

Summary
RNA helicase DDX21 plays vital roles in ribosomal RNA processing and the regulation of host innate immunity during virus infection. Here, we describe the optimized protocols for nucleic acid-free protein purification and crystallization of DDX21 in its different unwinding states. Rational design of the flexible region within the helicase core, and biophysical approach to characterize interactions between DDX21 and RNA, leads to successful crystallization of DDX21. This protocol can be applied to the crystallography of other DExD/H-box RNA helicases.
For complete details on the use and execution of this protocol, please refer to Chen et al. (2020).
Subject areas: Molecular Biology, Protein Biochemistry, Protein expression and purification, Structural Biology, X-ray Crystallography
Graphical abstract
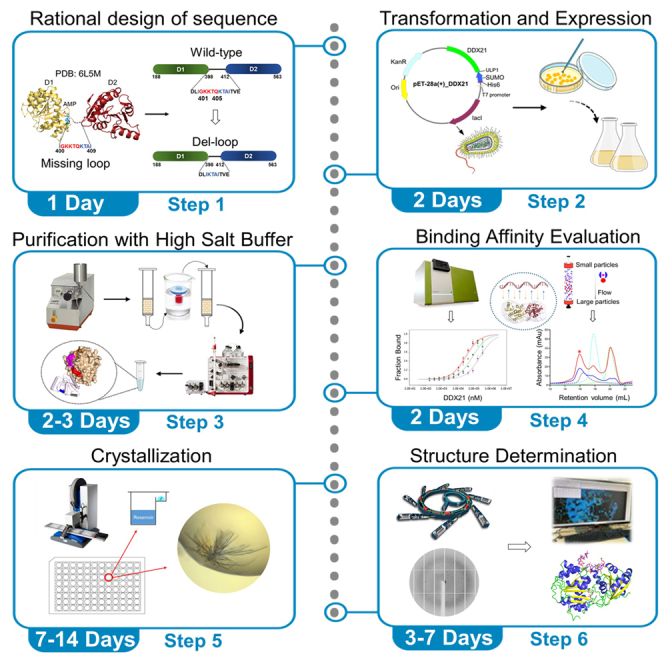
Highlights
-
•
Optimized protocols for nucleic acid-free DDX21 protein purification
-
•
Rational design of the flexible region within DDX21 helicase core
-
•
Biophysical approach to characterize interactions between DDX21 and RNA
-
•
Crystallization of ligand-free DDX21 or in complex with ATP analogs or RNA of interest
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
RNA helicase DDX21 plays vital roles in ribosomal RNA processing and the regulation of host innate immunity during virus infection. Here, we describe the optimized protocols for nucleic acid-free protein purification and crystallization of DDX21 in its different unwinding states. Rational design of the flexible region within the helicase core, and biophysical approach to characterize interactions between DDX21 and RNA, leads to successful crystallization of DDX21. This protocol can be applied to the crystallography of other DExD/H-box RNA helicases.
Before you begin
RNA helicases unwind their RNA substrates in an ATP-dependent manner and are central to all cellular processes involving RNA (Owttrim, 2013). Mammalian DExD/H-box RNA helicases sense viral infections and trigger host innate immunity during virus infection (Fullam and Schröder, 2013). They are also linked to neurological disorders, cancer, and aging processes, which render them to be attractive therapeutic targets and potential biomarkers (Zhang and Li, 2021). However, the difficulty of obtaining RNA helicase proteins with high purity and yield is a tremendous challenge for understanding their physiological and pathological functions. Despite numerous efforts, to date, only a limited number of RNA helicase structures have been determined, which is also due to their structural dynamics and in vitro instability. We were able to overcome these difficulties and solve several sets of RNA helicase structures of DDX21 in its different unwinding states. And more importantly, the protocol described here could be applied to other RNA helicases. We anticipate that this protocol will foster the crystal structure determination of a number of DExD/H-box RNA helicases, either in apo form or in complex with the substrates, such as different ATP analogs and RNAs of interest, e.g., the R-loops or guanine-quadruplexes (G4s).
This protocol described the whole process from protein purification to crystallization of human RNA helicase DDX21, which could serve as a practical guide to other crystallographers. The procedure is divided into three parts. In the first stage, we show how to express and purify RNA helicase from Escherichia coli cells. This is easily achieved by using a high-salt buffer during cell lysis and affinity purification steps, coupled to an extensive washing step to remove any tightly bound nucleic acid from RNA helicases. In the second stage, we describe the methodology for the identification of conditions promoting the interactions between the RNA helicase and the RNAs of interest using microscale thermophoresis (MST) and analytical size exclusion chromatography (SEC) co-elution binding assay. Finally, the third stage involves the crystallization screening of purified RNA helicases, including the apo form or in complex with different ATP analogs, or RNAs of interest. To begin with, we need to generate the expression plasmids by performing DNA cloning, mutagenesis and transformation, and prepare Ubiquitin-like-specific protease 1 (Ulp1).
Engineering two forms of RNA helicase DDX21 for crystallization
Timing: 1 day
-
1.
Obtain the human DDX21 gene sequence from the UniProt database (UniProt ID: Q9NR30) (UniProt, 2007).
-
2.
Use intrinsic protein disorder predictor GlobPlot (http://globplot.embl.de/) to analyze the globular domain and disorder region of DDX21.
Note: For this protocol, the boundary of DDX21 helicase core was determined using several online software, including the secondary structure prediction server JPred4 (https://www.compbio.dundee.ac.uk/jpred/), the Protein Model Portal (https://www.proteinmodelportal.org/; unavailable now), and the tertiary structure prediction sever Phyre2 (https://www.sbg.bio.ic.ac.uk/phyre2). In particular, we used MultAlin (http://multalin.toulouse.inra.fr/multalin) and ESPript (https://espript.ibcp.fr/ESPript/ESPript/index.php) to align sequences of DDX21 with several homologous sequences with known structures, including heat resistant RNA dependent ATPase (PDB: 4KBF), ATP-dependent RNA helicase vasa (PDB: 2DB3), and probable ATP-dependent RNA helicase MJ0669 (PDB: 1HV8). Based on all the analysis, residues 188–563 of DDX21 encompassing the conserved RecA-like helicase core domains D1 and D2 was chosen for expression.
-
3.
Construct a minimal active DDX21 construct, and add a stop codon at the C-terminus.
Note: Here, a His-sumo tag was fused to the N-terminus of the human helicase core D1D2 (amino acids (aa) 188–563) to increase protein expression and benefit for the following affinity purification.
-
4.
Delete part of the linker loop (GKKTQ; residues 401–405) to reduce the flexibility (Figure 1).
Note: The conserved RecA-like helicase core domains D1 and D2 of DDX21 are originally linked with a relatively long linker region (aa around 398–412). The design of the deletion region was based on sequence alignment with other homologous sequences with known structures as described above, and no other constructs were tested since this design was proved successful in subsequent studies.
-
5.
Use the wild-type DDX21 helicase core construct to crystallize the DDX21-RNA complex.
-
6.
Use the del-loop form for the crystallization of apo-formed DDX21 or DDX21 in complex with ATP analogs.
Figure 1.

Engineering a less flexible form of the minimal active construct of human DDX21
Schematic drawing of wild-type helicase core (right top) and its del-loop form (right bottom).
Del-loop: delete aa 401–405 (red color). The loop region (aa Ile400 – Ile409) in the crystal structure of DDX21-AMP (left) is invisible, which leads to the rational design of the del-loop construct to reduce the flexibility and facilitate crystallization.
Prepare the plasmids
-
7.
Use the pSMT3 vector (a generous gift from Dr. Chris Lima, Sloan Kettering Institute).
Note: The pSMT3 vector is modified from pET-28a backbone and encodes a N-terminal His6-SUMO tag followed along with a BamHI restriction site for inserting gene.
-
8.
Design the forward and reverse primers.
-
9.
Amplify DDX21 (188–563, WT) by PCR, using expression plasmid encoding human full length DDX21 (aa 1–783) (Chen et al., 2020) as template.
-
10.
Set up the standard PCR reactions in a 50 μL format as below.
PCR reaction master mix
| Reagent | Amount |
|---|---|
| DNA template (100 ng/μL) | 0.5 μL |
| KOD Hot Start Master Mix (Merk; 0.04 U/μL) | 25 μL |
| 10 μM DDX21 WT primer – forward | 1.5 μL |
| 10 μM DDX21 WT primer – reverse | 1.5 μL |
| ddH2O | 21.5 μL |
-
11.
Perform the PCR amplification as below in a T100 Thermal Cycler (Bio-Rad).
PCR cycling conditions
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 2 min | 1 |
| Denaturation | 95°C | 20 s | 30 cycles |
| Annealing | 56°C | 10 s | |
| Extension | 70°C | 15 s | |
| Hold | 4°C | forever | |
-
12.
