

Abstract
Alzheimer's disease is characterized by the accumulation in the brain of the amyloid β (Aβ) peptide in the form of senile plaques. According to the amyloid hypothesis, the aggregation process of Aβ also generates smaller soluble misfolded oligomers that contribute to disease progression. One of the mechanisms of Aβ oligomer cytotoxicity is the aberrant interaction of these species with the phospholipid bilayer of cell membranes, with a consequent increase in cytosolic Ca2+ levels, flowing from the extracellular space, and production of reactive oxygen species (ROS). Here we investigated the relationship between the increase in Ca2+ and ROS levels immediately after the exposure to misfolded protein oligomers, asking whether they are simultaneous or instead one precedes the other. Using Aβ42-derived diffusible ligands (ADDLs) and type A HypF-N model oligomers (OAs), we followed the kinetics of ROS production and Ca2+ influx in human neuroblastoma SH-SY5Y cells and rat primary cortical neurons in a variety of conditions. In all cases we found a faster increase of intracellular Ca2+ than ROS levels, and a lag phase in the latter process. A Ca2+-deprived cell medium prevented the increase of intracellular Ca2+ ions and abolished ROS production. By contrast, treatment with antioxidant agents prevented ROS formation, did not prevent the initial Ca2+ flux, but allowed the cells to react to the initial calcium dyshomeostasis, restoring later the normal levels of the ions. These results reveal a mechanism in which the entry of Ca2+ causes the production of ROS in cells challenged by aberrant protein oligomers.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-022-04513-w.
Keywords: NMDA receptors, AMPA receptors, Membrane destabilization, Calcium homeostasis, Oxidative stress, Protein misfolding, Neurodegenerative diseases
Introduction
Alzheimer’s disease (AD), which is the most common neurodegenerative disease and the most common form of dementia, is characterized by the extracellular deposition in the brain of the amyloid β (Aβ) peptide in the form of senile plaques [1] and by the intraneuronal formation of neurofibrillary tangles of the tau protein [2]. The aggregation process of Aβ generates a large variety of protein aggregates, such as oligomers, protofibrils and fibrils, all characterised by high levels of polymorphism [3]. According to the amyloid hypothesis, the small diffusible oligomers of Aβ are neurotoxic and are thought to contribute to AD development and progression [3–5]. Oligomer cytotoxicity appears to result, in its early phases, from the aberrant interactions of such species with a number of molecular targets on neurons, including the lipid bilayer of their cell membranes [1, 3, 5, 6]. This interaction results in the disruption of cell membranes, compromising its ability to maintain cellular homeostasis, and promoting two important early biochemical changes. The first is the uncontrolled increase in cytosolic calcium (Ca2+) levels flowing from the extracellular space into the cytosol [7–14], and the second is the accumulation of reactive oxygen species (ROS) [10, 11, 15, 16].
It is known that Aβ oligomers are able to interact and insert into the phospholipid bilayer of the cell membrane causing the passage through it of small molecules and ions, such as free Ca2+ ions [6, 7, 11, 17, 18], as well as permitting the activation of ionotropic glutamate receptors functioning as Ca2+ channels, including the N-methyl-d-aspartate (NMDA) receptors [9, 14, 15, 18–21] and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [14, 15, 18, 19, 22]. In particular, the rapid oligomer-induced activation of extrasynaptic NMDA/AMPA receptors is a crucial mechanism in the AD pathogenesis. This process takes place through the insertion of the oligomers in the bilayer, which changes the mechanical properties of the membrane that is transmitted down to the receptors that are therefore activated through their mechanosensitivity, without a direct interaction with the oligomers [14]. Other Ca2+ channels that seem to be involved in the Aβ-induced flux of Ca2+ ions are the transient receptor potential melastatin 2 (TRPM2) [23], the voltage-dependent Ca2+ channels (VDCCs) [24], and the transient receptor potential A1 (TRPA1) [25].
Another relevant early biochemical change resulting from the interaction of Aβ oligomers with cell membranes is oxidative stress, which is associated with the accumulation of ROS in the cytosol [10, 11, 15, 16] and represents an important determinant in AD pathogenesis [26, 27]. The elevation of ROS is caused by the activation of the oxidative metabolism to respond to the intracellular Ca2+ increase induced by the oligomers and the consequent increased need for ATP by the Ca2+ pumps, that try to restore the normal levels of intracellular Ca2+ [10, 28]. It was also observed that Aβ aggregation can induce oxidative stress though intra-mitochondrial mechanisms, with disruption of the electron transport chain that initiate ROS production [29–31], or with the suppression of α-ketoglutarate dehydrogenase [32]. On the other hand, some studies have proposed that the oxidative stress precedes Aβ accumulation and may therefore induce amyloid production [29–31]. Analysis performed in murine AD models with human overexpression of the amyloid precursor protein (APP) showed that lipid peroxidation and oxidative damage occurs before Aβ accumulation [33]. Moreover, using HEK293 human embryonic kidney cells, it was observed that ROS produced in mitochondria drove Aβ production [34]. Eventually, therefore, Aβ aggregation may be both a cause and an effect of oxidative stress.
It is not yet clear, however, if interactions and cause-and-effect relationships between ROS production and Ca2+ signalling induced by misfolded protein oligomers can be considered as bidirectional, or whether one of them is causative of the other. Oxidative stress has been considered, at least in part, a consequence of Ca2+ entry into cells, because of the increased need to produce ATP by mitochondria to pump out Ca2+ ions, which produces ROS itself [10, 28, 35]. On the other hand, ROS can significantly affect Ca2+ homeostasis in the cell and intracellular Ca2+ stores by oxidising multiple methionine residues within the Ca2+ signalling protein calmodulin (CaM) resulting in an inability to activate a range of target proteins, including the cell membrane Ca-ATPase involved in the maintenance of Ca2+ homeostasis [10, 36, 37]. The analysis reported here allowed to clarify how these two mechanisms are interconnected and whether a precise cause-and-effect relationship exists. The kinetics of these processes in neuroblastoma cells and primary rat cortical neurons were analysed under various conditions in which ROS production or Ca2+ influx induced by misfolded protein oligomers were specifically inhibited, showing that the lack of an influx of Ca2+ ions into the cytosol can reduce the ROS production, whereas the protection against ROS formation did not prevent the initial Ca2+ flux but allowed the cells to react, on a longer term, to the initial Ca2+ dyshomeostasis, restoring the normal levels of the ions.
Materials and methods
Preparation of HypF-N oligomers and Aβ42 ADDLs
Wild-type HypF-N from E. coli was prepared and purified as described [38], and stored at − 80 °C in 20 mM potassium phosphate buffer, pH 7.0, with 2 mM dithiothreitol (DTT). Type A oligomers (OAs) were prepared by incubating HypF-N at 48 μM, with 50 mM acetate buffer, 12% (v/v) trifluoroethanol (TFE), 2 mM DTT, pH 5.5, 25 °C, for 4 h without agitation, as previously described [38].
Lyophilised Aβ42 (Bachem, Bubendorf, Switzerland) was dissolved in HFIP to 1.0 mM and incubated for 1 h at room temperature to allow complete peptide monomerization. Aβ42-derived diffusible ligands (ADDLs) were prepared as described previously [39]. In particular, the HFIP was evaporated with a gentle flow of N2 and the dried protein was resuspended to 5 mM with DMSO and then diluted with F-12 HAM to 100 μM. The sample was then incubated at 4 °C for 24 h and centrifuged at 12,000g for 10 min to collect the supernatant.
Cell cultures
Human SH-SY5Y neuroblastoma cells (ATCC CRL-2266, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), F-12 HAM with 25 mM N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES) and NaHCO3 (1:1) and supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine and 1% antibiotics, as reported [40]. Cell cultures were maintained in a 5% CO2 humidified atmosphere at 37 °C and grown until they reached 80% confluence for a maximum of 20 passages. Primary rat cortical neurons (Thermo Fisher Scientific) were plated in 24-well plate at the density of 200,000 cells per well and maintained in neuronal basal plus medium (Thermo Fisher Scientific) supplemented with GlutaMAX (Gibco) at the concentration of 0.5 mM and 2% (v/v) B-27 serum-free complement (Gibco), in a 5% CO2 humidified atmosphere at 37 °C. Every 4 days the medium was partially replaced with fresh one. All the experiments were performed 12–16 days after plating.
