

Abstract
Monoclonal antibody therapies targeting tumor antigens drive cancer cell elimination in large part by triggering macrophage phagocytosis of cancer cells1–7. However, cancer cells evade phagocytosis using mechanisms that are incompletely understood. Here we develop a platform for unbiased identification of factors that impede antibody-dependent cellular phagocytosis (ADCP) using complementary genome-wide CRISPR knockout and overexpression screens in both cancer cells and macrophages. In cancer cells, beyond known factors such as CD47, we identify many regulators of susceptibility to ADCP, including the poorly characterized enzyme adipocyte plasma membrane-associated protein (APMAP). We find that loss of APMAP synergizes with tumour antigen-targeting monoclonal antibodies and/or CD47-blocking monoclonal antibodies to drive markedly increased phagocytosis across a wide range of cancer cell types, including those that are otherwise resistant to ADCP. Additionally, we show that APMAP loss synergizes with several different tumour-targeting monoclonal antibodies to inhibit tumour growth in mice. Using genome-wide counterscreens in macrophages, we find that the G-protein-coupled receptor GPR84 mediates enhanced phagocytosis of APMAP-deficient cancer cells. This work reveals a cancer-intrinsic regulator of susceptibility to antibody-driven phagocytosis and, more broadly, expands our knowledge of the mechanisms governing cancer resistance to macrophage phagocytosis.
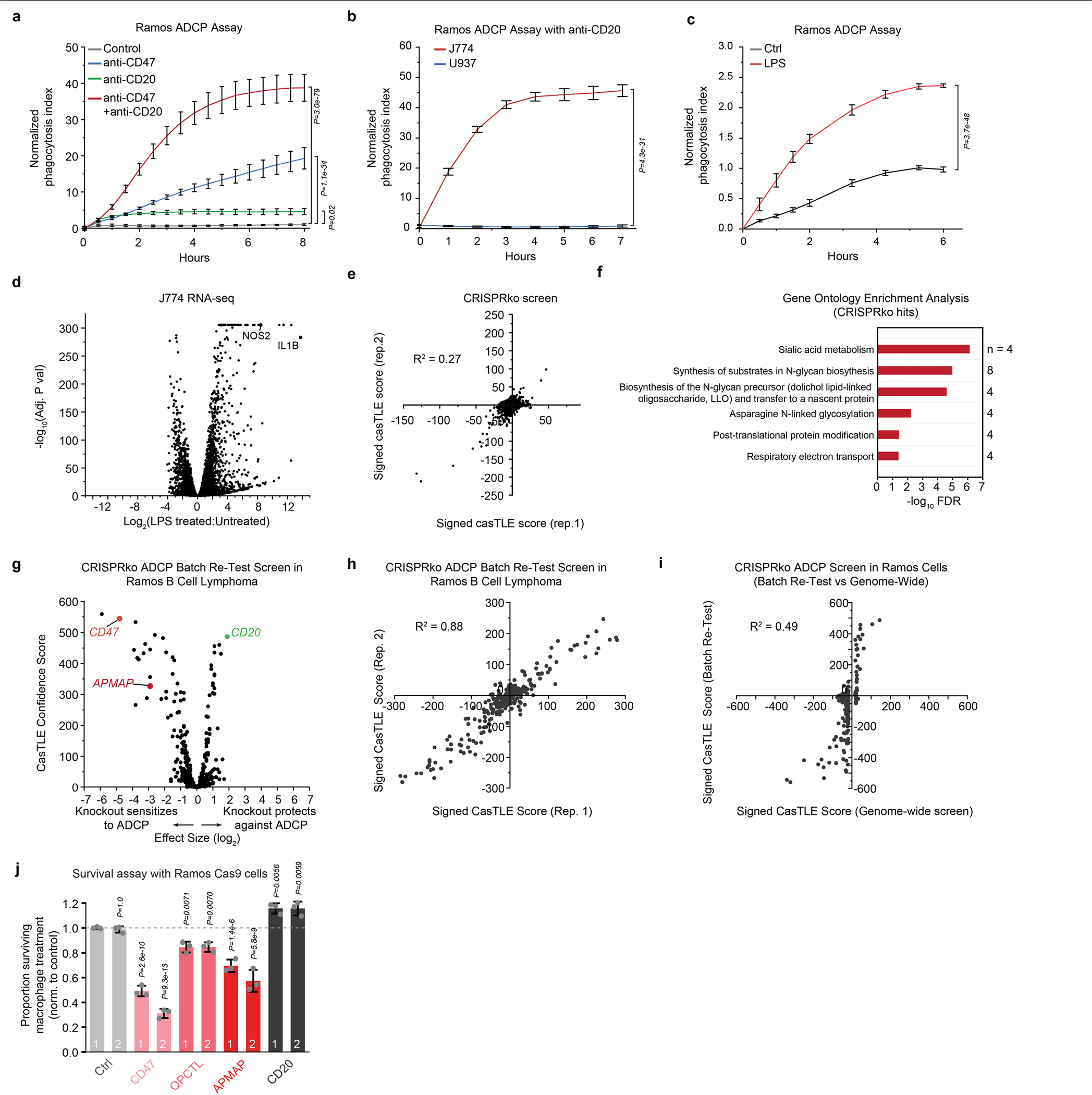
To identify cancer-intrinsic regulators of ADCP, we subjected Ramos lymphoma cells carrying a genome-wide CRISPR knockout library to multiple rounds of antibody-dependent killing by macrophages (Fig. 1a, Extended Data Fig. 1a–d, Supplementary Table 1, Supplementary Note 1). The top sensitizing hits in this screen included genes encoding the known anti-phagocytic factor CD473,8, the CD47-modifying enzyme QPCTL9 and several enzymes involved in synthesis of sialic acids, which are known to protect cancer cells against innate immune cell killing10 (Fig. 1b, Extended Data Fig. 1e, f), validating the ability of this screening platform to identify ADCP regulators. The target of the antibody used to drive ADCP, CD20 (also known as MS4A1), was recovered as the top protective hit (Fig. 1b). Beyond these known regulators, we identified many genes not previously linked to phagocytosis regulation, including the poorly characterized gene APMAP (Fig. 1b, Supplementary Table 2). We validated the phenotypes for these genes in both higher-coverage batch retest screens (Extended Data Fig. 1g–i, Supplementary Table 3) and, for several genes, in competitive growth assays in the presence of anti-CD20 and macrophages (Extended Data Fig. 1j).
Fig. 1 |. Genome-wide CRISPR screens reveal novel regulators of ADCP.
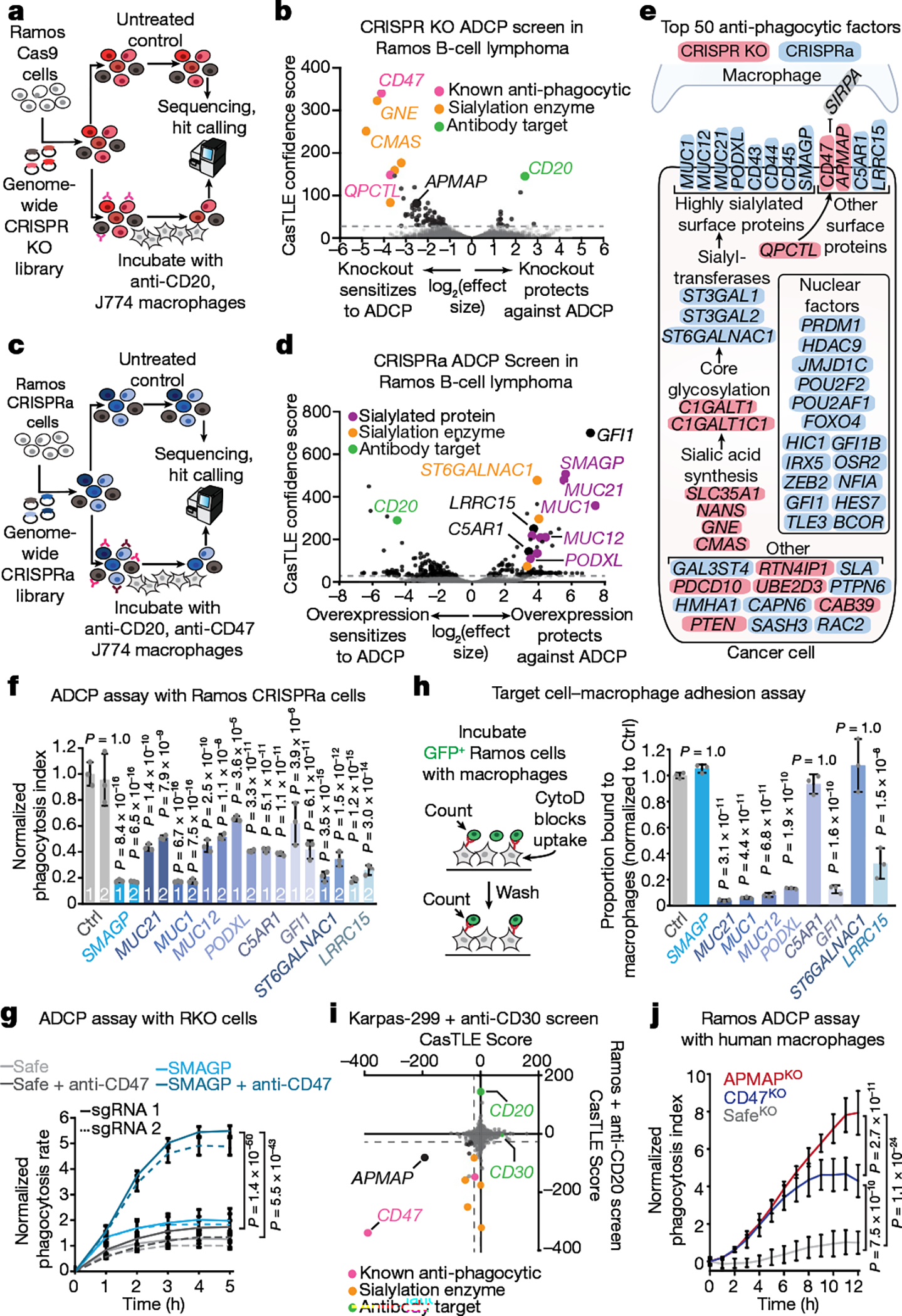
a, Schematic of the genome-wide CRISPR knockout (KO) screening strategy. b, Volcano plot of genome-wide screen in Ramos cells depicted in a. c, Schematic of the genome-wide CRISPR activation screening strategy. d, Volcano plot of genome-wide CRISPRa screen in Ramos cells depicted in c. e, Diagram of top 50 anti-phagocytic hits from CRISPR knockout (pink) and CRISPRa (blue) screens in Ramos cells. SIRPA, the macrophage receptor for CD47, is depicted in grey. f, Phagocytosis assay for uptake of pHrodo-Red-labelled Ramos CRISPRa cells expressing the indicated sgRNAs by J774 macrophages in the presence of anti-CD20 and anti-CD47 antibodies. The phagocytosis index was calculated as the total pHrodo Red signal (which increases in the low-pH environment of the lysosome) per well, normalized to signal in cells expressing a Safe-targeting control sgRNA (5 h timepoint) (n = 3 cell culture wells). Ctrl, control. g, Phagocytosis assay for uptake of pHrodo-labelled RKO Cas9 cells expressing indicated sgRNAs by J774 macrophages in the presence or absence of anti-CD47 antibodies, normalized to signal in control (SafeKO) cells at the 5 h timepoint (n = 4 cell culture wells). h, Left, schematic of target cell–macrophage adhesion assay. See Methods for details. The proportion of Ramos cells expressing indicated sgRNAs bound to macrophages is plotted, normalized to Ramos cells expressing Safe-targeting control sgRNA (n = 3 cell culture wells). CytoD, cytochalasin D. i, Comparison of Ramos–anti-CD20 and Karpas-299–anti-CD30 ADCP screens. j, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes by human U937 macrophages in the presence of anti-CD20 antibodies, normalized to control (SafeKO) cells expressing an sgRNA targeting a non-functional genomic locus40 (n = 3 cell culture wells). In b, d, i, dotted lines indicate 5% false discovery rate (FDR). In f–h, j, data are mean ± s.d. One-way analysis of variance (ANOVA) (f, h) or two-way ANOVA (g, j) with Bonferroni correction.
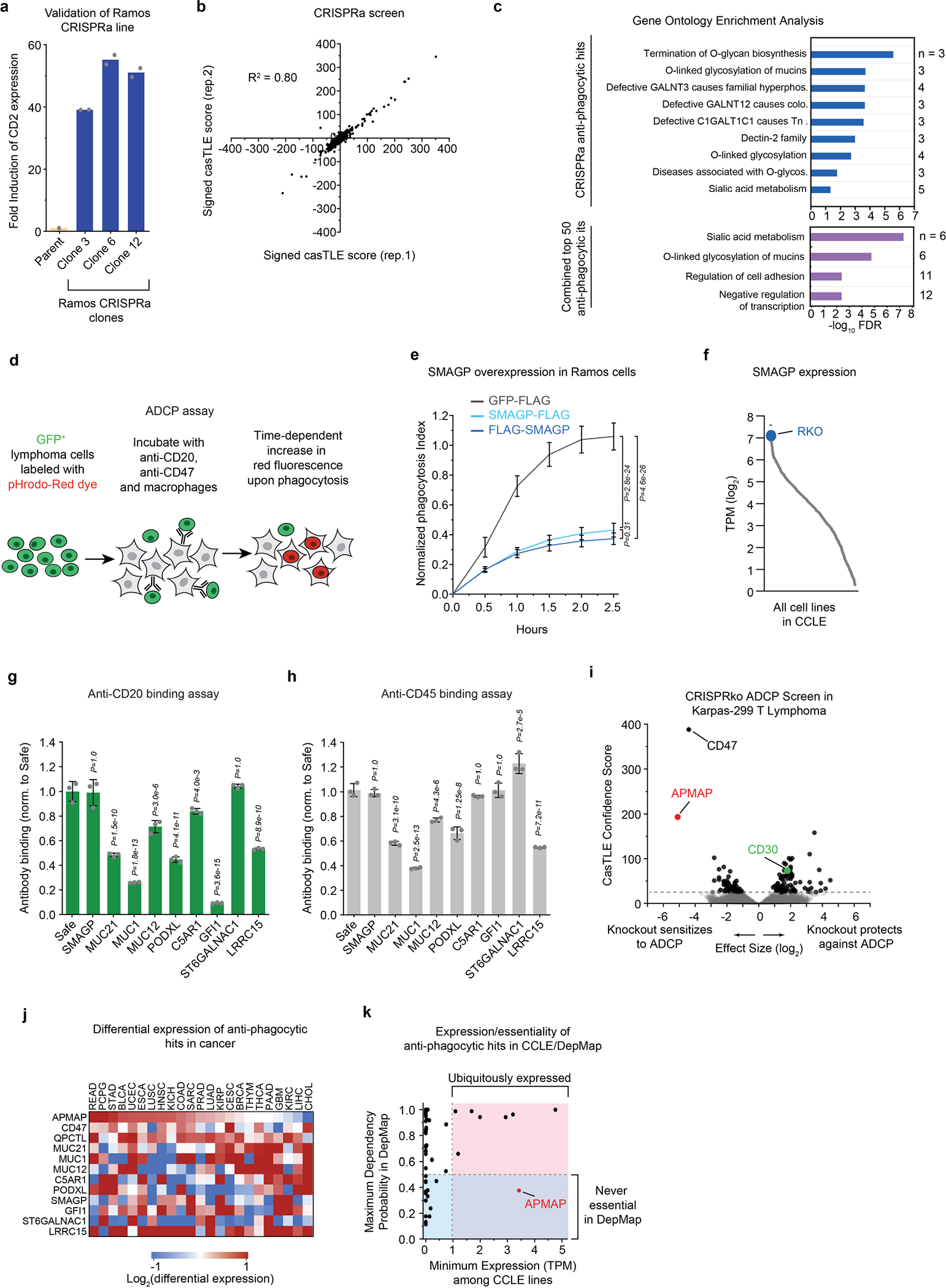
We next used CRISPR activation (CRISPRa) screens to uncover additional regulators of ADCP that are not endogenously expressed at high levels in Ramos cells (Fig. 1c, Extended Data Fig. 2a, Supplementary Note 2). As we had identified CD47 as the most potent anti-phagocytic factor expressed by Ramos cells (Fig. 1b), we also included anti-CD47 blocking antibodies (Extended Data Fig. 1a), reasoning that blocking CD47 would improve detection of additional anti-phagocytic factors (Fig. 1c). This screen revealed a large number of both anti-phagocytic hits (that is, genes whose overexpression increases cell survival upon incubation with macrophages and anti-CD20 and anti-CD47 antibodies) and pro-phagocytic hits (Fig. 1d, Extended Data Fig. 2b, Supplementary Table 4). As expected, CD20 scored as a strong pro-phagocytic hit (Fig. 1d). Gene ontology enrichment analysis of the anti-phagocytic hits from the CRISPRa screen, as well as the combined list of anti-phagocytic hits from both the CRISPR knockout and CRISPRa screens (Fig. 1e), revealed a strong enrichment for genes involved in sialic acid biosynthesis and/or protein sialylation (Extended Data Fig. 2c).
