

Abstract
Chemoprevention of hepatocellular carcinoma (HCC) is highly desirable in clinic. Berberine (BBR) is reported to play potential roles in cancer treatment and prevention. We studied the chemopreventive effect of BBR on hepatocellular carcinogenesis in an inflammation-driven mouse model, as it was enriched in liver after oral administration. Oral BBR significantly decreased the number and volume of visible nodular tumors, and prolonged the median overall survival by 9 and 8 weeks in the diethylnitrosamine (DEN)-injected male and female mice respectively. The nodular tumors were induced through activation of the lysophosphatidic acid (LPA) pathway in liver. LPA stimulated the abnormal leptin transcription through interacting with LPA receptor-2 (LPAR2) followed by p38 activation, and BBR inhibited carcinogenesis by suppressing the bioactivity of LPA. Specifically, BBR significantly reduced the expression of the LPA synthetase autotaxin (ATX) and LPAR2 in the nodular tumors of DEN-injected mice. Subsequently, BBR repressed the abnormal transcription of leptin stimulated by LPA-induced phosphorylation of p38 in hepatoma cells. In fact, BBR reduced the abnormal expression of leptin in livers of DEN-injected male mice throughout the course of an 8-month experiment. BBR might be a preventive agent for HCC, working at least partially through antagonizing the ATX-LPA-LPAR2-p38-leptin axis in liver.
Keywords: hepatocellular carcinoma, berberine, LPA receptor-2, autotaxin, leptin
Graphical abstract
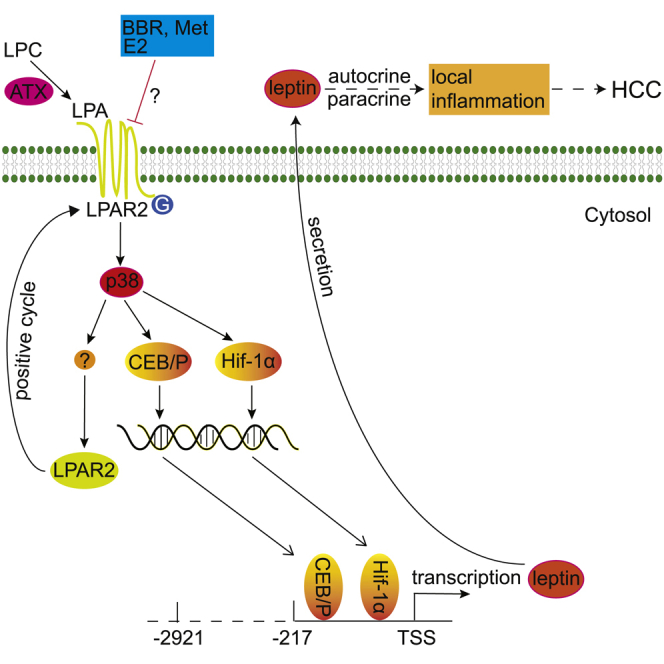
Berberine (BBR, supplemented in diet) significantly repressed liver tumorigenesis in the diethylnitrosamine (DEN)-induced mouse hepatoma model through antagonizing the ATX-LPA-LPAR2-p38-leptin axis. As BBR has been an OTC medicine in clinic for decades, it might be straightforwardly translated into clinical chemoprevention against hepatocellular carcinoma (HCC) in high-risk populations.
Introduction
Hepatocellular carcinoma (HCC) is one of the most commonly seen cancers worldwide. The causative risk factors of HCC could be hepatitis (caused by hepatitis B or hepatitis C virus), alcohol, and food contamination of aflatoxin B.1 The major process for hepatocellular carcinogenesis involves non-resolving inflammation (NRI) in liver with elevation of the pro-inflammatory cytokines, including interleukin (IL)-6, tumor necrosis factor (TNF), and leptin.2, 3, 4, 5 Recent investigations have identified the ATX-LPA-LPARs axis to be a central player in the carcinogenesis for almost all hepatomas with various risk factors, in which ATX stands for autotaxin (also known as ectonucleotide pyrophosphatase/phosphodiesterase family member 2, ENPP2), LPA for lysophosphatidic acid, and LPARs for lysophosphatidic acid receptors.6 LPA belongs to lysophospholipids (LPLs), which are a class of endogenous bioactive lipids specifically using the glycerol as its structural backbone.7 As the smallest LPL, LPA acts as an extracellular signaling molecule involved in inflammation and cancer.8,9 LPA can be biosynthesized in two routes, and the main pathway is regulated by ATX through its lysophospholipase D activity, which removes choline from lysophosphatidylcholine (LPC) in the extracellular space.9 LPA can elicit a variety of biological functions via signaling through G protein-coupled LPARs, and, to date, six LPAR members (LPAR1–LPAR6) have been discovered.9
In the ATX-LPA-LPARs axis, ATX acts as an enzyme for the synthesis of LPA, which then binds LPARs on cell surface and promotes cancer-related inflammation through inducing cytokine production thereafter.9 Researches have shown that high level of ATX and LPA positively associates with cancer development.8,10 As LPA also induces ATX expression, the ATX-LPA-LPARs axis is considered a typical positive feedback loop in carcinogenesis.8,11 All of the LPARs are seven-span trans-membrane proteins, which differ in tissue distribution and downstream effects. For example, binding of LPA with LPAR1 activates hepatic stellate cells to secrete collagen into the extracellular matrix and thus promotes liver cirrhosis.6,12
Berberine (BBR) is a botanic agent (Figure 1D, molecular weight [MW] 371.82) derived from medicinal plants such as Berberis vulgaris and Coptis chinensis. It is very safe in humans and has been used in China for decades as an over-the-counter (OTC) medicine for patients with bacterial-caused diarrhea. We have previously discovered BBR to be a safe drug efficiently lowering blood lipids (both total cholesterol and triglyceride levels) and glucose through novel mechanisms in patients with hyperlipidemia and type 2 diabetes mellitus respectively.13,14 BBR’s therapeutic effect has been widely confirmed in clinic since 2004.15 Furthermore, the cancer-prevention activity of BBR has been reported in the DEN-induced hepatoma of rodents.16, 17, 18 Recently, a clinical study on 891 patients (429 BBR versus 462 placebo) with colorectal adenoma has clearly demonstrated that oral BBR could reduce the risk of recurrence of colorectal adenoma by 11% (36% BBR versus 47% placebo).19 It has provoked further interest in its anticancer mechanism and future application. In this article, we exhibit the significant effect of BBR in reducing the development of DEN-induced hepatoma in mice. The mode of action of BBR appeared to link, at least in part, with its inhibitory effect on the ATX-LPA-LPARs pathway in hepatoma cells, resulting in a drop of the abnormal leptin expression in liver.
Figure 1.

Berberine inhibited DEN-induced hepatocellular carcinogenesis
(A) Nodular tumors were identified by H&E staining of liver sections from male mice 8 months after DEN injection, and are indicated by the black arrows. The images are of representative gross livers or H&E-stained samples from 24 mice per group with identical results. (B) The tumor numbers and volumes were reduced significantly by BBR and Met treatment in male mice 8 months after DEN injection, compared with the mice fed with normal chow diet. Data are expressed as the mean ± SEM (n = 24 for each group). (C) BBR decreased the serum AFP at the end of the indicated months in DEN-injected male mice. Values are mean ± SEM (n = 6 for each group). (D) The tumor numbers and volumes were examined from DEN-injected male mice, after BBR treatment for the first 2 weeks (0.2% BBR, n = 21), first 3 months (0.04% BBR, n = 10; 0.2% BBR, n = 11), last 5 months (0.2% BBR, n = 21), and full course of 8-month experiment (0.2% BBR, n = 24), or not (normal chow, n = 24). Data are expressed as the mean ± SEM. (E and G) BBR (0.2%, w/w) significantly prolonged the median overall survival of DEN-injected male and female mice by 9 and 8 weeks during the 80-week and 100-week study period, respectively. The number of mice per group and median survival (w, weeks) are indicated. Log-rank Mantel-Cox test was used. (F) The tumor numbers and volumes were examined from female mice 12 months after DEN injection. Values are mean ± SEM in each group (n = 22–24). (H) Leptin was found to be reduced in the groups treated with BBR as detected by Luminex system. Values are the mean ± SEM (n = 15–16 for 1 month, n = 14–15 for 4 months, n = 12–14 for 6 months, n = 23–24 for 8 months). ∗p < 0.05, ∗∗p < 0.01 versus that of the DEN model group fed with normal chow.
