

Abstract
High-throughput assay systems have had a large impact on understanding the mechanisms of basic cell functions. However, high-throughput assays that directly assess molecular functions are limited. Herein, we describe the “GigaAssay”, a modular high-throughput one-pot assay system for measuring molecular functions of thousands of genetic variants at once. In this system, each cell was infected with one virus from a library encoding thousands of Tat mutant proteins, with each viral particle encoding a random unique molecular identifier (UMI). We demonstrate proof of concept by measuring transcription of a GFP reporter in an engineered reporter cell line driven by binding of the HIV Tat transcription factor to the HIV long terminal repeat. Infected cells were flow-sorted into 3 bins based on their GFP fluorescence readout. The transcriptional activity of each Tat mutant was calculated from the ratio of signals from each bin. The use of UMIs in the GigaAssay produced a high average accuracy (95%) and positive predictive value (98%) determined by comparison to literature benchmark data, known C-terminal truncations, and blinded independent mutant tests. Including the substitution tolerance with structure/function analysis shows restricted substitution types spatially concentrated in the Cys-rich region. Tat has abundant intragenic epistasis (10%) when single and double mutants are compared.
Keywords: Tat, Transcription, High-throughput assay, Saturation mutagenesis, Protein structure, Intragenic epistasis
1. Introduction
High-throughput screening (HTS) technologies have transformed biomedical sciences and many of these technologies have sufficiently improved to have an impact on clinical care. Most high-throughput technologies identify cellular components such as DNA, RNA, or protein species, and some assess intermolecular interactions. CRISPR/Cas9 and RNAi genome-wide screens identify genes necessary for cellular or organismal processes. Pathways and networks are often predicted from the resulting data, but these experiments only indicate a role for a gene, and do not conclusively assess mechanisms of action.
Although many high-throughput screens have been developed, there is no platform to broadly assesses molecular functions and cell processes in the context of human or mammalian cells. [1,2] These functions are the key to understanding disease etiology and mechanism, and to the development of therapeutic drugs. Some assays have been developed to assess a subset of molecular functions. For example, phage display, yeast display, and yeast 1- or 2-hybrid screens assess molecular interactions. However, these methods to not assess interactions in living mammalian cells and lack native post-translational modifications. [3–6] Likewise, deep mutational scanning (DMS) has been used to assess specific enzymatic activities for phosphatase and tensin homolog (PTEN) or Arylsulfatases [7], and to examine G-protein-coupled receptor signaling. [8,9]
DMS lethality or toxicity selection screens of mutant libraries, including some screens in mammalian cells, identify candidate loss- or gain-of-function mutants. For example, cells with one of many different p53 mutants will not survive after two weeks of culture. [10] Variants that survive lethality selection are identified by comparing sequences from pre-screen and post-screen samples; negatives are inferred for those mutants that do not survive the screen. However, survival screens measure a cell response far downstream of many molecular functions and may contribute to a high rate of false positives.
Here, we demonstrate proof-of-principle for a new modular high-throughput assay system we call the GigaAssay. The GigaAssay system is a one-pot, single-cell assay for molecular activities in living mammalian cells, in which each cDNA molecule with a genetic variant is individually barcoded with a unique molecular identifier (UMI), assayed by a fluorescent readout, flow sorted into bins by fluorescent signal intensity, deep sequenced, and then the impact of each genetic variant on a specific activity is bioinformatically deconvolved. The GigaAssay is not a survival screen in which negatives are not directly measured. The GigaAssay measures both positives and negatives for nearly a million individually UMI-barcoded DNA molecules in human cells. Herein, we present results that demonstrate the GigaAssay is a highly accurate, adaptable, and reproducible assay.
The GigaAssay has several other advantages over previously developed high throughput assays and screens. It is flexibleand readily adapted to many cell processes and molecular function assays in living mammalian cells (Fig. 1A). For example, in this paper we present results measuring transcription driven by HIV Tat, but have also adapted the assay to measure activation of the MAPK pathway signaling reporter, phosphorylation of Her2, and autocrine activation of an interferon-sensitive response element signaling reporter. It is a high-throughput assay capable of measuring tens of thousands of reads for each of about a million individually UMI-barcoded cDNAs, in which different genotypes are pooled for each amino acid substitution. In this way, robust statistical probabilities and metrics can be calculated to determine the reliability of each measurement. The high throughput and the high reproducibility among both technical and biological replicate cell lines greatly increase the accuracy of the results. In this one-pot assay system, each DNA variant molecule is barcoded in a plasmid library, and after several steps in the GigaAssay, is bioinformatically deconvolved to determine its functional activity.
Fig. 1.
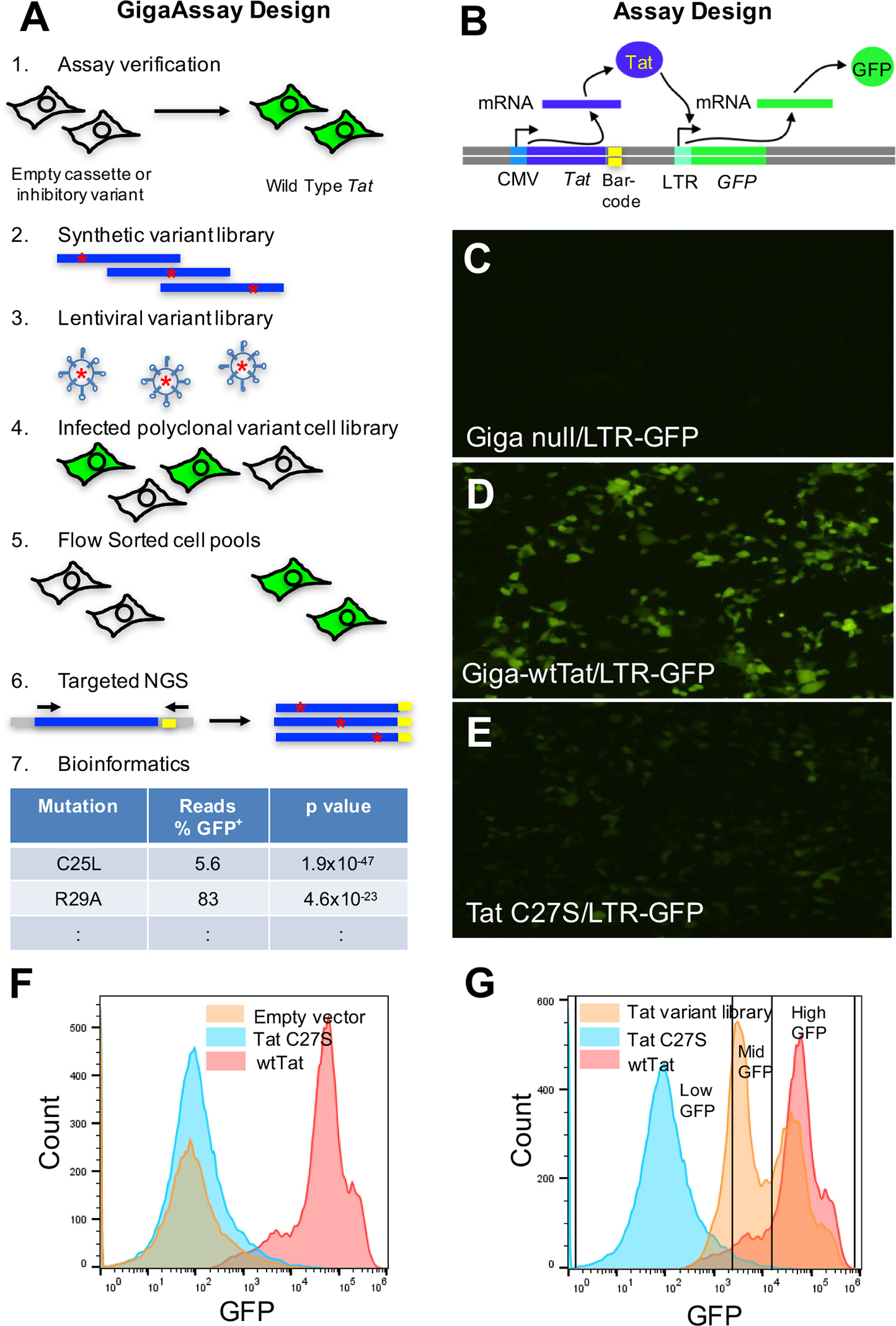
Design and implementation of the GigaAssay for Tat transcriptional activation. A. Design of GigaAssay system. Propagation of the recombined cells under poison selection. Cell sorting based on GFP reporter expression. gDNA is isolated, and a targeted Tat amplicon library is prepared and sequenced by NGS. Schematic representation of Tat dependent LTR transactivation inducing GFP expression. B.-D. Epifluorescence microscopic images of LentiX293T/LTR-GFP cells transfected with GigaAssay plasmids: Empty vector/LTR-GFP (B. - control); wtTat/LTR-GFP (C, + control); and an inhibitory mutant [12], C27S-Tat/LTR-GFP (D, − control). E. Flow cytometry of GigaAssay controls in LentiX293T/LTR-GFP cell to define gates. F. Flow cytometry sorting of GigaAssay LentiX293T/LTR-GFP cell library cells with gates defined by − and + controls.
2. Results
To test the GigaAssay, we assayed the HIV Tat transactivation of long terminal repeat (LTR)-driven green fluorescent protein (GFP) expression in LentiX293T/LTR-GFP reporter cells as a model system. The LTR is a long terminal repeat region in the HIV genome. This system has the advantages of having an established robust reproducible assay and the availability of abundant benchmark data for performance assessment. Furthermore, Tat is a small gene that is suitable for assay development and is of pathological significance for HIV infection and its exit from latency.
2.1. Development of the GigaAssay system
The first major goal was to build a functional GigaAssay system. After multiple rounds of testing and optimization, we arrived at the current GigaAssay approach for Tat-driven transcription. Induction of the reporter by the Tat transgene was compared to the empty vector and an inactivating mutation as controls to quantify basal reporter expression (Fig. 1A). Once the reporter system and cassette were verified, a barcoded plasmid library was generated from a synthetic saturating mutagenesis ds-DNA library. Each molecule in this library was randomly barcoded with a UMI and used to prepare a lentiviral variant library. A human cell line was transduced with the lentiviral library at a low multiplicity of infection (MOI; 0.1) to minimize double infections. A polyclonal cell library was selected for stable viral DNA integration into each cell with puromycin. Fluorescent and non-fluorescent cells were sorted into GFP+ and GFP− bins by flow cytometry. gDNA was purified from each bin and a targeted UMI-barcoded Tat amplicon was cloned to make a next-generation sequencing (NGS) library.
The resulting paired-end read sequences were analyzed with a bioinformatics pipeline including several custom scripts to group UMI-barcodes with sequencing errors, interpret variants, and calculate the transcriptional activity of each mutant in the library.
In the test system, a GigaAssay cassette encodes a constitutively expressed Tat translated from a UMI-barcoded mRNA (Fig. 1A). Tat binds to the CDK9/CyclinT1/AFF4 complex, which is then recruited to the HIV LTR element of LTR-GFP in the LentiX293T/ LTR-GFP reporter cell line. The Tat/CDK9/CyclinT1/AFF4 complex drives transcriptional elongation. [11] Binding of this complex to the TAR element of the HIV LTR drives GFP expression. The Tat transactivation system was tested by transiently transfecting individually prepared clones into separate LentiX293T/LTR-GFP cultures and visualized by epifluorescence microscopy. Cells transfected with empty vector had little detectable GFP fluorescence, while those containing wild-type (WT) Tat had high fluorescence as expected for Tat-driven GFP reporter expression (Fig. 1B, C). Cells transfected with a C27S mutant that inactivates Tat transactivation had low levels of florescence as expected (Fig. 1D). [12] Similar results were obtained when Jurkat cells were transduced with the same control viruses (see Data in Brief co-submission). Jurkat cells, which are derived from T cells, are a more suitable model for HIV protein studies.
