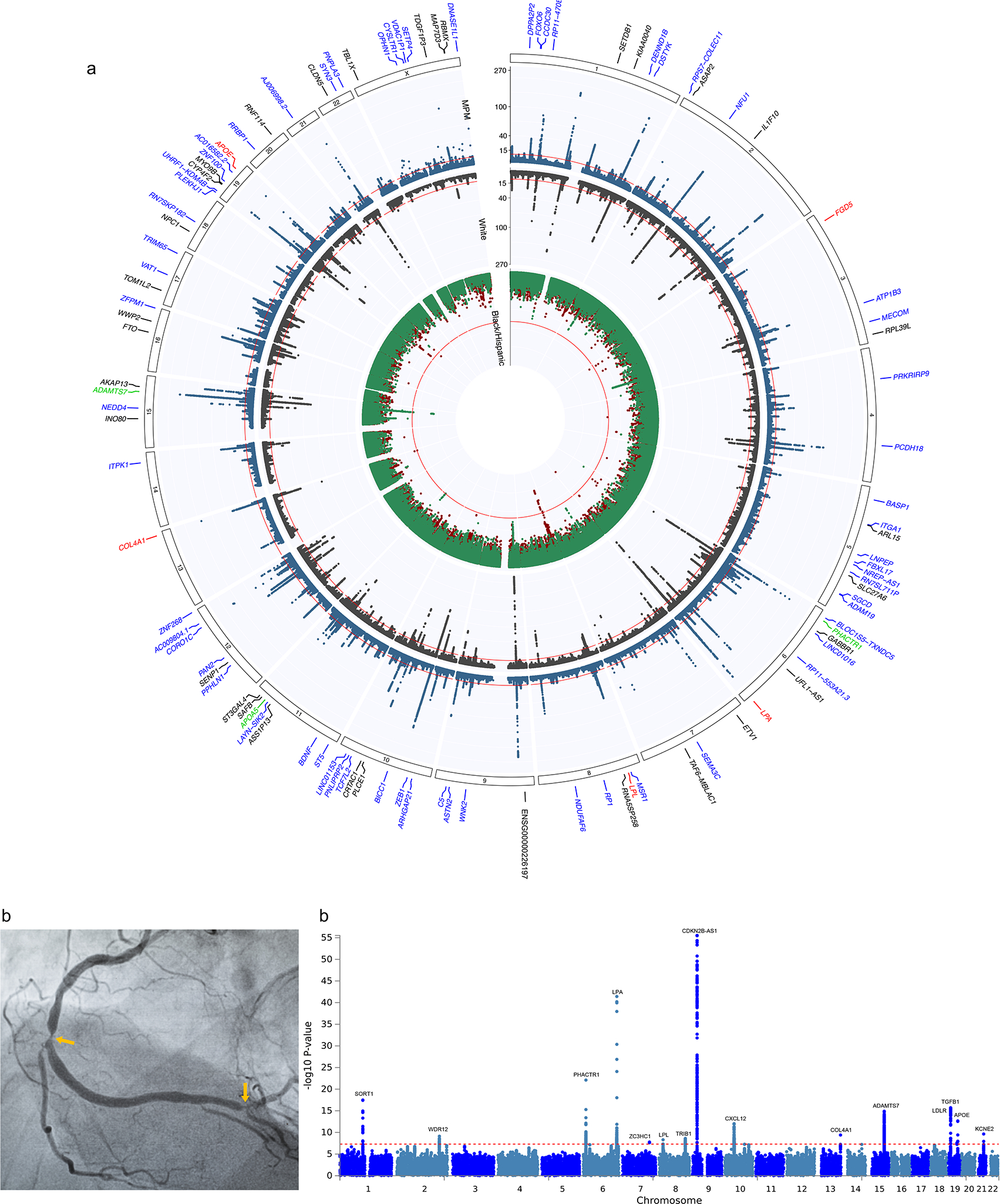
a, Circos plot indicating the −log10(P) for association with CAD for population-specific and multi-population GWAS meta-analyses. See Figure 1a for sample sizes. P values are derived from inverse variance weighted meta-analysis using METAL or GWAMA and are two-sided. The inner track plots the 2-stage meta-analysis association results for Blacks in red and Hispanics (HISP) in green, while the middle track plots the results for the meta-analysis of Whites in black and the multi-population metanalysis further incorporating the GWAS of MVP Blacks, MVP Hispanics, and of Biobank Japan in blue. The red line indicates genome-wide significance (GWS) (P = 5.0 × 10−8). The outer track lists the nearest mapped gene to the lead SNPs reaching GWS in each of these four meta-analyses including five loci in Blacks (red font), three loci in Hispanics (green font), 33 novel loci among Whites (black font), and 62 additional novel loci after the multi-population meta-analysis (blue font). b, Example of X-ray image from an angiogram of the right coronary artery used to estimate the burden of coronary atherosclerosis. The image shows 2 high-grade obstructions (arrows) as contrast agent is injected into the blood vessel (Adobe Stock FILE #: 413211903). Manhattan plot (right) of multi-population meta-analysis of GWAS (n=41,507) for burden of coronary atherosclerosis as estimated by the number of arteries with obstructions >50% on an angiogram. P values are derived from inverse variance weighted meta-analysis using METAL and are two-sided.
XPEB and two-stage joint analysis in Blacks and Hispanics
Our GWAS of Blacks and Hispanics in MVP did not yield any GWS loci that passed quality control within either population in isolation. We were unable to replicate findings among Blacks at CDK14, a locus reported as GWS about a decade ago20. The same region was entirely void of signal in our MVP Blacks and two SNPs in high LD (r2=1) with the previously reported lead SNP (rs1859023) had p-values near one (rs7792416, p=0.97; and rs10639151, p = 0.99). However, XPEB, an empirical Bayes mapping approach that adaptively incorporates cross-population evidence with an ‘auxiliary base GWAS’ (CAD meta-analysis in Whites), identified 37 SNPs at 16 loci in MVP Blacks and 157 SNPs at 38 loci in MVP Hispanics with a local False Discovery Rate (FDR) < 0.05 (Supplementary Table 5). All but one of the loci identified by XPEB were GWS in the base GWAS (meta-analysis in Whites).
We then extended our GWAS analysis of MVP Blacks and MVP Hispanics to include additional data from multiple external cohorts (Extended Data Table 3) for the most promising variants from our GWAS (P<1×10−5) and all SNPs identified by XPEB (Methods, Supplementary Text, Supplementary Tables 5–6). A two-stage joint meta-analysis of these SNPs yielded the first five GWS loci in Blacks and the first three in Hispanics (Fig. 2a, Supplementary Tables 7–8, Extended Data Table 4), all of which have been previously established in Whites14. Three out of five loci in Blacks (LPA, FGD5, and LPL) included GWS signals generated by low-frequency African specific genetic variation (Extended Data Fig. 1). The SNPs identified through XPEB and cross-population evidence include loci with more moderate allelic effects; therefore, a priori, we did not expect all of them to reach GWS in the much smaller two-stage meta-analysis of Blacks and Hispanics. However, this group of SNPs exhibited a significantly higher proportion of directional consistency and correlation of effect sizes between the MVP discovery cohort and the external cohorts, for both Blacks (13 out 15 loci with available data in external cohorts were directionally consistent, binomial P=0.0032, Pearson’s rho=0.82) and Hispanics (33 out of 36 loci directionally consistent, P=1.1×10−7, rho=0.80) (Supplementary Table 9).
GWAS of angiographically determined burden of CAD
We conducted the largest GWAS to date of angiographically determined burden of coronary atherosclerosis, defined by number of significantly obstructed (>50% of luminal diameter) coronary arteries. Analysis included 41,507 MVP participants: stratified GWAS was performed in 31,658 Whites, 7,313 Blacks, and 2,536 Hispanics followed by multi-population meta-analysis (Methods, Extended Data Tables 5–6) identified 15 GWS of which 12 also reached GWS in Whites alone and 1 (LPL) in Blacks alone (Fig. 2b, Supplementary Table 10). All 15 loci have been previously reported for clinical CAD, and eight (CDKN2B-AS1, SORT1, CXCL12, WDR12, PHACTR1, LDLR, KCNE2, ADAMTS7) were among the 12 earliest loci associated with clinical CAD by GWAS and all but TGFB1 were identified prior to 201314.
Credible set analysis of genome wide significant loci
We conducted a credible set analysis of all 188 known and novel loci reaching GWS within our meta-analysis of Whites to identify candidate causal variants, then compared results to the same analysis performed in our multi-population meta-analysis in the same regions (Supplementary Tables 11–12). Most loci (134/188, or 71%), had a reduction in the number of SNPs within their credible set when comparing the multi-population meta-analysis to that of Whites, with a 27.7% median and 34.1% mean percent reduction of SNPs per locus. A small fraction of loci (23/188, or 12%) had a modest increased number of SNPs among the multi-population meta-analysis credible set (median +19.0%, mean + 31%, respectively), mostly because of a second independent signal reaching a level of significance comparable to the initial region with the larger sample size including non-Whites. The remaining loci had no change in the number of SNPs per credible set.
We annotated all SNPs within the credible sets for our 95 novel loci with Ensembl Variant Effect Predictor (Supplementary Table 13). Protein coding genes with high +/− moderate impact variants within these sets include COQ10A, FBF1, GUF1, CYFIP2, MSR1, and FAM120AOS while genes with moderate impact genetic variants include DHDDS, ZMYND12, IL1F10, PRDM6, ADAM19, MCM7, TRAF1, C5, LOXL4, R3HCC1L, ST3GAL4, BDNF, ZNF268, ANKRD52, STAT2, AKAP13, LRRC48, MYO15A, COASY, MLX, TUBG2. CNTNAP1, TRIM65, ZNF100, RRBP1, PNPLA3, SAMM50, PLXNA3, UBL4A, ZNF100, and CYSLTR1.
Local ancestry and haplotype analysis at the 9p21 locus
The well-established susceptibility locus at 9p21 did not reach GWS among Blacks or Hispanics even after two-stage meta-analysis involving >27,000 and >12,100 CAD cases, respectively. A previously reported lead SNP at 9p21 in a meta-analysis of multiple African American cohorts was rs6475606 with a p-value of 6.4×10−4 21. The p-value in MVP Blacks for this SNP was 1.6×10−3.
We explored whether the ancestral origin of the high-risk haplotype block at 9p21 among Blacks influences the observed magnitude of association with CAD (Methods). Using RFMix, we stratified MVP Blacks into three subgroups based on whether they had inherited two (Black_AFR = +/+, 66.8%), one (Black_AFR = +/−, 29.6%), or zero (Black_AFR = −/−, 3.6%) chromosomal 9p21 segments from African (AFR) ancestry when compared to European (EUR) ancestry through admixture (Extended Data Fig. 2a). Only the first two of these three subgroups had adequate power to detect an association at 9p21. Between these two, we found notably stronger associations with CAD among Blacks with one AFR segment (Black_AFR = +/−, lowest P=6.4×10−7) despite a sample size of less than one half of Blacks with two AFR segments (Black_AFR = +/+, lowest P=1×10−3) (Extended Data Fig. 2b, Supplementary Table 14).
Haplotype analysis at 9p21 (Methods) revealed a largely non-overlapping set of haplotypes when comparing Whites to Blacks with zero 9p21 segments derived from EUR (Fig. 3a, Supplementary Table 15). Only 17 out of a possible 32 haplotypes were observed to any appreciable frequency. Two haplotypes (AACATT, GGTTCA) account for a large majority (87%) of observed haplotypes among Whites but these same two haplotypes are virtually absent (<0.5%) among the majority of Blacks with no EUR admixture at 9p21. Most of the remaining haplotypes are present to an appreciable frequency in Black_AFR+/+ but are virtually absent in Whites. Only one haplotype (AGTTCA) has appreciable frequency in both Whites (~5%) and Black_AFR+/+ (~10%). Our haplotype trend regression analysis suggests the second most common haplotype (GGTTCA) is associated with an increased risk for CAD when compared to the most common haplotype among Whites (AACATT, 47%) and these two haplotypes are largely responsible for the risk-stratifying potential of this locus within this group (Fig. 3b–c, Supplementary Table 15). However, the AACATT is unable to risk stratify among Blacks given it is virtually absent among Black_AFR+/+. Any signal among Blacks is dependent on the presence of this haplotype through local admixture with Whites, although analyses among the small subgroup Black_AFR−/− do not generate a reliable signal likely because of inadequate power.
Fig. 3: Local ancestry and haplotype analyses at the 9p21 susceptibility locus for CAD in the Million Veteran Program.
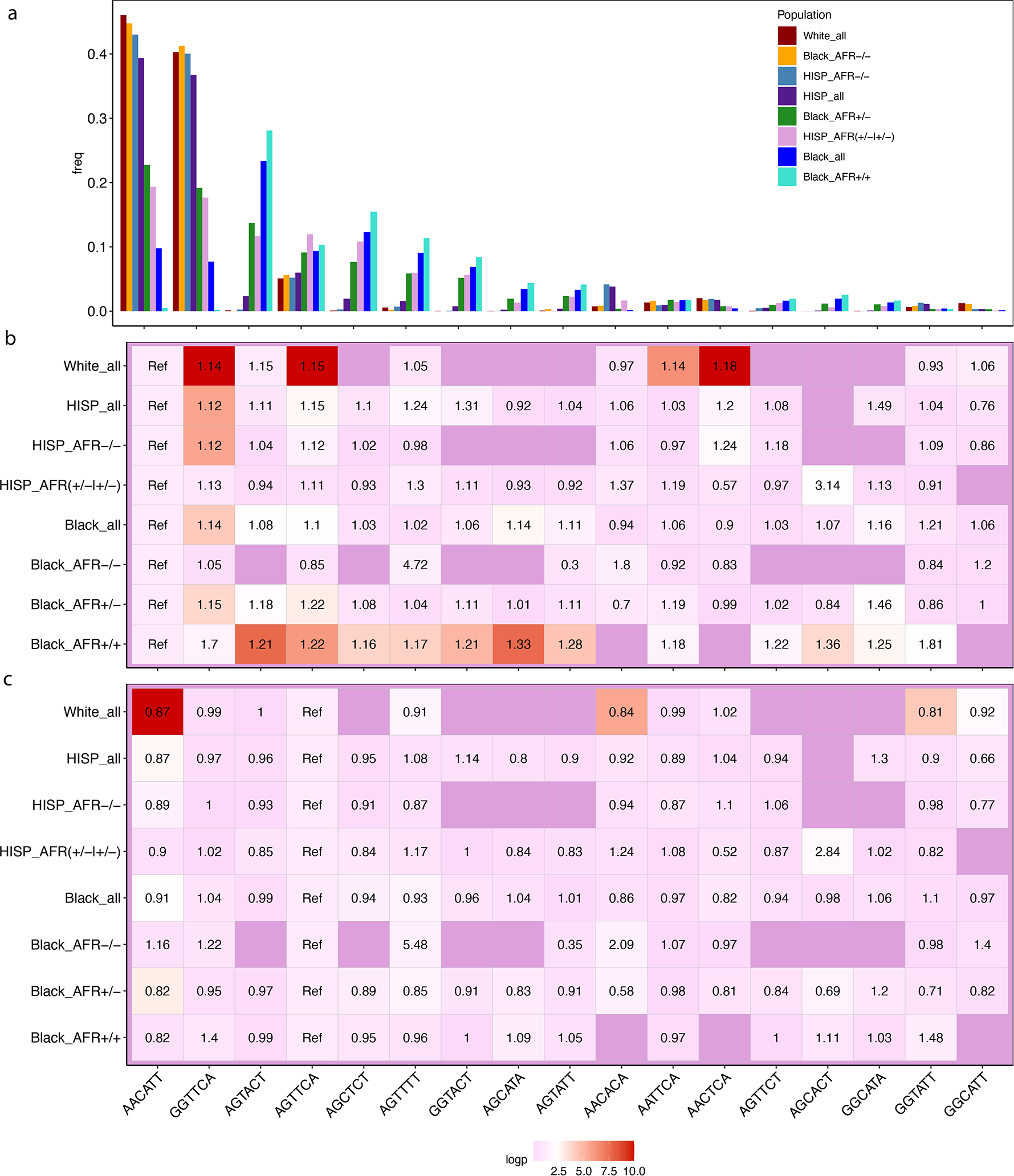
a-c, Black (n=17,247 cases / 60,578 controls) and Hispanic (n=6,388 / 24,479) MVP participants were stratified into groups based on the degree of African ancestry at the 9p21 locus for CAD as determined by RFMix. Whites (n=11,170 / 39,706) were analyzed as a single non-admixed group. The three subgroups among Blacks formed includes subjects with a high probability of having inherited two African (Black_AFR+/+, n=11,173 / 39,706) derived chromosomes in the 9p21 region, one African and one European (Black_AFR+/−, n=5,136 / 17,451), or two European chromosomes (Black_AFR−/−, n=654 / n=2,101). The two subgroups among Hispanics included those with high probability of having either 1 or 2 African chromosomes (Hisp_AFR+/−|+/+, n=985 / 3,943) vs. those without any African ancestry in this region (Hisp_AFR−/−, n=5,298 / 20,556). Among SNPs in the high-risk region of 9p21 that reached genome wide significance among Whites, six SNPs with a minor allele frequency >10% in Black_AFR+/+ were used to infer haplotypes in the region. Each column along the x-axis represents a haplotype, named by the alleles of the six defining SNPs. a, frequency of 17 observed haplotypes overall in each population and by subgroup of Blacks and Hispanics. b-c, odds ratio (OR) of CAD and −log10(p-value) obtained through a haplotype trend regression analysis where AACATT is the reference haplotype in b and AGTTCA is the reference haplotype in c.