Notably, several of the top hit genes are oncogenes (GFI111 and MUC1212), cancer antigens (for example, MUC113 and LRRC1514,15), drivers of metastasis (for example, MUC2116, PODXL17 and SMAGP18) or negative prognostic indicators in cancer (for example, C5AR119), but have unclear functions in tumorigenesis and have not been previously implicated in regulation of phagocytosis. We validated nine of these genes as regulators of susceptibility to ADCP by incubating pHrodo red-labelled Ramos CRISPRa cells expressing single guide RNAs (sgRNAs) targeting each gene and monitoring the time-dependent increase in red fluorescence upon target cell internalization and acidification (Fig. 1f, Extended Data Fig. 2d). For SMAGP, which encodes a poorly characterized cell-surface glycoprotein18 and was among the top hits in the CRISPRa screen, we additionally showed that expression of Flag-tagged SMAGP under the strong EF1α promoter was sufficient to protect Ramos cells against ADCP (Extended Data Fig. 2e). We next tested whether the Ramos CRISPRa screen dataset could be used to predict genes that protect other cancer cell lines against ADCP on the basis of their high expression. We noted that SMAGP is expressed at high levels in RKO colon cancer cells (Extended Data Fig. 2f). We transduced RKO cells with Cas9 and sgRNAs targeting SMAGP, and found that deletion of SMAGP strongly sensitized RKO-Cas9 cells to phagocytosis in the presence of anti-CD47 antibodies (Fig. 1g). Thus, our CRISPRa screening approach enables identification of anti-phagocytic factors that are expressed at high levels in only a subset of cell lines.
To determine the stage of antibody-dependent phagocytosis that is affected by overexpression of these hits, we developed an imaging-based cell adhesion assay to assess target cell binding in the absence of phagocytosis, by inhibiting actin polymerization with cytochalasin D (Fig. 1h). We found that overexpression of a subset of hits, including the genes encoding the mucins MUC1, MUC12 and MUC21, prevented macrophages from binding Ramos target cells in the presence of rituximab, consistent with the reported ability of mucin overexpression to sterically occlude the cancer cell surface20 (Fig. 1h). Overexpression of most of these genes also decreased binding of two distinct antibodies—anti-CD20 and anti-CD45—to the cell surface, whereas overexpression of GFI1—which encodes a transcriptional repressor—specifically reduced anti-CD20 binding (Extended Data Fig. 2g, h). By contrast, overexpression of several genes, including SMAGP and ST6GALNAC1, did not affect binding of target cells to macrophages or decrease antibody binding (Fig. 1h, Extended Data Fig. 2g, h). Thus our screening platform identified genes that enable cancer evasion of ADCP via diverse mechanisms.
APMAP loss sensitizes lymphoma cells to ADCP
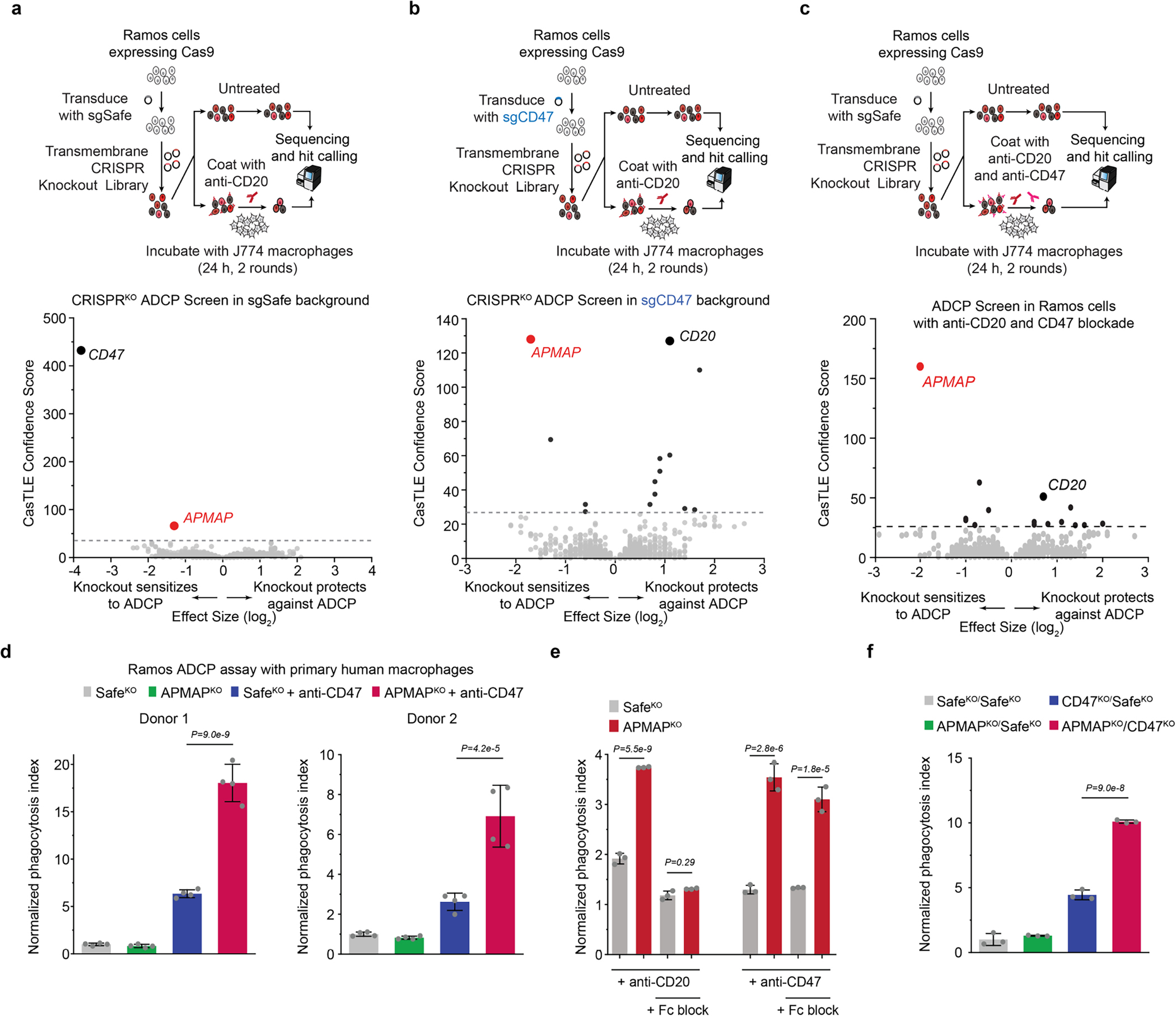
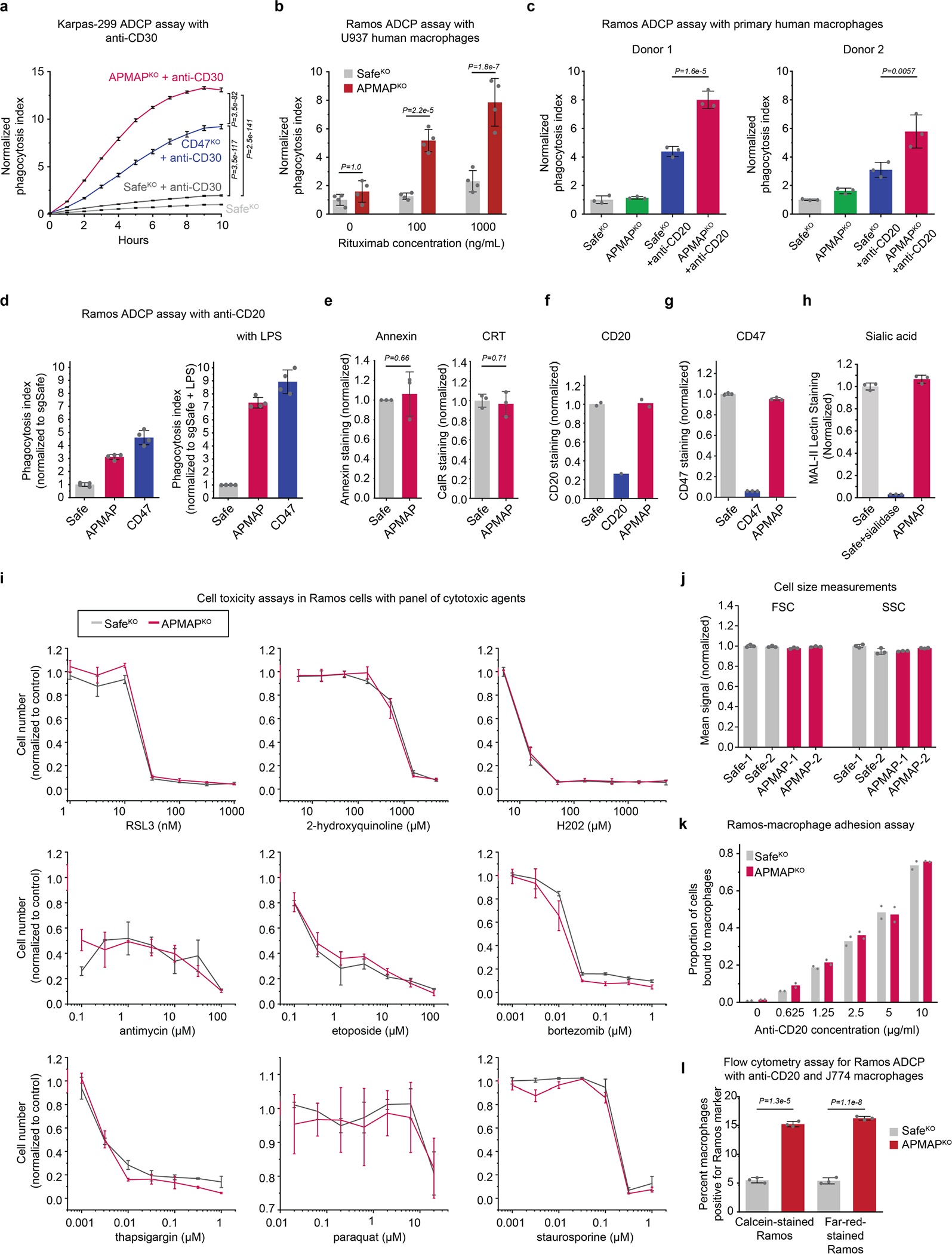
The poorly characterized gene APMAP was among the strongest modifiers of sensitivity to phagocytosis identified in both the Ramos CRISPR knockout screen (Fig. 1b) and a second, genome-wide ADCP screen we conducted in Karpas-299 T lymphoma cells using anti-CD30 antibodies (Fig. 1i, Extended Data Fig. 2i), suggesting an important role for APMAP in cancer evasion of phagocytosis. APMAP is ubiquitously expressed in human tissue types and in all cell lines of the Cancer Cell Line Encyclopedia, and is overexpressed in several malignancies21 (Extended Data Fig. 2j). Nonetheless, APMAP is not essential for growth under standard culture conditions in any of the 789 lines profiled in DepMap (Extended Data Fig. 2k) or for mouse viability22, implying that targeting APMAP would not exhibit broad toxicity. In individual assays, APMAP-knockout (APMAPKO) Ramos cells were significantly sensitized to ADCP in the presence of anti-CD20 and human macrophages, even more than CD47-knockout (CD47KO) cells (Fig. 1j). Unlike CD47KO cells, APMAPKO Ramos cells were not more susceptible to phagocytosis than SafeKO control cells in the absence of antibodies (Fig. 2a, b). APMAP loss in Ramos cells thus synergizes with tumour antigen-targeting antibodies to drive phagocytosis but does not increase uptake of unopsonized cells. We further tested whether APMAP would enhance the effects of CD47 blockade. Indeed, in two screens with a sublibrary containing approximately 3,100 genes enriched for membrane proteins, aimed at identifying genes that further sensitize cells to ADCP when CD47 is deleted or blocked, APMAP was the top hit (Extended Data Fig. 3a–c, Supplementary Table 5). Additionally, in a genome-wide screen for susceptibility to phagocytosis driven by anti-CD47 antibodies in Karpas-299 cells, APMAP was among the top hits (Fig. 2c). In quantitative live-cell imaging experiments, we confirmed that APMAP-deficient Ramos cells were significantly more sensitive to phagocytosis in the presence of CD47-blocking antibodies than control cells using either J774 macrophages (Fig. 2d) or primary human macrophages (Extended Data Fig. 3d). APMAP loss also enhanced ADCP induced by combined anti-CD20 and anti-CD47 treatment (Fig. 2e). In a series of additional experiments, we confirmed that APMAP loss specifically sensitized cancer cells to phagocytosis without affecting expression of known pro- or anti-phagocytic signals, binding of tumour-targeting antibodies or sensitivity to a panel of other cytotoxic agents (see Extended Data Figs. 3e, f, 4, Supplementary Note 3).
Fig. 2 |. APMAP loss synergizes with monoclonal antibodies and CD47 blockade to increase cancer cell susceptibility to phagocytosis.
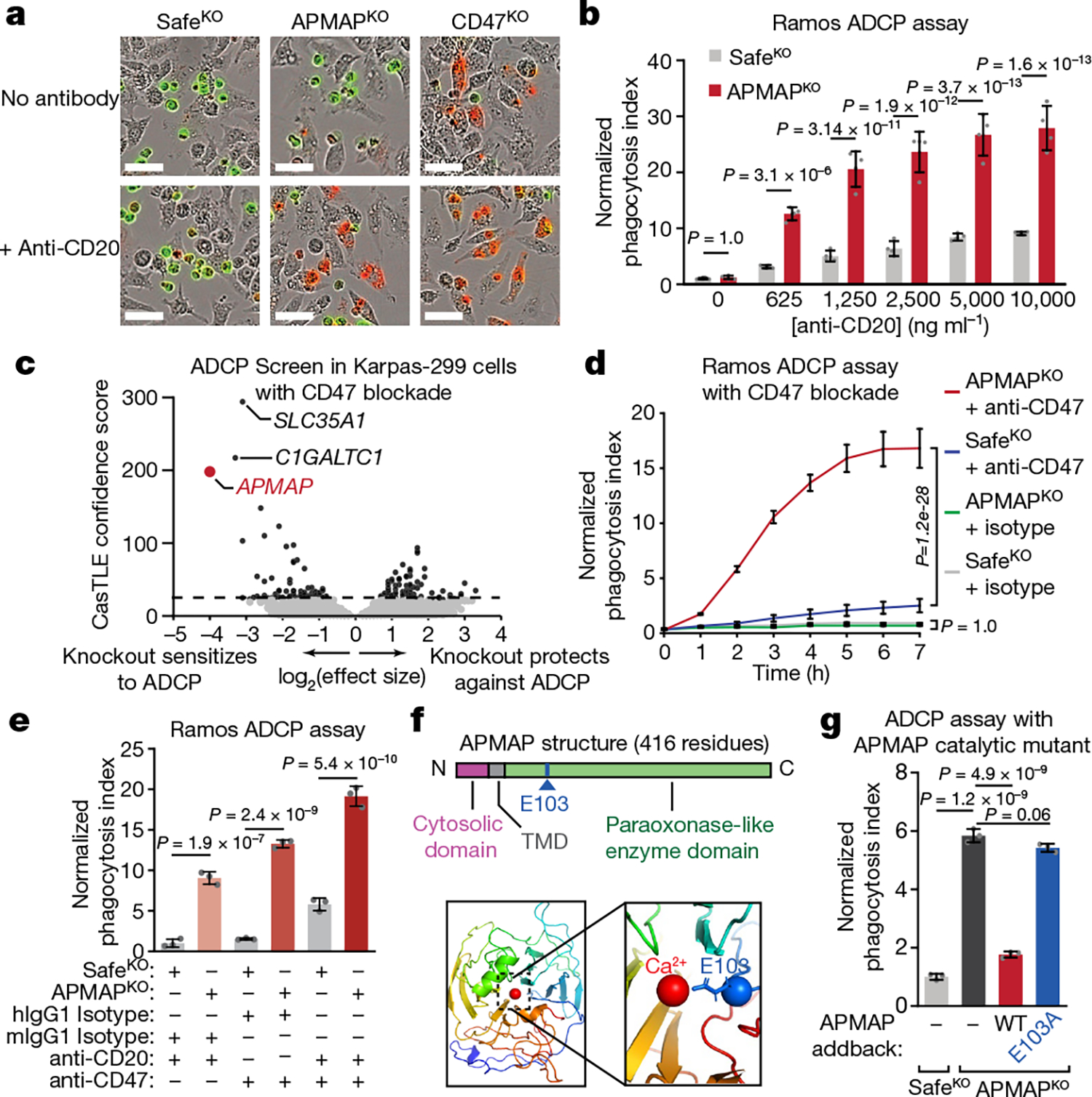
a, Images of pHrodo-labelled GFP+ Ramos cells of indicated genotypes after 12 h incubation with J774 macrophages with or without anti-CD20. Scale bars, 50 μm. Representative of three biologically independent experiments. b, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes by J774 macrophages with indicated concentrations of anti-CD20, normalized to control (SafeKO) cells in absence of anti-CD20 (n = 4 replicate wells). c, Volcano plot of genome-wide CRISPR knockout screen in Karpas299-Cas9 cells for resistance to treatment with anti-CD47 and macrophages. Dotted line indicates 5% FDR. d, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes following incubation with J774 macrophages and anti-CD47 antibodies or mouse isotype control (mIgG1) antibodies, normalized to control (SafeKO) cells with isotype control antibody (n = 3 cell culture wells). e, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells expressing indicated sgRNAs, incubated for 2 h with J774 macrophages and anti-CD20, anti-CD47, or human (hIgG1) or mouse (mIgG1) isotype control antibodies, normalized to signal in control (SafeKO) cells in anti-CD20 or mIgG1-isotype condition (n = 3 cell culture wells). f, Top, schematic of APMAP structure. Bottom left, APMAP homology model (residues 61–407) (Methods). Bottom right, magnified view of catalytic site of APMAP homology model, showing predicted positioning of residue E103 near the catalytic calcium ion. TMD, transmembrane domain. g, Phagocytosis assay for uptake of pHrodo-labelled indicated Ramos-Cas9 cells by J774 macrophages in the presence of anti-CD20 antibodies, normalized to control (SafeKO) cells (n = 3 cell culture wells). In b, d, e, g, data are mean ± s.d. One-way ANOVA (g) or two-way ANOVA (b, d, e) with Bonferroni correction.
