

Abstract
Individuals with schizophrenia (SZ) exhibit cognitive performance below expected levels based on familial cognitive aptitude. One such cognitive process, working memory (WM), is robustly impaired in SZ. These WM impairments, which emerge over development during the premorbid and prodromal stages of SZ, appear to reflect alterations in the neural circuitry of the dorsolateral prefrontal cortex (DLPFC). Within the DLPFC, a microcircuit formed by reciprocal connections between excitatory layer 3 pyramidal neurons (PNs) and inhibitory parvalbumin basket cells (PVBCs) appears to be a key neural substrate for WM. Postmortem human studies indicate that both layer 3 PNs and PVBCs are altered in SZ, suggesting that levels of excitation and inhibition are lower in the microcircuit. Studies in monkeys indicate that features of both cell types exhibit distinctive postnatal developmental trajectories. Together, the results of these studies suggest a model in which 1) genetic and/or early environmental insults to excitatory signaling in layer 3 PNs give rise to cognitive impairments during the prodromal phase of SZ and evoke compensatory changes in inhibition that alter the developmental trajectories of PVBCs, and 2) synaptic pruning during adolescence further lowers excitatory activity to a level that exceeds the compensatory capacity of PVBC inhibition, leading to a failure of the normal maturational improvements in WM during the prodromal and early clinical stages of SZ. Findings that support as well as challenge this model are discussed.
Keywords: GABA, gamma oscillations, glutamate, cognition, working memory, schizophrenia
Cognitive impairments: A core and early clinical feature of schizophrenia
Cognitive deficits are considered one of the core features of schizophrenia (SZ)(1). In most individuals with SZ, cognitive performance falls below expectations based on familial cognitive aptitude(2), and the magnitude of the premorbid cognitive deficit directly predicts the risk of psychosis(3). Furthermore, cognitive deficits are the best predictor of long-term functional outcomes in persons with SZ(4).
Among the cognitive domains that have been well-studied, working memory (WM), the ability to transiently maintain and manipulate a limited amount of information to guide thought or behavior(5), is robustly impaired in individuals with SZ. Multiple studies have demonstrated that SZ is associated with WM deficits of large effect sizes across various types of WM tasks(6). These WM deficits cannot be explained by effects of IQ, duration of illness, or use of antipsychotic medications(6). Unaffected siblings of persons with SZ also exhibit lower than expected performance on WM tasks(7–10), suggesting a shared genetic and/or environmental liability for WM impairments(11). Finally, some studies indicate that WM deficits might be a core contributing factor to disturbances in other cognitive domains in SZ(12).
WM deficits likely have a neurodevelopmental basis, as they are present at the first psychotic episode(13) and are also detectable before psychosis onset(14). In individuals who are later diagnosed with SZ, some cognitive domains are impaired in early childhood, whereas impairments in other domains emerge during adolescence(15–17). For example, between 18 months and 8 years of age, individuals later diagnosed with SZ exhibited a decline in IQ, which continued to worsen through age 20(18). These individuals also progressively fell behind their peers in WM performance during their teen years(17,18). These developmental impairments in cognitive function were not found in individuals later diagnosed with major depression(18), suggesting that they are specific to the disease process of SZ.
These findings suggest that understanding both the neural circuitry basis for WM impairments in SZ, and the developmental trajectories of the components of this neural circuitry, might reveal which components are vulnerable to the disease process of SZ and when they are most susceptible.
Role of a local neural circuit in the dorsolateral prefrontal cortex in WM
Working memory is dependent on a distributed cortical neural network which includes a key node in the dorsolateral prefrontal cortex (DLPFC)(19). Indeed, multiple lines of evidence indicate that alterations in the DLPFC are central to impaired WM performance in people with SZ(20). Within the primate DLPFC, gamma frequency (30–80 Hz) oscillations, which reflect the synchronous activity of large groups of neurons in cortical layers 2–3(21,22), appear to be a neural corollary of mental representation during WM. For example, the power of frontal gamma oscillations increases in proportion to WM load(23,24), and individuals with SZ fail to increase this power in response to greater WM demands(25–27).
Consistent with this association between WM and gamma oscillations, studies in monkeys and other experimental systems suggest that the same neural circuit involving DLPFC layer 3 pyramidal neurons (PNs) mediates both processes. These PNs furnish a principal axon which gives rise to both local axon collaterals (which equally target PN dendritic spines and GABA-containing dendrites) and horizontally-spreading axon collaterals (most of which target the dendritic spines of other layer 3 PNs(28–30)). In experimental and computational modeling studies, these PN-PN connections form a local circuit(31) that could support recurrent excitatory activity among subsets of DLPFC layer 3 PNs during the delay period of WM tasks(31–34). The delay period activity of these PNs is, in turn, shaped by inhibitory inputs from neighboring GABA neurons(35–37).
Among cortical GABA neuron subtypes, parvalbumin (PV)-containing basket cells (PVBCs) are suspected to be the most involved in regulating PN activity important for WM. PVBCs have extensive axonal arborizations(38) that provide fast synaptic inhibition onto the perisomatic region of multiple PNs(39) and might coordinate the activity of PN subsets(40) that are responsive to a particular stimulus(41). When this inhibition ends, the innervated PNs share a high probability of firing synchronously(42). Given the predominance of α1 subunit-containing GABAA receptors at PVBC synapses(43), the decay kinetics of this inhibition results in a firing rate of PNs at gamma frequency(44). Indeed, computational modeling and experimental studies in the hippocampus support a central role for PVBCs in entraining gamma frequency activity of PNs(45). Finally, in rodent neocortex, stimulation or inhibition of PV neurons can induce or suppress gamma oscillations, respectively(46–48). However, a causal role of PVBCs in the generation of gamma oscillations in the primate cortex has not been experimentally demonstrated.
The potential role of PVBCs in gamma oscillation generation is not shared by PV-containing chandelier cells. Although chandelier cells synapse on the axon initial segment of PNs(49,50) and powerfully veto action potential generation(51), they lack the synaptic contacts with other chandelier neurons that are essential for organizing oscillatory activities(52). Furthermore, chandelier neuron synapses on PN axon initial segments are enriched for GABAA receptors containing α2 subunits with decay kinetics that are likely too slow to generate gamma frequency oscillations.
DLPFC layer 3 microcircuitry alterations in SZ and potential neurodevelopmental origins
The findings reviewed above suggest that the PN-PVBC microcircuit in DLPFC layer 3 plays a significant role in gamma oscillations and WM. Altered development of one or more microcircuit components could contribute to WM dysfunction during the premorbid, prodromal, and clinical stages of SZ. In the following sections, we first present data highlighting alterations to components of this microcircuit in postmortem studies of the DLPFC from individuals with SZ. We then summarize key findings from studies of the postnatal development of these same circuitry components in the monkey DLPFC. Finally, we compare these findings to suggest when the circuitry alterations present in SZ might arise during development.
Layer 3 PNs
Alterations of layer 3 PNs in SZ.
DLPFC layer 3 PNs exhibit numerous morphological anomalies in people with SZ, including smaller somal volumes, shorter and less complex dendritic arbors, and lower dendritic spine density(53–57). Because dendritic spines receive most of the excitatory inputs to PNs(58), the combination of shorter dendrites and lower spine density suggests that DLPFC layer 3 PNs receive a lower number of excitatory synapses in SZ. In contrast, these morphological alterations are not present in layer 5 and 6 PNs(59) that are not involved in gamma oscillations or WM delay period activity.
