

Abstract
The use of radiotherapy for hypopharyngeal cancer (HC) treatment is increasing, and it is currently the primary treatment option for this cancer. However, radioresistance occurs in a proportion of patients. Here, we found that radiation increased proteasomal gene expression and that proteasome assembly was dependent on the induction of transcription factor NRF1 in HC. Through screening assays, we identified a mechanism by which proteasome-mediated degradation of DEP domain-containing mTOR-interacting protein (DEPTOR) contributes to the elevation of mTORC1 signaling after radiation. Therefore, after treatment with proteasome inhibitors (PIs), stabilization of DEPTOR inhibited mTORC1 signaling elevated by radiation and ultimately sensitized HC to radiotherapy. Mechanically, PIs not only interrupted the deubiquitination and degradation of DEPTOR but also suppressed the ubiquitination of DEPTOR mediated by β-TrCP. Clinically, the high levels of DEPTOR in HC cells were associated with sensitivity to radiotherapy and favorable prognosis. Stabilizing DEPTOR through targeting proteasome-mediated degradation is a potential strategy for sensitizing HC to radiotherapy.
Keywords: hypopharyngeal cancer, DEPTOR, proteasome, mTORC1, radioresistance
Graphical abstract
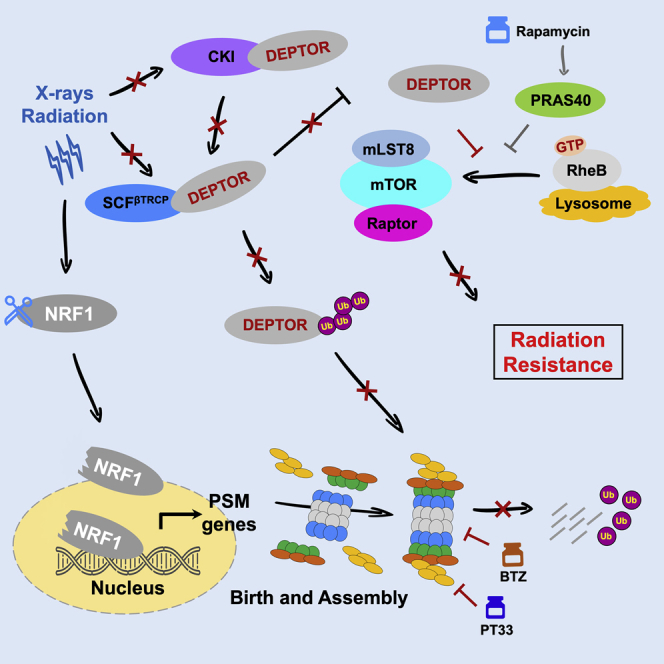
Wang et al. found that high DEPTOR levels in hypopharyngeal cancer (HC) cells were associated with the sensitivity to radiotherapy and favorable prognosis. Stabilizing DEPTOR through targeting proteasome-mediated degradation is a potential strategy for sensitizing HC to radiotherapy.
Introduction
Hypopharyngeal cancer (HC) is caused by abnormal growth of cells in the hypopharynx (also known as the laryngopharynx), where the larynx and esophagus meet, including the pyriform sinus, postcricoid area, and posterior pharyngeal wall. HC is relatively rare, accounting for approximately 3%–5% of all head and neck malignancies, but presents the worst prognosis of all head and neck cancer subsites.1, 2, 3 Traditional laryngopharyngectomy is difficult due to submucosal and lymphatic spread, resulting in worse quality of life than organ-preserving treatments. Treatment based on radiotherapy, driven by constant technological advances, is increasing and has been the major treatment strategy for HC.4, 5, 6, 7 Even so, the overall survival (OS) rates of HC are poor, usually under 50% at 3–5 years after treatment, and radioresistance still occurs in a proportion of patients.1,8 Thus, understanding the mechanisms of radioresistance will help develop novel sensitizing drug targets for future therapeutic intervention, reduce recurrence after radiotherapy, and prevent metastasis in HC.9,10
The ubiquitin-proteasome system (UPS) is the main mechanism by which cancer cells regulate protein degradation in response to numerous stresses or physiological processes.11,12 Currently, the correlation between the radiotherapeutic response and the capacity for protein degradation in HC cells is poorly understood. Here, we show that the radiation-mediated increase in proteasome gene expression and cellular proteasome content is dependent on the induction of the transcription factor nuclear factor erythroid-derived 2-related factor 1 (NRF1). As expected, targeting the proteasome markedly enhanced the sensitivity to radiotherapy in HC.
Mammalian target of rapamycin (mTOR) is an evolutionarily conserved serine/threonine kinase that integrates growth factors or stress signals to regulate multiple processes.13 It is increasingly apparent that regulation of the mTOR pathway occurs in the radiotherapeutic response, even in radioresistance.14, 15, 16, 17 An emerging concept in the field is that there is continuous cross-talk between mTORC1 signaling and proteasomal activity;18,19 however, little is known about how such coordination is achieved in radioresistance. Our results indicated that radiation indeed caused the enhancement of mTORC1 activity in HC, but more important, proteasome inhibition obviously suppressed radiation-induced elevation of mTORC1 signaling. Accordingly, it is necessary to identify and understand the proteasome-mTOR signaling regulatory network that mediates radioresistance.
Here, through the screen, we found that DEP domain-containing mTOR-interacting protein (DEPTOR) was rapidly degraded by the proteasome after radiation in HC cells. DEPTOR, whose stability is governed by the 26S proteasome, is an inhibitor of mTORC1 and mTORC2. DEPTOR is a physiological substrate of SCFβ-TrCP E3 ligase for targeted degradation. In response to growth signals, mTOR-dependent signaling, in collaboration with casein kinase I (CKI), phosphorylates DEPTOR and facilitates its binding to β-transducin repeats-containing proteins (β-TrCP) for subsequent degradation.20,21 In this way, mTOR generates an auto-amplification loop to protect cancers.22 In this work, we found that two kinds of proteasome inhibitors (PIs) maintained the stability of DEPTOR via interrupting β-TrCP-mediated degradation, and then inhibited radiation-elevated mTORC1 signaling to sensitize HC to radiotherapy. Furthermore, a higher DEPTOR level was found to potentially confer radiosensitivity and to be associated with a favorable prognosis in HC patients receiving chemoradiotherapy (CRT).
Results
Radiation induces the increase in HC cellular proteasome content
The proteasome degrades most cellular proteins in a controlled and tightly regulated manner, thereby controlling many processes, including signaling, trafficking, and protein quality control. To determine whether proteasomal degradation is associated with radiotherapy resistance, we studied the radiation-induced mechanisms that regulate the degradation activity in HC. To this end, we generated two-dimensional (2D) adherent monolayers and 3D spheroid suspensions models to measure proteasomal activity with X-ray radiation (XR) treatment in vitro (Figure 1A). Using a 26S proteasome hydrolysis activity assay, we observed two phenomena: (1) in both models, XR-treated FaDu cells possessed significantly higher hydrolysis activity (26S) than untreated cells collected on the same day, and (2) the corresponding hydrolysis activity significantly increased with the prolonging of culture time in 2D models or with sustained radiation treatment in 3D models (Figures 1B and S1). Cells tend to control the level of proteasome assembly to alter the proteasomal abundance, which is a highly regulated process that finely responds to cellular stress. Thus, we performed genome-wide transcriptomic analysis of XR-treated and untreated cells in 2D models, which revealed that XR treatment enhanced the expression of proteasomal subunit genes, such as PSMB5, PSMB10 (encoding 20S core particle [CP] components), PSMD9–11, PSMD14 (encoding 19S regulatory particle [RP] components; PSMD9/10 also belong to RP assembly chaperones [RACs]), NRF1 (encoding a common transcription factor of 26S proteasome subunits), PSMG1, PSMG2, and PAAF1 (encoding CP and RP assembly chaperones) (Figure 1C). Next, validation experiments were performed to confirm whether the overexpression (OE) of some factors was associated with the response to radiotherapy in FaDu cells. We observed that only NRF1 OE could lead to radioresistance in 2D adherent models, whereas PSMB5 (encoding a CP component) and PSMD14 (encoding an RP component) co-OE had no effect on overall radiosensitivity (Figures 1D and S2). NRF1 directly binds to the promoters of global PSM genes containing shared regulatory elements, raising the possibility of increasing 26S levels. Indeed, exogenous OE of NRF1 led to elevated levels of intact proteasomes, unlike PSMB5 and/or PSMD14 OE (Figure 1E). Strikingly, XR enhanced the number of NRF1-Myc foci in the nucleus in NRF1-Myc OE Fadu cells. That is to say, XR-mediated induction of NRF1 resulted in a marked increase in the processed isoform (p110) of NRF1Myc-tag located in the nucleus in FaDu cells (Figures 1F and 1G).
Figure 1.