Cell treatments
SH-SY5Y cells were plated in 6-well plates containing coverslips at a density of 40,000 cells per well at 37 °C. After 24 h, they were washed with PBS and treated with HypF-N OAs diluted in cellular medium at the monomer equivalent concentration of 12 µM for 5, 10, 15, 30 and 60 min, or with Aβ42 ADDLs diluted in cellular medium at the monomer equivalent concentration of 1 µM for 5, 10, 15, 30, 60, 90, 120 and 180 min. In other sets of experiments, before the treatment with HypF-N OAs or Aβ42 ADDLs, SH-SY5Y cells were pre-treated for 1 h with 5 µM CNQX, or 10 µM memantine, or both. In other sets of experiments, cells were pre-treated with 2 µM l-α-lysophosphatidylcholine (LPC) for 2 h, with 30 µM Trolox for 1 h, or in a medium without Ca2+ for 1 h. In another set of experiments, the SH-SY5Y cells were treated for 10 min with 1 mM NMDA or 50 µM AMPA, with or without pre-treatment with 30 µM Trolox for 1 h.
Primary rat cortical neurons were plated in 24-well plates containing glass coverslips coated with poly-D-lysine at a density of 200,000 cells per well at 37 °C. 12–16 days after plating, they were washed with PBS and treated with Aβ42 ADDLs diluted in cellular medium at the monomer equivalent concentration of 1 µM for 10 and 60 min. In other sets of experiments, before the treatment with Aβ42 ADDLs, the cells were pre-treated for 1 h with 5 µM CNQX, or 10 µM memantine, or 30 µM Trolox.
Measurement of cytosolic free Ca2+ levels and intracellular ROS production
Cytosolic Ca2+ levels were measured in living SH-SY5Y cells and primary rat cortical neurons after the different treatments, or after adding 1 µM ionomycin for 1 h as a positive control. The cells were then washed with PBS and loaded with 4 µM Fluo-4 AM (Thermo Fisher Scientific) for 10 min. The Ca2+ levels for untreated cells were evaluated in SH-SY5Y cells at each time point, from 0 to 180 min, changing the cellular medium at the different time lengths, washing with PBS and loading the Fluo-4 AM probe for 10 min. Considering the absence of changes in basal Ca2+ levels, all data are reported relative to untreated cells at time 0. ROS levels were measured in living SH-SY5Y cells and primary rat cortical neurons after the different treatments, or after adding 250 µM H2O2 for 1 h, as a positive control, and then by loading 5 µM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) in the last 15 min of the different treatments. ROS levels for untreated cells were evaluated in SH-SY5Y cells at the different time points, changing the cell medium and loading the probe in the last 15 min of the treatment. Considering the absence of significant variation, all data are reported relative to untreated cells at 15 min, which is the probe incubation time and therefore the shortest time that can be analysed. Both Ca2+ and ROS levels were then evaluated after excitation at 488 nm by a TCS SP8 scanning confocal microscopy system equipped with an argon laser source (Leica Microsystems).
In another set of experiments, cytosolic Ca2+ and ROS levels were measured in living SH-SY5Y cells by loading 5 µM X-Rhod-1 AM (Thermo Fisher Scientific) in the last 20 min and 5 µM CellRox™ Deep Red Reagent (Thermo Fisher Scientific) in the last 30 min of the different treatments, respectively. Ca2+ and ROS levels were then evaluated after excitation at 561 and 633 nm, respectively, by the same TCS SP8 scanning confocal microscopy system described above.
In all cases, a series of 1 µm thick optical sections (1024 × 1024) was taken through the cell depth for each sample using a Leica Plan Apo 63 × oil immersion objective and projected as a single composite image by superimposition (Leica Microsystems). Three different experiments were carried out and 10–22 cells were analysed in each experiment, in both Ca2+ and ROS analyses, using Image J software. Values were averaged over the 10–22 cells in each experiment and the mean and standard error of the mean (SEM) were determined from the averaged values of the three experiments (n = 3). All data were normalized to the positive control value, obtained with ionomycin and H2O2 respectively, which were attributed 100%.
Statistical analysis
All data were expressed as means ± SEM (standard error of the mean). Comparisons between the different groups were performed by Student’s t-test. The single (*;§), double (**;§§) and triple (***;§§§) symbols refer to p values < 0.05, < 0.01 and < 0.001, respectively.
Results
Toxic HypF-N oligomers increase intracellular Ca2+ levels and ROS production
We started our analysis using model oligomers formed by the protein HypF-N (named type A oligomers, or OAs), which were previously found to have effects similar to those of Aβ42 oligomers in cell and animal models [10, 11, 38, 41, 42]. These HypF-N OAs are highly stable, versatile, easy to isolate and have a non-toxic counterpart (known as type B oligomers, or OBs), which is useful as a negative control [38].
In a previous work it was shown that the HypF-N OAs, similarly to the Aβ42 ADDLs, are able to cause a progressive increase of intracellular Ca2+ levels on SH-SY5Y cells by activating rapidly extrasynaptic NMDA receptors and, to a lower extent, AMPA receptors [14]. We therefore started our analysis by evaluating whether the freshly formed toxic HypF-N OAs prepared for this study confirmed this effect. The treatment over time of SH-SY5Y cells with HypF-N OAs (12 μM monomer equivalents) showed a gradual increase of intracellular Ca2+ levels, which was evident already after 5 min and reached the maximum level after 30 min of treatment (images in Fig. 1a, histograms in Fig. 1b and corresponding kinetic plot in Fig. 1c). When cells were pre-treated with CNQX, an AMPA receptor competitive antagonist, or with memantine, a NMDA receptor uncompetitive inhibitor, a slight reduction of the OA-induced cytoplasmic Ca2+ increase was observed in the early stages, up to 10 min of treatment, which was more significant with memantine (Fig. 1a–c). This reduction was followed by a gradual increase of the intracellular Ca2+ concentration, until normal levels reached after 60 min of treatment (Fig. 1a–c). Overall, these pre-treatments cause a deceleration of the increase of intracellular Ca2+ (Fig. 1c). Moreover, after a pre-treatment with both CNQX and memantine, the intracellular Ca2+ levels remained similar to untreated cells up to 15 min of treatment, and significantly lower than those of cells without inhibitor pre-treatment up to 60 min of oligomer treatment (Fig. 1a,b), further slowing down the kinetics of OA-induced Ca2+ increase (Fig. 1c).
Fig. 1.

Toxic HypF-N oligomers increase intracellular Ca2+ levels and ROS production. a Representative confocal scanning microscopy images of free Ca2+ levels in SH-SY5Y cells following the treatment with no inhibitors (first row), 5 µM CNQX (second row), 10 µM memantine (third row), and both inhibitors (fourth row), and analysed after 5, 10, 15, 30 and 60 min of treatment with 12 µM (monomer equivalents) HypF-N OAs. A positive control of Ca2+ influx following treatment with 1 µM ionomycin for 1 h is shown at the bottom. b Semi-quantitative analysis of intracellular free Ca2+-derived fluorescence. The Ca2+ levels for untreated cells were not found to vary with time (Fig. S1a–c) and for simplicity the value recorded at time 0 min was reported, here and in other figures. c Kinetic plots showing the fluorescence versus time as reported in panel b. d Representative confocal scanning microscopy images of intracellular ROS levels in SH-SY5Y cells following the treatment with no inhibitors (first row), 5 µM CNQX (second row) 10 µM memantine (third row) and both inhibitors (fourth row), and analysed after 5, 10, 15, 30 and 60 min of treatment with 12 µM (monomer equivalents) HypF-N OAs. A positive control of ROS production following treatment with 250 µM H2O2 for 1 h is shown at the bottom. e Semi-quantitative analysis of intracellular ROS-derived fluorescence. The ROS levels for untreated cells were not found to vary with time (Fig. S1d–f) and for simplicity the value recorded at time 15 min was reported, here and in other figures. f Kinetic plots showing the fluorescence versus time as reported in panel e. Three different experiments were carried out, with 10–22 cells each, for each condition. Data are represented as mean ± SEM (n = 3). The single (*), double (**) and triple (***) asterisks refer to p values < 0.05, < 0.01 and < 0.001, respectively, relative to untreated cells. The single (§), double (§§) and triple (§§§) symbols refer to p values < 0.05, < 0.01 and < 0.001, respectively, relative to HypF-N OAs without inhibitors at corresponding time points
We then focused our attention on ROS production in SH-SY5Y cells with the same conditions of oligomer treatment and inhibitor pre-treatment described above. The treatment with HypF-N OAs (12 μM monomer equivalents) showed a slow and gradual increase of ROS production, which was slower than that observed by monitoring Ca2+ concentration, clearly detectable after 15 min, and reaching the maximum level after 30–60 min of treatment (images in Fig. 1d, histograms in Fig. 1e and corresponding kinetic plot in Fig. 1f). Cellular pre-treatment with CNQX or memantine determined again a reduction of cytoplasmic ROS levels in the early stages, up to 30 min of oligomer treatment, followed by an increase, until almost normal levels were reached after 60 min of treatment (Fig. 1d,e). Overall, these pre-treatments caused a deceleration of the increase of ROS levels (Fig. 1f), which was even more marked than that detected by monitoring intracellular Ca2+ levels. Moreover, after a pre-treatment with both CNQX and memantine, the ROS levels remained significantly lower than those of cells without inhibitor pre-treatment, up to 60 min of oligomer treatment (Fig. 1d,e).