The same control cells were sorted by flow cytometry, and the sorting profiles were used to set gates for sorting the cells transduced with a Tat mutant library. Cells expressing the C27S loss-of-function (LOF) mutant produced a low GFP expression that was not different from the expression in control cells transduced with an empty vector lacking a Tat cDNA [13,14]. Cells expressing the reporter system with WT Tat had high GFP fluorescence (Fig. 1E). These microscopy and flow cytometry sorting experiments reproduced previous results obtained for low-throughput assays and thus verify the assay reporter system. [12,15,16]
A saturating mutagenesis ds-DNA Tat library (Tat accession number: AAK08486.1) was extended with synthetic 32-bp random UMI-barcodes in the 3’ UTR with DNA polymerase. The library was then subcloned into a lentiviral vector. NGS and subsequent bioinformatic analyses of the plasmid library with multiple barcoded cDNAs showed no dropout for any of the 1615 possible single amino acid substitutions (85 amino acids × 19 possible substitutions). A lentiviral library was prepared by co-transfection of lentiviral vectors encoding the library of mutant Tat cDNAs into LentiX293T cells.
LentiX293T/LTR-GFP cells were transduced with the lentiviral library, and after poison selection for cells with an integrated virus, GFP−, mid-GFP, and GFP+ cells were each sorted into bins by flow cytometry (Fig. 1F). The gating thresholds were based on GFP fluorescent intensities determined from negative and positive control cell samples expressing empty vector or WT Tat (Fig. 1E). gDNA was isolated from each sorted bin. Targeted NGS libraries were constructed for the UMI-barcoded Tat cDNA. The complete Tat cDNA was sequenced by NGS on an Illumina platform producing overlapping 2 × 250-bp paired-end reads. Samples were then analyzed with a custom NGS analysis pipeline (see Methods).
Summary statistics for the different stages of the GigaAssay pipeline are shown in Table 1. After transduction of recombinant viruses, each cell was uniquely barcoded with a UMI. During selection, these cells divided to form clonal UMI-barcoded cell groups. For the different samples and cell lines there were 179,763 unique UMI-barcoded cell groups after filtering. Each mutant in each replicate sample had an average of 102 independent UMIs. The transcriptional activity for each barcode group was calculated from the GFP− and GFP+ reads. Each UMI-barcoded cell group had an average of 273 reads after filtering, while each mutant with multiple UMI-barcodes had an average of 25,662 reads for each replicate; approximately 2000–90,000 reads were sequenced for each mutant (Data in Brief co-submission). The variants were called, and Tat transactivation activities were calculated from these reads.
Table 1.
Summary pipeline statistics for next generation sequencing of GigaAssay libraries.
| Step | Program | Runtime | Input reads | Total yield (Step) | Barcodes | Mutants |
|---|---|---|---|---|---|---|
|
| ||||||
| Fastqc | Fastqc | 226 min | 399,525,219 | 100.0% | ||
| Flash | Flash | 210 min | 399,525,219 | 92.2% | ||
| Trimmomatic | Trimmomatic | 175 min | 368,408,071 | 88.9% | ||
| Adapter Trimming | Cutadapt | 86 min | 327,536,831 | 98.8% | ||
| Barcode Extraction | Cutadapt | ~1 h | 323,651,918 | 99.8% | ||
| Barcode Grouping | Starcode | ~30 min | 323,163,108 | 100.0% | ||
| Demultiplexing | globalDemuxer.py | ~1 1/2 h | 323,163,108 | 91.2% | ||
| Variant Calling | caller.py | ~9 1/2 days | 294,692,904 | 100.0% | Total:561,000 | |
| Data formatting | phenoModeler.py | ~5 min | 294,692,904 | 55.9% | Unique:180,091 | 1774 |
| Data filtration | goldenStandard.py | <1 min | N/A | N/A | Unique: 179,763 | 1685 |
This design with many UMIs per variant can withstand a small percentage of incorrect barcodes or variant calls, which is important because NGS has a significant error rate. Even though we used a low MOI (0.1) for lentiviral transduction, it is possible to have double insertions of lentiviral DNA in the same cell, which could produce erroneous results for a small percentage of barcodes.
To estimate the error rate of double insertions arising from double lentivirus infections of the same cell, we generated lentiviruses that constitutively express either the zsGreen or mScarlet fluorescent protein under control of the CMV promoter. LentiX293T cells were transduced with equal amounts of each virus with MOIs ranging from 0.025 to 5. The percentage of cells expressing both markers would then reflect double integration events that were quantified by flow cytometry.
Five percent of the cells with a combined MOI of 0.1 expressed both markers. Even with double insertions, approximately half of these cells will not produce an erroneous measurement of activity when the mutant library is analyzed because if both mutants in the same cell have either WT or LOF levels of activity, the measured activity for these UMI barcodes will not be erroneous. An error in a UMI will occur only when one mutant has WT activity and the other has LOF. Thus, assuming a 50%/50% mixture of mutants in the variant library with WT and LOF activity levels, the estimated error rate among UMI barcodes will be approximately 2.5%, half of the measured 5% error rate. This should not have a significant impact on the assessment of Tat mutant activities because the average number of barcodes for each mutant was 102, and on average only 2–3 UMIs will have an erroneous activity measurement. Double insertions are thus expected to have only a minor impact on the statistics for each mutant. If we did not use UMI barcodes, 2.5% would be a false positive rate, and if double insertions were not assessed by a different method, the false positive rate could be much higher.
2.2. Impact of single Tat point mutations on GFP reporter transcription
Analysis of 179,763 UMI barcodes in 561,000 reads in the different flow-sorted bins (Table 1) reveals no drop out. Activities were measured for all possible amino acid substitutions at all positions excluding mutants of the start Met codon at position 1. Most substitutions (64%) had activities like those of the WT (meta p < 0.05 under Fisher’s method), demonstrating a general robustness for mutation tolerance in transcriptional transactivation (Fig. 2A). Approximately 18% of the mutants had activities matching a set of known Tat LOF mutations (meta p < 0.05 under Fisher’s method), indicating that a significant number of substitutions inactivate Tat-driven transcription. Of those with reduced activity, 35% had reduced activity when compared to WT Tat. The distribution of Tat mutant activities relative to WT Tat (Fig. 2B) shows a bimodal distribution with most mutants having a digital (on/off) transcriptional response.
Fig. 2.
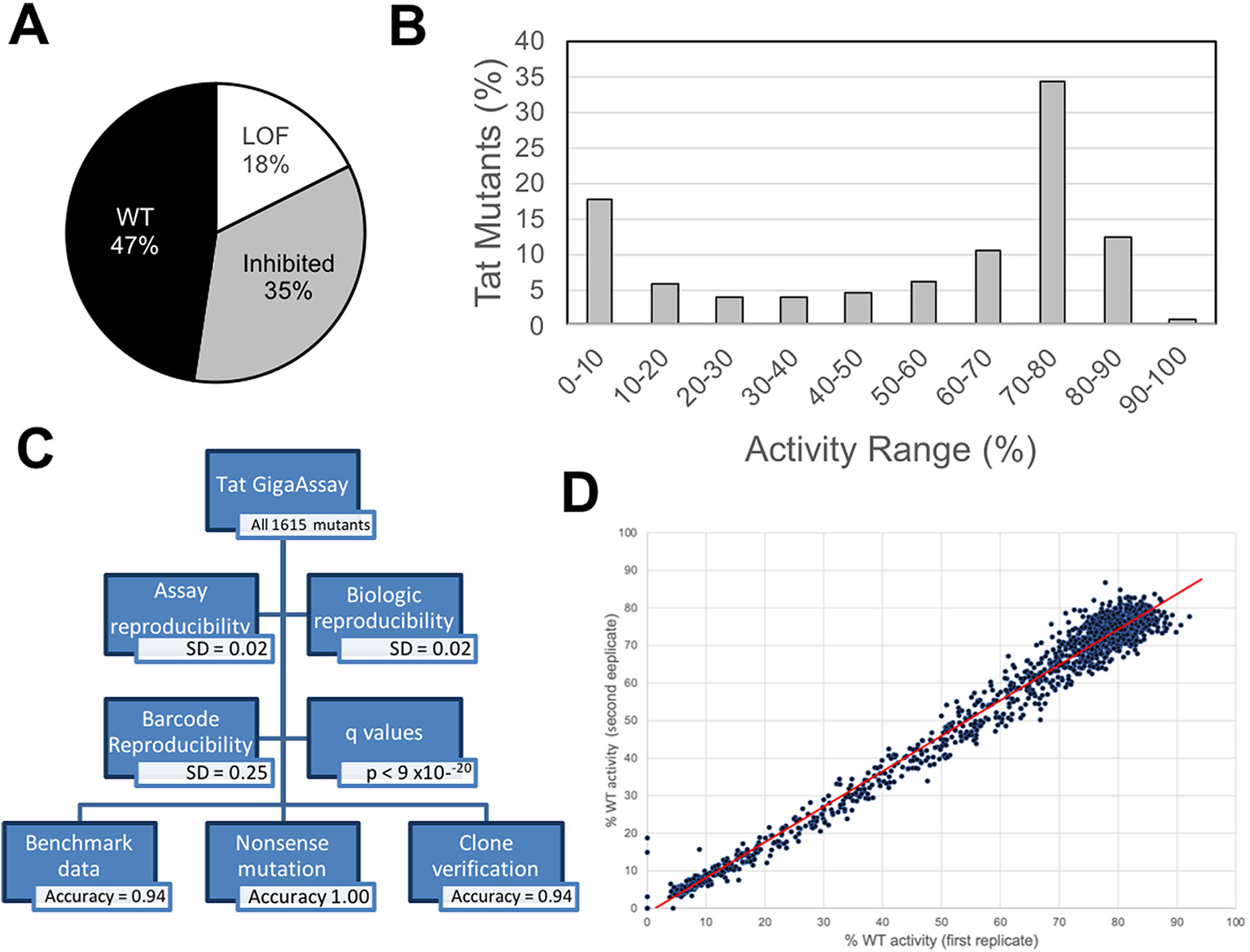
Summary of Tat mutant transcriptional activities and GigaAssay verification. Tat transactivation activity for a saturating mutagenesis GigaAssay. The activity represents the level of Tat transactivation activity score measured by GFP+ / (GFP+ + GFP−) reads for each UMI-barcode averaged for each mutant. A. Pie graph showing percentage of mutants with activities similar to known WT and LOF activities. B. Bin plot showing range of activities for Tat mutants (n = 1,615). C. Assay reproducibility and verification summary. D. Scatter plots for technical replicates. Transcriptional activity [GFP+/(GFP− + GFP+)] correlation among replicate GigaAssays (R2 = 0.99).
We compared each mutant’s activity to the activity reported for 1) sets of true positive mutants with established WT activity and 2) sets of true negative mutants with greatly reduced activity (Fig. 3 and Data in Brief co-submission). Most substitutions had either WT activity (58%, p < 0.05) or reduced activity (20%, p < 0.05). Approximately 22% of the mutants were moderately inhibited.
Fig. 3.

Heatmaps of p values for Tat mutant transcriptional activities in LentiX293T/LTR-GFP cells. q values for comparison of Tat mutant activity to sets of mutants with WT (A) or LOF activity (B). Keys for q value colors are shown.
Fig. 4 shows a heatmap of the activities for each single-site amino acid substitution at each position in Tat. For example, the T20W mutant has a medium level of activity. The figure includes a map of other functional sites and structural features for comparison to the GigaAssay data.
Fig. 4.

Heatmap showing Tat-induced transcriptional activity for a saturating mutagenesis GigaAssay. Heatmap for mutated amino acid for each position in Tat. The color gradient represents the level of Tat transactivation activity score measured by GFP+ / (GFP+ + GFP−) reads for each UMI-barcode averaged for each mutant. Black boxes are the WT amino acids and grey boxes are null values. A color key is shown. Abbreviations are LOF = loss-of-function, SS = secondary structure, Surface – solvent accessible surface, PTM – post-translational modification.