APMAP contains a short cytosolic domain, a single transmembrane domain, and a predicted extracellular or endoplasmic reticulum-lumenal domain with sequence homology to the paraoxonase family of antioxidant enzymes23 (Fig. 2f). To assess the contributions of each domain to the function of APMAP in protecting cancer cells against phagocytosis, we transduced APMAPKO Ramos cells with constructs encoding a series of Flag-tagged APMAP alleles. Full-length Flag-tagged wild-type APMAP restored the ability of APMAPKO Ramos cells to suppress phagocytosis (Fig. 2g, Extended Data Fig. 5a, b). Truncation of the cytosolic domain, replacement of the transmembrane domain with that of unrelated endoplasmic reticulum proteins, or mutation of the two N-glycosylated residues of APMAP did not affect APMAP function in regulation of ADCP (Extended Data Fig. 5c–f). By contrast, mutation of a predicted calcium-binding residue expected to be required for catalysis on the basis of homology to PON1 (Fig. 2f) abrogated APMAP function in ADCP (Fig. 2g). Nonetheless, this mutant exhibited a similar expression level (Extended Data Fig. 5b) and localization pattern (Extended Data Fig. 5a) as wild-type APMAP. Thus, the predicted catalytic activity of APMAP, contained in its endoplasmic reticulum-lumenal or extracellular domain, is required for suppression of phagocytosis.
APMAP limits ADCP of diverse cancer cell lines
To investigate whether the role for APMAP in protecting cancer cells against phagocytosis is conserved in other tumour types, we examined the sensitivity to phagocytosis of eight additional cancer cell lines derived from diverse lineages, each of which expresses APMAP and CD47 at moderate levels (Extended Data Fig. 6a, b). For each line, we found expression of APMAP-targeting sgRNAs greatly sensitized cells to phagocytosis driven by anti-CD47 (Fig. 3a, Extended Data Fig. 6c, d). This is notable, as several of the cell lines (for example, HCT-116 and RKO) were highly resistant to CD47 blockade-induced phagocytosis24. Additionally, APMAP loss sensitized four EGFR+ cell lines to ADCP driven by anti-EGFR (cetuximab), including cell lines (NCI-H23 and OVCAR8) that were otherwise insensitive to cetuximab (Extended Data Fig. 6e, f).
Fig. 3 |. APMAP loss sensitizes diverse tumour types to monoclonal antibodies in vitro and in mice.
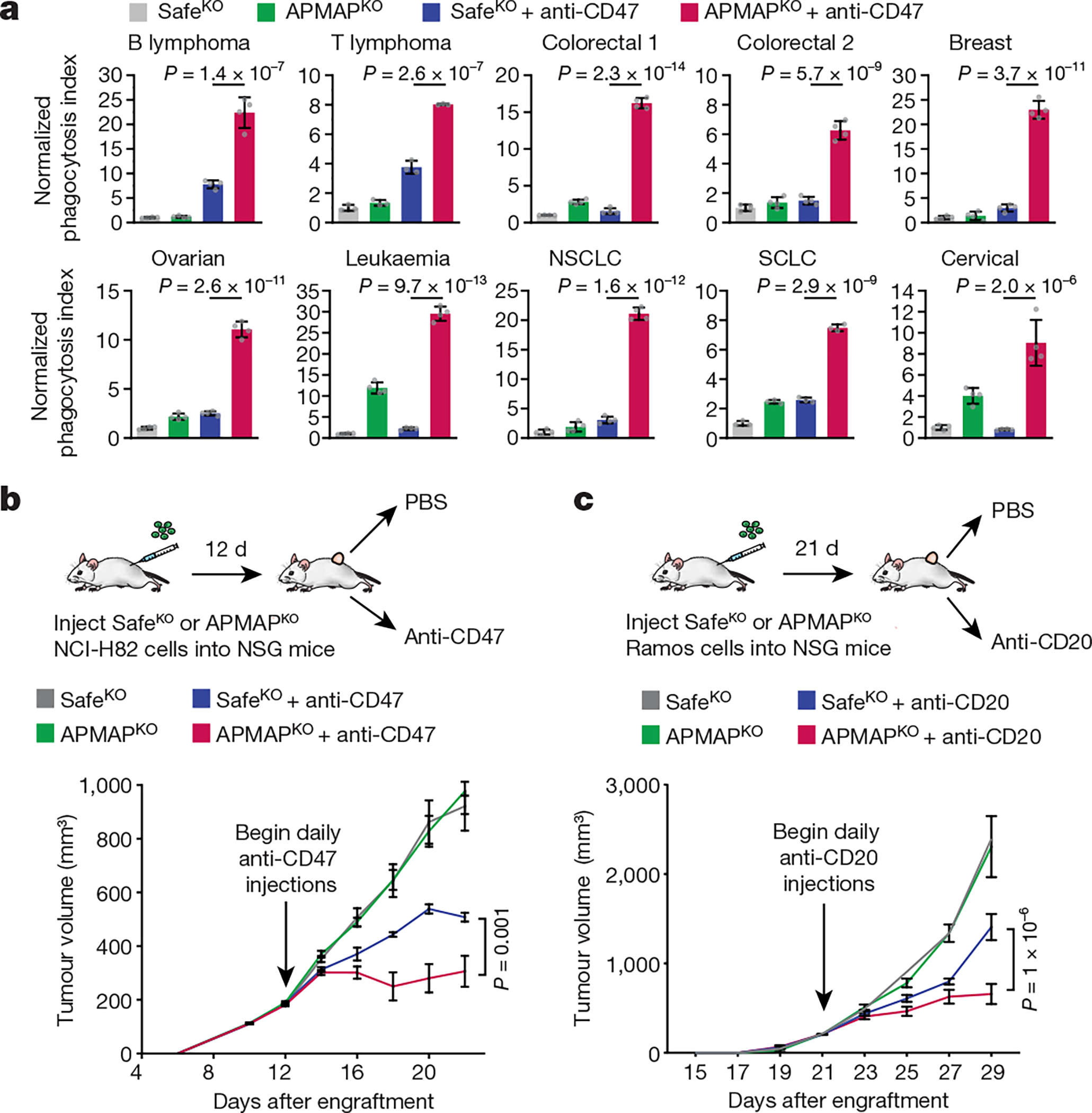
a, Phagocytosis assay for uptake of pHrodo-labelled cells for indicated Cas9-expressing cell lines—Ramos (B cell lymphoma), Karpas-299 (T cell lymphoma), HCT-116 (colorectal 1), RKO (colorectal 2), SKBR3 (breast), OVCAR8 (ovarian), K562 (leukaemia), NCI-H23 (non-small-cell lung carcinoma (NSCLC)), NCI-H82 (small-cell lung carcinoma (SCLC)) and HeLa (cervical)—expressing indicated sgRNAs by J774 macrophages in the presence or absence of anti-CD47 antibodies. Phagocytosis index normalized to control (SafeKO) cells without anti-CD47. Data are mean ± s.d. (n = 4 cell culture wells for all but Karpas-299 and NCI-H82, for which n = 3). One-way ANOVA with Bonferroni correction. b, SafeKO or APMAPKO NCI-H82 cells were transplanted into NSG mice and allowed to form tumours. Mice were treated with anti-CD47 or PBS daily starting 12 d after transplantation, and tumour size was measured every 2 d. Data are mean ± s.e.m. (n = 5). Two-way ANOVA with Tukey correction. c, SafeKO or APMAPKO Ramos cells were transplanted into NSG mice and allowed to form tumours. Mice were treated with anti-CD20 or PBS daily starting 21 d after transplantation, and tumour size was measured every 2 d. Data are mean ± s.e.m. (n = 6 (control groups) or 7 (antibody-treated groups)). Two-way ANOVA with Tukey correction.
APMAP synergy with tumour antigen antibodies
We examined the effect of APMAP loss on tumour growth in four mouse models. In the first two models, Ramos lymphoma or NCI-H82 small cell lung cancer cells were injected into the flanks of NSG mice, which lack adaptive immune cells but contain functional macrophages. After allowing tumours to develop, mice were treated with CD47-blocking antibodies or phosphate-buffered saline (PBS) control (Fig. 3b, Extended Data Fig. 6g). As expected, CD47 blockade inhibited development of both Ramos (Extended Data Fig. 6g–i) and NCI-H82 (Fig. 3b, Extended Data Fig. 6i) tumours. Of note, APMAP deficiency enhanced the effect of CD47 blockade on inhibition of the growth of both tumour types, but had no effect in the absence of CD47 blockade (Fig. 3b, Extended Data Fig. 6g). In a third model, we found that APMAP loss both increased macrophage infiltration (Extended Data Fig. 6j) and enhanced the effect of rituximab on Ramos tumours (Fig. 3c). Finally, we injected mouse B16-F10 melanoma cells into syngeneic C57BL/6 mice and treated with anti-TRP1 tumour-targeting antibodies25. Tumours lacking APMAP were significantly sensitized to anti-TRP1 treatment (Extended Data Fig. 6lk–m). Thus, while further studies are required to assess the therapeutic potential of targeting APMAP, these experiments demonstrated that APMAP loss enhances the effects of multiple tumour-targeting antibodies on inhibition of tumour growth in mice.
Macrophage GPR84 promotes ADCP of APMAPKO cells
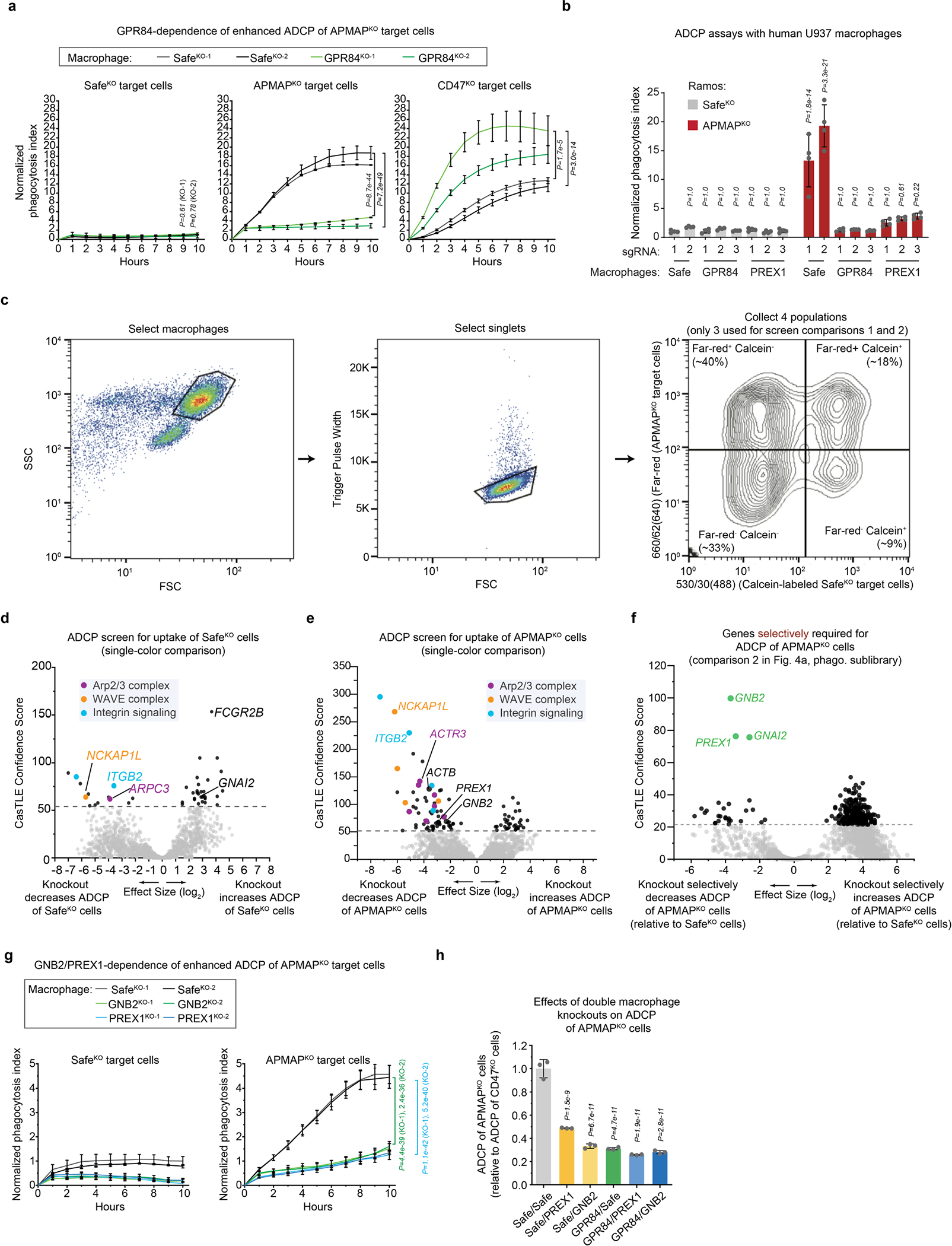
We hypothesized that APMAP loss enhances cancer cell uptake by modulating phagocytic signalling through a macrophage receptor. To identify candidate receptors, we conducted a complementary fluorescence-activated cell sorting (FACS)-based screen, this time with the CRISPR knockout library in macrophages, and used cancer cells as phagocytic targets to identify both general regulators of ADCP and specific mediators of enhanced ADCP of APMAPKO cells (Supplementary Note 4 on optimization of macrophage CRISPR screening, Extended Data Fig. 7, Supplementary Table 6). After incubating the genome-wide knockout macrophage pool with anti-CD20-opsonized SafeKO and APMAPKO Ramos cells (each bearing a distinct fluorescent label), we separated macrophages that had phagocytosed each target cell type (Fig. 4a). We deep sequenced the sgRNAs in each sorted population and performed two key comparisons. First, by comparing the double-negative and far-red single-positive populations, we identified genes required for uptake of APMAPKO cells, which included key actin-regulators and signalling factors required for phagocytosis (Fig. 4b, Supplementary Table 7). Second, we compared the two single-positive populations to identify genes selectively required for uptake of APMAPKO cells. This analysis revealed two genes required specifically for phagocytosis of APMAPKO cells: GPR84 and GNB2 (Fig. 4c, Supplementary Table 8). G-protein-coupled receptor 84 (GPR84) is a macrophage-specific26 pro-inflammatory G-protein-coupled receptor previously shown to stimulate phagocytosis when activated by lipid agonists27,28, and G protein subunit-β2 (GNB2) is a heterotrimeric Gβ protein subunit. Across several recent single-cell RNA-sequencing studies of human tumours, we found that GPR84 was expressed specifically in tumour-associated macrophages (Extended Data Fig. 8).
Fig. 4 |. GPR84 mediates enhanced uptake of APMAPKO cancer cells.