As a sensitivity analysis, we repeated haplotype analysis using more stringent thresholds for assigning homozygous local ancestry (probability of 1 for AFR +/+ and probability of 0 for AFR −/−) and found results to be virtually unchanged (details not shown).
Analyses of the frequency of the same haplotypes in the 1000G populations suggest that these two haplotypes likely provide most of the risk-stratifying potential in all but West African populations where both haplotypes are virtually absent (Supplementary Table 16). Supporting these observations, we found that a single SNP (rs1333050) reaches GWS among Hispanics when GWAS analysis is restricted to the subgroup of Hispanics with no AFR admixture at 9p21 despite a very substantial reduction in sample size (Supplementary Table 17, Extended Data Fig. 3).
Pleiotropy assessment of novel loci
We explored the potential mechanisms of action of our novel loci by performing an extended phenome-wide association study (PheWAS) in MVP of all 95 lead novel SNPs (Methods). All but two (98%) of these SNPs were associated with one or more non-CAD phenotypes at an FDR<0.05. A total of 55 (58%) were associated with ≥ 1 traditional risk factor (TRF) for CAD, defined by blood lipid levels/hyperlipidemia (36 loci), blood pressure/hypertension (24 loci), diabetes mellitus (15 loci), body mass index (BMI)/obesity (12 loci), and/or smoking/tobacco use disorder (seven loci) (Fig. 4, Supplementary Table 18–20). Of these 55 loci, 33 (53% of TRF loci, 31% overall) were also associated with one or more TRFs even after excluding CAD cases. The five most pleiotropic loci (TCF7L2, FTO, PNPLA3, CDK12, and TDGF1P3) were linked to a range of 74 to 198 phenotypes while four additional loci (DSTYK, NPC1, IL1F10, and WWP2) were associated with >40 phenotypes. Of these 10 highly pleiotropic loci, five (FTO, IL1F10, PNPLA3, TCF7L2, TDGF1P3) were linked to a family history of the same dominant TRF even among MVP participants without CAD. Other phenotypes found to associate frequently with our novel loci included white blood cell related counts (23 loci), cancer (17 loci), renal function (15 loci), platelets (12 loci) and height (12 loci).
Fig. 4: Pleiotropic assessment of 95 novel loci through extended phenome wide association of lead SNPs.

Network plot of genotype-phenotype associations reaching significance at FDR<0.05 among 194,022 White participants in MVP without CAD for the lead SNPs in the 95 novel loci. Nodes are labelled either with the mapped gene for a lead SNP (purple font) or a phenotype tested in the PheWAS (black font). To highlight most pleiotropic SNPs and facilitate interpretation, the plot is restricted to lead SNPs associated with at least three distinct phenotypes. Distinct colors of nodes and edges represent a group of genotypes and phenotypes in the same dominant network. The thickness of the edges is correlated with the strength of the SNP-phenotype association (z-score). The size of the labels is dictated by the number of connections to phenotypes or genes and the strength of association. Network plot was created using Yifan Yu proportional and Atlas 2 layout algorithms as implemented in Gephi software.
Colocalization analysis between CAD and TRFs (Methods) for these loci further confirmed a strong link between our novel signals for CAD and analogous signals among TRFs which likely mediate the risk of CAD at many of these loci. We found strong evidence of the same causal variant for CAD and TRF for 6 loci with the strongest signals identified for ABCA1 with multiple lipid and blood pressure traits, TCF7L2 with diabetes, and NBC1, FBXL17, and FTO with BMI (Supplementary Table 21). Evidence for colocalization with different causal variants was present for an additional 20 loci.
Gene and pathway-based association analyses
Almost all genes implicated by four gene-based analyses (Methods) fell within or very near previously or our newly implicated loci that have reached GWS (Supplementary Tables 22–24). Comparing the DEPICT analyses before and after the addition of MVP GWAS of Whites, we found a large majority (95.6%) of the 19,460 genes tested were not implicated in either analysis. Among the 437 genes at FDR<0.05 in the previously published analysis9, 73% had a similar or lower FDR after the addition of MVP data while the remainder had a higher FDR or were no longer implicated. Adding MVP data also implicated 189 new genes at FDR<0.05. While the probability of a gene being implicated within a tissue relevant to CAD in our predicted gene expression analyses (MetaXcan) increased in tandem with the fraction of the remaining three algorithms that implicated the gene, the proportion was still very low with only 9.3% of the 321 genes implicated by DEPICT, MAGMA, and RSS-E also being implicated by MetaXcan. We annotated all implicated genes by providing hyperlinks of the genes to three gene-based portals (Supplementary Table 25).
Gene-set enrichment analyses using MAGMA, RSS-E and DEPICT highlight the involvement of many of the same pathways identified through similar analyses in previous large-scale GWAS of CAD (Supplementary Tables 26–28). A sizable fraction of the most significant curated gene-sets tested by MAGMA, RSS-E, as well as the protein-protein interaction subnetworks tested by DEPICT involve basic cellular processes/gene networks responsible for cell cycle, division/replication, and growth. For at least some of these gene-sets/networks, the ‘hub gene’ includes a gene mapped to either one of our novel loci (e.g., CDKN1A) or within previously established loci (e.g., TCF21).
We implemented MAGMA and DEPICT to prioritize cells and systems/tissues based on our GWAS meta-analysis of Whites (Methods, Fig. 5, Supplementary Tables 29–32). MAGMA identified 15 of 54 (27%) GTEx tissues, 95 of 729 (13%) Mouse Atlas cell types, 27 of 119 (22%) Tubula Muris FACS, and 19 out of 75 (25%) Tubula Muris Droplet cells as enriched in the expression of genes associated with CAD. A total of 35 out of 209 tissues/cell types reached an FDR<0.05 in DEPICT. MAGMA gene property analyses of a wide range of single-cell RNA datasets from mice as well as a more restricted set of cell types in humans highlight the relevance of the endothelial, stromal/fibroblast, and smooth muscle cells in the pathogenesis of CAD (Fig. 5a–b) with DEPICT reinforcing these findings and further delivering strong signals for hepatocytes and adipocytes (Fig 5d). The most significant system/tissue for both algorithms involved arteries, with MAGMA producing a top signal specifically for the ‘coronary artery’, a tissue almost exclusively made up of endothelial, stromal/fibroblast, and smooth muscle cells (Fig. 5c, f). In DEPICT, these findings were supported by significant associations in related vasculature (e.g., veins, portal system). Additional tissues prioritized across both algorithms included: i. components of the female reproductive system rich in smooth muscles (e.g., uterus, cervix, and the fallopian tube) with DEPICT implicating the myometrium specifically, ii. the esophagus and the sigmoid colon (MAGMA) as well as other components of the upper GI track including the liver and the pancreas (DEPICT), iii. the steroidogenic endocrine tissues of the ovary (MAGMA) and the adrenal cortex (DEPICT), iv. the lung, v. the bladder, and vi. multiple sources and types of adipose tissue (DEPICT). Findings unique to DEPICT include a signal involving the ‘aortic valve’ second only to ‘arteries’ in strength, the spleen, and a cluster of four signals involving joint related tissues.
Fig. 5: Downstream analyses to prioritize systems, pathways, tissues, and cells relevant to CAD.
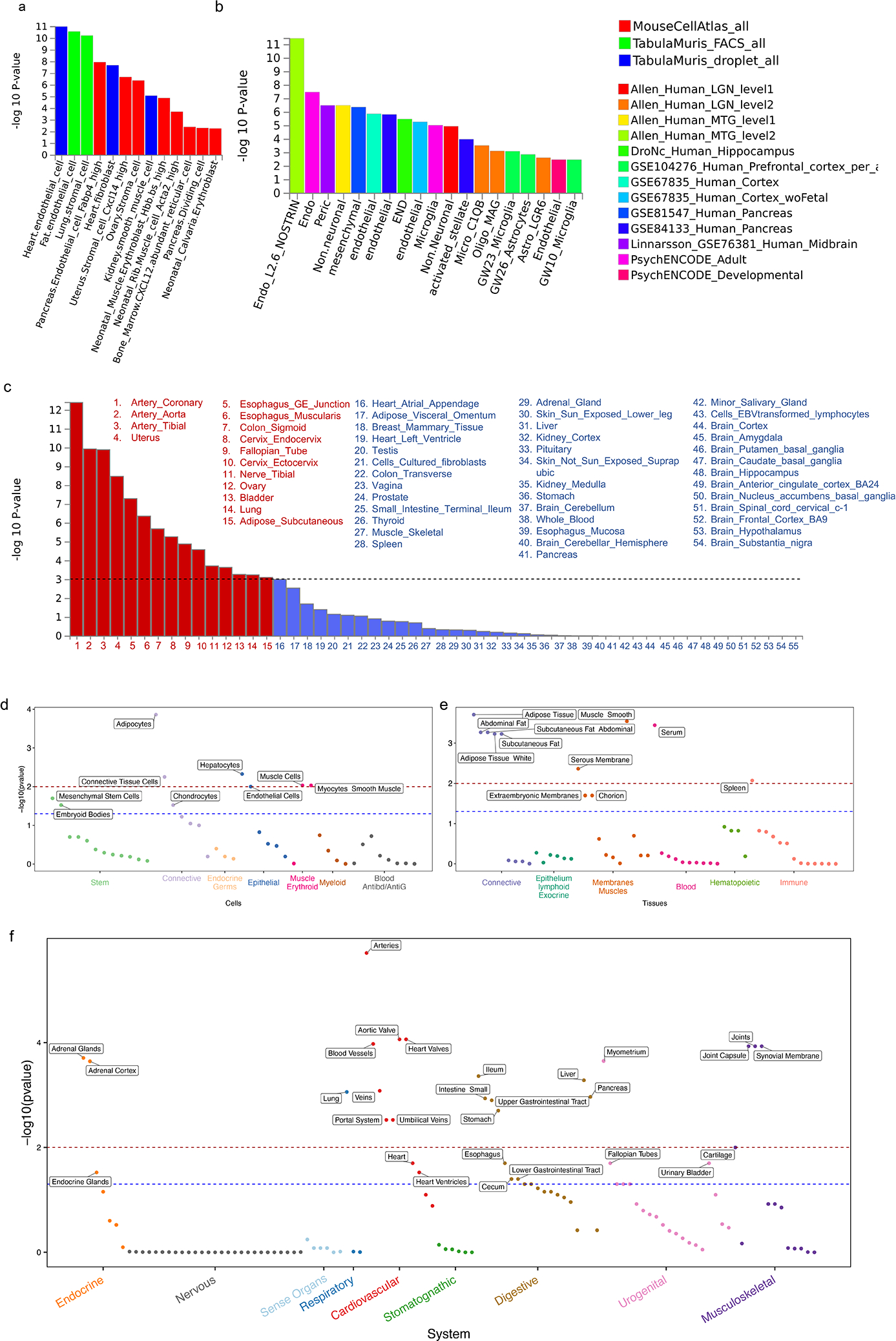
a-c, MAGMA gene-property analyses to test relationship between expressed genes in specific cells or tissues and genetic associations (meta-analysis of Whites) as implemented in FUMA. The gene-property analysis is based on the regression model, Z∼β0+EtβE+AβA+BβB+ϵ where Z is a gene-based Z-score converted from the gene-based P-value, B is a matrix of technical confounders, Et is the gene expression value of a testing tissue type c and A is the average expression across tissue types in a data set. A one-sided test (βE>0) is performed testing the positive relationship between tissue specificity and genetic association of genes. Data in a are restricted to three mouse single-cell RNA-seq (sc-RNA) datasets involving a broad range of cell types/organs while data in b are restricted to human datasets mostly involving the brain but also the pancreas and blood. Results show only independent cell-type associations based on within-dataset conditional analyses ordered by p-value across datasets. Data in c shows results for 54 specific tissue from the GTEx RNA-seq dataset v8 in order of p-value significance with red bars and font highlighting statistically significant tissues after adjusting for multiple testing (horizontal black dashed line) while remaining tissues are in blue. d-f, DEPICT following standard algorithm on the same GWAS used for MAGMA analyses in a-c. A tissue/cell type expression matrix was constructed by averaging gene expression levels of microarray samples with the same Medical Subject Heading tissue and cell type annotation. In this matrix, each column includes relative and normalized expression values of genes across 209 tissue/cell types. Enrichment in a tissue/cell type is then quantified by summing z-scores of the expression of genes with variants reaching genome wide significance in our meta-analysis of Whites. Z-scores are adjusted for confounding factors using 200 precomputed null GWAS in the Diabetes Genetics Initiative (DGI). Type 1 error rates were calculated by replacing null GWAS in DGI with simulated GWAS with positive signals but no underlying biological basis. DEPICT results are separated into d, cells e, tissues, and f, systems. −log10(p-value) for a false discovery rate (FDR) of <0.05 is demarcated by red dashed line while the FDR <0.2 threshold is shown in blue. Only cells/tissues reaching an FDR<0.2 are labelled.
Performance of externally validated polygenic scores in MVP
Four polygenic risk scores (PRS) of CAD previously derived and validated in datasets of primarily European-ancestry populations external to MVP (Methods) predicted clinical CAD status in all populations in MVP (Fig. 6a, Extended Data Table 7, Supplementary Table 33). The LDPred22 and metaGRS23 PRSs generated the highest odds ratios (ORs) per standard deviation (SD) increase of PRS with differences between the four scores least evident among Blacks. ORs were higher among the subset of cases with EHR evidence of myocardial infarction and/or a revascularization procedure and subjects with an age of onset of CAD below the median. The former subgroup also allowed for a direct comparison of the performance of the LDPred and the metaGRS PRS to that observed in the validation cohorts in the UK Biobank Whites. Based on the ratio of the log ORs, this comparison demonstrated a relative efficiency of the PRS of 75% to 80% when transferred to MVP Whites and as low as ~30–35% when transferred to Blacks consistent with prior studies23,24. ORs were notably lower among the subset of cases with first evidence of CAD after enrollment in MVP (i.e., incident cases) as compared to cases with first event prior to enrollment (i.e., prevalent), a finding that is also consistent with prior studies23,24. The four PRSs were also near linearly associated with burden of CAD among Whites with a similar ranking of performance to that observed for clinical events (Fig. 6b). Overall, we found the metaGRS slightly but consistently outperformed LDPred PRS based on the point estimate of the OR with the most notable difference between the two observed among Hispanics.
Fig. 6: Testing of externally derived polygenic risk scores and new multi-population scores in the Million Veteran Program.