2.3. GigaAssay performance verification
Since the GigaAssay is a new assay system, we rigorously assessed its performance. The reproducibility and accuracy of the GigaAssay was examined with five independent verification tests (Fig. 2C). These tests yielded high average accuracy = 0.95; sensitivity = 0.9; specificity = 0.96; positive predictive value (PPV) = 0.98; and negative predictive value (NPV) = 0.94. The first test compared GigaAssay results to benchmark data from previous reports for Tat mutant transcriptional transactivation. Initially we annotated activities for 442 Tat mutants from 43 papers. We removed mutants that had ambiguous activity reports or had multiple missense mutations or INDELs yielding a final list of 107 mutants from 28 papers (see Data in Brief co-submission). The GigaAssay results were first compared to these benchmark mutants, in which mutants with activities (GFP+/(GFP+ + GFP−−) greater or less than 50% were classified as WT activity or reduced activity, respectively. Data in different samples were normalized to reads per million (rpm), and variants with a threshold below 2.5 rpm were discarded. This threshold was selected after optimizing performance metrics obtained from testing multiple thresholds. For this test the GigaAssay performance statistics were: accuracy = 0.93; sensitivity = 0.94; specificity = 0.89; PPV = 0.95; and NPV = 0.89 when results were compared to true positives and true negatives from independently published benchmark data (Table 2, Data in Brief co-submission). [17]
Table 2.
Summary performance statistics for next generation sequencing of GigaAssay libraries.
| Cells | Verification method | Accuracy | Sensitivity | Specificity | PPV | NPV |
|---|---|---|---|---|---|---|
|
| ||||||
| LentiX293T | Benchmark data | 0.93 | 0.95 | 0.89 | 0.95 | 0.89 |
| LentiX293T | Nonsense mutations | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| LentiX293T | Verified clones | 0.94 | 0.75 | 1.0 | 1.0 | 0.92 |
| LentiX293T | All (average) | 0.95 | 0.9 | 0.96 | 0.98 | 0.94 |
The second verification test was based upon an independent source of true negatives and positives measured in the GigaAssay. Tat exon 1, which encodes the first 58 amino acids of Tat, the minimal region required for Tat transactivation activity. [18–20] Although we did not intentionally include missense mutants in the oligonucleotide library, errors in oligonucleotide synthesis produced several Tat truncation mutants. Truncation mutants less or more than 58 amino acids long (n = 70 and n = 8, respectively) were expected to be negatives or positives, respectively. For this test, the GigaAssay the GigaAssay performance statistics were: accuracy = 1.0; sensitivity = 1.0, specificity = 1.0; PPV = 1.0; and NPV = 1.0 (Fig. 2C. Table 2, Data in Brief co-submission). This analysis indicates very high accuracy.
The third verification was based on a comparison to independent testing of a set of pre-tested Tat mutant clones. Prior to the experiment, we randomly selected 18 Tat mutants, made stable LentiX293T/LTR-GFP cell lines expressing these mutants, and measured transcription activation of the LTR-GFP reporter by flow cytometry (see Data in Brief co-submission). We were blinded to the true positive and true negative results until the GigaAssay was complete. We then compared to the GigaAssay results to these true negatives and positives. For this test, the performance statistics were: accuracy = 0.94; sensitivity = 0.75; specificity = 1.00; PPV = 1.00; and NPV = 0.92 (Table 2), verifying the high accuracy measured by the previous two approaches.
The fourth verification approach assessed the reproducibility of the GigaAssay between two technical replicate samples, in which the steps after the viral library preparation were completed in duplicate. The LentiX293/LTR-GFP cells were transduced, selected, flow sorted, sequenced, and analyzed separately in duplicate. The global standard deviations (SDs) for Tat mutant activities between duplicate samples were very low (SD = 0.02). For example, a mutant in one assay had 98% activity, while the replicate had 97% activity. Mutant activities for replicate samples were highly correlated (R2 = 0.99) in both cell lines, indicating high reproducibility (Fig. 2D, Data in Brief co-submission).
The fifth verification test compared the variability of Tat mutant activities between biological replicates for two different cell lines (LentiX293/LTR-GFP and Jurkat/LTR-GFP cells). Similar results for the performance statistics, reproducibility, and mutant activities were observed for Jurkat/LTR-GFP cells (Data in Brief co-submission). There were only minor differences in transcriptional activities for each mutant between the cell lines for each mutant (Data in Brief co-submission; R2 = 0.93). The major differences were for Tat mutants that had activities intermediate between those of the WT and LOF mutants.
The high number of barcoded single cDNAs for each mutant in this GigaAssay experiment, produces reliable activities for each mutant with confidence metrics. We first tested the hypothesis that the percentage of GFP+ reads was different from 0.5 (50% activity) for each mutant (null model percentage GFP+ = 0.5). The p value for each mutant in each cell with their distributions are reported in (Data in Brief, co-submission). Most p values for mutants in both cell lines (95%) reached statistical significance and some p values for Tat mutants ranged as low as 10−271. Many p values indicated a higher confidence due to the large number of UMI-barcode replicates with a high average (n = 102) for each mutant replicate sample.
We tested the hypotheses that the transcriptional activity of each mutant is 1) like that of WT Tat, and 2) like that of LOF Tat mutants. The association test showed that most substitutions had either WT (60%, p < 0.05) or reduced activity (23%, p < 0.05). The q values for each mutant are shown in Fig. 3. Approximately 26% of the mutants were moderately inhibited.
As further validation of the GigaAssay, the tolerance data for each position was generally consistent with the Shannon entropy score for amino acid variability among Tat clinical isolate sequences in the Los Alamos HIV sequence database. [21] We conclude that the GigaAssay experimental design, in which each individual variant cDNA has a separate random UMI-barcode that is tracked through the experiment, produced exceptional performance for a high-throughput assay. Very few high-throughput screens or assays have this level of accuracy.
2.4. Structure/function/tolerance of Tat mutants
The saturation mutagenesis landscape heatmap of Tat protein (Fig. 4) shows the variable impact of mutants on Tat transactivation activity. This mutation landscape enables an improved interpretation of the mutation tolerance of secondary structure elements, post-translational modification (PTM) sites and protein-protein interaction (PPIs) sites on Tat activity. We suggest that the typical structure/function analyses of proteins be expanded to include amino acid substitution tolerance, capturing the chemistries of amino acid substitutions that preserve or inactivate function. Since our experiments are relevant to the interpretation and/or confirmation of the hundreds of previously published reports on Tat mutants, we limit the scope of comparisons to a few examples. However, the results of the Tat tolerance analysis can assist with interpretation of the many published studies of Tat mutants.
The Tat secondary structure is mostly random coil with one helix and three turns. Several secondary structure positions are sensitive to mutations. Mutations were generally well tolerated in the first turn, but not in the second or third turns (Fig. 4). The only mutations in the first turn (7R-L8) with low activity were R7P, L8P, and L8G. The second turn starting at K28 has the sequence 28KKCCF32 (Fig. 4). No mutations at C30 were tolerated and only C31A and C31S with small volume amino acid substitutions at position 31 retained activity, supporting the steric hindrance constraints of the ϕ and ψ angles for amino acids located in turns. Only conservative large hydrophobic substitutions and some small aliphatic substitutions of F21 retained activity. Mutations in K41 and A42 in the third turn were generally not tolerated, although A42G and A42C had some residual activity. Scattered mutations in the helices and the random coil regions had reduced activities, and were more tolerant of mutations, especially after position 46 in the C-terminus. Notably, no substitutions were tolerated at K41 or in six C residues in the Cys-rich domain as previously reported. [14] C31 did tolerate substitutions of S, T, or small aliphatic amino acids and C31S, a natural variant in clade C Tat proteins, was previously shown to be active. [22]
We examined whether mutation of any of the residues covalently modified by PTMs affected Tat activity (Fig. 4). Tat has 18 reported PTMs of six different types: acetylation, proteolysis, methylation, ADP ribosylation, phosphorylation and ubiquitylation. [23,24] Hardly any of the mutations in positions modified by a PTM (positions 19,46,49–53,57,58,62,64,68,71,78, and 86) affected transcriptional activity. We provide a couple of examples of how tolerance can aid in the interpretation of PTM sites.
Tat is ADP ribosylated at E2, E9, and E86. Tat remains active, even when these positions are substituted for amino acids that lack a function group that links to ADP ribose, suggesting that ADP ribosylation is not necessary for Tat-driven transcription in agreement with a previous report. [25] This conclusion cannot be conclusively resolved from previous published work without the new tolerance data. K28 is acetylated and is required for Tat activation. K28 acetylation increases the affinity and stability of Tat–CycT1–TAR complexes. [26] Mutation of K28 to other amino acids (K28P, K28C, K28R, K28V, and K28A) should eliminate acetylation at position 28, but preserves transcriptional activity when mutated, indicating that K28 acetylation is not an absolute requirement. Thus, other mechanisms may increase the affinity for the transcriptional complex. Two other explanations suggested by the heatmap data in Fig. 4 are that K28 is in turn 2, a secondary structure element that is prone to loss of activity when mutated and that K28 is part of the p73 binding site. However, while K28R was inactive in the published report, it is possible that different Tat genetic backgrounds have epistatic interactions that explain the observed difference. Nevertheless, this example shows how GigaAssay results can aid in identifying which PTMs are essential for activity.
Tat has known binding sites for about 18 proteins (Fig. 4), half of which have at least one substitution in the binding site that inhibits Tat-driven transcriptional activity. Most of these PPI binding sites are in a hotspot (residues 29–60). [24,27] The PPI sites that Tat activity is most sensitive to are sites that interact with Cyclin T1 and Importinβ. In the Tat-CyclinT1 complex crystal structure, CyclinT1 makes contact with these 15 amino acids, most of which are in the core region of Tat. [28] Our results show that for 13 of the 15 positions, there is at least one mutant that blocks Tat transactivation. CyclinT1 is essential for Tat activity because it is needed to recruit RNA polymerase for transcript elongation. [11,29–31] On the other hand, CDK9, which forms a complex with Tat and Cyclin T1, binds the C-terminal region of Tat, which the presents results show is not essential for Tat-driven transcription.
Tat translocates to the nucleus through a critical interaction with Importinβ. Single substitutions in the Importinβ interaction site (50KKRRQRRRAHQ60) did not greatly affect Tat activity. Our results indicate that acidic amino acid substitutions in positions 50–56 mildly inhibited activity. These residues also overlap the RNApol2 binding site (52RRQRRRA57). The fact that double mutants in this site (50–60) disrupt Tat activity (see below) may reflect its importance in the recruitment of key Importinβ and CyclinT1/CDK9 complexes. [26,28,30,32,33] The RNApol2 binding site also overlaps with the P53 and SWI/SNF binding sites, which may also affect the impact of mutants in this region (Fig. 4). These results show that the GigaAssay can aid in identifying the PPIs that have the largest impact on function.
We further examined the impact of mutations on transcriptional activity by coloring the surface of the positions that most strongly affect Tat activity on the surface of the 3D structure of Tat, which can then be spatially compared to other positions, regions, and secondary structures of Tat (Fig. 5). [34] Ala scanning mutagenesis is an accepted approach to identify positions important for different functions. [35,36] Ala scanning identified 18 positions with LOF mutations scattered across the N-terminal half of the protein (Figs. 4, 5). Scanning with Pro or Asp was more sensitive than Ala scanning, identifying 23–24 LOF mutations. Cys scanning was less sensitive, identifying only 9 LOF positions, which happen to be a subset of Ala scanning. Gly scanning identified additional positions, probably because Gly, the smallest amino acid, has less constraints with more flexibility in dihedral angles (Figs. 4, 5). These results are consistent with an earlier report comparing Ala and Gly scanning mutagenesis. [37]
Fig. 5.