These morphological alterations in PNs have been suggested(60) to stem from disturbances in signaling pathways that regulate the actin dynamics essential for dendritic arbor and spine formation and maintenance(61). In SZ, genetic risk factors implicate gene products involved in regulating actin dynamics signaling pathways(62). Indeed, the expression of critical molecules that regulate actin dynamics is altered in the DLPFC of people with SZ. For example, transcript levels of Rho GTPase cell division cycle 42 (CDC42), which regulates the actin polymerization required for spine maturation(61), and of kalirin, a Rho guanine exchange factor that regulates spine integrity through CDC42 signaling pathways(63), are lower in the DLPFC of subjects with SZ(64,65). These pathways may be selective for, or at least enriched in, layer 3 PNs of the DLPFC, as levels of CDC42 and kalirin mRNAs predicted spine density only on layer 3 PNs(60,64,65).
Development of DLPFC layer 3 PNs.
In both monkeys and humans, the densities of dendritic spines on DLPFC layer 3 PNs and layer 3 excitatory synapses exhibit a characteristic developmental trajectory (Figure 1): these densities increase rapidly during the postnatal period, remain at a stable plateau through the prepubertal period, and then decline by ~50% during the peripubertal period and early adulthood until stable levels are achieved(66–69). For dendritic spines, this characteristic developmental trajectory is most pronounced on layer 3 PNs which exhibit greater overproduction and pruning of dendritic spines relative to PNs in layers 5 or 6(67,68).
Figure 1.
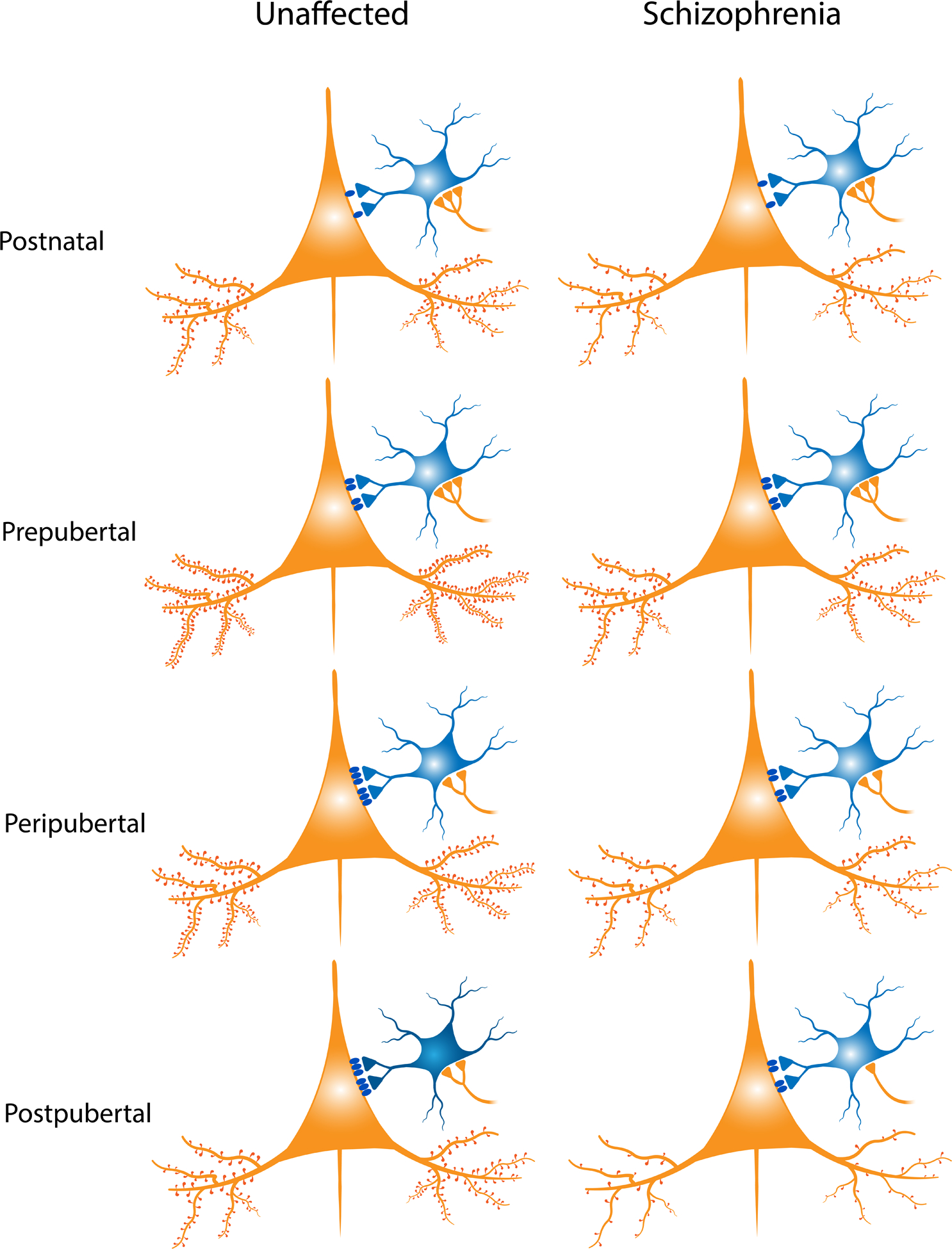
Schematic summary of the DLPFC layer 3 pyramidal neuron (PN) – PV basket cell (PVBC) circuit across monkey development (left panel) and in schizophrenia (SZ) (right panel). During postnatal development in monkeys, the density of dendritic spines on the basilar dendrites of layer 3 PNs (orange) increases during the immediate postnatal period and through the prepubertal period; spine density then reaches a plateau until synaptic pruning is initiated during the peripubertal period(66). In PVBCs (blue), levels of PV protein in the cell bodies(113) and axon terminals(109, 110) increase (depicted here as increasing color saturation), with adult levels of the protein achieved in the postpubertal period. Expression levels of the GABAA α1 receptor subunit (blue ovals), which is enriched at PVBC inputs, increase in layer 3 PNs between the pre- and postpubertal periods(149), coincident with the maturation of PVBC inputs. Levels of proteins that index the strength of excitatory inputs (orange triangles) onto PVBCs increase between the pre- and postpubertal periods, whereas the density of these inputs declines between the prepubertal and peripubertal periods(113), coinciding with the pruning of spines on layer 3 PNs.
In our proposed model of SZ, alterations in components of this circuitry are present at every developmental stage. First, the density of dendritic spines is predicted to be lower early in postnatal development due to impaired spinogenesis, with this deficit becoming greater during adolescence due to elevated spine pruning. Second, the lower PV levels in PVBC boutons present in adult individuals with SZ are predicted to be a consequence of PV levels not increasing during the peripubertal period(98, 99). Third, as part of the compensatory response to deficient excitatory drive from fewer spines, layer 3 PNs do not exhibit the peripubertal increase in GABAA α1 receptor subunit mRNA levels(143) (Note: these findings have not been demonstrated at the protein level as depicted here). Finally, although the protein levels of pre- and postsynaptic indices of excitatory inputs onto PVBCs are not altered at any developmental stage in SZ, the density of excitatory inputs is predicted to decline across the peripubertal and postpubertal periods(97), coincident with the predicted excessive excitatory synapse pruning onto layer 3 PNs across these periods.