Comparing the kinetics of cytosolic Ca2+ and ROS increases induced by HypF-N OAs in the absence of NMDA/AMPA inhibitors, the ROS time course appears to be slower, particularly in the first minutes (Fig. 2a). A lag time appears to be present only in the ROS time course, suggesting that the increase of ROS levels follows that in Ca2+ (Fig. 2a). Comparing the times courses in the presence of CNQX, memantine, or both, the ROS time course appears again to be slower than the corresponding time course of Ca2+ (Fig. 2a–c). Interestingly, we also observed a longer delay in ROS level increase following pre-treatment with CNQX (Fig. 2a, orange dotted line), or memantine (Fig. 2b, blue dotted line) or both (Fig. 2c, yellow dotted line) compared to Ca2+ levels following the same pre-treatment (Fig. 2a–c, orange, blue and yellow line, respectively), suggesting that inhibition of AMPA and NMDA receptors, with the consequent reduction of the early Ca2+ influx, allowed the cells to postpone the production of ROS even more markedly. Overall, all the kinetic data indicate that an increase of intracellular Ca2+ levels precedes ROS production.
Fig. 2.
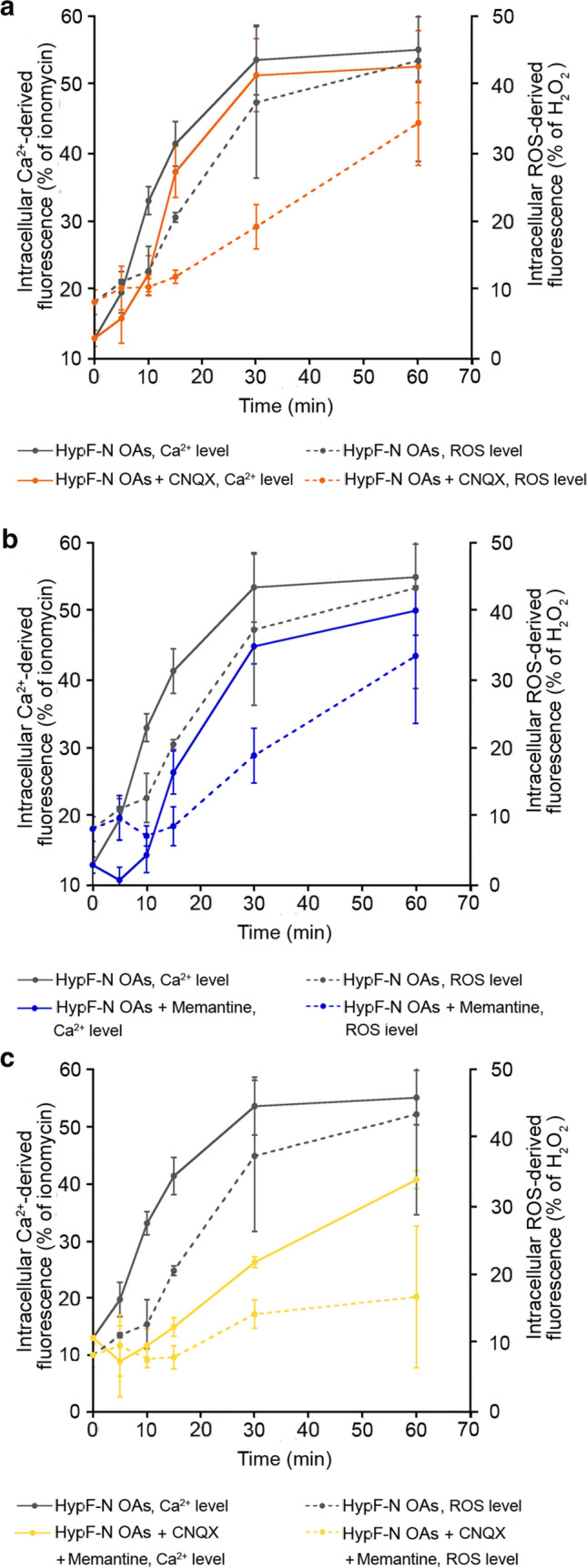
Increase of intracellular Ca2+ levels precedes ROS production. a–c Kinetic plots showing the fluorescence associated with intracellular Ca2+ and ROS versus time after treatment with HypF-N OAs. The time courses refer to Ca2+ levels (solid lines) and ROS levels (dotted lines) without inhibitors (grey), with CNQX (orange), with memantine (blue) and with both CNQX and memantine (yellow)
Lysophosphatidylcholine enrichment reduces both Ca2+ level increase and ROS production
It was previously shown that the enrichment of SH-SY5Y cell membranes with 2 μM lysophosphatidylcholine (LPC) inhibits the OA-induced Ca2+ flow mediated by the mechanosensitive NMDA receptors, suggesting that the opposing force exerted by LPC (compression) effectively inhibits the mechanical signal (stretching) generated by the action of the oligomers onto the membrane [14]. To investigate whether this inhibition is also effective on ROS production, we pre-treated SH-SY5Y cells with 2 μM LPC for 2 h, and then we treated them with HypF-N OAs (12 μM monomer equivalents) for 5, 10, 15, 30 and 60 min, monitoring both intracellular Ca2+ and ROS levels. In the presence of LPC, the Ca2+ levels remained similar to those of untreated cells up to 15 min; they then increased, but remained significantly lower than those recorded without LPC at corresponding time points, even after 60 min of treatment (Fig. 3a,b).
Fig. 3.

Lysophosphatidylcholine (LPC) enrichment reduces both the Ca2+ level increase and ROS production. a Representative confocal scanning microscopy images of intracellular free Ca2+ levels in SH-SY5Y cells following no treatment (first row) and treatment with 2 µM LPC (second row), and analysed after 5, 10, 15, 30, and 60 min of treatment with 12 µM (monomer equivalents) HypF-N OAs. b Semi-quantitative analysis of intracellular Ca2+-derived fluorescence. The value for untreated cells refers to 0 min and did not change with time. c Representative confocal scanning microscopy images of intracellular ROS levels in SH-SY5Y cells following no treatment (first row) and treatment with 2 µM LPC (second row), and analysed after 5, 10, 15, 30, and 60 min of treatment with 12 µM (monomer equivalents) HypF-N OAs. d Semi-quantitative analysis of intracellular ROS-derived fluorescence. The value for untreated cells refers to 15 min and did not change with time. Three different experiments were carried out, with 10–22 cells each, for each condition. Data are represented as mean ± SEM (n = 3). The single (*), double (**) and triple (***) asterisks refer to p values < 0.05, < 0.01 and < 0.001, respectively, relative to untreated cells. The single (§), double (§§) and triple (§§§) symbols refer to p values < 0.05, < 0.01 and 0.001, respectively, relative to HypF-N OAs without treatment with LPC at corresponding time points
The ROS levels also remained constant for 15 min and then increased, but remained significantly lower than the corresponding values in the absence of LPC pre-treatment, up to 60 min (Fig. 3c,d). These two time courses suggest that the LPC-mediated membrane compression is able to inhibit the OA-induced Ca2+ flow through NMDA receptors for a prolonged time and the subsequent rise of ROS levels. They also confirm the presence of an interconnection between the two mechanisms.