Radiation induces the enhancement of proteasomal activity in HC cells
(A) Schematic illustration showing how 2D adherent monolayers and 3D spheroid suspensions models were treated with XR at the indicated times. (B) Relative 26S proteasome hydrolysis activity in 2D adherent and 3D spheroid FaDu cells treated with XR/no radiation (NR) as in (A). Data were normalized to the control group (n = 3). (C) Heatmap of transcriptomic analysis results for proteasomal genes in FaDu cells treated with a single fraction of XR (6-Gy) and cultured as normal for 7 days or not as in (A) (n = 3). (D) Images of 2D adherent colony formation assays in FaDu cells expressing vector or exogenous NRF1, PSMB5, PSMD14, or PSMB5 plus PSMD14 treated for 48 h with or without a single 2-Gy dose of XR (n = 3). (E) (Top) Cell survival (%) of 2D colony formation assays related to (D). (Bottom) Relative proteasomal activity in 2D adherent FaDu cells expressing vector or exogenous NRF1, PSMB5, PSMD14, or PSMB5 plus PSMD14 treated for 48 h with a single 2-Gy dose of XR, calculated as the AMC fluorescence increment (fold change) per minute (from the data in Figure S1) and normalized to that of the vector group. (F) Western blot analysis of the levels of NRF1-Myc in FaDu cells treated with or without a single 6-Gy dose for 48 h. (G) Immunofluorescence analysis of localization of exogenous NRF1-Myc in FaDu cells treated with or without a single 6-Gy dose for 48 h. (Top) Representative images. (Bottom) The quantitative analysis: The number of NRF1 foci per cell nucleus. (H) Immunofluorescence analysis of colocalization of exogenous PSMD14-EGFP and endogenous PSMB5 in FaDu cells treated with or without a single 6-Gy dose for 48 h. (Top) Representative images. (Bottom) The quantitative analysis: the number of co-localization foci per cell. (B and E) The statistical data are presented as means ± standard deviation (SD), 1-way ANOVA. (G and H) The statistical analyses of the tail moment of all views from 3 independent experiments, 2-sample t test. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Furthermore, we observed that XR markedly increased the number of foci of colocalized exogenous FLAG-PSMD14 and endogenous PSMB5 (Figure 1H). In brief, these data indicated that XR enhanced proteasome-mediated protein degradation by promoting the assembly of the proteasome and the expression of NRF1 functioning downstream as a transcription factor. On this basis, we speculated that this was related to radioresistance in HC.
Proteasome inhibitors sensitize HC cells to radiation
To determine whether the inhibition of the proteasome was functionally associated with the response to XR, we evaluated the sensitization effects of two kinds of PIs targeting proteasome 20S CP subunits—PSMB5 (Bortezomib [BTZ]) or 19S RP-associated deubiquitinases (DUBs) (PT33) using HC cells. We performed 2D colony formation assays. The radiation dose-survival curves revealed that both BTZ and PT33 pretreatment significantly reduced the colony formation of FaDu cells after XR (Figure 2A). To describe the synergistic effects of combined treatments, sensitivity enhancement ratios (SERs) for estimated survival rates of 50%, 30%, and 10% were calculated in the combined groups in FaDu cells. SER shows the factor by which the radiotherapy dose must be multiplied to achieve the same estimated survival rate in the absence of the PIs. In accordance with the clonogenic survival rates, SERs of 50%, 30%, and 10% were 2.15, 1.92, and 1.57, respectively (BTZ), or 2.07, 1.87, and 1.58, respectively (PT33). All SERs >1, which means PIs caused radiosensitization (Figure 2A). Given that the clone-forming ability of Detroit 562 cells was weak, colony formation assays were not performed in Detroit 562 cells. Then, cell viability assays were used to further confirm the synergistic effects of XR and PIs. The combination index (CI), which indicates synergism, was calculated using the Chou-Talalay method, where CI > 1, CI = 1, or CI < 1 indicates an antagonistic, additive, or synergistic effect, respectively. In Figures 2B and 2D, we observed that combination treatment with XR and BTZ/PT33 synergistically suppressed FaDu and Detroit 562 cell viability under four inhibitory effects (CI < 1). Moreover, in conditional 3D spheroid culture models (Figure S3A), the combined administration of XR and PIs inhibited oncosphere formation of FaDu cells much more effectively than XR or either of the single agents alone, yielding fewer and smaller oncospheres, starting either on day 4 (forming spheroids, Figure 2C) or on day 1 (before forming spheroids, Figure S3B) after planting FaDu cells. Of note, when intervention started on day 4, both doses of BTZ (5 or 2.5 nM) or PT33 (10 or 5 nM) synergistically enhanced the inhibitory effects of XR on oncosphere formation (all Q > 1.15, Figure 2E). Meanwhile, when intervention started on day 1, BTZ (2.5 nM) or PT33 (5 nM) exerted similar synergistic effects (Q > 1.15, Figure S3C). Together, these results indicate that PIs and radiation in combination synergistically kill HC cells in vitro.
Figure 2.

Proteasome inhibitors sensitize HC cells to radiation
(A) Two-dimensional colony formation assays in FaDu cells treated with PIs (BTZ: 5 nM, PT33: 10 nM) for 12 h and subsequently with a single dose of 0–4 Gy. (Left) Representative images. (Right) Survival curves and relative SER estimated survival rates of 50%, 30%, and 10%, shown as means ± SDs from 3 independent experiments (1-way ANOVA, DMSO as a vehicle control). (B and D) The combination index (CI) of XR (indicated doses) and PIs (indicated concentrations) on FaDu cells based on the results of cell viability assays using CCK-8 in (B) FaDu and (D) Detroit 562 cells. (C and E) Conditional 3D spheroid formation assays for FaDu cells treated with PIs (DMSO as vehicle control) plus XR (NR as control) starting on day 4 as in Figure S3A. (C) Representative images. (E) Numbers of spheres (diameter ≥75 μm) from 3 independent experiments, shown as means ± SDs (1-way ANOVA). Q was calculated as follows: Q = IAB/[IA + IB (1 − IA)], where IA, IB, and IAB indicate the inhibitory rates of PIs, XR, and PIs + XR, respectively. Q < 0.85, 0.85 ≤ Q ≤ 1.15, and Q > 1.15 indicate antagonistic, additive, and synergistic effects, respectively. (F) Annexin V/PI double-staining assays with flow cytometry to clarify the percentage of apoptotic cells in FaDu and Detroit 562 cells at 72 h after XR treatment. Cells were pretreated with PIs (BTZ: 5 nM, PT33: 10 nM; DMSO as vehicle control) for 12 h before XR. The statistical data are presented as the means ± SDs of at least 3 independent experiments (1-way ANOVA). (G) Western blot analysis of the levels of PARP (pro-/cleaved) and caspase 3 (pro-/cleaved) in FaDu and Detroit 562 cells at 72 h after XR treatment, PIs (BTZ: 5 nM, PT33: 10 nM; DMSO as vehicle control) were pretreated for 12 h before XR. (H and I) The xenograft tumor model in nude mice showed that PIs enhanced the sensitivity of HC to radiotherapy in vivo. FaDu xenografts formed in the right flank of BALB/c nude mice were treated alternately with PIs (BTZ 0.5 mg/kg × 7 or PT33 1 mg/kg × 7; normal saline [NS] was used as negative control; purple arrows) and X-rays (2-Gy every other day × 7; orange arrows). (H) Xenograft tumors were dissected and weighed in the NS, BTZ, and PT33 groups (n = 8 tumors/group). (I) The tumor growth curves were drawn by measuring the tumor volume in different groups, shown as means ± SDs (1-way ANOVA). Q value in (F) and (H) indicates the combination effect of XR treatment and PIs (see the legend for E). All statistical data: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; N.S., not significant.
We next used flow cytometry to examine whether the combination synergistically triggered apoptosis. In agreement with the role of PIs in increasing radiation sensitivity, both BTZ and PT33 significantly increased XR-induced apoptosis in FaDu (the average apoptotic ratios increased from 23.29% to 37.16% [BTZ] or 37.78% [PT33]) or Detroit 562 cells (the ratios increased from 13.61% to 21.30% [BTZ] or 19.77% [PT33]). We also calculated that combination treatment with XR and PIs synergistically promoted apoptosis (all Q > 1.15) (Figures 2F and S3D). Consistent with this, the levels of cleaved poly (ADP-ribose) polymerase PARP and cleaved caspase 3 were increased in the combined (BTZ/PT33 plus XR) treatment groups compared to the XR or BTZ/PT33 alone groups in FaDu and Detroit 562 cells (Figure 2G). In contrast, treatment with PT33 or BTZ alone had almost no effects on apoptosis, suggesting that PIs promote radiation-inducible apoptosis in HC cells.
To investigate the potential role of PIs in HC cell sensitivity to XR in vivo, we next tested the inhibitory effects of combinations of XR and PIs in FaDu cell xenograft mouse models. Tumors were established by subcutaneous transplantation of diced tumor before the initiation of treatment. The treatment period started on day 8 and ended on day 21 after transplantation. We observed that tumor growth was almost not reduced by single-drug therapy; tumors in mice treated with 0.5 mg/kg/day × 7 BTZ or 1 mg/kg/day × 7 PT33 grew as rapidly as those in vehicle-treated mice. In contrast, combination treatment of XR and PIs dramatically decreased tumor growth relative to XR plus vehicle treatment, with essentially no increase in tumor size (Figures 2H and 2I). Notably, the tumor inhibition ratios were markedly improved to 86.63% or 86.26% by the combination of XR and PT33 or BTZ, respectively, compared with 51.21% of inhibition by XR plus vehicle treatment (Figure S3E). The combination of PIs and XR displayed a synergistic effect (Q = 1.61 or 1.46 for BTZ or PT33, respectively; Figure 2H). None of these treatments reduced murine weight or induced observable toxicity. In brief, these results revealed that PIs sensitized HC cells to XR treatment in vitro and in vivo.