In this model, the insufficient formation and subsequent excessive pruning of dendritic spines in SZ results in a progressive reduction in excitatory drive to layer 3 PNs. This reduction in activity evokes compensatory downregulation of inhibition from PVBCs, but later developmental declines in excitation exceeds the capacity of the inhibitory system to compensate further. Together, these alterations are manifest as lower levels of excitation and inhibition which are insufficient to maintain the neural activity necessary for proper WM function during the prodromal and early clinical stages of SZ.
Whether the lower dendritic spine density in SZ reflects an early failure to form the normal complement of spines, a later excessive pruning of spines, or an imbalance of both processes throughout development remains an open question(70,71). The appearance of WM dysfunction during the early teen years in individuals later diagnosed with SZ(18) might reflect an ‘unmasking’ of alterations that occurred earlier in development(72). For example, maternal immune activation might increase the risk for SZ in offspring(73) by increasing cellular stress, and inflammatory markers could initiate a pathogenic process that disrupts vital functions in the regulation of actin dynamics critical for spine development. Indeed, markers of actin dynamics that are altered in DLPFC layer 3 PNs in SZ(60) typically exhibit a pronounced shift in expression in monkey layer 3 PNs during the early postnatal period(74) when spines are massively overproduced. Therefore, maternal immune activation could interfere with the normal developmental trajectory of molecular markers regulating actin dynamics and potentially impair the genesis of dendritic spines. Fewer dendritic spines would be expected to reduce the volume of neuropil. Indeed, maternal immune activation in monkeys was associated with smaller prefrontal gray matter volumes and altered cognitive development in the prepubertal period of offspring(75).
However, it is unclear if the evidence of elevated immune-related markers in the DLPFC of individuals with SZ(76,77) reflects the persistence of such a prenatal insult, a later elevation of immune-related transcript expression, or some combination of both(78). For example, postnatal immune dysfunction might interact with peripubertal stress(79) to produce the alterations in inflammatory markers observed in SZ(77); in animal models, these inflammatory processes can directly impact dendritic spine morphogenesis and plasticity in a developmentally-mediated fashion(80,81). Thus, the genetic and environmental liabilities of SZ could impair spine development early in development.
Alternatively, the spine deficit in SZ might represent over-pruning during adolescence, a view supported by the finding that the genetic locus with the strongest statistical linkage to SZ harbors the major histocompatibility complex/complement component 4A (C4A)(82,83). Higher levels of C4A gene copy numbers in SZ(84) have been suggested to result in greater complement activity, microglial-mediated synaptic engulfment(85), and a reduction in spine density during the SZ prodrome. Supporting this idea, elevated levels of plasma C4-anaphylatoxin, a C4 protein fragment released upon C4 protein activation, have been reported in SZ(86). However, a recent analysis of large-scale genetic and transcriptomic data sets did not find enrichment of complement system genes in SZ and suggested that synaptic pathways are a more prominent factor in SZ risk(87).
The pronounced developmental trajectory of layer 3 PN dendritic spines and their preferential vulnerability relative to SZ might reflect unique molecular properties of these spines. As reviewed elsewhere(88), dendritic spines on layer 3 PNs in monkey DLPFC exhibit high cAMP-PKA protein levels, promoting the concentration of cytosolic calcium levels. Moderate levels of cytosolic calcium are thought to contribute to the strengthening of synapses, but pathologically high levels of cytosolic calcium in dendritic spines could induce an inflammatory response and promote higher levels of pruning by microglia. Thus, the distinctive molecular properties of dendritic spines on DLPFC layer 3 PNs might render these spines more susceptible to pruning during the premorbid or prodromal phases of SZ.
Finally, it might be that spines with immature excitatory inputs are preferentially vulnerable to excessive pruning in SZ; if so, then understanding when these inputs obtain their mature state might suggest the developmental period(s) of heightened vulnerability. Studies in monkey DLPFC suggest that the excitatory inputs to layer 3 PNs are functionally mature well before puberty. For example, during the early postnatal period, excitatory inputs to primate DLPFC layer 3 PNs have immature functional properties, such as lower α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/N-methyl-D-aspartate (NMDA) receptor ratio and high probability of glutamate release, but these functional properties achieve adult levels prior to the onset of puberty(89). Similarly, adult levels of postsynaptic molecular determinants of glutamate neurotransmission (e.g., AMPA receptor subunit GluA1 and NMDA receptor subunit GluN1) are reached prior to puberty in DLPFC layer 3 PNs in monkeys(60).These findings suggest that the peripubertal refinement of excitatory connections in primate DLPFC involves the elimination of mature rather than immature synapses. Thus, presynaptic factors (e.g., the source of the excitatory input) might tag a mature subset of synapses for pruning during the peripubertal period(89). Indeed, intrinsic axon collaterals of DLPFC layer 3 PNs undergo much greater pruning during the peripubertal period than do associational axons arising from PNs in other cortical regions(90). Thus, the deficit in dendritic spines on layer 3 PNs in SZ might reflect the pathological pruning of a specific source(s) of excitatory inputs.
In concert, the available data suggest that the deficiency of dendritic spines in SZ likely reflects some combination of both insufficient spine formation early in development and a later over-pruning of mature synapses (Figure 1)
PV basket cells
Alterations of PVBCs in SZ.
Multiple studies in different cohorts of subjects have shown that levels of activity-dependent PV mRNA are lower in the DLPFC of subjects within SZ(91–95). Consistent with these findings, lower levels of PV protein immunoreactivity have been found in PVBC cell bodies(96,97) and axon terminals(98,99). However, neither the density nor morphology of PV neurons in the DLPFC is altered in SZ(93,96,97,99–101) (Figure 1).
PV neuron functional capacity, which can be indexed by quantifying levels of transcripts and proteins involved in GABA signaling, appears lower in SZ. For example, transcript levels of the 67 kilodalton form of glutamic acid decarboxylase (GAD67), the enzyme responsible for most cortical GABA synthesis, have been consistently lower in the DLPFC in multiple studies of SZ(94,102–106). In particular, SZ is associated with lower levels of GAD67 mRNA in PV cell bodies(93) and of GAD67 protein in PV axon terminals of PVBCs, but not in the axon cartridges of PV chandelier cells(99,105,107). In addition, the density of excitatory inputs to PV neurons is lower in the DLPFC in SZ(97).
Development of layer 3 PVBCs.
In monkey DLPFC, the density of cell bodies with PV immunoreactivity reaches mature levels at the end of the postnatal period(108). In contrast, the density of PV-positive axonal boutons, presumably from PVBCs, rises steadily from the postnatal to postpubertal period(109). The latter findings reflect an increase in PV protein levels per bouton between prepubertal and postpubertal monkeys without a developmental change in the density of PVBC boutons(110,111). These findings—consistent with evidence from Golgi studies that the axonal arbors of PVBCs do not change over this same period of development(112)—suggest that PVBC axons mature morphologically early in development but undergo progressive changes in their biochemical properties across postnatal development. These developmental patterns appear to be cell type-specific as the axon terminals of PV-positive chandelier cells show the opposite pattern of development: the mean number of PV-positive boutons per axon cartridge declines with age, but PV protein levels per bouton do not(110).
The density of excitatory synapses (identified as the overlap of presynaptic vesicular glutamate transporter 1 (VGLUT1) and postsynaptic PSD95 immunoreactive puncta) on DLPFC PV neurons was higher in prepubertal than postpubertal monkeys(113). In contrast, VGLUT1 and PSD95 protein levels across puncta were higher in postpubertal monkeys and positively predicted cellular PV levels(113). Together, these findings suggest that although excitatory inputs to PV neurons are pruned during development, the remaining synapses show indices of greater synaptic strength that could contribute to an activity-dependent increase in PVBC PV levels during the same developmental time frame.