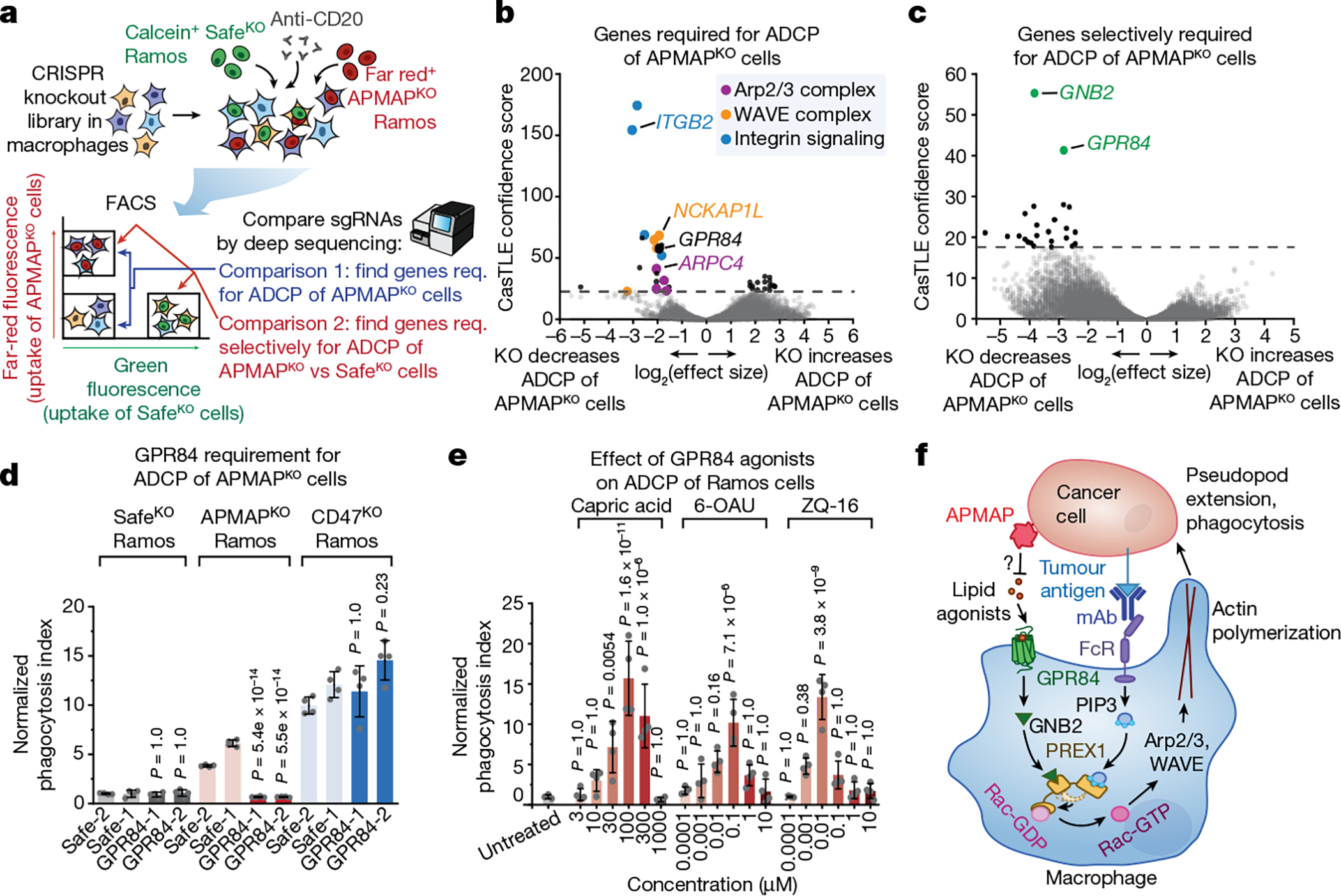
a, Schematic of macrophage screen, using a genome-wide CRISPR knockout library in J774 macrophages, for uptake of calcein+ SafeKO cells and far red+ APMAPKO Ramos cells. Req., required. b, Volcano plot of all genes required for uptake of APMAPKO cells (comparison 1 of screen depicted in a). c, Volcano plot of genes selectively required for uptake of APMAPKO cells relative to SafeKO cells (comparison 2 of screen depicted in a). d, Phagocytosis assay for uptake of pHrodo-labelled Ramos-Cas9 cells expressing indicated sgRNAs, incubated with J774 macrophages expressing indicated sgRNAs, in the presence of anti-CD20 antibodies, normalized to Safe-1 macrophages with SafeKO Ramos cells (n = 4 culture wells). P values are for comparisons with Safe-1 macrophages for each Ramos cell type. e, Phagocytosis assay for uptake of pHrodo-labelled SafeKO Ramos cells, incubated with U937 macrophages, anti-CD20 antibodies and indicated GPR84 agonists at indicated concentrations, normalized to untreated cells (n = 4 cell culture wells). P values are for comparisons with untreated cells. f, Model for APMAP-mediated regulation of macrophage phagocytosis through GPR84, GNB2 and PREX1. The role of PREX1 as a coincidence detector for Gβ and phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) in Rac activation is indicated; the Rac-GEF domain of PREX1 is synergistically released from an autoinhibited state upon binding of these two ligands at distinct sites. The question mark indicates a putative role of APMAP enzymatic activity in preventing GPR84-dependent phagocytosis activation. FcR, Fc receptor; mAb, monoclonal antibody. Dotted lines in b, c indicate 5% FDR. In d, e, data are mean ± s.d. One-way ANOVA with Bonferroni correction.
We validated that GPR84 is required for enhanced ADCP of APMAPKO cells using time-lapse imaging assays; deletion of GPR84 abolished enhanced uptake of APMAPKO cells by human or mouse macrophages, but did not diminish uptake of SafeKO cells or CD47KO cells (Fig. 4d, Extended Data Fig. 9a, b). To identify additional genes required for pro-phagocytic signalling downstream of GPR84, we conducted a higher-coverage screen using a 2,208-gene phagocytosis regulator-enriched sublibrary, which revealed several additional genes selectively required for uptake of APMAPKO cells: GNAI2, which encodes a Gαi subunit, and PREX1, which encodes phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchanger 1, a Rac guanine-exchange factor (GEF)29 (Extended Data Fig. 9c–f, Supplementary Table 8). We validated that both GNB2 and PREX1 are required for enhanced ADCP of APMAPKO cells (Extended Data Fig. 9g). Deletion of GNB2 or PREX1 in cells already lacking GPR84 only modestly decreased ADCP of APMAP-knockout cells, consistent with their function in a common pathway with GPR84 (Extended Data Fig. 9h). Stimulation of GPR84 using either the medium-chain fatty acid capric acid, which was previously reported to activate GPR8428, or two synthetic agonists, ZQ-16 and 6-OAU, stimulated antibody-dependent phagocytosis of SafeKO cells (Fig. 4e, Extended Data Fig. 10a), but not APMAPKO cells (Extended Data Fig. 10b). We observed inhibition of uptake in APMAPKO target cells at high concentrations of GPR84 agonists (Extended Data Fig. 10b), which may reflect GPR84 desensitization, as previously observed28. Finally, we tested whether GPR84 stimulation—like APMAP loss—exhibits synergy with monoclonal antibodies in driving cancer cell phagocytosis, and found that GPR84 agonists stimulated Ramos phagocytosis in the presence of either anti-CD20 (Extended Data Fig. 10c, d) or anti-CD47 (Extended Data Fig. 10e). By contrast, GPR84 agonists had no effect in the absence of monoclonal antibodies (Extended Data Fig. 10c–e) or on GPR84-knockout (GPR84KO) macrophages (Extended Data Fig. 10f). Together, our combined chemical and genetic analyses reveal an unexpected role for G-protein-coupled receptor signalling in enhanced ADCP of APMAP-deficient target cells.
Discussion
Monoclonal antibody therapies have been described as ‘magic bullets’ in cancer therapy because, in principle, they enable customizable elimination of cancer cells by macrophages, which are abundant in most tumours5,6,30. However, low phagocytic activity of macrophages in the tumour microenvironment and expression of anti-phagocytic factors by cancer cells remain key obstacles to fulfilling the promise of monoclonal antibody therapies in the treatment of diverse cancers31–34. Here we developed a functional genomic approach to study tumour–macrophage interactions by conducting complementary genome-wide screens for ADCP regulators in both cancer cells and macrophages. We identified a large number of genes that enhance or suppress ADCP, which help to illuminate the basic biology of macrophage–tumour interactions and may serve to identify novel potential therapeutic targets.
Our identification of APMAP as a cancer-intrinsic anti-phagocytic factor is notable because of the broad conservation of this function across different cancer types. We evaluated the effect of APMAP in 11 different cancer cell lines treated with 5 tumour-targeting antibodies, using both primary human macrophages and mouse tumour models; in each case we found that APMAP loss significantly enhanced the effects of tumour-targeting antibodies. Using complementary CRISPR screens in macrophages, we identified GPR84 as essential for enhanced uptake of APMAP-deficient cells. These findings raise the question of how GPR84 ‘recognizes’ APMAP-deficient cells. One possibility, suggested by homology between APMAP and the paraoxonase family of lipid hydrolases35,36, is that APMAP hydrolyses and/or prevents the accumulation of endogenous lipids that function as paracrine agonists of GPR84 (Fig. 4f, Supplementary Note 5). Our identification of the Rac-GEF PREX1 as an additional macrophage requirement for enhanced uptake of APMAPKO cells suggests the mechanistic basis for the synergy we observe between APMAP loss and monoclonal antibodies in ADCP. PREX1 is synergistically activated by Gβ proteins (a class of proteins that includes GNB2) and phosphatidylinositol-3,4,5-trisphosphate, which convey signals downstream of G-protein-coupled receptor and Fc receptor activation, respectively37, and may thus serve as a ‘coincidence detector’ that triggers Rac-induced actin polymerization only upon receipt of two distinct activating signals.
Although some immunosuppressive molecules inhibit multiple immune cell types, with, for example, CD47 found to confer resistance to T cell killing38,39, most of the genes we identify—including APMAP—were not detected by previous screens for regulators of T cell killing. For APMAP, this might be because GPR84 expression is restricted to macrophages26 and because the sensitizing effect of APMAP loss appears to be specific to monoclonal-antibody-induced cancer cell killing. A distinct advantage of our inter-cellular screening platform is that it facilitates identification of target-specific immune receptors. Our finding that GPR84 is a substrate-selective macrophage receptor for APMAP-deficient cancer cells provides a proof of principle for this approach, which may now be applied to other orphan anti-phagocytic factors. Overall, our work provides both a rich resource of candidate cancer-intrinsic regulators of phagocytosis and establishes a platform for rapidly identifying their cognate receptors on macrophages.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-021-03879-4.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment. Statistical analyses were performed as indicated in the legends using GraphPad PRISM (version 9).
Cell culture
Ramos, Karpas-299, NCI-H82 and K562 cells were maintained in suspension culture in T-150 flasks for library propagation and tissue culture plates for single-gene knockout lines, all in RPMI-1640 supplemented with 2 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% fetal calf serum (FCS). U937 cells were maintained in suspension culture in RPMI-1640 supplemented with 2 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% heat-inactivated FCS. J774 cells were cultured in 15-cm plates for screens and library preparation in DMEM supplemented with 2 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% heat-inactivated FCS and were passaged when nearing confluency by scraping. HCT-116, RKO, SKBR3 and NCI-H23 cells were cultured in RPMI supplemented with 2 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% FCS and passaged with Accutase. HeLa cells and OVCAR8 cells were cultured in DMEM supplemented with 2 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% FCS and passaged with Accutase. B16-F10 cells were cultured in DMEM supplemented with 2 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% FCS and passaged with trypsin. All cells were cultured in a humidified 37 °C incubator set at 5% CO2 and passaged 2–3 times weekly. To generate frozen aliquots, cells were pelleted by centrifugation (300g, 5 min, room temperature), suspended in 90% FCS and 10% dimethylsulfoxide (DMSO), and frozen in cell-freezing containers at −80 °C overnight before transfer to liquid nitrogen for long-term storage. Cell lines from ATCC were authenticated by the vendor using short tandem repeat (STR) profiling to distinguish between individual human cell lines and rule out intra-species contamination. No further authentication was performed. Cell lines were tested and confirmed negative for mycoplasma contamination by PCR.
Genome-wide and sublibrary CRISPR screens in Ramos cells
For the CRISPR knockout screen in Ramos cells, a genome-wide, 10 sgRNA per gene CRISPR deletion library was synthesized, cloned and infected into Cas9-expressing Ramos cells as previously described40. In brief, about 300 million Ramos cells stably expressing SFFV-Cas9-BFP were infected with the CRISPR knockout library at a multiplicity of infection (MOI) of about 0.2. Cells expressing sgRNAs were selected using puromycin (1 μg ml−1) for 3 d such that >90% of cells were mCherry-positive as measured by flow cytometry. Selected cells were then allowed to recover and expand in puromycin-free medium for up to 7 d. For the CRISPRa screen, a clonal dCas9–VPR-expressing line was first constructed by transducing wild-type Ramos cells with a lentiviral construct expressing dCas9–VPR-t2a-GFP. Cells expressing low levels of GFP were sorted into single-cell clones and active clones were identified on the basis of induction of CD2 expression following transduction with an sgRNA targeting the CD2 transcription start site. One of the active clones was selected for subsequent experiments based on the degree of CD2 induction as well as its similar growth rate and phagocytosis susceptibility compared to the parental cell population. As with the CRISPR knockout screen, a genome-wide, 10-sgRNA-per-gene library41 was synthesized, cloned and infected into CRISPRa-expressing Ramos cells and selected with puromycin as above.
For the screens, cells were split into two conditions, each in duplicate: an untreated control group and an ADCP-treated group. In the ADCP group, for each round of treatment, Ramos cells were incubated with J774 macrophages, which had been seeded into 15-cm plates 48 h earlier (at 5 million cells per plate) and treated with lipopolysaccharide (LPS) (100 ng ml−1) starting 24 h earlier, in the presence of anti-CD20 antibodies (hcd20-mab1, Invivogen) (50 ng ml−1) (CRISPR knockout screen) or both anti-CD20 antibodies (50 ng ml−1) and anti-CD47 antibodies (clone B6.H12, BioXCell) (100 ng ml−1) (CRISPRa screen). 25 million Ramos cells were added to each plate and incubated with J774 macrophages for 24 h, and were then removed from the macrophage-containing plates via two rounds of washing with complete RPMI medium. Ramos cells were then allowed to recover in T-150 flasks for 24–72 h before the next round of treatment. At the end of the screen, 300 million cells were recovered from each condition and pelleted by centrifugation. Genomic DNA of each condition was extracted using a DNA Blood Maxi kit (Qiagen, catalogue no. 51194). The sgRNA sequences were amplified and sequenced using an Illumina NextSeq with about 40 million reads per condition; about 200× coverage per library element). Analysis and comparison of guide composition of ADCP treated versus untreated conditions were performed using casTLE as previously described42. The complete genome-wide Ramos screen results are presented in Supplementary Tables 2, 4. Sublibrary screens in Extended Data Fig. 3a–c were conducted similarly, but in the indicated genetic backgrounds and with indicated antibodies, and using a 3,124-gene sublibrary enriched for transmembrane proteins. Batch re-test screens were conducted similarly, but at 2,000× coverage of the library, and using a 730-gene sublibrary that contained the top 250 genes from the Ramos CRISPR knockout screen and 480 genes with positive effect size from the Ramos CRISPRa screen. In the batch re-test screen, because the library was highly enriched for hits, genes were noted as hits when their combination effect score at 95% credible interval did not include zero, as described previously43,44. Sublibraries were synthesized as previously described40. The complete sublibrary Ramos CRISPR screen results are presented in Supplementary Tables 3, 5.
Genome-wide CRISPR screens in Karpas-299 cells
Genome-wide CRISPR knockout screens in Karpas-299 cells were performed similarly to the genome-wide screens in Ramos cells. In Extended Data Fig. 2i, anti-CD30 (clone Hu8, R and D Systems) was included in place of anti-CD20, and in Fig. 2c, anti-CD47 (clone B6.H12) was included in place of anti-CD20.
Genome-wide and sublibrary macrophage screens
For the CRISPR knockout screen in J774 cells, a genome-wide, 10-sgRNA-per-gene CRISPR mouse deletion library was synthesized and cloned as previously described40 and infected into Cas9-expressing J774 cells. Following puromycin selection, the genome-wide library was propagated for 7–10 d before use in screens. For the genome-wide bead screen in Extended Data Fig. 7, J774 macrophages were plated in 15-cm tissue culture dishes at a starting density of 10 million cells per plate. After 24 h, macrophages were incubated with Protein G Dynabeads (about 2 beads per cell, Thermo Fisher) that were opsonized by successive incubations with mouse anti-Flag (Sigma) and goat anti-mouse antibodies (Thermo Fisher Scientific). Cells were lifted by scraping, washed in PBS, and exposed to a DynaMag magnet (Thermo Fisher Scientific). After a series of four washes, magnet-bound cells were collected and collected for genomic DNA extraction. The unbound fraction and the first wash were collected and re-exposed to the magnet for 10 min before collection for genomic DNA extraction. PCRs, sequencing and hit identification (by comparing the bound and unbound cell fractions) with CasTLE were performed as previously described45. For the genome-wide ADCP screen in Fig. 4, macrophages were plated in 15-cm tissue culture dishes at a starting density of 5 million cells per plate. After 24 h, medium was replaced with DMEM containing 100 ng ml−1 LPS. After a further 24 h, macrophages were incubated with a mixture of calcein-labelled SafeKO Ramos cells and CellTrace far-red-labelled APMAPKO macrophages for 24 h. Unphagocytosed Ramos cells were removed by extensive washing with PBS. Macrophages were lifted by scraping, washed twice in PBS, concentrated by centrifugation, and separated into four populations on an Aria (BD) flow cytometer. In total, about 80 million cells were collected for each replicate across the 4 gated populations and used for genomic DNA extractions, PCRs, next-generation sequencing and subsequent hit identification using CasTLE, as above. The complete genome-wide screen results are presented in Supplementary Tables 6, 7. Sublibrary screens in Extended Data Fig. 9 were conducted similarly, but using a 2,208-gene sublibrary enriched for phagocytosis regulators, derived from the J774 IgG bead genome-wide screen hits and the literature. The complete sublibrary J774 CRISPRko FACS screen results are presented in Supplementary Table 8.
Gene ontology enrichment analysis
Gene ontology enrichment analyses were performed using gProfiler46 (version e.96_eg.43) as ordered queries.
Generation of cell lines
For generating individual sgRNA-expressing cell lines, Ramos, J774, U937, Karpas-299, HeLa, HCT-116, NCI-H23, RKO, K562, SKBR3 and NCI-H82 cells that express Cas9 or dCas9–VPR were infected with lentiviral constructs expressing a given sgRNA along with puromycin or blasticidin resistance cassette. At 3 d after infection, cells were selected with 1–2 μg ml−1 puromycin or 10 μg ml−1 blasticidin for an additional 3 d, and allowed to recover for 2–3 d before use in assays. B16-F10 knockout cells were constructed by transfecting cells with two plasmids expressing Cas9–BFP and sgRNA/GFP and isolating GFP/BFP-positive cells by FACS. Sequences of sgRNAs used for validation are listed in Supplementary Table 9.