Inhibition of the proteasome suppresses XR-induced elevated mTORC1 signaling
Given the functional inhibition of the proteasome in sensitizing HC to radiotherapy, emerging evidence suggests that proteasome-mediated degradation activity enhancement induced by XR may activate several signaling pathways, which fight against radiation-caused damage and then enable cells to survive radiation treatment in HC. Thus, we performed a screen to identify the elevated signaling pathway induced by XR, which could be interrupted by PIs (designed as shown in Figure S4A). Single (XR or BTZ/PT33) and combined treatments (XR plus BTZ/PT33) were given to FaDu cells (Figure S4B), and then western blot (WB) analysis was conducted to detect the activities of multiple critical signaling pathways. Our results demonstrated that XR-mediated enhancement of mTORC1 activity was markedly attenuated by PIs. The phosphorylated levels of mTOR and downstream substrates, including ribosomal protein S6 kinase (S6K), ribosomal protein S6 (S6), eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), and Unc-51-like autophagy activating kinase 1 (ULK1), were significantly decreased in the PI-treated groups with or without XR, even if XR induced markedly the elevation of these phosphorylated levels (Figure 3A). However, when cells were treated with PIs and/or XR, no effects were observed on the epidermal growth factor receptor (EGFR), mitogen-activated protein/extracellular signal-regulated kinase 1/2 (MAPK/ERK), nuclear factor κB (NF-κB), JAK/STAT, stress-activated protein kinase/Jun amino-terminal kinase (SAPK/JNK), Wnt/β-catenin, and Hippo signaling pathways (Figure S4C). Based on this, we next investigated whether PIs affected the XR-induced activation of mTORC1 and found that mTORC1 signaling was upregulated in a time-dependent manner during XR treatment, but interestingly, PT33 or BTZ pretreatment markedly interfered with this upregulation (Figures 3B and S5).
Figure 3.

Inhibition of proteasome suppresses XR-induced elevation in mTORC1 signaling
(A) Western blot analysis of the levels of mTORC1 signaling pathway components in FaDu cells treated with PIs plus XR as in Figure S4B. (B) Western blot analysis of mTORC1 signaling activation in FaDu cells treated with PT33 followed by XR at indicated times. (C) Western blot analysis of the influence of XR on mTORC1 signaling at indicated times in FaDu cells at a high cell density. (D) Inhibition of mTORC1 signaling by PIs for various time intervals in FaDu cells before treatment with XR at a medium/high cell density. (E) Western blot analysis of the levels of mTORC1 signaling pathway components in FaDu cells treated with XR followed by rapamycin or PT33 for the indicated time, respectively. For all assays, 10 nM of BTZ, 20 nM of PT33, and 100 nM of rapamycin were used; DMSO or NR was used as vehicle control; RAPA: rapamycin; and the XR dose was 6-Gy.
We next determined the pathway or contributing factor by which PIs disrupt mTORC1 signaling. Previous studies showed that mTORC1 signaling activity was reduced when the cell density increased.23 Consistently, we found that increasing the cell density indeed resulted in reduced mTORC1 signaling in FaDu cells (Figure 3D). Thus, we further examined whether low mTORC1 kinase activity (high cell density) was affected by XR and found that mTORC1 signaling, which was inhibited by high cell density, was restored in cells exposed to XR (Figure 3C). However, treatment of cells with PIs also significantly inhibited mTOR signaling, which was restored at 8–12 h after XR treatment in high cell density and medium cell density (Figure 3D). These results indicated that PIs interrupt the process of mTORC1 signaling activation induced by XR.
Given the role of mTORC1 in the process of radioresistance, we compared the inhibitory effects on mTORC1 activity of PIs and mTORC1 inhibitor-rapamycin after XR. Both control (no radiation [NR]) and XR-treated FaDu cells displayed rapamycin-sensitive inhibition of mTORC1 signaling within 8 h, whose inhibitory effect was markedly faster than that of PT33 treatment (Figures 3E and S6). However, with increasing treatment time, unlike PT33, XR caused the reactivation of mTORC1 signaling, which meant that rapamycin failed to sustain that inhibitory effect (Figure 3E). These results indicated that the inhibitory mechanism of PT33 on mTORC1 signaling is radically different from that of rapamycin.
Furthermore, we investigated whether PIs inhibited mTOR signaling by activating autophagy or protein phosphatase 2A (PP2A) in FaDu cells. We observed that the addition of an autophagy inhibitor (bafilomycin A1) had little effect on PI-mediated suppression of elevated mTORC1 signaling after XR (Figure S7). Meanwhile, we found that the PP2A inhibitor LB100 also failed to reverse PI-mediated suppression of elevated mTORC1 signaling after XR; actually, even if LB100 resulted in increased phosphorylation levels of S6, it did not affect the top-down mTORC1 signaling inhibited by PIs (Figure S8). On the whole, these results indicated that the primarily inhibitory effect of PIs on mTORC1 signaling after XR was independent of autophagy or PP2A through an as yet unidentified mechanism.
PIs inhibit XR-elevated mTORC1 signaling via stabilizing DEPTOR
We further assessed whether XR induced the degradation of mTORC1 components or regulators in response to stress. Our results showed that the level of DEPTOR was markedly decreased after XR treatment in two HC cells (Figure 4A). This phenomenon was unique to DEPTOR, as no apparent fluctuation was observed in the expression levels of other components or regulators we examined (Figure S9). Furthermore, XR induced a decrease of endogenous DEPTOR or exogenous FLAG-tagged DEPTOR in a time-dependent manner (Figure S10). However, PIs efficiently maintained the stability of DEPTOR protein after XR treatment in FaDu and Detroit 562 cells, which decreased the mRNA levels of DEPTOR as a feedback regulation (Figures 4B and S11). Consistent with this, cycloheximide (CHX) chase analysis revealed that the increase in DEPTOR abundance following PT33 but not rapamycin treatment is primarily due to the persistently increased DEPTOR half-life after XR (Figures 4C and S12). To determine whether the increased DEPTOR turnover rate after PI plus XR treatment depended on proteasome-mediated degradation, FLAG-DEPTOR stable OE FaDu cells were transfected with hemagglutinin (HA)-tagged K48-specific ubiquitination. Then, we performed immunoprecipitation (IP)/WB assays to observe the level of K48-linked polyubiquitinated DEPTOR and found that the K48- ubiquitinated DEPTOR accumulated and reached a peak at 12 h after XR treatment and then decreased gradually, indicating a unidirectional process of ubiquitination and then degradation of DEPTOR by UPS. As expected, PT33 as a PI inversely interrupted that process and retained the levels of K48-ubiquitinated and non-ubiquitinated DEPTOR (Figures 4D and 4E). These results reveal that K48-ubiquitinated DEPTOR increased in FaDu cells treated with PT33 plus XR compared to DMSO plus XR group, which means PT33 inhibited the degradation of DEPTOR by ubiquitination (Figure 4E).
Figure 4.

PIs inhibit XR-elevated mTORC1 signaling via stabilizing DEPTOR
(A) Western blot analysis of the levels of mTORC1 signaling pathway components and regulators in FaDu and Detroit 562 cells treated with PIs (DMSO as vehicle control) plus XR (NR as control) as in Figure S4B. (B, D–F, H) FaDu cells were treated with PT33 (20 nM), BTZ (10 nM), rapamycin (RAPA, 100 nM), or PT33 (20 nM) + rapamycin (100 nM) for 12 h (DMSO as vehicle control) and subsequently exposed to XR (6-Gy). (B) Western blot analysis of DEPTOR levels in FaDu and Detroit 562 cells treated with BTZ (top) or PT33 (bottom), followed by XR for indicated times. (Left) Representative images; (right) DEPTOR grayscale intensity normalized to GAPDH at the indicated times after XR treatment. (C) Western blot analysis of the DEPTOR levels in FaDu and Detroit 562 cells, at 2 h with CHX (50 μM) and PT33 (20 nM) treatment, subsequently exposed to XR, and collected cell lysis for the indicated times. (Left) Representative images. (Right) DEPTOR grayscale intensity normalized to GAPDH at indicated times after XR treatment. (D and E) Co-IP and Western blot analyses for detecting the K48-linked polyubiquitinated levels of DEPTOR. (D) Cells were transfected with HA-ubiquitin (K48-specific) and FLAG-DEPTOR followed by XR treatment, and collected cell lysis for the indicated times. IP with FLAG, IB with HA. (E) Cells were transfected with HA-ubiquitin (K48 specific) followed by XR treatment, and collected cell lysis for the indicated times. IP with HA, IB with endogenous DEPTOR. (F) Co-IP and Western blot analyses of the interaction between β-TrCP/CKI/mTOR and DEPTOR in FLAG-DEPTOR or Myc-β-TrCP overexpression cells treated with XR, and collected cell lysis for 6 h, NR as control. (G) 2D adherent colony formation assays in DEPTOR-KO and TSC2-KO cells exposed to a single 3- or 6-Gy dose of XR (mock or NC cells were used as control). (Top) Cell survival rates. (Bottom) The ratio of DEPTOR-KO or TSC2-KO to NC colony number. (H) Western blot analysis of the levels of mTORC1 signaling pathway components in DEPTOR-KO and TSC2-KO cells pretreated with BTZ, PT33, or rapamycin and subsequently exposed to XR, and collected cell lysis for 6 h. (I) 2D adherent colony formation assays for cell survival rate in DEPTOR-KO or WT FaDu cells pretreated with PT33 (10 nM), and consequently exposed to a single 3-Gy dose of XR treatment. (J) Cell viability was measured using the CCK-8 assay, and half-maximal inhibitory concentration (IC50) values were calculated in DEPTOR-KO or WT FaDu cells treated with PT33 plus XR-3Gy (NR as control). All statistical data are presented as means ± SDs (n = 3; B, G, and J: 1-way ANOVA; C and I: 2-sample t test). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; N.S., not significant.