Intracellular Ca2+ influx and ROS production induced by HypF-N OAs are connected
As observed in the previous experiments, the intracellular rise of free Ca2+ is associated with the elevation of ROS following treatment with HypF-N OAs. The kinetic traces described so far lead to the hypothesis that the second event is caused by the first, rather than being independent of it, based on the observation that: (i) the former is more rapid than the latter, (ii) the rise of ROS has a lag time and follows the lag-independent rise of Ca2+, (iii) the lag times are longer in the ROS time courses than in the corresponding Ca2+ time courses in the presence of NMDA/AMPA inhibitors, and (iv) the effects caused by inhibitors of the Ca2+ flow (CNQX and memantine) are even larger on the time-dependent increase of ROS than Ca2+.
Since these suggestions are only kinetic and, therefore, not definitive, we further investigated if the two processes are linked to each other in our system with a clear cause-and-effect relationship between them. For this purpose, we treated the SH-SY5Y cells with HypF-N OAs over time, after a 1 h pre-treatment with 30 µM Trolox, which is a highly soluble and membrane-unbound antioxidant analogue of Tocopherol [43]. An increase of cytosolic Ca2+ concentration was observed in the early stages up 10 min, followed by a reduction down to levels observed in untreated cells (Fig. 4a,b, green bars). By contrast, the levels of ROS did not increase and were similar to the untreated cells at all time points, up to 60 min of OAs treatment (Fig. 4c,d, green bars). These results indicate that Trolox acted correctly as an antioxidant preventing ROS production in cells very effectively within the time frame explored here, but it did not prevent the early rise of Ca2+ mediated by AMPA and NMDA receptors. However, the presence of the antioxidant in the medium allowed the cells to re-establish the normal Ca2+ homeostasis that had been initially lost as a consequence of the activation of NMDA and AMPA receptors, indicating that the lack of ROS production allows the cells to face effectively the stress induced by the HypF-N OAs and the Ca2+ flow across the cell membrane.
Fig. 4.

Intracellular Ca2+ influx and ROS production induced by HypF-N OAs are connected. a Representative confocal scanning microscopy images of intracellular free Ca2+ levels in SH-SY5Y cells following no treatment (first row), pre-treatment with 30 µM Trolox (second row), and in a medium without Ca2+ (third row), and analysed after 5, 10, 15, 30, and 60 min of treatment with 12 µM (monomer equivalents) HypF-N OAs. b Semi-quantitative analysis of intracellular Ca2+-derived fluorescence. The value for untreated cells refers to 0 min and did not change with time. c Representative confocal scanning microscopy images of intracellular ROS levels in SH-SY5Y cells following no treatment (first row), pre-treatment with 30 µM Trolox (second row), and in a medium without Ca2+ (third row), and analysed after 5, 10, 15, 30, and 60 min of treatment with 12 µM (monomer equivalents) HypF-N OAs. d Semi-quantitative analysis of intracellular ROS-derived fluorescence. The value for untreated cells refers to 15 min and did not change with time. Three different experiments were carried out, with 10–22 cells each, for each condition. Data are represented as mean ± SEM (n = 3). The single (*), double (**) and triple (***) asterisks refer to p values < 0.05, < 0.01 and < 0.001, respectively, relative to untreated cells. The single (§), double (§§) and triple (§§§) symbols refer to p values < 0.05, < 0.01 and 0.001, respectively, relative to HypF-N OAs without treatment with Trolox or Ca2+-deprived medium at corresponding time points
In a control experiment, to assess whether Trolox interferes directly with the AMPA and NMDA receptor opening, we activated the two receptors using their specific agonists, AMPA and NMDA, respectively, after a 1 h pre-treatment with 30 µM Trolox, finding that both agonists are able to induce an increase of the intracellular Ca2+ levels independently of the presence of the antioxidant agent (Fig. S2).
With the same purpose of investigating the cause-and-effect link between Ca2+ and ROS level increases, we treated the SH-SY5Y cells with HypF-N OAs over time, in a Ca2+-free medium (Fig. 4, light blue bars). In this case, the OA-induced increase of cytosolic Ca2+ was fully inhibited, up to 60 min (Fig. 4a,b, light blue bars), confirming previous demonstrations that the source of such intracellular Ca2+ ions is the extracellular medium rather than intracellular organelles [10]. It is interesting to note that a complete inhibition of ROS production was also observed, again up to 60 min (Fig. 4c,d, light blue bars).
Taken together, the kinetic data obtained with Trolox and the Ca2+-free medium indicate that the lack of an influx of Ca2+ ions from the extracellular space into the cytosol is able to reduce ROS production, whereas the protection against ROS formation does not prevent an initial rise of intracellular Ca2+ concentration, underling the consequential nature of ROS formation relative to Ca2+ influx. They also provide evidence on the existence of an oxidative metabolism required to restore Ca2+ homeostasis and responsible for ROS accumulation, which does not allow an effective pumping of Ca2+ ions outside the cells, unless an antioxidant environment maintains the levels of ROS under control, allowing the cells to restore Ca2+ homeostasis effectively (see “Discussion” for further details).
Aβ42 ADDLs oligomers increase intracellular Ca2+ levels and ROS production
We then extended the analysis carried out with the model HypF-N OAs to Aβ oligomers, using Aβ42-derived diffusible ligands (Aβ42 ADDLs) [39] at the concentration of 1 μM. In previous works it was shown that Aβ42 ADDLs, similarly to HypF-N OAs, are able to cause a progressive increase of the intracellular Ca2+ levels in SH-SY5Y cells by activating rapidly extrasynaptic NMDA and AMPA receptors [14]. We therefore prepared freshly formed Aβ42 ADDLs oligomers and evaluated whether they maintained this effect. The treatment over time of SH-SY5Y cells with Aβ42 ADDLs showed a gradual increase of the intracellular Ca2+ levels, which was clearly detectable already after 5 min and reached a plateau after 180 min of treatment (images in Fig. 5a, histograms in Fig. 5b and corresponding kinetic plot in Fig. 5c). When cells were pre-treated with CNQX, with memantine, or with both CNQX and memantine, a slight reduction of the Aβ42 ADDLs-induced cytoplasmic Ca2+ increase was observed in the early stages, up to 10 min of treatment (Fig. 5a,b). With Aβ42 ADDLs, a combination of both inhibitors showed kinetics similar to the memantine treatment. This reduction was followed by a gradual increase of the intracellular Ca2+ concentration, until normal levels were reached after prolonged treatment (Fig. 5a,b). Overall, these pre-treatments cause a deceleration of the intracellular Ca2+ increase at early time points (Fig. 5c).
Fig. 5.

Aβ42 ADDLs oligomers increase intracellular Ca2+ levels and ROS production in SH-SY5Y cells. a Representative confocal scanning microscopy images of free Ca2+ levels in SH-SY5Y cells following the treatment with no inhibitors (first row), 5 µM CNQX (second row), 10 µM memantine (third row), and both inhibitors (fourth row), and analysed after 5, 10, 15, 30, 60, 90, 120 and 180 min of treatment with 1 µM (monomer equivalents) Aβ42 ADDLs oligomers. b Semi-quantitative analysis of intracellular free Ca2+-derived fluorescence. The value for untreated cells refers to 0 min and did not change with time. c Kinetic plots showing the fluorescence versus time as reported in panel b. d Representative confocal scanning microscopy images of intracellular ROS levels in SH-SY5Y cells following the treatment with no inhibitors (first row), 5 µM CNQX (second row), 10 µM memantine (third row), and both inhibitors (fourth row), and analysed after 5, 10, 15, 30, 60, 90, 120 and 180 min of treatment with 1 µM (monomer equivalents) Aβ42 ADDLs oligomers. e Semi-quantitative analysis of intracellular ROS-derived fluorescence. The value for untreated cells refers to 15 min and did not change with time. f Kinetic plots showing the fluorescence versus time as reported in panel e. Three different experiments were carried out, with 10–22 cells each, for each condition. Data are represented as mean ± SEM (n = 3). The single (*), double (**) and triple (***) asterisks refer to p values < 0.05, < 0.01 and < 0.001, respectively, relative to untreated cells. The single (§), double (§§) and triple (§§§) symbols refer to p values < 0.05, < 0.01 and 0.001, respectively, relative to Aβ42 ADDLs oligomers without inhibitors at corresponding time points
The treatment of SH-SY5Y cells with Aβ42 ADDLs under the same conditions also showed a gradual increase of ROS production, which was evident after 15 min up to 180 min, and hence slower than that observed by monitoring Ca2+ concentration (Fig. 5d–f). Interestingly, such increase appeared to occur more rapidly than that observed with HypF-N OAs, which can be attributed to the known oxidative potential of Aβ42 ADDLs through Ca2+-independent mechanisms [44–46]. Cellular pre-treatment with CNQX or memantine, or both inhibitors, determined again a reduction of ROS levels in the early stages, up to 30 min for CNQX and 60 min for memantine and both inhibitors together, followed by a gradual increase, until normal levels were reached after 90 min (Fig. 5d–f). These pre-treatments, therefore, caused a deceleration of the ROS increase mediated by the oligomers (Fig. 5f), which was again more marked than that detected by monitoring intracellular Ca2+ levels. All these results confirmed the observation with the HypF-N OAs.