Digest the PCR product and the pSMT3 vector with BamHI (NEB) and HindIII (NEB) restriction endonucleases.
-
13.
Set up the reaction as follows:
| Component | Amount |
|---|---|
| PCR product or pSMT3 vector | 1 μg |
| 10 × NEBuffer r3.1 | 5 μL |
| BamHI | 1 μL |
| HindIII | 1 μL |
| Milli-Q water | up to 50 μL |
-
14.
Incubate the reaction mixture at 37°C for 1 h.
-
15.
Run both the digested PCR product and pSMT3 vector by a 1% agarose gel.
-
16.
Purify the gel slices using the QIAquick Gel Extraction Kit (QIAGEN) according to the manufacturer’s protocol.
-
17.
Prepare a 10 μL reaction mix by combining the following reagents.
| Reagent | Amount |
|---|---|
| T4 DNA Ligase Buffer (10×) (NEB) | 1 μL |
| digested pSMT3 vector | 20 ng |
| digested PCR product | 100 ng |
| T4 DNA Ligase (NEB) | 1 μL |
| Milli-Q water | up to 10 μL |
Note: The optimal insert to vector DNA ratio, is usually between 2:1 and 10:1. Higher concentrations of DNA reaction components will result in a higher rate of reaction.
-
18.
Mix thoroughly and incubate at 16°C for 12–16 h.
-
19.
When the incubation is completed, place the ligation product on ice or store at –20°C.
-
20.Transform the ligation product into DH5α competent cells (Thermo Fisher Scientific).
-
a.Thaw competent cells on ice.
-
b.Add 1 μL of ligation product into DH5α cells.
-
c.Carefully flick the tube 4–5 times to mix cells and DNA.
-
d.Place the tube on ice for 30 min.
-
e.Heat shock at 42°C for 45 s.
-
f.Immediately put the competent cells back on ice for 2 min.
-
g.Add 900 μL LB media and shake at 145 rpm in New Brunswick Innova 44 shaker (Eppendorf) at 37°C for 1 h.
-
h.Spread 100 μL of cell suspension on a LB agar plate containing 50 μg/mL kanamycin by using a commercial sterile spreader.
-
i.Invert plates (agar side up) and incubate at 37°C for 12–16 h.
-
a.
-
21.
Pick a single colony with a sterile pipette tip or toothpick from the LB agar plate and inoculate into 3 mL LB media.
-
22.
Incubate at 37°C in New Brunswick Innova 44 shaker (Eppendorf) with shaking at 250 rpm for 14–18 h.
-
23.
Extract the plasmids from the cells by a QIAprep Spin Miniprep Kit (QIAGEN) according to the manufacturer’s protocol.
-
24.
Sent a small aliquot of the plasmid for DNA sequencing.
-
25.Generate the DDX21 (188–563, del 401–405) construct by mutagenesis using QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies).
-
a.Design the mutagenesis primers using QuikChange Primer Design server by Agilent (https://www.agilent.com/store/primerDesignProgram.jsp).
-
b.Prepare the reaction mixture as below.PCR reaction master mix
Reagent Amount DNA plasmid template (5–50 ng/μL) 1 μL PfuUltra HF DNA polymerase (2.5 U/μL) 1 μL DDX21 del-loop primer – forward (100 ng/μL) 1.25 μL DDX21 del-loop primer – reverse (100 ng/μL) 1.25 μL DNTP mix 1 μL 10 × reaction buffer 5 μL ddH2O 39.5 μL -
c.Perform the PCR reaction in a T100 Thermal Cycler (Bio-Rad) using the cycling parameters outlined in below table.PCR cycling conditions
Steps Temperature Time Cycles Initial Denaturation 95°C 30 s 1 Denaturation 95°C 30 s 18 cycles Annealing 55°C 1 min Extension 68°C 6 min Final Extension 68°C 10 min 1 Hold 4°C forever -
d.Dpn I digestion of the amplification products.
-
i.Add 1 μL of the Dpn I restriction enzyme (10 U/μL) directly to the amplification reaction.
-
ii.Gently and thoroughly mix the reaction mixture by pipetting the solution up and down several times.
-
iii.Spin down the reaction mixtures in a microcentrifuge for 1 min.
-
iv.Immediately incubate each reaction at 37°C for 1 h to digest the parental template.
-
i.
-
e.Transformation of XL1-Blue supercompetent Cells.
-
i.Gently thaw the XL1-Blue supercompetent cells on ice.
-
ii.Transfer 1 μL of the Dpn I-treated DNA to the XL1-Blue supercompetent cells.
-
iii.Swirl the transformation reactions gently to mix and incubate the reactions on ice for 30 min.
-
iv.Heat pulse the transformation reactions for 45 s at 42°C and then place the reactions on ice for 2 min.
-
v.Add 1 mL of LB broth and incubate the transformation reactions at 37°C in New Brunswick Innova 44 shaker (Eppendorf) for 1 h with shaking at 225–250 rpm.
-
vi.Plate 100 μL of the transformation reaction on agar plates containing 50 μg/mL kanamycin.
-
vii.Incubate the transformation plates at 37°C for 16 h.
-
i.
-
a.
-
26.
Pick a single colony with a sterile pipette tip or toothpick from the LB agar plate.
-
27.
Inoculate into 3 mL LB-Ampicillin broth.
-
28.
Incubate at 37°C in New Brunswick Innova 44 shaker (Eppendorf) with shaking at 250 rpm for 14–16 h.
-
29.
Extract the plasmids from the cells by a QIAprep Spin Miniprep Kit (QIAGEN: cat# 27106X4) according to the manufacturer’s protocol.
-
30.
Send a small aliquot of the plasmid for DNA sequencing.
Prepare the antibiotics and IPTG stocks
-
31.
Prepare kanamycin, chloramphenicol, and isopropyl-beta-d-thiogalactopyranoside (IPTG) stock solutions for E. coli BL21-CodonPlus (DE3) RIL (Agilent Technologies) culture.
Note: BL21-CodonPlus (DE3) RIL is chloramphenicol-resistant and engineered to contain extra copies of genes that encode the tRNAs that most frequently limit the translation of heterologous proteins in E. coli. It is ideal for difficult protein expression, especially when codon bias is a problem. For more details please see manufacturer’s instruction manual (https://www.agilent.com/cs/library/usermanuals/public/230240.pdf).
-
32.
Weigh out 2.25 g of kanamycin and dissolve it in 45 mL MilliQ ultrapure water. Stock concentration is 50 mg/mL.
-
33.
Sterilize with a 0.22 μm syringe filter and store at −20°C.
-
34.
Weigh out 340 mg of chloramphenicol.
-
35.
Transfer it to a 15 mL screw-capped tube.
-
36.
Add 70% ethanol and mix until all chloramphenicol dissolves completely in a final volume of 10 mL. Stock concentration is 34 mg/mL.
-
37.
Store chloramphenicol stock at −20°C.
-
38.
Dissolve 2.38 g of IPTG in 10 mL MilliQ ultrapure water.
-
39.
Sterilize with a 0.22 μm syringe filter. Stock concentration is 1 M.
-
40.
Store IPTG in 1 mL aliquots at −20°C.
Prepare LB agar plates
-
41.
Weigh out 10 g LB agar, Miller (Thermo Fisher Scientific) into a 250 mL Glass Erlenmeyer flask (Thermo Fisher Scientific).
-
42.
Add Milli-Q water up to 200 mL.
-
43.
Swirl to mix and cover the top of the flask with aluminum foil and label with autoclave tape.
-
44.
Autoclave at 121°C for 15 min.
-
45.
Allow the agar solution to cool to 50°C.
-
46.
Add 200 μL of kanamycin stock and 200 μL of chloramphenicol stock respectively into the LB agar and swirl to mix.
CRITICAL: Antibiotics must be added after the LB agar cooling down as they degrade at high temperatures.
-
47.
Pour ∼20 mL of LB agar into each plate.
-
48.
Place the lids on the plates and allow them to cool for 30–60 min (until solidified).
-
49.
Label the bottom of plates with antibiotic and date. Seal the plates with parafilm and store at 4°C.
Note: Storage time will vary depending on antibiotic added, but plates are generally good for 1–2 months at 4°C.
Prepare LB broth
-
50.
Weigh out 25 g LB Broth, Miller (Thermo Fisher Scientific) per liter into a 2 L Glass Narrow-Mouth Erlenmeyer Flask (Thermo Fisher Scientific).
-
51.
Add MilliQ ultrapure water until the total volume reaches 1 L.
-
52.
Cover of the flask with aluminum foils.
-
53.
Place autoclave tapes on top of the foil.
-
54.