Since β-TrCP as an E3 ubiquitin ligase targets DEPTOR explicitly for degradation, we then asked whether β-TrCP may be involved in the ubiquitination of DEPTOR for degradation. For this, we conducted co-IP/WB assays with exogenous expression of FLAG-DEPTOR or Myc-β-TrCP, respectively. Our results showed XR indeed enhanced the β-TrCP-DEPTOR interaction; concordantly, XR abrogated the bind of DEPTOR to mTOR. In addition, CKI as a kinase of DEPTOR facilitates DEPTOR binding to β-TrCP, and we found that XR induced the enhancement of the interaction of CKI and DEPTOR. By contrast, PT33 absolutely abolished the β-TrCP-DEPTOR and CKI-DEPTOR interactions and inversely enhanced the DEPTOR-mTOR interaction with or without XR. Notably, unlike PT33, rapamycin had no apparent effect on the interaction between β-TrCP and DEPTOR. This is probably due to rapamycin failed to affect the formation of CKI-TrCP-DEPTOR complex after XR (Figure 4F). In addition, after treatment with a growth factor (insulin), we noted that the β-TrCP level was increased, coupled with decreased DEPTOR levels, and elevated mTORC1 signaling (Figure S13A). Consistent with this, both exogenous and endogenous β-TrCP were upregulated in response to XR, which can be attenuated by PT33 (Figure S13B), probably because PT33 inhibited the normal ubiquitination process of DEPTOR and β-TrCP as E3 ubiquitin ligase was controlled by feedback inhibition. Collectively, these results indicated that UPS-mediated degradation of DEPTOR induced by XR treatment can be arrested by abrogating proteasomal activity.
It has been well established that the functional abrogation of proteasome arrests XR-induced elevated mTORC1 signaling, which ultimately renders HC cells sensitive to radiotherapy. Given that both DEPTOR and TSC2 reduce mTORC1 activity, we next examined whether DEPTOR or TSC2 deficiency affects the mTORC1-governed radioresistance. Notably, using a 2D adherent colony formation assay, we found that both DEPTOR-knockout (KO) and TSC2-KO cells were resistant to moderate-dose XR (3-Gy); however, unlike TSC2-KO cells, which were sensitive to high-dose XR (6-Gy), DEPTOR-KO cells were also resistant to high-dose XR (Figures 4G and S14). These results suggested that DEPTOR deficiency resulted in more robust activation of mTORC1 signaling in response to XR treatment. These effects were likely to be associated with radiotherapeutic resistance.
Given that proteasome inhibition resulted in the accumulation of DEPTOR, we further examined whether inactivation of mTORC1 signaling mediated by PIs depended on DEPTOR. Indeed, treatment of DEPTOR-deficient cells with PIs failed to inhibit mTORC1 signaling after XR treatment. However, in TSC2-depleted cells, the PI-induced inhibition of mTORC1 signaling was not reversed by the loss of TSC2 (Figure 4H). In addition, our results illustrated that the inhibitory effect of mTORC1 signaling caused by rapamycin was gradually eliminated after XR treatment, which was likely concomitant with the decrease in DEPTOR levels. This did not happen in TSC2-deficient cells (Figure 4H). Functionally, in DEPTOR-depleted cells, PT33 not only almost lost the ability of radiosensitization as determined by the 2D adherent colony formation assay but it also failed to enhance the sensitivity to XR to kill cells as determined by assessing cell viability (Figures 4I, 4J, and S15). Taken together, PIs inhibited XR-elevated mTORC1 signaling in a DEPTOR-dependent manner, and the stability of DEPTOR was indispensable to repress mTORC1 signaling.
High DEPTOR levels are associated with sensitivity to radiotherapy and favorable prognosis
Analysis of publicly available gene expression data (Databases: The Cancer Genome Atlas [TCGA]: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga and Oncomine: www.oncomine.org/resource/login.html) showed that DEPTOR levels were significantly decreased in head and neck squamous cell carcinoma (HNSCC) samples compared with normal samples (Figure S16), which suggests that DEPTOR is involved in the development of HNSCC. There are few studies on the role of DEPTOR in radiotherapy resistance, so exploiting new applications for this factor is of clinical importance. To identify the potential role of DEPTOR in the response to radiotherapy, we conducted immunohistochemical staining. We obtained the immunoreactive scores (IRSs) on a total of 63 clinically annotated pretreatment HC biopsies of tumor specimens from The First Affiliated Hospital, Sun Yat-sen University (SYSUFAH). Among them, 60 patients underwent pre- and post-CRT magnetic resonance imaging (MRI). Of these, 15 patients had a complete response (CR), 16 patients had a partial response (PR), stable disease (SD) was observed in 24 patients, and progressive disease (PD) was observed in 5 patients according to the Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 guidelines for solid tumors after CRT treatment.24 CR and PR were defined as objective response (OR), which means that these patients were sensitive to CRT.
Analysis revealed that DEPTOR levels were significantly higher in tumors from OR patients than in those from non-OR patients (Figure 5A); there were fewer patients with OR in the DEPTOR-low group (8/27, 29.6%) than in the DEPTOR-high group (23/33, 69.7%). Notably, higher DEPTOR levels were significantly associated with lower p-S6 levels (p = 0.049), OR (p = 0.002), and favorable prognosis (p = 0.002) (Table 1). In addition, the levels of DEPTOR were negatively correlated with p-S6 levels in pretreatment biopsies of tumor specimens (r = −0.685, p < 0.001, Figure 5B). As expected, we verified this correlation in nude mice bearing subcutaneous FaDu xenografts, accordingly, and PIs reduced p-S6 levels and enhanced DEPTOR levels (Figure S17).
Figure 5.

DEPTOR is highly expressed in HC patients who are sensitive to radiation and associated with favorable prognosis
(A) MRI of patients before/after CRT and representative images of DEPTOR and p-S6 IHC staining in tumors before CRT in HC. (B) IHC scores showing the correlation between DEPTOR and p-S6 levels in HC before CRT. Kaplan-Meier analysis of (C) overall survival and (D) progression-free survival in the patients from SYSUFAH separated by DEPTOR IRS. Kaplan-Meier analysis of (E) overall survival and (F) progression-free survival in the patients from SYSUFAH separated by p-S6 IRS.
Table 1.
Correlation between levels of DEPTOR and clinicopathological parameters for pre-CRT samples
| Variables | No. patients (%) | DEPTOR levels |
Hazard ratio (95% CI) | p | |
|---|---|---|---|---|---|
| Low (n, %) | High (n, %) | ||||
| Gender | 0.065 | ||||
| Male | 59 (93.7) | 28 (44.4) | 31 (49.2) | 1.129 (1.002–1.272) | |
| Female | 4 (6.4) | 0 | 4 (6.35) | – | |
| Age, ya | 0.176 | ||||
| ≤58 | 33 (52.4) | 12 (19.0) | 21 (33.3) | 0.714(0.431–1.185) | |
| >58 | 30 (47.6) | 16 (25.4) | 14 (22.2) | 1.429(0.852–2.396) | |
| Smoke | 0.575 | ||||
| No | 18 (28.6) | 7 (11.1) | 11 (17.5) | 0.795(0.355–1.783) | |
| Yes | 45 (71.4) | 21 (33.3) | 24 (38.1) | 1.094(0.802–1.491) | |
| Drink | 0.344 | ||||
| No | 22 (34.9) | 8 (12.7) | 14 (22.2) | 0.714(0.350–1.456) | |
| Yes | 41 (65.1) | 20 (31.7) | 21 (33.3) | 1.190(0.832–1.703) | |
| cT | 0.374 | ||||
| I-II | 17 (27.0) | 6 (9.5) | 11 (17.5) | 0.682 (0.288–1.614) | |
| III-IV | 46 (73.0) | 22 (34.9) | 24 (38.1) | 1.146 (0.852–1.541) | |
| cN | 0.948 | ||||
| 0–1 | 16 (25.4) | 7 (11.1) | 9 (14.3) | 0.972 (0.414–2.283) | |
| 2–3 | 47 (74.6) | 21 (33.3) | 26 (41.3) | 1.010 (0.756–1.348) | |
| cTNM | 0.575 | ||||
| II-III | 18 (28.6) | 9 (14.3) | 9 (14.3) | 1.250 (0.574–2.724) | |
| IVA-IVB | 45 (71.4) | 19 (30.2) | 26 (41.3) | 0.913 (0.663–1.259) | |
| Pathologic grade | 0.441 | ||||
| Low/intermediate | 23 (46.0) | 11 (22.0) | 12 (24.0) | 1.266 (0.698–2.295) | |
| High | 27 (54.0) | 10 (20.0) | 17 (34.0) | 0.812 (0.472–1.398) | |
| Induced chemotherapy | 0.419 | ||||
| No | 4 (6.3) | 1 (1.6) | 3 (5.0) | 0.417 (0.046–3.790) | |
| Yes | 59 (93.7) | 27 (42.9) | 32 (50.8) | 1.055 (0.932–1.194) | |
| Induced chemotherapy regimens | 0.239 | ||||
| TPF | 25 (42.4) | 11 (18.6) | 14 (23.7) | – | |
| TP | 19 (32.2) | 6 (10.2) | 13 (22.0) | – | |
| PF | 12 (20.3) | 8 (13.6) | 4 (6.8) | – | |
| TPF/TP + PDL1 | 3 (5.1) | 2 (3.4) | 1 (1.7) | – | |
| Concurrent chemotherapy | 0.565 | ||||
| No | 6 (9.5) | 2 (3.2) | 4 (6.3) | 0.625 (0.123–3.168) | |
| Yes | 57 (90.5) | 26 (41.3) | 31 (49.2) | 1.048 (0.896–1.227) | |
| Tumor size, cm | 0.006 | ||||
| ≤2 | 17 (27.0) | 5 (7.9) | 12 (19.0) | – | |
| >2–≤4 | 31 (49.2) | 11 (17.5) | 20 (31.7) | – | |
| >4 | 15 (23.8) | 12 (19.0) | 3 (4.8) | – | |
| RECIST | 0.002 | ||||
| CR/PR | 31 (51.7) | 8 (13.3) | 23 (38.3) | 0.425 (0.228–0.793) | |
| SD/PD | 29 (48.3) | 19 (31.7) | 10 (16.7) | 2.322 (1.310–4.116) | |
| p-S6 | <0.001 | ||||
| IRS<8 | 32 (50.8) | 5 (7.9) | 27 (42.9) | 0.231 (0.103–0.523) | |
| IRS≥8 | 31 (49.2) | 23 (36.5) | 8 (22.9) | 3.594 (1.909–6.766) | |
| Status | 0.002 | ||||
| Alive | 44 (69.8) | 14 (22.2) | 30 (47.6) | 0.583 (0.393–0.865) | |
| Dead | 19 (30.2) | 14 (22.2) | 5 (7.9) | 3.500 (1.434–8.540) | |
Boldface values are statistically significant.