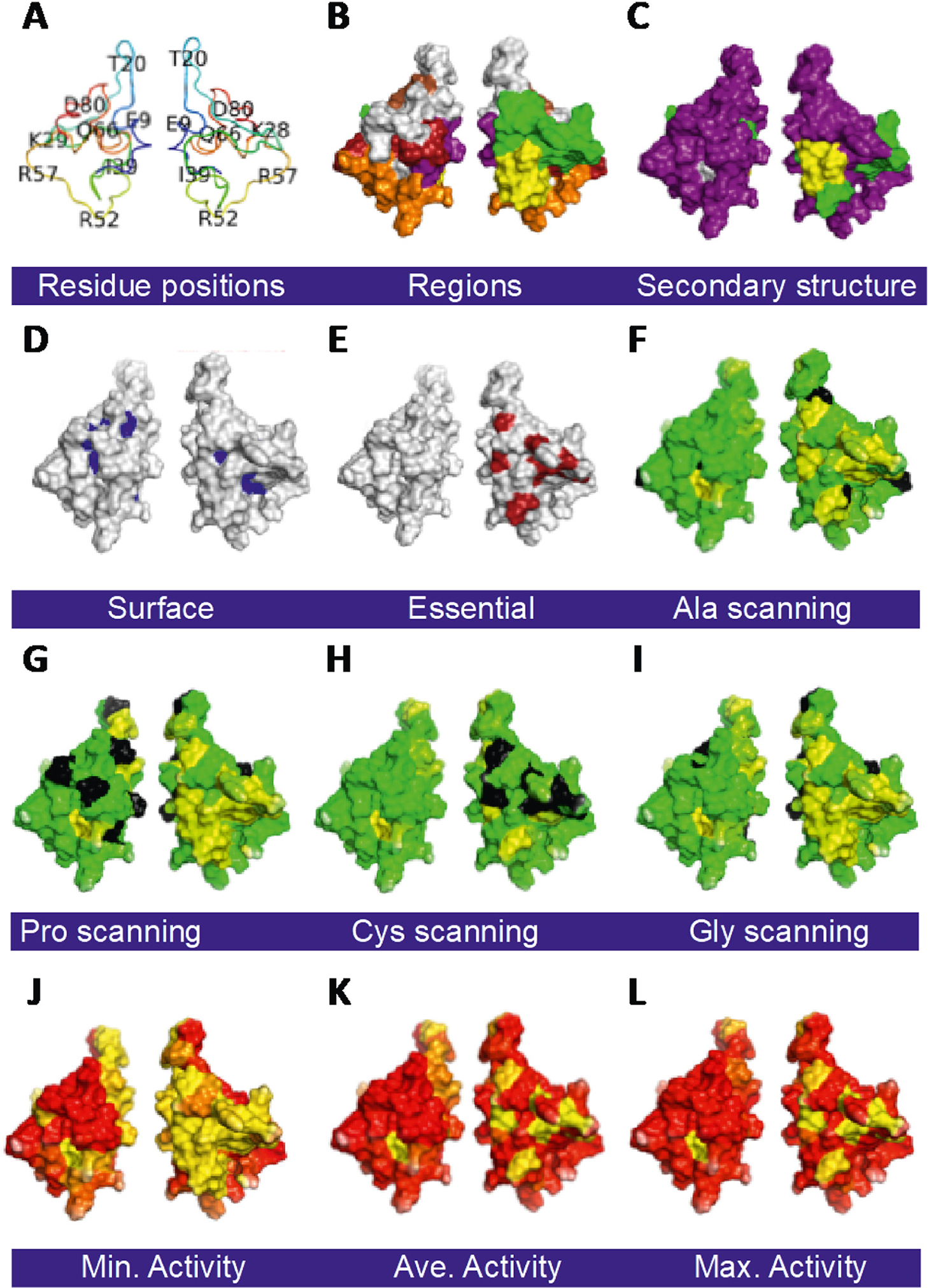
Tat mutant impact on structure/function. All surface maps are on the WT Tat 3D structure (PDB: 1TEV) with one member of each pair rotated 180o about the Y axis: A. Amino acid positions on Tat backbone. B. Regions of Tat [20]. C. Secondary structures. D. Solvent assessable surfaces are with residues with <10% solvent exposure colored blue. E. Tat positions that do not tolerate any substitution (C25, C27, C30, C33, C34, C37, and K41; red). F. Ala scanning substitutions. E. Pro scanning substitutions. F. Cys scanning substitutions. G. Gly scanning substitutions. F.-I. Residues colored black are for reference amino acids that match the type of scanning. A gradient of yellow with no activity to green with full activity is shown. Minimum (J), average (K), and maximum (L) transactivation activity heatmap for all substitutions. A gradient of red with WT activity to yellow with no activity is shown. Abbreviations are: Single letter amino acid code. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Heatmaps do not efficiently and intuitively present all of the substitution tolerance information gained from saturation mutagenesis. A novel approach that better summarizes the additional information gained from saturation mutagenesis, is to score positions for physiochemical groups with similar side chain properties (small aliphatic, large hydrophobic, polar noncharged or charged, negatively charged, positively charged). Here, positions were scored with the Mathews Correlation Coefficient (MCC). This approach better segregates the substitution tolerance for each position, whereas scanning with alanine or other amino acids does not capture this specificity. The heatmap with MCC scoring for groups of substitutions (Data in Brief co-submission) identifies positional tolerance: F32 only tolerates a large hydrophobic residue, G15 only tolerates a polar-noncharged, and C31 only tolerates amino acids with smaller volumes. Most of the residues with specific tolerances were located in the Cys-rich region.
Surface plots of MCC heatmaps for different physiochemical properties reveal spatial relationships of tolerance not captured in heatmaps. There is little specificity for amino acid mutation tolerance over most of the protein, including residues S46-E86, a random coil region that tolerates nearly all substitutions (Figs. 4, 5). However, the specificity for different groups of substitutions is clustered in the Cys-Rich and Core regions. These regions 1) have seven residues that do not tolerate any substitution, 2) have reduced activity in Ala, Pro, and Gly scanning mutagenesis, 3) do not contain buried residues, and 4) include key binding sites for CyclinT1, Importinβ, and several other PPIs (Figs. 4–6). The new physiochemical tolerance surface plots based on MCCs identify the residue tolerance of each position and their relative spatial locations, as well as surface accessibility (Figs. 4, 5).
Fig. 6.
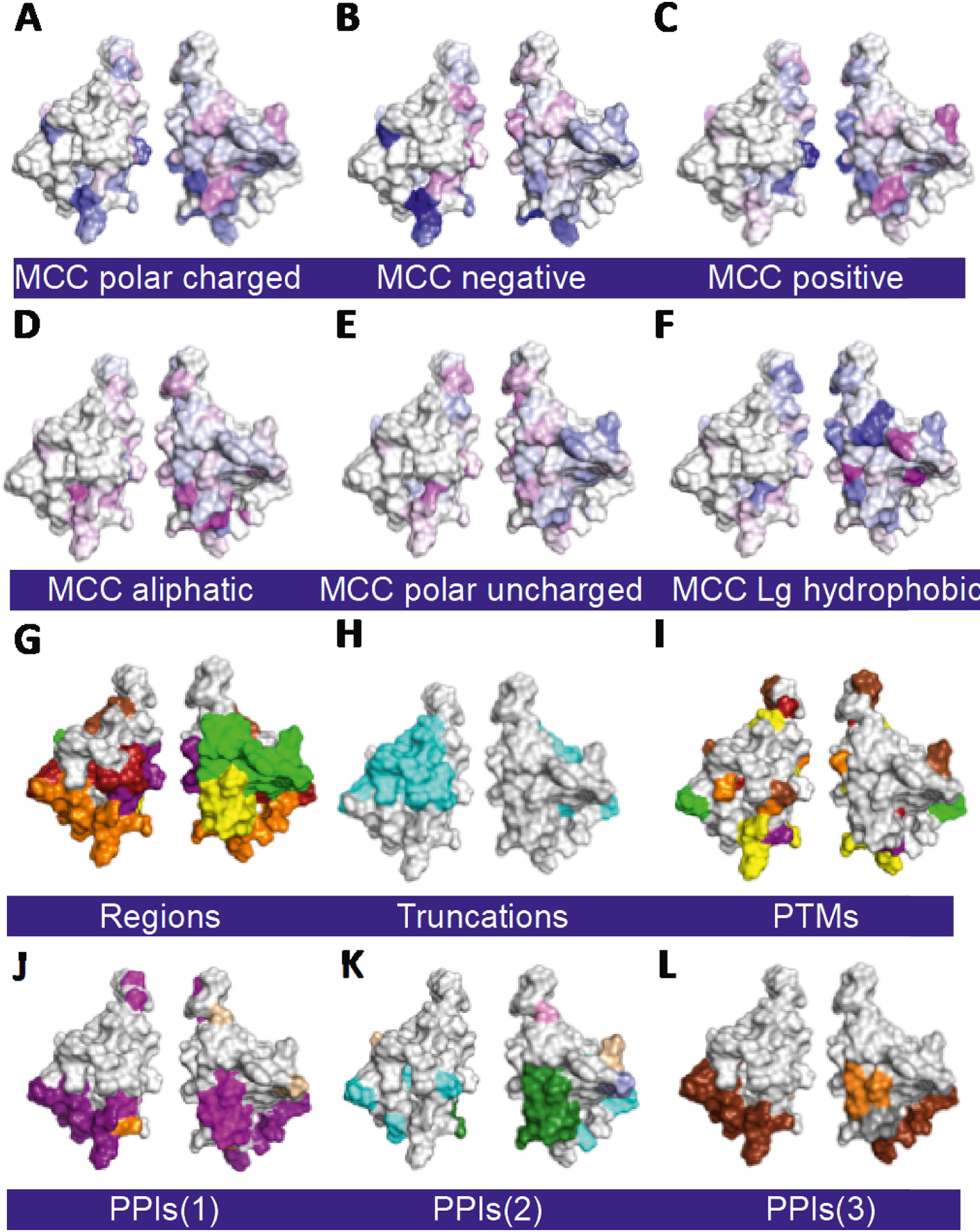
3D structure surface plots of different properties and function of Tat. All surface maps are on WT Tat 3D structure (PDB: 1TEV): A-F. Physiochemical tolerance surface plots for polar charged amino acids, those separated by positively and negatively charged amino acids, small aliphatic, polar uncharged, and large hydrophobic amino acids, respectively (see Methods). MCC = Mathews Correlation Coefficient. A gradient of blue to white to magenta ranging from lower to higher MCC scores for each position for the class of amino acids indicated is shown. Panel G is repeated from Fig. 4B here for visual comparison. H. Regions of Tat truncation and missense mutants that lose (cyan) or retain (light grey) activity. I. Tat PTMs. J.-L. Tat PPIs in 3 groups. The color key for regions, secondary structure, PTMs, PPIs, PPVs, and Tat activity are as in Fig. 4. Abbreviations are: PTM = post translational modification; PPI = protein-protein interaction. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We reorganized the heatmap data in Fig. 4 by side chain volume, to show how mutation tolerance is constrained by the volume of sidechains (Data in Brief co-submission). Some positions (Y26, F32, and F38) prefer large amino acids, while others (E9, L10, G15, S16, T23, C31, M39, and A42) prefer small amino acids, and others (D5, C25, L43, and I45) favor medium sized amino acids. In conclusion, some positions do not tolerate substitutions, while others tolerate substitutions with similar side chain volumes. Furthermore, the tolerances of some other positions are due to a combination of secondary structure and/or physiochemical properties of sidechains.
2.5. Intragenic epistasis of Tat double mutants
Analysis of high throughput GigaAssay results revealed interdependencies between positions of Tat double mutants, a phenomenon called intragenic epistasis. Tat double mutants arose from random errors introduced by oligonucleotide synthesis on single mutants. Since the double mutants were the result of synthesis errors, they were less frequently observed than single mutations (averaging about 2 UMIs/double mutant). UMIs for these double mutants were identified and analyzed. A total of 3429 double mutants were observed among replicates for both the LentiX293T cells and Jurkat cells (Data in Brief co-submission).