Plasmids
To generate APMAP expression constructs to reconstitute in APMAP knockout cell lines, the human APMAP open reading frame, with a 3×Flag tag appended to the C terminus, and with synonymous mutations to block targeting with APMAP-targeting sgRNAs, was synthesized (Twist Biosciences) and cloned into a pCR-Blunt backbone vector. Point mutations (E103A in Fig 2g, N160A and N196A in Extended Data Fig. 5e, f) were installed using site-directed mutagenesis, and the APMAP–Flag region was then subcloned into pMCB394, a lentiviral expression vector. For the Δcyto truncation in Extended Data Fig. 5c, the region encoding APMAP residues 62–416 was amplified with a new start codon and cloned into pMCB394. The chimeric constructs in Extended Data Fig. 5d were created by amplifying the regions encoding the cytoplasmic and transmembrane domains of two single-pass transmembrane proteins with similar topology to APMAP–POMK (residues 1–43) and TFRC (residues 1–88, with the first 6 residues encoded in the primer to change them from the endogenous sequence to MSRRRS, mutations that were shown to retain TFRC primarily in the endoplasmic reticulum47) and fusing them via overlap extension PCR to the region encoding APMAP residues 62–416. The TFRCRR allele was used in place of TFRCWT, as the TFRCWT–APMAP–Flag chimera was not detectable by western blotting.
Immunoblotting
Cleared cell extracts prepared in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1× cOmplete protease inhibitor cocktail (Roche) were heated in SDS loading buffer and separated by SDS–PAGE, transferred to nitrocellulose, blotted and imaged using an Odyssey CLx (LI-COR Biosciences) or Supersignal West Femto Maximum Sensitivity Substrate with a Chemidoc System (Bio-Rad). In Extended Data Fig. 6a, cell pellets were resupended directly in SDS loading buffer, sonicated, and separated by SDS–PAGE. In Extended Data Fig. 5e, extracts were first treated with PNGase F (NEB) following the manufacturer’s instructions. The following antibodies were used: mouse monoclonal anti-APMAP (OTI4F6, Origene, 1:2,000 dilution), mouse monoclonal anti-Flag (clone M2, F1804, Sigma, 1:2,000 dilution), mouse monoclonal anti-GAPDH (clone 6C5, AM4300, Fisher, 1:5,000 dilution), and rabbit polyclonal anti-β-actin (ab8227, Abcam, 1:2,000 dilution).
Flow cytometry
Ramos cells were analysed by flow cytometry using the following antibodies: anti-CD20-APC (clone 2H7, Biolegend), anti-CD2-APC (clone REA972, Mitenyi Biotech), anti-CD47 (clone B6.H12, BioXCell), and anti-Calreticulin-DyLight-488 (FMC 75, Enzo Life Sciences), and with Annexin-V FITC (A13199, Thermo Fisher), or with biotinylated Maackia amurensis lectin II (MAL-II) (B-1265, VectorLabs) followed by streptavidin-APC (Thermo Fisher). Sialidase (neuraminidase from Vibrio cholerae, Sigma 11080725001) treatment was performed by incubating cells with 30 nM enzyme in dPBS at a cell concentration of 1 × 106 ml−1 for 1 h at 37 °C. In Extended Data Fig. 6j, single-cell suspensions were prepared from diced tumours and fixed in paraformaldehyde as described previously. Cells were stained with Zombie-NIR viability dye (BioLegend), anti-CD45 (clone 30-F11, BioLegend), anti-F4/80 (clone BM8, BioLegend) and anti-CD11b (clone M1/70, BioLegend) and analysed using an Acea Novocyte Quanteon flow cytometer and FlowJo software (version 10.6.1). Live CD45+ cells were counted using gates that separated the CD45− and CD45+ populations, and macrophages were counted using a single gate applied equally to all samples.
Isolation of human peripheral blood mononuclear cell derived macrophages
Primary human donor-derived macrophages were isolated and cultured following a previously described protocol48 with minor modifications. Leukocyte reduction chambers were obtained from de-identified healthy donors from the Stanford Blood Center. The protocol was approved by the Stanford University Administrative Panel on Human Subjects in Medical Research and all donors provided informed consent. Cells were separated using Ficoll–Paque gradient centrifugation as previously described48. Monocytes were then isolated via their adhesion to tissue culture plastic in serum-free RPMI-1640, and subsequently differentiated into macrophages in RPMI-1640 containing 20% heat-inactivated FCS containing 20 ng ml−1 human M-CSF (Peprotech) for 5 d, then lifted with Accutase and manual scraping, and allowed to adhere overnight in 24 well plates. Macrophages were then treated for 48 h with 100 ng ml−1 LPS before use in phagocytosis assays.
Time-lapse microscopy assay for phagocytosis
For use in phagocytosis assays, U937 cells were cultured and differentiated with phorbol myristate acetate (PMA) as described previously45, and then plated for 3 d after initiating differentiation at a density of 100,000 cells per well in 24-well tissue culture plates, then treated with 100 ng ml−1 LPS (Sigma) for 48 h before use in assays. J774 cells were lifted by scraping, counted, and plated in 24-well tissue culture plates at a density of 100,000 cells per well in 0.5 ml medium 48 h before the start of the experiment. At 24 h after plating, medium was aspirated and replaced with fresh medium containing 100 ng ml−1 LPS (Sigma) (that is, in all instances other than Extended Data Figs. 1c, 4d (left)). On the day of the experiment, target cells that had been transduced with sgRNAs and selected with puromycin or blasticidin as described above were counted, washed three times in dPBS before incubation in dPBS containing 100 nM pHrodo-Red succinimidyl ester (Thermo Fisher) at a cell concentration of 1 million ml−1 for 30 min at 37 °C, pelleted, and resuspended in DMEM supplemented with 2 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 10% heat-inactivated FCS, and containing anti-CD20, anti-CD30, anti-EGFR (clone Hu1, R and D Systems), anti-CD47 antibodies, anti-TRP1 (clone TA99, BioXCell), and/or isotype control antibodies, as indicated, at a cell concentration of 1 × 106 ml−1. Antibodies were used at the following concentrations in ADCP assays unless otherwise indicated: anti-CD20 (500 ng ml−1), anti-CD30 (500 ng ml−1), anti-EGFR (500 ng ml−1), anti-CD47 (10 μg ml−1), anti-TRP1 (10 μg ml−1). Cell suspensions (200,000–500,000 cells per well of a 24-well plate for suspension cell lines, or 50,000 cells per well for adherent cell lines) were then added to each well and allowed to settle. Where indicated, GPR84 agonists (capric acid (C1875, Sigma), 6-OAU (17687, Cayman), ZQ-16 (SML1646, Sigma)) or other compounds (acetic acid (BP2400500, Fisher), palmitic acid (10006627, Cayman), docosanoic acid (9000338, Cayman)) were included at indicated concentrations. In Extended Data Fig. 3e, where indicated, macrophages were pre-incubated with mouse Fc receptor-blocking antibodies (50 μg ml−1,101319, BioLegend) for 45 min at 4 °C before addition of target cells. Plates were transferred to an incubator and imaged every 30 or 60 min using an Incucyte (Essen). Total red intensity for each well, averaged over 9–16 images per well, was calculated after applying a threshold for red intensity that excluded most or all cells at the first time point using top-hat background subtraction. Reported values represent the mean total red fluorescence intensity at each timepoint (Fig. 1f, 5 h; Fig. 2b, 6.5 h; Fig. 2e, 3 h; Fig. 2g, 14 h; Fig. 3a: Ramos, 4.5 h; Karpas-299, 4 h; HCT-116, 3.5 h; RKO, 3 h; SKBR3, 6 h; OVCAR8, 20 h; K562, 3 h; NCI-H23, 8 h; NCI-H82, 4 h; HeLa; 3h; Fig. 4d, 8 h; Fig. 4e, 9 h; Extended Data Fig. 3d, 3.5 h; Extended Data Fig. 3e, 4 h; Extended Data Fig. 3f, 3 h; Extended Data Fig. 4b, 12 h; Extended Data Fig. 4c, 6 h; Extended Data Fig. 4d, 2 h; Extended Data Fig. 5c, 4 h; Extended Data Fig. 5d, 7.5 h; Extended Data Fig. 5f, 4 h; Extended Data Fig. 6d: 8 h; Extended Data Fig. 6e: HCT-116, 6 h, OVCAR8, 8 h, NCI-H23, 7 h, HeLa 4 h; Extended Data Fig. 6k, 6 h; Extended Data Fig. 9b, 24 h; Extended Data Fig. 10a, b, 2.5 h; Extended Data Fig. 10c, 5 h; Extended Data Fig. 10d, e, 4 h; Extended Data Fig. 10f, 2 h. In some panels with GFP+ target cells, red intensity was normalized to total GFP signal at t = 0 h to account for minor variation in cell abundance, and the total red fluorescence at the initial timepoint was subtracted (to remove signal from occasional autofluorescent debris), of duplicate, triplicate or quadruplicate cell culture wells, as indicated in the legend.
Flow cytometry assays for phagocytosis
In Extended Data Fig. 4l, Ramos target cells were stained with calcein, AM (500 ng ml−1, Thermo Fisher) or CellTrace Far Red dyes (1:2,000 dilution of stock prepared according to the manufacturer’s instructions, C34564, Thermo Fisher) for 10 min in PBS at 1 × 106 cells per ml, washed twice with DMEM containing 10% FCS, and incubated for 24 h with LPS-treated J774 macrophages prior to analysis by flow cytometry (BD Accuri C6). In Extended Data Fig. 9h, Ramos CD47KO target cells were stained with calcein, AM (500 ng ml−1, Thermo Fisher) and APMAPKO cells were stained with CellTrace Far Red dyes (1:2,000 dilution of stock prepared according to the manufacturer’s instructions) for 10 min in PBS at 1 × 106 cells per ml, washed twice with DMEM containing 10% FCS, mixed together with anti-CD20 (500 ng μl−1), and incubated for 24 h with LPS-treated J774 macrophages before analysis by flow cytometry (BD Accuri C6).
Target cell–macrophage adhesion assay
To measure antibody-dependent binding between J774 macrophages and Ramos target cells, macrophages were plated in 24 well tissue culture plates at a density of 100,000 cells per well in 0.5 ml medium 48 h before the start of the experiment and stimulated with LPS 24 h before the experiment as above. On the day of the experiment, medium was aspirated and replaced with DMEM containing 1 μg ml−1 cytochalasin D (Sigma C8273), and incubated for 10 min at room temperature. Ramos cells (200–500,000) were then added to wells and incubated for 2 h at 37 °C. Plates were imaged before and after three washes with PBS to determine the abundance of GFP+ Ramos cells (as measured by integrated green intensity) using an Incucyte (Essen) using a 10× objective.
Cell survival analyses
To measure cell survival during co-culture with macrophages, two methods were used. In Extended Data Fig. 1j, mCherry+ Ramos cells expressing indicated sgRNAs were mixed with an equal number of GFP+ Ramos control cells, and incubated with LPS-treated J774 macrophages in the presence of anti-CD20. The percentage of mCherry+ Ramos cells out of all live Ramos cells was quantified for each cell line, and normalized to the percentage of mCherry+ Ramos control cells, which expressed a non-targeting sgRNA. In Extended Data Fig. 6c, f, survival was measured by quantifying the total GFP signal per well before and after incubation of GFP+ target cells with LPS-treated J774 macrophages (GFP fluorescence is quenched following uptake of target cells and delivery to the lysosome).
Transcriptome analysis
J774 cells were seeded in triplicate at a density 1 × 106 cells per 10-cm plate. Twenty-four hours later, cells were either collected (untreated condition) or medium was replaced with medium containing 100 ng ml−1 LPS for 24 h. At collection, cells were lysed in RLT buffer and RNA was isolated using the RNeasy Micro Kit (Qiagen). cDNA libraries were prepared using a TruSeq Stranded mRNA Kit (Illumina) and sequenced using an Illumina NextSeq with about 25 million reads per condition. Transcripts were mapped using STAR (v2.7.0)49 and gene-level counts were generated using HTSeq (v.0.13.5)50, followed by differential gene expression analysis using DESeq2 (1.28.1)51.
Viability measurements
In Extended Data Fig. 4i, Ramos cells were plated in 96-well plates in triplicate at a starting density of 50,000 cells per well. Drugs (Sigma) were then added to wells at indicated final concentrations. Cell viability was determined by measuring the phase confluence at 72 h using an Incucyte (Essen). In Extended Data Fig. 6l, B16-F10 cells were seeded in 24-well plates and phase confluence was measured every 8 h for 6 d.
Confocal microscopy
HeLa cells were transduced with a lentiviral construct co-expressing APMAP–Flag and BlastR, selected with blasticidin and cultured in glass-bottom 24-well plates. Cells were fixed with paraformaldehyde and probed with anti-Flag (clone M2, Sigma F1804, 1:100 dilution) and anti-calnexin (Abcam Technologies ab22595, 1:1,000 dilution) primary antibodies, and Alexa Fluor 488 goat anti-mouse (A-11029, Thermo Fisher) and APC goat-anti-rabbit (A-10931, Thermo Fisher) secondary antibodies, and imaged using an inverted Nikon Eclipse Ti-E spinning disk confocal microscope using NIS-Elements software (v4.4, Nikon) and an Andor Ixon3 EMCCD camera using an oil-immersion 100× objective (NA 1.45). Images were assembled and adjusted for brightness and contrast in Photoshop CS6 (Adobe).
Analysis of differential expression in TCGA
Differential gene expression in 23 TCGA tumour studies52 was calculated for each gene by comparing gene expression in each tumour study to normal tissue and calculating the fold change with DESeq2.
Analysis of single-cell RNA sequencing studies
Analysis of GPR84 expression in human tumour samples was performed using four single-cell RNA-sequencing datasets53–56. scRNAseq pre-processing, cell type assignment, t-distributed stochastic neighbour embedding (t-SNE) (Extended Data Fig. 8a–d), and gene expression quantification were performed as described previously53–56.
Homology model of APMAP
Using Swiss-Model57 (template library SMTL version 2020–07-22, PDB release 2020–07-17) with Uniprot accession code for APMAP (Q9DHC9) as input, 50 template structures were retrieved up to a GMQE* score of 0.25. A parallel search was done using FUGUE58 (version 2.0). Swiss-Model was used with strictosidine synthase from Rauvolfia serpentina (STR1; sequence similarity = 34%; PDB accession ID: 6nv5, unpublished) and serum paraoxonase-1 (PON1; sequence similarity = 29 %; PDB accession ID: 3sre59) first as the highest scoring template structures. Modelling was done on the soluble region, residues 61–407 of APMAP, using the PON1 structure. The automatically generated model was completed by assigning it the 30 N-terminal residues from Drp35 from Staphylococcus aureus (PDB accession ID: 2dg160). The modelling resulted in a 6-blade beta-propeller structure, which was manually inspected in COOT61 (version 0.7 (revision 4459)), and the coordinates refined against reference parameters with REFMAC562 (version 5.8.0135) until deviation from ideal bond length, bond angle, planar restraints, chiral volume, reached convergence and Ramachandran outliers were minimized (1.7% (5 out of 347) of the total residues were outliers).
Mouse tumour xenografts
For mouse tumour experiments, tumour xenografts were established by injection of SafeKO and APMAPKO Ramos or NCI-H82 cells into the flank of male and female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice, purchased at 8–12 weeks of age (Jackson Laboratories). In brief, 4 × 106 Ramos cells or 1 × 106 NCI-H82 cells were mixed with Matrigel (1:1, BD Biosciences) and injected into mouse flanks. Tumours were allowed to form for 12–21 d, and then mice were injected intraperitoneally with PBS or antibody daily (anti-CD47: 400 μg; B6H12, BioXCell; anti-CD20 (Rituxan): 200 μg, Stanford Pharmacy), and tumour size was measured using calipers. In the NCI-H82 xenograft experiment, tumours in all groups were subjected to irradiation (3 Gy/1 fr) on day 12. Animal irradiation was performed using a PXi X-rad SmART cabinet irradiator (Precision X-ray). Tumour size was measured once every 2 days until the end of the experiment. The number of mice represented in the final analysis was 5–7 for all groups. No statistical tests were used to calculate sample size. Following injection of SafeKO or APMAPKO tumour cells into 14–17 mice, the mice that failed to develop tumours were excluded, and the remaining mice were randomly assigned into two treatment groups for PBS or antibody injection. Fold change in tumour volume was statistically analysed for indicated comparisons using two-way ANOVA with Tukey’s correction. Blinding was not possible because the experiments were performed by a single researcher. All mouse experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committees (IACUC) of Stanford University. Mice were euthanized once the largest xenograft tumours reached 1,500 mm3. No mouse exhibited severe loss of body weight (>15%) or evidence of infections or wounds. Full data are included as Source Data.