Median; cT: clinical T stage; cN: clinical N stage.
For the total population, higher DEPTOR levels were significantly correlated with longer OS (p < 0.001) and progressive-free survival (PFS, p < 0.001) (Figures 5C and 5D). Reciprocally, lower p-S6 levels were significantly correlated with longer OS (p < 0.001) and PFS (p < 0.001) (Figures 5E and 5F). Furthermore, the significance was unique to DEPTOR; both univariate and multivariate Cox proportional hazard regression analysis revealed that higher DEPTOR levels were correlated with longer OS and PFS, and DEPTOR could serve as an independent prognostic factor (Table S1).
To clarify whether chemotherapy also contributed to DEPTOR-mediated sensitivity to CRT, we investigated the change of DEPTOR levels in FaDu cells treated with chemotherapy drugs, including paclitaxel, cisplatin, and 5-fluorouracil in vitro. These chemotherapeutic drugs failed to affect the level of DEPTOR (Figure S18). These findings suggest that high DEPTOR expression confers sensitivity to radiotherapy but not to chemotherapy in CRT. In summary, DEPTOR was found to potentially confer radiosensitivity in HC; thus, higher DEPTOR levels could predict favorable prognosis in HC patients receiving CRT.
Discussion
Recent studies implicated that either cellular or purified proteasome may be a direct target of irradiation, and proteasome activity was inhibited immediately after irradiation,25, 26, 27 perhaps because the irradiation had a destructive effect on the 26S complex. Accordingly, in this study, we observed that multiple low-dose XR reduced proteasome gene expression and proteasome assembly, and the cellular proteasome contents were dependent on the induction of the transcription factor NRF1 in HC. On this basis, we speculate that proteasome damage caused by radiation is instantaneous; subsequently, the induction of proteasomal gene expression and proteasome assembly can restore the activity of 26S rapidly. Hence, proteasome-mediated degradation is probably necessary for radiotherapeutic response or resistance. However, the precise mechanisms of this function are not well understood.
Through the screening assays, we identified a previously uncharacterized mechanism by which UPS-mediated degradation of DEPTOR contributes to the elevation of mTORC1 signaling after radiation, ultimately leading to radioresistance in HC. As is known, mTOR directly interacts with DEPTOR via its PDZ domain, phosphorylates DEPTOR, and promotes its degradation via binding β-TrCP.21,22 Little is known about the other function of DEPTOR, which always appears to be dedicated to negatively regulating the mTORC1 signaling network.28,29 Consistent with this, in response to radiation, the elevated effect was unique to mTORC1 signaling, but not other pathways, accompanied by the unique elimination of DEPTOR, as no noticeable fluctuation of other components or regulators of mTORC1 signaling were observed. These data support a model whereby radiation leads to an increase in new proteasome content, which preferentially targets a specific substrate to enhance outputs of mTORC1 signaling.
Ubiquitination involves a complex interplay of ubiquitinating and DUBs, and E3 ubiquitin ligase is an extremely important element to control the types of substrates.30 Indeed, we found that β-TrCP as E3 ubiquitin ligase of DEPTOR was similarly upregulated in a time-dependent manner after radiation or growth factor stimulation treatment, coupled with decreased DEPTOR levels and elevated mTORC1 signaling. In this case, the ubiquitination of DEPTOR mediated by β-TrCP may represent another critical response after radiation. Together, the elevated activity of both DEPTOR ubiquitination and the proteasome jointly promote the elimination of DEPTOR. However, we found that targeting the proteasome using PIs abrogated not only proteasomal activity but also the inhibition of ubiquitination mediated by β-TrCP to maintain the stability of DEPTOR as a feedback loop, inhibiting elevated mTORC1 signaling caused by radiation. Thus, consistent with previous reports,31, 32, 33 PIs can also efficiently enhance the sensitivity to radiation in HC. Notably, BTZ, the first 20S CP inhibitor, is used to treat multiple myeloma in the clinic; however, resistance almost inevitably arises.34 Interestingly, the defects of BTZ can be overcome, for example, by targeting 19S RP with PT33, a novel 19S RP-associated DUB inhibitor.35 Thus, regardless of the drugs used, effectively inhibiting proteasomal activity could be a promising strategy to maintain DEPTOR abundance.
In addition, several studies investigated the cross-talk between the proteasome and mTORC1 signaling at the levels of protein degradation and cellular metabolism. It has recently been shown that the inhibition of mTORC1 increases proteasome abundance to enhance the activity of degradation.36, 37, 38 Another study reported that the uncontrolled activation of mTORC1 in TSC2-depleted cells increased the level of active proteasome in an NRF1-dependent manner.18 In the present study, we revealed that the inhibition of proteasomal activity suppresses radiation-induced elevated mTORC1 signaling in HC. Nevertheless, we do not yet know the exact cause-effect relationship between the proteasome and mTORC1 in response to radiation. One possibility is that radiation-induced enhancement of proteasome degradation activates mTORC1. On the basis of this, we speculated that targeting the proteasome had advantages over directly targeting mTORC1 to interrupt radiation-elevated mTORC1 signaling. Indeed, in our study, we found that unlike PIs, rapamycin as an mTORC1-specific inhibitor failed to persistently sustain the inhibitory effect on mTORC1 signaling after radiation in HC cells. As in our results, rapamycin had no apparent effects on the formation of the CKI-TrCP-DEPTOR complex, which is key to the degradation of DEPTOR via SCFβ-TrCP E3 ligase. Besides, DEPTOR might as an indispensable factor for rapamycin to exert its effects. This may mean that rapamycin does not affect mTOR signaling by regulating the degradation of DEPTOR.
Here, we have described a mechanism in which DEPTOR functions as a molecular linker between the UPS and mTORC1 pathways. Our results provide a basis for the development of new strategies to improve radiotherapeutic intervention in HC.
Materials and methods
Cell culture and reagents
FaDu and Detroit 562 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). For the conventional 2D adherent culture, the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, C11995500BT, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum at 37°C in a 5% CO2 atmosphere. All of the cell lines were authenticated by short tandem repeat analysis at the China Center for Type Culture Collection (Wuhan, China). The absence of mycoplasma contamination was verified using a PCR detection kit (Shanghai Biothrive Science & Technology, Shanghai, China). Cells were frozen in liquid nitrogen and used for experiments at passages 3 to 10 after thawing.
BTZ (5043140001, Sigma-Aldrich, St. Louis, MO, USA), PT33 (designed and synthesized in Prof. Xianzhang Bu’s laboratory at the School of Pharmaceutical Sciences, SYSU), rapamycin (no. T1527, Topscience, Shanghai, China), bafilomycin A1 (Baf-A1, no. T6740, Topscience), and LB-100 (no. T2068, Topscience) were dissolved according to the manufacturer’s instructions.
X-ray radiation treatment
At room temperature, X-ray exposure at a dose rate of 1.18 Gy/min (160 kV, 25 mA, 0.3 mm Cu filter) was applied to cells in the logarithmic growth phase using an RS 2000 X-ray biological irradiator (Rad Source Technologies). Cells were irradiated with doses as indicated (see Results section).
Lentiviral transduction and transient transfection
The coding sequences of full-length human PSMD14, β-TrCP, DEPTOR, and K48-specific ubiquitin were inserted into pCMV-C-EGFP, pCDNA4.0 with a Myc tag, pCDNA3.0 with a FLAG tag, and pCDNA3.0 with an HA tag, respectively. NRF1, PSMB5, PSMD14, mCherry, and DEPTOR cDNAs were inserted into the pCDH-EF1-MCS-T2A-Puro vector (tagged with 3× FLAG at the N terminus). Lentiviruses were produced by co-transfection of plasmids pCDH-EF1-MCS-T2A-Puro carrying the corresponding gene, psPAX2, and Pmd2.G into HEK293T cells. The cells stably expressing related proteins were obtained after transduction with lentivirus, followed by puromycin selection. Transient transfection was performed using Lipofectamine 3000 transfection reagent (no. L3000008, Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction.39
Generation and validation of CRISPR-KO cell lines
The CRISPR-KO cell lines were constructed by cloning single-guide RNAs (sgRNAs) targeting DEPTOR (sg1: 5′-CACCGTCGCAAAAGAACTGATTGAC-3′; sg2: 5′-CACCGGCCGCACGGCCCTAAAACCA-3′), TSC2 (sg1: 5′-CACCGCGTCTGCGACTACATGTACG-3′; sg2: 5′-CACCGACCTCGACGAGTACATCGCA-3′) into the lentiCRISPRv2 backbone (Addgene [Watertown, MA, USA] plasmid no. 52961).40 Then, cells were co-transfected with a mix of lentiCRISPRv2 carrying a gRNA and psPAX2 and Pmd2.G plasmids. After that, cells were cultured in selection medium containing 1–2 μg/mL puromycin for 3 days. Single cells sorted by fluorescence-activated cell sorting (FACS) were seeded onto 96-well plates at 1 cell per well, and then clones expanded from single cells. After collecting cells, genomic DNA was extracted with a DNA Extraction Kit (Tiangen, Beijing, China). Sanger sequencing was performed across the sgRNA target sites following PCR amplification, and successful gene KO was identified following sequence analysis. All of the edited cell lines were validated for KO efficiency by WB.