The transcriptional activities of double mutants were compared to their corresponding single mutants and assigned as positive (1% of the double mutants), negative (9%), or no intragenic epistasis (90%). In a separate experiment in Jurkat cells, 2% of the mutants had positive epistasis and 13% had negative epistasis (Data in Brief co-submission). Considering that the double mutants were independently sampled from two different cell lines, each with replicate samples, only 51 ambiguous epistatic types were observed between the cell lines, which places the lower bound of the error rate at 1.5%. This error rate was consistent with the double viral insertion rate estimated from a control experiment in which cells were cotransduced with zsGreen and mScarlet lentiviruses and then those cells expressing both fluorophores were scored by flow cytometry. The observed rate of intragenic epistasis (10–15%) was less than other recent estimates (32%–74%), but these results are for transcription in living human cells, whereas previous intragenic epistasis studies of HSP90, TEM-1 β-lactamase, and ΦX174 focused on fitness and were measured in a bacterium, a yeast, and a bacteriophage. [38–40]
To further explore intragenic epistasis for a specific molecular function, we examined the nuclear localization sequence (NLS) in Tat (positions 50–60) that binds to Importinβ. The Importinβ binding site is also of interest based on Tat truncation mutants. As previously mentioned, the activities of 78 truncation mutants were measured from nonsense mutants across both cell lines. Seventy of the mutants were truncated before position 58, while the other 8 mutants were truncated after position 58. The former had little or no detectable activity, whereas the latter had WT activity. Recall that truncations before position 58 are known to be inactive, whereas those after 58 are active The near-perfect accuracy, PPV and NPV for this analysis are consistent with the presence of a protease cleavage site between residues 57–58, which is also an exon boundary between Tat’s two exons, and many previous observations support a similar truncation tolerance for longer truncations. [18–20,41,42]
Almost any missense mutation in amino acids S46-E86 was tolerated (Fig. 4). However, several truncation mutants between S46 and R57 are LOF mutants, which appears inconsistent with the missense mutation results. The region of difference (S46-R57) contains the NLS and binding sites for Importinβ, and P53. The most likely explanation is that truncations remove all or part of the NLS, thereby eliminating localization of Tat to the nucleus and its transcriptional activity. [13,43–45] In the presence of any single mutation in this region, Tat is still localized to the nucleus, which likely reflects the evolutionary robustness of the NLS. The hypothesis that the NLS of Tat is robust is supported by GigaAssay results showing that substitution of all positively charged residues in the NLS with negatively charged residues inhibited but did not block the transcriptional activity.
The discrepancy between truncations in the S46-R57 region being inactive, but missense mutations in the same NLS region being active, is further resolved by analysis of intragenic epistasis. Others have mutagenized and tested the NLS and found that only double mutants in the NLS effectively blocked localization to the nucleus. [13,43–45] We identified 16 negative epistatic interactions in the NLS with double mutations (R49M/K50H, R49M/K50Q, R49S/K50H, R49E/R52P, R49Y/R55Q, Q50/54E, R49W/R55L, K50Q/K51N, K50N/K51T, K51N/R52I, K51F/R56L, K51W/R56Q, R52L/R53V, R53T/R55L, R53S/R55L, and R53W/R55L). None of these interactions had positive epistasis in the NLS, and these intragenic epistatic interactions cover the entire NLS. These results strongly support the hypothesis that the NLS has evolved an evolutionary robustness in which single acidic-to-basic amino acid substitutions mildly inhibit activity, but double mutations in the same region eliminate activity. This analysis shows the value of examining intragenic epistasis in assessing molecular functions.
3. Discussion
In this demonstration of the GigaAssay, we assayed nearly a million barcodes, but the primary library had considerably more clones, As the throughput of sequencing technology grows, we expect it will not be long before we routinely assay a billion barcodes (hence, GigaAssay). In the present case, we focused on a transcriptional function.
An assay is a procedure for qualitatively assessing or measuring the presence, amount, or functional activity of a target entity with high accuracy. Another term commonly confused with assay is screen. Screens and assays are very similar but have different goals. The goal of a screen is to select candidates from a population that have some property, whereas the goal of an assay is to measure a specific property accurately.
Herein, we demonstrate proof-of-principle of a GigaAssay prototype, testing the transcriptional activity of a library of HIV Tat mutants with an LTR-GFP transcriptional reporter assay in human cells. This experiment produced one of the most detailed and accurate functional maps for a protein in human cells reported so far. Our assay of >561,000 UMI-barcoded mutants in living cells represents four orders of magnitude enhancement over routine low-throughput cell-based assays. For example, in a previous low-throughput study, mutation of six Pro residues in a PxxP motifs were used to measure the impact of SH3 binding motifs on guanine nucleotide exchange factor activity. [46] Because individual molecules are barcoded with UMIs, we can sum Tat activities from hundreds of independent cells for thousands of mutants in one experiment.
In the GigaAssay, all mutants are assayed under standardized conditions, in the same cells, and with the same genetic background, which together produce highly consistent results. In contrast other mutant studies are often examined in multiple labs with different assay systems, genetic backgrounds, and conditions, which introduce variability and ambiguities that make comparisons challenging.
The GigaAssay is not a unique technique. Rather, it is a platform that couples established techniques of saturation mutagenesis, FACS, barcoding, and NGS to produce a powerful tool for assessing protein functions and mechanisms in human cells. It is a direct assay, in which all mutations are directly and reliably measured to an average accuracy of ~95% and average PPV of 98%, as measured by three global and independent methods. In comparison, lethality screens using many of the same techniques have the limitation of stochastic clonal growth that can produce numerous false positives and do not directly measure negatives. Furthermore, all such experiments in yeast lack the PTMs and PPIs native to mammalian cells.
A technique similar to the GigaAssay that has gained a relatively wide acceptance is deep mutational scanning (DMS) screens, e.g. [47]. DMS screens try to infer the function of mutants that survive a lethality screen. Accuracies generally cannot be reported because negatives are not measured. Two other mutant assays that resemble the Tat GigaAssay are a transcriptional assay called FACS-seq and an assay of GCN4 in yeast. [6,48] However, these assays analyze far less mutants, do not assess mutants at the UMI barcode level, and do not yield accuracy, PPV and other performance metrics. An advantage of the GigaAssay is that errors arising from oligonucleotide synthesis are readily identified and properly grouped, which cannot be done without UMI barcodes.
The GigaAssay is flexible and can be readily adapted to measure any cell process or molecular function as long as we can generate a fluorescent reporter signal with a good dynamic range. In addition, the mutation libraries can be adapted to different types of libraries such as loss- or gain-of-function mutations, single mutations, and haplotypes. In this capacity, the GigaAssay could be used to systematically dissect the molecular mechanisms of variant effects. Separate assays could be set up to assess mutations that impact a variety of processes such as post translational modifications, protein trafficking, protein-protein interactions, etc. In a separate unpublished study, we adapted the GigaAssay to examine Her2 receptor signaling with phospho- Her2 antibodies to assess receptor activation, and phosphor-Erk antibodies as a proxy for stimulation of the MAPK pathway. The GigaAssay can also likely be adapted to investigate variable RNA or DNA libraries, such as RNAi screening libraries.
The GigaAssay has a few limitations. One limitation is that assay development and optimization are time consuming. The GigaAssay requires a fluorescent readout of activity. Setup and verification of the fluorescent reporter system can take several months. This involves testing different variables to produce and optimize the separation of cell populations by flow cytometry. While in vivo experiments or those with primary cells are preferred, the GigaAssay needs to assess millions of single cells, so cell lines are the most suitable model. For many assays, WT endogenous molecules may contribute to the basal assay signal, and cells may need to be engineered by gene editing to reduce their background fluorescence. This was not the case for Tat as it is an exogenous viral transcription factor and the LTR-GFP reporter has a low background signal. In the Tat experiment, the GigaAssay was semiquantitative with read frequencies counted from cells that were sorted into three bins. A better approach to quantify activity was used in the abovementioned yeast transcriptional reporter assay called FACS-seq, which sorts cells into 20 bins of graded fluorescence. [48] This approach could be adapted to future implementations of the GigaAssay.
NGS, while providing robustness and high throughput to the GigaAssay, has several limitations. [49] NGS techniques often identify and filter reads. However, since the read frequency in bins is the basis for quantifying activity in the GigaAssay, the sources of error and error rates for reads that are normally filtered must be identified and quantitated. Some sources of these errors are groupings of reads with sequencing errors in the barcodes, reads with poor PHRED scores, and orphaned reads. Another major NGS analysis category is variant calling. Variant calling in the Tat GigaAssay relies on read depths >2000×, but this does not rule out potential errors due to variants in the barcode or index hopping. [50]
A limitation specific to Tat is that its C-terminus overlaps with the coding region of another HIV protein, Rev. [51] Although modifying Tat’s C-terminus may have little or no effect on Tat, the mutants could affect Rev. function. A viral fitness test examining Tat mutants identified many deleterious substitutions in the last 45 amino acids of Tat that are tolerated in our transcriptional assay, implying that these substitutions may be more relevant to Rev, than to Tat function. [52] Consistent with this hypothesis, this region in Rev did not tolerate many different substitutions in the fitness assay. However, our analysis of double mutant substitutions and epistasis identified haplotypes in the nuclear localization sequence that are not well tolerated. Thus, differing genetic backgrounds can be an alternative explanation.
Ala scanning, which is a standard approach for mapping binding sites or identifying functional elements, can identify key positions in Tat, but it lacks sensitivity (Fig. 4, Fig. 5F). Some missed positions were only detected with saturating Pro and Gly scanning mutagenesis analysis (Fig. 4, Fig. 5G, I). [53–58] Scanning with these alternative amino acids has been tested before, but not at all positions in a protein. Cys scanning (Fig. 4, Fig. 5H), also previously used, is less sensitive than Ala scanning, but has the advantage that Cys can be crosslinked or readily covalently modified. [59,60]
We propose to expand structure-activity relationship (SAR) studies that are currently limited to Ala scanning to include determining the mutation tolerance at each position. We designate this new approach as Structure/Activity/Tolerance Relationships (SATR). We noticed in the GigaAssay results that some substitutions were only allowed or prevented depending on a specific chemical property or amino acid side chain volume. For example, position 15 in Tat favored amino acids with a hydroxyl in the side chain (Ser or Thr) and position 32 favored large hydrophobic amino acids. We created MCC scores as a metric of substitution tolerance. For an amino acid with a specific physiochemical type, a positive score indicates the degree to which it is required, and a negative score indicates the degree to which it needs to be excluded. Surface plots of these values clearly identify those regions and pockets of the protein that have stricter requirements for substitutions and aid in interpretation of PTMs, PPIs or other functional activities (Figs. 5, 6). For example, positions 49, 50, 52, and 53 in the NLS had MCC values supporting any substitution that excluded an acidic amino acid.
Nearly all amino acid substitutions in PPIs and PTMs sites in Tat are tolerated and thus these sites can be considered robust. However, key structural elements or substitutions that alter structure (e.g. R6P or L7P) are not as well tolerated. When considering the role of the CDK9/CyclinT1/AFF5 complex with Tat and TAR to promote transcriptional elongation, only residues in the Cyclin T1 contact site are essential for transcription (Fig. 4). [29,30] Although Tat binds the HIV RNA TAR element and CDK9, these contacts are not necessary for Tat’s effect on transcriptional elongation. By comparing the binding sites of Cyclin T1 in a 3D crystal structure to the SAR and mutation tolerance profiles for these amino acids, we can better determine which components of the complex are essential for Tat’s activity. [30] Several of the mutation tolerances/intolerances help better understand the requirements of Cyclin T1 for binding. For example, P3 cannot be a basic residue, S16 must be small, positions 18, 37, and 41 cannot be changed, V36 cannot be acid, and position 43 must be a small aliphatic. With these requirements, in addition to spatial relationships, it is easy to see why the Tat:Cyclin T1 interaction is specific. Furthermore, a holistic view of all binding sites for all interactors helps to identify Cyclin T1 as the most important Tat interactor. However, we must also consider that the mutations could affect the stability, folding, and/or expression of Tat, and not directly impact its interactions or other molecular functions. Additional GigaAssays could probe these facets effecting Tat expression levels.
Even though we did not design the Tat saturating mutagenesis library to include double mutants, we were able to determine transcriptional activities of thousands of Tat double mutants as well as some truncation mutants. This is an advantage of the GigaAssay, in which each individual cDNA for each mutant is randomly barcoded with a UMI, making it possible to identify and separately analyze single barcode mutants. This approach allowed us to identify oligonucleotide synthesis errors. By comparing the transcriptional activities of double mutants to their corresponding single mutants, we were able to estimate the percentage of mutants with intragenic epistasis for Tat transcriptional activity. Most previous epistasis experiments testing intragenic epistasis examined organismal or viral fitness. We are not aware of other intragenic epistasis experiments that tested a molecular function.
Genetic testing usually focuses on single substitutions and generally does not account for epistasis. Our results indicate that epistasis may have a significant effect on interpretation of mutagenesis experiments. We observed intragenic epistasis for 10–15% of double mutants that were tested in the Tat GigaAssay (n = 3429 in two cell lines). A better understanding of intragenic epistatic could help to explain current puzzles in human genetics such as missing heritability, variant effects in different genetic backgrounds, low penetrance, and differential expressivity. However, additional experiments will be needed to verify this as there are few UMIs for each double mutation in the present experiment. If the above intragenic epistasis rate is typical of other genes (as has already been observed in some fitness assays [38,39]), the potential clinical impact upon genetic tests and companion diagnostics, as well as the impact on patient care cannot be overstated.