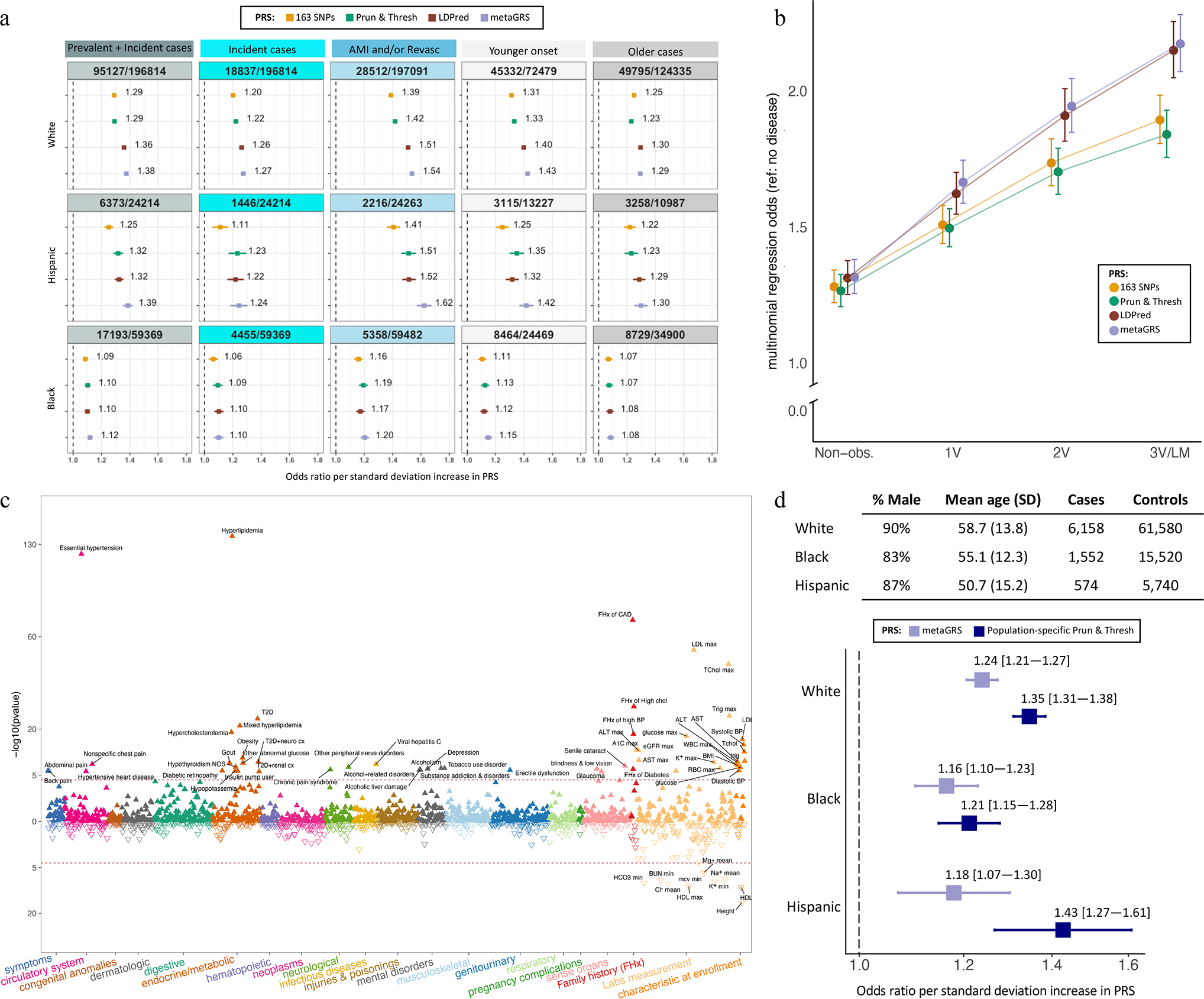
a, Performance of four externally derived and previously validated polygenic risk scores (PRS) in Whites, Blacks, and Hispanics, respectively, included in the MVP GWAS (see Fig. 1a for sample sizes of the three cohorts and methods for details on the origins of these PRS). Odds ratios and 95% confidence intervals per standard deviation (SD) increase in PRS are shown derived from logistic regression. In addition to all cases combined, subgroups of incident only cases (after enrollment), severe cases with evidence of either a myocardial infarction (AMI) and/or a revascularization (Revasc) procedure, and younger vs older onset cases (divided by median age of onset) were tested. b, externally derived PRS were tested for burden of coronary atherosclerosis among 25,600 Whites who underwent coronary angiography using multinomial logistic regression. Subjects with normal coronaries on angiography serve as the reference group and are compred to each of four progressively higher burdens of disease including non-obstructive disease (‘Non-obs.’), 1-vessel disease (1V), 2-vessel disease (2V), and 3-vessel or left main disease (3V/LM). Odds ratio and 95% confidence intervals are reported per SD increase in PRS. c, The best performing score in a and b, the metaGRS, was tested for association with Phecodes, clinical labs and anthropomorphic measures, as well as selected components of the baseline questionnaires among up to 164,534 Whites with no EHR evidence of atherosclerosis related complications at the end of EHR follow up. P-value are derived from a t-test implemented in the GLM and LM functions in R and are two-sided. d, New multi-population PRSs were developed using the pruning and thresholding strategy applied to the multi-population meta-analysis. These PRSs were tuned on an independent set of prevalent cases and controls in MVP, using population-specific tuning. Score performance of each score is shown in an independent set of incident cases and controls. Odds ratio and 95% confidence intervals are reported per SD increase in PRS and compared to performance of the metaGRS.
Performance of a new multi-population polygenic score in MVP
We derived new CAD PRSs using a pruning and thresholding (P+T) approach applied to our multi-population meta-analysis. We performed population-specific tuning to identify optimal window size, LD r2, and p-value thresholds for each PRS. The tuning cohorts consisted of prevalent cases and controls independent of the GWAS. For each population, the score with the highest OR (Supplementary Table 34) was then tested in a validation cohort. The validation cohorts consisted of incident cases and controls independent of both the GWAS and the tuning cohorts. We observed numerically higher OR for the population-specific P+T PRS compared to metaGRS across all populations, though the confidence intervals overlap within the Black and Hispanic groups (Fig. 6d).
Phenome-wide association study of PRS among controls in MVP
We explored factors through which a PRS mediates CAD susceptibility by conducting a PheWAS of the metaGRS among MVP participants. To minimize ascertainment bias of risk factors, the PheWAS was restricted to MVP White controls with further exclusion of subjects with evidence of peripheral arterial disease (PAD) or ischemic stroke (IS). After excluding only subjects with CAD, we found evidence that a higher PRS of CAD was associated with a higher risk of non-coronary related atherosclerosis complications (stroke, PAD, abdominal aneurysm, erectile dysfunction) and all TRFs including smoking (Supplementary Table 35). When further excluding subjects with PAD or IS, associations with all TRFs were sustained (Fig. 6c, Supplementary Table 36).
Extending the PheWAS to self-reported family history revealed not only an association with a family history of CAD but also with a family history of high cholesterol, hypertension, and diabetes. Extending the PheWAS to physical exam measures and laboratory measurements not only reinforced our Phecode findings through robust associations with analogous quantitative traits but also linked the PRS to renal function. Additional non-TRF associations included three lab indices derived from a complete blood count and several other commonly measured chemistries as well as hypothyroidism, viral hepatitis C, multiple common disorders of the eyes (cataract, glaucoma, blindness/low vision), and shorter height.
In addition to ‘tobacco use disorder’, we found evidence of a more widespread predisposition to substance abuse through associations with Phecodes ‘alcoholism’, ‘alcohol-related disorder’, and ‘substance addiction disorder’. These three codes were found to be modestly correlated with tobacco use disorder (Pearson r = 0.29, 0.29, and 0.32, all p<2.2×10−308). We were able to replicate all four of these associations in an independent set of 92,242 White subjects without evidence of clinical CAD (p-value <.001, Supplementary Table 37).
Discussion
We report the largest multi-population GWAS for CAD to date incorporating nearly a quarter of a million cases from four populations and increasing the total number of GWS loci for CAD by ~50% through the identification of 95 new loci reaching genome wide significance including the first nine on the X-chromosome. While several of these loci have already been strongly implicated through large scale consortium studies of causal risk factors (e.g., FTO, TCF7L2, TDGF1P3)25,26, exome sequencing association studies (PNPLA3)27, and subgroup analyses restricted to cases with documented myocardial infarction (IL1F10, UFL1-AS1)28, our analysis of multiple populations provides important insights on the genetic architecture of CAD.
First, we document a largely equivalent degree of heritability of CAD across multiple ancestries using a uniform and unbiased approach of estimation among unrelated individuals. Our absolute estimates of heritability are somewhat lower than the range previously reported in twin studies for fatal CAD7,8 but in line with an estimate of heritability derived in the UK Biobank using BOLT-LMM9. The remaining heritability may be captured through future large-scale whole genome sequencing association of cohorts capturing the full spectrum of CAD including both fatal and non-fatal presentations29.
Second, the CAD susceptibility loci of populations with a high proportion of either African and/or Indigenous American ancestry are likely to overlap substantially with those identified to date in other populations, as the first eight loci reaching GWS in Black and Hispanic populations have all been previously identified. Further supporting the presence of such overlap is the number of established loci implicated by XPEB and the degree of replication/correlation observed for these loci in our external Stage-2 Black and Hispanic cohorts. As these cohorts expand in size, many of the XPEB loci may reach GWS.
Third, GWAS in admixed populations may be leveraged to better understand the source of heterogeneity of effects across populations at some CAD loci. We show this for the widely replicated susceptibility locus at 9p2130 where common SNPs in the same haplotype block are GWS in South and East Asians12,13 but not in Blacks or Hispanics. Taking advantage of admixed populations, we provide compelling evidence for the presence of a protective haplotype at this locus which is common in all but African descent chromosomes where it is virtually absent. Further, the presence of an association signal among Blacks and Hispanics at 9p21 is dependent on the inheritance of non-African haplotypes in the region. Thus, the 9p21 locus is unlikely to ever serve as a key risk stratifying locus among populations with a high proportion of African ancestry at this locus, in stark contrast to its prominent risk-stratifying role in all other ancestral populations.
The degree to which genetic variation underlies sex differences in the incidence of CAD remains unclear. Initial GWAS of CAD did not detect sex differences in the magnitude of effects of autosomal susceptibility loci between men and women31 but more recent GWAS of adiposity-related traits such as waist-to-hip ratio as well as a study examining a PRS of CAD in the UK biobank have identified compelling sex differences32,33. While gonadal hormones undoubtedly serve as a major determinant of sex-differences in obesity and related traits, the X-chr may further contribute to sex differences in the rates of CAD through dosage effects on adiposity, lipid level and inflammation-related traits34. Determining the contribution, if any, of the novel and X-chr loci to sex-differences in the rates of CAD will require the study of additional very large populations of females with CAD.
Our GWAS of angiographically derived burden of coronary atherosclerosis did not identify novel CAD loci. Larger sample sizes may prove more fruitful, and our current results suggest that a large fraction of the initial loci uncovered for CAD increase risk of clinical disease by promoting coronary plaque rather than predisposing to plaque rupture or thrombosis35.
PheWAS for our lead novel SNPs continue to suggest that about one half of CAD loci influence risk through known risk factors9–11. We note a more prominent role of highly pleiotropic loci operating through the obesity, insulin resistance, and diabetes risk axis among our novel loci including the top GWAS signals for obesity (FTO)25, diabetes (TCF7L2)26, and non-alcoholic fatty liver disease (PNPLA3)36, as well as the previously known lipid loci TDGF1P3 and NPC1, which are also associated with metabolic indices37,38. Furthermore, we note the appearance of loci associated with smoking status. These findings for single novel SNPs were consistent with our PheWAS of the externally derived metaGRS23, which now provides evidence that a genome-wide PRS for CAD incorporates a strong readout for predisposition to every well-established TRF including a family history of not only CAD but also risk factors for CAD. In the PheWAS of the metaGRS, we also found evidence that the PRS predisposes to alcohol and substance addiction disorders. While these associations may be at least partially mediated by the co-morbid use of tobacco, the chronic use of other addictive substances may also independently contribute to the formation of coronary atherosclerosis, plaque rupture, vasospasm, and/or hypertension39.
Our gene-based association analyses expand on prior efforts to identify the most likely causal gene within a susceptibility locus. Despite substantially larger sample sizes and an improvement in analytic methods, it remains a challenge to unambiguously identify a causal gene within susceptibility loci. Our results highlight the need for integrative and orthogonal genomic methods to reliably identify the most likely causal gene and its putative mechanism within specific tissues40.
Our gene-set enrichment analyses continue to highlight well-established relevant biology in CAD but also point to an enrichment of pathways related to basic cellular processes/gene networks responsible for cell cycle, division/replication, and growth. This observation is buttressed by our PheWAS findings which link nearly one third of the novel loci to either a cancer or to height. Intriguingly, a shared biology between atherosclerosis and oncogenesis has long been hypothesized and others have recently documented the genetic basis of longstanding epidemiologic correlations between height, CAD, and cancer41,42. Expanding on this relationship, we note that Breast Cancer 1 gene (BRCA1) falls within one of our novel loci. The plausibility of BRCA1 as a CAD gene is supported by recent evidence of a genetic correlation between CAD and breast cancer43,44. Further, BRCA1 overexpression has been shown to protect against atherosclerosis and improve endothelial function45. We also identify ZEB1 as a candidate novel causal gene. ZEB1 is an oncogene and master regulatory of the epithelial-mesenchymal transition (EMT) that is well-established in breast cancer pathophysiology, and its expression may be dependent on BRCA146,47. BRCA1 suppresses EMT during tumorigenesis48. Other key EMT genes, including ZEB2, TWIST1, and SNAI1 are all previously identified CAD loci with established roles in cancer biology and recent experimental work suggests that EMT genes may impact CAD risk through the regulation of smooth muscle cell transitions in atherosclerotic plaques49–51. Overall, we suspect that these links reflect the prominence of these processes in tissues and cell types most relevant to CAD such as the de-differentiation, proliferation, and migration of endothelial cells, vascular smooth muscle cells, fibroblasts, and fibromyocytes within the vascular wall in response to the development of coronary atherosclerosis40,52,53.
Cell types prioritized for CAD include endothelial cells, fibroblasts, smooth muscle cells, hepatocytes, and adipocytes using two independent analytic algorithms. The first three comprise the vast majority of the cells in the normal vasculature54 consistent with top tissue signals observed for these tissues as well as the vessel rich lung. Strong signals involving the aortic valve, joints, joint capsule, synovial membrane, and cartilage may reflect shared gene networks expressed in these subtypes of connective tissue54. Signals involving the female reproductive tract, the GI tract, and the bladder may reflect the smooth muscle cell make up in these tissues54 with signals in the pancreas and the small intestine possibly further amplified by the key role these tissues play in the digestion and absorption of dietary lipids and cholesterol55. Lastly, strong signals in the liver, adrenal gland, and serum likely reflect the dominance of cholesterol-related gene networks within these tissues.
Our testing of externally derived PRSs of CAD in multi-population MVP participants confirms previously observed patterns with unprecedented precision and provides some additional insights. First, genome-wide PRSs of CAD substantially outperform genetic risk scores restricted to GWS loci. Second, higher ORs are observed for prevalent vs. incident, younger vs. older onset, and more severe (e.g., acute myocardial infarction and/or revascularization procedure) vs. less severe manifestations of CAD. These patterns likely reflect a higher average burden of CAD in one subgroup of cases when compared to the other with a proportional increase in the mean PRS for that subgroup. This hypothesis is supported by the strong linear relationship we observed between the same PRSs and the number of obstructed coronary arteries, a proxy for burden. Third, we observe a reduction in predictive performance of PRSs derived and validated externally among largely European participants when these scores are transferred to MVP most evident in Blacks and consistent with previous validation reports in smaller multi-population EHR cohorts24,56. While our newly constructed and validated multi-population PRS for CAD improved risk prediction across all populations, it did not decrease the performance gap between populations. Overall, our results underscore the pressing need to produce more data among non-white populations and develop more sophisticated analytic methods to eradicate such differences in performance and minimize the potential for exacerbating existing health disparities as PRSs are implemented into clinical practice5.