3D spheroid generation and culture
Cell lines were grown in their standard culture conditions, harvested, and dissociated into single-cell suspensions for the spheroid generation. Cells were seeded at 1,000–3,000 cells per well in their optimal conditions in low-attachment 6-well plates (CLS3471, Corning, Corning, NY, USA) in sphere culture medium (DMEM, 100 μg/mL EGF [AF-100-15, PeproTech, Cranbury, NJ, USA], 10 μg/mL VEGF [AF-100-20A, PeproTech], and 10 μg/mL B-27, made fresh every time). Plates were incubated at 37°C in 5% CO2 and spheroids were maintained by performing 500 μL medium replenishments every 3–4 days.41 Spheroids were allowed to grow for 7–14 days, and images were captured using an inverted fluorescence microscope. The spheroid number and diameter were assessed by ImageJ (NIH). Synergism was determined by the Q value with Zheng-Jun Jin’s method, where Q < 0.85, 0.85 ≤ Q ≤ 1.15, and Q > 1.15 indicate antagonistic, additive, and synergistic effects, respectively.42
26S proteasome hydrolysis activity assay
Two-dimensional adherent or 3D spheroid cells were irradiated with XR (Figure 1) or left untreated. Cell lysates were collected, 5 μL of nuclear lysate (NL) or whole-cell lysate (WCL) was incubated in assay buffer (50 mM Tris, 150 mM NaCl, 10% [v/v] glycerol, and 0.03% [w/v] SDS) in 96-well plates, and then Suc-LLVY-AMC (fluorogenic substrate) was added to 1 mM (final reaction volume, 100 μL). The plates were subjected to fluorescence density measurements on a BioTek Epoch Multi-Mode Microplate Reader at an excitation wavelength of 380 nm and an emission wavelength of 460 nm at indicated times. The proteasome hydrolysis activities were calculated as the fluorescence increment (fold change) per minute, indicating the hydrolysis rate of substrate Suc-LLVY-AMC.
Genome-wide transcriptomic analysis
FaDu cells were treated with a single fraction of 6-Gy and cultured as usual for 7 days. Total RNA was isolated from cells using TRIzol (Invitrogen). RNA sequencing was performed by Novogene Bioinformatics Institute (Beijing, China) on an Illumina HiSeq 4000 platform.
Immunofluorescence imaging using confocal microscopy
To evaluate colocalization of exogenous PSMD14-EGFP and endogenous PSMD5, we seeded FaDu cells expressing PSMD14-EGFP in confocal dishes before treatment with or without XR (single fractions of 6-Gy). Cells were fixed with 4% paraformaldehyde for 20 min at room temperature and then blocked and permeabilized with blocking buffer (1× PBS, 3% bovine serum albumin [BSA], and 0.5% Triton X-100) for 1 h. Next, the cells were incubated with primary antibody diluted 1:200–250 with antibody dilution solution (1× PBS, 3% BSA, and 0.5% Triton X-100) overnight at 4°C, washed, and incubated with fluorogenic secondary antibody at 1:500 dilution for 1 h at room temperature. The nuclei were visualized by staining with DAPI. Images were taken using an LSM880 with Airyscan FAST Confocal microscopy (Zeiss, Oberkochen, Germany).43 The quantitative analysis can be carried out by counting the number of foci per cell/cell nucleus.
WB and IP assay
Cells were lysed with IP buffer (9803, Cell Signaling Technology, Danvers, MA, USA) supplemented with protease and phosphatase inhibitor cocktail (78442, ThermoFisher Scientific). Normalized protein lysates were separated by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (03001, Sigma-Aldrich), which were subjected to immunoblotting with primary antibodies. Subsequently, the membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG. Protein bands were visualized using ECL (WP20005, TFS), and signals were captured by a ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA). Quantitative analysis of grayscale images was performed using ImageJ.
For co-IP, cell lysates were incubated with M2 anti-FLAG agarose overnight at 4°C or primary antibodies overnight at 4°C plus protein G agarose beads (sc-2003, Santa Cruz Biotechnology, Dallas, TX, USA) for 2 h at 4°C. After washing, the pulldown products were examined by WB, as mentioned above.
Colony formation assay
Cells were seeded in 6-well plates. After 24 h, the cells were exposed to a single dose of 0-, 2-, 4-, or 6-Gy of XR and treated with drugs (5 nM BTZ or 10 nM PT33). After 10–14 days, the cells were stained with 0.5% crystal violet for 30 min at room temperature. Stained colonies of at least 50 cells were visually selected and manually counted. Plating efficiency (PE) and the survival fraction (SF) were calculated using the following equations: PE = number of colonies formed/number of cells seeded × 100%; SF = number of colonies formed after irradiation/(number of cells seeded × PE). Moreover, the impact of co-treatment was assessed by calculating SER. SER is defined as the quotient of the radiation dose in the DMSO group and of that in the BTZ/PT33 group, where each radiation dose results in the same clonogenic survival rates x: SER = radiation dose (survival x in DMSO group)/radiation dose (survival x in BTZ/PT33 group). The radiation doses were estimated by linear quadratic regression derived from clonogenic survival results and SERs calculated for isoeffective surviving fractions x of 50%, 10%, and 1% for BTZ and PT33.44
CCK-8 assays and CI analyses
Cells (at a density of 2500 cells per well) were seeded into 96-well plates and allowed to adhere for 24 h before supplementation with varying concentrations of BTZ or PT33 and XR. After 72 h, the medium was removed, and the cells were treated with 10% CCK-8 (HY-K0301, MCE, Monmouth Junction, NJ, USA). Absorbance at 450 nm was measured.
The CI analyses of XR and PIs in FaDu cells were based on the results of the CCK-8 assay using the Chou-Talalay method.45 CI for x% average inhibition was calculated as follows: CI = C/Cx + D/Dx + C × D/(Cx × Dx), where C and D are XR doses and PIs for an average inhibition of x% by combined treatment, and Cx or Dx is the dose of X-rays or PIs for an average inhibition of x% by treatment with PIs or XR alone. CI > 1, CI = 1, or CI < 1 indicated antagonistic, additive, or synergistic effects, respectively.46,47
Annexin V/PI double-staining assay
To measure apoptosis, we treated FaDu cells with PIs plus XR. Then cells were stained with fluorescein isothiocyanate (FITC)-conjugated annexin V and PI using a commercially available kit (BestBio, Shanghai, China). The non-adherent cells were collected, and the adherent cells were washed with PBS and detached using trypsin. Subsequently, cells were stained with annexin V-FITC/PI according to the manufacturer’s instructions and quantified by a CytoFLEX flow cytometer.
Quantitative reverse transcriptase-PCR (qRT-PCR)
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) and reverse transcribed into cDNA using an M-MLV Reverse Transcriptase Kit (Promega, Madison, WI, USA). Quantitative real-time-PCR was performed using iTaq SYBR Green Mix (Bio-Rad) and the primers listed in Table S2.39,48 Each sample was examined at least in triplicate. PCR product specificity was confirmed by melting-curve analysis. The relative mRNA expression was calculated using the 2−ΔΔCt method. Glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH) was used as an internal control.
In vivo studies
Animal experiments were approved by the Institutional Ethics Committee for Clinical Research and Animal Trials of SYSUFAH, following the Animal Welfare and Rights in China. Female BALB/c nude mice (4–5 weeks, 15–18 g; SLRC Laboratory Animal Company, Shanghai, China) were used. The in vivo cell line-derived xenografts were generated by subcutaneous transplantation of diced tumor (approximately 5 mm3) from FaDu cell xenografts into the right flank. Once their tumors reached approximately 150 mm3 in volume, mice were treated with PIs, XR, or both. The mice were irradiated in lead boxes. A diamond cutout on the top of the box permitted the tumors to be directly exposed to XR. The tumor volumes were monitored weekly. They were calculated using the following formula: tumor volume = 0.52 × width2 × length. After all treatments for 2 weeks, the mice were euthanized, and then xenografts were removed and weighed.
Human samples
This research was approved by the Institutional Ethics Committee for Clinical Research and Animal Trials of SYSUFAH and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each patient. In total, 63 formalin-fixed and paraffin-embedded tumor biopsy specimens before treatment were collected from patients pathologically diagnosed with HC at this hospital between 2016 and 2020. All of them received radical radiotherapy (volumetric modulated arc therapy techniques, 68-Gy, 2-Gy/day, 5 days/week). Among them, 59 patients received induced chemotherapy, and 57 patients received concurrent radio- and chemotherapy. After CRT, the tumor response was assessed according to RECIST version 1.1 criteria, performed by two board-certified radiologists.
Histology and immunohistochemistry (IHC) assays
Tissue specimens were fixed in buffered 10% formalin for 24 h followed by paraffin embedding. Next, 4-μm-thick sections were prepared for histology and IHC assays. Tissue sections were deparaffinized and then rehydrated before antigen retrieval and endogenous peroxidase inactivation. The specimens were incubated overnight at 4°C with primary antibodies diluted 1:100–200 after blocking specimens in blocking buffer (1× PBS, 10% normal goat serum, and 0.3% Triton X-100) for 1 h. The next day, sections were incubated for 1 h at room temperature with an HRP-conjugated secondary antibody, visualized with an Envision Detection Kit (K5007, Dako, Santa Clara, CA, USA), and counterstained with hematoxylin.