Lastly, standard deviations between barcodes were very low for technical (Fig. 2D) and biological replicates (see Data in Brief co-submission), but the variance among about 100 UMI barcodes for each mutant was significantly higher at 25%. The transcriptional activity tended to be digital (on/off)with transcription being on or off for each barcode. However, while a majority of barcodes, were on or off for different mutants, we did observe a minor fraction of barcodes with the opposite digital output. The high standard deviation among barcodes for the same mutant reflects this observation. Future studies will need to investigate the source of the observed variance among UMI barcodes for the same mutants.
The present Tat results should lead to new interpretation of previous studies, more accurate interpretation of future studies, and a better understanding of HIV latency. With this demonstration, we expect that the GigaAssay will be useful for addressing the structure-activity-tolerance relationships of many other genes.
4. Materials an methods
4.1. Cloning
All primers and synthetic oligonucleotides used for cloning and PCR are in Supplementary File S1.
The plasmid pLjm1_mcs was made by introducing compatible EcoRI, SalI, and AsiSI restriction enzyme sites in the pLjm1-Empty (Addgene) vector for cloning of the Tat variant library. Tat or mutant Tat encoding a C27S mutation was PCR amplified from pNL4–3 as a template with Q5® High-Fidelity DNA Polymerase (New England Biolabs) and cloned into EcoRI/SalI digested pLjm1_mcs1. For generating a LentiX293T/LTR-GFP reporter cell line, a plasmid harboring LTR-GFP and blasticidin S resistance was constructed. The LTR-GFP cassette and Blasticidin S resistance (bsr) gene were amplified by PCR with pNL4–3 (NIH AIDS reagent program), pEGFP and LentiCRISPR-v2 Blast (Addgene) as templates. LTR, GFP, and bsr amplicons were fused by inverse PCR using Q5® High-Fidelity DNA Polymerase. The fused amplicons were cloned into pAAVS1-Puro-DNR (Origene) previously digested with SpeI and EcoRI. For lentiviral constructs to test doubles insertions, a NheI-KpnI fragment containing ZsGreen1-DR-PPT-PGK promoter-HygB and a similar NheI-MluI fragment containing mScarlet were separately cloned into pJLM1-MCS to create lentiviral vectors with CMV promoters expressing ZsGreen1-DR and mScarlet, respectively.
4.2. Generation of UMI-barcoded variant plasmid libraries
A double stranded (ds) DNA library containing HIV-1 Tat cDNAs with sequences for all the possible single amino acid mutant mutants (n = 1615 Tat mutants) was synthesized by Twist Bioscience (San Francisco, CA). The ds-DNA from each well of 96-well plates were pooled and a single round of overlap PCR extension appended random 32mers oligonucleotides to the 3′ untranslated region. The synthesized ds-DNA library has a 3′-overhang sequence after the stop codon that overlaps with the 5′ overhang sequence upstream of the 32mers random oligonucleotide sequence. The pooled ds-DNA library and the random oligomer were mixed in 1:10 M ratio, denatured, and annealed. Hybridized DNA was extended with the Q5® High-Fidelity DNA Polymerase (New England Biolabs) for one cycle of PCR. The 50 μl of PCR reaction mix was then treated with 2 μl of Exonuclease I (New England Biolabs), incubated at 37 °C for 15 min, and DNA was purified by PCR cleanup kit (Macherey-Nagel).
The purified DNA was digested with EcoRI-HF (New England Biolabs) and AsiSI (New England Biolabs) for 3 h at 37 °C and ligated into EcoRI-HF/AsiSI digested pLjm1_mcs plasmid (molar ratio vector: insert = 1:3) with electroligase (New England Biolabs). Ligation reactions (12) were pooled, purified with a PCR cleanup kit, and drop dialyzed on MF-Millipore® Membrane Filter, 0.025 μm pore size (Millipore Sigma). The purified ligation reaction mixture was electroporated into E. cloni® 10G ELITE electrocompetent cells (Lucigen), plated on prewarmed LB ampicillin plates, and incubated for 18 h at 37 °C. Transformants were scrapped and plasmid library from the pooled cell suspension was isolated using EndoFree Plasmid Mega kit (Qiagen).
4.3. Production and titration of lentiviral libraries
Lentiviral libraries were produced in LentiX293T cells (Takara). Approximately 3 million LentiX293T cells were seeded in 100 mm petri dish and grown in 10 ml complete DMEM media [(DMEM+10% Fetal Calf serum), Gibco] for 24 h. Plasmids pLjm1_Twist Tat Library (8.5 μg); pMDLG/pRRE (Addgene, 7.6 μg); pRSV/pRev (Addgene, 4.0 μg); pMD2. G (Addgene, 4.0 μg) were diluted to a final volume to 613 μl in a 15 ml conical tube. CaCl2 (87 μl of 2 M) was added to plasmid mixture. 2XHBS (700 μl) was added dropwise to the above transfection mix with gentle stirring in a circular motion. The transfection mix was incubated for 15 min and added dropwise to the cells in a 100 mm petri dish. The cells were incubated at 37 °C for 12 h in a CO2 incubator at 37 °C with a 5% CO2 atmosphere. Post-transfection (12h), the calcium phosphate-containing medium was replaced with 7 ml complete media (DMEM+10%FBS) and incubated for 48 h in CO2 incubator at 37 °C with a 5% CO2 atmosphere. Spent media from confluent transfected LentiX293T cells was filtered through a 0.45 μm Uniflow syringe filter (Cytiva Whatman). Aliquots of the filtered spent media with the lentivirus (100 μl to 5 ml) were stored in at −80 °C.
Lentiviral vectors for specific clones were produced in LentiX293T cells. Briefly, the 0.6 million LentiX293T cells was seeded in a well of a 6-well plate. After 24 h, cells were co-transfected with pLjm1-mcs, pLjm1-Tat, or pLjm1-TatC27S (1 μg); pMDLG/pRRE (1.0 μg), and pRsv-Rev (0.5 μg) and pMD2.G (0.5 μg) transfecting with Lipofectamine LTX (Invitrogen) at a 1:3 ratio [DNA(μg): Transfection reagent(μl)]. After 6 h of incubation, media was replaced, and cells were cultured in complete media for an additional 48 h. Cell supernatants containing lentivirus were collected, filtered through a 0.45 μm syringe filter (Millipore), and stored at −80 °C.
Lentiviruses were titered by seeding10,000 cells/well in 96 well plate and cultured in 200 μl of complete DMEM media (DMEM+10% FBS). After 24 h, 100 μl of serial dilutions of lentivirus were added after removing majority of the spent media from the wells and incubated 4 h. Complete DMEM media (100 μl) was added and incubated 24 h. Spent media (100 μl) was removed, replaced with DMEM media containing puromycin (Invitrogen, 1.5 μg/ml final concentration), and incubated for 96–120 h. The cells were inspected for viability under the microscope and colonies were counted to calculate the infectious unit/ml.
4.4. Experiments testing virus double integrations
Prior to viral transduction (24 h), 100,000 LentiX HEK293T cells were seeded into 6 well plates in DMEM. After 24 h, the media was replaced with DMEM containing a dilution series of matched MOIs of CMV-ZsGreen1-DR and CMV-mScarlet viral supernatants (2.5–200 μL), supplemented with 4 μg/mL polybrene (Millipore). After transduction (72 h), cells were harvested by trypsinization and reporter expression in each cell was quantitated on a Sony SH800Z flow cytometer, with 20,000 events captured in the FL-1 (ZsGreen1-DR) and FL-2 (mScarlet) channels. The percentage of positive cells for either reporter was then calculated based on an un-transfected control and viral titers (IFU/mL) with the following equation [(cells seeded × % positive cells)/mL viral supernatant].
4.5. Generation of LentiX293T/LTR-GFP cell line
LentiX293T cells (0.6 million) were seeded in the well of a 6-well plate and grown in 3 ml of complete DMEM media. After 24 h, a GFP reporter plasmid (1.5 μg) carrying LTR-GFP and the blasticidin S-resistance (BSR) gene was transfected in LentiX293T cells and incubated for 48 h. Transfected cells were selected for blasticidin S [(5 μg/ml), Invitrogen] resistance for 14 days, exchanging DMEM media with the poison every 3 days. Cells were trypsinized and 100,000 cells were serially diluted in 96-well plates. After 14 days of incubation, single colonies were screened after expansion.
For confirming lentiviral integration, gDNA was isolated, Tat was amplified with GFP-FP and GFP-RP primers, amplicons were subcloned, and sequenced. Tat transcriptional activity was measured in a subculture of each clonal cell line. Cells culture in 96-well plate were transfected with 50 ng of WT Tat expression vector and cultured for 48 h. Transactivation-induced GFP expression was evaluated by Nikon TE2000E epifluorescence microscopy. The clonal reporter cell lines were propagated and stored at −80 °C.
4.6. Stable cell libraries and cell lines
LentiX293T/LTR-GFP cells (33 million) were transduced with the Tat variant lentiviral library at a multiplicity of infection (MOI) of 0.1. After 24 h of infection, cells were cultured and maintained in complete DMEM media supplemented with puromycin (1.5 μg/ml). After 5 days, confluent cells were harvested, counted, and washed once with 1× PBS before fixing and isolating gDNA for NGS of the Tat amplicon.
For performance evaluation of the GigaAssay, 18 random mutants of Tat, as well as empty vector and WT Tat were stably expressed in LentiX293T/LTR-GFP cells. Approximately 0.15 million cells were seeded in a well of a 24 well plate and incubated for 24 h. Cells were transduced with lentivirus, selected, and maintained in complete DMEM media with puromycin (1.5 μg/ml) for 96 h. Cells were harvested and sorted by flow cytometry to assess for LTR transactivated GFP expression. Selected clones for empty vector, WT Tat, and Tat C27S were stored at −80 °C.
4.7. Flow sorting of cells and deep sequencing
One fourth of the LentiX293T/ LTR-GFP cells were harvested, gDNA isolated using Qiagen DNeasy Blood & Tissue Kit, and sequenced to evaluate library representation before Flow Sorting. The remaining cells were fixed in 2% paraformaldehyde/PBS for 10 min, washed twice with 1× PBS and resuspended in 1× PBS for analysis by flow sorting (Sony 800S Cell sorter). Cells were sorting into three bins of GFP signal intensity (low-GFP, mid-GFP and high-GFP) gated with threshold determined for cells stably expressing WT Tat for maximal transactivation of LTR-GFP, and cells stable expressing a Tat C27S mutant or empty vector for low background of basal transactivation of the LTR-GFP.
For deep sequencing, primers were designed to flank the Tat targeted region from gDNA and incorporate the NGS sequencing adaptors. gDNA was amplified by PCR with NEBNext Q5 Hot Start HiFi PCR Master Mix. The PCR protocol denatured strands at 98°C for 30 s only in the first cycle followed by: denaturation at 98°C for 10s, annealing at 58°C for 15s, elongation at 72°C for 30s, and a final elongation for 2min. NGS libraries for each sample category used 10 NGS library forward primers and 1 NGS library reverse primer. The forward primers were common for all the sample categories and the reverse primer being unique for each sample. The Tat amplicons were pooled and 20 μl of the sample was purified by gel extraction with Ampure-XP beads (Beckman Coulter). All the samples were pooled and sequenced with a Novaseq 6000 sequencing platform at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. This SP flow cell produces approximately 2 × 250 bp paired-end reads. 18 samples were sequenced (synthetic ds-DNA Tat variant library, plasmid library, selected cell libraries in LentiX293T (in duplicate), Flow sorted low-GFP, mid-GFP, and high-GFP cells (in duplicate).