Autoclave the LB meida at 121°C for 20 min.
-
55.
Store the sterilized LB media in a 4°C fridge once the flask cools down.
Purification of ubiquitin-like-specific protease 1 (Ulp1)
-
56.
Transform 10 ng pFGET19_Ulp1 plasmid (Addgene) (Guerrero et al., 2015) that encodes the N-terminal His6 tagged Ulp1 into BL21-CodonPlus (DE3) RIL chemically competent cells (Agilent Technologies).
-
57.
Grow colonies on LB agar plates containing 50 μg/mL kanamycin and 34 μg/mL chloramphenicol.
-
58.
Inoculate a single colony into 50 mL Luria-Bertani (LB) media supplemented with 50 μg/mL kanamycin and 34 μg/mL chloramphenicol, in a 250 mL Glass Narrow-Mouth Erlenmeyer Flask (Thermo Fisher Scientific).
-
59.
Grow the cells in a New Brunswick Innova 44 shaker (Eppendorf) at 37°C with shaking at 250 rpm for 14–18 h.
-
60.
Dilute 10 mL of cell culture into 1 L of LB media in a 2 L Glass Narrow-Mouth Erlenmeyer Flask (Thermo Fisher Scientific).
-
61.
Grow cells in a New Brunswick Innova 44 shaker (Eppendorf) at 37°C with shaking at 250 rpm, until the OD600 reaches 0.6–0.8.
-
62.
Protein expression is induced by adding IPTG at a final concentration of 0.1 mM, along with agitation at 180 rpm and incubation at 30°C for 4 h.
-
63.
Centrifuge the cells in Sorvall™ RC 6 Plus Centrifuge (Thermo Scientific) using a F10-6 × 500y Fixed-Angle Rotor (Thermo Fisher Scientific) at 5,000 × g for 10 min at 4°C, and then discard the supernatant.
-
64.
Resuspend the cell pellets in lysis buffer A with a ratio of 10 mL buffer per gram (wet weight) cell pellet, by using an ULTRA-TURRAX T10 Basic Disperser (IKA).
-
65.
Resuspend the cells homogeneously with intervals of 30 s on and 10 s off on ice.
-
66.
Lyse the resuspended cells by using an APV Laboratory Homogenizers (SPX FLOW), under 800–1,500 bar pressure and disrupt the cells for 3–4 cycles.
Note: Keep the lysate at a low temperature with 4°C cold circulation water.
Alternatives: Sonication could be used to lyse the E.coli cells. For details also refer to Liu et al. (2020).
-
67.
Centrifuge the cell lysate using a SS-34 Fixed-Angle Rotor (Thermo Fisher Scientific) in Sorvall™ RC 6 Plus Centrifuge (Thermo Scientific) at 15,000 × g for 30 min.
-
68.
For purification from 1 L culture, incubate the supernatant with 1 mL Ni-NTA resin (QIAGEN) in a 50 mL Falcon tube and rocking on a Tube Roller Mixer (AliExpress) at 4°C for 1 h.
Note: The capacity of Ni-NTA resin (QIAGEN) is up to 50 mg/mL and 1 mL of this resin is enough for 1 L purification for Ulp1.
-
69.
Wash the Ni-NTA resin with 100 mL lysis buffer A containing 20 mM imidazole.
-
70.
Elute Ulp1 protein with 10 mL lysis buffer A containing 500 mM imidazole.
-
71.
Dialyze the eluate to 2 L lysis buffer A using a D-Tube Dialyzers (15 mL, MWCO 3.5 kDa; Millipore) for 16 h at 4°C.
-
72.
Concentrate Ulp1 protein to 5 mg/mL using a 10 kDa MWCO Amicon® Ultra-15 Centrifugal Filter Unit (Millipore).
-
73.
Centrifuge at 3,000 × g using an A-4-62 rotor (Eppendorf) in Eppendorf 5810R Centrifuge (Eppendorf).
-
74.
Aliquot and store the Ulp1 protein at −80°C.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| BL21-CodonPlus (DE3)-RIL Competent Cells | Agilent Technologies | Cat#230245 |
| MAX Efficiency DH5α Competent Cells | Thermo Fisher Scientific | Cat#18258012 |
| Chemicals, peptides, and recombinant proteins | ||
| Dithiothreitol | Gold Biotechnology | Cat#DTT10 |
| TCEP | Sigma | Cat#646547 |
| BSA | Sigma | Cat#A7030 |
| Kanamycin | GoldBio | Cat#K-120 |
| Chloramphenicol | GoldBio | Cat#C-105 |
| LB Broth, Miller | Thermo Fisher Scientific | Cat#BP1426-2 |
| LB Agar, Miller | Thermo Fisher Scientific | Cat#BP9724-500 |
| IPTG | GoldBio | Cat#I2481 |
| Imidazole | Sigma | Cat#I5513 |
| ADP | Sigma | Cat#A2754 |
| AMPPNP | Roche | Cat#10102547001 |
| NP-40 (alternative) | Millipore | Cat#492016 |
| Quick Start™ Bradford 1× Dye Reagent | Bio-Rad | Cat#500-0205 |
| Ni-NTA beads | QIAGEN | Cat#30210 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#11836170001 |
| Tris-HCI Buffer, pH 7.5 (1 M) | Thermo Fisher Scientific | Cat#15567027 |
| BamHI | NEB | Cat#R0136S |
| HindIII | NEB | Cat#R0104S |
| KOD Hot Start Master Mix | Merck | Cat#71842-3 |
| Ulp1 protease | This paper | N/A |
| Critical commercial assays | ||
| JCSG + suite kit | NeXtal | Cat#130920 |
| PEG ion kit | Hampton | Cat#HR2-139 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent Technologies | Cat#200524 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat#28706X4 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat#27106X4 |
| Deposited data | ||
| Crystal structure of DDX21-AMPPNP-ssRNA | This paper | PDB: 6L5N |
| Crystal structure of DDX21-apo | This paper | PDB: 6L5L |
| Crystal structure of DDX21-ADP | This paper | PDB: 6L5O |
| Crystal structure of DDX21-AMP | Chen et al. (2020) | PDB: 6L5M |
| Oligonucleotides | ||
| DDX21 WT primer – forward 5′-gagaaca gattggtggatccttctctaattttcccatatctg-3′ |
This paper | N/A |
| DDX21 WT primer – reverse 5′-ggccgcaag cttgtcgacggagctcttatattcgtttgaacttaattc-3′ |
This paper | N/A |
| DDX21 del-loop primer – forward 5′-tgaacag gtggacctgattaaaacggcaataactgtgg-3′ |
This paper | N/A |
| DDX21 del-loop primer – reverse 5′-ccacagttattgccgttttaatcaggtccacctgttca-3′ |
This paper | N/A |
| U10 RNA: 5′-UUUUUUUUUU-3′ | This paper | N/A |
| U15 RNA: 5′-UUUUUUUUUUUUUUU-3′ | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pET28a(+)_DDX21 (188–563, WT) | This paper | N/A |
| Plasmid: pET28a(+)_DDX21 (188–563, del 401–405) | This paper | N/A |
| pFGET19_Ulp1 | Guerrero et al. (2015) | Addgene Plasmid #64697 |
| pSMT3 vector | Gift from Dr. Chris Lima, Sloan Kettering Institute | N/A |
| Software and algorithms | ||
| XDS | Kabsch (2010) | https://xds.mr.mpg.de/ |
| HKL2000 | Otwinowski and Minor (1997) | https://www.hkl-xray.com/download-instructions-hkl-2000 |
| PHASER | McCoy et al. (2007) | https://www.phaser.io |
| REFMAC5 | Murshudov et al. (2011) | https://www.ccp4.ac.uk/html/refmac5.html |
| Phenix | Adams et al., 2010 | http://www.phenix-online.org/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Other | ||
| Amicon® Ultra-15 Centrifugal Filter Unit (10 KDa MWCO) | Millipore | Cat#UFC901096 |
| Amicon® Ultra-15 Centrifugal Filter Unit (30 KDa MWCO) | Millipore | Cat#UFC903096 |
| Nanodrop 2000 | Thermo Scientific | ND-2000 |
| Novex™ 4%–20% Tris-Glycine Mini Gels | Thermo Fisher Scientific | Cat#EC6021BOX |
| Econo-Column Chromatography Column | Bio-Rad | Cat#7374151 |
| D-Tube Dialyzers (15 mL, MWCO 3.5 kDa) | Millipore | Cat#71742 |
| 250 mL Glass Narrow-Mouth Erlenmeyer Flask | Thermo Fisher Scientific | Cat#FB500250 |
| 2 L Glass Narrow-Mouth Erlenmeyer Flask | Thermo Fisher Scientific | Cat#FB5006000 |
| New Brunswick Innova 44 shaker | Eppendorf | Cat#M1282-0004 |
| APV Laboratory Homogenizers | SPX FLOW | APV-1000 |
| Tube Roller Mixer | AliExpress | Cat#MX-T6-S |
| Magnetic stirrer | Corning | PC-410 |
| AKTA pure 25 M protein purification system | Cytiva | Cat. #29018226 |
| Monolith NT.Automated Premium Capillary Chips | NanoTemper Technologies | Cat#MO-AK005 |
| Monolith NT.Automated | NanoTemper Technologies | https://nanotempertech.com/zh_cn/monolith-automated/ |
| Superdex 200 10/300 GL | Cytiva | Cat#17517501 |
| HiLoad 16/600 Superdex 200 prep grade | Cytiva | Cat#28989335 |
| T100 Thermal Cycler | Bio-Rad | Cat#1861096 |
| Eppendorf 5810R Centrifuge | Eppendorf | Cat#5811000065 |
| Eppendorf 5430R Centrifuge | Eppendorf | Cat#022620509 |
| Sorvall™ RC 6 Plus Centrifuge | Thermo Scientific | Cat#46910 |
| SS-34 Fixed-Angle Rotor | Thermo Fisher Scientific | Cat#28020TS |
| F10-6 × 500y Fixed-Angle Rotor | Thermo Fisher Scientific | Cat#78510TS |
| A-4-62 Rotor | Eppendorf | Cat#022638203 |
| FA-45-30-11 Rotor | Eppendorf | Cat#022638009 |
Materials and equipment
Lysis buffer A
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH7.5 (1 M) | 50 mM | 50 mL |
| NaCl (5 M) | 500 mM | 100 mL |
| DTT (1 M) | 0.2 mM | 0.2 mL |
| ddH2O | N/A | 849.8 mL |
| Total | N/A | 1,000 mL |
Store at 4°C within 14 days.