Results
BBR inhibited carcinogenesis in the DEN-induced hepatoma model
To explore the preventive effect of BBR in liver tumorigenesis, 15-day-old mice were subjected to the widely used protocol of DEN. This hepatoma model was chosen for the study because DEN causes 100% of the male mice to develop hepatoma, and the genetic signature of this tumor model is similar to that of human HCCs with poor survival rate.2
Our experiment was first done in male mice. Macroscopic examination and pathological analysis revealed that BBR reduced HCC development, showing decreased whitish tumor nodules notably on liver lobes of male mice 8 months after DEN injection (Figure 1A). Compared with the DEN-injected mice fed with normal chow diet, BBR significantly decreased the number of visible nodular tumors by 76% and 57%, and reduced the volumes of these tumors by 83% and 88%, after being supplemented in diet at the dose of 0.04% and 0.2% (w/w) respectively (p < 0.01 for both; BBR versus normal chow; Figure 1B).
Consistent with this, the serum alpha-fetoprotein (AFP) level was reduced by 89% and 94% in mice fed with 0.04% and 0.2% BBR in diet respectively at the end of the 8-month experiment (p < 0.01 for both; BBR versus normal chow; Figure 1C). Note that serum AFP was decreased by 87% and 91% at the end of 6 months after DEN injection in the mice treated with 0.04% and 0.2% BBR respectively (p < 0.01 for both; BBR versus normal chow; Figure 1C), although at this time point the inhibitory effect of BBR was not significant on the number and volume of nodular tumors (data not shown). Even at the end of 4 months, when the tumor nodules could be hardly seen, AFP was reduced by 74% and 76% respectively in the 0.04% and 0.2% BBR-treated mice, indicating AFP is a sensitive biomarker for the early evaluation of potential chemopreventive agents in the hepatoma model (Figure 1C).
When we treated the DEN-injected male mice with BBR for only the first 2 weeks or first 3 months after DEN injection, the cancer-prevention effect of BBR almost vanished, suggesting that it might be insufficient for BBR to interrupt the tumor development through control of the early stage of carcinogenesis (Figure 1D). On the other hand, BBR treatment for the last 5 months (from the beginning of the fourth month after DEN injection to the end of 8-month experiment) significantly decreased the tumor number by 44% (p < 0.01 versus normal chow; Figure 1D), and the best preventive efficacy was seen when BBR was given in the full course of the 8-month experiment (p < 0.01 versus normal chow; Figure 1D).
Generally speaking, at the end of the 8-month experiment, the male mice treated with BBR were in a more healthy condition than those of the DEN model group, in view of their body weight, liver weight, liver enzymes, blood lipids, and glucose (Table 1). For survival analysis, the experiment was extended from 8 months (34 weeks) to 18 months (80 weeks) for male mice. BBR at the high dose (0.2% supplement in diet, w/w) prolonged the median overall survival by 9 weeks (p = 0.0038 versus normal chow) and improved the survival rate by 2.7-fold at the 70-week time point, compared with the DEN model group (Figure 1E).
Table 1.
BBR reduced body weight, liver weight, fasting blood glucose, serum ALT, and total cholesterol level in male C57/BL6 mice 8 months after DEN injection
| Examinations | Sham | Normal chow | 0.04% BBR | 0.2% BBR | 0.1% Met |
|---|---|---|---|---|---|
| Body weight (g) | 36.9 ± 6.3 | 36.8 ± 3.9 | 33.8 ± 4.9∗ | 33.3 ± 4.4∗ | 32.4 ± 2.9∗∗ |
| Liver weight (g) | 1.32 ± 0.19 | 1.84 ± 0.81## | 1.3 ± 0.29∗∗ | 1.34 ± 0.2∗∗ | 1.46 ± 0.21∗ |
| ALT (IU/L) | 36.8 ± 3.4 | 125.5 ± 161.8 | 43.3 ± 11.2∗ | 43.2 ± 9.2∗ | 52 ± 19.3 |
| Total cholesterol (mmol/L) | 3.24 ± 0.18 | 4.95 ± 2.39# | 3.37 ± 0.87∗ | 3.17 ± 0.74∗∗ | 3.67 ± 0.93∗ |
| Fasting glucose (mmol/L) | 5.8 ± 0.3 | 7.0 ± 1.8## | 5.5 ± 1.4∗∗ | 5.6 ± 0.6∗∗ | 5.7 ± 0.9∗∗ |
| Fed glucose (mmol/L) | 9.6 ± 0.7 | 9.4 ± 1.1 | 8.4 ± 1.2∗ | 8.7 ± 1.0 | 9.2 ± 1.1 |
Values are mean ± SDs. ∗p < 0.05, ∗∗p < 0.01, compared with that of DEN model group fed with normal chow. #p < 0.05, ##p < 0.01, compared with the sham-treated group. The sham-treated group: n = 22 for the body/liver weight, n = 4 for the other indexes. BBR- (0.04% and 0.2%, w/w) and Met (0.1%, w/w)-treated groups: n = 24 for the body/liver weight and fasting/fed glucose, n = 14 for the other indexes.
The cancer-preventive effect of BBR was also observed in female mice, in which the tumor nodules could be visible 12 months after DEN injection. Although the tumor incidence was also 100% at that time in this model, the average tumor number in the female mice was only 27% that of the male mice 8 months after DEN injection (Figures 1F and 1B). Estradiol (E2) might be the factor to protect female mice from HCC in the DEN model.2 In the female mice, we show that BBR-containing diet could significantly reduce the tumor volumes (p < 0.05 for both; BBR versus normal chow), but not the tumor number, with respect to the DEN model group fed with normal chow (Figure 1F). BBR at the high dose (0.2% supplement in diet, w/w) prolonged the median overall survival by 8 weeks (p = 0.014 versus normal chow), and improved the survival rate by 1.8-fold at the 80-week time point, with respect to the DEN model group in females (Figure 1G).
Metformin (Met) was reported to prevent DEN-induced liver tumorigenesis in C57BL/KsJ-+Leprdb/+Leprdb (db/db) obese and diabetic mice,20 and hence was used as a reference control in the present study. Our results showed that Met exhibited a cancer-preventive effect similar to that of BBR in DEN-injected C57BL/6 mice of both sexes (Figures 1B and 1F). Met also reduced body/liver weight gain and improved serum biochemical indexes as BBR did (Table 1).
It has been reported that the DEN-caused hepatoma was closely related to the abnormal level of inflammatory cytokines.2, 3, 4 To explore which cytokines were responsible for the preventive effect of BBR on the DEN-induced hepatoma, the Luminex system was used to monitor the change of serum levels of 12 major cytokines and metabolic hormones associated with HCC. Of the inflammatory biomarkers, leptin was the only one continuously reduced by BBR, from the early stage of DEN injection to the end of the experiment (Figure 1H; Tables S1–S4), synchronized with that of AFP, as shown in Figure 1C. It suggested leptin is a potential key target for BBR’s action.
To learn the cancer-preventive effect of BBR on other tumors, we used the azoxymethane (AOM)-induced colon tumor model in mice for this study as well. This model was reported to reflect the inflammation-driven tumor progression,21 similar to that by DEN. As shown in Figure S1, BBR significantly inhibited colon carcinogenesis after oral administration (0.4% supplement in diet, w/w) for 10 weeks, further confirming the results of BBR’s effect on DEN-induced hepatoma.
BBR reduced ATX and LPAR2 expression to repress liver tumorigenesis
As we have mentioned, the ATX-LPA-LPARs axis is crucial for HCC. Thus, this signal pathway was among the targets of investigation. ATX expression in tissue and serum was elevated in the DEN-injected mice, and reduced by BBR or Met treatment as demonstrated in western blot, real-time RT-PCR, and ELISA assay (Figures 2A–2C). First, BBR decreased ATX protein expression in nodular tumors as well as non-tumor liver tissues of the male mice 6 and 8 months after DEN injection, respectively (Figure 2A).