Potential relationships between alterations in layer 3 PNs and PVBCs
The developmental trajectories of layer 3 PNs and PVBCs in the primate DLPFC suggest potential sensitive periods for the emergence of alterations in both cell types, and subsequent impairments in gamma oscillation power and WM performance, in individuals with SZ. Below, we posit a developmental model of how PN and PVBC alterations might arise in SZ.
According to this model (Figure 1), genetically-driven, cell-autonomous, molecular alterations in layer 3 PNs produce a deficit in dendritic spines early in development, an associated reduction in excitatory inputs, and a resulting hypoactivity of DLPFC layer 3 PNs, which is worsened during the peripubertal period by microglial-mediated pruning of dendritic spines. This idea of a hypoactive state is supported by findings that DLPFC layer 3 PNs in SZ exhibit lower levels of transcripts that are activity-dependent(114,115) and a coordinated downregulation of mitochondrial markers(116,117), an expected finding in neurons under a lower level of demand for energy production(118). The alternative view, that a primary problem in mitochondrial energy production leads to dendritic spine deficits, seems less likely for the following reasons: 1) Genetic studies have not implicated nuclear genes regulating mitochondrial function as risk factors for SZ(83,119–121). 2) Our cell type-specific studies in schizophrenia revealed larger alterations in mitochondrial-related gene expression in layer 3 PNs than in PVBCs(122), whereas a primary problem in mitochondrial energy production might have produced the opposite findings, given that PVBCs are more active and require greater ATP production than PNs(123–127). 3) Genetic manipulations that impaired mitochondrial function in PV neurons were associated with greater excitatory inputs to PV neurons and greater gamma band power(128), the opposite of findings in SZ(97,129).
Because PVBCs in cortical layers 3–4 are a major recipient of excitatory inputs from layer 3 PNs(130), hypoactivity of layer 3 PNs in SZ would be expected to produce activity-dependent alterations in PVBCs, such as lower levels of PV and GAD67. Indeed, the magnitude of the deficit in excitatory inputs onto PV cells was inversely associated with GAD67 and PV transcript levels in SZ(97). This compensatory downregulation of inhibition from PVBCs could restore some level of homeostasis in the circuit(129).
How might this chain of pathogenic events transpire developmentally? The available data suggests that it begins as early as the postnatal period. For example, an increase in spine density on DLPFC layer 3 PNs(66–69) during the postnatal period is temporally concordant with marked shifts in the expression of genes regulating actin dynamics(74). This association raises the possibility that alterations to these molecular shifts could give rise to an early deficit in excitatory activity in DLPFC circuitry that contributes to the deficit in IQ seen during early childhood in individuals who are later diagnosed with SZ(18).
The compensatory changes in PVBCs might not appear until later, given the delayed maturational trajectory of GABA neurotransmission relative to similar indices of glutamate neurotransmission(131). Thus, early alterations in excitatory signaling might alter later developmental trajectories of PVBC cell bodies and axon terminals(108,110). For example, because PV and GAD67 mRNA expression are activity-dependent(132,133), weaker excitatory signaling onto PVBCs might blunt the normal developmental increase in PV and GAD67 mRNAs, contributing to the lower DLPFC levels of these transcripts in SZ.
This downregulation of PVBC-mediated inhibition in SZ might provide a sufficient compensatory response to partially minimize the negative impact of layer 3 PN hypoactivity on the function of the DLPFC WM circuit during the prepubertal period. However, the onset of excessive excitatory synapse pruning during the peripubertal period might exceed the compensatory capacity of downregulated PVBC signaling and give rise to the progressively more severe WM impairments observed in individuals later diagnosed with SZ(18) (Figure 1).
An alternative model that alterations in PVBCs are primary in the SZ disease process also bears consideration. The lower levels of GAD67 mRNA in PV neurons could be due to disease-related alterations in upstream factors that regulate expression of the GAD1 gene. For example, a GAD1 gene variant was putatively associated with greater risk for schizophrenia(134) and altered chromatin structures at the GAD1 promoter(135) were associated with lower GAD67 expression in schizophrenia; however, replications of these findings have not been reported. If replicated, such alterations might explain GAD67 mRNA deficits in laminar locations (e.g., layer 2-superficial 3) where PVBCs are relatively sparse(102).
PVBC dysfunction in schizophrenia could also be mediated by alterations in molecular cascades intrinsic to these neurons. For example, excitatory synapse number on PV interneurons is regulated, at least in part, by erb-b2 receptor tyrosine kinase 4 (ErbB4), whose function is influenced by alternative splicing(136), which has been suggested to be a ‘primary mechanism’ in SZ(137). Indeed, alternative splicing of ErbB4 is altered in SZ(138), and this shift in splicing was associated with both fewer excitatory inputs to PV neurons and the down-regulation of PV expression in SZ(97,113,139) and during normal pubertal development in monkeys(113). Furthermore, conditional deletion of ErbB4 in murine PV neurons produced cellular, physiological, and behavioral abnormalities suggestive of SZ, including deficits in dendritic spine density(140). Thus, alterations in ErbB4 splicing intrinsic to PVBCs could cause a cell-autonomous decrease in the number of excitatory inputs to PVBCs and the constellation of molecular alterations in PVBCs found in SZ. However, the experimental production of GAD67 deficits in PV neurons fails to produce other alterations observed in SZ(141), arguing against a primary GABA deficit in the illness.
In the face of a primary deficit in inhibition from PVBCs, postsynaptic GABAA receptors, such as those containing the GABAA α1 subunit, would be expected to exhibit a compensatory upregulation as they are critically involved in cortical plasticity, especially during developmental critical periods(142). However, in SZ, mRNA levels of the GABAA α1 receptor subunit are lower in layer 3 PNs in SZ(143). Although it is unknown if the lower expression of the α1 subunit is present in early development as a primary pathology or a later compensation for PN hypoactivity, current experimental evidence supports the latter hypothesis(144). For example, suppression pyramidal neuron activity in layers 2–3 of mouse visual cortex via overexpression of an inwardly-rectifying potassium channel (Kir2.1) results in a proportional downregulation of PV-mediated inhibition(144).
Of course, the current data do not exclude other scenarios. It is possible that 1) the disease process of SZ involves alterations in layer 3 PNs and PVBCs that are co-primary, or 2) the alterations in PVBCs are secondary deleterious consequences (as opposed to compensations) of the PN abnormalities, or 3) both PN and PVBC alterations are secondary to a shared (but currently unknown) upstream disturbance. Discriminating among these possibilities requires proof-of-concept studies capable of converting associations observed in the disease state into evidence of causal mechanisms. In this regard, recent findings using dynamic causal modeling of in vivo neuroimaging and electroencephalography data from individuals with SZ support the hypothesis that deficits in PN signaling contribute to the alterations in inhibitory neurons and not vice versa(145).
Conclusions
Alterations in excitatory and inhibitory signaling in the layer 3 PN-PVBC microcircuit of the DLPFC are likely key contributors to impaired gamma oscillations and WM in people with SZ. Based on the available data, a parsimonious model posits that early impairments in excitatory signaling among DLPFC layer 3 PNs contribute to the cognitive deficits that emerge during childhood in individuals who are later diagnosed with SZ. These impairments in excitatory signaling evoke compensatory changes in the developmental trajectory of inhibitory signaling from PVBCs. However, these changes in inhibition are inadequate to compensate for the further reduction in excitatory signaling associated with the peripubertal pruning of excitatory synapses, impairing the normal maturation of WM function during adolescence and young adulthood.