In conclusion, our large-scale multi-population GWAS provides important new insights into the genetic basis of CAD and brings us closer to precision medicine approaches for CAD across the diversity spectrum, but follow-up studies are needed to improve the transferability of PRS for CAD, to identify and understand mechanisms of causal genes, and to develop cross-population and population-specific novel therapies based on this understanding.
Online Methods
Design
Active users of the Veterans Health Administration (VA) of any age have been recruited from more than 75 VA Medical Centers nationwide since 2011 with current enrollment at >885,00057. Informed consent is obtained from all participants to provide blood for genomic analysis and access to their full EHR within the VA prior to and after enrollment including inpatient International Classification of Diseases (ICD9/10) diagnosis codes, Current Procedural Terminology (CPT) codes, clinical laboratory measurements, and reports of diagnostic imaging modalities. The EHR is continuously being integrated with MVP genomic data and access to these linked coded data is provided to approved investigators. All participants are also asked to optionally complete two short surveys, the Baseline and Lifestyle questionnaires, designed to augment data contained in the EHR. The study received ethical and study protocol approval from the VA Central Institutional Review Board.
Genetic Data and Quality Control
We genotyped 468,961 participants who enrolled in MVP between 2011 and 2017 with a customized Affymetrix Axiom array in two batches. The first batch including 359,964 participants and the second batch including 108,997 participants. The genotyping data generated underwent extensive quality control (QC)58. We initially imputed to the 1000 Genomes phase 3 version 5 reference panel (1000G)59 in each batch of genotyped data separately using EAGLE v2.360 and Minimac361 before joint imputation was performed in the two batches combined using EAGLE v2.4 and Minimac4. Prior to imputation, variants that were poorly called (genotype missingness > 5%) or that deviated from their expected allele frequency observed in the reference data (1000G) were excluded. Genotyped SNPs were interpolated into the imputation file.
Assignment of Populations
We assigned population membership to participants using HARE62, an algorithm that integrates genetically inferred ancestry with self-identified race/ethnicity. HARE assigned >98% of participants with genotype data to one of four non-overlapping groups: non-Hispanic Whites (Europeans), non-Hispanic Blacks (Africans), Hispanics, and non-Hispanic Asians. The sample size of Non-Hispanic Asians was too small for discovery and was excluded from further analyses62.
Additional Quality Control for X-chromosome
We implemented additional QC steps for analyses involving the X-chr to minimize risk of false positive associations due to sex-specific genotype calling errors. First, we excluded subjects with suspected XXY (n = 350) and XYY (n = 850) karyotypes based on an analysis of the median logR ratios of nonPAR X and Y chromosome SNP intensities. Second, we quarantined 6,707 out of 17,809 genotyped X-chr SNPs that met one or more of the following criteria: i. out of Hardy-Weinberg equilibrium among females (P<1×10−6); ii. demonstrated differential missingness between cases and controls and/or between males and females (P<1×10−6); iii. demonstrated differential minor allele frequencies between males and females (P<1×10−6); iv. high homology to another chromosome (mostly for the Y-chr within the pseudo-autosomal 3 region). Lastly, we phase and re-imputed the X-chr across all genotyped subjects combined using only the remaining 11,102 SNPs before proceeding with association analyses.
Phenotype
Clinical CAD
We used inpatient and outpatient ICD diagnostic and CPT procedure codes to identify subjects with clinical CAD in MVP. EHR data was available retrospectively before enrollment going back to October 1999 and prospectively after enrollment until mid-August 2018. An individual was classified as a case if he or she had: 1) any admission to a VA hospital with a discharge diagnosis of acute myocardial infarction (AMI) or 2) any procedure code for revascularization of the coronary arteries, or 3) two or more ICD codes for CAD (410 to 414) in at least two different encounters. Individuals with only one ICD code for CAD in a single encounter and no discharge diagnoses for AMI or revascularization procedures were excluded from the analyses. The remaining subjects were classified as controls.
We accessed individual level genetic and phenotypic data for the UK Biobank and implemented the same case-control definitions for clinical CAD used by others to conduct association analyses involving the X-chr.
Angiographic burden of CAD based on number of obstructed vessels
We linked MVP participants to the Veterans Affairs Clinical Assessment, Reporting, and Tracking (CART) Program, a national quality and safety organization for invasive cardiac procedures, to reliably estimate the burden of atherosclerosis among participants who had undergone at least one coronary angiogram by October 201863. Data were available retrospectively starting in 2004 in select sites and from all sites by 201064. A total of 31,658 non-Hispanic White, 7,313 non-Hispanic Black, and 2,536 Hispanic participants, a majority of which were subjects with clinical CAD, were found to have at least one evaluation of the degree of angiographically defined coronary atherosclerosis. For each angiogram, we classified an individual’s extent of disease to one of the following categories of disease of the native vessels: normal, non-obstructive, 1 vessel, 2 vessel, 3 vessel and/or left main coronary artery disease. Obstructive disease of a native vessel was defined as the presence of at least one lesion >50% or a prior revascularization procedure involving that vessel. Non-obstructive disease of a native vessel was defined as a vessel with at least one stenosis >20% of luminal diameter but no lesion >50%. We modified a previously validated algorithm to derive these classifications by decreasing the threshold of significant disease in a vessel from at least one lesion >70% to one lesion >50%65. Entries were filtered to remove those where disease severity was missing or listed as “other”, then subjects were removed if they were missing a HARE assignment, date of birth, sex, or had previously received a cardiac transplant. For subjects with multiple angiograms over follow up where at least one reported disease, we assigned severity based on the procedure reporting the most advanced disease. If more than one angiogram reported the same advanced disease, we used the earliest one. Age was calculated on the date of the cardiac catheterization with the most severe disease for cases and the last normal angiogram for controls.
Statistical Analysis
Genetic Relatedness
We used KING, version 2.0, to identify 20,881 related participants at a 3rd degree or closer58. Among these individuals, we preferentially retained 5,289 unrelated cases and 4,909 unrelated non-cases in analyses and excluded the remaining individuals (1,023 cases and 9,601 non-cases).
Analyses of heritability across populations
We used GREML-LDMS-I as implemented in Genome-wide Complex Trait Analysis (GCTA) 1.93.0beta to estimate the multicomponent narrow sense heritability of CAD in our three HARE-defined MVP groups and in the Biobank Japan dataset66. GREML-LDMS-I is one of the most accurate heritability estimation methods when considering common factors that may bias such estimates67. To minimize the confounding effects of admixture, we identified minimally admixed subsets of individuals in each of the HARE groups by performing a combined PCA of MVP data and 1000G data, then selecting White, Black, and Hispanic MVP subjects who clustered most closely with the 1000G European, African and Peruvian populations, respectively. Restricted by computing memory requirements, we next randomly selected 19,395 of least-admixed Hispanic participants (our smallest group) to run through GREML-LDMS-I68,69. To minimize the influence of differences in the severity of the cases and the age of controls between populations on the final estimates of heritability, we then matched an approximately equal number of MVP Blacks (n=19,392), MVP Whites (n=19,392), and Japanese from Biobank Japan (n=18,747) to the Hispanic group using case-control status, EHR-based estimated age of onset of CAD, the type of case (MI/revascularization versus other), and the age of controls as factors. These sample sizes provided us with >80% power to detect a heritability of at least 7% on the liability scale and 100% of at least 11% assuming a prevalence of disease of 8%70. We then estimated heritability within each group after applying identical QC procedures. First, SNP dosages used for all GWAS were converted to hard-call genotypes using the default settings in PLINK 2.0 described under section “Standard data input/dosage import settings”. SNPs that were multi-allelic, had MAC < 3, or hard call-rate < 95% were removed. Since CAD case status is a binary trait, SNPs with p < 0.05 for Hardy-Weinberg equilibrium or differential missingness in cases vs controls were also removed68,69. LD scores were computed on each autosome using GCTA default settings with an r2 cutoff of 0.01, and the genome-wide LD score distribution was used to assign SNPs to 1 of 4 LD quartile groups, where groups 1–4 represented SNPs with progressively higher LD scores. Within each LD group, SNPs were further stratified into 6 MAF bins ([0.001, 0.01], [0.01, 0.1], [0.1, 0.2], [0.2, 0.3], [0.3, 0.4], [0.4, 0.5]) and a genetic relatedness matrix (GRM) was constructed from each bin, ultimately creating 24 GRMs. Finally, GCTA --reml was used to fit a model of CAD case status based on the 24 GRMs, with age and sex as covariates. Total observed heritability estimates were transformed to estimate disease liability4 across a range of presumed CAD prevalence estimates in the general population.
Genome-wide association study in MVP
We performed a GWAS of autosomes for clinical CAD and for coronary angiographic burden of disease within each of the three HARE groups using logistic and linear regression, respectively, implemented in PLINK 2.0 alpha. Models assumed an additive genetic effect adjusted for sex and the respective first 10 ancestry-specific principal components (PCs). For burden of disease, we further adjusted models for age at the time of angiography. Association tests were performed within each HARE group and across 2 tranches of MVP genotyped data. Thus, six GWAS were performed for each phenotype. Each set of results was filtered separately using PLINK and EasyQC. First, we removed SNPs with i. population-specific imputation r2 < 0.4, ii. OR, p-value and/or SE missing value as well as SNPs with absolute(beta) >4; iii. multi-allelic SNPs, and iv. SNPs with minor allele count (MAC) <6. Second, we filtered all SNPs with a minor allele frequency < 0.01 in non-Whites and less than 0.001 in Whites. Third, we filtered any SNP that was not in HWE among controls as defined by deviation from HWE with a p<1×10−6. METAL71 was then used to apply a genomic control to each input dataset and meta-analyze GWAS results across genotype releases within each HARE group. For Whites, we also ran METAL with genomic control turned off to create a dataset suitable for LD score regression72.
X-chr association testing in MVP for both phenotypes was conducted stratified by sex in addition to HARE group. In the UK Biobank, X-chr analyses were restricted to unrelated subjects of White/European descent (34,541 CAD cases and 261,984 controls). We implemented a standard logistic regression model using plink with no X-chr inactivation assumption (males coded as 0/1, females as 0/1/2). We then used GWAMA for meta-analysis of male and female within each ancestry group and tested for difference in effect between sex as well as sex-interaction.
Meta-analysis with external datasets
We used METAL to conduct two fixed-effect inverse variance weighted meta-analyses for the clinical CAD phenotype. The first involved the MVP Whites with the CARDIoGRAMplusC4D 1000G study and the UK Biobank CAD study and the second further incorporated the MVP Blacks, MVP Hispanics, and Biobank Japan. Genomic control was applied to each input dataset by METAL. This second multi-population meta-analysis was also performed using MR-MEGA73. METAL and MR-MEGA were also used to conduct a multi-population meta-analysis of the CART derived burden of CAD with the MVP datasets. For the X-chr, we used METAL to conduct a meta-analysis of the X-chr data in MVP Whites with the UK Biobank and the X-chr study by CARDIoGRAMplusC4D74. Lastly, we used MR-MEGA to conduct a multi-population meta-analysis of the X-Chr through further inclusion of the MVP Blacks, MVP Hispanics, and Biobank Japan.
Credible set analyses
We generated a list of credible sets of SNPs at all loci, known and novel, reaching GWS in our meta-analysis of Whites using a Bayesian approach for credible set analysis assuming a single causal variant per locus75. Briefly, we first calculated approximate Bayes factors for each variant within a 1MB region centered on the lead SNP using the beta, standard error, and sample size from the METAL meta-analysis of Whites. We then estimated the posterior probability of each SNP being causal using the Bayesian factor. Lastly, a credible set was defined as the smallest set of SNPs for which the sum of posterior probability reached 99%. We also generated credible sets for the exact same genomic regions using Bayes factors derived from MR-MEGA in our multi-population meta-analysis.
Definition of a locus including parameters for lead and candidate genetic variants
We used FUMA76 to define genomic risk loci including independent, lead, and candidate variants. First, independent genetic variants were identified as variants with a P below a specific threshold and not in substantial linkage disequilibrium (LD) with each other (r2 < 0.6). Second, variants in LD (r2 ≥ 0.6) with an independent variant and with p < 0.05 were retained as candidate variants to form an LD block. Third, LD blocks within 500kb of each other were merged into one locus. Lastly, a second clumping of the independent variants was performed to identify the subset of lead SNPs with LD r2 < 0.1 within each locus. For our meta-analyses of Whites alone and our multi-population meta-analyses, we used a UK Biobank release 2b EUR reference panel of genotype data imputed to the UK10K/1000G SNPs created by FUMA including ~17 million SNPs. This panel includes a random subset of 10,000 unrelated subjects among all subjects with genotype data mapped to the 1000G populations based on the minimum Mahalanobis distance. We used the 1000G AFR reference panel of 661 subjects with ~43.7 million SNPs for our Blacks, and the AMR reference panel of 347 subjects with ~29.5 million SNPs for our Hispanics.
Two-stage joint analysis of most promising findings in non-Europeans
We sought replication of all promising genomic risk loci in our MVP Black and MVP Hispanic GWAS for clinical CAD in multiple external datasets. Replication was attempted not only for all lead SNP(s) with P<1×10−5 but also for all other independent and candidate genetic variant members of these loci. In the same external datasets, we also sought replication for all SNP with local FDR < 0.05 from our XPEB analyses as described below.
Definition of a significant and novel locus and annotation
A locus was considered GWS if at least one lead genetic variant within it reached a P<5×10−8 in any of the terminal meta-analyses. For meta-analyses involving METAL, the variant also had to lack any significant heterogeneity (P>5×10−8 for test of heterogeneity). A GWS locus was considered novel if none of its lead, independent, or candidate SNPs (as defined above) overlapped with a SNP that has previously reached GWS in the setting of a GWAS meta-analysis or two-stage analysis for clinical CAD. Novel GWS loci were identified at three stages: i. after the meta-analysis of all GWAS available among Whites, ii. after combining genome-wide summary statistics in Blacks and Hispanics, respectively, with external replication data limited to promising loci, and iii. after multi-population meta-analyses of all summery statistics of GWAS (i.e., not including 2nd-stage data in Blacks and Hispanics). For the multi-population meta-analysis, we first identified novel loci with lead SNPs with no significant heterogeneity using METAL and supplemented these with any additional non-overlapping genome-wide findings identified with MR-MEGA. We annotated the lead SNP(s) at each novel locus by creating URL hyperlinks to five variant-base portals: OpenTargets, QTLbase, Common Metabolic Disease Knowledge, Open GWAS, and PhenoScanner.
Cross-population empirical Bayes method
We implemented the cross-population empirical Bayes method, XPEB77, for the clinical CAD phenotype. XPEB takes as input p-value summary statistics from two GWAS, a target-GWAS that is typically a smaller non-European population of primary interest and a base-GWAS that is typically a much larger GWAS of Europeans and adaptively reprioritizes variants in the target population to compute local false discovery rates. We ran XPEB with the MVP Blacks as the target GWAS and the meta-analysis of MVP Whites, CARDIoGRAMplusC4D, and the UK Biobank as the base-GWAS. We then ran it a second time with the MVP Hispanics as the target GWAS. For both runs, analyses were restricted to genotyped SNPs in the target populations.
Calculation of and testing externally derived Polygenic Risk Scores (PRS) of CAD in MVP
We calculated four externally derived and previously validated PRS for CAD of increasing complexity in each participant included in the MVP GWAS of Whites, Blacks, and Hispanics. The four scores included: i. a weighted PRS restricted to a curated list of up to 163 independent SNPs having reached GWS among predominantly populations of European ancestry as of 2019, ii. the best performing weighted PRS in the UK Biobank calculated from a standard pruning & thresholding method of the CARDIoGRAMPplusC4D 1000G summary statistics involving 1.5 million SNPs, iii. the metaGRS, a 1.7 million-SNP PRS consisting of a weighted average of three standardized risk scores followed by LD pruning; and iv. the best performing PRS in the UK Biobank derived from applying the LDPred algorithm onto the CARDIoGRAMPplusC4D 1000G summary statistics involving 6.6 million SNPs but assuming 0.1% of SNPs are causal. All scores were standardized to a mean of zero and standard deviation (SD) of one within each HARE group.