Comparing the Ca2+ and ROS kinetics without NMDA/AMPA inhibitors, the ROS time course appears to be slower in the first minutes (Fig. 6a), suggesting that the increase of the intracellular Ca2+ levels anticipates ROS production. Moreover, comparing the times courses in the presence of CNQX or memantine, or both, the ROS time courses appear again to be slower than the corresponding time courses of Ca2+ (Fig. 6a–c). Also with this type of oligomers we observed a longer delay in ROS level increase following pre-treatment with CNQX (Fig. 6a, orange dotted line) or memantine (Fig. 6b, blue dotted line) or both (Fig. 6c, yellow dotted line), compared to the Ca2+ kinetics following the same pre-treatment (Fig. 6a,b, orange, blue and yellow line, respectively), confirming that the reduction of the early Ca2+ influx, observed by inhibiting the NMDA and AMPA receptors, allowed the cells to delay the production of ROS.
Fig. 6.
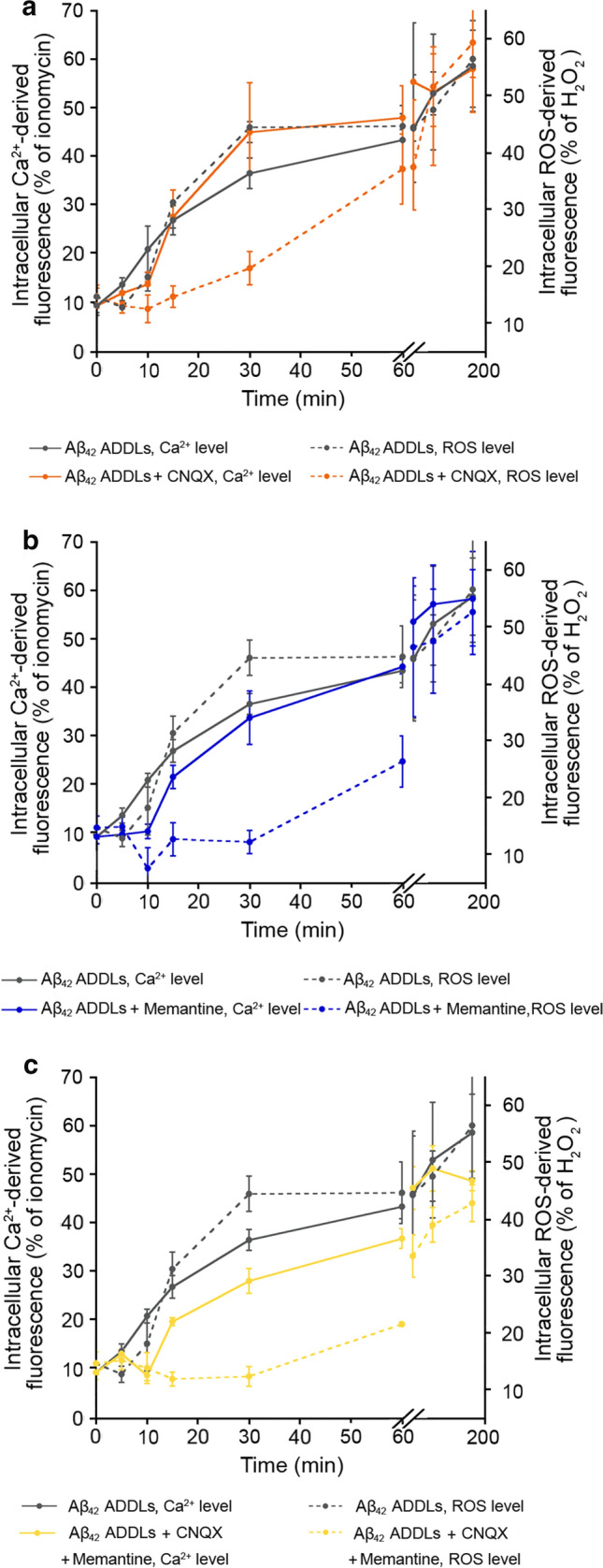
Increase of intracellular Ca2+ levels anticipates ROS production in SH-SY5Y cells. a–c Kinetic plots showing the fluorescence associated with intracellular Ca2+ and ROS versus time after treatment with Aβ42 ADDLs. The time courses refer to Ca.2+ levels (solid lines) and ROS levels (dotted line) without inhibitors (grey), with CNQX (orange), with memantine (blue) and with both CNQX and memantine (yellow)
When repeated on primary rat cortical neurons, the Aβ42 ADDLs had a similar effect. After 10 min of treatment, the Aβ42 ADDLs induced an increase of the intracellular Ca2+ levels, which further increased after 60 min of treatment (Fig. 7a,b). When the cells were pre-treated with CNQX or memantine, a significant reduction of the Aβ42 ADDLs-induced cytoplasmic Ca2+ levels was observed after 10 min of treatment with the oligomers, confirming the involvement of the receptors in the Ca2+ influx (Fig. 7a,b). After 60 min of treatment with ADDLs, the levels of Ca2+ in the presence of pre-treatment went back to the levels observed in its absence (Fig. 7a,b). Moreover, Aβ42 ADDLs also induced an increase of ROS levels after 10 min and a further increase after 60 min of treatment (Fig. 7c,d), with the former being significantly reduced with CNQX or memantine (Fig. 7c,d).
Fig. 7.

Aβ42 ADDLs oligomers increase intracellular Ca2+ levels and ROS production in primary rat cortical neurons. a Representative confocal scanning microscopy images of intracellular free Ca2+ levels in primary rat cortical neurons treated with no inhibitors (first row), 5 µM CNQX (second row) and 10 µM memantine (third row), and analysed after 10 and 60 min of treatment with 1 µM (monomer equivalents) Aβ42 ADDLs oligomers. b Semi-quantitative analysis of intracellular free Ca2+-derived fluorescence. c Representative confocal scanning microscopy images of intracellular ROS levels in primary rat cortical neurons treated with no inhibitors (first row), 5 µM CNQX (second row) and 10 µM memantine (third row), and analysed after 10 and 60 min of treatment with 1 µM (monomer equivalents) Aβ42 ADDLs oligomers. d Semi-quantitative analysis of intracellular ROS-derived fluorescence. Three different experiments were carried out, with 10–22 cells each, for each condition. Data are represented as mean ± SEM (n = 3). The single (*) and double (**) asterisks refer to p values < 0.05 and < 0.01, respectively, relative to untreated cells. The single (§) and double (§§) symbols refer to p values < 0.05 and < 0.01, respectively, relative to Aβ42 ADDLs oligomers without inhibitors at corresponding time points
Intracellular Ca2+ influx and ROS production induced by Aβ42 ADDLs are connected
We then treated SH-SY5Y cells with Aβ42 ADDLs in the presence and absence of a pre-treatment for 1 h with the antioxidant Trolox. In the presence of Trolox, an initial increase of cytosolic Ca2+ concentration was observed, particularly after 10–30 min of treatment with the oligomers, followed by a reduction at180 min (Fig. 8a,b). These results confirm that the maintenance of a redox balance allowed the cells to react to the initial Ca2+ flux induced by the Aβ42 ADDLs and normalize Ca2+ homeostasis, initially lost because of the action of the oligomers.
Fig. 8.

Intracellular Ca2+ influx and ROS production induced by Aβ42 ADDLs are connected in SH-SY5Y cells. a Representative confocal scanning microscopy images of intracellular free Ca2+ levels in SH-SY5Y cells following no treatment (first row), and pre-treatment with 30 µM Trolox (second row), and analysed after 5, 10, 15, 30, 60, 90, 120 and 180 min of treatment with 1 µM (monomer equivalents) Aβ42 ADDLs oligomers. b Semi-quantitative analysis of intracellular Ca2+-derived fluorescence. The value for untreated cells refers to 0 min and did not change with time. c Representative confocal scanning microscopy images of intracellular ROS levels in SH-SY5Y cells following no treatment (first row), and treatment in a medium without Ca2+ (second row), and analysed after 5, 10, 15, 30, 60, 90, 120 and 180 min of treatment with 1 µM (monomer equivalents) Aβ42 ADDLs oligomers. d Semi-quantitative analysis of intracellular ROS-derived fluorescence. The value for untreated cells refers to 15 min and did not change with time. Three different experiments were carried out, with 10–22 cells each, for each condition. Data are represented as mean ± SEM (n = 3). The double (**) and triple (***) asterisks refer to p values < 0.01 and < 0.001, respectively, relative to untreated cells. The single (§), double (§§) and triple (§§§) symbols refer to p values < 0.05, < 0.01 and < 0.001, respectively, relative to Aβ42 ADDLs oligomers without treatment with Trolox or Ca2+-deprived medium at corresponding time points
With the same purpose, ROS production in SH-SY5Y cells was evaluated after treatment with Aβ42 ADDLs over time, with or without Ca2+ in the cell medium. The absence of extracellular Ca2+ determined levels of ROS similar to those observed in untreated cells up to 180 min of treatment with the oligomers, without any initial increase at early time points (Fig. 8c,d), indicating that the cells without any Ca2+ influx and dyshomeostasis did not undergo any oxidative stress, despite the treatment with toxic Aβ42 ADDLs in the absence of antioxidants (Fig. 8c,d). These results emphasise that while the suppression of the Ca2+ influx in the cells suppresses the oxidative stress for the entire length of time of the analysis, the cellular protection by a reducing agent does not suppress the initial oligomer-induced Ca2+ influx.
To confirm these results with different probes of intracellular Ca2+ and ROS, we repeated the experiments with ADDLs after 10 and 60 min of treatment, with or without Trolox and with or without Ca2+ in the cell medium, using the X-Rhod-1 AM and the CellRoxTM Deep Red Reagent to monitor Ca2+ and ROS levels, respectively. The results confirm that the presence of the antioxidant allowed the cells to react to and normalise the initial Ca2+ influx observed after 10 min of treatment, which reached the levels of untreated cells after 60 min of treatment (Fig. S3a,b), and that ROS levels remained constant and similar to those of untreated cells when the treatment was performed in a medium without Ca2+ (Fig. S3c,d).
The effect of Trolox was also tested on primary rat cortical neurons. The cells were treated with Aβ42 ADDLs for 10 or 60 min, with or without the 1 h pre-treatment with Trolox. Similarly to SH-SY5Y cells, the presence of Trolox did not prevent a slight increase of the intracellular Ca2+ concentration, but caused lower levels of Ca2+ after both 10 and 60 min of treatment with Aβ42 ADDLs relative to cells pre-treated with Trolox (Fig. 9). This suggests that also in this cellular system the oxidative stress reduction allows the cells to counteract the initial Ca2+ influx across the membrane and restore the normal levels of Ca2+.
Fig. 9.
Intracellular Ca2+ influx and ROS production induced by Aβ42 ADDLs are connected in primary rat cortical neurons. a Representative confocal scanning microscopy images of intracellular free Ca2+ levels in primary rat cortical neurons with no treatment (first row), and pre-treatment with 30 µM Trolox (second row), and analysed after 10 and 60 min of treatment with 1 µM (monomer equivalents) Aβ42 ADDLs oligomers. b Semi-quantitative analysis of intracellular free Ca2+-derived fluorescence. Three different experiments were carried out, with 10–22 cells each, for each condition. Data are represented as mean ± SEM (n = 3). The single (*) and double (**) asterisks refer to p values < 0.05 and < 0.01, respectively, relative to untreated cells. The single (§) symbol refers to p values < 0.05 relative to Aβ42 ADDLs oligomers without pre-treatment with Trolox at corresponding time points
Discussion
Dysregulation of Ca2+ signalling and excessive production of intracellular ROS are common early features of neurodegenerative disorders, in particular AD [13, 15, 47, 48]. Several studies have shown that the passage from the extracellular space into the cytosol of small molecules and ions, such as Ca2+ ions, is mediated by the interaction of Aβ oligomers, characteristic of AD, with the lipid bilayer [7, 11, 14, 17, 18]. Our results confirm these observations. They also confirm that this early modification is associated with the increase of cytosolic ROS levels. Interestingly, the increase of ROS production appears to occur more rapidly following the treatment with Aβ42 ADDLs than HypF-N OAs, which can be attributed to the known oxidative potential of Aβ42 ADDLs through Ca2+-independent mechanisms [44–46].
The maintenance of the Ca2+ gradient across the cell membrane, where the Ca2+ concentration is 50–100 nM inside the cell and 1.1 mM outside, represents a great energetic expense, because the plasma membrane Ca2+-ATPase (PMCA) and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) need ATP to pump out the ions from the cytosol and restore homeostasis [6, 28, 36, 37]. Therefore, the increased need for ATP caused by the oligomer-induced Ca2+ dyshomeostasis activates the Krebs cycle, electron transport chain and oxidative phosphorylation in mitochondria, which determines the mitochondrial generation of ROS through the increased O2 reduction [28, 37]. ROS can also be produced by extramitochondrial enzymes, such as NADPH oxidase, xanthine oxidase, cytochrome P450, myeloperoxidase, cyclooxygenase, lipoxygenase and uncoupled nitric oxide synthase, all of which are modulated by Ca2+ [37]. This explains the association between Ca2+ dysregulation and increased ROS production.
The kinetic results presented here show that the delay in ROS production, which is evident as a lag phase and slower overall process in both time courses of ROS production following Aβ42 ADDLs and HypF-N OAs addition, is suggestive, albeit not a demonstration per se, that the ROS increase follows, and is caused by, that in Ca2+. To address further the cause-and-effect relationship between these two events, we took into consideration the data obtained with inhibitors of the Ca2+ influx and the known relationship between the two processes. Indeed, the extracellular-to-cytosol influx of Ca2+ induced by misfolded protein oligomers arises, at least in its early stages, from the passage of the ions through the AMPA and NMDA receptors [9, 15, 18, 19], which are mechanically activated following the modification of the phospholipid bilayer induced by the oligomers [14]. The pharmacological inhibition of the two glutamatergic receptors, with CNQX and memantine, respectively, delayed transiently the Ca2+ influx induced by these oligomers, with no significant increase within the first minutes of treatment. The delay mediated by CNQX and memantine, however, did not only involve the Ca2+ influx, but also ROS production. We also observed a delay in ROS levels increase following the pre-treatment with the inhibitors and this delay was even larger than that observed for Ca2+ levels. These results suggest that the inhibition of AMPA and NMDA receptors, with the consequent reduction of the early Ca2+ influx, allowed the cells to postpone ROS production. At later time points, intracellular Ca2+ levels increase despite the persistent inactivation of the two receptors, reaching the same levels observed in the absence of any inhibition, because it is caused by the direct passage of the ions through the cell membrane after the interaction of the oligomers with the lipid bilayer and a consequent destabilization and perforation [17, 18]. In addition, the Ca2+ pumps are inhibited by ROS, contributing to increase Ca2+ levels at later time points (see below). In the same way, ROS production increases, while continuing to maintain this slight delay because of the inactivation of the AMPA/NMDA receptors. The kinetic data, in particular, indicate that the use of either CNQX or memantine, or both, results in a lag time of the increase in Ca2+ levels, followed by the extension of the lag phase in ROS production. Other Ca2+ channels are probably involved in the oligomer-mediated Ca2+ influx, such as TRPM2 [23], VDCCs [24] and TRPA1 [25], but these have not been found previously to be involved significantly in our system [14]. The delay caused by CNQX and Memantine on Ca2+ and ROS kinetics was lower on cells treated with Aβ42 ADDLs, compared to cells treated with HypF-N OAs, probably because the damaging action of the first type of oligomer on the membrane is stronger, and, after its interaction with the lipid belayer and the consequent destabilization and perforation, the direct passage of the ions through the cell membrane is more pronounced compared to that of HypF-N OAs.