Further testing of this model requires a consideration of the other components of cortical circuitry that contribute to WM function. For example, somatostatin-containing GABA neurons influence excitatory inputs to layer 3 PNs, and levels of somatostatin mRNA in the DLPFC are lower in SZ(92,94). Similarly, inputs from other brain regions (e.g., hippocampus, thalamus, and dopamine neurons from the midbrain) influence WM and are altered in SZ(146). Thus, studies to determine the nature of the schizophrenia-associated alterations in these elements of cortical circuitry are needed to fully understand the neural basis for WM dysfunction in SZ.
Studies that examine the effect of PN and PVBC alterations at different stages of development are also needed. For example, ablation of NMDARs in PV interneurons early in postnatal development produced more prominent behavioral alterations than the same manipulation in adult mice(147). In addition, compensatory strategies in response to PN hypoactivity appear to differ across development. For example, in mouse visual cortex, manipulations early in postnatal development that lower neuronal activity elicit greater PN intrinsic excitability without changes in inhibition, whereas the same manipulation in adulthood results in lower inhibition without changes in PN intrinsic excitability(148).
Further testing and refinements of the model proposed in this review will hopefully provide the groundwork for novel therapeutic interventions directed at preempting the emergence of cognitive symptoms in individuals at risk for SZ.
Acknowledgements
We thank Mary Brady and Anneliese Murphree for assistance in preparing the figures. Supported by NIH grants MH051234 and MH043784.
Financial Disclosures
Dr. Lewis receives research funding from Merck. The other authors report no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kahn RS, Keefe RS (2013): Schizophrenia is a cognitive illness: time for a change in focus. JAMA Psychiatry. 70:1107–1112. [DOI] [PubMed] [Google Scholar]
- 2.Keefe RSE, Kahn RS (2017): Cognitive Decline and Disrupted Cognitive Trajectory in Schizophrenia. JAMA Psychiatry. 74:535–536. [DOI] [PubMed] [Google Scholar]
- 3.Kendler KS, Ohlsson H, Mezuk B, Sundquist JO, Sundquist K (2016): Observed Cognitive Performance and Deviation From Familial Cognitive Aptitude at Age 16 Years and Ages 18 to 20 Years and Risk for Schizophrenia and Bipolar Illness in a Swedish National Sample. JAMA Psychiatry. 73:465–471. [DOI] [PubMed] [Google Scholar]
- 4.Green MF (1996): What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 153:321–330. [DOI] [PubMed] [Google Scholar]
- 5.Baddeley A (1992): Working memory. Science. 255:556–559. [DOI] [PubMed] [Google Scholar]
- 6.Forbes NF, Carrick LA, McIntosh AM, Lawrie SM (2009): Working memory in schizophrenia: a meta-analysis. Psychol Med. 39:889–905. [DOI] [PubMed] [Google Scholar]
- 7.Horan WP, Braff DL, Nuechterlein KH, Sugar CA, Cadenhead KS, Calkins ME, et al. (2008): Verbal working memory impairments in individuals with schizophrenia and their first-degree relatives: findings from the Consortium on the Genetics of Schizophrenia. Schizophr Res. 103:218–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park S, Holzman PS, Goldman-Rakic PS (1995): Spatial working memory deficits in the relatives of schizophrenic patients. Arch Gen Psychiatry. 52:821–828. [DOI] [PubMed] [Google Scholar]
- 9.Pirkola T, Tuulio-Henriksson A, Glahn D, Kieseppa T, Haukka J, Kaprio J, et al. (2005): Spatial working memory function in twins with schizophrenia and bipolar disorder. Biol Psychiatry. 58:930–936. [DOI] [PubMed] [Google Scholar]
- 10.Seidman LJ, Meyer EC, Giuliano AJ, Breiter HC, Goldstein JM, Kremen WS, et al. (2012): Auditory working memory impairments in individuals at familial high risk for schizophrenia. Neuropsychology. 26:288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velthorst E, Mollon J, Murray RM, de Haan L, Germeys IM, Glahn DC, et al. (2021): Cognitive functioning throughout adulthood and illness stages in individuals with psychotic disorders and their unaffected siblings. Mol Psychiatry. 26:4529–4543. [DOI] [PubMed] [Google Scholar]
- 12.Silver H, Feldman P, Bilker W, Gur RC (2003): Working memory deficit as a core neuropsychological dysfunction in schizophrenia. Am J Psychiatry. 160:1809–1816. [DOI] [PubMed] [Google Scholar]
- 13.Mesholam-Gately RI, Giuliano AJ, Goff KP, Faraone SV, Seidman LJ (2009): Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology. 23:315–336. [DOI] [PubMed] [Google Scholar]
- 14.Bora E, Murray RM (2014): Meta-analysis of cognitive deficits in ultra-high risk to psychosis and first-episode psychosis: do the cognitive deficits progress over, or after, the onset of psychosis? Schizophr Bull. 40:744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gur RC, Calkins ME, Satterthwaite TD, Ruparel K, Bilker WB, Moore TM, et al. (2014): Neurocognitive growth charting in psychosis spectrum youths. JAMA Psychiatry. 71:366–374. [DOI] [PubMed] [Google Scholar]
- 16.MacCabe JH, Wicks S, Lofving S, David AS, Berndtsson A, Gustafsson JE, et al. (2013): Decline in cognitive performance between ages 13 and 18 years and the risk for psychosis in adulthood: a Swedish longitudinal cohort study in males. JAMA Psychiatry. 70:261–270. [DOI] [PubMed] [Google Scholar]
- 17.Reichenberg A, Caspi A, Harrington H, Houts R, Keefe RS, Murray RM, et al. (2010): Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry. 167:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mollon J, David AS, Zammit S, Lewis G, Reichenberg A (2018): Course of Cognitive Development From Infancy to Early Adulthood in the Psychosis Spectrum. JAMA Psychiatry. 75:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller EK, Cohen JD (2001): An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 24:167–202. [DOI] [PubMed] [Google Scholar]
- 20.Smucny J, Dienel SJ, Lewis DA, Carter CS (2022): Mechanisms underlying dorsolateral prefrontal cortex contributions to cognitive dysfunction in schizophrenia. Neuropsychopharmacology. 47:292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bastos AM, Loonis R, Kornblith S, Lundqvist M, Miller EK (2018): Laminar recordings in frontal cortex suggest distinct layers for maintenance and control of working memory. Proc Natl Acad Sci U S A. 115:1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheeringa R, Fries P (2019): Cortical layers, rhythms and BOLD signals. Neuroimage. 197:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howard MW, Rizzuto DS, Caplan JB, Madsen JR, Lisman J, Aschenbrenner-Scheibe R, et al. (2003): Gamma oscillations correlate with working memory load in humans. Cereb Cortex. 13:1369–1374. [DOI] [PubMed] [Google Scholar]
- 24.Roux F, Uhlhaas PJ (2014): Working memory and neural oscillations: alpha-gamma versus theta-gamma codes for distinct WM information? Trends Cogn Sci. 18:16–25. [DOI] [PubMed] [Google Scholar]
- 25.Zilles D, Gruber E, Falkai P, Gruber O (2010): Patients with schizophrenia show deficits of working memory maintenance components in circuit-specific tasks. Eur Arch Psychiatry Clin Neurosci. 260:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minzenberg MJ, Firl AJ, Yoon JH, Gomes GC, Reinking C, Carter CS (2010): Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology. 35:2590–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho RY, Konecky RO, Carter CS (2006): Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci U S A. 103:19878–19883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kritzer MF, Goldman-Rakic PS (1995): Intrinsic circuit organization of the major layers and sublayers of the dorsolateral prefrontal cortex in the rhesus monkey. J Comp Neurol. 359:131–143. [DOI] [PubMed] [Google Scholar]
- 29.Melchitzky DS, Gonzalez-Burgos G, Barrionuevo G, Lewis DA (2001): Synaptic targets of the intrinsic axon collaterals of supragranular pyramidal neurons in monkey prefrontal cortex. J Comp Neurol. 430:209–221. [DOI] [PubMed] [Google Scholar]