We then estimated the increase in risk of clinical CAD associated with a 1 SD increase in PRS for each of the four PRSs within each of the three HARE groups using logistic regression adjusting for imputation release batch, age, sex and the first 10 HARE specific PCs where age was defined as the age at the time of first ICD code for cases and age at the time of last visit to the VA for controls. Similarly, we estimated the increase in the burden of disease per one SD increase in PRS using linear regression where age was defined as age at time of coronary angiography.
Derivation and validation of a new multi-population polygenic risk score in MVP
We constructed new PRSs using a pruning and thresholding approach implemented in PRSice2 and applied to our multi-population meta-analysis78. We used a recently genotyped independent MVP cohort (release 4) of Whites, Blacks and Hispanics to tune and validate the PRS we constructed with the remaining MVP participants included in our multi-population meta-analysis (release 3). From the independent MVP cohort, we set aside all subjects who had their first ever CAD event after enrollment along with 10 random controls. The prevalent cases and remaining controls were used for tuning the PRSs. Thus, the GWAS cohort used for the derivation of the new multi-population PRS, the tuning cohort, and the validation cohort were independent.
We used a cosmopolitan cohort of randomly selected MVP participants as the LD reference panel. Multiple LD pruning (R2<0.2, R2<0.4, and R2<0.8); distances between pruning region (250kb and 500kb), as well as p-value thresholds were used to create PRS that were then tested in the tuning cohort to identify the best performing PRS as estimated by the odds ratio per SD increase in the score. We then tested this best PRS in the validation set and compared to the performance of the best performing externally derived PRS.
Phenome-wide association study of novel loci and best performing externally derived PRS of CAD in MVP
We conducted a PheWAS for each of the lead SNPs at all novel loci, for the 163 SNP PRS, and for the externally derived genome-wide PRS with the highest OR for CAD in MVP. We adopted the standard PheWAS protocol79,80 and augmented this basic approach by including phenotypes derived from the physical exam (e.g., measured weight, height, blood pressure, and heart rate), laboratory results (e.g., blood cell counts and biochemistries), and select variables derived from the MVP questionnaires (family history, smoking status, and alcohol use). For individual novel SNPs, we ran the PheWAS in each HARE group separately in both cases and controls combined and controls alone, with associations considered significant if their FDR was < 0.05 by the Benjamini-Hochberg method. For the PheWAS PRS, we restricted association analyses to Whites and ran analyses in i. all subjects; ii. after excluding CAD cases; and iii. after further excluding subjects with other manifestations of atherosclerosis including peripheral arterial disease and ischemic stroke. For select Phecodes, we attempted to replicate significant associations in a newly genotyped independent set of 92,242 White MVP participants (release 4).
We generated a network plot with the Yifan Yu proportional multi-level layout and Atlas 2 layout algorithms implemented in Gephi Software using the subset of significant individual novel SNP PheWAS associations. The node size was defined using the weighted in-degree network statistic with the directionality from SNP to phenotype. The edge size was defined by the number of connections between two nodes (SNPs and phenotypes) and only include associations between SNP and phenotype represent by the z-score statistic of the SNP-phenotype association. The size of the label of the node was proportional to the weighted degree statistic. The color of the edges was define using the modularity matrix, a network statistic for unfolding communities in large network.
Colocalization analysis
We assessed for the presence of colocalization of genetic association signals between novel loci for CAD and associations in analogous regions for traditional risk factors (TRFs) using COLOC81. For these analyses, we input results from our meta-analysis for CAD in Whites as well as recent large scale genetic studies of traditional risk factors independent of our MVP dataset including GWAS of BMI, lipids, blood pressure, smoking, and type 2 diabetes82–86. Evidence of colocalization at a locus with the same causal variant shared between CAD and the TRF was defined as a posterior probability Bayesian factor H4 (PP.H4.abf) > 0.7 while evidence of colocalization with a different variant was defined as defined as a posterior probability Bayesian factor H3 (PP.H3.abf) > 0.7.
Local ancestry inference and haplotype analysis at susceptibility loci of interest
We used RFMix87 to derive the most likely ancestral origin of the chromosomal segment encompassing loci of interest in MVP Blacks and Hispanics. The YRI, MEL and IBR populations from the 1000G project as the African reference, and the GBR, CEU and TSI populations as the European reference to infer the most likely sequence of ancestry within the locus. The results allowed us to subdivide the MVP Blacks into three groups: i. subjects with a high probability of African ancestry on both chromosomes (homozygote Africans), ii. subjects with high probability of one African and one European ancestry chromosome (heterozygotes), and iii. subjects with a high probability of European ancestry on both chromosomes. For haplotype analyses within loci of interest, we identified all common (MAF>10%) SNPs in linkage equilibrium (r2<0.05) in our homozygote Africans Blacks among all SNPs reaching GWS (P<5×10−8) in our meta-analysis of Whites and used these SNPs to construct haplotypes and perform a haplotype trend regression of this region using the EM algorithm implemented in the R package haplo.stats.
Downstream analyses to prioritize genes, pathways, cells, and tissues/systems relevant to CAD
We conducted downstream analyses to prioritize genes, pathways, and tissues involved in the pathogenesis of CAD based on the results of our meta-analyses. We applied four analytic algorithms to the summary statistics including Multi-marker Analysis of GenoMic Annotation (MAGMA) v1.09 for gene, gene-set, and gene-property analysis, as implemented in FUMA76,88,89, a model-based enrichment method for GWAS summary data using biological pathways to define gene-sets, Regression with Summary Statistics exploiting Enrichments (RSS-E)90, Data-driven Expression Prioritized Integration for Complex Traits (DEPICT)91, and MetaXcan92. Gene and cell/tissue/system specificity/prioritization analyses incorporating gene-expression data into their algorithms were restricted to Whites given a majority of the gene-expression data incorporated into these analyses are derived from Whites. We combined results from MAGMA, RSS-E, DEPICT, and MetaXcan, at the gene level and compared to the gene level DEPICT analyses performed on the CARDIoGRAMplusC4D and UK Biobank meta-analysis alone. We annotated implicated genes by creating URL hyperlinks to information on these genes in three gene-based portals: Mouse Genome Informatics, Online Mendelian Inheritance of Man, Therapeutic Target Database. MAGMA gene-set analyses were run on 10,678 gene sets (curated gene sets: 4,761, GO terms: 5,917) from MSigDB v6.2 while gene-property analyses were conducted on GTEx V8 and multiple single cell RNA-seq databases incorporated into the FUMA bioinformatic pipeline including the Mouse Cell Atlas, the Tabula Muris dataset (FACS and droplet) and several datasets of human brain, pancreas, and blood. For RSS-E, gene-sets were derived from nine databases (BioCarta, BioCyc, HumanCyc, KEGG, miRTarBase, PANTHER, PID, Reactome, WikiPathways) that are archived by four repositories: Pathway Commons v7, NCBI Biosystems, PANTHER (v3.3), and BioCarta. We downloaded preprocessed pathway and gene data from http://doi.org/10.5281/zenodo.1473807 on October 29, 2018 and used a list of 3,803 pathways that contains between 2 to 400 autosomal protein-coding genes per pathway in the present study.
URLs
CARDIoGRAMplusC4D http://www.cardiogramplusc4d.org;
Japanese ENcyclopedia of GEnetic associations by Riken: http://jenger.riken.jp/en/result
R statistical software, www.R-project.org;
PLINK 2.0: https://www.cog-genomics.org/plink/; https://www.cog-genomics.org/plink/2.0/input#dosage_import_settings;
LDSC: https://github.com/bulik/ldsc;
Gephi: https://gephi.org/;
FUMA, http://fuma.ctglab.nl/;
PheWAS, https://github.com/PheWAS/PheWAS;
RFMixv2: https://github.com/slowkoni/rfmix;
GCTA, http://cnsgenomics.com/software/gcta/#Overview;
METAL: https://genome.sph.umich.edu/wiki/METAL;
GWAMA: https://genomics.ut.ee/en/tools/gwama;
MAGMA: https://ctg.cncr.nl/software/magma;
DEPICT: https://data.broadinstitute.org/mpg/depict/;
RSS-E: https://github.com/stephenslab/rss;
MetaXcan: https://github.com/hakyimlab/MetaXcan;
OpenTargets: https://genetics.opentargets.org/;
QTLbase: http://www.mulinlab.org/qtlbase;
Common Metabolic Disease Knowledge Portal: https://hugeamp.org/;
Open GWAS: https://gwas.mrcieu.ac.uk/;
PhenoScanner: http://www.phenoscanner.medschl.cam.ac.uk/;
Mouse Genome Informatics: http://www.informatics.jax.org/;
Online Mendelian Inheritance of Man: https://www.omim.org/;
Therapeutic Target Database: http://db.idrblab.net/ttd/
Data availability
Summary statistics for the Biobank Japan study were obtained from http://jenger.riken.jp/en/result. Summary statistics for the CARDIoGRAMplusC4D study were obtained from http://www.cardiogramplusc4d.org. Summary statistics for the UK Biobank study for CAD were obtained from https://www.cardiomics.net/download-data.
The full summary level association data from the individual population association analyses in MVP as well as the multi-population meta-analysis from this report will be available through dbGaP, with accession number phs001672 at the time of publication in a peer reviewed journal. This research has been conducted using the UK Biobank Resource under Application Numbers 13721 & 19416.
Consortia
Regeneron Genetics Center, Biobank Japan, CARDIoGRAMplusC4D, The VA Million Veteran Program
Extended Data
Extended Data Fig. 1. LocusZoom plots of loci reaching genome wide significance in Blacks and Hispanics.
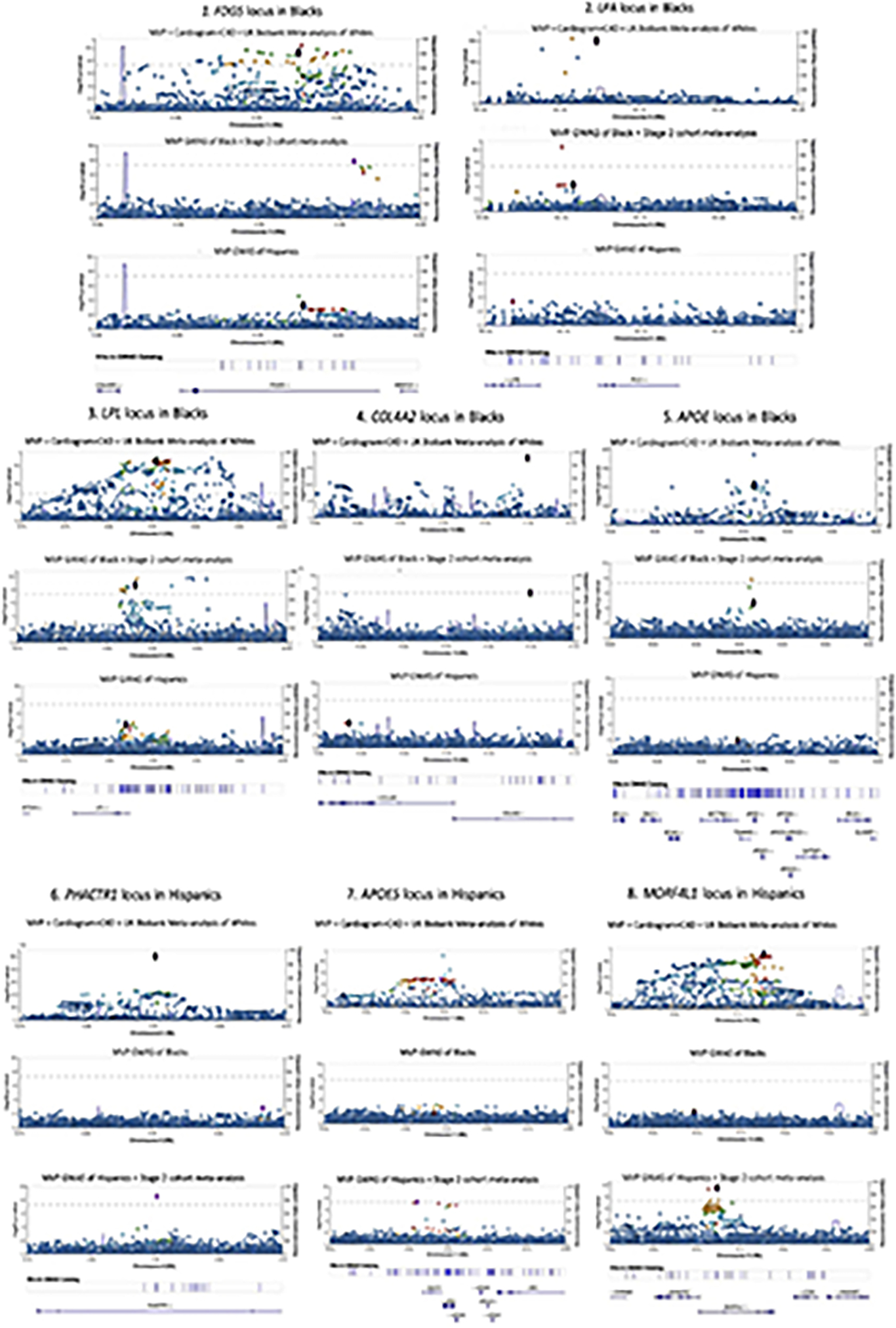
Sets of LocusZoom plots for five loci in Blacks and 3 loci in Hispanics reaching genome wide significance after two-stage meta-analysis with external cohorts. Each set of plots show the association results for a locus for all three populations using the same chromosome location scale (x-axis) but not the same p-value scale (y-axis). P values are derived from inverse variance weighted meta-analysis using METAL and are two-sided.
Extended Data Fig. 2. Allele frequencies and association results at the 9p21 locus among Black in the Million Veteran Program stratified by local ancestry status.