The IRS was used for analysis of the expression of IHC markers. In brief, the percentage of protein-positive cells was divided into four categories (<10%, 11%–50%, 51%–80%, and >80%, with corresponding scores of 1, 2, 3, and 4, respectively), and the staining intensity was given a value between 0 (no detectable immunostaining) and 3 (strong immunostaining). The IRS (0–12) was then calculated by multiplying the score values.49 If an examined sample stained heterogeneously for IHC markers, then each staining intensity was scored independently, and the results were summed.50
Antibodies
Primary to antibodies to phospho-S6 ribosomal protein (Ser235/236; no. 2211), phospho-p70 S6 kinase (Thr389; D5U1O; no. 97596), phospho-mTOR (Ser2448, D9C2, no. 5536), phospho-4E-BP1(Ser65, no. 9451), 4E-BP1 (53H11, no. 9644), DEPTOR/DEPDC6 (D9F5, no. 11816), GβL (86B8, no. 3274), phospho-Jak1 (Tyr1034/1035, E9Y7V, no. 66245), phospho-Jak2 (Tyr1007, D15E2, no. 4406), Jak1 (6G4, no. 3344), Jak2 (D2E12, no. 3230), Stat3 (124H6, no. 9139), MEK1/2 (D1A5, no. 8727), phospho-MEK1/2 (Ser217/221, 41G9, no. 9154), cleaved caspase-3 (Asp175, 5A1E, no. 9664), LC3A/B (D3U4C, no. 12741), FLAG-tag (9A3, no. 8146), Myc-tag (9B11, no. 2276), phospho-EGF receptor (Tyr1068, no.2234), EGFR (D38B1, no. 4267), phospho-p38 MAPK (Thr180/Tyr182, D3F9, no. 4511), p38 MAPK (D13E1, no. 8690), phospho-p44/42 MAPK (Erk1/2, Thr202/Tyr204, D13.14.4E, no. 4370), p44/42 MAPK (Erk1/2, 137F5, no. 4695), phospho-ULK1 (Ser757, D7O6U, no. 14202), ULK1 (D8H5, no. 8054), phospho-Akt1 (Ser473, D7F10, Akt1 specific, no. 9018), Akt1 (D9R8K, no. 75692), phospho-Akt2 (Ser474, D3H2, Akt2 specific, no. 8599), Akt2 (L79B2, no. 5239), IκBα (L35A5, no. 4814), phospho-NF-κB p65 (Ser536, 93H1, no. 3033), NF-κB p65 (D14E12, no .8242), phospho-Stat1 (Tyr701, 58D6, no. 9167), Stat1 (D1K9Y, no. 14994), phospho-Stat3 (Tyr705, D3A7, no. 9145), phospho-SAPK/JNK (Thr183/Tyr185, G9, no. 9255), SAPK/JNK (no. 9252), β-catenin (D10A8, no. 8480), phospho-YAP (Ser127, D9W2I, no. 13008), YAP (D8H1X, no. 14074), Rheb (E1G1R, no. 13879), RagA (D8B5, no. 4357), RagB (D18F3, no. 8150), RagC (D8H5, no. 9480), RagD (no. 4470), tuberin/TSC2 (D93F12, no. 4308), phospho-tuberin/TSC2 (Ser1387, D2R3A, no. 23402), phospho-tuberin/TSC2 (Thr1462, 5B12, no. 3617), CKI (no. 2655), GAPDH (no. 2118), actin (no. 3700), normal mouse IgG (no. 5415), HRP-linked secondary antibody to rabbit IgG (no. 7074), mouse IgG (no. 7076; WB 1:1,000) from Cell Signaling Technology; antibody to PSMB5/MB1 (ab3330), ubiquitin linkage-specific K48 (no. ab14601), and HA tag (no. ab18181) from Abcam (Cambridge, UK).
The fluorogenic secondary antibody comprised goat anti-rabbit Alexa Fluor 488, anti-rabbit Alexa Fluor 594, goat anti-mouse Alexa Fluor 488, anti-mouse Alexa Fluor 594 (nos. A11034, A11037, A32723, and A11032, respectively) from Invitrogen. M2 anti-FLAG agarose (no. A2220) came from Sigma-Aldrich.
Statistical analysis
Statistical analysis was conducted to support the main conclusions in this study. Unless otherwise specified, all of the experiments were performed at least three times. All of the values were reported as means ± SDs. The data were analyzed using GraphPad Prism 7.0 (GraphPad, San Diego, CA, USA) or SPSS 20.0 (IBM SPSS Statistics, Armonk, NY, USA) software. Comparisons between two or among more groups were evaluated with a two-tailed Student’s t test (2-sample t test) or analysis of variance (ANOVA), respectively. Relationships between clinical categorical variables were analyzed by the chi-square test. The survival times of particular groups of patients were analyzed with the log rank test, Kaplan-Meier curves, and the Cox proportional hazards model. p < 0.05 was considered significant.
Data availability
The data that support the findings of this study are available from the corresponding authors.
Acknowledgments
This work was financially supported by the China Postdoctoral Science Foundation, China (nos. 2020TQ0379 and 2020M673006 to X.W.); the National Natural Science Foundation of China (NSFC), China (no. 82003218 to X.W., no. 81903037 to S.H., and no. 82072029 to Z.P.), the Guangdong Basic and Applied Basic Research Foundation, China (no. 2021A1515011182 to Y.C. and no. 2021A1515012496 to X.Y.). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Author contributions
Study concept and design, Y.C., Z.P., and R.-Y.L.; clinical data and sample collection and preparation, X.W., L.L., C.W., Y.W., M.Z., and X.Z.; IHC score assessment, D.J. and T.L.; in vitro functional studies and data analysis, X.W., Z.C., X.Y., T.L., G.W., W.W., L.L., and Z.W.; animal experiments and data analysis, X.W., Z.C., X.Y., and T.L.; critical cellular tools generation, T.L., W.W., and Z.W.; mechanistic studies and data analysis, X.Y., X.W., T.L., and Z.C.; statistical analysis, X.Y., T.L., X.W., L.L., and M.J.; data supervision, X.B. and Y.C.; data analysis and interpretation and writing of the manuscript, X.Y., Z.P., and X.W.; funding acquisition, X.W., X.Y., Z.P., Y.C., and S.H. All of the authors read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.08.002.
Contributor Information
Ran-yi Liu, Email: liury@sysucc.org.cn.
Zhenwei Peng, Email: pzhenw@mail.sysu.edu.cn.
Yong Chen, Email: chenyong@mail.sysu.edu.cn.
Supplemental information
References
- 1.Kwon D.I., Miles B.A. Hypopharyngeal carcinoma: do you know your guidelines? Head Neck. 2019;41:569–576. doi: 10.1002/hed.24752. [DOI] [PubMed] [Google Scholar]
- 2.Newman J.R., Connolly T.M., Illing E.A., Kilgore M.L., Locher J.L., Carroll W.R. Survival trends in Hypopharyngeal cancer: a population-based review. Laryngoscope. 2015;125:624–629. doi: 10.1002/lary.24915. [DOI] [PubMed] [Google Scholar]
- 3.Takes R.P., Strojan P., Silver C.E., Bradley P.J., Haigentz M., Wolf G.T., Shaha A.R., Hartl D.M., Olofsson J., Langendijk J.A., et al. Current trends in initial management of hypopharyngeal cancer: the declining use of open surgery. Head Neck. 2012;34:270–281. doi: 10.1002/hed.21613. [DOI] [PubMed] [Google Scholar]
- 4.Chung E.-J., Jeong W.-J., Jung Y.H., Kwon S.K., Kwon T.-K., Ahn S.-H., Sung M.-W., Keam B., Heo D.-S., Kim J.H., et al. Long-term oncological and functional outcomes of induction chemotherapy followed by (chemo)radiotherapy vs definitive chemoradiotherapy vs surgery-based therapy in locally advanced stage III/IV hypopharyngeal cancer: multicenter review of 266 cases. Oral Oncol. 2019;89:84–94. doi: 10.1016/j.oraloncology.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 5.Cui J., Wang L., Piao J., Huang H., Chen W., Chen Z., Yang H., Tan X., Li J., Liu G. Initial surgical versus non-surgical treatments for advanced hypopharyngeal cancer: a meta-analysis with trial sequential analysis. Int. J. Surg. 2020;82:249–259. doi: 10.1016/j.ijsu.2020.04.059. [DOI] [PubMed] [Google Scholar]
- 6.Kılıç S., Kılıç S.S., Hsueh W.D., Eloy J.A., Baredes S., Woo Park R.C., Mahmoud O.J.H. Radiotherapy modality as a predictor of survival in hypopharyngeal cancer. Head Neck. 2018;40:2441–2448. doi: 10.1002/hed.25360. [DOI] [PubMed] [Google Scholar]
- 7.Kim Y.-J., Lee R. Surgery vs. radiotherapy for locally advanced hypopharyngeal cancer in the contemporary era: a population-based study. Cancer Med. 2018;7:5889–5900. doi: 10.1002/cam4.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sewnaik A., Hoorweg J.J., Knegt P.P., Wieringa M.H., van der Beek J.M.H., Kerrebijn J.D.F. Treatment of hypopharyngeal carcinoma: analysis of nationwide study in the Netherlands over a 10-year period. Clin. Otolaryngol. 2005;30:52–57. doi: 10.1111/j.1365-2273.2004.00913.x. [DOI] [PubMed] [Google Scholar]
- 9.Liu C., Liao K., Gross N., Wang Z., Li G., Zuo W., Zhong S., Zhang Z., Zhang H., Yang J., Hu G. Homologous recombination enhances radioresistance in hypopharyngeal cancer cell line by targeting DNA damage response. Oral Oncol. 2020;100:104469. doi: 10.1016/j.oraloncology.2019.104469. [DOI] [PubMed] [Google Scholar]
- 10.Smith L., Qutob O., Watson M.B., Beavis A.W., Potts D., Welham K.J., Garimella V., Lind M.J., Drew P.J., Cawkwell L. Proteomic identification of putative biomarkers of radiotherapy resistance: a possible role for the 26S proteasome? Neoplasia. 2009;11:1194–1207. doi: 10.1593/neo.09902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nandi D., Tahiliani P., Kumar A., Chandu D. The ubiquitin-proteasome system. J. Biosci. 2006;31:137–155. doi: 10.1007/bf02705243. [DOI] [PubMed] [Google Scholar]
- 12.Bard J.A.M., Goodall E.A., Greene E.R., Jonsson E., Dong K.C., Martin A. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018;87:697–724. doi: 10.1146/annurev-biochem-062917-011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoncu R., Efeyan A., Sabatini D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei F., Liu Y., Guo Y., Xiang A., Wang G., Xue X., Lu Z. miR-99b-targeted mTOR induction contributes to irradiation resistance in pancreatic cancer. Mol. Cancer. 2013;12:81. doi: 10.1186/1476-4598-12-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eshleman J.S., Carlson B.L., Mladek A.C., Kastner B.D., Shide K.L., Sarkaria J.N. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002;62:7291–7297. [PubMed] [Google Scholar]