Figure 2.

Berberine decreased ATX and LPAR2 expression in DEN model
(A) ATX protein expression was detected by western blot of tumor and non-tumor tissues respectively from male mice 6 and 8 months after DEN injection, and normalized to that of ACTB (β-actin). The ratio of ATX/ACTB was set as 1 in sham-treated group. Data are expressed as the mean ± SEM (n = 2–3). (B and E), ATX and LPAR2 mRNA expression was detected by real-time RT-PCR in tumor and non-tumor tissues of DEN-injected male mice, which was normalized to ACTB and presented as fold of that in the sham-treated group. Data are expressed as the mean ± SEM (n = 9). (C) Serum ATX was assayed by ELISA of the indicated groups 8 and 12 months after DEN injection for male and female mice respectively. Data were expressed as the mean ± SEM (n = 9–10). (D), serum LPA was examined by ELISA in female mice 12 months after DEN injection. Data are expressed as the mean ± SEM (n = 6). (F) HepG2 cells were subjected to 20 μM LPA combined with BBR (0.1 μM, 0.5 μM, 2.5 μM) and Met (250 μM) respectively for 18 h, after being starved overnight and pretreatment of BBR or Met for 4 h. LPAR2 mRNA was detected by real-time RT-PCR, which was normalized to ACTB and presented as fold of that in the DMSO group without LPA treatment. Values are the mean ± SEM of three independent experiments. ∗p < 0.05, ∗∗p < 0.01 versus that of DEN or LPA model group.
Moreover, ATX mRNA expression was much higher in the tumor than that in the non-tumor liver tissues (Figure 2B), and BBR at doses of 0.04% and 0.2% in diet (w/w) significantly reduced ATX mRNA by 46% and 54% in the tumor tissues respectively, suggesting that BBR might act on the transcription stage (Figure 2B). Finally, in both the male and female DEN-injected mice, serum ATX reached 3–18 ng/mL at the end of experiment; however, ATX level was almost undetectable in the BBR- and Met-treated mice, and the level was significantly decreased by 78%–98% (p < 0.01 for all the BBR and Met-treated groups versus DEN model group; Figure 2C).
Serum LPA was also elevated in the DEN-injected mice, but it was not reduced by BBR or Met (Figure 2D). As reported, LPA could also be produced by another route using the enzymes phospholipase D (PLD) and secreted phospholipase A2 (sPLA2).9 Therefore, it was speculated that BBR and Met might have no influence on their expression and hence did not change the serum level of LPA, which would be studied in the future.
It has been reported that LPAR2 and LPAR6 were expressed and played important roles in hepato-carcinogenesis.22,23 Of the two subtypes of LPARs, we found that only LPAR2 mRNA was upregulated in the DEN group, and it was significantly reduced by 33% and 38% in tumors, and 44% and 55% in non-tumor tissues of the mice treated with 0.04% and 0.2% BBR, respectively (p < 0.01 for both; BBR versus the DEN model group; Figure 2E). Thus, it seems that BBR might prevent tumorigenesis in liver through reducing the expression of ATX and LPAR2, the two key factors in the LPA pathway.
In the HepG2 cell tests, we found that LPA increased LPAR2 transcription, and the stimulatory effect was significantly inhibited by pretreatment of the cells with BBR (0.5 and 2.5 μM) or Met (250 μM) (Figure 2F). The result seems in agreement with the report that LPA increased the expression of its own synthetase ATX in an autocrine manner.11
BBR also reduced the leptin level in tumor tissues of the DEN model mice
ELISA assay shows that serum leptin was largely reduced by BBR in male mice at the endpoint of the experiment (Figure 3A); for the female mice, serum leptin was also substantially decreased by BBR at 9 and 12 months after DEN injection (Figure 3B). The result was consistent with that obtained in the Luminex assay (Figure 1H). We then investigated whether the elevated leptin in serum was originated from the livers of DEN-injected mice. Our results showed that leptin expression was elevated both in the non-tumor liver tissues and tumor nodules of the DEN-injected mice, and was significantly reduced by BBR and Met (Figure 3C).
Figure 3.

BBR reduced leptin expression in DEN model
(A and B) Serum leptin was reduced by BBR in male and female mice respectively, as detected by ELISA. Values are the mean ± SEM (male, n = 6 for each group in A; female, n = 5 for each group of 1 month; n = 8 for sham, n = 11 for the other groups of 6 months; n = 11 for each group of 9 and 12 months in B). (C) Leptin expression was detected by western blot of tumor and non-tumor liver tissues from male mice 6 and 8 months after DEN injection respectively, and normalized to that of ACTB. The ratio of leptin/ACTB was set as 1 in sham-treated group. Values are the mean ± SEM (n = 2–3). (D–F) Leptin expression was detected by ELISA in tumor and non-tumor tissues of the mouse livers from male mice sacrificed at 1, 4, 6, and 8 months after DEN injection, respectively. Values are the mean ± SEM (n = 5 for non-tumor tissues in D and E, n = 6 for nodular tumors in F). (G) Leptin mRNA was detected of tumor and liver tissues from male mice 8 months after DEN injection by real-time RT-PCR. The result was normalized to ACTB and presented as fold of that in the sham-treated group. Data are expressed as the mean ± SEM (n = 9). ∗p < 0.05, ∗∗p < 0.01 versus that of DEN model group. #p < 0.05 versus the group treated with 0.04% BBR.
Also, ELISA quantitative detection showed that the leptin content in livers and tumors elevated progressively over the time. On average, the leptin content was increased from 1,738 pg/g tissue at the first month time point to 11,613 pg/g tissue at the eighth month in non-tumor liver tissues of the DEN model group (Figure 3D), and it reached 14,671 pg/g tissue and 18,583 pg/g tissue at the 6- and 8-month time points respectively in tumor nodules of the DEN model group (Figure 3E). It seems that there was more leptin in tumor nodules than in non-tumor liver tissues, consistent with the case of ATX and LPAR2 (Figures 2B and 2E). Compared with the DEN model group fed with normal chow diet, 0.04% and 0.2% BBR significantly reduced the leptin content in the non-tumor liver tissues and nodular tumors respectively (p < 0.05 or p < 0.01 versus normal chow in Figures 3D and 3E). The leptin level was not significantly influenced by BBR in the liver tissues of sham-injected C57BL/6 mice (p > 0.05 versus sham; Figure S2), suggesting that BBR did not reduce the basic expression of leptin in mice.
Last, leptin mRNA was significantly reduced by BBR and Met, both in liver and tumor tissues of the DEN-injected mice (Figure 3F). It seems that BBR and Met decreased leptin expression at the transcription level, similar to their actions on LPAR2 and ATX genes (Figures 2B and 2E).
BBR inhibited LPA-induced p38 phosphorylation and caused leptin reduction
Our next question was whether the effect of BBR on ATX-LPA-LPARs axis was connected to its inhibitory effect on leptin expression. To learn this, HepG2 cells were exposed to LPA after pretreatment with BBR or Met. The results showed that addition of LPA increased cellular leptin expression at both the protein and mRNA levels in cultured cells, and pretreatment with BBR at a dose of 0.1 μM significantly suppressed the bioactivity of LPA (Figures 4A and 4B). Met showed a very good inhibition of leptin expression as well, but its concentration (250 μM) was higher than that of BBR.
Figure 4.