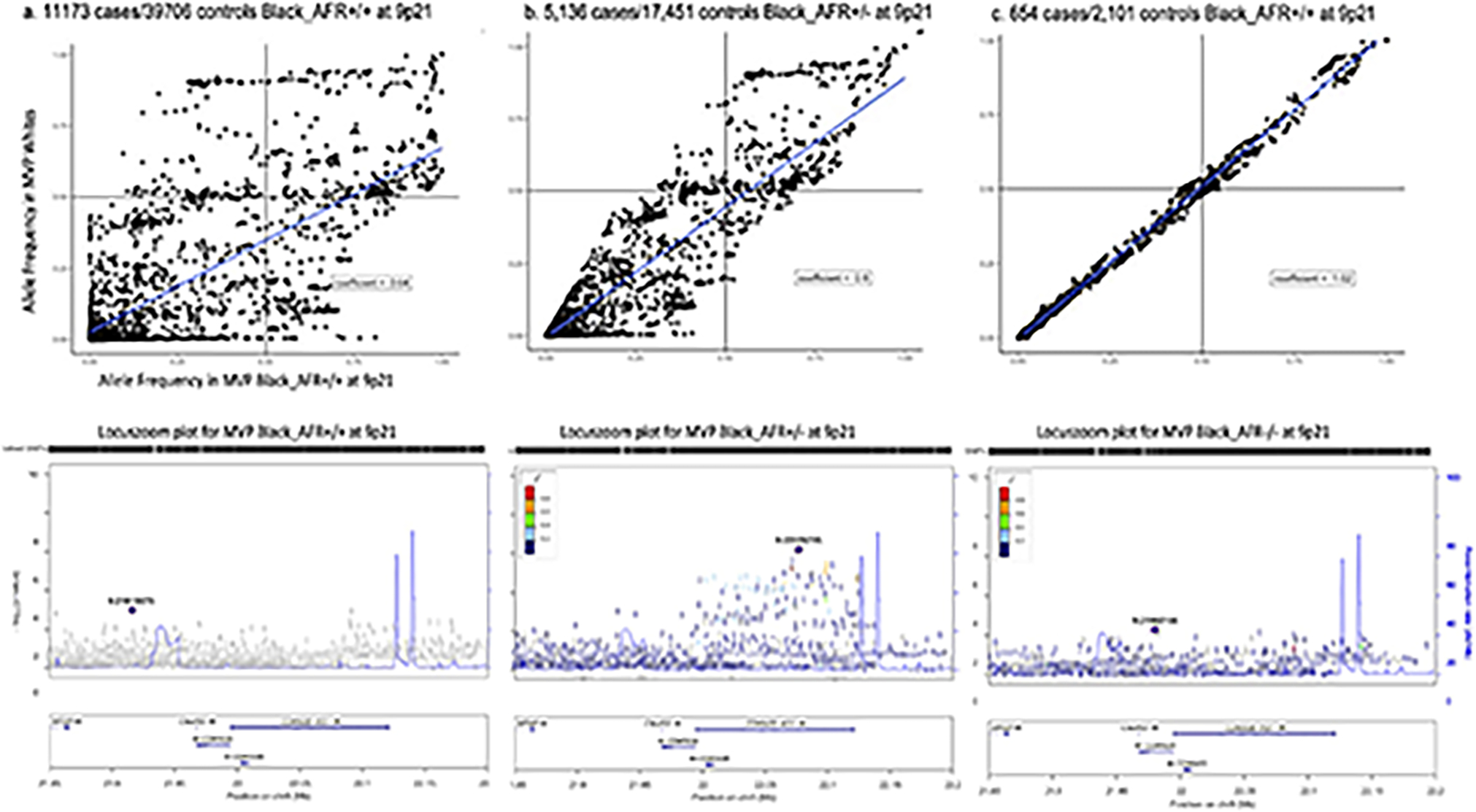
Top panels show plots of corresponding allelic frequencies at the 9p21 susceptibility locus observed in MVP Whites vs. subgroups of MVP Blacks including those with a. two African chromosomes (chr), b. one African chr, and c. no African chr at the locus. Corresponding LocusZoom plots for each group are in the panels immediately below. Association testing was performed using logistic regression with adjustment on sex and principal component as implemented in PLINK. P values were derived from a Wald test and are two-sided.
Extended Data Fig. 3. LocusZoom plots of SNP association at the 9p21 susceptibility locus for CAD.
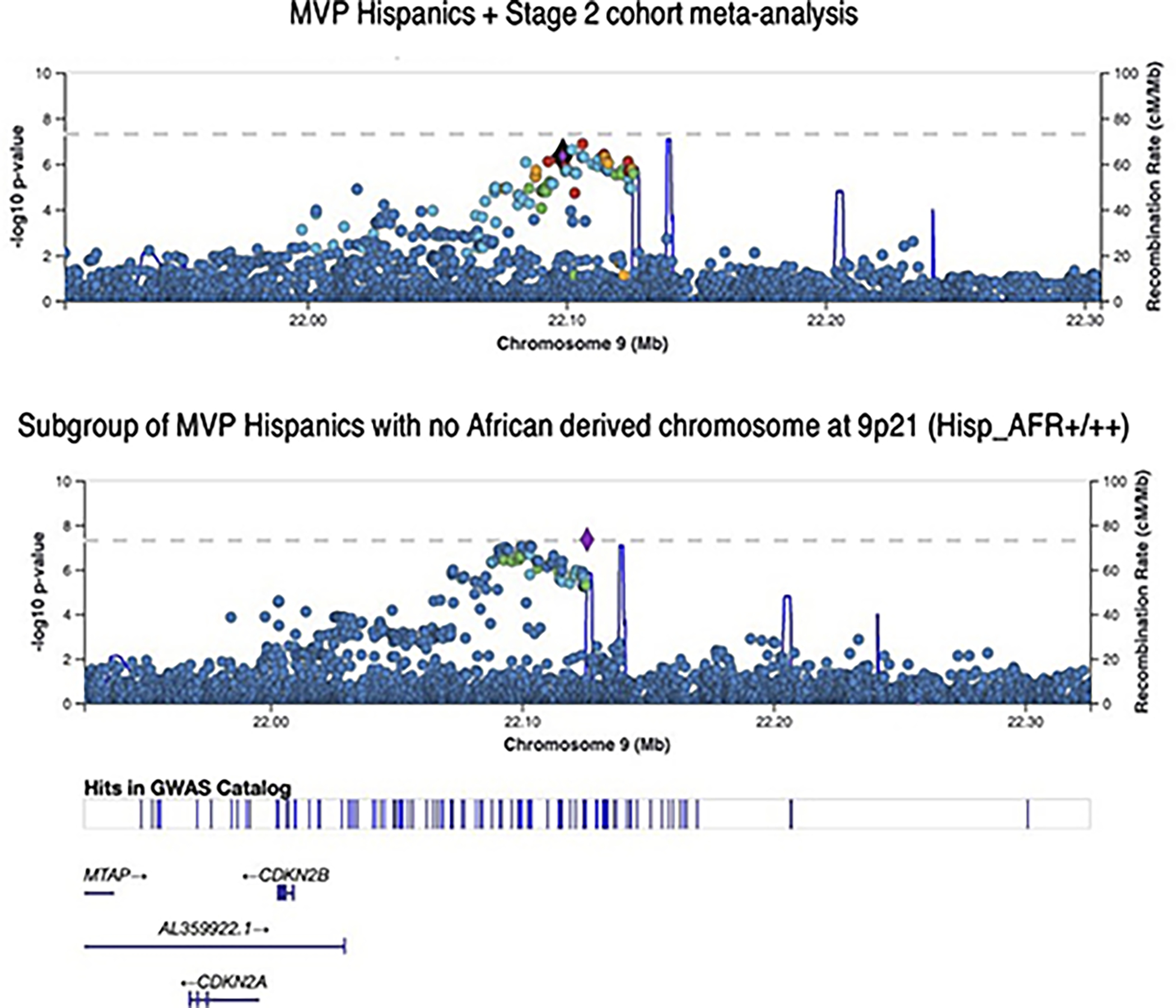
Top panel plots the results for MVP GWAS of all Hispanics + Stage 2 cohort meta-analysis. P values are derived from inverse variance weighted meta-analysis using METAL and are two-sided. Bottom panel plots the subset of MVP Hispanics with no African derived chromosomes at 9p21 based on local ancestry assessment using RFMix (5,298 cases / 20,556 controls). Association testing was performed using logistic regression with adjustment on sex and principal component as implemented in PLINK. P values were derived from a Wald test and are two-sided.
Extended Data Table 1.
Demographic characteristics of the Million Veteran Program participants included in the genome wide association study analyses of the clinical coronary artery disease phenotype.
| Cases (118,731) | Non-cases (281,064) | Excluded (28,148) | |
|---|---|---|---|
|
| |||
| Calendar Year of Enrollment: count (%) | |||
| 2011 | 5,162 (4.3) | 8,102 (2.9) | 1,079 (3.8) |
| 2012 | 27,011 (22.7) | 50,591 (18.0) | 6,262 (22.3) |
| 2013 | 27,279 (23.0) | 59,534 (21.2) | 6,278 (22.3) |
| 2014 | 23,218 (19.6) | 57,873 (20.6) | 5,665 (20.1) |
| 2015 | 18,160 (15.3) | 51,121 (18.2) | 4,593 (16.3) |
| 2016 | 9,267 (7.8) | 27,080 (9.6) | 2,168 (7.7) |
| 2017 | 8,634 (7.3) | 26,763 (9.5) | 2,102 (7.5) |
| 2018 | -- | -- | 1 (0.0) |
| Mean Age at enrollment: mean (SD) | 69.40 (9.8) | 59.70 (14.2) | 65.50 (11.1) |
| Male Sex count (%) | 115,606 (97.4) | 250,509 (89.2) | 26190 (93.0) |
| Case subgroups | |||
| AMI on discharge summary | 19,341 (16.3) | ||
| Revascularization procedure (CABG or PCI) | 29,439 (24.8) | ||
| AMI and/or revascularization procedure | 36,086 (30.4) | ||
| chronic CAD (no AMI or revascularization) | 83,070 (70.0) | ||
| HARE ancestry assignment counts (%) | |||
| white/European | 95,151 (80.2) | 197,287 (70.2) | 19,909 (70.7) |
| black/African American | 17,202 (14.5) | 59,507 (21.2) | 6,283 (22.3) |
| Hispanic | 6,378 (5.3) | 24,270 (8.6) | 1,956 (7.0) |
| Cases diagnosed before enrollment count (% of all cases) | 101,861 (85.6) | -- | -- |
| Age at earliest CAD code mean (SD) | 63.3 (13.0) | -- | -- |
| Age at earliest CAD code median (quartiles) | 65 (56.7) | -- | -- |
| years of follow up after first CAD code: mean (SD) | |||
| prevalent cases only | 11.5 (4.9) | -- | -- |
| incident cases only | 2.5 (1.6) | -- | -- |
| all cases | 10.0 (5.5) | -- | -- |
| Age at last visit day mean (SD) | 73.3 (9.5) | 63.3 (14.3) | 69.3 (11.0) |
SD: standard deviation. AMI: acute myocardial infarction. HARE: Harmonizing Genetic Ancestry and Self-identified Race/Ethnicity algorithm. CAD: coronary artery disease.
Extended Data Table 2.
Demographic characteristics of the White, Black, and Hispanic participants from the Million Veteran Program and the Japanese from Biobank Japan used in GREML-LDMS-I heritability analyses.
| Stratum | MVP Whites | MVP Blacks (high proportion of African ancestry) | MVP Hispanics (high proportion of Native American ancestry) | Japanese from Biobank Japan | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Status | Sex | N | Mean | SD | N | Mean | SD | N | Mean | SD | N | Mean | SD |
|
| |||||||||||||
| CONTROL | Females | 1520 | 46.17 | 13.31 | 1684 | 44.92 | 12.6 | 1520 | 46.14 | 13.3 | 1518 | 46.3 | 13.3 |
| CONTROL | Males | 13791 | 57.54 | 15.87 | 13627 | 60.85 | 14.48 | 13794 | 57.55 | 15.9 | 13143 | 58.9 | 15.1 |
| HARD CASES | Females | 20 | 58.25 | 10.24 | 55 | 51.85 | 7.86 | 22 | 57.64 | 11.19 | 23 | 58.2 | 11.1 |
| HARD CASES | Males | 1400 | 60.49 | 9.04 | 1365 | 58.5 | 9.12 | 1398 | 60.53 | 9.06 | 1402 | 60.5 | 9.1 |
| SOFT CASES | Females | 55 | 57.16 | 11.01 | 42 | 55.55 | 9.36 | 58 | 55.19 | 12.96 | 60 | 55.2 | 12.6 |
| SOFT CASES | Males | 2606 | 61.96 | 9.59 | 2619 | 61.98 | 9.42 | 2603 | 61.99 | 9.54 | 2601 | 61.9 | 9.5 |
N: number in stratum. SD: standard deviation.
Extended Data Table 3.
Characteristics of Stage 1 and Stage 2 Black and Hispanic cohorts.
| Cases | Controls | |||||
|---|---|---|---|---|---|---|
| N | % male | Mean age (sd) | N | % male | Mean age (sd) | |
| Stage 1 COHORT: BLACK | ||||||
| MVP | 17202 | 92.2 | 59.1 (9.8) | 59507 | 75.3 | 59.8 (12.3) |
| Stage 2 COHORTS: BLACKS | ||||||
| PAGE Cohorts | 5225 | 20702 | ||||
| ARIC | 615 | 48 | 54.7 (5.6) | 2207 | 34 | 53.0 (5.8) |
| BioME all | 656 | 43 | 61.9 (11.8) | 6458 | 37 | 48.8(14.8) |
| BioME MEGA | 553 | 43 | 61.9 (11.4) | 4613 | 37 | 51.0(14.2) |
| BioME non-MEGA | 103 | 43 | 61.9 (13.5) | 1845 | 36 | 43.3 (14.9) |
| MEC all | 2640 | 3361 | ||||
| PAGE 2 MEGA diabetes | 914 | 33 | 71.2 (7.4) | 530 | 27 | 67.7 (8.1) |
| PAGE 2 MEGA controls | 426 | 31 | 71.2 (7.7) | 1147 | 24 | 66.8 (8.2) |
| Breast CA study - cases | 176 | 0 | 71.1 (8.4) | 275 | 0 | 66.4 (9.4) |
| Breast CA study - controls | 199 | 0 | 70.9 (8.4) | 347 | 0 | 64.8 (10.0) |
| Prostate CA study - cases | 455 | 100 | 71.1 (6.6) | 475 | 100 | 69.1 (7.4) |
| Prostate CA study - controls | 470 | 100 | 69.5 (7.4) | 587 | 100 | 66.7 (8.4) |
| WHI | 1314 | 0 | 63.8 (7.0) | 8676 | 0 | 61.2 (7.0) |
| MEGA | 938 | 0 | 63.8 (7.1) | 5891 | 0 | 59.4 (6.5) |
| SHARE | 354 | 0 | 63.6 (6.7) | 2690 | 0 | 65.1 (6.5) |
| GARNET | 22 | 0 | 63.9 (7.4) | 95 | 0 | 61.0 (7.5) |
| Other | 4594 | 22096 | ||||
| BioVU | 151 | 49.1 | 69.9 (11.8) | 1719 | 31.6 | 57.5 (16.1) |
| CHS | 419 | 38.2 | 73.1 (5.7) | 401 | 36.4 | 72.6 (5.7) |
| Health ABC | 322 | 51.3 | 73.6 (3.0) | 783 | 40.1 | 73.4 (2.9) |
| HANDLS | 84 | 32.6 | 47.1 (8.4) | 854 | 45.1 | 48.5 (9.0) |
| JHS | 54 | n/a | n/a | 1400 | n/a | n/a |
| eMERGE | 1820 | 42.1 | 58.8 (12.8) | 7286 | 30.3 | 48.8 (17.0) |
| Penn Biobank | 1395 | 51.5 | 67.5 (12.2) | 4276 | 31.3 | 53.2 (16.1) |
| UK-Biobank | 349 | 55 | 66.8 (7.8) | 5377 | 58.3 | 60.5 (7.9) |
| TOTAL Stage 2 cohorts | 9819 | 42798 | ||||
| OVERALL Stage 1+2 TOTALS | 27021 | 102305 | ||||
| Stage 1 COHORT: HISPANICS | ||||||
| MVP | 6378 | 94.7 | 60.9 (9.8) | 24270 | 77.6 | 56.1 (15.6) |
| Stage 2 COHORTS: HISPANICS | ||||||
| BioME all | 1230 | 50 | 65.1 (11.4) | 8793 | 35 | 49.9 (15.8) |
| BioME MEGA | 702 | 50 | 65.0 (11.2) | 4860 | 35 | 50.3 (15.9) |
| BioME non-MEGA | 528 | 52 | 65.2 (11.7) | 3933 | 36 | 49.4 (15.8) |
| MEC all | 3174 | 4530 | ||||
| PAGE2 MEGA diabetes | 516 | 46 | 70.1 (7.1) | 394 | 36 | 65.6 (6.6) |
| PAGE2 MEGA controls | 250 | 50 | 70.3 (6.3) | 781 | 38 | 67.0 (6.3) |
| Breast CA study - cases | 145 | 0 | 69.0 (6.8) | 292 | 0 | 64.5 (7.5) |
| Breast CA study - controls | 144 | 0 | 67.3 (7.0) | 288 | 0 | 62.5 (7.7) |
| Prostate CA study - cases | 404 | 100 | 71.0 (6.6) | 453 | 100 | 69.0 (7.0) |
| Prostate CA study - controls | 363 | 100 | 71.7 (6.8) | 418 | 100 | 67.7 (7.6) |
| T2D 2.5M study - cases | 760 | 46 | 69.5 (6.5) | 638 | 44 | 66.6 (6.8) |
| T2D 2.5M study - controls | 460 | 50 | 70.9 (6.6) | 1026 | 40 | 67.1 (6.8) |
| Hecht Smokers | 132 | 62 | 66.6 (6.4) | 240 | 49 | 64.5 (6.1) |
| WHI (MEGA) | 397 | 0 | 62.5 (6.8) | 4229 | 0 | 60.1 (6.7) |
| eMERGE | 938 | 44.7 | 61.0 (12.7) | 3031 | 30.6 | 49.4 (17.0) |
| TOTAL Stage 2 cohorts | 5739 | 20583 | ||||
| OVERALL Stage 1+2 TOTALS | 12117 | 44853 | ||||
PAGE: Population Architecture through Genomics and Environment Study (funded by the National Institutes of Health - NHGRI). ARIC: Atherosclerosis Risk in Communities Study (funded by the National institutes of Health - NHLBI). MEC: Multiethnic Study (funded by the National Cancer Institute). WHI: Women’s Health Initiative study (funded by the National institutes of Health - NHLBI). MEGA (WHI): PAGE substudy in the WHI genotyped with the Illumina Multi-Ethnic Genotyping Array. SHARE: SNP Health Association Resource substudy in WHI genotyped using Affymetrix 6.0 array. GARNET: Genomics and Randomized Trials Network substudy in WHI genotyped using Illumina HumanOmni1-Quad v1–0 B. BioVU: Vanderbilt’s biorepository of DNA extracted from discarded blood collected during routine clinical testing and linked to de-identified medical records in the Synthetic Derivative. CHS: Cardiovascular Health Study (funded by the National institutes of Health - NHLBI). Health ABC: The Health, Aging and Body Composition Study (funded by the National institutes of Health - NIA). HANDLS: The Healthy Aging in Neighborhoods of Diversity across the Life Span study (funded by the National Institutes of Health - NIA). JHS: The Jackson Heart Study (funded by the National Institutes of Health - NHLBI and NIMHD). eMERGE: The electronics Medical records and Genomics consortium (funded by the National Institutes of Health - NHGRI). Penn Biobank: The Penn Medicine BioBank (Institute for Translational Medicine and Therapeutics at the University of Pennsylvania). UK-Biobank: The UK Biobank Study. BioME: The BioME Biobank Program (The Institute for Personalized Medicine at the Icahn School of Medicine at Mount Sinai).