Note: Right before purification, add cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche) into the lysis buffer A.
Lysis buffer B
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH7.5 (1 M) | 20 mM | 20 mL |
| NaCl (5 M) | 1 M | 200 mL |
| Glycerol (100%) | 5% | 50 mL |
| DTT (1 M) | 0.2 mM | 0.2 mL |
| ddH2O | N/A | 729.8 mL |
| Total | N/A | 1,000 mL |
Store at 4°C within 14 days.
Dialysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH7.5 (1 M) | 20 mM | 20 mL |
| NaCl (5 M) | 0.5 M | 100 mL |
| Glycerol (100%) | 5% | 50 mL |
| DTT (1 M) | 0.2 mM | 0.2 mL |
| ddH2O | N/A | 829.8 mL |
| Total | N/A | 1,000 mL |
Store at 4°C within 14 days.
Imidazole stock solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Imidazole | 5 M | 340.4 g |
| ddH2O | N/A | Up to 1,000 mL |
| Total | N/A | 1,000 mL |
Adjust pH to 7.0 and store at 25°C within 1 year.
Note: Mix proper volume of imidazole stock solution with lysis buffer to make a desirable concentration of imidazole solutions, for preparing Ni-NTA wash buffer or elution buffer.
Gel filtration buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH7.5 (1 M) | 20 mM | 20 mL |
| NaCl (5 M) | 100 mM | 100 mL |
| TCEP (0.5 M) | 1 mM | 2 mL |
| ddH2O | N/A | 878 mL |
| Total | N/A | 1,000 mL |
Store at 4°C within 14 days.
MST binding buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH7.5 (1 M) | 20 mM | 20 mL |
| KCl | 50 mM | 0.373 g |
| MgCl2 | 2 mM | 0.019 g |
| NP-40 | 0.01% | 10 μL |
| TCEP (0.5 M) | 1 mM | 2 mL |
| BSA | 0.05 mg/mL | 0.005 g |
| AMPPNP | 1 mM | 0.051 g |
| ddH2O | N/A | Up to 100 mL |
| Total | N/A | 100 mL |
Store at 4°C within 14 days.
Size exclusion chromatography co-elution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH7.5 (1 M) | 20 mM | 20 mL |
| NaCl (5 M) | 100 mM | 100 mL |
| TCEP (0.5 M) | 1 mM | 2 mL |
| ddH2O | N/A | 878 mL |
| Total | N/A | 1,000 mL |
Store at 4°C within 14 days.
Note: Add AMPPNP to DDX21-RNA mixture to reach a final concentration of 50 μM, and incubate for 1 h before loading to gel-filtration column.
Step-by-step method details
Transformation and expression of DDX21
Transform the expression plasmids into the BL21-CodonPlus (DE3) RIL cells and express DDX21 protein with IPTG. Collect the cell pellets for future purification.
The plasmids and corresponding antibiotic resistance are summarized as bellow:
| Plasmids | Antibiotic resistance |
|---|---|
| pET28a(+) DDX21 (188–863, WT) | Kanamycin |
| pET28a(+) DDX21 (188–863, del 401–405) | Kanamycin |
Note: BL21-CodonPlus (DE3) RIL Competent Cells (Agilent Technologies) are chloramphenicol-resistant.
-
1.Resuspension of plasmid.
-
a.Spin lyophilized commercial custom plasmid (from GenScript) down at 10,000 × g for 2 min in an Eppendorf 5430R Centrifuge with an FA-45-30-11 rotor (Eppendorf).
-
b.Add 40 μL MilliQ ultrapure water directly to the bottom of the tube containing 100 μg plasmid.
-
c.Vortex it for 10 s to make a DNA plasmid stock of 40 ng/μL final concentration.
-
d.Spin it down at 10,000 × g for 2 min in Eppendorf 5430R Centrifuge with a FA-45-30-11 rotor (Eppendorf).
-
e.Store the plasmid at −80°C until use.
-
a.
-
2.
Take BL21 (DE3) RIL competent cells (Agilent Technologies) cells from −80°C and place them on ice.
-
3.
Thaw both the competent cells and the plasmid on ice for 30 min.
-
4.
Add 1 μL of the plasmid into 50 μL of competent cells.
-
5.
Mix the plasmid gently through the bacterial solution by pipetting.
-
6.
Incubate the cells for 30 min on ice.
-
7.
Perform heat shock at 42°C for 90 s.
-
8.
Put the cells back on ice for 2 min.
-
9.
Add 500 μL of pre-warmed (37°C) LB medium into the Eppendorf tube that contains transformed competent cells.
-
10.
Incubate the culture tube in an incubator at 37°C for 60 min.
-
11.
Spread 100 μL of cell suspension using a commercial sterile spreader into kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL)-containing agar plate that is preheated to 37°C.
-
12.
Put the plate upside down in a 37°C incubator and incubate for 18 h.
-
13.
Inoculate a single colony from the plate into 50 mL LB media supplemented with 34 μg/mL chloramphenicol and 50 μg/mL kanamycin.
-
14.
Culture the cells in New Brunswick Innova 44 shaker (Eppendorf) at 37°C and 250 rpm for around 16 h.
-
15.
Inoculate the overnight culture 1:100 (v/v) in 1 L LB media supplemented with 50 μg/mL kanamycin, in a 2 L Erlenmeyer flask (Thermo Fisher Scientific).
-
16.
Grow the cells in New Brunswick Innova 44 shaker (Eppendorf) with an agitation speed of 250 rpm at 37°C until the optical density of OD600 reaches around 0.8.
-
17.
Switch the shaker temperature to 16°C and the speed to 180 rpm, while cooling down the cell culture in a 4°C refrigerator for 15 min.
-
18.
Put the cell culture back to the shaker, and induce protein expression by adding IPTG at a final concentration of 0.1 mM, along with shaking at 180 rpm at 16°C for 20 h.
-
19.
Harvest cells by centrifugation using a F10-6 × 500y Fixed-Angle Rotor (Thermo Fisher Scientific) in Sorvall™ RC 6 Plus Centrifuge (Thermo Scientific) at 5,000 × g for 10 min at 4°C, and then discard the supernatant.
Pause point: Pellets can be stored at −80°C for up to 12 months.
Purification of DDX21 with a high salt buffer
Purify nucleic acid-free DDX21 protein using a high-salt buffer during both the loading and washing steps (Figure 2). High salt interrupts the electrostatic interactions between protein and nucleic acid. It is easy, cheap, fast, and sufficient to remove bound nucleic acid in general, and is the only way to purify nucleic acid-binding proteins in one step.
Note: Both DDX21 constructs (WT and del-loop) were purified using the same procedure.
-
20.
Resuspend the cell pellets with lysis buffer B with a ratio of 10–20 mL buffer per gram (wet weight) cell pellet, by using an ULTRA-TURRAX T10 Basic Disperser (IKA), and resuspend cell homogeneously with intervals of 30 s on and 30 s off on ice.