- 30.Levitt JB, Lewis DA, Yoshioka T, Lund JS (1993): Topography of pyramidal neuron intrinsic connections in macaque monkey prefrontal cortex (areas 9 and 46). J Comp Neurol. 338:360–376. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez-Burgos G, Barrionuevo G, Lewis DA (2000): Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex. 10:82–92. [DOI] [PubMed] [Google Scholar]
- 32.Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, et al. (2013): NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 77:736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Compte A, Brunel N, Goldman-Rakic PS, Wang XJ (2000): Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical network model. Cereb Cortex. 10:910–923. [DOI] [PubMed] [Google Scholar]
- 34.Camperi M, Wang XJ (1998): A model of visuospatial working memory in prefrontal cortex: recurrent network and cellular bistability. J Comput Neurosci. 5:383–405. [DOI] [PubMed] [Google Scholar]
- 35.Sawaguchi T, Matsumura M, Kubota K (1989): Delayed response deficits produced by local injection of bicuculline into the dorsolateral prefrontal cortex in Japanese macaque monkeys. Exp Brain Res. 75:457–469. [DOI] [PubMed] [Google Scholar]
- 36.Rao SG, Williams GV, Goldman-Rakic PS (2000): Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 20:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Constantinidis C, Williams GV, Goldman-Rakic PS (2002): A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci. 5:175–180. [DOI] [PubMed] [Google Scholar]
- 38.Conde F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA (1994): Local circuit neurons immunoreactive for calretinin, calbindin D-28k or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol. 341:95–116. [DOI] [PubMed] [Google Scholar]
- 39.Krimer LS, Zaitsev AV, Czanner G, Kroner S, Gonzalez-Burgos G, Povysheva NV, et al. (2005): Cluster analysis-based physiological classification and morphological properties of inhibitory neurons in layers 2–3 of monkey dorsolateral prefrontal cortex. J Neurophysiol. 94:3009–3022. [DOI] [PubMed] [Google Scholar]
- 40.Williams SM, Goldman-Rakic PS, Leranth C (1992): The synaptology of parvalbumin-immunoreactive neurons in the primate prefrontal cortex. J Comp Neurol. 320:353–369. [DOI] [PubMed] [Google Scholar]
- 41.Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS (2004): Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. ProcNatlAcadSci USA. 101:1368–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buzsaki G, Wang XJ (2012): Mechanisms of gamma oscillations. Annu Rev Neurosci. 35:203–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mody I, Pearce RA (2004): Diversity of inhibitory neurotransmission through GABA(A) receptors. Trends Neurosci. 27:569–575. [DOI] [PubMed] [Google Scholar]
- 44.Whittington MA, Cunningham MO, LeBeau FE, Racca C, Traub RD (2011): Multiple origins of the cortical gamma rhythm. Dev Neurobiol. 71:92–106. [DOI] [PubMed] [Google Scholar]
- 45.Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH (2000): Inhibition-based rhythms: experimental and mathematical observations on network dynamics. Int J Psychophysiol. 38:315–336. [DOI] [PubMed] [Google Scholar]
- 46.Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. (2007): Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 53:591–604. [DOI] [PubMed] [Google Scholar]
- 47.Sohal VS, Zhang F, Yizhar O, Deisseroth K (2009): Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 459:698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. (2009): Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 459:663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeFelipe J, Hendry SH, Jones EG, Schmechel D (1985): Variability in the terminations of GABAergic chandelier cell axons on initial segments of pyramidal cell axons in the monkey sensory-motor cortex. J Comp Neurol. 231:364–384. [DOI] [PubMed] [Google Scholar]
- 50.Szentagothai J (1975): The ‘module-concept’ in cerebral cortex architecture. Brain Res. 95:475–496. [DOI] [PubMed] [Google Scholar]
- 51.Dudok B, Szoboszlay M, Paul A, Klein PM, Liao Z, Hwaun E, et al. (2021): Recruitment and inhibitory action of hippocampal axo-axonic cells during behavior. Neuron. 109:3838–3850 e3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang XJ, Buzsaki G (1996): Gamma oscillation by synaptic inhibition in a hippocampal interneuronal network model. J Neurosci. 16:6402–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pierri JN, Volk CL, Auh S, Sampson A, Lewis DA (2001): Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry. 58:466–473. [DOI] [PubMed] [Google Scholar]
- 54.Rajkowska G, Selemon LD, Goldman-Rakic PS (1998): Neuronal and glial somal size in the prefrontal cortex: a postmortem morphometric study of schizophrenia and Huntington disease. Arch Gen Psychiatry. 55:215–224. [DOI] [PubMed] [Google Scholar]
- 55.Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, et al. (1998): Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 65:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glantz LA, Lewis DA (2000): Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 57:65–73. [DOI] [PubMed] [Google Scholar]
- 57.Konopaske GT, Lange N, Coyle JT, Benes FM (2014): Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry. 71:1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuste R (2011): Dendritic spines and distributed circuits. Neuron. 71:772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kolluri N, Sun Z, Sampson AR, Lewis DA (2005): Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 162:1200–1202. [DOI] [PubMed] [Google Scholar]
- 60.Datta D, Arion D, Corradi JP, Lewis DA (2015): Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biol Psychiatry. 78:775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hotulainen P, Hoogenraad CC (2010): Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 189:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paskus JD, Herring BE, Roche KW (2020): Kalirin and Trio: RhoGEFs in Synaptic Transmission, Plasticity, and Complex Brain Disorders. Trends Neurosci. 43:505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cahill ME, Xie Z, Day M, Photowala H, Barbolina MV, Miller CA, et al. (2009): Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc Natl Acad Sci U S A. 106:13058–13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ide M, Lewis DA (2010): Altered cortical CDC42 signaling pathways in schizophrenia: implications for dendritic spine deficits. Biol Psychiatry. 68:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hill JJ, Hashimoto T, Lewis DA (2006): Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 11:557–566. [DOI] [PubMed] [Google Scholar]
- 66.Anderson SA, Classey JD, Conde F, Lund JS, Lewis DA (1995): Synchronous development of pyramidal neuron dendritic spines and parvalbumin-immunoreactive chandelier neuron axon terminals in layer III of monkey prefrontal cortex. Neuroscience. 67:7–22. [DOI] [PubMed] [Google Scholar]
- 67.Petanjek Z, Judas M, Simic G, Rasin MR, Uylings HB, Rakic P, et al. (2011): Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A. 108:13281–13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bourgeois JP, Goldman-Rakic PS, Rakic P (1994): Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cereb Cortex. 4:78–96. [DOI] [PubMed] [Google Scholar]
- 69.Huttenlocher PR, Dabholkar AS (1997): Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 387:167–178. [DOI] [PubMed] [Google Scholar]
- 70.Feinberg I (1982): Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 17:319–334. [DOI] [PubMed] [Google Scholar]
- 71.McGlashan TH, Hoffman RE (2000): Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry. 57:637–648. [DOI] [PubMed] [Google Scholar]
- 72.Marenco S, Weinberger DR (2000): The neurodevelopmental hypothesis of schizophrenia: following a trail of evidence from cradle to grave. Dev Psychopathol. 12:501–527. [DOI] [PubMed] [Google Scholar]