In AD brains, high levels of intracellular ROS were found to react with several macromolecules, such as proteins, polysaccharides, nucleic acids and lipids, causing their oxidation and leading to the production of reactive ketone/aldehyde moieties and other carbonyl derivatives [48]. An important deleterious effect of ROS in the brain is lipid peroxidation, which directly damages biological membranes [44, 49]. Moreover, high levels of ROS cause the oxidation of multiple methionine residues of the Ca2+ signalling protein calmodulin (CaM), with its consequent inability to activate its target proteins, including the PMCA, which is important for the maintenance of Ca2+ homeostasis [36, 50, 51]. High levels of ROS also result in tyrosine and cysteine oxidation of the SERCA [52–54]. Indeed, upon treatment with the antioxidant agent Trolox in our experiments, which completely inhibits the increase of ROS levels and prevents its damaging effects, it is likely that the cells are able to restore Ca2+ homeostasis effectively, as a result of the lack of ROS-mediated oxidation of the PMCA and SERCA, among other cellular factors.
The selective oxidation and inactivation of the Ca2+ regulatory proteins mediated by ROS may represent an adaptive response to the oxidative stress, because it down-regulates ATP production through the mitochondrial electron transport chain and the inevitable generation of ROS associated with it [28].
Further evidence of the importance of Ca2+ influx in ROS production occurs in the treatment with the oligomers in a medium without Ca2+ (to inhibit Ca2+ influx) and with an antioxidant agent (to inhibit ROS production). The absence of Ca2+ in the extracellular medium fully inhibits the increase of the intracellular Ca2+ levels that normally flow from the extracellular space, but at the same time fully inhibits ROS production, confirming that ROS result from the need to restore Ca2+ homeostasis. By contrast, treatment with the antioxidant agent Trolox leads the restoration of Ca2+ homeostasis, but only at prolonged time points. This latter analysis showed that Ca2+ ions enter the cells in the first minutes, because the antioxidant agent inhibits only ROS production and is not able to inhibit the oligomer-mediated activation of AMPA and NMDA receptors that occurs within the first minutes of interaction of the oligomers with the cell membrane. This rapid increase of intracellular Ca2+ is followed by a decrease, suggesting that the cells are able to pump out Ca2+ and restore homeostasis as they benefit from an effective antioxidant capacity induced by Trolox and absence of any direct ROS-induced inhibition of the PMCA, SERCA, other pumps and possibly other cellular factors involved in these processes.
Conclusions
Vicious cycles, or positive feedback loops, exist between Ca2+ signalling and ROS production [28, 37], and even between Aβ production and Ca2+ signalling [55] and between Aβ production and ROS production [55], where the various events sustain each other. However, a precise cause-and-effect relationship between increased levels of intracellular Ca2+ and cytosolic ROS production at the very early stages of the overall dysregulation induced by misfolded protein oligomers emerges from our results by three distinct lines of evidence, namely: (i) a lag time observed in the time course of oligomer-induced ROS production (but not in Ca2+ increase), (ii) an ability of AMPA/NMDA receptor inhibitors to retard ROS production even more effectively than Ca2+ influx and (iii) an inability of antioxidant agents to inhibit the early Ca2+ influx, while fully maintaining the redox status of the cells, whereas a Ca2+ deprived medium inhibits fully and effectively both Ca2+ influx and ROS production. Hence, the oligomers cause the entry of Ca2+ ions in the cells, determining the formation of ROS due to the increased demand of ROS-generating ATP production by mitochondria; ROS in turn prevent the cells from pumping back Ca2+ ions into the extracellular space and from restoring the normal Ca2+ homeostasis, indicating a positive feedback on Ca2+ dyshomeostasis on the longer time scale.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
GF: conceptualization, investigation, validation, formal analysis, visualisation, writing—original draft, writing—review and editing. CELT: investigation, formal analysis. RC: conceptualization, writing—review and editing. CC: conceptualization, writing—review and editing, funding acquisition. MV: conceptualization, writing—review and editing. FC: conceptualization, writing—review and editing, supervision, project administration, funding acquisition.
Funding
Open access funding provided by Università degli Studi di Firenze within the CRUI-CARE Agreement. This work was supported by Regione Toscana (FAS-Salute 2018), Project PRAMA (GF, RC, CC, FC), by Ministero dell’Istruzione, dell’Università e della Ricerca, Project Dipartimento di Eccellenza (CC) and by Università degli Studi di Firenze, Fondi di Ateneo (RC, CC and FC).
Data availability
Data will be made available on reasonable request.
Declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Michele Vendruscolo, Email: mv245@cam.ac.uk.
Fabrizio Chiti, Email: fabrizio.chiti@unifi.it.
References
- 1.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stancu IC, Vasconcelos B, Terwel D, Dewachter I. Models of β-amyloid induced Tau-pathology: the long and “folded” road to understand the mechanism. Mol Neurodegener. 2014;9:51. doi: 10.1186/1750-1326-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68. doi: 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- 4.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 5.Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 6.Cascella R, Cecchi C. Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. Int J Mol Sci. 2021;22:4914. doi: 10.3390/ijms22094914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 8.Diaz JC, Simakova O, Jacobson KA, Arispe N, Pollard HB. Small molecule blockers of the Alzheimer Abeta calcium channel potently protect neurons from Abeta cytotoxicity. Proc Natl Acad Sci USA. 2009;106:3348–3353. doi: 10.1073/pnas.0813355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decker H, Jürgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST. N-methyl-d-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-β peptide oligomers. J Neurochem. 2010;115:1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x. [DOI] [PubMed] [Google Scholar]
- 10.Zampagni M, Cascella R, Casamenti F, Grossi C, Evangelisti E, Wright D, Becatti M, Liguri G, Mannini B, Campioni S, Chiti F, Cecchi C. A comparison of the biochemical modifications caused by toxic and non-toxic protein oligomers in cells. J Cell Mol Med. 2011;15:2106–2116. doi: 10.1111/j.1582-4934.2010.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evangelisti E, Cecchi C, Cascella R, Sgromo C, Becatti M, Dobson CM, Chiti F, Stefani M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J Cell Sci. 2012;125:2416–2427. doi: 10.1242/jcs.098434. [DOI] [PubMed] [Google Scholar]
- 12.Cascella R, Conti S, Mannini B, Li X, Buxbaum JN, Tiribilli B, Chiti F, Cecchi C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins in vitro. Biochim Biophys Acta. 2013;1832:2302–2314. doi: 10.1016/j.bbadis.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Tong BC, Wu AJ, Li M, Cheung KH. Calcium signaling in Alzheimer’s disease & therapies. Biochim Biophys Acta Mol Cell Res. 2018;1865:1745–1760. doi: 10.1016/j.bbamcr.2018.07.018. [DOI] [PubMed] [Google Scholar]
- 14.Fani G, Mannini B, Vecchi G, Cascella R, Cecchi C, Dobson CM, Vendruscolo M, Chiti F. Aβ oligomers dysregulate calcium homeostasis by a mechanosensitive activation of AMPA and NMDA receptors. ACS Chem Neurosci. 2021;12:766–781. doi: 10.1021/acschemneuro.0c00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 16.García F, Lobos P, Ponce A, Cataldo K, Meza D, Farías P, Estay C, Oyarzun-Ampuero F, Herrera-Molina R, Paula-Lima A, Ardiles ÁO, Hidalgo C, Adasme T, Muñoz P. Astaxanthin counteracts excitotoxicity and reduces the ensuing increases in calcium levels and mitochondrial reactive oxygen species generation. Mar Drugs. 2020;18:335. doi: 10.3390/md18060335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sepúlveda FJ, Fierro H, Fernandez E, Castillo C, Peoples RW, Opazo C, Aguayo LG. Nature of the neurotoxic membrane actions of amyloid-β on hippocampal neurons in Alzheimer’s disease. Neurobiol Aging. 2014;35:472–481. doi: 10.1016/j.neurobiolaging.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 18.Cascella R, Evangelisti E, Bigi A, Becatti M, Fiorillo C, Stefani M, Chiti C, Cecchi C. Soluble oligomers require a ganglioside to trigger neuronal calcium overload. J Alzheimers Dis. 2017;60:923–938. doi: 10.3233/JAD-170340. [DOI] [PubMed] [Google Scholar]