Alternatives: HL-SAN nonspecific endonuclease (ArcticZymes) or equal product of nuclease which could endure high salt conditions, could be added into the lysis buffer B to digest nucleic acid.
Alternatives: Polyethyleneimine (PEI) can be used to precipitate nucleic acids, but itmay cause protein loss from co-precipitation, and must be handled with care.
Note: Please keep the cell on ice for resuspension, to avoid protein aggregation or precipitation caused by heating.
Note: 1 tablet of EDTA-free protease inhibitor cocktail (Roche) per 100 mL buffer should be added to inhibit the protease activity.
-
21.
Lyse the resuspended cells by using APV Laboratory Homogenizers (SPX FLOW), under 800–1,500 bar pressure and disrupt the cells for 3–4 cycles.
Note: Keep the lysate at a low temperature with 4°C cold circulation water.
Alternatives: Sonication can be used to lyse the cell.
-
22.
Centrifuge the cell lysate at 15,000 × g using a SS-34 Fixed-Angle Rotor (Thermo Fisher Scientific) for 30 min to remove cell debris and insoluble fractions.
-
23.
Equilibrate 10 mL Ni-NTA resin (QIAGEN) with lysis buffer B.
-
24.
Incubate the supernatant with the pre-equilibrated Ni-NTA resin in 50 mL Falcon tubes, rocking on a Tube Roller Mixer (AliExpress) at 4°C overnight.
Alternatives: A pre-packed Ni-affinity column can be used along with an AKTA system. But overnight batch binding with Ni-NTA beads significantly increased the yield of affinity purification in our lab.
-
25.
Apply the suspension with the protein-bound Ni-NTA resin to an Econo-Column Chromatography Column (Bio-Rad).
-
26.
To remove impurities and nucleic acid, wash the resin extensively with at least 500 mL lysis buffer B that contains 20 mM imidazole.
Note: Quick Start™ Bradford 1× Dye Reagent (Bio-Rad) can be used to roughly evaluate the protein concentration of the wash or elution fractions from a gravity column. Add 10 μL of the fractions to 100 μL of Bradford 1× Dye Reagent and observe the depth of the blue color, to roughly estimate the protein concentration.
-
27.
Elute his-sumo tagged DDX21 protein with lysis buffer B containing 500 mM imidazole.
-
28.
Add the pre-prepared Ulp1 enzyme (5 mg/mL, 200 μL) to the elution fractions to cleave between the His-sumo tag and DDX21.
Note: In general Ulp1 could efficiently cleave sumo tagged protein at a ratio between 1: 500 and 1: 1,000 (w/w) for a over-night cleavage at 4°C, with a proper buffer condition. Since high concentration of salt and imidazole will reduce the cleavage efficiency of Ulp1, we recommend to add an excess amount of Ulp1 to ensure complete cleavage. Here 200 μL of 5 mg/mL (1 mg total) Ulp1 enzyme could efficiently cleave at least 100 mg of DDX21 in this study.
-
29.
Transfer the reaction mixture to several D-Tube Dialyzers (15 mL, MWCO 3.5 kDa; Millipore).
-
30.
Immerse the D-Tube Dialyzers into 4 L dialysis buffer, stirring with a magnetic stirrer (Corning) and incubate for 16 h at 4°C to cleave the His6-SUMO tag.
Alternatives: Other high-performance dialysis products, such as Slide-A-Lyzer Dialysis Cassettes (Thermo Scientific), could also be used for dialysis to remove imidazole at this step.
-
31.
Analyze a 15 μL sample of the Ulp1 cleavage reaction by SDS polyacrylamide gel electrophoresis (SDS-PAGE) using a Novex™ 4%–20% Tris-Glycine Mini Gel (Thermo Fisher Scientific). Troubleshooting 1.
-
32.
Once the desirable extent of the cleavage reaction is achieved (90%–100%), load the reaction mixture back onto an Econo-Column Chromatography Column (Bio-rad) containing 10 mL Ni-NTA resin, which is pre-equilibrated with lysis buffer B containing 20 mM imidazole.
Note: Calculate the final concentration of imidazole in the reaction mixture after dialysis, and add imidazole to reach a final concentration of 20 mM in the reaction mixture, before loading onto the “substractive” Ni-NTA column.
-
33.
Collect the flow-through.
-
34.
Wash the Ni-NTA column with lysis buffer B containing 20 mM imidazole.
-
35.
Analyze a 15 μL sample of each purification step by SDS Polyacrylamide gel electrophoresis (SDS-PAGE) using a Novex™ 4%–20% Tris-Glycine Mini Gel (Thermo Fisher Scientific). Troubleshooting 2.
-
36.
Combine the Ni-NTA column flow-through and wash fractions that contain the tag-free DDX21.
-
37.
Add DTT to a 5 mM final concentration.
-
38.
Concentrate the protein to be with 3–5 mL final volume, using a 30 kDa MWCO Amicon Ultra-15 Centrifugal Filter Unit (Millipore) and centrifuge at 3,000 × g and 4°C in an Eppendorf 5810 R Centrifuge with an A-4-62 rotor (Eppendorf).
Note: For DDX21(188–563, WT) and DDX21(188–563, del-loop) constructs, the solubility limit of the two proteins is around 40 mg/mL.
-
39.
Transfer the concentrated protein into several 1.5 mL centrifuge tubes.
-
40.
Centrifuge at 16,000 × g and 4°C in an Eppendorf 5430R Centrifuge with an FA-45-30-11 rotor (Eppendorf) for 5 min.
-
41.
Collect the supernatant by pipetting.
-
42.
Inject the concentrated protein onto a HiLoad 16/600 Superdex ® 200 prep grade (Cytiva) pre-equilibrated with gel-filtration buffer.
-
43.
Set the flow rate at 0.5 mL/min. DDX21 protein elutes at ∼90 mL on this column. Troubleshooting 3.
-
44.
Analyze a 15 μL sample of each gel-filtration fraction by SDS polyacrylamide gel electrophoresis (SDS-PAGE) using a Novex™ 4%–20% Tris-Glycine Mini Gel (Thermo Fisher Scientific).
-
45.
Pool protein-containing fractions, and concentrate the protein to 30 mg/mL using a 30 kDa MWCO Amicon Ultra-15 Centrifugal Filter Unit (Millipore) at 3,000 × g in an Eppendorf 5810R Centrifuge with an A-4-62 rotor (Eppendorf).
Note: Use Nanodrop (Thermo Fisher Scientific) to determine the OD280 value, and calculate the protein concentration using the Beer-Lambert law equation with molar extinction coefficient, which could be obtained from online tools such as ExPASy Protparam tool (https://web.expasy.org/protparam/). For the two DDX21 constructs used here, 1 A[280] is correlated to 1.7 mg/mL for WT construct and 1.67 for del-loop construct. For example, for WT construct when the absorbance value of the protein solution at 280 nm is 1, then its concentration is 1.7 mg/mL.
Alternatives: The Quick Start™ Bradford 1× Dye Reagent (Bio-Rad) could also be used to quantify the protein concentration. Use the BSA standard to make a standard curve. Then measure and calculate the concentration of your protein with the standard curve.
Note: Also use Nanodrop (Thermo Fisher Scientific) to determine the OD260/OD280 ratio, a ratio less than 0.60 is a good indication of pure protein with minimal nucleic acid contamination.
-
46.
Transfer the concentrated protein into several 1.5 mL centrifuge tubes.
-
47.
Centrifuge at 16,000 × g and 4°C in an Eppendorf 5430R Centrifuge with an FA-45-30-11 rotor (Eppendorf) for 10 min.
-
48.
Collect the supernatant by pipetting.
-
49.
Aliquot the protein into 1.5 mL centrifuge tubes (50 μL per tube).
-
50.
Snap-freeze in liquid nitrogen, and store at −80°C for further use.
Figure 2.

Diagram of the procedure for expression and purification of DDX21
(A) Preparation of cells expressing DDX21.
(B) Affinity purification and removal of the His-SUMO tag from DDX21.
(C) Further purification by size exclusion chromatography.
MST binding assay
Measure the binding affinity of the interaction between DDX21 and RNA using the microscale thermophoresis (MST), which is a rapid and precise method to quantify protein-nucleic acid interactions in solution (Mueller et al., 2017), especially for RNA that tends to degrade (Figure 3).