Syngeneic mouse model
For syngeneic mouse tumour experiments, tumours were established by injection of either SafeKO or APMAPKO B16-F10 cells into the flank of 8- to 12-week-old female C57BL/6J mice (Jackson Laboratories). In brief, 5 × 105 cells were injected in a 100 μl suspension, consisting of 25% Matrigel Basement Membrane Matrix (Corning) and 75% RPMI (Life Technologies). Tumours were established for 5 d, and on day 5, mice were randomized into treatment and isotype control groups. Starting on Day 5 post-engraftment, mice were injected intraperitoneally with either anti-TRP1 (TA99) or isotype control (mouse IgG2a) antibodies (BioXCell, 250 μg) every other day, following a dosing regimen used previously63. Engraftment outliers were removed as determined by Graphpad Prism Outlier Calculator. Tumour size was measured in two dimensions using precision calipers twice weekly for the duration of the experiment. Tumour volumes were calculated by approximating tumours as ellipsoids with two radii, x and y, in which x is the largest measurable dimension of the tumour and y is the dimension immediately perpendicular to x, using the formula: volume = 4/3π × (x/2)2 × (y/2). The number of mice represented in the final analysis was 6–7 for all groups. No statistical tests were used to calculate sample size. Starting sample size was n = 7–8 for each treatment group to account for differences in tumour engraftment and growth, and to ensure recovery of a sufficient quantity of mice with tumours of approved size at each time point of the study. Data are presented up to day 20 (when sufficient mice were still alive to analyse the full randomized cohort), and full data (until day 25) are included as Source Data. Change in tumour volume was evaluated using two-way ANOVA in GraphPad Prism. Blinding was not possible because the experiments were primarily performed by a single researcher. All mouse experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committees (IACUC) of Stanford University. Mice were euthanized once several of the largest xenograft tumours had reached 2,000 mm3. No mouse exhibited severe loss of body weight (>15%) or evidence of infections or wounds. Full data are included as Source Data.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
CRISPR screen and RNA-seq raw sequencing data are available under BioProject accession number PRJNA748551. All other primary data for all figures and supplementary figures are available from the corresponding author upon request. Gene dependency data from the Cancer Dependency Map are publicly available at www.depmap.org. Cancer expression data from The Cancer Genome Atlas are available at https://gdc.cancer.gov. CCLE data are available at https://sites.broadinstitute.org/ccle/. Source data are provided with this paper.
Extended Data
Extended Data Fig. 1 |. CRISPR knockout screening platform for regulators of ADCP in cancer cells and batch retest validation screen results.
a, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells by J774 macrophages in the presence or absence of anti-CD20 and/or anti-CD47 antibodies. Normalized phagocytosis index was calculated as average total pHrodo Red signal per well, normalized to signal in untreated control condition at final timepoint. Data represent mean +/− s.d. (n = 4). Two-way ANOVA with Bonferroni correction. b, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells by J774 or U937 macrophages in the presence of anti-CD20. Normalized phagocytosis index was calculated as average total pHrodo Red signal per well, normalized to signal in U937 cells at final timepoint. Data represent mean +/− s.d. (n = 3). Two-way ANOVA with Bonferroni correction. c, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells by J774 macrophages with or without 24 h pre-treatment with 100 ng ml-1 LPS. Normalized phagocytosis index was calculated as average total pHrodo Red signal per well, normalized to signal in untreated control condition at final timepoint. Data represent mean +/− s.d. (n = 4). Two-way ANOVA with Bonferroni correction. d, Differential expression analysis of J774 macrophages before and after treatment (24 h) with 100 ng ml-1 LPS, showing induction of classic LPS-activated M1 macrophage markers NOS2 and IL1B. e, Replicates for CRISPRko screen in Ramos cells for susceptibility to ADCP driven by anti-CD20 antibodies. f, Gene ontology enrichment analysis for negative Ramos CRISPRko ADCP hits (cutoff of CasTLE score > 50). n indicates number of genes among query gene list annotated with indicated term. g, Batch re-test screen for ADCP sensitivity in Ramos Cas9 cells. Library comprised top 250 hits (both positive and negative effect sizes) from genome-wide CRISPRko screen and top 480 anti-phagocytic hits from CRISPRa screen. Hits were defined based on 95% confidence interval of CasTLE effect size (see Supplementary Table 3). h, Replicates of Batch re-test screen in Ramos Cas9 cells. i, Comparison of Ramos batch re-test and genome-wide ADCP CRISPRko screens for genes that were hits in the CRISPRko screen. j, Survival assay for Ramos Cas9 cells subjected to treatment with macrophages and anti-CD20, expressing indicated sgRNAs (2 distinct sgRNAs per gene). GFP+ Ramos Cas9 cells expressing negative control sgRNA were mixed with an equal number of mCherry+ cells expressing indicated sgRNAs and cultured in the presence of J774 macrophages and anti-CD20 antibodies. Plotted is the mean percentage of surviving Ramos cells that were mCherry+ after 2 d, normalized to control (Ctrl) Ramos cells that expressed an empty vector) (n = 3 cell culture wells, data represent mean +/− s.d.). One-way ANOVA with Bonferroni correction.
Extended Data Fig. 2 |. CRISPR activation screening platform development and analysis of anti-phagocytic hits.
a, Validation of Ramos CRISPRa clones. Single-cell derived CRISPRa clones were constructed as described in the Methods and transduced with sgRNAs targeting CD2 (using a lentiviral vector co-expressing GFP). Indicated clones and parent Ramos cells were stained with anti-CD2-APC antibodies. Mean APC signal in the GFP+ population is plotted (n = 2 technical replicates, mean is shown). Clone #6 was used for screening. b, Replicates for CRISPRa screen in Ramos cells for susceptibility to ADCP driven by anti-CD20 and anti-CD47 antibodies. c, Gene ontology enrichment analysis for positive Ramos CRISPRa ADCP hits (cutoff of CasTLE score > 50) (top) and top 50 anti-phagocytic factors (bottom). n indicates number of genes among query gene list annotated with that term. d, Schematic of time-lapse imaging assay for ADCP. pHrodo-Red fluorescence intensity increases in low-pH conditions, such as in the lysosome following internalization of the target cell. e, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells, stably expressing indicated constructs, by J774 macrophages in the presence of anti-CD20 and anti-CD47 antibodies. Normalized phagocytosis index was calculated as average total pHrodo Red signal per well, normalized to signal in GFP-FLAG cells at final timepoint. Data represent mean +/− s.d. (n = 4 cell culture wells). Two-way ANOVA with Bonferroni correction. f, Expression (TPM) of SMAGP in 1304 cell lines in CCLE. g, h, Flow cytometry assays for anti-CD20 and anti-CD45 binding to Ramos CRISPRa cells expressing indicated sgRNAs. Data represent mean +/− s.d. (n = 3 independently stained samples). One-way ANOVA with Bonferroni correction. i, Volcano plot of screen in Karpas-299 cells conducted in presence of anti-CD30 antibodies. Dotted line indicates 5% FDR. j, Heatmap of differential expression for 12 selected anti-phagocytic genes in 23 tumour types compared to normal tissue. Tumour type abbreviations are listed here: https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations. k, Minimum expression (TPM) across all cell lines in CCLE is plotted against maximum probability of essentiality in all cell lines profiled in DepMap for 50 top anti-phagocytic hits shown in Fig. 1e.
Extended Data Fig. 3 |. Screens for cancer cell regulators of ADCP in the presence or absence of CD47 and evaluation of importance of antibodies and Fc receptor for APMAP effect.
a, Schematic and volcano plot of CRISPR screen in Ramos Cas9 cells for sensitivity to macrophage phagocytosis in the presence of anti-CD20 in cells expressing an sgRNA targeting a Safe locus. Dotted line indicates 5% FDR. A transmembrane gene-enriched sublibrary containing 3,124 genes was used. b, Schematic and volcano plot of CRISPR screen in Ramos Cas9 cells for sensitivity to macrophage phagocytosis in the presence of anti-CD20 in cells expressing an sgRNA targeting the CD47 locus. Dotted line indicates 5% FDR. A transmembrane gene-enriched sublibrary containing 3,124 genes was used. c, Schematic and volcano plot of CRISPRko screen in Ramos Cas9 cells for sensitivity to macrophage phagocytosis in the presence of anti-CD20 and anti-CD47 in cells expressing an sgRNA targeting a Safe locus. Dotted line indicates 5% FDR. A transmembrane gene-enriched sublibrary containing 3,124 genes was used. d, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes by human primary peripheral blood-derived macrophages, from two independent healthy de-identified human donors, in the presence or absence of anti-CD47 antibodies. Phagocytosis index normalized to control (SafeKO) cells without anti-CD47. Data represent mean +/− s.d. (n = 4 cell culture wells). One-way ANOVA with Bonferroni correction. e, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes by J774 macrophages in the presence of anti-CD20 or anti-CD47 antibodies. Where indicated, J774 macrophages were pre-incubated with Fc-blocking antibodies for 45 min on ice. Phagocytosis index normalized to control (SafeKO) cells without antibody analysed in parallel (condition not shown). Data represent mean +/− s.d. (n = 3 cell culture wells). Two-way ANOVA with Bonferroni correction. f, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes by J774 macrophages in the absence of antibodies. Phagocytosis index normalized to control (SafeKO/SafeKO) cells. Data represent mean +/− s.d. (n = 3 cell culture wells). One-way ANOVA with Bonferroni correction.
Extended Data Fig. 4 |. APMAP loss sensitizes cells to ADCP in a highly specific manner and without affecting surface levels of other pro- and anti-phagocytic factors.
a, Phagocytosis assay for uptake of pHrodo-labelled Karpas-299 Cas9 cells expressing indicated sgRNAs by J774 macrophages in the presence or absence of anti-CD30 antibodies. Normalized phagocytosis index was calculated as average total pHrodo Red signal per well, normalized to signal in untreated control condition at final timepoint. Data represent mean +/− s.d. (n = 4 cell culture wells). Two-way ANOVA with Bonferroni correction. b, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes by human U937 macrophages in the presence or absence of anti-CD20 (rituximab) antibodies at indicated concentrations. Phagocytosis index normalized to control (SafeKO) Ramos cells without anti-CD20. Data represent mean +/− s.d. (n = 4 cell culture wells). Two-way ANOVA with Bonferroni correction. c, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells with indicated genotypes by human primary peripheral blood-derived macrophages, from two independent healthy de-identified human donors, in the presence or absence of 10 ng ml−1 anti-CD20 antibodies. Phagocytosis index normalized to control (SafeKO) Ramos cells without anti-CD20. Data represent mean +/− s.d. (n = 3 cell culture wells). One-way ANOVA with Bonferroni correction. d, Phagocytosis assay for uptake of pHrodo-labelled Ramos Cas9 cells expressing indicated sgRNAs by J774 macrophages with or without 24 h pre-treatment with 100 ng ml-1 LPS. Normalized phagocytosis index was calculated as average total pHrodo Red signal per well, normalized to signal in untreated control condition at final timepoint. Data represent mean +/− s.d. (n = 4 cell culture wells). e, Ramos Cas9 cells expressing indicated sgRNAs were incubated with Annexin V-FITC or anti-Calreticulin-DyLight-488 and analysed by flow cytometry. CRT, calreticulin. Data represent mean +/− s.d. (n = 3 independently stained samples). P-values were from two-tailed t-tests. f, Flow-cytometry based measurement of cell surface levels of CD20 in Ramos Cas9 cells expressing indicated sgRNAs. Data represent mean (n = 2 independently stained samples, except cells expressing CD20 sgRNA (n = 1)). g, Flow-cytometry based measurement of cell surface levels of CD47 in Ramos Cas9 cells expressing indicated sgRNAs. Data represent mean +/− s.d. (n = 3 independently stained samples). h, Flow-cytometry based measurement of cell surface levels of sialic acid in Ramos Cas9 cells expressing indicated sgRNAs. Where indicated, cells were treated with sialidase as a positive control. Data represent mean +/− s.d. (n = 3 independently stained samples). i, Viability assays (measured as cell confluence after 72 h on Incucyte, normalized to untreated SafeKO control cells) of indicated Ramos cells in the presence of indicated concentrations of 9 drugs. Data represent mean +/− s.d. (n = 3 cell culture wells). j, Flow-cytometry based measurement of forward scatter (FSC) and side scatter (SSC) in Ramos Cas9 cells expressing indicated sgRNAs. Data represent mean +/− s.d. (n = 3 independently analysed samples). k, Ramos-J774 adhesion assay in the presence of indicated antibody concentrations, using indicated GFP+ Ramos Cas9 knockout cells. Data represent mean +/− s.d. (n = 2 cell culture wells). l, Flow-cytometry based measurement of ADCP of Ramos Cas9 cells expressing indicated sgRNAs and stained with either calcein or CellTrace-Far-Red dye before incubation with J774 macrophages and anti-CD20 for 24h. Data represent mean +/− s.d. (n = 3 cell culture wells). Two-tailed t-tests were used to compare SafeKO and APMAPKO cells within each labeling condition.
Extended Data Fig. 5 |. APMAP localizes to the endoplasmic reticulum and its cytosolic domain, transmembrane domain, and N-glycosylation are not required for its function in ADCP.
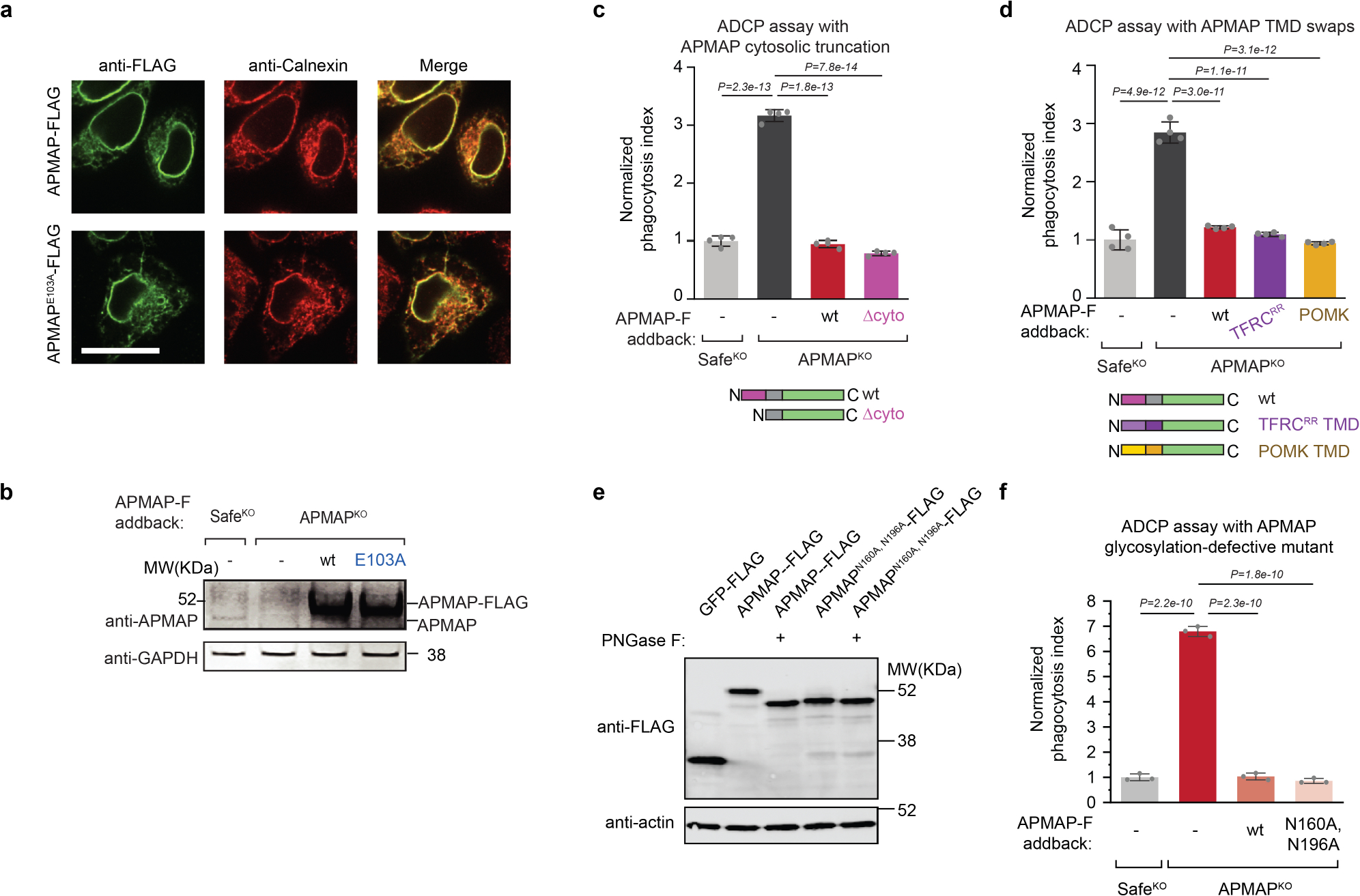
a, Localization of APMAP-FLAG and APMAPE103A-FLAG to the endoplasmic reticulum in HeLa cells. Scale bar, 20 μm. Calnexin is used as a marker of the endoplasmic reticulum. FLAG staining was representative of two independent experiments. b, Immunoblotting of cell extracts derived from Ramos cells of indicated genotypes expressing indicated APMAP-FLAG constructs. GAPDH served as loading control. Experiment was performed twice. c, d, Phagocytosis assay for uptake of pHrodo-labelled Ramos-Cas9 cells with indicated genotypes by J774 macrophages in the presence of anti-CD20 antibodies. APMAP-F, APMAP-FLAG. TFRCRR, mutant allele of TFRC that localizes primarily to the endoplasmic reticulum47. Phagocytosis index normalized to control (SafeKO) cells. Data represent mean +/− s.d. (n = 4 cell culture wells). One-way ANOVA with Bonferroni correction. e, Immunoblotting of cell extracts that were treated, where indicated, with PNGase F to remove N-glycosylation. Actin served as loading control. Experiment was performed once. f, Phagocytosis assay for uptake of pHrodo-labelled Ramos Cas9 cells expressing indicated sgRNAs and indicated addback constructs by J774 macrophages in the presence of anti-CD20 antibodies. Normalized phagocytosis index was calculated as average total pHrodo Red signal at 5 h for each well, normalized to signal in SafeKO cells at 5 h timepoint. Data represent mean +/− s.d. (n = 3 cell culture wells). One-way ANOVA with Bonferroni correction. For gel source data, see Supplementary Figure 1.