4.8. Processing NGS data with a bioinformatics pipeline
Paired-end reads were processed with a multistep bioinformatic pipeline, BaseSpace, and resulting reads in bcl files were converted into FASTQ files with BCL2FASTQ; read quality is assessed with FASTQC. [61] Individual paired end reads (250 bp each) have expected overlap of 30–39 bp. Paired end reads for all samples are merged with FLASH to build complete Tat contigs of average length 465 bp. [62] Contigs were quality filtered with Trimmomatic such that any contigs containing 4 consecutive bp with average PHRED score below 16 were removed. [63] Adapters are trimmed and discarded with CutAdapt, leaving only the Tat encoding region, a small 3′ extension, and the 32 bp UMI-barcode. [64] Barcodes are then extracted using CutAdapt. Reads across all samples are pooled to perform global barcode grouping through Starcode. [65] The sequence reads are demultiplexed into subsets of read sequences for each cell clone based on UMI-barcode groups with a custom Python script that processes the output of Starcode. Resulting reads are then aligned to the Tat cDNA with BWA MEM. [66] The BAM file with nucleotide variants are called for each subset of Tat contigs (cell clones) and output as a VCF file with BCFtools (mpileup). [67] Custom Python scripts are used to identify the amino acid substitution for the VCFs, the number of reads for each UMI-barcode in each sample, and the barcodes groups for cells with the same amino acid substitutions. The PyVCF library was used in scripts that gathered the information for each variant from the VCF files. [68] Barcode data are multiplexed corresponding to which amino acid variant is identified. Read counts are normalized to each flow sort group into reads per million (RPM), and amino acid substitutions which have <2.5 total RPM are filtered out. Reads for each amino acid variant are compared and the activity is calculated as the percentage of GFP positive RPM over GFP positive plus GFP negative RPM.
4.9. Data analysis, statistics, and figure preparation
Statistics are calculated for each mutation. We assume there are n cell lines (biological replicates) and each cell line has m technical replicates. For each barcode (group) in a sample, we calculate the percentage of the number of reads in the GFP+ group vs the total number of reads in both GFP+ and GFP− groups, denoted as h ratio (h ∈ [0,1]). We expect a high h percentage for WT, while a low h percentage suggests a mutant. Then for each mutant, we calculate the averaged h ratio for all the UMI-barcodes assigned to the same mutant, denoted as a mutant level summary score. We use a one sample t-test to evaluate 1) whether the mutant has a significantly different number of reads in the GFP+ group compared with the GFP− group within a technical replicate, and 2) whether the mutant has a significantly different number of reads in the GFP+ group compared with the GFP− group among different cell lines based on biological replicates (null hypothesis H0: h =0.5).
In addition to the t-test comparing the GFP+ ratio among the mutants, we also devised an association test between the genotype (Variant/WT) and GFP expression (binary variable GFP+ or GFP−. We used a mixed effect logistic regression, with random intercepts for UMI-barcodes and replicates to model the nested structure in our experimental design. For the WT control populations, we used the cells with no variant calls (sequences identical to the reference). Each variant was compared against the common WT control population. The model M1 with genotype included as fixed effects was compared to a null model M0 without genotype in a likelihood ratio test (LRT). Similar to Genome-Wide Association Studies (GWAS), a significant result indicates that the variant/WT is associated with the percentage of GFP+ cells. For variants where the model fit was singular, we simplified the model by dropping the random effects. p-values were false discovery rate (FDR)-adjusted using Storey’s q-values.
Tests were done at the replicate level with models:
Tests were done at the cell type level with models:
We classify mutants with high h percentage as WT and a low h percentage as a LOF mutant. To estimate type I error for the classification, we compiled a list of true mutants with WT transcriptional activity and true LOF mutants with low activity (Data in Brief co-submission). Then we fit their h percentages with a beta distribution as the null distribution. Specifically, for the WT detection, we use the true mutant as the null, and vice versus, for the mutant detection, we use the WT as the null. Moment estimators are used for estimating the model parameters. The p values for different cell lines are combined using Fisher’s method into a global test p value. Performance metrics of accuracy, sensitivity, specificity, positive predictive value and negative value are based upon standard formulas. [17]
Figures were prepared with PowerPoint, Excel, FlowJo, R, and Pymol. Bin, Bar, and Pie plots, as well as saturating mutagenesis heatmaps were generated with Excel. Values for saturating mutagenesis heatmaps and 3D surfaces plots were generated with custom python scripts. 3D physiochemical tolerance surface plots for the amino acid tolerance at each position are based upon MCCs for physiochemical properties and colored with gradients from blue to white to magenta. Magenta is the highest MCC and blue is the lowest MCC. MCC is calculated for groups of amino acids with similar physiochemical properties. [17] Solvent accessible surface area (SASA) was calculated for the Tat structure (1TIV) with the Accessible Surface Area and Accessibility Tool. [69] Residues are considered buried if <10% of surface area is exposed to solvent (Figs. 4, 5).
The MCC formula is calculated with the following data definitions for large hydrophobic amino acids, at a position in Tat as an example: If either Phe, Tyr, or Trp have >50% activity they are true positives and if the other amino acids have <50% activity they are true negatives. If either Phe, Tyr, or Trp have <50% activity they are false positives and if the other amino acids have >50% activity they are false negatives. We also considered the WT amino acid to be a true positive when it was in the physiochemical group, and as a true negative when it was not. The MCC captures the tolerance for types of amino acids at each position and when mapped the surface of the 3D structure, is a new visual mining approach to reveal the spatial relationships of amino acids tolerances and their relevance to other Tat functions.
Supplementary Material
Acknowledgments
We thank Drs. Edwin Oh, and Richard Tillet from the UNLV Nevada Institute of Personalized Medicine Genome Acquisition and Analysis Core for access to a flow cytometer sorter and help with some NGS sequencing and interpretation for GigaAssay development. We Thanks Drs. Jefferson Kinney (University of Nevada, Las Vegas) and Tom Metzger (Roseman University) for use of their flow cytometer. We wish to acknowledge the help of Dr. Nora Caberoy with electroporation. We appreciate the discussions we had with Drs. Qing Wu and Michael F. Lin about statistical assessment of the GigaAssay results. We thank Dr. James Raymond for help with editing the manuscript. NIH: R21AI116411, R15GM107983, R21AI078708, R56AI109156, P20GM121325, COBRE and the Governor’s Office of Economic Development (Grant Number: 1547526), and the Prabhu endowed professorship. We also acknowledge the UNLV College of Science for a grant to develop the GigaAssay.
Footnotes
Declaration of Competing Interest
Part of this technology is owned by the University of Nevada Las Vegas and is part of a pending patent application with the United States Patent and Trademark Office [Patent No: PCT/US2017/042179 Canadian PCT-CA (0445–02)]. MRS LV, LB, and CJG are employees of Heligenics which has licensed the technology from UNLV and is pursuing commercial interest. UNLV manages a conflict-of-interest management plan for Principal Investigator, MRS. ZW is contracted by Heligenics to build and implement a part of a statistical model for the GigaAssay.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygeno.2022.110439.
Data availability
All data is shared as part of aData in Brief co-submission
References
- [1].Picot J, Guerin CL, Le Van Kim C, Boulanger CM, Flow cytometry: retrospective, fundamentals and recent instrumentation, Cytotechnology. 64 (2012) 109–130, 10.1007/s10616-011-9415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zheng GXY, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, Mudivarti P, Wyatt PW, Bharadwaj R, Makarewicz AJ, Li Y, Belgrader P, Price AD, Lowe AJ, Marks P, Vurens GM, Hardenbol P, Montesclaros L, Luo M, Greenfield L, Wong A, Birch DE, Short SW, Bjornson KP, Patel P, Hopmans ES, Wood C, Kaur S, Lockwood GK, Stafford D, Delaney JP, Wu I, Ordonez HS, Grimes SM, Greer S, Lee JY, Belhocine K, Giorda KM, Heaton WH, McDermott GP, Bent ZW, Meschi F, Kondov NO, Wilson R, Bernate JA, Gauby S, Kindwall A, Bermejo C, Fehr AN, Chan A, Saxonov S, Ness KD, Hindson BJ, Ji HP, Haplotyping germline and cancer genomes with high-throughput linked-read sequencing, Nat. Biotechnol. 34 (2016) 303–311, 10.1038/nbt.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Smith GP, Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface, Science. 228 (1985) 1315–1317. [DOI] [PubMed] [Google Scholar]
- [4].Fields S, Song O, A novel genetic system to detect protein-protein interactions, Nature. 340 (1989) 245–246, 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- [5].Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B, A generic protein purification method for protein complex characterization and proteome exploration, Nat. Biotechnol. 17 (1999) 1030–1032, 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- [6].Wilson TE, Fahrner TJ, Johnston M, Milbrandt J, Identification of the DNA binding site for NGFI-B by genetic selection in yeast, Science. 252 (1991) 1296–1300, 10.1126/science.1925541. [DOI] [PubMed] [Google Scholar]
- [7].Bayer CD, van Loo B, Hollfelder F, Specificity effects of amino acid substitutions in promiscuous hydrolases: context-dependence of catalytic residue contributions to local fitness landscapes in nearby sequence space, Chembiochem. 18 (2017) 1001–1015, 10.1002/cbic.201600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bill A, Rosethorne EM, Kent TC, Fawcett L, Burchell L, van Diepen MT, Marelli A, Batalov S, Miraglia L, Orth AP, Renaud NA, Charlton SJ, Gosling M, Gaither LA, Groot-Kormelink PJ, High throughput mutagenesis for identification of residues regulating human prostacyclin (hIP) receptor expression and function, PLoS One 9 (2014), e97973, 10.1371/journal.pone.0097973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jones EM, Lubock NB, Venkatakrishnan A, Wang J, Tseng AM, Paggi JM, Latorraca NR, Cancilla D, Satyadi M, Davis JE, Babu MM, Dror RO, Kosuri S, Structural and functional characterization of G protein–coupled receptors with deep mutational scanning, ELife. 9 (2020), e54895, 10.7554/eLife.54895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A, Hopf TA, Marks DS, Oren M, Segal E, A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation, Mol. Cell 71 (2018) 178–190.e8, 10.1016/j.molcel.2018.06.012. [DOI] [PubMed] [Google Scholar]
- [11].Mbonye U, Wang B, Gokulrangan G, Shi W, Yang S, Karn J, Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation, J. Biol. Chem. 293 (2018) 10009–10025, 10.1074/jbc.RA117.001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ulich C, Dunne A, Parry E, Hooker CW, Gaynor RB, Harrich D, Functional domains of Tat required for efficient human immunodeficiency virus type 1 reverse transcription, J. Virol. 73 (1999) 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ruben S, Perkins A, Purcell R, Joung K, Sia R, Burghoff R, Haseltine WA, Rosen CA, Structural and functional characterization of human immunodeficiency virus tat protein, J. Virol. 63 (1989) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sadaie MR, Mukhopadhyaya R, Benaissa ZN, Pavlakis GN, Wong-Staal F, Conservative mutations in the putative metal-binding region of human immunodeficiency virus tat disrupt virus replication, AIDS Res. Hum. Retrovir. 6 (1990) 1257–1263, 10.1089/aid.1990.6.1257. [DOI] [PubMed] [Google Scholar]
- [15].Dorsky DI, Wells M, Harrington RD, Detection of HIV-1 infection with a green fluorescent protein reporter system, J Acquir Immune Defic Syndr Hum Retrovirol. 13 (1996) 308–313, 10.1097/00042560-199612010-00002. [DOI] [PubMed] [Google Scholar]