-
51.Assay setup pretests: before you start, make sure that you are using the optimal concentration of the labeled molecule, the correct capillary type and a buffer composition in which your sample is homogeneous.Note: A proper buffer recipe should be used to ensure the binding between the RNA of interest and the RNA helicase protein.Note: Before you start, compare the Monolith systems to find the suitable one. Monolith NT.115 gives the most flexibility when it comes to labeling strategy with multiple fluorescent channels, while Monolith NT. Labelfree utilizes intrinsic tryptophan fluorescence and may have less sensitivity. Furthermore, Monolith NT. Automated can help you with your screening projects with higher throughput, detection versatility, and automation option (used in this study).Note: For more details on the MST assay to quantify protein-nucleic acid interaction, also refer to Huang and Li (2022).
-
a.Fluorescence check.
-
i.Fill the labeled RNA in a standard capillary and place the capillary on the tray.
-
ii.Insert the tray in the instrument and close the door.
-
iii.Select the “LED Color” fitting the dye that you are using in the box “LED Settings” (Blue color for Fam fluorescent dye).
-
iv.Select the “LED Power” as 50% in the box of “Capillary Scanning”.
-
v.Press the “Start Cap Scan” button and the instrument will automatically scan the sample tray by measuring the fluorescence.
-
vi.The “Capillary Scan” will start from the last capillary and move to the first position.Note: Make sure the reading is between 200–1,500 fluorescence counts. To achieve this, sample concentration can be adjusted, or the LED power can be varied between 15% and 95%.
-
vii.Choose a concentration of the labeled RNA to achieve optimal fluorescence counts. Typically, 5–100 nM labeled RNA is used.
-
i.
-
b.Capillary Check.
-
i.Prepare the labeled RNA at the concentration you want to use in the assay.
-
ii.Fill four standard treated capillaries, four hydrophilic capillaries, and four hydrophobic capillaries with the RNA.
-
iii.Put these 12 capillaries on the tray.
-
iv.Insert them into the instrument and start a capillary scan.
-
v.Pick the capillary that gives a symmetrical fluorescence peak.Note: Avoid using the capillary that gives the fluorescence peak having shoulders or even clear double peak.Note: If the fluorescence peak has shoulders or even a clear double peak, it means the labeled sample was slightly or firmly adsorbed to the surface of the capillary. In this case, try another type of capillary. In the unlikely case that the sample is sticking to all types of capillaries, you can also try different buffers (e.g., containing detergent, BSA, casein, or other additives). Additionally, try to improve buffer conditions by adjusting pH and ionic strength.
-
i.
-
c.Sample check.
-
i.Dilute the labeled RNA in 100 μL of the the MST binding buffer.
-
ii.Fill into the suitable type of capillary determined.
-
iii.Start a capillary scan with the predetermined setting, and measure the labeled RNA sample at 40% MST power with an on-time of 30 s.
-
iv.Load and analyze the results in the MO. Affinity Analysis software.Note: If there is no bumpiness of the MST curves and a decimal appears on the left axis of the plot, the quality of the sample is good (Huang and Li, 2022).
-
i.
-
a.
-
52.MST binding test: Setting up the MST experiment with a fixed concentration of the fluorescent-labeled RNA, titrated with the unlabeled DDX21 protein.Note: We used polyU in the MST binding test because it represents ssRNA and is the RNA we used for co-crystallization with DDX21. Other RNA sequences can be tested using MST with similar protocol.Note: Binding of RNA helicase with RNA of interest may need additional co-factors (i.e., Mg2+ ion and ATP analogs), and a suitable pH value of the buffer is important to retain the stability and activity of your proteins. In addition, high salt (i.e., 500 mM NaCl) will tend to disrupt the binding between them, so in general please avoid using high salt buffer condition in this MST binding test.Note: The del-loop construct still binds to polyU RNA with comparable binding affinity.
-
a.Fill 20 μL of the highest concentration of the unlabeled DDX21 protein in the first well of a 384-well low attachment microplate (Corning).Alternatives: Instead of the microplate, micro reaction tubes can be used.CRITICAL: It is crucial to ensure that the correct protein amount is pipetted because there is no possibility of normalizing protein concentrations.
-
b.Fill 10 μL of the MST binding buffer in the wells 2–12.
-
c.Transfer 10 μL solution from well 1 to well 2.
-
d.Mix thoroughly by pipetting up and down for several times without making any bubbles.
-
e.Repeat to get a serial dilution.
-
f.Finally, remove 10 μL from well 12 after mixing.Note: Protein samples were serially diluted to obtain a concentration range spanning from ∼0.1 × Kd to ∼10 × Kd, where possible. In this study we do the two-fold serial dilution of DDX21 starting from 340 μM.
-
g.Prepare a stock of the fluorescent-labeled RNA at double the concentration determined in the MST binding buffer.
-
h.Mix 10 μL of 100 nM RNA with 10 μL of the titrated DDX21 protein and so the final concentration of RNA in the mixture is 50 nM.
-
i.Mix well by pipetting up and down several times without making any bubbles.
-
j.Incubate at 25°C in the dark for 30 min before loading them into the capillaries.
-
k.To fill a capillary, dip Monolith NT. Automated Premium Capillary Chips (NanoTemper Technologies) directly into the reaction samples from the 384-well plate.Note: All steps, including the work with capillaries, should be performed with powder-free gloves to prevent impurities and adverse effects on the glass surface, because the powder used in laboratory gloves can fluoresce and scatter light and hence, impair the measurement. In addition, touch only the ends but not the middle part of the capillaries where the observation field is located (Zillner et al., 2012).
-
l.Start MST analysis. Measure at 3 different MST powers starting from the lowest MST power (e.g., 20%, 40%, 80%), and compare the results.
-
m.Choose the power that yields a good signal to noise ratio as low as possible (Huang and Li, 2022).
-
a.
-
53.Perform data interpretation.
-
a.Open MO.Affinity Analysis software.
-
b.Create a new analysis and load raw data.
-
c.Click Initial Fluorescence button in the View Panel to check.Note: Typically, the variation among the 12 capillaries should be lower than 10% between different capillaries. Troubleshooting 4.
-
d.The KD can be determined by selecting the KD fit model with the default setting in the Dose Response Fit window. Troubleshooting 5.Alternatives: Data can be exported and loaded to GraphPad Prism for binding analysis if preferred.
-
e.The Compare Results screen allows for a side-by-side comparison of MST runs.
-
f.The Export button in the Compare Results screen allows for exporting the dose-response chart as either a figure or raw data format.
-
g.The Generate Full Report button in the Compare Result screen allows for generating a full PDF report.
-
a.
Figure 3.

Characterization of the interaction between DDX21 and RNA by MST
RNAs are labeled with 5′-FAM and DDX21 is titrated. Data are presented as mean ± SD. U15 RNA has a higher affinity (lower KD value) than U10 RNA when binding to DDX21, and Both AMPPNP and MgCl2 co-factors are necessary for the interaction of DDX21 with RNA.
Size exclusion chromatography co-elution binding assay
Evaluate the interaction between DDX21 and RNA and the stability of the DDX21-RNA complex using a size exclusion chromatography (SEC) co-elution binding assay. A prepacked Superdex 200 10/300 GL gel filtration column (Cytiva) is used to separate the RNA-DDX21 complex from individual components (Figure 4).
-
54.
Dilute DDX21 protein to 1 mg/mL in a 1.5 mL centrifuge tube with size exclusion chromatography (SEC) co-elution buffer supplemented with 100 μM AMPPNP in a final volume of 200 μL.
-
55.
Dilute RNA for analysis in another 1.5 centrifuge tube with MST binding buffer to a final volume of another 200 μL.
Note: Make sure that the molar ratio of RNA: protein is around 1:1.
-
56.
Mix the diluted DDX21 protein and RNA and incubate the mixture at 4°C for 1 h.
-
57.
Centrifuge the DDX21-RNA mixture at 16,000 × g and 4°C in an Eppendorf 5430R Centrifuge with an FA-45-30-11 rotor (Eppendorf) for 5 min.
-
58.
Load the supernatant into Superdex 200 10/300 GL gel filtration column (Cytiva).
-
59.
Run sample with a flow rate of 0.5 mL/min.
-
60.
Set up control of individual components including DDX21 alone, RNA alone, and AMPPNP alone.
Note: Run 200 μg DDX21 protein, or 15 μM RNA of interest, or 100 μM AMPPNP respectivtly in the same gel filtration column with a flow rate of 0.5 mL/min. Pre-dilute the three samples respectively with size exclusion chromatography (SEC) co-elution buffer in a final volume of 400 μL, before loading into the column.
-
61.
Superimpose the gel-filtration profiles.
-
62.
Compare the retention volume of DDX21-RNA mixture with individual components.
Note: Left shift of the peak of the RNA-DDX21 mixture from gel-filtration indicates the binding of RNA and DDX21 protein. Troubleshooting 5.
Note: AMPPNP has very strong absorbance at OD260, and elutes with a retention volume of around 20 mL.