Extended Data Table 4.
Loci reaching genome wide significance after two-stage meta-analysis in Blacks and Hispanics.
| Genomic locus | locus | rsID | chr | pos | EA | NEA | EAF | OR | P | SNP annotation | Distance from nearest gene | Gene symbol |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Blacks | ||||||||||||
|
| ||||||||||||
| 1 | 3p25.1 | rs76838170 | 3 | 14959209 | T | C | 0.92 | 0.9 | 3.32E-08 | intronic | 0 | FGD5 |
| 2 | 6q26.3 | rs575962368 | 6 | 1.61E+08 | T | G | 0.02 | 1.28 | 4.28E-11 | intergenic | 13591 | LPA |
| 3 | 8p21.3 | rs13702 | 8 | 19824492 | T | C | 0.46 | 1.06 | 4.60E-09 | UTR3 | 0 | LPL |
| 3 | 8p21.3 | rs58625286 | 8 | 19894289 | C | G | 0.07 | 0.87 | 1.50E-10 | intergenic | 71175 | LPL |
| 8p22* | rs7012408 | 8 | 13624936 | A | G | 0.85 | 0.89 | 3.93E-09 | intergenic | 24646 | DLC1, SGCZ | |
| 4 | 13q34 | rs9515203 | 13 | 1.11E+08 | T | C | 0.72 | 1.06 | 4.81 E-08 | intronic | 0 | COL4A1 |
| 5 | 19q13.3 | rs72654473 | 19 | 45414399 | A | C | 0.17 | 0.93 | 1.73E-08 | intergenic | 1750 | APOE |
|
| ||||||||||||
| Hispanics | ||||||||||||
|
| ||||||||||||
| 6 | 15q25 | rs7164479 | 15 | 79123054 | T | C | 0.53 | 1.11 | 1.44E-10 | intergenic | 19282 | ADAMTS7 |
| 7 | 11q23.3 | rs9326246 | 11 | 1.17E+08 | C | G | 0.16 | 1.13 | 4.84E-08 | intergenic | 100000 | APOA5 |
| 8 | 6q24.1 | rs9349379 | 6 | 12903957 | A | G | 0.66 | 0.90 | 1.72E-09 | intronic | 0 | PHACTR1 |
The 8p22 locus among Blacks was carefully examined for the possibility of a false positive association as was not even a hint of a genetic signal in this region among Whites despite more than adequate power due to much larger sample size and a substantially higher frequency of the lead SNPs. There was no obvious link between the signal and the status of the inversion in the immediately adjacent 8p23 region which we called using principal component analyses of SNPs within the inversion site. The region reached genome wide significance in MVP Blacks based solely on imputed genotypes. We determined that neighboring genotypes in the region were not reliably called due to an unrecognized African specific deletion in the region subsequently reported by the gnoMAD consortium. The deletion also affected the reliability of the imputed SNPs. Even after recalling the genotypes in this region taking into consideration the presence of the deletion, no genotyped SNPs were genome wide significant for CAD. P values are derived from inverse variance weighted meta-analysis using METAL and are two-sided.
Extended Data Table 5.
Derivation of the VA Clinical Assessment Reporting and Tracking (CART) sub cohort for genome wide association study.
| Severity of disease | Normal | Non-Obstructive | 1V Obstructive | 2 V Obstructive | 3V/LM Obstructive | Missing | Other | Total |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| all procedures | 9804 | 17806 | 18800 | 12578 | 17053 | 2101 | 475 | 78617 |
| remove “Missing” or “Other” | 9804 | 17806 | 18800 | 12578 | 17053 | 0 | 0 | 76041 |
| restrict to procedures from individuals with genetic data and age/sex covariates | 7366 | 13449 | 14385 | 9642 | 13138 | 0 | 0 | 57980 |
| subjects with 1 proc only | 5806 | 8413 | 7466 | 4671 | 6089 | 0 | 0 | 32445 |
| subjects with >1 proc but severity same | ||||||||
| all Normal* | 281 | 0 | 0 | 0 | 0 | 0 | 0 | 281 |
| all not normal** | 0 | 659 | 859 | 497 | 1329 | 0 | 0 | 3344 |
| subjects with >1 proc but severity NOT same | ||||||||
| single instance of most severe disease | 0 | 344 | 1101 | 1488 | 2172 | 0 | 0 | 5105 |
| >1 instance of most severe disease*** | 0 | 46 | 251 | 292 | 708 | 0 | 0 | 1297 |
| # of individuals in each category of severity after assignment to 1 category | 6087 | 9462 | 9677 | 6948 | 10298 | 0 | 0 | 42472 |
| remaining subjects after removal of subjects with a history of cardiac transplant, age discrepancy, or undefined HARE assessment**** | 5957 | 9304 | 9534 | 6819 | 10124 | 0 | 0 | 41738 |
| HARE assignment white/European | 3704 | 6767 | 7496 | 5470 | 8221 | 0 | 0 | 31658 |
| HARE assignment black/African American | 1867 | 1952 | 1454 | 908 | 1132 | 0 | 0 | 7313 |
| HARE assignment Latino/Hispanic | 359 | 551 | 534 | 399 | 693 | 0 | 0 | 2536 |
| HARE assignment East/South Asian | 27 | 34 | 50 | 42 | 78 | 0 | 0 | 231 |
|
| ||||||||
| FINAL cohort for GWAS after removing Asian HARE category due to small numbers | 5930 | 9270 | 9484 | 6777 | 10046 | 0 | 0 | 41507 |
1V = 1 vessel >50% obstruction, 2V = 2 vessels with >50% obstruction, 3V/LM = 3 vessels and/or left main disease >50% obstruction.
age assigned to last procedure.
age assigned to earliest procedure.
age assigned to the earliest procedure showing the most severe disease.
n with history of a cardiac transplant = 137, major age discrepancy between CART and CDW derived age = 2, and undefined HARE assignment = 595.
Extended Data Table 6.
Age and sex characteristics of VA Clinical Assessment Reporting and Tracking (CART) subcohort by Harmonizing Genetic Ancestry and Self-identified Race/Ethnicity algorithm (HARE) assigned populations and severity of disease.
| Severity of disease | Normal | Non-Obstructive | 1V Obstructive | 2V Obstructive | 3V/LM Obstructive | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Count (%) | age (SD) | Count (%) | age (SD) | Count (%) | age (SD) | Count (%) | age (SD) | Count (%) | age (SD) | |
|
|
||||||||||
| HARE Whites | ||||||||||
| Males | 3335 (90) | 62 (10) | 6503 (96.1) | 66.1 (8.7) | 7331 (97.8) | 66.6 (8.6) | 5390 (98.5) | 67.3 (8.4) | 8149 (99.1) | 67.9 (8.4) |
| Females | 369 (10) | 57.7 (9.6) | 264 (3.9) | 62.5 (10) | 165 (2.2) | 63.4 (9.2) | 80 (1.5) | 63.9 (10.2) | 72 (0.9) | 67.9 (11) |
| HARE Blacks | ||||||||||
| Males | 1637 (87.7) | 59.1 (9.2) | 1834 (94) | 62.3 (8.9) | 1409 (96.9) | 63.3 (8.8) | 880 (96.9) | 63.3 (8.5) | 1113 (98.3) | 64.3 (8.5) |
| Females | 230 (12.3) | 54.5 (8.2) | 118 (6) | 56.6 (7.6) | 45 (3.1) | 56.1 (8) | 28 (3.1) | 57.9 (8.4) | 19 (1.7) | 63.1 (12) |
| HARE Hispanics | ||||||||||
| Males | 329 (91.6) | 58.7 (10.9) | 540 (98) | 63.2 (8.8) | 528 (98.9) | 64.2 (9.3) | 397 (99.5) | 64.6 (9) | 687 (99.1) | 65.9 (8.2) |
| Females | 30 (8.4) | 52.7 (8.2) | 11 (2) | 60.7 (9.9) | 6 (1.1) | 54.5 (5.7) | 2 (0.5) | 66.8 (1.7) | 6 (0.9) | 63.4 (6.1) |
| HARE Asians* | ||||||||||
| Males | 27 (100) | 54.6 (11.7) | 34 (100) | 62.7 (10.2) | 48 (96) | 61.4 (8.6) | 42 (100) | 64.6 (9) | 77 (98.7) | 69.3 (10.3) |
| Females | 0(0) | -- | 0(0) | -- | 2(4) | 61.8 (5.2) | 0(0) | -- | 1 (1.3) | -- |
did not proceed with genetic association in this group because of low numbers.
Extended Data Table 7.
Odds ratio of CAD per standard deviation increase of the externally derived LDPred and metaGRS scores in the MVP White, Black, and Hispanic cohorts followed by relative efficiency estimates based on ratios of betas.
| Prevalent disease (at enrollment) + Incident (post-enrollment) | Incident (post enrollment) | Estimated Relative efficiency**** compared to | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Cohort | Race/Ethnic | Outcome*** | PRS | non-Cases | Cases | OR (95%CI) | Cases | OR (95%CI) | UK Biobank | MVP Whites |
|
| ||||||||||
| UK Biobank* | white | AMI / Revasc | LDPred | 116317 | 3963 | 1.72 (1.67–1.78) | not reported | not reported | ||
|
| ||||||||||
| MVP | white | AMI / Revasc | LDPred | 197091 | 28512 | 1.51 (1.49–1.53) | 9320 | 1.46 (1.43–1.49) | 0.76 | 1 |
| MVP | Hispanic | AMI / Revasc | LDPred | 24263 | 2216 | 1.52 (1.45–1.59) | 725 | 1.49 (1.38–1.61) | 0.77 | 1.01 |
| MVP | Afr. Amer. | AMI / Revasc | LDPred | 59482 | 5358 | 1.17 (1.14–1.21) | 2121 | 1.15 (1.1–1.2) | 0.29 | 0.39 |
| MVP | white | All CAD | LDPred | 197091 | 95151 | 1.36 (1.35–1.37) | 18831 | 1.26 (1.24–1.28) | n/a | n/a |
| MVP | Hispanic | All CAD | LDPred | 24263 | 6378 | 1.32 (1.28–1.36) | 1451 | 1.22 (1.15–1.29) | n/a | n/a |
| MVP | Afr. Amer. | All CAD | LDPred | 59482 | 17202 | 1.1 (1.08–1.12) | 4454 | 1.1 (1.07–1.14) | n/a | n/a |
|
| ||||||||||
| UK Biobank** | white | AMI / Revasc | metagrs | 460387 | 22242 | 1.71 (1.68–1.73) | 12513 | 1.58 (1.55–1.61) | ||
|
| ||||||||||
| MVP | white | AMI / Revasc | metagrs | 197091 | 28512 | 1.54 (1.52–1.56) | 9320 | 1.47(1.44–1.5) | 0.8 | 1 |
| MVP | Hispanic | AMI / Revasc | metagrs | 24263 | 2216 | 1.62 (1.54–1.71) | 725 | 1.5 (1.38–1.63) | 0.9 | 1.13 |
| MVP | Afr. Amer. | AMI / Revasc | metagrs | 59482 | 5358 | 1.2 (1.17–1.24) | 2121 | 1.17 (1.12–1.22) | 0.35 | 0.43 |
| MVP | white | All CAD | metagrs | 197091 | 95151 | 1.38 (1.36–1.39) | 18831 | 1.27 (1.25–1.29) | n/a | n/a |
| MVP | Hispanic | All CAD | metagrs | 24263 | 6378 | 1.39 (1.34–1.43) | 1451 | 1.24 (1.17–1.32) | n/a | n/a |
| MVP | Afr. Amer. | All CAD | metagrs | 59482 | 17202 | 1.12 (1.1–1.14) | 4454 | 1.1 (1.07–1.14) | n/a | n/a |
LDPred score for CAD as previously described22
metaGRS for CAD as previously described23
AMI / Revasc: subset of cases with evidence of discharge diagnosis of acute myocardial infarction or revascularization procedure in the EHR. All CAD: further include subjects with CAD codes that are not AMI or revascularization
Relative efficiency: ratio of log ORs (beta coefficients) between MVP and UK Biobank. n/a: not applicable as this broader phenotype not reported in the LDpred and metaGRS reports. glm function in R for logistic regression covariates: age, sex, genotyping batch and top 10 genotype-based PCs. p-value corresponding to the z ratio based on a Standard Normal reference distribution, 2-sided.
Supplementary Material
Acknowledgements
This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by Veterans Administration awards I01–01BX003362, I01-BX004821 (K-M. C., P.S.T), I01-BX003340 (K. C., P.W.F.W) and VA HSR RES 13–457 (VA Informatics and Computing Infrastructure). The content of this manuscript does not represent the views of the Department of Veterans Affairs or the United States Government.
The eMERGE Network was initiated and funded by NHGRI through the following grants: Phase III: U01HG8657 (Kaiser Permanente Washington/University of Washington); U01HG8685 (Brigham and Women’s Hospital); U01HG8672 (Vanderbilt University Medical Center); U01HG8666 (Cincinnati Children’s Hospital Medical Center); U01HG6379 (Mayo Clinic); U01HG8679 (Geisinger Clinic); U01HG8680 (Columbia University Health Sciences); U01HG8684 (Children’s Hospital of Philadelphia); U01HG8673 (Northwestern University); U01HG8701 (Vanderbilt University Medical Center serving as the Coordinating Center); U01HG8676 (Partners Healthcare/Broad Institute); and U01HG8664 (Baylor College of Medicine) Phase II: U01HG006828 (Cincinnati Children’s Hospital Medical Center/Boston Children’s Hospital); U01HG006830 (Children’s Hospital of Philadelphia); U01HG006389 (Essentia Institute of Rural Health, Marshfield Clinic Research Foundation and Pennsylvania State University); U01HG006382 (Geisinger Clinic); U01HG006375 (Group Health Cooperative/University of Washington); U01HG006379 (Mayo Clinic); U01HG006380 (Icahn School of Medicine at Mount Sinai); U01HG006388 (Northwestern University); U01HG006378 (Vanderbilt University Medical Center); and U01HG006385 (Vanderbilt University Medical Center serving as the Coordinating Center). U01HG004438 (CIDR) and U01HG004424 (the Broad Institute) serving as Genotyping Centers. And/or The PGRNSeq dataset (eMERGE PGx), please also add U01HG004438 (CIDR) serving as a Sequencing Center. Phase I: U01-HG-004610 (Group Health Cooperative/ University of Washington); U01-HG-004608 (Marshfield Clinic Research Foundation and Vanderbilt University Medical Center); U01-HG-04599 (Mayo Clinic); U01HG004609 (Northwestern University); U01-HG-04603 (Vanderbilt University Medical Center, also serving as the Administrative Coordinating Center); U01HG004438 (CIDR) and U01HG004424 (the Broad Institute) serving as Genotyping Centers.