BBR inhibited LPA-induced p38 phosphorylation and leptin transcription
(A) BBR reduced the leptin expression induced by LPA. HepG2 cells were subjected to 20 μM LPA combined with BBR (0.1 μM, 0.5 μM, 2.5 μM) and Met (250 μM) respectively for 18 h, after being starved overnight and pretreatment with BBR or Met for 4 h. Cellular total proteins were extracted for western blot analysis, which were normalized to that of ACTB. (B) Leptin mRNA expression was detected by real-time RT-PCR, normalized to ACTB, and presented as fold of the DMSO group without LPA treatment. (C) BBR and Met significantly decreased the abnormal activity of leptin promoter induced by LPA in HepG2 cells transfected with the pGL4.17-LP1 plasmid. After transfection, cells were screened using 0.4 mg/mL G418. Stable transfected cells were then treated as above. The promoter activity was detected as in section “methods.” (D) The promoter region (247 bp) was sufficient for LPA to induce the transcription of leptin gene. HepG2 cells were co-transfected with pGL4.74 and four pGL4.17-LP plasmids containing full-length and truncated 5′ regulatory region of the leptin gene respectively and treated as above. (E) BBR inhibited LPA-induced p38 phosphorylation at Thr180/Tyr182. HepG2 cells were treated as above. Western blot of P-p38 and p38 was conducted as described in section “methods,” and normalized to that of ACTB. (F) BBR inhibited p38 activation in tumor and liver tissues from male mice 8 months after DEN injection. The ratio of P-p38/ACTB was set as 1 in sham-treated group. (G and H) The LPA-induced activity of leptin promoter was reduced by the inhibitors of p38 (SB 203580 and SB 202190) and HIF-1α (KC7F2 and BAY 87-2243). Transfected cells were subjected to LPA (20 μM) combined with BBR (2.5 μM), Met (250 μM), SB 202190 (5 μM), SB 203580 (5 μM), KC7F2 (40 μM), and BAY 87-2243 (10 μM) as above. The luminescence was set as 1 in the vehicle group treated with DMSO. (I) BBR decreased the mRNA expression of CEBPA and HIF1A. HepG2 cells were treated as above. Results were normalized to ACTB and presented as fold of the DMSO group without LPA treatment. Values were the mean ± SEM of three independent experiments. Data are expressed as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01 compared with the LPA- or S1P-stimulated group.
Furthermore, LPA increased leptin transcription by stimulating the promoter activity of leptin gene (Figure 4C). In this experiment, the HepG2 cells were transfected with the PGL4.17 luciferase reporter plasmid containing the 5′ regulation region of leptin gene as described in the section “methods.” BBR treatment significantly suppressed the activity of leptin promoter stimulated by LPA in the transfected cells by 21%, 30%, and 44%, at the doses of 0.1 μM, 0.5 μM, and 2.5 μM respectively (Figure 4C). Agreeing with the results of tissue tests in the DEN-injected mice (Figure S2), BBR only reduced the abnormal expression of leptin stimulated by LPA, but not the basic leptin mRNA expression or promoter activity (Figures 4B and 4C).
Sphingolipids are another important class of bioactive lipids with sphingosine as the backbone.7 Sphingosine 1-phosphate (S1P), the most studied sphingolipid, also increased the activity of leptin promoter, but its potency is less than that of LPA (Figure 4C). BBR, but not Met, significantly deceased the leptin promoter activity induced by S1P (Figure 4C). However, LPC, a precursor of LPA, did not increase leptin expression (data not shown).
The leptin promoter region (247 bp) was sufficient to mediate the effect of LPA, revealed by gradual truncations of the 5′ regulatory region (2,951 bp) of the leptin gene (Figure 4D). BBR also significantly inhibited the leptin promoter activation mediated by all the truncated and full-length sequences (Figure 4D). It appeared that the promoter region might be the key response element of the leptin gene for LPA.
Moreover, p38 phosphorylation at Thr180/Tyr182 was increased by LPA in HepG2 cells, but inhibited by BBR and Met pretreatment (Figure 4E). Indeed, the in vivo experiments showed that BBR and Met decreased p38 phosphorylation in liver and tumor tissues of the DEN-treated mice, respectively (Figure 4F). Interestingly, the leptin promoter activity elevated by LPA could be attenuated by the selective p38 inhibitors SB 203580 and SB202190 (p < 0.01 for both p38 inhibitors versus the LPA-stimulated group; Figure 4G). The results suggested p38 is an upstream event for the leptin transcription, and inhibition of p38 could suppress the promoter activity of leptin induced by LPA.
Finally, CCAAT enhancer binding protein alpha (CEBPA) and hypoxia inducible factor 1 subunit alpha (HIF-1α) were reported to be the key factors regulating leptin gene transcription.24,25 Our experiments revealed that the leptin promoter activity was significantly decreased by the HIF-1α inhibitors BAY 87-2243 and KC7F2 (Figure 4H). In fact, BBR decreased CEBPA and HIF-1α mRNA transcription promoted by LPA (Figure 4I). It is suggested that BBR inhibited the LPA-induced leptin transcription, at least in part, through reducing p38 phosphorylation as well as the expression of transcription factors CEBPA and HIF-1α.
E2 inhibited the p38 and leptin promoter activation induced by LPA
As mentioned above, female mice were somehow resistant to the DEN-induced hepatoma, and the answer could be related to the level of E2 in the animals.2 Indeed, in the present study, E2 treatment reduced the LPA-induced leptin promoter activity (Figure 5A), indicating that E2 might decrease the leptin expression. For the inhibitory effect of BBR and E2 on the leptin promoter activation induced by LPA, BBR might have an activity stronger than E2 did; the group treated with either BBR or BBR plus E2 got better inhibition than that of E2 alone (Figure 5A). In addition, E2 reduced p38 phosphorylation at Thr180/Tyr182 stimulated by LPA (Figure 5B).
Figure 5.

E2 inhibited the leptin promoter activity and p38 phosphorylation stimulated by LPA
(A) pGL4.17-LP1 transfected cells were treated as above, BBR as 2.5 μM, and E2 at a dose of 100 pM. The luminescence was set as 1 in the vehicle group treated with DMSO. Values are expressed as mean ± SEM, ∗∗p < 0.01 versus the LPA-stimulated group treated with DMSO. ##p < 0.01 versus the LPA-stimulated group treated with E2. (B) E2 (100 pM) inhibited LPA-induced p38 phosphorylation at Thr180/Tyr182. HepG2 cells were treated and western blot was conducted as described above. (C) Schematic diagram illustrates BBR restrained liver tumorigenesis through antagonizing the ATX-LPA-LPAR2-p38-leptin axis, and reducing LPA-stimulated abnormal leptin transcription in hepatoma cells.
In summary, LPA could activate p38 and trigger the abnormal leptin transcription via stimulating its promoter activity, and thus promote hepatocellular carcinogenesis (Figure 5C). BBR (and Met) may suppress liver tumorigenesis through antagonizing the ATX-LPA-LPAR2-p38-leptin axis, showing the decreased expression of ATX, LPAR2, and leptin in hepatoma cells.
Discussion
As an OTC drug, berberine is used for treating bacterial-caused diarrhea in China. The dose is 0.1–0.3 g three times a day (0.3–0.9 g/day). It is 5–15 mg/kg for humans weighing 60 kg, and 61.5–184.5 mg/kg for mice after conversion.26 In the DEN model, 0.04% and 0.2% (w/w, drug/diet) were equal to 50 mg/kg and 250 mg/kg of berberine for mice weighing 20 g, as the amount of food intake was about 2.5 g/day/mouse; 50 mg/kg (0.04%) was set for studying the minimal dose for BBR’s action, and 250 mg/kg (0.2%) was chosen according to the previous reports in diabetic mice.27, 28, 29
In addition, 0.1% metformin (w/w, drug/diet) was reported to improve lifespan in mice.30 As the MW of berberine chloride (BBR; MW, 371.81) is about twice that of metformin hydrochloride (Met; MW, 165.62), 0.2% BBR approximated 0.1% Met on the molecular level.