Alternatives: Collect each peak fraction and run SDS-PAGE gel to check, especially when the interaction is not very strong and the RNA-protein complex is not stable enough. A partial left shift of the gel filtration fractions in the SDS-PAGE gel still indicates the interaction between RNA and protein, but is not recommended to be used for co-crystallization.
Figure 4.

Characterization of the interaction between DDX21 and RNA by SEC co-elution
U15 RNA forms a stable complex with DDX21 while U10 RNA does not.
(A) Graphical representation of the principle of the gel filtration technique in which the separation of components is based on differences in their stokes radius (also called hydrodynamic radius), which is influenced by both molecular weight and shape (Hagel, 2011). SEC employs a gel matrix with pores of a specific size range and separates on the basis of whether or not a protein of a particular size or shape can enter the pore or is excluded from the pore.
(B) Superimposition of the gel-filtration profiles among DDX21, U15, AMPPNP, and the DDX21-U15-AMPPNP mixture. DDX21-U15-AMPPNP forms a stable complex in SEC (the peak marked with asterisk). U15 RNA: DDX21=1:1.3 (molar ratio).
(C) Superimposition of the gel-filtration profiles among DDX21, U10, AMPPNP, and the DDX21-U10-AMPPNP mixture. DDX21-AMPPNP and U10 could not form a stable complex in SEC.
Crystallization and structure determination of DDX21
Generate the crystals of DDX21-apo (with DDX21 aa 188–563, del 401–405), DDX21-ADP (with DDX21 aa 188–563, del 401–405) and DDX21-AMPPNP-U15 RNA (with DDX21 aa 188–563, WT). Collect X-ray data sets of these crystals and determine the structures (Figure 5).
-
63.
Perform the crystallization screening by using a crystal screening robot of Phoenix (ARI), with commercially available JCSG + suite kit (NeXtal) and pEG ion kit (Hampton).
-
64.
Store the INTELLI-PLATE 96 Well plates (Art Robbins Instruments) in Rockimager (Formulatrix) to record the formation of crystals.
-
65.
After initial crystal hits are obtained, optimize crystals by using a 24-well sitting drop plate with a grid search strategy, to evaluate appropriate pH, salt, and precipitant concentrations.
-
66.
Grow crystals by equilibrating a mixture containing 1 μL protein solution and 1 μL reservoir solution against 500 μL reservoir solution at 20°C using sitting drop vapor diffusion methods. Troubleshooting 6.
-
67.
Mix 20 mg/mL of DDX21 (188–563, del 401–405) with an equal volume of reservoir solution containing 20% PEG3350 and 0.2 M Na2HPO4) to grow DDX21-apo crystals at 20°C.
-
68.
Mix 20 mg/mL of DDX21 (188–563, del 401–405) protein supplemented with 5 mM ADP and 10 mM MgCl2 with an equal volume of reservoir solution containing 20% pEG3350, 0.2 M Magnesium acetate, to grow DDX21-ADP crystals at 20°C.
-
69.
Dilute purified DDX21 (188–563) to 6–8 mg/mL and incubate with a U15 RNA at a protein to RNA ratio of 1:1.1, in 20 mM Tris-HCl (pH 7.5) buffer containing 100 mM NaCl, 1 mM TCEP, 1.5 mM AMPPNP and 3 mM MgCl2.
-
70.
Then mix 1 μL of the DDX21-U15 RNA mixture sample with 1 μL of reservoir solution comprised of 7% v/v 2-Methyl-2,4-pentanediol (MPD) and 0.1 M Bicine, pH8.5, to grow DDX21-AMPPNP-U15 RNA crystals at 20°C.
Alternatives: Purifying the DDX21-RNA complex by SEC may help to obtain the complex with correct stoichiometric ratio, and remove excess individual components. But normally this step will cause protein loss and is unnecessary, especially when the correct stoichiometric ratio is pre-determined from SEC co-elution.
-
71.
Plate-shaped crystals of the dimensions of ∼ 100 × 50 × 5 mm3 for DDX21-ADP and DDX21-AMPPNP-U15 complex appear within 7–14 days.
-
72.
Plate-shape crystals of the dimensions of ∼ 60 × 20 × 5 mm3 for DDX21-apo are formed within 14 days.
-
73.
Loop the crystals using appropriate loops under the optical microscope.
-
74.
Cryoprotect the crystals with 30%–35% glycerol before freezing in liquid nitrogen. Troubleshooting 8.
-
75.
Collect X-ray diffraction data at the synchrotron (Shanghai Synchrotron Radiation Facility beamlines).
-
76.
Use the HKL2000 program (Otwinowski and Minor, 1997) and the XDS program package (Kabsch, 2010) for indexing, integrating, and scaling of crystallographic data.
-
77.
Perform crystal structure determinations by molecular replacement, using the pre-solved DDX21-AMP structure (PDB: 6L5M) (Chen et al., 2020) as a searching model with PHASER (McCoy et al., 2007), and one molecule was used to search for in the asymemetric unit.
-
78.
Perform structure refinement with REFMAC5 (Murshudov et al., 2011) and PHENIX (Adams et al., 2010).
-
79.
Perform iterative rounds of the model building using COOT (Emsley et al., 2010).
Note: The RNA was not part of the search during molecular replacement.
-
80.
Deposit data in Worldwide Protein Data Bank OneDep SYSTEM (https://deposit-2.wwpdb.org/deposition/).
Note: Timing of crystallization and structure determination may vary depending on the protein and X-ray data quality.
Figure 5.

Crystallization and structure determination of DDX21
Scale bar, 100 μm.
Expected outcomes
The difficulty of obtaining high purity and enough RNA helicase proteins is a tremendous challenge for understanding their physiological and pathological functions. Despite numerous efforts, to date, only a limited number of RNA helicase structures have been determined. We were able to overcome these difficulties and develop a general method to solve several sets of RNA helicase DDX21 structures in different unwinding states. Protocols for RNA helicase protein purification and crystallography are most often found accompanying the publication of specific targets. While this methodology was developed to study the helicase core domain of DDX21, with some variations it could be broadly used to purify and crystallize other RNA helicases in general. In our study, we expect to obtain 20–30 mg of pure DDX21 helicase core protein from 1 L bacterial culture. Subsequent interaction analysis between protein and RNA is performed with MST and SEC co-elution method, and then the stable protein-RNA complex is used to generate a co-crystal structure. Other apo structure or ADP-bound structure are obtained with rational engineering construct. The protocol could serve as a practical guide to direct crystallographers at the bench.
Quantification and statistical analysis
The crystal structures of DDX21 were determined using materials and software listed in the key resources table. Statistics generated from X-ray crystallography data processing, refinement, and structure validation is described in (Chen et al., 2020).
MST binding assays were measured in triplicates. The raw data is processed with MO. Affinity Analysis software as described in the method details. The resulting signals from thermophoresis were analyzed using the affinity analysis software (NanoTemper Technologies) and fitted to the Kd model with the equation:
where f(cligand) is the Fnorm value with a given protein concentration cligand; Unbound is the Fnorm signal of the target alone; Bound is the Fnorm signal of the complex; Kd is the dissociation constant or binding affinity; and ctarget is the final concentration of RNA in the assay.
Limitations
The key limitation of the protocol is due to the crystallization complexity; thus far, there are no set rules or 'magic bullets' that will guarantee the success of good crystals. Here, we provided a general protocol to obtain high purity and enough amount of RNA helicase DDX21 protein, and the growth of diffraction quality crystals. However, the process of protein crystallization is highly variable, and obtaining good crystals might be difficult in some cases. Screening as more crystallization kits as possible, seeding or micro matrix seeding (MMS), and additive screen are all useful to improve the quality of the crysatls. In addition, screening more constructs with carefully designed boundaries is essential to obtain the initial hits.
Our sequence analysis focuses on the linker loop length between the two RecA domains and is based on in silico analysis or prediction, thus lacking experimental guidance, such as the technology of hydrogen-deuterium exchange mass spectrometry (HDX MS). In addition, for those helicase families with big auxiliary domains other than the helicase core, it may need more sequence analysis or rational mutagenesis, such as for surface residues, flexible tails, or interdomain regions, to make the proteins more amenable to crystallization. In addition, an entire unwinding cycle by DExD/H box helicase also includes the pre-unwound state. However, a structure of DDX21 in complex with dsRNA for this state is not available yet. Therefore, the dsRNA binding condition and the design of a proper DDX21 construct are still awaited.
Another limitation is that the E. coli system is used in this protocol, while some other RNA helicases may need a baculovirus or mammalian expression system, which retains the necessary machinery to ensure post-translational modifications or correct folding, and may represent the more authentic conformational information and the interaction mode with RNA. However, we expect that our protocol is adaptable to switch to those protein expression systems, using the similar purification conditions and general considerations provided in this protocol.