Extended Data Fig. 6 |. Evaluation of APMAP role in ADCP across diverse cancer cell lines and in syngeneic mice.
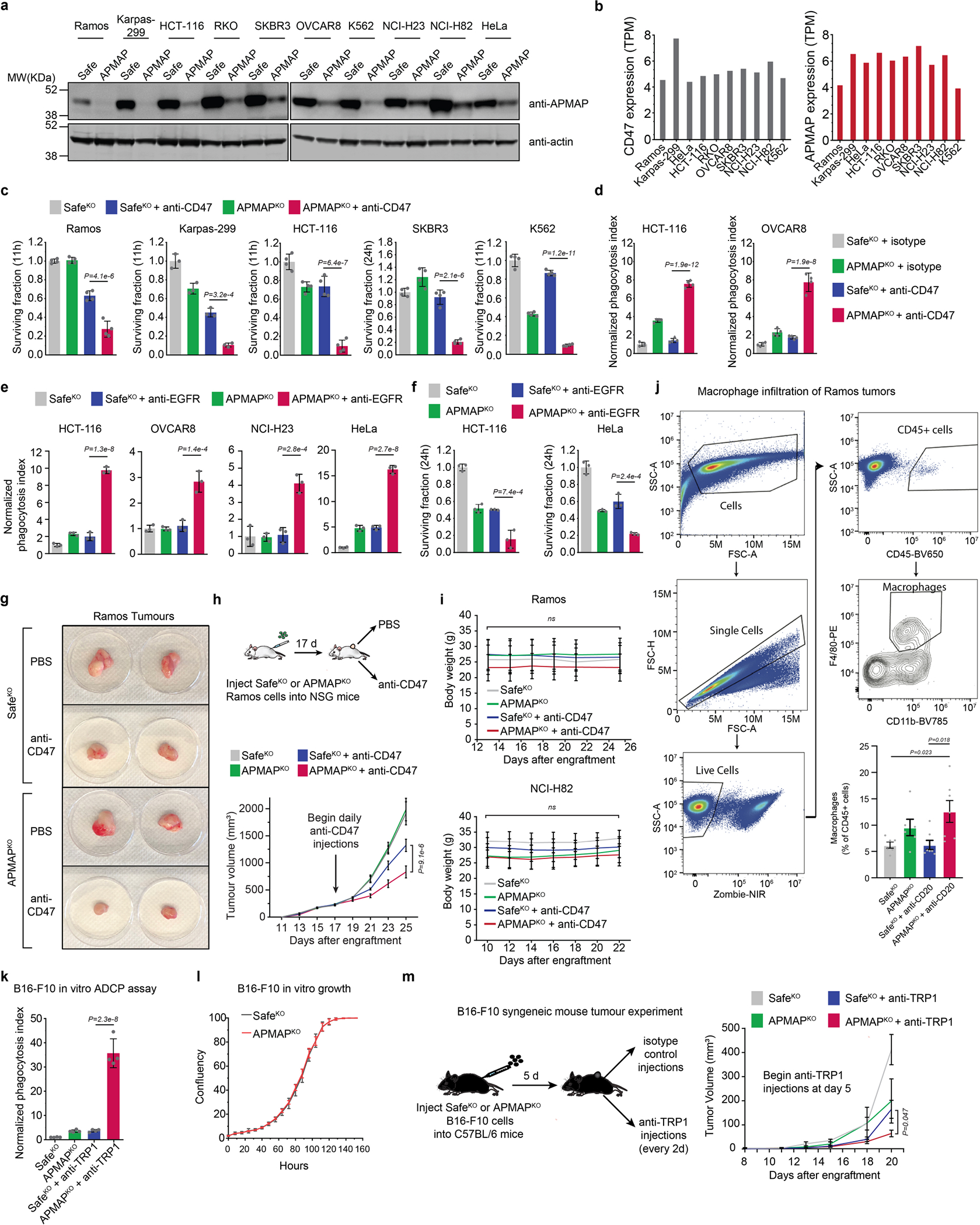
a, Levels of APMAP in ten cell lines measured by Western blot. All cell lines stably express Cas9 and were transduced with indicated sgRNAs. Actin served as loading control. Western blot to confirm knockout across all ten cell lines on one gel was performed once. For gel source data, see Supplementary Figure 1. b, Expression levels (TPM) of CD47 and APMAP in ten cell lines (data from CCLE). c, Survival measurements of selected (GFP+) cell lines in Fig. 3a, measured as percentage of GFP remaining after indicated number of hours of incubation with J774 macrophages in presence or absence of anti-CD47. Data represent mean +/− s.d. (n = 4 cell culture wells, except Karpas-299 (n = 3)). One-way ANOVA with multiple comparisons correction. d, Phagocytosis assays as in Fig. 3a, but with isotype control antibodies. Data represent mean +/− s.d. (n = 4 cell culture wells). One-way ANOVA with Bonferroni correction. e, Phagocytosis assay for uptake of pHrodo-labelled cells for indicated Cas9-expressing cell lines expressing indicated sgRNAs by J774 macrophages in the presence or absence of anti-EGFR/cetuximab antibodies. Phagocytosis index normalized to control (SafeKO) cells without anti-EGFR. Data represent mean +/− s.d. (n = 3 cell culture wells). One-way ANOVA with Bonferroni correction. f, Survival measurements of selected (GFP+) cell lines in Extended Data Fig. 6e, measured as percentage of GFP remaining after indicated number of hours of incubation with J774 macrophages in presence or absence of anti-EGFR. Data represent mean +/− s.d. (n = 3 cell culture wells). One-way ANOVA with Bonferroni correction. g, Representative photographs depicting Ramos tumours of indicated genotype extracted from NSG mice at 25 d following transplantation. h, SafeKO or APMAPKO Ramos cells were transplanted into NSG mice and allowed to form tumours. Mice were treated with anti-CD47 (B6H12, BioXCell) or PBS daily starting 17 d following transplantation, and tumour size was measured every 2 d. Data represent mean +/− s.e.m. (n = 5 (SafeKO groups) and 6 (APMAPKO groups)). Two-way ANOVA with Tukey correction (comparison between SafeKO/anti-CD47 and APMAPKO/anti-CD47 for final timepoint is shown). I, Mouse weights in Ramos (top) and NCI-H82 (bottom) xenograft experiments (Extended Data Fig. 6h, Fig. 3b). Data represent mean +/− s.d. Two-way ANOVA with Bonferroni correction (n = 5 (all NCI-H82 groups and Ramos SafeKO groups) and 6 (Ramos APMAPKO groups)). P-values are reported for the interaction between treatment groups. j, Single-cell suspensions were prepared from SafeKO or APMAPKO Ramos tumours treated with PBS or anti-CD20 (from experiment in Fig. 3c) and analysed for the presence of macrophages (CD45+/F4–80+/Cd11b+) as a percentage of all CD45+ cells. Gating strategy is shown (top/left). Data (bottom right) represent mean +/− s.e.m. (n = 6 (PBS groups) and 7 (antibody-treated groups)). One-way ANOVA with Tukey correction. k, Phagocytosis assay for uptake of pHrodo-labelled B16-F10 cells with indicated genotypes by J774 macrophages in the presence or absence of anti-TRP1 antibodies. Phagocytosis index normalized to control (SafeKO) cells without antibody. Data represent mean +/− s.d. (n = 4 cell culture wells). One-way ANOVA with Bonferroni correction. l, In vitro growth of B16-F10 cells of indicated genotypes, measured using time-lapse microscopy as total confluence per well over 6 d. Data represent mean +/− s.d. (n = 4 cell culture wells). m, SafeKO or APMAPKO B16-F10 cells were transplanted into syngeneic C57BL/6 mice and allowed to form tumours. Mice were treated with anti-TRP1 or mouse IgG2a isotype control antibody daily starting 5 d following transplantation, and tumour size was measured every 2 d. Data represent mean +/− s.e.m. (n = 7 for SafeKO groups, n = 6 for both APMAPKO groups). Two-way ANOVA with Tukey correction (comparison between SafeKO/anti-TRP1 and APMAPKO/anti-TRP1 for final timepoint is shown).
Extended Data Fig. 7 |. Genome-wide magnetic screen in J774 macrophages for phagocytosis of IgG-coated beads.
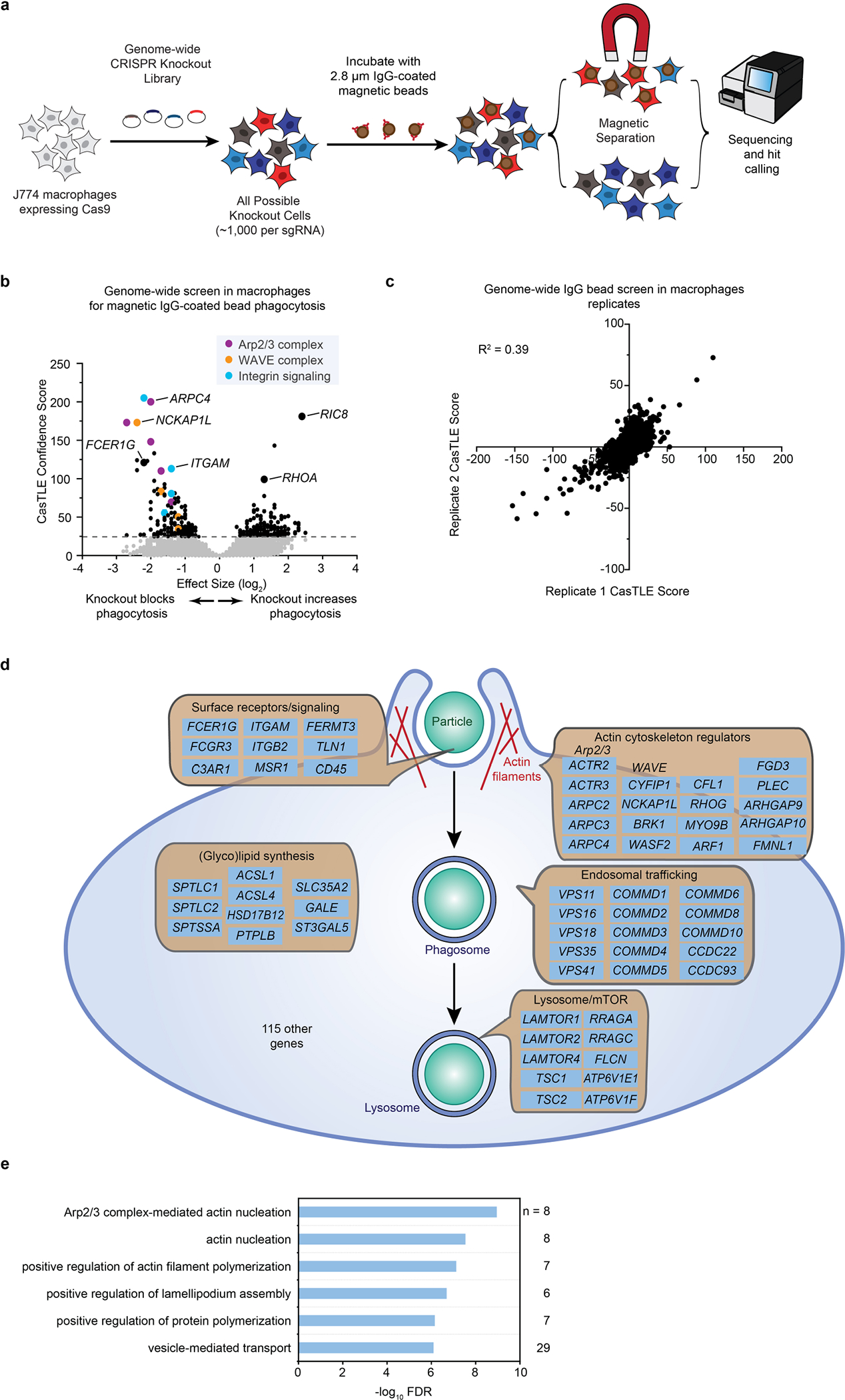
a, Schematic of genome-wide screen in J774 macrophages for phagocytosis of 2.8 micron IgG-coated magnetic beads. b, Volcano plot of screen diagrammed in a. Dotted line indicates 5% FDR. c. Replicates of screen diagrammed in a. d, Diagram of hits with negative effect size (i.e. required for phagocytosis) from genome-wide screen for IgG bead phagocytosis in J774 macrophages. e, Gene ontology enrichment analysis for macrophage IgG bead screen hits with negative effect size (required for phagocytosis) (5% FDR). Selected terms shown. n indicates number of genes among hits annotated with indicated term.
Extended Data Fig. 8 |. GPR84 is expressed in tumour associated macrophages.
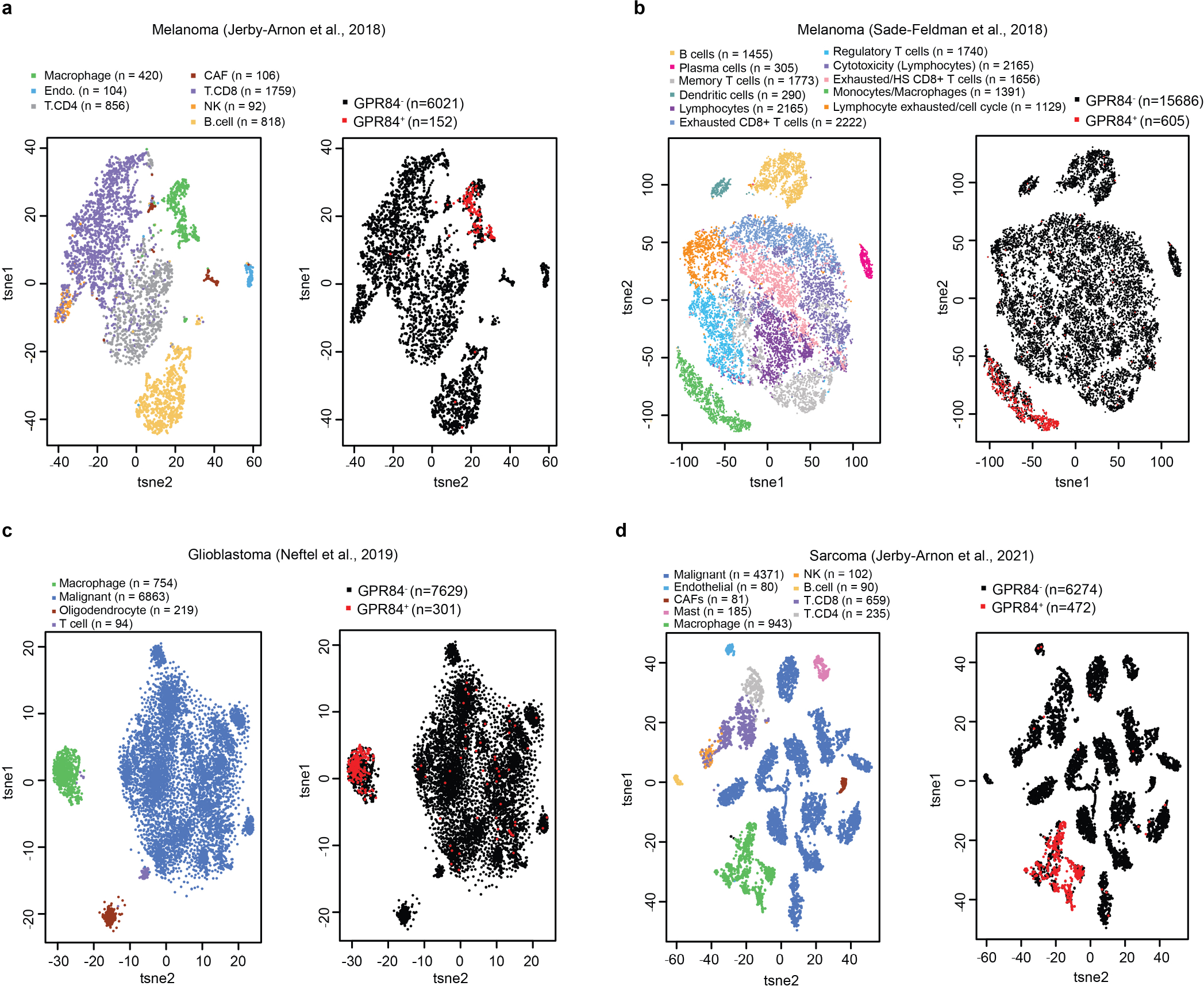
a–d, Single-cell RNA-seq analyses of human tumours from patients with melanoma54,55 (a, b), patients with glioblastoma56 (c) and patients with sarcoma53 (d), showing cell type annotations (left) and detection of GPR84 (right). GPR84+ and GPR84− denote TPM > 0 and = 0, respectively; n denotes the number of cells shown.