The Population Architecture Using Genomics and Epidemiology (PAGE) program is funded by the National Human Genome Research Institute (NHGRI) with co-funding from the National Institute on Minority Health and Health Disparities (NIMHD), supported by U01HG007416 (CALiCo), U01HG007417 (ISMMS), U01HG007397 (MEC), U01HG007376 (WHI), and U01HG007419 (Coordinating Center).
The MultiEthnic Study (MEC) was supported by U01 CA164973.
The Women’s Health Initiative (WHI) program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C. Scientific Computing Infrastructure at Fred Hutch funded by ORIP grant S10OD028685. Funding support for the “Exonic variants and their relation to complex traits in minorities of the (WHI) study is provided through the NHGRI PAGE program (U01HG004790).
The Atherosclerosis Risk in Communities (ARIC) Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C).
The Cardiovascular Health Study (CHS) was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, 75N92021D00006; and NHLBI grants U01HL080295, R01HL085251, R01HL087652, R01HL105756, R01HL103612, R01HL120393, and U01HL130114 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
BioBank Japan (BBJ) was supported by the Tailor-Made Medical Treatment Program of the Ministry of Education, Culture, Sports, Science, and Technology and Japan Agency for Medical Research (AMED) under grant numbers JP17km0305002 and JP17km0305001.
Healthy Aging in Neighborhoods of Diversity across the Life Span (HANDLS): was funded by the Interlaboratory Proposal Funding of the Intramural Research Program of the National Institute on Aging (NIA), the National Institutes of Health (NIH), Baltimore, Maryland. Funding number: [AG000989].
X.Z. was supported by the Stein Fellowship from Stanford University and Institute for Computational and Data Sciences Seed Grant from The Pennsylvania State University. S.M.D., J.A.L., and K.M.L. were supported by the U.S. Department of Veterans Affairs (IK2-CX001780). Y.L. is supported by NIH R56HL150186. S. Koyama and K. Ito were supported by AMED under Grant Numbers JP20km0405209 and JP20ek0109487. K.E.N. is supported by NIH R01HL142302. R.D. is supported by NIH R35GM124836 & R01HL139865. F.C. is supported by NCI T32CA229110. B.F.V. was supported by the NIH R01DK101478 and a Linda Pechenik Montague Investigator Award. PN is supported by grants from the NIH/NHLBI (R01HL142711, R01HL148050, R01HL127564, R01HL151152), NIH/NHGRI (U01HG011719), Fondation Leducq (TNE-18CVD04), and Massachusetts General Hospital (Fireman Chair).
Support for title page creation and format was provided by AuthorArranger, a tool developed at the National Cancer Institute. We thank Carlos D. Bustamente for his review and feedback of specific cross-population analyses involving the 9p21 region. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Ethics declarations - Competing interests
A.B. and L.A.L. are employees of Regeneron Pharmaceuticals. R.D. has received grants from AstraZeneca, grants and nonfinancial support from Goldfinch Bio, being a scientific co-founder, consultant and equity holder for Pensieve Health and being a consultant for Variant Bio. T. M. M. is an employee of the Healthcare Innovation Lab at BJC HealthCare / Washington University School of Medicine, an advisor of Myia Labs, and a compensated director the J.F Maddox Foundation in New Mexico. S.K. is an is an employee of Verve Therapeutics, holds equity in Verve Therapeutics and Maze Therapeutics, and has served as a consultant for Acceleron, Eli Lilly, Novartis, Merck, Novo Nordisk, Novo Ventures, Ionis, Alnylam, Aegerion, Haug Partners, Noble Insights, Leerink Partners, Bayer Healthcare, Illumina, Color Genomics, MedGenome, Quest, and Medscape. D.J.R. is on the Scientific Advisory Board of Alnylam, Novartis, and Verve Therapeutics. M.D.R. is on the scientific advisory board for Goldfinch Bio and Cipherome. C.J.O became an employee of Novartis after initial submission of manuscript. P.N. reports investigator-initiated grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Invitae, Foresite Labs, Novartis, Roche / Genentech, is a co-founder of TenSixteen Bio, is a shareholder of geneXwell, TenSixteen Bio, and Vertex, scientific advisory board member of geneXwell and TenSixteen Bio, and spousal employment at Vertex, all unrelated to the present work. S.M.D. receives research support from RenalytixAI to his institution and consulting fees from Calico Labs. A.G.B. is a scientific co-founder and equity holder in TenSixteen Bio. The remaining authors declare no competing interests.
Peer review information:
Primary Handling editor: Michael Basson, in collaboration with the Nature Medicine team.
Peer review information:
Nature Medicine thanks Riyaz Patel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Consortium Members
Regeneron Genetics Center
Aris Baras65, Luca A. Lotta65
65Regeneron Genetics Center, Tarrytown, NY, USA,
A full list of members and their affiliations appears in the Supplementary Information.
CardiogramplusC4D
Satoshi Koyama17, Iftikhar J. Kullo52, Kaoru Ito17, Kazuyoshi Ishigaki60, Yoichiro Kamatani60,61
Aris Baras65, Luca A. Lotta65, Sekar Kathiresan67,23,68,69, Christopher J. O’Donnell18,33, Themistocles L Assimes1,2,88,87
A full list of members and their affiliations appears in the Supplementary Information.
Million Veteran Program
Elizabeth R. Hauser70,71, Kelly Cho18,33, J. Michael Gaziano18,33, Kyong-Mi Chang20,21, Peter W. F. Wilson83,84, Yan V. Sun85,86, Philip S. Tsao1,74,87
A full list of members and their affiliations appears in the Supplementary Information.
Biobank Japan
Satoshi Koyama17, Kaoru Ito17, Kazuyoshi Ishigaki60, Yoichiro Kamatani60,61
A full list of members and their affiliations appears in the Supplementary Information.
References
- 1.Roth GA et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J Am Coll Cardiol 76, 2982–3021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Statistics;, N.C.f.H. Health, United States Spotlight: Racial and Ethnic Disparities in Heart Disease (Centers for Disease Control and Prevention, 2019). [Google Scholar]
- 3.Churchwell K et al. Call to Action: Structural Racism as a Fundamental Driver of Health Disparities: A Presidential Advisory From the American Heart Association. Circulation 142, e454–e468 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Popejoy AB & Fullerton SM Genomics is failing on diversity. Nature 538, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin AR et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarke SL, Assimes TL & Tcheandjieu C The Propagation of Racial Disparities in Cardiovascular Genomics Research. Circ Genom Precis Med 14, e003178 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zdravkovic S et al. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med 252, 247–54 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Wienke A, Holm NV, Skytthe A & Yashin AI The heritability of mortality due to heart diseases: a correlated frailty model applied to Danish twins. Twin Res 4, 266–74 (2001). [DOI] [PubMed] [Google Scholar]
- 9.van der Harst P & Verweij N Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res 122, 433–443 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koyama S et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet 52, 1169–1177 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Webb TR et al. Systematic Evaluation of Pleiotropy Identifies 6 Further Loci Associated With Coronary Artery Disease. J Am Coll Cardiol 69, 823–836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 43, 339–44 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Lu X et al. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet 44, 890–894 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Assimes TL & Roberts R Genetics: Implications for Prevention and Management of Coronary Artery Disease. J Am Coll Cardiol 68, 2797–2818 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.National Center for Health Statistics (U.S.). Crude percentages of coronary heart disease for adults aged 18 and over, United States, 2015–2018. National Health Interview Survey. in National Center for Health Statistics, National Health Interview Survey, 2015–2018 (2020). [Google Scholar]
- 17.Institute for Health Metrics and Evaluation (IHME). GBD Compare Data Visualization. Seattle, WA: IHME, University of Washington, 2020. Available from http://vizhub.healthdata.org/gbd-compare. (Accessed 2020) [Google Scholar]
- 18.Nikpay M et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 47, 1121–1130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishigaki K et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat Genet 52, 669–679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbalic M et al. Genome-wide association analysis of incident coronary heart disease (CHD) in African Americans: a short report. PLoS Genet 7, e1002199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lettre G et al. Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet 7, e1001300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inouye M et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J Am Coll Cardiol 72, 1883–1893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dikilitas O et al. Predictive Utility of Polygenic Risk Scores for Coronary Heart Disease in Three Major Racial and Ethnic Groups. Am J Hum Genet 106, 707–716 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Speliotes EK et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42, 937–948 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vujkovic M et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 52, 680–691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu DJ et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet 49, 1758–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartiala JA et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur Heart J 42, 919–933 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wainschtein P et al. Recovery of trait heritability from whole genome sequence data. bioRxiv, 588020 (2021). [Google Scholar]
- 30.McPherson R et al. A common allele on chromosome 9 associated with coronary heart disease. Science 316, 1488–91 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.CARDIoGRAMplusC4D Consortium et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 45, 25–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shungin D et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y et al. Sexual Differences in Genetic Predisposition of Coronary Artery Disease. Circ Genom Precis Med 14, e003147 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zore T, Palafox M & Reue K Sex differences in obesity, lipid metabolism, and inflammation-A role for the sex chromosomes? Mol Metab 15, 35–44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salfati E et al. Susceptibility Loci for Clinical Coronary Artery Disease and Subclinical Coronary Atherosclerosis Throughout the Life-Course. Circ Cardiovasc Genet 8, 803–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Speliotes EK et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 7, e1001324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Natarajan P et al. Chromosome Xq23 is associated with lower atherogenic lipid concentrations and favorable cardiometabolic indices. Nat Commun 12, 2182 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fletcher R et al. The role of the Niemann-Pick disease, type C1 protein in adipocyte insulin action. PLoS One 9, e95598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghuran A, van Der Wieken LR & Nolan J Cardiovascular complications of recreational drugs. BMJ 323, 464–6 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wirka RC et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med 25, 1280–1289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson CP et al. Genetically determined height and coronary artery disease. N Engl J Med 372, 1608–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ong JS et al. Height and overall cancer risk and mortality: evidence from a Mendelian randomisation study on 310,000 UK Biobank participants. Br J Cancer 118, 1262–1267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clarke SL et al. Broad Clinical Manifestations of Polygenic Risk for Coronary Artery Disease in the Women’s Health Initiative. medRxiv, 2021.06.15.21258993 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao B et al. Inference of causal relationships based on the genetics of cardiometabolic traits and conditions unique to females in >50,000 participants. medRxiv, 2022.02.02.22269844 (2022). [Google Scholar]
- 45.Singh KK et al. BRCA1 is a novel target to improve endothelial dysfunction and retard atherosclerosis. J Thorac Cardiovasc Surg 146, 949–960 e4 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Wu HT et al. Oncogenic functions of the EMT-related transcription factor ZEB1 in breast cancer. J Transl Med 18, 51 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ibrahim N et al. BRCA1-associated epigenetic regulation of p73 mediates an effector pathway for chemosensitivity in ovarian carcinoma. Cancer Res 70, 7155–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bai F et al. BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development. Cancer Res 74, 6161–72 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Fardi M, Alivand M, Baradaran B, Farshdousti Hagh M & Solali S The crucial role of ZEB2: From development to epithelial-to-mesenchymal transition and cancer complexity. J Cell Physiol (2019). [DOI] [PubMed] [Google Scholar]
- 50.Soini Y et al. Transcription factors zeb1, twist and snai1 in breast carcinoma. BMC Cancer 11, 73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng P et al. ZEB2 Shapes the Epigenetic Landscape of Atherosclerosis. Circulation 145, 469–485 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabas I, Garcia-Cardena G & Owens GK Recent insights into the cellular biology of atherosclerosis. J Cell Biol 209, 13–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagao M et al. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ Res 126, 517–529 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowrie DJ Jr. Histology : an essential textbook (Thieme Publishers, New York, 2020). [Google Scholar]
- 55.Ko CW, Qu J, Black DD & Tso P Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol 17, 169–183 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Fahed AC et al. Transethnic Transferability of a Genome-Wide Polygenic Score for Coronary Artery Disease. Circ Genom Precis Med 14, e003092 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaziano JM et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol (2015). [DOI] [PubMed] [Google Scholar]
- 58.Hunter-Zinck H et al. Genotyping Array Design and Data Quality Control in the Million Veteran Program. Am J Hum Genet 106, 535–548 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loh PR et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Das S et al. Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fang H et al. Harmonizing Genetic Ancestry and Self-identified Race/Ethnicity in Genome-wide Association Studies. Am J Hum Genet 105, 763–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Byrd JB et al. Data quality of an electronic health record tool to support VA cardiac catheterization laboratory quality improvement: the VA Clinical Assessment, Reporting, and Tracking System for Cath Labs (CART) program. Am Heart J 165, 434–40 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Maddox TM et al. A national clinical quality program for Veterans Affairs catheterization laboratories (from the Veterans Affairs clinical assessment, reporting, and tracking program). Am J Cardiol 114, 1750–7 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Maddox TM et al. Nonobstructive coronary artery disease and risk of myocardial infarction. JAMA 312, 1754–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang J et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet 47, 1114–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Evans LM et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat Genet 50, 737–745 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee SH, Wray NR, Goddard ME & Visscher PM Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet 88, 294–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee SH et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet 44, 247–50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Visscher PM et al. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet 10, e1004269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Magi R et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum Mol Genet 26, 3639–3650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loley C et al. No Association of Coronary Artery Disease with X-Chromosomal Variants in Comprehensive International Meta-Analysis. Sci Rep 6, 35278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Graham SE et al. The power of genetic diversity in genome-wide association studies of lipids. Nature (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coram MA et al. Leveraging Multi-ethnic Evidence for Mapping Complex Traits in Minority Populations: An Empirical Bayes Approach. Am J Hum Genet (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choi SW & O’Reilly PF PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Denny JC et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu P et al. Mapping ICD-10 and ICD-10-CM Codes to Phecodes: Workflow Development and Initial Evaluation. JMIR Med Inform 7, e14325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Giambartolomei C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Global Lipids Genetics Consortium et al. Discovery and refinement of loci associated with lipid levels. Nat Genet 45, 1274–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Evangelou E et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 50, 1412–1425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Erzurumluoglu AM et al. Meta-analysis of up to 622,409 individuals identifies 40 novel smoking behaviour associated genetic loci. Mol Psychiatry 25, 2392–2409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yengo L et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet 27, 3641–3649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xue A et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun 9, 2941 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maples BK, Gravel S, Kenny EE & Bustamante CD RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet 93, 278–88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Leeuw CA, Stringer S, Dekkers IA, Heskes T & Posthuma D Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat Commun 9, 3768 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhu X & Stephens M Large-scale genome-wide enrichment analyses identify new trait-associated genes and pathways across 31 human phenotypes. Nat Commun 9, 4361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pers TH et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun 6, 5890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barbeira AN et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun 9, 1825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics for the Biobank Japan study were obtained from http://jenger.riken.jp/en/result. Summary statistics for the CARDIoGRAMplusC4D study were obtained from http://www.cardiogramplusc4d.org. Summary statistics for the UK Biobank study for CAD were obtained from https://www.cardiomics.net/download-data.
The full summary level association data from the individual population association analyses in MVP as well as the multi-population meta-analysis from this report will be available through dbGaP, with accession number phs001672 at the time of publication in a peer reviewed journal. This research has been conducted using the UK Biobank Resource under Application Numbers 13721 & 19416.