- 19.Alberdi E, Sánchez-Gómez MV, Cavaliere F, Pérez-Samartín A, Zugaza JL, Trullas R, Domercq M, Matute C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47:264–272. doi: 10.1016/j.ceca.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 20.Texidó L, Martín-Satué M, Alberdi E, Solsona C, Matute C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium. 2011;49:184–190. doi: 10.1016/j.ceca.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 21.Sinnen BL, Bowen AB, Gibson ES, Kennedy MJ. Local and use-dependent effects of β-amyloid oligomers on NMDA receptor function revealed by optical quantal analysis. J Neurosci. 2016;36:11532–11543. doi: 10.1523/JNEUROSCI.1603-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tozaki H, Matsumoto A, Kanno T, Nagai K, Nagata T, Yamamoto S, Nishizaki T. The inhibitory and facilitatory actions of amyloid-beta peptides on nicotinic ACh receptors and AMPA receptors. Biochem Biophys Res Commun. 2002;294:42–45. doi: 10.1016/S0006-291X(02)00429-1. [DOI] [PubMed] [Google Scholar]
- 23.Ostapchenko VG, Chen M, Guzman MS, Xie YF, Lavine N, Fan J, Beraldo FH, Martyn AC, Belrose JC, Mori Y, MacDonald JF, Prado VF, Prado MA, Jackson MF. The transient receptor potential melastatin 2 (TRPM2) channel contributes to β-Amyloid oligomer-related neurotoxicity and memory impairment. J Neurosci. 2015;35:15157–15169. doi: 10.1523/JNEUROSCI.4081-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quan QK, Li X, Yuan HF, Wang Y, Liu WL. Ginsenoside Rg1 inhibits high-voltage-activated calcium channel currents in hippocampal neurons of beta-amyloid peptide-exposed rat brain slices. Chin J Integr Med. 2016 doi: 10.1007/s11655-015-2301-4. [DOI] [PubMed] [Google Scholar]
- 25.Bosson A, Paumier A, Boisseau S, Jacquier-Sarlin M, Buisson A, Albrieux M. TRPA1 channels promote astrocytic Ca2+ hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-β peptide. Mol Neurodegener. 2017;12:53. doi: 10.1186/s13024-017-0194-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, Smith MA. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59:290–294. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Squier TC. Oxidative stress and protein aggregation during biological aging. Exp Gerontol. 2001;36:1539–1550. doi: 10.1016/s0531-5565(01)00139-5. [DOI] [PubMed] [Google Scholar]
- 29.Rhein V, Baysang G, Rao S, Meier F, Bonert A, Müller-Spahn F, Eckert A. Amyloid-β leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell Mol Neurobiol. 2009;29:1063–1071. doi: 10.1007/s10571-009-9398-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spuch C, Ortolano S, Navarro C. New insights in the amyloid-β interaction with mitochondria. J Aging Res. 2012;2012:324968. doi: 10.1155/2012/324968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bobba A, Amadoro G, Valenti D, Corsetti V, Lassandro R, Atlante A. Mitochondrial respiratory chain complexes I and IV are impaired by β-amyloid via direct interaction and through complex I-dependent ROS production, respectively. Mitochondrion. 2013;13:298–311. doi: 10.1016/j.mito.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Gibson GE, Blass JP, Beal MF, Bunik V. The α-ketoglutarate-dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- 33.Practicò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leuner K, Schütt T, Kurz C, Eckert SH, Schiller C, Occhipinti A, Mai S, Jendrach M, Eckert GP, Kruse SE, Palmiter RD, Brandt U, Dröse S, Wittig I, Willem M, Haass C, Reichert AS, Müller WE. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid β formation. Antioxid Redox Signal. 2012;16:1421–1433. doi: 10.1089/ars.2011.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordeeva AV, Zvyagilskaya RA, Labas YA. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry (Mosc) 2003;68:1077–1080. doi: 10.1023/a:1026398310003. [DOI] [PubMed] [Google Scholar]
- 36.Squier TC, Bigelow DJ. Protein oxidation and age-dependent alterations in calcium homeostasis. Front Biosci. 2000;5:D504–526. doi: 10.2741/squier. [DOI] [PubMed] [Google Scholar]
- 37.Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, Chiti F. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. 2010;6:140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 39.Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL. Vaccination with soluble Aβ oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79:595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- 40.Capitini C, Conti S, Perni M, Guidi F, Cascella R, De Poli A, Penco A, Relini A, Cecchi C, Chiti F. TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS One. 2014;9:e86720. doi: 10.1371/journal.pone.0086720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tatini F, Pugliese AM, Traini C, Niccoli S, Maraula G, Ed Dami T, Mannini B, Scartabelli T, Pedata F, Casamenti F, Chiti F. Amyloid-β oligomer synaptotoxicity is mimicked by oligomers of the model protein HypF-N. Neurobiol Aging. 2013;34:2100–2109. doi: 10.1016/j.neurobiolaging.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 42.Evangelisti E, Cascella R, Becatti M, Marrazza G, Dobson CM, Chiti F, Stefani M, Cecchi C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci Rep. 2016;6:32721. doi: 10.1038/srep32721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui L, McClements DJ, Decker EA. Impact of phosphatidylethanolamine on the antioxidant activity of α-tocopherol and trolox in bulk oil. J Agric Food Chem. 2015;63:3288–3294. doi: 10.1021/acs.jafc.5b00243. [DOI] [PubMed] [Google Scholar]
- 44.Zampagni M, Evangelisti E, Cascella R, Liguri G, Becatti M, Pensalfini A, Uberti D, Cenini G, Memo M, Bagnoli S, Nacmias B, Sorbi S, Cecchi C. Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. J Mol Med. 2010;88:597–608. doi: 10.1007/s00109-010-0603-8. [DOI] [PubMed] [Google Scholar]
- 45.Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:316523. doi: 10.1155/2013/316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tönnies E, Trushina E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimers Dis. 2017;57:1105–1121. doi: 10.3233/JAD-161088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakroborty S, Stutzmann GE. Calcium channelopathies and Alzheimer’s disease: insight into therapeutic success and failures. Eur J Pharmacol. 2014;739:83–95. doi: 10.1016/j.ejphar.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 48.Gella A, Durany N. Oxidative stress in Alzheimer disease. Cell Adh Migr. 2009;3:88–93. doi: 10.4161/cam.3.1.7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montine KS, Quinn JF, Zhang J, Fessel JP, Robert LJ, II, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Gao J, Yao Y, Squier TC. Oxidatively modified calmodulin binds to the plasma membrane Ca-ATPase in a nonproductive and conformationally disordered complex. Biophys J. 2001;80:1791–1801. doi: 10.1016/S0006-3495(01)76149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zaidi A. Plasma membrane Ca-ATPases: targets of oxidative stress in brain aging and neurodegeneration. World J Biol Chem. 2010;1:271–280. doi: 10.4331/wjbc.v1.i9.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schöneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem J. 1999;340:657–669. doi: 10.1042/bj3400657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viner RI, Williams TD, Schöneich C. Peroxynitrite modification of protein thiols: oxidation, nitrosylation, and S-glutathiolation of functionally important cysteine residue(s) in the sarcoplasmic reticulum Ca-ATPase. Biochemistry. 1999;38:12408–12415. doi: 10.1021/bi9909445. [DOI] [PubMed] [Google Scholar]
- 54.Sharov VS, Dremina ES, Galeva NA, Williams TD, Schöneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J. 2006;394:605–615. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanabria-Castro A, Alvarado-Echeverría I, Monge-Bonilla C. Molecular pathogenesis of Alzheimer's disease: an update. Ann Neurosci. 2017;24:46–54. doi: 10.1159/000464422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on reasonable request.