- 73.Muller N (2018): Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr Bull. 44:973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dienel SJ, Bazmi HH, Lewis DA (2017): Development of transcripts regulating dendritic spines in layer 3 pyramidal cells of the monkey prefrontal cortex: Implications for the pathogenesis of schizophrenia. Neurobiol Dis. 105:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vlasova RM, Iosif AM, Ryan AM, Funk LH, Murai T, Chen S, et al. (2021): Maternal Immune Activation during Pregnancy Alters Postnatal Brain Growth and Cognitive Development in Nonhuman Primate Offspring. J Neurosci. 41:9971–9987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. (2013): Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 18:206–214. [DOI] [PubMed] [Google Scholar]
- 77.Volk DW, Chitrapu A, Edelson JR, Lewis DA (2015): Chemokine receptors and cortical interneuron dysfunction in schizophrenia. Schizophr Res. 167:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Volk DW, Lewis DA (2013): Prenatal ontogeny as a susceptibility period for cortical GABA neuron disturbances in schizophrenia. Neuroscience. 248C:154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. (2013): Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. 339:1095–1099. [DOI] [PubMed] [Google Scholar]
- 80.Dietz DM, Sun H, Lobo MK, Cahill ME, Chadwick B, Gao V, et al. (2012): Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nat Neurosci. 15:891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hering H, Sheng M (2001): Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2:880–888. [DOI] [PubMed] [Google Scholar]
- 82.Mokhtari R, Lachman HM (2016): The Major Histocompatibility Complex (MHC) in Schizophrenia: A Review. J Clin Cell Immunol. 7:479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. (2016): Schizophrenia risk from complex variation of complement component 4. Nature. 530:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prasad KM, Chowdari KV, D’Aiuto LA, Iyengar S, Stanley JA, Nimgaonkar VL (2018): Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients-a pilot study. Translational psychiatry. 8:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yilmaz M, Yalcin E, Presumey J, Aw E, Ma M, Whelan CW, et al. (2021): Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat Neurosci. 24:214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kalinowski A, Liliental J, Anker LA, Linkovski O, Culbertson C, Hall JN, et al. (2021): Increased activation product of complement 4 protein in plasma of individuals with schizophrenia. Translational psychiatry. 11:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim M, Haney JR, Zhang P, Hernandez LM, Wang LK, Perez-Cano L, et al. (2021): Brain gene co-expression networks link complement signaling with convergent synaptic pathology in schizophrenia. Nat Neurosci. 24:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arnsten AFT, Datta D, Wang M (2021): The genie in the bottle-magnified calcium signaling in dorsolateral prefrontal cortex. Mol Psychiatry. 26:3684–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gonzalez-Burgos G, Kroener S, Zaitsev AV, Povysheva NV, Krimer LS, Barrionuevo G, et al. (2008): Functional maturation of excitatory synapses in layer 3 pyramidal neurons during postnatal development of the primate prefrontal cortex. Cereb Cortex. 18:626–637. [DOI] [PubMed] [Google Scholar]
- 90.Woo TU, Pucak ML, Kye CH, Matus CV, Lewis DA (1997): Peripubertal refinement of the intrinsic and associational circuitry in monkey prefrontal cortex. Neuroscience. 80:1149–1158. [DOI] [PubMed] [Google Scholar]
- 91.Bitanihirwe BK, Woo TU (2014): Transcriptional dysregulation of gamma-aminobutyric acid transporter in parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. Psychiatry Res. 220:1155–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fung SJ, Webster MJ, Sivagnanasundaram S, Duncan C, Elashoff M, Weickert CS (2010): Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry. 167:1479–1488. [DOI] [PubMed] [Google Scholar]
- 93.Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. (2003): Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 23:6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Volk DW, Sampson AR, Zhang Y, Edelson JR, Lewis DA (2016): Cortical GABA markers identify a molecular subtype of psychotic and bipolar disorders. Psychol Med. 46:2501–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chung DW, Chung Y, Bazmi HH, Lewis DA (2018): Altered ErbB4 splicing and cortical parvalbumin interneuron dysfunction in schizophrenia and mood disorders. Neuropsychopharmacology. 43:2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Enwright JF, Sanapala S, Foglio A, Berry R, Fish KN, Lewis DA (2016): Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia. Neuropsychopharmacology. 41:2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chung DW, Fish KN, Lewis DA (2016): Pathological Basis for Deficient Excitatory Drive to Cortical Parvalbumin Interneurons in Schizophrenia. Am J Psychiatry. 173:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Glausier JR, Fish KN, Lewis DA (2014): Altered parvalbumin basket cell inputs in the dorsolateral prefrontal cortex of schizophrenia subjects. Mol Psychiatry. 19:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fish KN, Rocco BR, DeDionisio AM, Dienel SJ, Sweet RA, Lewis DA (2021): Altered Parvalbumin Basket Cell Terminals in the Cortical Visuospatial Working Memory Network in Schizophrenia. Biol Psychiatry. 90:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Woo TU, Miller JL, Lewis DA (1997): Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry. 154:1013–1015. [DOI] [PubMed] [Google Scholar]
- 101.Tooney PA, Chahl LA (2004): Neurons expressing calcium-binding proteins in the prefrontal cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 28:273–278. [DOI] [PubMed] [Google Scholar]
- 102.Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA (2000): Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 57:237–245. [DOI] [PubMed] [Google Scholar]
- 103.Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney JWE, et al. (1995): Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Archives of General Psychiatry. 52:258–266. [DOI] [PubMed] [Google Scholar]
- 104.Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, et al. (2011): Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 31:11088–11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Curley AA, Arion D, Volk DW, Asafu-Adjei JK, Sampson AR, Fish KN, et al. (2011): Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am J Psychiatry. 168:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Duncan CE, Webster MJ, Rothmond DA, Bahn S, Elashoff M, Shannon Weickert C (2010): Prefrontal GABA(A) receptor alpha-subunit expression in normal postnatal human development and schizophrenia. J Psychiatr Res. 44:673–681. [DOI] [PubMed] [Google Scholar]
- 107.Rocco BR, Lewis DA, Fish KN (2016): Markedly Lower Glutamic Acid Decarboxylase 67 Protein Levels in a Subset of Boutons in Schizophrenia. Biol Psychiatry. 79:1006–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Condé F, Lund JS, Lewis DA (1996): The hierarchical development of monkey visual cortical regions as revealed by the maturation of parvalbumin-immunoreactive neurons. Developmental Brain Research. 96:261–276. [DOI] [PubMed] [Google Scholar]
- 109.Erickson SL, Lewis DA (2002): Postnatal development of parvalbumin- and GABA transporter-immunoreactive axon terminals in monkey prefrontal cortex. J Comp Neurol. 448:186–202. [DOI] [PubMed] [Google Scholar]
- 110.Fish KN, Hoftman GD, Sheikh W, Kitchens M, Lewis DA (2013): Parvalbumin-containing chandelier and basket cell boutons have distinctive modes of maturation in monkey prefrontal cortex. J Neurosci. 33:8352–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gonzalez-Burgos G, Miyamae T, Pafundo DE, Yoshino H, Rotaru DC, Hoftman G, et al. (2015): Functional Maturation of GABA Synapses During Postnatal Development of the Monkey Dorsolateral Prefrontal Cortex. Cereb Cortex. 25:4076–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lund JS, Lewis DA (1993): Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J Comp Neurol. 328:282–312. [DOI] [PubMed] [Google Scholar]