- [16].Siekevitz M, Feinberg MB, Holbrook N, Wong-Staal F, Greene WC, Activation of interleukin 2 and interleukin 2 receptor (Tac) promoter expression by the trans-activator (tat) gene product of human T-cell leukemia virus, type I, Proc. Natl. Acad. Sci. U. S. A. 84 (1987) 5389–5393, 10.1073/pnas.84.15.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Baratloo A, Hosseini M, Negida A, El Ashal G, Part 1: simple definition and calculation of accuracy, sensitivity and specificity, Emerg (Tehran). 3 (2015) 48–49. [PMC free article] [PubMed] [Google Scholar]
- [18].Link RW, Mele AR, Antell GC, Pirrone V, Zhong W, Kercher K, Passic S, Szep Z, Malone K, Jacobson JM, Dampier W, Wigdahl B, Nonnemacher MR, Investigating the distribution of HIV-1 Tat lengths present in the Drexel medicine CARES cohort, Virus Res. 272 (2019), 197727, 10.1016/j.virusres.2019.197727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mele AR, Marino J, Dampier W, Wigdahl B, Nonnemacher MR, HIV-1 tat length: comparative and functional considerations, Front. Microbiol. 11 (2020) 444, 10.3389/fmicb.2020.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kuppuswamy M, Subramanian T, Srinivasan A, Chinnadurai G, Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis, Nucleic Acids Res. 17 (1989) 3551–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kamori D, Ueno T, HIV-1 tat and viral latency: what we can learn from naturally occurring sequence variations, Front. Microbiol. 8 (2017), 10.3389/fmicb.2017.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chopard C, Tong PBV, Tóth P, Schatz M, Yezid H, Debaisieux S, Mettling C, Gross A, Pugnière M, Tu A, Strub J-M, Mesnard J-M, Vitale N, Beaumelle B, Cyclophilin a enables specific HIV-1 tat palmitoylation and accumulation in uninfected cells, Nat. Commun. 9 (2018) 2251, 10.1038/s41467-018-04674-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sargeant D, Deverasatty S, Luo Y, Baleta AV, Zobrist S, Rathnayake V, Russo JC, Vyas J, Muesing MA, Schiller MR, HIVToolbox, an integrated web application for investigating HIV, PLoS One 6 (2011), e20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sargeant DP, Deverasetty S, Strong CL, Alaniz IJ, Bartlett A, Brandon NR, Brooks SB, Brown FA, Bufi F, Chakarova M, David RP, Dobritch KM, Guerra HP, Hedden MW, Kumra R, Levitt KS, Mathew KR, Matti R, Maza DQ, Mistry S, Novakovic N, Pomerantz A, Portillo J, Rafalski TF, Rathnayake VR, Rezapour N, Songao S, Tuggle SL, Yousif S, Dorsky DI, Schiller MR, The HIVToolbox 2 web system integrates sequence, structure, function and mutation analysis, PLoS One 9 (2014), e98810, 10.1371/journal.pone.0098810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kameoka M, Tanaka Y, Ota K, Itaya A, Yamamoto K, Yoshihara K, HIV-1 tat protein is poly(ADP-ribosyl)ated in vitro, Biochem. Biophys. Res. Commun. 261 (1999) 90–94, 10.1006/bbrc.1999.0964. [DOI] [PubMed] [Google Scholar]
- [26].D’Orso I, Frankel AD, Tat acetylation modulates assembly of a viral-host RNA-protein transcription complex, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 3101–3106, 10.1073/pnas.0900012106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sarmady M, Dampier W, Tozeren A, HIV protein sequence hotspots for crosstalk with host hub proteins, PLoS One 6 (2011), e23293, 10.1371/journal.pone.0023293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH, Crystal structure of HIV-1 tat complexed with human P-TEFb, Nature. 465 (2010) 747–751, 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schulze-Gahmen U, Hurley JH, Structural mechanism for HIV-1 TAR loop recognition by Tat and the super elongation complex, Proc. Natl. Acad. Sci. U. S. A. 115 (2018) 12973–12978, 10.1073/pnas.1806438115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schulze-Gahmen U, Echeverria I, Stjepanovic G, Bai Y, Lu H, Schneidman-Duhovny D, Doudna JA, Zhou Q, Sali A, Hurley JH, Insights into HIV-1 proviral transcription from integrative structure and dynamics of the tat:AFF4:P-TEFb:TAR complex, Elife. 5 (2016), 10.7554/eLife.15910 pii: e15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kao S-Y, Calman AF, Luciw PA, Peterlin BM, Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product, Nature. 330 (1987) 489–493, 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- [32].Gu W-G, Genome editing-based HIV therapies, Trends Biotechnol. 33 (2015) 172–179, 10.1016/j.tibtech.2014.12.006. [DOI] [PubMed] [Google Scholar]
- [33].D’Orso I, Jang GM, Pastuszak AW, Faust TB, Quezada E, Booth DS, Frankel AD, Transition step during assembly of HIV tat:P-TEFb transcription complexes and transfer to TAR RNA, Mol. Cell. Biol. 32 (2012) 4780–4793, 10.1128/MCB.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kuppuswamy M, Subramanian T, Srinivasan A, Chinnadurai G, Multiple functional domains of tat, the trans-activator of HIV-1, defined by mutational analysis, Nucleic Acids Res. 17 (1989) 3551–3561, 10.1093/nar/17.9.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schullek JR, Ruf W, Edgington TS, Key ligand interface residues in tissue factor contribute independently to factor VIIa binding, J. Biol. Chem. 269 (1994) 19399–19403. [PubMed] [Google Scholar]
- [36].Gray VE, Hause RJ, Luebeck J, Shendure J, Fowler DM, Quantitative missense variant effect prediction using large-scale mutagenesis data, Cell Syst. 6 (2018) 116–124.e3, 10.1016/j.cels.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Genevaux P, Schwager F, Georgopoulos C, Kelley WL, Scanning mutagenesis identifies amino acid residues essential for the in vivo activity of the Escherichia coli DnaJ (Hsp40) J-domain, Genetics. 162 (2002) 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bank C, Hietpas RT, Jensen JD, Bolon DNA, A systematic survey of an intragenic epistatic landscape, Mol. Biol. Evol. 32 (2015) 229–238, 10.1093/molbev/msu301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gonzalez CE, Ostermeier M, Pervasive pairwise intragenic epistasis among sequential mutations in TEM-1 β-lactamase, J. Mol. Biol. 431 (2019) 1981–1992, 10.1016/j.jmb.2019.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Poon A, Chao L, The rate of compensatory mutation in the DNA bacteriophage phiX174, Genetics. 170 (2005) 989–999, 10.1534/genetics.104.039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bertrand SJ, Aksenova MV, Mactutus CF, Booze RM, HIV-1 tat protein variants: critical role for the cysteine region in synaptodendritic injury, Exp. Neurol. 248 (2013) 228–235, 10.1016/j.expneurol.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Frankel AD, Biancalana S, Hudson D, Activity of synthetic peptides from the tat protein of human immunodeficiency virus type 1, Proc. Natl. Acad. Sci. U. S. A. 86 (1989) 7397–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Meredith LW, Sivakumaran H, Major L, Suhrbier A, Harrich D, Potent inhibition of HIV-1 replication by a tat mutant, PLoS One 4 (2009), e7769, 10.1371/journal.pone.0007769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Truant R, Cullen BR, The arginine-rich domains present in human immunodeficiency virus type 1 tat and rev function as direct importin β-dependent nuclear localization signals, Mol. Cell. Biol. 19 (1999) 1210–1217, 10.1128/MCB.19.2.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hauber J, Malim MH, Cullen BR, Mutational analysis of the conserved basic domain of human immunodeficiency virus tat protein, J. Virol. 63 (1989) 1181–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schiller MR, Chakrabarti K, King GF, Schiller NI, Eipper BA, Maciejewski MW, Regulation of RhoGEF activity by intramolecular and intermolecular-SH3 interactions, J.Biol.Chem. 281 (2006) 17774–17786. [DOI] [PubMed] [Google Scholar]
- [47].Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, Shendure J, Accurate classification of BRCA1 variants with saturation genome editing, Nature. 562 (2018) 217–222, 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Noderer WL, Flockhart RJ, Bhaduri A, Diaz de Arce AJ, Zhang J, Khavari PA, Wang CL, Quantitative analysis of mammalian translation initiation sites by FACS-seq, Mol. Syst. Biol. 10 (2014) 748, 10.15252/msb.20145136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pfeiffer F, Gröber C, Blank M, Händler K, Beyer M, Schultze JL, Mayer G, Systematic evaluation of error rates and causes in short samples in next-generation sequencing, Sci. Rep. 8 (2018) 10950, 10.1038/s41598-018-29325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Farouni R, Djambazian H, Ferri LE, Ragoussis J, Najafabadi HS, Model-based analysis of sample index hopping reveals its widespread artifacts in multiplexed single-cell RNA-sequencing, Nat. Commun. 11 (2020) 2704, 10.1038/s41467-020-16522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rosen CA, Tat and rev: positive modulators of human immunodeficiency virus gene expression, Gene Expr. 1 (1991) 85–90. [PMC free article] [PubMed] [Google Scholar]
- [52].Fernandes JD, Faust TB, Strauli NB, Smith C, Crosby DC, Nakamura RL, Hernandez RD, Frankel AD, Functional segregation of overlapping genes in HIV, Cell. 167 (2016) 1762–1773.e12, 10.1016/j.cell.2016.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Carvajal-Rondanelli P, Aróstica M, Álvarez CA, Ojeda C, Albericio F, Aguilar LF, Marshall SH, Guzmán F, Understanding the antimicrobial properties/activity of an 11-residue Lys homopeptide by alanine and proline scan, Amino Acids 50 (2018) 557–568, 10.1007/s00726-018-2542-6. [DOI] [PubMed] [Google Scholar]
- [54].Hyun S, Choi Y, Jo D, Choo S, Park TW, Park S-J, Kim S, Lee S, Park S, Jin SM, Cheon DH, Yoo W, Arya R, Chong YP, Kim KK, Kim YS, Lee Y, Yu J, Proline hinged amphipathic α-helical peptide sensitizes gram-negative bacteria to various gram-positive antibiotics, J. Med. Chem. 63 (2020) 14937–14950, 10.1021/acs.jmedchem.0c01506. [DOI] [PubMed] [Google Scholar]
- [55].Orzáez M, Salgado J, Giménez-Giner A, Pérez-Payá E, Mingarro I, Influence of proline residues in transmembrane helix packing, J. Mol. Biol. 335 (2004) 631–640, 10.1016/j.jmb.2003.10.062. [DOI] [PubMed] [Google Scholar]
- [56].Mori H, Sakashita S, Ito J, Ishii E, Akiyama Y, Identification and characterization of a translation arrest motif in VemP by systematic mutational analysis, J. Biol. Chem. 293 (2018) 2915–2926, 10.1074/jbc.M117.816561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Weinglass AB, Smirnova IN, Kaback HR, Engineering conformational flexibility in the lactose permease of Escherichia coli: use of glycine-scanning mutagenesis to rescue mutant Glu325–>Asp, Biochemistry. 40 (2001) 769–776, 10.1021/bi002171m. [DOI] [PubMed] [Google Scholar]
- [58].Weinglass AB, Sondej M, Kaback HR, Manipulating conformational equilibria in the lactose permease of Escherichia coli, J. Mol. Biol. 315 (2002) 561–571, 10.1006/jmbi.2001.5289. [DOI] [PubMed] [Google Scholar]
- [59].Frillingos S, Ujwal ML, Sun J, Kaback HR, The role of helix VIII in the lactose permease of Escherichia coli: I. Cys-scanning mutagenesis, Protein Sci. 6 (1997) 431–437, 10.1002/pro.5560060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Markham KJ, Tikhonova EB, Scarpa AC, Hariharan P, Katsube S, Guan L, Complete cysteine-scanning mutagenesis of the salmonella typhimurium melibiose permease, J. Biol. Chem. 297 (2021), 101090, 10.1016/j.jbc.2021.101090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].FASTX-toolkit, Cold Spring Harbour Laboritories. http://hannonlab.cshl.edu/fastx_toolkit/index.html, 2021. [Google Scholar]
- [62].Magoc T, Salzberg SL, FLASH: fast length adjustment of short reads to improve genome assemblies, Bioinformatics. 27 (2011) 2957–2963, 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics. 30 (2014) 2114–2120, 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet J. 17 (2011) 10, 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- [65].Zorita E, Cuscó P, Filion GJ, Starcode: sequence clustering based on all-pairs search, Bioinformatics. 31 (2015) 1913–1919, 10.1093/bioinformatics/btv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Li H, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, in: ArXiv Preprint ArXiv:1303.3997, 2013. [Google Scholar]
- [67].Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R, BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data, Bioinformatics. 32 (2016) 1749–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].pyVCF library. https://pyvcf.readthedocs.io/en/latest/API.html, 2022.
- [69].Accessible Surface Area and Accessibility Tool, Center for Informational Biology, Ochanomizu University, 2022. http://cib.cf.ocha.ac.jp/bitool/ASA/index.html. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is shared as part of aData in Brief co-submission