It is reported that the serum concentration of BBR was about 0.02 μM (6.99 ng/mL) in humans, and BBR concentrated in liver after oral administration.31 There was an increase of 50- to 70-fold (1–1.4 μM) in the ratio of the area under the concentration-time curve value for berberine in liver compared with plasma.31,32 Hence, it was estimated that 0.1 μM and 0.5 μM could be achieved in liver cells, which was the low and medium dose of BBR for in vitro experiments in the present study. As the high dose for cell treatment, 2.5 μM was chosen, according to the previous report.33
It has been reported that BBR had an in vitro anticancer effect through induction of apoptosis at a concentration ranging from 20 to 100 μM (7–37 μg/mL), depending on cell types.34, 35, 36 The concentration used in those previous reports is 1,000 times higher than that in blood,31,37 suggesting its unknown mechanism against cancer. Pharmacokinetic study revealed two key features of BBR in body distribution; first, the compound is poorly absorbed in intestine after oral administration, and, second, the small portion of BBR taken up from the intestine is concentrated in liver.31,37
The present study investigated chemoprevention effect of BBR against hepatoma in the DEN model, which was initiated based on the observation that BBR is enriched in liver after oral administration. It showed that, in the DEN-injected mice (male and female), oral BBR in daily food could significantly reduce liver tumorigenesis. It appeared that the mechanism, at least partially, links to the inhibitory effect of BBR on the ATX-LPA-LPAR2-p38-leptin pathway. In this action, BBR significantly reduced LPAR2 expression, leading to a decline of the interaction between LPA and LPAR2, and subsequent p38 activation. This decline caused reduction of leptin expression through decreasing its promoter activity, and also might in turn downregulate ATX production by intervention of the positive feedback loop of LPA-LPARs-ATX.11
However, as mentioned earlier, BBR regulates a variety of factors related to energy metabolism. The therapeutic efficacies of BBR on metabolic disorders in humans have been widely confirmed, and intensive mechanistic studies have identified BBR to be a multiple target drug,38 active at least in upregulating LDLR and insulin receptor (InsR) expression, activating AMPK, and inhibiting PCSK9. These activities might also be helpful in preventing cancer recurrence, among which AMPK activation is well known to be an inhibitory factor in cancer development.39 In addition, we recently found that BBR treatment could significantly downregulate the expression level of galectin-3,40 which also closely relates to carcinogenesis.41,42 Therefore, in synergy with its action on the ATX-LPA-LPAR2-p38-leptin axis against liver cancer, the chemoprevention effect of BBR might be its systemic effects above for other cancers. For example, we found that oral BBR significantly inhibited AOM-induced colon carcinogenesis in mice. Consistent with this, in a clinical study, oral BBR prevented recurrence of colorectal adenoma.19 This study was considered of high value, but the mechanism remained largely unclear, which will be among the targets of our investigation in the future.
Reduplicative treatment of DEN is needed for adult mice/rats to form HCC, but would be toxic to the animals and reduce their life span. Hence, no survival data were provided in the previous reports.16, 17, 18 In our present study, 15-day-old calf mice were used for DEN modeling by single-injection strategy. The single-injection strategy demonstrated the role of chronic inflammation in HCC formation,2 and was considered suitable for evaluating the efficacy of candidate agents against HCC.43 Using this strategy, we found BBR significantly prolonged the life span of DEN-injected mice of both sexes.
DEN injection caused abnormal expression of several important cancer-related cytokines, as described in the section “Introduction”.Leptin caught our attention because BBR continuously reduced its serum level in the DEN-injected mice throughout the course of the experiment, and the chemoprevention efficacy of BBR in the partial-course treatment regimens (first 2 weeks, first 3 months, or last 5 months after DEN injection) failed to compare with that of the full-course BBR treatment. It suggested that leptin might be important in the DEN-induced hepatoma and agreed with the role of leptin in hepato-carcinogenesis, as reported before.4,44
Leptin is mainly expressed from white adipose tissue (WAT), and the serum level of leptin is proportional to WAT size. As an endocrine hormone, leptin regulates food intake to control body weight by acting on the hypothalamus.45 Recently, leptin was detected as a pro-inflammatory factor by promoting local inflammation in an autocrine/paracrine manner, and hence increased incidence of osteoarthritis, breast cancer, and hepatoma.4,46,47
The hepatoma incidence in men is two to four times higher than that in women, and E2 plays an important role in preventing HCC in the DEN-injected female mice.2 Indeed, it is reported that ovariectomy increased HCC induction in female mice,2,48 and this is consistent with the clinical observation that oophorectomy increased the risk of HCC in women.49,50 We show here that treating HepG2 cells with E2 could moderately reduce leptin promoter activity induced by LPA, but the potency was less than that of BBR. The result was consistent with the animal experiment, in which BBR was effective in female mice after DEN injection.
Endogenous bioactive lipids, including lysophopholipids and sphingolipids, were reported to be involved in NRI.7 Among these lipids, LPA was found to enhance cancer progression in lung, breast, ovary, liver, and colon through promoting cell survival, proliferation, migration, invasion, and angiogenesis, as well as inducing an enabling inflammatory setting in the tumor environment.9 Recently, the ATX-LPA-LPARs axis has been considered a key for hepato-carcinogenesis,6,8 further demonstrating the role of LPA in cancer development. Regulation of this pathway might be of significance. Although BBR was active in reducing ATX, LPAR2, and leptin expression, as well as p38 phosphorylation, the action of BBR on LPA-induced LPAR2 expression might be the key step because it could reduce the contact between LPA and LPAR2, terminate the autocrine pathway, and thus lead to an interruption of the ATX-LPA-LPARs axis. Also, it could reduce the p38 phosphorylation, which seems to be a downstream event of the ATX-LPA-LPARs axis, and activate the promoter of leptin gene. Still, there could be other kinases participating in this axis, which would be studied in the future.
LPA (LPL) and S1P (sphingolipid) are well-studied endogenous bioactive lipids. Our experiments revealed that both LPA and S1P stimulated leptin promoter activation. BBR significantly reduced the leptin promoter activity induced by both LPA and S1P, but Met did not (Figure 4C). It is suggested that BBR might be more effective than Met in patients with HCC, through inhibiting the aberrant leptin production induced by endogenous bioactive lipids in vivo. Also, as an OTC drug, BBR is more tolerated by patients and more convenient to get. Hence, BBR might be the better choice for clinical chemoprevention against HCC.
Taken together, LPA might bind LPAR2, activate p38, and stimulate leptin transcription through C/EBP and Hif-1α in hepatoma cells, thus promoting HCC development. BBR and Met appeared to repress liver tumorigenesis through antagonizing the ATX-LPA-LPAR2-p38-leptin axis and subsequently decreasing the abnormal leptin expression in livers of DEN-injected model mice. The action might help to prevent HCC in vivo. Currently, the 5-year survival rate for HCC is about 20%, and over 70% of the patients relapse in 5 years after resection, indicating the importance of chemoprevention after conventional therapy.1 As BBR has been an OTC drug in the past half century, it could be tested immediately for its chemoprevention effect against HCC recurrence in clinic.
Methods
Regents and kits
N-nitrosodiethylamine (also known as diethylnitrosamine [DEN], purity over 99%, #73861), dimethyl sulfoxide (DMSO, D2438), berberine (BBR, purity over 98%, B3251), metformin (Met, purity over 97%, D150959), activated charcoal (C6241), lysophosphatidic acid (LPA, purity over 98%, L7260), and S1P (purity over 98%, #73914) were purchased from Sigma-Aldrich (St. Louis, MO). Geneticin Selective Antibiotic (G418 Sulfate, #10131027), minimum essential medium (MEM, #11095080), fetal bovine serum (FBS, #10100154), Lipofectamine 3000 Transfection Reagent (L3000075), Opti-MEM Reduced Serum Medium (#31985088), protease and phosphatase inhibitor cocktail (#78442), M-PER Mammalian Protein Extraction Reagent (#78505), T-PER Tissue Protein Extraction Reagent (#78510), and the kit for total protein quantification (BCA assay, #23225) were purchased from Thermo Fisher Scientific (Shanghai, China). Fatty acid free bovine serum albumin (BSA, MB0094) was purchased from Meilun Biotechnology (Dalian, China). Estradiol (E2, purity over 99%, HY-B0141), the p38 inhibitors SB 203580 (purity over 99%, HY-10256), SB 202190 (purity over 99%, HY-10295), the HIF-1α inhibitors BAY 87-2243 (purity over 99%, HY-15836), and KC7F2 (purity over 99%, HY-18777) were purchased from MedChemExpress (Shanghai, China). The antibody against autotaxin (ATX, 14243-1-AP) was purchased from Proteintech Group (Rosemont, PA). The antibody against leptin (ab16227) was purchased from Abcam (Cambridge, MA). The antibodies against Phospho-p38 MAPK (#4511), p38 MAPK (#9212), and β-actin (ACTB) (#4970) were purchased from Cell Signaling Technology (Danvers, MA). The pGL4.17 [luc2/Neo] plasmid (E6721), pGL4.74 [hRluc/TK] plasmid (E6921), Dual-Glo Luciferase Assay System (E2940), and ONE-Glo Luciferase Assay System (E6120) were purchased from Promega Corporation (Madison, WI). RNeasy Mini Kit (#74106) was purchased from Qiagen (Germantown, TN) for total RNA extraction. Kits for RT (RR037A) and quantitative RT-PCR (RR820A) were purchased from Takara Bio (Shiga, Japan). Mouse alpha-Fetoprotein/AFP Quantikine ELISA Kit (MAFP00) and Mouse/Rat Leptin Quantikine ELISA kit (MOB00) were purchased from R&D Systems (Minneapolis, MN). Mouse ENPP2/Autotaxin ELISA kit (LS-F16526) and Lysophosphatidic Acid ELISA kit (LS-F25111) were purchased from LifeSpan BioScience (Seattle, WA). The kits were purchased from Bio-Rad Laboratories (Hercules, CA) for Luminex analysis of mouse serum cytokines and hormones.
Plasmid construction
The DNA sequences were synthesized commercially in the LEP 5' regulatory region (NCBI: 106728418. DNA sequence at the front of leptin gene: −2921~+30, 2951 bp; named LP1) and its truncated fragments, which included LP2 (−1415~+30, 1445 bp), LP3 (−469~+30, 499 bp), and LP4 (−217~+30, 247 bp; the promoter region of leptin gene). These sequences were then constructed into pGL4.17 [luc2/Neo] vector expressing the firefly luciferase, to establish the plasmids for monitoring the leptin promoter (LP) activity. These four plasmids were named from pGL4.17-LP1 to pGL4.17-LP4.
Cell culture, transfection, and drug treatment
The human HepG2 hepatoma cells were obtained from the Cell Culture Center of Peking Union Medical College (Beijing, China), and cultured in MEM plus 10% FBS at 37°C in a humidified atmosphere with 5% CO2. One day before transfection, cells were trypsinized with 0.25% trypsin containing EDTA and seeded onto six-well plates with 1 × 106/well. Lipofectamin 3000 (Thermo Fisher) was used to transfect the four pGL4.17-LP plasmids into HepG2 cells respectively.
The pGL4.74 [hRluc/TK] vector expresses the Renilla luciferase. For transient transfection, the pGL4.74 vector was used as a control reporter and co-transfected with the four pGL4.17-LP plasmids respectively at a ratio of 1:20. Then, 24 h after transfection, the cells were trypsinized and seeded onto the white-bottom 96-well plates with 3 × 104/well in culture with MEM plus 10% charcoal-treated FBS (FBSC) for 24 h. The FBSC was obtained as previously reported.51 Briefly, FBS was mixed with the activated charcoal at a ratio of 25 mL:1 g and rotated slowly at 4°C overnight, and then filtered using the 0.22 μM sterile syringe filter. The transfected cells were then starved in MEM medium without FBS overnight, and treated with LPA (dissolved as a stock solution of 5 mM in 0.1% BSA, fatty acid free) and BBR (dissolved in DMSO as a 10 mM stock solution) for the indicated time. The luminescence for the firefly and Renilla luciferases was detected respectively using the Dual-Glo Luciferase Assay System kit (Promega), and the ratio (firefly:Renilla) was calculated.
In addition, the plasmid pGL4.17-LP1 was transfected into HepG2 cells containing the full-length 5' regulatory region (2,951 bp) of the leptin gene. Stably expressed cells were selected in medium containing 0.4 mg/mL G418 for 2 months, and sowed onto the white-bottom 96-well plates with 3 × 104/well and cultured with MEM plus 10% FBSC for 24 h. After being starved overnight and drug treated, the cells were lysed and the firefly luminescence was detected by the ONE-Glo Luciferase Assay System kit (Promega).
Animal studies
C57BL/6 mice were purchased from Vital River Lab Animal Technology (Beijing, China). To generate DEN-induced hepatoma, mice of both sexes were subjected to single intraperitoneal injection with DEN (25 mg kg−1, Sigma-Aldrich) on the 15th day after birth. These mice were randomly grouped and fed with either normal chow diet (Keao Xieli Feed, Beijing, China) or the diet supplement with BBR (0.04% and 0.2%, w/w) and Met (0.1%, w/w) respectively from the day of DEN injection. Met was used as a positive reference in this study.20 One cohort was sacrificed to observe BBR effect on hepato-carcinogenesis at the end of the indicated months (1, 4, 6, and 8 months for male, as shown in Figure 1D, and 1, 6, 9, and 12 months for female) after DEN injection, and the other cohort was followed to assess BBR’s efficiency on the survival of mice with hepatoma. All visible nodular tumors were counted on the surface of liver tissues, and those bigger than 0.5 mm in diameter were measured using a vernier caliper. The tumor volume was calculated based on the formula (v = 0.5ab,2 in which “v” represents tumor volume, “a” stands for the long diameter, and “b” for the short diameter of the nodular tumor). Half of the largest liver lobe was fixed in formalin, embedded with paraffin, and sectioned. The sections were used for H&E staining, and the images were obtained by using the Pannoramic Scanner and CaseViewer software (3DHISTECH, Hungary). Other liver and tumor samples were snap frozen in liquid nitrogen immediately after assessment of tumor numbers and volume. Centrifugally separated serum was frozen in a −80°C freezer for the Luminex and ELISA assay.
All animal experiments were conducted in accordance with the guidelines of the Institutional Committee for the Ethics of Animal Care and Treatment in Biomedical Research of Chinese Academy of Medical Sciences and Peking Union Medical College. The animal studies were consistent with the ARRIVE guidelines.52
Luminex and ELISA assay
The Luminex assay was performed with kits (Bio-Rad) following the manufacturer’s instructions, by using the Bio-plex system (Bio-Rad, Hercules, CA). Essentially, this assay was an immunoassay based on fluorescently dyed magnetic beads with distinct spectral addresses and a cytometer with lasers. The magnetic beads coupled to the specific capture antibodies, which interacted with different biomarkers (e.g., cytokines and hormones) of interest in serum samples. The cytometer then sorted the beads and measured the level of these biomarkers using lasers. Serum alpha-fetoprotein (AFP) (R&D), leptin (R&D), ATX (Lifespan), and LPA (Lifespan) were detected using ELISA kits according to the venders’ instructions. Serum total cholesterol and alanine aminotransferase (ALT) were measured by commercial kits (Biosino, China). Blood glucose was measured by a commercial meter (Roche).
Western blot
After separation, the liver and tumor tissues were ground respectively using the TissueLyzer device (Qiagen, Germantown, TN) and lysed in the extraction reagent containing protease and phosphatase inhibitors. After drug treatment, the cells cultured in six-well plates were rinsed with PBS and lysed in the extraction buffer. Total proteins were extracted and quantified by the BCA assay kit. For each sample, about 20 μg of proteins were subjected to 10% SDS-PAGE, and protein bands were transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA) through a Wet Transfer Cell (Bio-Rad, Hercules, CA). For the detection of target protein levels, the membranes were probed with specific antibodies using β-actin as an internal control. After washing and incubation with appropriate secondary antibodies, the bands were visualized with a horseradish peroxidase (HRP) Kit (Millipore).
Real-time RT-PCR
The liver and tumor tissues were fragmented and lysed by using the TissueLyzer device, and the total RNAs were isolated using a kit (Qiagen). The cultured cells were seeded onto six-well plates, and total cellular RNAs were extracted after drug treatment. Total RNAs were reverse transcribed into cDNAs as described previously.53 Quantitative real-time PCR was performed with these cDNAs using the SYBR Green method (Takara). Relative quantification was conducted by using ACTB as an internal control. Normalized levels of target genes were plotted as fold of the untreated control. The primer sequences are listed in Table S5.
Statistical analysis
The statistical significance of two groups was determined by the unpaired t test with Welch's correction. For comparisons of more than two groups, the results were examined by ANOVA. For analysis of survival curves, the log-rank (Mantel-Cox) test was used to evaluate the significance, and p < 0.05 was considered to be statistically significant.
Data availability
All raw data are available upon request.
Acknowledgments
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1–009), and the National Natural Science Foundation of China (81302823 and 81621064).
Author contributions
G.R., Methodology, investigation, formal analysis, data curation, and writing – original draft preparation;. J.-H.G., methodology, visualization, and investigation; C.-L.F., investigation; Y.-W.D., investigation; B.D., investigation; Y.-X.H., resources; Y.-H.L., validation, visualization; L.-L.W., investigation, project administration, resources; J.-D.J., conceptualization, methodology, formal analysis, writing – review & editing, supervision, funding acquisition.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.08.001.
Contributor Information
Gang Ren, Email: imbrengang@126.com.
Lu-Lu Wang, Email: wanglulu@imm.ac.cn.
Jian-Dong Jiang, Email: jiang.jdong@163.com.
Supplemental information
References
- 1.Villanueva A. Hepatocellular carcinoma. N. Engl. J. Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 2.Naugler W.E., Sakurai T., Kim S., Maeda S., Kim K., Elsharkawy A.M., Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 3.Park E.J., Lee J.H., Yu G.Y., He G., Ali S.R., Holzer R.G., Osterreicher C.H., Takahashi H., Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma D., Wang J., Fu P.P., Sharma S., Nagalingam A., Mells J., Handy J., Page A.J., Cohen C., Anania F.A., Saxena N.K. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology. 2010;52:1713–1722. doi: 10.1002/hep.23892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu L.X., Ling Y., Wang H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018;2:6. doi: 10.1038/s41698-018-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakagawa S., Wei L., Song W.M., Higashi T., Ghoshal S., Kim R.S., Bian C.B., Yamada S., Sun X., Venkatesh A., et al. Precision Liver Cancer Prevention Consortium Molecular liver cancer prevention in cirrhosis by Organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell. 2016;30:879–890. doi: 10.1016/j.ccell.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiurchiù V., Leuti A., Maccarrone M. Bioactive lipids and chronic inflammation: managing the fire within. Front. Immunol. 2018;9:38. doi: 10.3389/fimmu.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopane C., Agosti P., Gigante I., Sabbà C., Mazzocca A. Implications of the lysophosphatidic acid signaling axis in liver cancer. Biochim. Biophys. Acta Rev. Cancer. 2017;1868:277–282. doi: 10.1016/j.bbcan.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Valdés-Rives S.A., González-Arenas A. Autotaxin-lysophosphatidic acid: from inflammation to cancer development. Mediators Inflamm. 2017;2017:9173090. doi: 10.1155/2017/9173090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaffe E., Katsifa A., Xylourgidis N., Ninou I., Zannikou M., Harokopos V., Foka P., Dimitriadis A., Evangelou K., Moulas A.N., et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology. 2017;65:1369–1383. doi: 10.1002/hep.28973. [DOI] [PubMed] [Google Scholar]
- 11.Cao P., Aoki Y., Badri L., Walker N.M., Manning C.M., Lagstein A., Fearon E.R., Lama V.N. Autocrine lysophosphatidic acid signaling activates beta-catenin and promotes lung allograft fibrosis. J. Clin. Invest. 2017;127:1517–1530. doi: 10.1172/JCI88896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erstad D.J., Tager A.M., Hoshida Y., Fuchs B.C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell. Oncol. 2017;4:e1311827. doi: 10.1080/23723556.2017.1311827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong W., Wei J., Abidi P., Lin M., Inaba S., Li C., Wang Y., Wang Z., Si S., Pan H., et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat. Med. 2004;10:1344–1351. doi: 10.1038/nm1135. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H., Wei J., Xue R., Wu J.D., Zhao W., Wang Z.Z., Wang S.K., Zhou Z.X., Song D.Q., Wang Y.M., et al. Berberine lowers blood glucose in type 2 diabetes mellitus patients through increasing insulin receptor expression. Metabolism. 2010;59:285–292. doi: 10.1016/j.metabol.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 15.Lan J., Zhao Y., Dong F., Yan Z., Zheng W., Fan J., Sun G. Meta-analysis of the effect and safety of berberine in the treatment of type 2 diabetes mellitus, hyperlipemia and hypertension. J. Ethnopharmacol. 2015;161:69–81. doi: 10.1016/j.jep.2014.09.049. [DOI] [PubMed] [Google Scholar]
- 16.Anis K.V., Rajeshkumar N.V., Kuttan R. Inhibition of chemical carcinogenesis by berberine in rats and mice. J. Pharm. Pharmacol. 2001;53:763–768. doi: 10.1211/0022357011775901. [DOI] [PubMed] [Google Scholar]
- 17.Sengupta D., Chowdhury K.D., Sarkar A., Paul S., Sadhukhan G.C. Berberine and S allyl cysteine mediated amelioration of DEN+CCl4 induced hepatocarcinoma. Biochim. Biophys. Acta. 2014;1840:219–244. doi: 10.1016/j.bbagen.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 18.Zhao X., Zhang J.J., Wang X., Bu X.Y., Lou Y.Q., Zhang G.L. Effect of berberine on hepatocyte proliferation, inducible nitric oxide synthase expression, cytochrome P450 2E1 and 1A2 activities in diethylnitrosamine- and phenobarbital-treated rats. Biomed. Pharmacother. 2008;62:567–572. doi: 10.1016/j.biopha.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y.X., Gao Q.Y., Zou T.H., Wang B.M., Liu S.D., Sheng J.Q., Ren J.L., Zou X.P., Liu Z.J., Song Y.Y., et al. Berberine versus placebo for the prevention of recurrence of colorectal adenoma: a multicentre, double-blinded, randomised controlled study. Lancet Gastroenterol. Hepatol. 2020;5:267–275. doi: 10.1016/S2468-1253(19)30409-1. [DOI] [PubMed] [Google Scholar]
- 20.Ohno T., Shimizu M., Shirakami Y., Baba A., Kochi T., Kubota M., Tsurumi H., Tanaka T., Moriwaki H. Metformin suppresses diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-+Leprdb/+Leprdb mice. PLoS One. 2015;10:e0124081. doi: 10.1371/journal.pone.0124081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neufert C., Heichler C., Brabletz T., Scheibe K., Boonsanay V., Greten F.R., Neurath M.F. Inducible mouse models of colon cancer for the analysis of sporadic and inflammation-driven tumor progression and lymph node metastasis. Nat. Protoc. 2021;16:61–85. doi: 10.1038/s41596-020-00412-1. [DOI] [PubMed] [Google Scholar]
- 22.Mazzocca A., Dituri F., De Santis F., Filannino A., Lopane C., Betz R.C., Li Y.Y., Mukaida N., Winter P., Tortorella C., et al. Lysophosphatidic acid receptor LPAR6 supports the tumorigenicity of hepatocellular carcinoma. Cancer Res. 2015;75:532–543. doi: 10.1158/0008-5472.CAN-14-1607. [DOI] [PubMed] [Google Scholar]
- 23.Dai X., Ding L., Liu H., Xu Z., Jiang H., Handelman S.K., Bai Y. Identifying interaction clusters for MiRNA and MRNA pairs in TCGA network. Genes (Basel) 2019;10:E702. doi: 10.3390/genes10090702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambrosini G., Nath A.K., Sierra-Honigmann M.R., Flores-Riveros J. Transcriptional activation of the human leptin gene in response to hypoxia. Involvement of hypoxia-inducible factor 1. J. Biol. Chem. 2002;277:34601–34609. doi: 10.1074/jbc.M205172200. [DOI] [PubMed] [Google Scholar]
- 25.Miller S.G., De Vos P., Guerre-Millo M., Wong K., Hermann T., Staels B., Briggs M.R., Auwerx J. The adipocyte specific transcription factor C/EBPalpha modulates human ob gene expression. Proc. Natl. Acad. Sci. USA. 1996;93:5507–5511. doi: 10.1073/pnas.93.11.5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janhavi P., Divyashree S., Sanjailal K.P., Muthukumar S.P. DoseCal: a virtual calculator for dosage conversion between human and different animal species. Arch. Physiol. Biochem. 2022;128:426–430. doi: 10.1080/13813455.2019.1687523. [DOI] [PubMed] [Google Scholar]