- 113.Chung DW, Wills ZP, Fish KN, Lewis DA (2017): Developmental pruning of excitatory synaptic inputs to parvalbumin interneurons in monkey prefrontal cortex. Proc Natl Acad Sci U S A. 114:E629–E637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, et al. (2005): Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J Neurosci. 25:372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kimoto S, Zaki MM, Bazmi HH, Lewis DA (2015): Altered Markers of Cortical gamma-Aminobutyric Acid Neuronal Activity in Schizophrenia: Role of the NARP Gene. JAMA Psychiatry. 72:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Arion D, Corradi JP, Tang S, Datta D, Boothe F, He A, et al. (2015): Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry. 20:1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arion D, Huo Z, Enwright JF, Corradi JP, Tseng G, Lewis DA (2017): Transcriptome Alterations in Prefrontal Pyramidal Cells Distinguish Schizophrenia From Bipolar and Major Depressive Disorders. Biol Psychiatry. 82:594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Glausier JR, Enwright JF 3rd, Lewis DA (2020): Diagnosis- and Cell Type-Specific Mitochondrial Functional Pathway Signatures in Schizophrenia and Bipolar Disorder. Am J Psychiatry. 177:1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. (2014): De novo mutations in schizophrenia implicate synaptic networks. Nature. 506:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. (2014): A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 506:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. (2017): Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 49:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Enwright Iii JF, Huo Z, Arion D, Corradi JP, Tseng G, Lewis DA (2018): Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol Psychiatry. 23:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kann O, Papageorgiou IE, Draguhn A (2014): Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J Cereb Blood Flow Metab. 34:1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gulyas AI, Buzsaki G, Freund TF, Hirase H (2006): Populations of hippocampal inhibitory neurons express different levels of cytochrome c. Eur J Neurosci. 23:2581–2594. [DOI] [PubMed] [Google Scholar]
- 125.Nie F, Wong-Riley MT (1995): Double labeling of GABA and cytochrome oxidase in the macaque visual cortex: quantitative EM analysis. J Comp Neurol. 356:115–131. [DOI] [PubMed] [Google Scholar]
- 126.Glausier JR, Roberts RC, Lewis DA (2017): Ultrastructural analysis of parvalbumin synapses in human dorsolateral prefrontal cortex. J Comp Neurol. 525:2075–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fitzgerald ML, Chan J, Mackie K, Lupica CR, Pickel VM (2012): Altered dendritic distribution of dopamine D2 receptors and reduction in mitochondrial number in parvalbumin-containing interneurons in the medial prefrontal cortex of cannabinoid-1 (CB1) receptor knockout mice. J Comp Neurol. 520:4013–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Inan M, Zhao M, Manuszak M, Karakaya C, Rajadhyaksha AM, Pickel VM, et al. (2016): Energy deficit in parvalbumin neurons leads to circuit dysfunction, impaired sensory gating and social disability. Neurobiol Dis. 93:35–46. [DOI] [PubMed] [Google Scholar]
- 129.Gonzalez-Burgos G, Cho RY, Lewis DA (2015): Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry. 77:1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Melchitzky DS, Lewis DA (2003): Pyramidal neuron local axon terminals in monkey prefrontal cortex: differential targeting of subclasses of GABA neurons. Cereb Cortex. 13:452–460. [DOI] [PubMed] [Google Scholar]
- 131.Hoftman GD, Bazmi HH, Ciesielski AJ, Dinka LA, Chen K, Lewis DA (2021): Postnatal Development of Glutamate and GABA Transcript Expression in Monkey Visual, Parietal, and Prefrontal Cortices. Cereb Cortex. 31:2026–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Patz S, Grabert J, Gorba T, Wirth MJ, Wahle P (2004): Parvalbumin expression in visual cortical interneurons depends on neuronal activity and TrkB ligands during an Early period of postnatal development. Cereb Cortex. 14:342–351. [DOI] [PubMed] [Google Scholar]
- 133.Lau CG, Murthy VN (2012): Activity-dependent regulation of inhibition via GAD67. J Neurosci. 32:8521–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Straub RE, Lipska BK, Egan MF, Goldberg TE, Callicott JH, Mayhew MB, et al. (2007): Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Molecular Psychiatry. 12:854–869. [DOI] [PubMed] [Google Scholar]
- 135.Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, et al. (2007): Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 27:11254–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Veikkolainen V, Vaparanta K, Halkilahti K, Iljin K, Sundvall M, Elenius K (2011): Function of ERBB4 is determined by alternative splicing. Cell cycle (Georgetown, Tex). 10:2647–2657. [DOI] [PubMed] [Google Scholar]
- 137.Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, et al. (2018): Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 359:693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS, et al. (2014): The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry. 19:486–494. [DOI] [PubMed] [Google Scholar]
- 139.Chung DW, Volk DW, Arion D, Zhang Y, Sampson AR, Lewis DA (2016): Dysregulated ErbB4 Splicing in Schizophrenia: Selective Effects on Parvalbumin Expression. Am J Psychiatry. 173:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Del Pino I, Garcia-Frigola C, Dehorter N, Brotons-Mas JR, Alvarez-Salvado E, Martinez de Lagran M, et al. (2013): Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron. 79:1152–1168. [DOI] [PubMed] [Google Scholar]
- 141.Georgiev D, Yoshihara T, Kawabata R, Matsubara T, Tsubomoto M, Minabe Y, et al. (2016): Cortical Gene Expression After a Conditional Knockout of 67 kDa Glutamic Acid Decarboxylase in Parvalbumin Neurons. Schizophr Bull. 42:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fagiolini M, Fritschy JM, Low K, Mohler H, Rudolph U, Hensch TK (2004): Specific GABA(A) circuits for visual cortical plasticity. Science. 303:1681–1683. [DOI] [PubMed] [Google Scholar]
- 143.Glausier JR, Lewis DA (2011): Selective pyramidal cell reduction of GABA(A) receptor alpha1 subunit messenger RNA expression in schizophrenia. Neuropsychopharmacology. 36:2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xue M, Atallah BV, Scanziani M (2014): Equalizing excitation-inhibition ratios across visual cortical neurons. Nature. 511:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Adams RA, Pinotsis D, Tsirlis K, Unruh L, Mahajan A, Horas AM, et al. (2021): Computational Modeling of Electroencephalography and Functional Magnetic Resonance Imaging Paradigms Indicates a Consistent Loss of Pyramidal Cell Synaptic Gain in Schizophrenia. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lewis DA, Lieberman JA (2000): Catching up on schizophrenia: natural history and neurobiology. Neuron. 28:325–334. [DOI] [PubMed] [Google Scholar]
- 147.Mallien AS, Pfeiffer N, Vogt MA, Chourbaji S, Sprengel R, Gass P, et al. (2021): Cre-Activation in ErbB4-Positive Neurons of Floxed Grin1/NMDA Receptor Mice Is Not Associated With Major Behavioral Impairment. Front Psychiatry. 12:750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wen W, Turrigiano GG (2021): Developmental Regulation of Homeostatic Plasticity in Mouse Primary Visual Cortex. J Neurosci. 41:9891–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Datta D, Arion D, Lewis DA (2015): Developmental Expression Patterns of GABAA Receptor Subunits in Layer 3 and 5 Pyramidal Cells of Monkey Prefrontal Cortex. Cereb Cortex. 25:2295–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]