Extended Data Fig. 9 |. Macrophage screens for genes required for enhanced uptake of APMAPKO cancer cells.
a, Phagocytosis assay for uptake of pHrodo-labelled Karpas-299-Cas9 cells expressing indicated sgRNAs, incubated with J774 macrophages expressing indicated sgRNAs, in the presence of anti-CD30 antibodies. Phagocytosis index (arbitrary units) corresponds to the total pHrodo Red signal per well, normalized to SafeKO Karpas-299 cells fed to SafeKO macrophages. Data represent mean +/− s.d. (n = 4 cell culture wells). Two-way ANOVA with Bonferroni correction. b, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells expressing indicated sgRNAs, incubated with U937 macrophages expressing indicated sgRNAs (three independent sgRNAs per gene), in the presence of anti-CD20 antibodies. Phagocytosis index (arbitrary units) corresponds to the total pHrodo Red signal per well, normalized to SafeKO Ramos cells fed to SafeKO−1 macrophages. Data represent mean +/− s.d. (n = 4 cell culture wells). Two-way ANOVA with Bonferroni correction, P values for comparisons to SafeKO Ramos/SafeKO−1 U937 macrophages shown. c, Gating strategy for collecting single-positive and double-negative macrophage populations, corresponding to macrophages that phagocytosed calcein+ SafeKO or Far-red+ APMAPKO cells. d, Volcano plot for macrophage screen for genes required for uptake of SafeKO Ramos cells, using 2,208-gene sublibrary (enriched for phagocytosis genes, but lacking GPR84), conducted in J774 macrophages. Dotted line indicates 5% FDR. e, Volcano plot for macrophage screen for genes required for uptake of APMAPKO Ramos cells, using 2,208-gene sublibrary (enriched for phagocytosis genes, but lacking GPR84) in J774 macrophages. Dotted line indicates 5% FDR. f, Volcano plot for macrophage screen for genes required selectively for uptake of APMAPKO cells, following screen design in Fig. 4a (comparison 2), but using 2,208-gene sublibrary (enriched for phagocytosis genes, but lacking GPR84) in J774 macrophages, for uptake of calcein+ SafeKO cells and far-red+ APMAPKO Ramos cells. Dotted line indicates 5% FDR. g, Phagocytosis assay for uptake of pHrodo-labelled Ramos cells expressing indicated sgRNAs, incubated with J774 macrophages expressing indicated sgRNAs, in the presence of anti-CD20 antibodies. Phagocytosis index (arbitrary units) corresponds to the total pHrodo Red signal per well, normalized to SafeKO Ramos cells fed to SafeKO macrophages. Data represent mean +/− s.d. (n = 3 cell culture wells). Two-way ANOVA with Bonferroni correction. P-values correspond to comparisons to SafeKO-1. h, FACS-based phagocytosis assay for uptake of CellTrace Far-Red-labelled APMAPKO cells and calcein-labelled CD47KO Ramos cells by J774-Cas9 macrophages expressing indicated sgRNAs. Ratio of macrophages that phagocytosed APMAPKO Ramos cells to macrophages that phagocytosed CD47KO Ramos cells, normalized to SafeKO/SafeKO J774 macrophages, following 24 h co-incubation with anti-CD20 antibodies is plotted. Data represent mean +/− s.d. (n = 3 cell culture wells). One-way ANOVA with Bonferroni correction, P-values for comparisons to SafeKO/SafeKO J774 macrophages shown.
Extended Data Fig. 10 |. GPR84 agonists stimulate uptake of antibody-opsonized cancer cells.
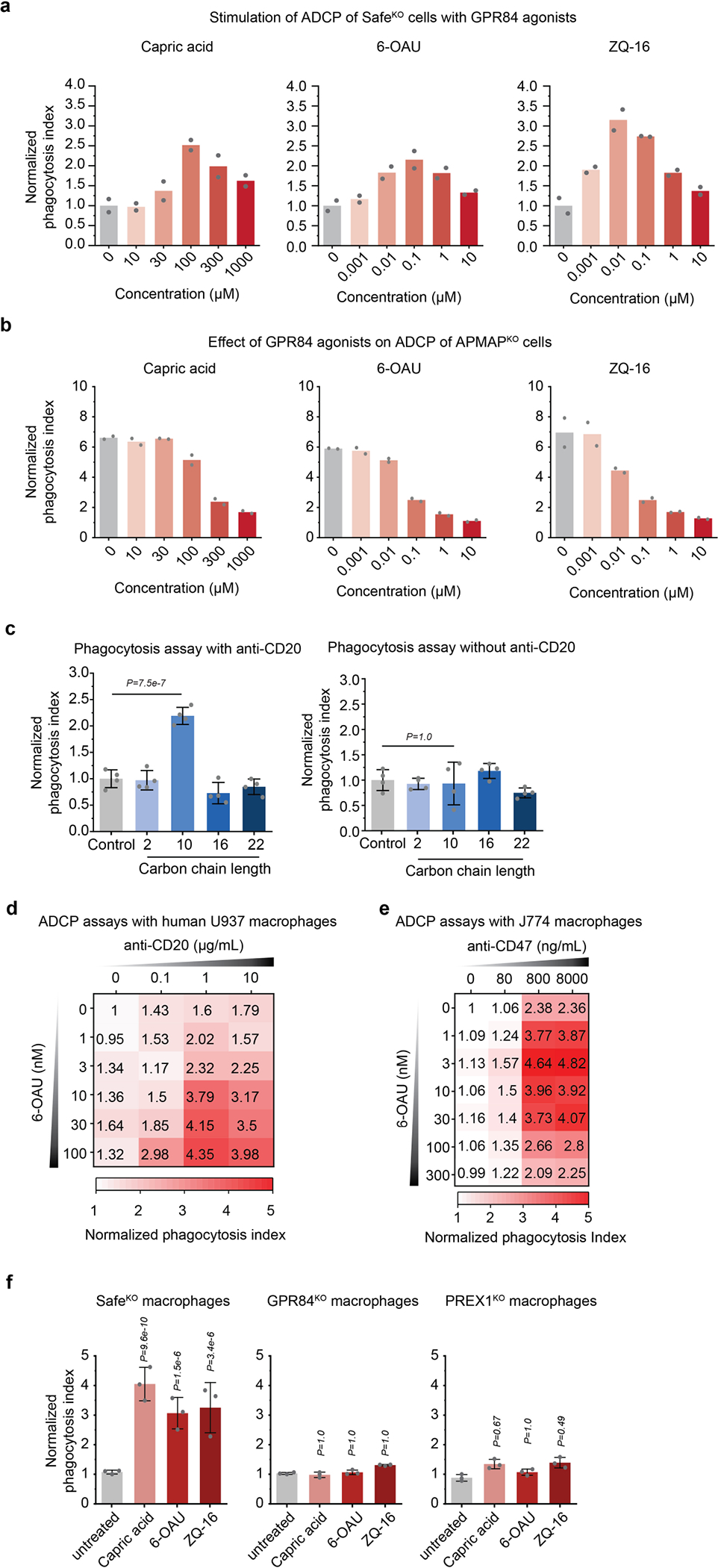
a, Phagocytosis assay for uptake of pHrodo-labelled Ramos Cas9 cells expressing Safe-targeting sgRNAs by J774 macrophages in the presence of anti-CD20 antibodies and indicated concentrations of GPR84 agonists. Data represent mean (n = 2 cell culture wells). b, Phagocytosis assay for uptake of pHrodo-labelled Ramos Cas9 cells expressing APMAP-targeting sgRNAs by J774 macrophages in the presence of anti-CD20 antibodies and indicated concentrations of GPR84 agonists. Data represent mean (n = 2 cell culture wells). c, Phagocytosis assay for uptake of pHrodo-labelled SafeKO Ramos Cas9 cells by J774 macrophages in the presence (left) or absence (right) of anti-CD20 antibodies and 100 μM saturated fatty acids of indicated carbon chain length (n = 2, acetic acid; n = 10, capric acid; n = 16, palmitic acid; n = 22, docosanoic acid). Data represent mean +/− s.d. (n = 4 cell culture wells). One-way ANOVA with Bonferroni correction. d, Heatmap of normalized phagocytosis index of Ramos cells incubated with U937 macrophages expressing indicated sgRNAs, in the presence of indicated concentrations of 6-OAU and anti-CD20. Data represent mean (n = 4 cell culture wells). e, Heatmap of normalized phagocytosis index of Ramos cells incubated with J774 macrophages expressing indicated sgRNAs, in the presence of indicated concentrations of 6-OAU and anti-CD47. Data represent mean (n = 4 cell culture wells). f, Phagocytosis assay for uptake of pHrodo-labelled Ramos Cas9 cells expressing Safe-targeting sgRNAs by J774 macrophages in the presence of anti-CD20 antibodies and GPR84 agonists (100 μM capric acid, 100 nM 6-OAU, 10 nM ZQ-16). Data represent mean +/− s.d. (n = 3 cell culture wells). One-way ANOVA with Bonferroni correction. P-values are for comparison to untreated condition for each macrophage genotype.
Supplementary Material
Acknowledgements
We thank R. Levy, S. Levy, C. Bertozzi, J. Long, S. Dixon, P. Jackson, M. Smith, T. Wyss-Coray, A. Drainas, A. Derry, B. Smith, C. Delaveris, J. Shon, S. Wisnovsky, J. Donnelly, E. Zhang, T. Raveh, D. Vorselen, J. Carozza, M. Ellenberger, J. Chan, L. Jiang, R. Jian, M. Snyder, K. McNamara, R. Chen and members of the Bassik laboratory, including G. Hess, R. Levin, K. Han, K. Tsui, M. Haney, D. Morgens, J. Tycko, M. Dubreuil, K. Aloul, B. Ego and A. Li, for helpful discussions and experimental advice; and A. Sil for providing the J774 Cas9 line. Cell sorting for this project was done on instruments in the Stanford Shared FACS Facility, including an instrument purchased by the Parker Institute for Cancer Immunotherapy. This research was supported by an NIH Director’s New Innovator award (1DP2HD084069-01) to M.C.B., by the Ludwig Institute for Cancer Research (J.S. and I.W.), the NIH (grants CA213273 and CA231997 to J.S., R35CA220434-05 and 1R01AI143889-01A1 to I.W.), the JSPS (JSPS overseas research fellowship to Y.N.), a Stanford School of Medicine Dean’s Postdoctoral Fellowship to R.A.K. and a Jane Coffin Childs Postdoctoral Fellowship to R.A.K. L.J.A. is a Chan Zuckerberg Biohub Investigator and holds a Career Award at the Scientific Interface from BWF.
Footnotes
Competing interests R.A.K. and M.C.B., through the Office of Technology Licensing at Stanford University, have filed a patent application on the methods and findings in this manuscript. I.W. is an inventor on several patents in the field of ADCP induced by blockade of several don’t-eat-me signals such as CD47, CD24, beta-2-microglobulin, and PDL1, and their macrophage cognate receptors, respectively, SIRPα, Siglec-10, LILRB1, and PD1. These have been licensed to several companies. I.W. is not currently affiliated with these companies and does not hold stock in them. He is, however, engaged in the formation of one or more start-up companies in the field. J.S. licensed a patent to Forty Seven Inc./Gilead on the use of CD47 blocking strategies in SCLC.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-03879-4.
Peer review information Nature thanks Ross Levine and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Scott AM, Wolchok JD & Old LJ Antibody therapy of cancer. Nat. Rev. Cancer 12, 278–287 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Sliwkowski MX & Mellman I Antibody therapeutics in cancer. Science 341, 1192–1198 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Weiskopf K & Weissman IL Macrophages are critical effectors of antibody therapies for cancer. mAbs 7, 303–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsao L-C et al. CD47 blockade augmentation of trastuzumab antitumor efficacy dependent on antibody-dependent cellular phagocytosis. JCI Insight 4, e131882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodsky FM Monoclonal antibodies as magic bullets. Pharm. Res. 5, 1–9 (1988). [DOI] [PubMed] [Google Scholar]
- 6.Maleki LA, Baradaran B, Majidi J, Mohammadian M & Shahneh FZ Future prospects of monoclonal antibodies as magic bullets in immunotherapy. Hum. Antibodies 22, 9–13 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Glennie MJ, French RR, Cragg MS & Taylor RP Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol. Immunol. 44, 3823–3837 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Chao MP et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Logtenberg MEW et al. Glutaminyl cyclase is an enzymatic modifier of the CD47–SIRPα axis and a target for cancer immunotherapy. Nat. Med. 25, 612–619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macauley MS, Crocker PR & Paulson JC Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 14, 653–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Northcott PA et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao S et al. The oncogenic role of MUC12 in RCC progression depends on c-Jun/TGF-β signalling. J. Cell. Mol. Med. 24, 8789–8802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor-Papadimitriou J et al. MUC1 and the immunobiology of cancer. J. Mammary Gland Biol. Neoplasia 7, 209–221 (2002). [DOI] [PubMed] [Google Scholar]
- 14.O’Prey J, Wilkinson S & Ryan KM Tumor antigen LRRC15 impedes adenoviral infection: implications for virus-based cancer therapy. J. Virol. 82, 5933–5939 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purcell JW et al. LRRC15 is a novel mesenchymal protein and stromal target for antibody–drug conjugates. Cancer Res. 78, 4059–4072 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Itoh Y et al. Identification and expression of human epiglycanin/MUC21: a novel transmembrane mucin. Glycobiology 18, 74–83 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Snyder KA et al. Podocalyxin enhances breast tumor growth and metastasis and is a target for monoclonal antibody therapy. Breast Cancer Res. 17, 46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarbé NG, Rio M-C, Hummel S, Weidle UH & Zöller M Overexpression of the small transmembrane and glycosylated protein SMAGP supports metastasis formation of a rat pancreatic adenocarcinoma line. Int. J. Cancer 117, 913–922 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Ajona D et al. Blockade of the complement C5a/C5aR1 axis impairs lung cancer bone metastasis by CXCL16-mediated effects. Am. J. Respir. Crit. Care Med. 197, 1164–1176 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollingsworth MA & Swanson BJ Mucins in cancer: protection and control of the cell surface. Nat. Rev. Cancer 4, 45–60 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Jiang S et al. Cholesterol induces epithelial-to-mesenchymal transition of prostate cancer cells by suppressing degradation of EGFR through APMAP. Cancer Res. 79, 3063–3075 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Gerber H et al. The APMAP interactome reveals new modulators of APP processing and beta-amyloid production that are altered in Alzheimer’s disease. Acta Neuropathol. Commun. 7, 13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ilhan A et al. Localization and characterization of the novel protein encoded by C20orf3. Biochem. J. 414, 485–495 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Barkal AA et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 19, 76–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clynes R, Takechi Y, Moroi Y, Houghton A & Ravetch JV Fc receptors are required in passive and active immunity to melanoma. Proc. Natl Acad. Sci. USA 95, 652–656 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lattin JE et al. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res. 4, 5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Recio C et al. Activation of the immune-metabolic receptor GPR84 enhances inflammation and phagocytosis in macrophages. Front. Immunol. 9, 1419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Wu X, Simonavicius N, Tian H & Ling L Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J. Biol. Chem. 281, 34457–34464 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Gont A, Daneshmand M, Woulfe J & Lorimer I PREX1 integrates G protein-coupled receptor and phosphoinositide 3-kinase signaling to promote glioblastoma invasion. Eur. J. Cancer 61, S171–S172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noy R & Pollard JW Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunha LD et al. LC3-associated phagocytosis in myeloid Cells Promotes Tumor Immune Tolerance. Cell 175, 429–441.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su S et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 175, 442–457.e23 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Pathria P, Louis TL & Varner JA Targeting tumor-associated macrophages in cancer. Trends Immunol. 40, 310–327 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Ruffell B & Coussens LM Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hicks MA et al. The evolution of function in strictosidine synthase-like proteins. Proteins Struct. Funct. Bioinf. 79, 3082–3098 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khersonsky O & Tawfik DS Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry 44, 6371–6382 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Flannagan RS, Jaumouillé V & Grinstein S The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Manguso RT et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawson KA et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgens DW et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 8, 15178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horlbeck MA et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgens DW, Deans RM, Li A & Bassik MC Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 34, 634–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu N et al. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553, 228–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeng EE et al. Systematic identification of host cell regulators of Legionella pneumophila pathogenesis using a genome-wide CRISPR screen. Cell Host Microbe 26, 551–563.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haney MS et al. Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat. Genet. 50, 1716–1727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reimand J et al. g:Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44, W83–W89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schutze M-P, Peterson PA & Jackson MR An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J. 13, 1696–1705 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delaveris CS, Chiu SH, Riley NM & Bertozzi CR Modulation of immune cell reactivity with cis-binding Siglec agonists. Proc. Natl Acad. Sci. USA 118, e2012408118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anders S, Pyl PT & Huber W HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jerby-Arnon L et al. Opposing immune and genetic mechanisms shape oncogenic programs in synovial sarcoma. Nat. Med. 27, 289–300 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jerby-Arnon L et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sade-Feldman M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neftel C et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waterhouse A et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi J, Blundell TL & Mizuguchi K FUGUE: sequence–structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310, 243–257 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Ben-David M et al. Catalytic versatility and backups in enzyme active sites: the case of serum paraoxonase 1. J. Mol. Biol. 418, 181–196 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Tanaka Y et al. Structural and mutational analyses of Drp35 from Staphylococcus aureus: a possible mechanism for its lactonase activity. J. Biol. Chem. 282, 5770–5780 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Murshudov GN et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sockolosky JT et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl Acad. Sci. USA 113, E2646–E2654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CRISPR screen and RNA-seq raw sequencing data are available under BioProject accession number PRJNA748551. All other primary data for all figures and supplementary figures are available from the corresponding author upon request. Gene dependency data from the Cancer Dependency Map are publicly available at www.depmap.org. Cancer expression data from The Cancer Genome Atlas are available at https://gdc.cancer.gov. CCLE data are available at https://sites.broadinstitute.org/ccle/. Source data are provided with this paper.