- 27.Shan Y.Q., Ren G., Wang Y.X., Pang J., Zhao Z.Y., Yao J., You X.F., Si S.Y., Song D.Q., Kong W.J., Jiang J.D. Berberine analogue IMB-Y53 improves glucose-lowering efficacy by averting cellular efflux especially P-glycoprotein efflux. Metabolism. 2013;62:446–456. doi: 10.1016/j.metabol.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Qin X., Jiang M., Zhao Y., Gong J., Su H., Yuan F., Fang K., Yuan X., Yu X., Dong H., Lu F. Berberine protects against diabetic kidney disease via promoting PGC-1alpha-regulated mitochondrial energy homeostasis. Br. J. Pharmacol. 2020;177:3646–3661. doi: 10.1111/bph.14935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia Q.S., Wu F., Wu W.B., Dong H., Huang Z.Y., Xu L., Lu F.E., Gong J. Berberine reduces hepatic ceramide levels to improve insulin resistance in HFD-fed mice by inhibiting HIF-2alpha. Biomed. Pharmacother. 2022;150:112955. doi: 10.1016/j.biopha.2022.112955. [DOI] [PubMed] [Google Scholar]
- 30.Martin-Montalvo A., Mercken E.M., Mitchell S.J., Palacios H.H., Mote P.L., Scheibye-Knudsen M., Gomes A.P., Ward T.M., Minor R.K., Blouin M.J., et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H.M., Xia M.F., Wang Y., Chang X.X., Yao X.Z., Rao S.X., Zeng M.S., Tu Y.F., Feng R., Jia W.P., et al. Efficacy of berberine in patients with non-alcoholic fatty liver disease. PLoS One. 2015;10:e0134172. doi: 10.1371/journal.pone.0134172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y.T., Hao H.P., Xie H.G., Lai L., Wang Q., Liu C.X., Wang G.J. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab. Dispos. 2010;38:1779–1784. doi: 10.1124/dmd.110.033936. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z., Zhang H., Li B., Meng X., Wang J., Zhang Y., Yao S., Ma Q., Jin L., Yang J., et al. Berberine activates thermogenesis in white and brown adipose tissue. Nat. Commun. 2014;5:5493. doi: 10.1038/ncomms6493. [DOI] [PubMed] [Google Scholar]
- 34.Yang X., Huang N. Berberine induces selective apoptosis through the AMPKmediated mitochondrial/caspase pathway in hepatocellular carcinoma. Mol. Med. Rep. 2013;8:505–510. doi: 10.3892/mmr.2013.1506. [DOI] [PubMed] [Google Scholar]
- 35.Lu Y., Han B., Yu H., Cui Z., Li Z., Wang J. Berberine regulates the microRNA-21-ITGBeta4-PDCD4 axis and inhibits colon cancer viability. Oncol. Lett. 2018;15:5971–5976. doi: 10.3892/ol.2018.7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Q., Li L., Zou X., Xu L., Yi P. Berberine attenuated proliferation, invasion and migration by targeting the AMPK/HNF4alpha/WNT5A pathway in gastric carcinoma. Front. Pharmacol. 2018;9:1150. doi: 10.3389/fphar.2018.01150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan X.S., Ma J.Y., Feng R., Ma C., Chen W.J., Sun Y.P., Fu J., Huang M., He C.Y., Shou J.W., et al. Tissue distribution of berberine and its metabolites after oral administration in rats. PLoS One. 2013;8:e77969. doi: 10.1371/journal.pone.0077969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong W.J., Vernieri C., Foiani M., Jiang J.D. Berberine in the treatment of metabolism-related chronic diseases: a drug cloud (dCloud) effect to target multifactorial disorders. Pharmacol. Ther. 2020;209:107496. doi: 10.1016/j.pharmthera.2020.107496. [DOI] [PubMed] [Google Scholar]
- 39.Umezawa S., Higurashi T., Nakajima A. AMPK: therapeutic target for diabetes and cancer prevention. Curr. Pharm. Des. 2017;23:3629–3644. doi: 10.2174/0929867324666170713150440. [DOI] [PubMed] [Google Scholar]
- 40.Wang C., Wang Y., Ma S.R., Zuo Z.Y., Wu Y.B., Kong W.J., Wang A.P., Jiang J.D. Berberine inhibits adipocyte differentiation, proliferation and adiposity through down-regulating galectin-3. Sci. Rep. 2019;9:13415. doi: 10.1038/s41598-019-50103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nangia-Makker P., Hogan V., Raz A. Galectin-3 and cancer stemness. Glycobiology. 2018;28:172–181. doi: 10.1093/glycob/cwy001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruvolo P.P. Galectin 3 as a guardian of the tumor microenvironment. Biochim. Biophys. Acta. 2016;1863:427–437. doi: 10.1016/j.bbamcr.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Jühling F., Hamdane N., Crouchet E., Li S., El Saghire H., Mukherji A., Fujiwara N., Oudot M.A., Thumann C., Saviano A., et al. Targeting clinical epigenetic reprogramming for chemoprevention of metabolic and viral hepatocellular carcinoma. Gut. 2021;70:157–169. doi: 10.1136/gutjnl-2019-318918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mittenbuhler M.J., Sprenger H.G., Gruber S., Wunderlich C.M., Kern L., Bruning J.C., Wunderlich F.T. Hepatic leptin receptor expression can partially compensate for IL-6Ralpha deficiency in DEN-induced hepatocellular carcinoma. Mol. Metab. 2018;17:122–133. doi: 10.1016/j.molmet.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu J., Bartolome C.L., Low C.S., Yi X., Chien C.H., Wang P., Kong D. Genetic identification of leptin neural circuits in energy and glucose homeostases. Nature. 2018;556:505–509. doi: 10.1038/s41586-018-0049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tenvooren I., Jenks M.Z., Rashid H., Cook K.L., Muhlemann J.K., Sistrunk C., Holmes J., Wang K., Bonin K., Hodges K., et al. Elevated leptin disrupts epithelial polarity and promotes premalignant alterations in the mammary gland. Oncogene. 2019;38:3855–3870. doi: 10.1038/s41388-019-0687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scotece M., Mobasheri A. Leptin in osteoarthritis: focus on articular cartilage and chondrocytes. Life Sci. 2015;140:75–78. doi: 10.1016/j.lfs.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 48.Goldfarb S., Pugh T.D. Ovariectomy accelerates the growth of microscopic hepatocellular neoplasms in the mouse: possible association with whole body growth and fat deposition. Cancer Res. 1990;50:6779–6782. [PubMed] [Google Scholar]
- 49.Yu M.W., Chang H.C., Chang S.C., Liaw Y.F., Lin S.M., Liu C.J., Lee S.D., Lin C.L., Chen P.J., Lin S.C., Chen C.J. Role of reproductive factors in hepatocellular carcinoma: impact on hepatitis B- and C-related risk. Hepatology. 2003;38:1393–1400. doi: 10.1016/j.hep.2003.09.041. [DOI] [PubMed] [Google Scholar]
- 50.McGlynn K.A., Sahasrabuddhe V.V., Campbell P.T., Graubard B.I., Chen J., Schwartz L.M., Petrick J.L., Alavanja M.C., Andreotti G., Boggs D.A., et al. Reproductive factors, exogenous hormone use and risk of hepatocellular carcinoma among US women: results from the Liver Cancer Pooling Project. Br. J. Cancer. 2015;112:1266–1272. doi: 10.1038/bjc.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benesch M.G.K., Zhao Y.Y., Curtis J.M., McMullen T.P.W., Brindley D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 2015;56:1134–1144. doi: 10.1194/jlr.M057661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kilkenny C., Browne W., Cuthill I.C., Emerson M., Altman D.G., NC3Rs Reporting Guidelines Working Group Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br. J. Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ren G., Guo J.H., Qian Y.Z., Kong W.J., Jiang J.D. Berberine improves glucose and lipid metabolism in HepG2 cells through AMPKalpha1 activation. Front. Pharmacol. 2020;11:647. doi: 10.3389/fphar.2020.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available upon request.