Troubleshooting
Problem 1
Cleavage reaction is incomplete (step 31).
Potential solution
While the high salt condition in both lysis buffer and the dialysis buffer might be critical to retain the stability of DDX21 or other RNA helicases, it may reduce the cleavage efficiency of the Ulp1 protease. In addition, imidazole concentration higher than 150 mM can adversely affect the activity of the Ulp1 protease. So dialysis during Ulp1 cleavage step is necessary to increase the cleavage efficiency. If an incomplete cleavage reaction is still observed, try to add more Ulp1 protease and keep cleavage for a longer time, or try to cleave at room temperature for several hours. Another option is to do the cleavage step after dialysis. If your protein is stable at a low salt concentration (i.e., 150 mM NaCl), then change your dialysis buffer recipe to a lower salt concentration, since Ulp1 functions optimally in a reaction mixture containing 150 mM NaCl. However, conditions may be optimized by varying the NaCl concentration from 100 mM to 300 mM.
Problem 2
His-sumo tag is not removed completely (step 35).
Potential solution
Use enough volume of fresh Ni-NTA resin and incubate the cleavage product with Ni-NTA resin for a while (i.e., 0.5–1 h), then continue the following flow-through and wash steps. Or repeat the “substractive” Ni-NTA purification step, by pooling the flow-through and washing fractions and loading again back to the Ni-NTA column, until the his-sumo tag is completely removed. Ion exchange might be another option to further polish the protein purity along with His-sumo tag removal.
Problem 3
Abnormal OD260/OD280 value is observed. OD260/OD280 higher than 0.6 (i.e., OD260/OD280=1) indicates the nucleic acid bound with protein is not removed completely (step 43).
Potential solution
Make sure extensive washing with high salt buffer is done during the Ni-NTA column affinity chromatography step. In addition, the Heparin column could be used to remove bound nucleic acid. Or try to use gel filtration buffer with higher salt concentration (i.e., 1 M NaCl), which may help to further get rid of nucleic acid. Try to collect the peak fractions with an acceptable OD260/OD280 ratio and discard those with higher ratios.
Problem 4
The aberration of the fluorescence changes is more than 10% (step 53).
Potential solution
Since all samples should contain the same concentration of fluorescently labeled RNA, individual differences in intensity should be maximally 10%. In case the fluorescence signals of single capillaries are highly divergent, there are several points that can be considered. First, solutions were not adequately mixed, and care should be taken next time to merge all solutions efficiently. Second, the capillary might have touched the surface of the tube and thus, fluorescence that stuck onto the cup was sucked in. Third, it is also possible that the pipette is imprecise, and another one should be tried in next experiments. However, a gradual increase in the signal with higher protein amounts indicates already a binding event and is not due to technical errors. Also, if there are stronger random variations, pay more attention to pipetting and use low-absorbance capillary (the Premium Capillary or Premium Capillary Chips).
Problem 5
No binding or weak binding is observed (step 53 and step 62).
Potential solution
Binding of DDX21 or other RNA helicases to RNA may need the assistance of co-factor including ATP analogs and Mg2+ ion. Try screening with all kinds of ATP analogs, such as ADP, ADP-BeF3 or AMPPNP etc. And an optimal protein construct with correct boundary and region needs to be carefully designed and tried. Furthermore, if RNA binding is sequence specific, then screening different RNA sequences is also necessary. Specifically, the binding of RNA with protein is sensitive to salt concentration and generally, high salt will disrupt the interaction between them. In our case, salt concentration higher than 200 mM NaCl will definitely abolish the binding between RNA and DDX21.
Problem 6
Protein does not crystallize (step 66).
Potential solution
Check the protein quality such as the protein purity and the homogeneity of monodisperse state. When trying to screen crystals for protein-RNA complex, make sure the binding between RNA and protein is strong enough. Try adding all kinds of co-factors that may help crystal formation, i.e., ADP, ADP-BeF3, ADPCP or AMPPNP and MgCl2. Screen as many conditions as possible, and try altering temperature (i.e., try 4°C condition). Screen for different protein constructs with rational design or mutation on the sequence. In addition, cross-seeding with existing crystal seeds might be helpful. Micro-seeds derived from previously obtained crystals of DDX21 can be used for cross-seeding.
Problem 7
Heavy precipitation is observed when mixing RNA and protein (step 70).
Potential solution
DDX21 binds to RNA along with dramatic conformational change. Make sure to pre-dilute both of RNA and protein, and pre-equilibrate them to room temperature before mixing together, which is helpful in avoiding precipitation. If precipitation still happens, try reducing the concentration of protein or RNA, or try different molar ratio between them (evaluated firstly by MST or SEC co-elution). Increasing salt concentration or divalent cations may help to improve the solubility of DDX21 protein, but also increases the risk of disrupting the interaction between protein and RNA, and thus should be evaluated by MST or SEC co-elution in advance.
Problem 8
Crystals dissolve during looping (step 74).
Potential solution
DDX21-AMPPNP-U15 co-crystals is very sensitive to subtle environment change of the solution. Therefore, prepare the cryoprotectant with the reservoir buffer supplemented with not only 30%–35% glycerol, but also with 1.5 mM AMPPNP and 3 mM MgCl2, to avoid crystals destabilization during looping.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jixi Li (lijixi@fudan.edu.cn).
Materials availability
All reagents generated in this study will be made available on reasonable request.
Acknowledgments
The authors thank the staff in the Shanghai Synchrotron Radiation Facility beamlines BL17U1, BL18U1, BL19U1, and BL19U2 for helping with X-ray and SAXS data collection. This work was supported by grants from the National Natural Science Foundation of China (32161160323, 82071782), the National Key Research and Development Program of China (2021YFC2301500), and the Shanghai Committee of Science and Technology (20XD1400800) to J.L., and from the National Natural Science Foundation of China/Research Grant Council of Hong Kong Joint Research Scheme under Project N_HKUST635/21 to J.H.
Author contributions
Conceptualization, J.L. and J.H.; methodology and writing, Z.C. and J.L.; supervision, J.L.; data collection, Z.C.; funding acquisition, J.L. and J.H.
Declaration of interests
The authors declare no competing interests.
Data and code availability
Coordinates and structure factors have been deposited in the Protein Data Bank under ID codes 6L5N, 6L5O, and 6L5L.
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Li Z., Hu X., Xie F., Kuang S., Zhan B., Gao W., Chen X., Gao S., Li Y., et al. Structural basis of human helicase DDX21 in RNA binding, unwinding, and antiviral signal activation. Adv. Sci. 2020;7:2000532. doi: 10.1002/advs.202000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullam A., Schröder M. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta. 2013;1829:854–865. doi: 10.1016/j.bbagrm.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero F., Ciragan A., Iwaï H. Tandem SUMO fusion vectors for improving soluble protein expression and purification. Protein Expr. Purif. 2015;116:42–49. doi: 10.1016/j.pep.2015.08.019. [DOI] [PubMed] [Google Scholar]
- Hagel L. Gel filtration: size exclusion chromatography. Methods Biochem. Anal. 2011;54:51–91. doi: 10.1002/9780470939932.ch3. [DOI] [PubMed] [Google Scholar]
- Huang Y., Li Y. Microscale thermophoresis assay: a powerful method to quantify protein-nucleic acid and protein-protein interactions. Methods Mol. Biol. 2022;2400:21–31. doi: 10.1007/978-1-0716-1835-6_3. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Xue Z., Vann K.R., Shi X., Kutateladze T.G. Protocol for biochemical analysis and structure determination of the ZZ domain of the E3 Ubiquitin ligase HERC2. STAR Protoc. 2020;1:100155. doi: 10.1016/j.xpro.2020.100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller A.M., Breitsprecher D., Duhr S., Baaske P., Schubert T., Längst G. MicroScale thermophoresis: a rapid and precise method to quantify protein-nucleic acid interactions in solution. Methods Mol. Biol. 2017;1654:151–164. doi: 10.1007/978-1-4939-7231-9_10. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Owttrim G.W. RNA helicases: diverse roles in prokaryotic response to abiotic stress. RNA Biol. 2013;10:96–110. doi: 10.4161/rna.22638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt C. The universal protein resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillner K., Jerabek-Willemsen M., Duhr S., Braun D., Längst G., Baaske P. Microscale thermophoresis as a sensitive method to quantify protein: nucleic acid interactions in solution. Methods Mol. Biol. 2012;815:241–252. doi: 10.1007/978-1-61779-424-7_18. [DOI] [PubMed] [Google Scholar]
- Zhang L., Li X. DEAD-box RNA helicases in cell cycle control and clinical therapy. Cells. 2021;10:1540. doi: 10.3390/cells10061540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under ID codes 6L5N, 6L5O, and 6L5L.