- 16.Contessa J.N., Hampton J., Lammering G., Mikkelsen R.B., Dent P., Valerie K., Schmidt-Ullrich R.K. Ionizing radiation activates Erb-B receptor dependent Akt and p70 S6 kinase signaling in carcinoma cells. Oncogene. 2002;21:4032–4041. doi: 10.1038/sj.onc.1205500. [DOI] [PubMed] [Google Scholar]
- 17.Murphy J.D., Spalding A.C., Somnay Y.R., Markwart S., Ray M.E., Hamstra D.A. Inhibition of mTOR radiosensitizes soft tissue sarcoma and tumor vasculature. Clin. Cancer Res. 2009;15:589–596. doi: 10.1158/1078-0432.ccr-08-1019. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., Nicholatos J., Dreier J.R., Ricoult S.J.H., Widenmaier S.B., Hotamisligil G.S., Kwiatkowski D.J., Manning B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513:440–443. doi: 10.1038/nature13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutter G., Zimmermann Y., Rieken M., Hartmann E., Rosenwald A., Hiddemann W., Dreyling M. Proteasome inhibition leads to dephosphorylation and downregulation of protein expression of members of the Akt/mTOR pathway in MCL. Leukemia. 2012;26:2442–2444. doi: 10.1038/leu.2012.118. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y., Xiong X., Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(βTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell. 2011;44:304–316. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao D., Inuzuka H., Tan M.K.M., Fukushima H., Locasale J.W., Liu P., Wan L., Zhai B., Chin Y.R., Shaik S., et al. mTOR drives its own activation via SCF(βTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol. Cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan S., Skaar J.R., Kuchay S., Toschi A., Kanarek N., Ben-Neriah Y., Pagano M. mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Mol. Cell. 2011;44:317–324. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dayan F., Bilton R.L., Laferrière J., Trottier E., Roux D., Pouyssegur J., Mazure N.M. Activation of HIF-1alpha in exponentially growing cells via hypoxic stimulation is independent of the Akt/mTOR pathway. J. Cell. Physiol. 2009;218:167–174. doi: 10.1002/jcp.21584. [DOI] [PubMed] [Google Scholar]
- 24.Eisenhauer E.A., Therasse P., Bogaerts J., Schwartz L.H., Sargent D., Ford R., Dancey J., Arbuck S., Gwyther S., Mooney M., et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 25.McBride W.H., Iwamoto K.S., Syljuasen R., Pervan M., Pajonk F. The role of the ubiquitin/proteasome system in cellular responses to radiation. Oncogene. 2003;22:5755–5773. doi: 10.1038/sj.onc.1206676. [DOI] [PubMed] [Google Scholar]
- 26.Pajonk F., McBride W.H. Ionizing radiation affects 26s proteasome function and associated molecular responses, even at low doses. Radiother. Oncol. 2001;59:203–212. doi: 10.1016/S0167-8140(01)00311-5. [DOI] [PubMed] [Google Scholar]
- 27.Kim K., Brush J.M., Watson P.A., Cacalano N.A., Iwamoto K.S., McBride W.H. Epidermal growth factor receptor vIII expression in U87 glioblastoma cells alters their proteasome composition, function, and response to irradiation. Mol. Cancer Res. 2008;6:426–434. doi: 10.1158/1541-7786.mcr-07-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caron A., Briscoe D.M., Richard D., Laplante M. DEPTOR at the nexus of cancer, metabolism, and immunity. Physiol. Rev. 2018;98:1765–1803. doi: 10.1152/physrev.00064.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson T.R., Laplante M., Thoreen C.C., Sancak Y., Kang S.A., Kuehl W.M., Gray N.S., Sabatini D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rousseau A., Bertolotti A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018;19:697–712. doi: 10.1038/s41580-018-0040-z. [DOI] [PubMed] [Google Scholar]
- 31.Mofers A., Pellegrini P., Linder S., D’Arcy P. Proteasome-associated deubiquitinases and cancer. Cancer Metastasis Rev. 2017;36:635–653. doi: 10.1007/s10555-017-9697-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillessen S., Groettup M., Cerny T. The proteasome, a new target for cancer therapy. Onkologie. 2002;25:534–539. doi: 10.1159/000068624. [DOI] [PubMed] [Google Scholar]
- 33.Delic J., Masdehors P., Ömura S., Cosset J.M., Dumont J., Binet J.L., Magdelénat H. The proteasome inhibitor lactacystin induces apoptosis and sensitizes chemo-and radioresistant human chronic lymphocytic leukaemia lymphocytes to TNF-α-initiated apoptosis. Br. J. Cancer. 1998;77:1103–1107. doi: 10.1038/bjc.1998.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acosta-Alvear D., Cho M.Y., Wild T., Buchholz T.J., Lerner A.G., Simakova O., Hahn J., Korde N., Landgren O., Maric I., et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. eLife. 2015;4:e08153. doi: 10.7554/eLife.08153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue X., Zuo Y., Ke H., Luo J., Lou L., Qin W., Wang Y., Liu Z., Chen D., Sun H., et al. Identification of 4-arylidene curcumin analogues as novel proteasome inhibitors for potential anticancer agents targeting 19S regulatory particle associated deubiquitinase. Biochem. Pharmacol. 2017;137:29–50. doi: 10.1016/j.bcp.2017.04.032. [DOI] [PubMed] [Google Scholar]
- 36.Rousseau A., Bertolotti A. An evolutionarily conserved pathway controls proteasome homeostasis. Nature. 2016;536:184–189. doi: 10.1038/nature18943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo T., Fu J., Xu A., Su B., Ren Y., Li N., Zhu J., Zhao X., Dai R., Cao J., et al. PSMD10/gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy. 2016;12:1355–1371. doi: 10.1080/15548627.2015.1034405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao J., Zhai B., Gygi S.P., Goldberg A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA. 2015;112:15790–15797. doi: 10.1073/pnas.1521919112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X.C., Yue X., Zhang R.X., Liu T.Y., Pan Z.Z., Yang M.J., Lu Z.H., Wang Z.Y., Peng J.H., Le L.Y., et al. Genome-wide RNAi screening identifies RFC4 as a factor that mediates radioresistance in colorectal cancer by facilitating nonhomologous end joining repair. Clin. Cancer Res. 2019;25:4567–4579. doi: 10.1158/1078-0432.Ccr-18-3735. [DOI] [PubMed] [Google Scholar]
- 40.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roper S.J., Linke F., Scotting P.J., Coyle B. 3D spheroid models of paediatric SHH medulloblastoma mimic tumour biology, drug response and metastatic dissemination. Sci. Rep. 2021;11:4259. doi: 10.1038/s41598-021-83809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin Z.J. About the evaluation of drug combination. Acta Pharmacol. Sin. 2004;25:146–147. [PubMed] [Google Scholar]
- 43.Kim Y.-M., Krantz S., Jambusaria A., Toth P.T., Moon H.-G., Gunarathna I., Park G.Y., Rehman J. Mitofusin-2 stabilizes adherens junctions and suppresses endothelial inflammation via modulation of β-catenin signaling. Nat. Commun. 2021;12:2736. doi: 10.1038/s41467-021-23047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naumann P., Liermann J., Fortunato F., Schmid T.E., Weber K.J., Debus J., Combs S.E. Sulforaphane enhances irradiation effects in terms of perturbed cell cycle progression and increased DNA damage in pancreatic cancer cells. PLoS One. 2017;12:e0180940. doi: 10.1371/journal.pone.0180940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou T.-C., Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 46.Zhao L., Au J.L.S., Wientjes M.G. Comparison of methods for evaluating drug-drug interaction. Front. Biosci. 2010;2:241–249. doi: 10.2741/e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang L., Jiang Y., Chen Y. Predicting drug combination index and simulating the network-regulation dynamics by mathematical modeling of drug-targeted EGFR-ERK signaling pathway. Sci. Rep. 2017;7:40752. doi: 10.1038/srep40752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui D., Dai X., Gong L., Chen X., Wang L., Xiong X., Zhao Y. DEPTOR is a direct p53 target that suppresses cell growth and chemosensitivity. Cell Death Dis. 2020;11:976. doi: 10.1038/s41419-020-03185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D'Arcy V., Abdullaev Z.K., Pore N., Docquier F., Torrano V., Chernukhin I., Smart M., Farrar D., Metodiev M., Fernandez N., et al. The potential of BORIS detected in the leukocytes of breast cancer patients as an early marker of tumorigenesis. Clin. Cancer Res. 2006;12:5978–5986. doi: 10.1158/1078-0432.Ccr-05-2731. [DOI] [PubMed] [Google Scholar]
- 50.Krajewska M., Krajewski S., Epstein J.I., Shabaik A., Sauvageot J., Song K., Kitada S., Reed J.C. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am. J. Pathol. 1996;148:1567–1576. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors.