

Abstract
The environmental role in disease progression has been appreciated for decades; however, the understanding of how airborne toxicant exposure can affect organs beyond the lungs is an underappreciated area of scientific inquiry. Particulate matter (PM) includes various gases, liquids, and particles in suspension and is produced by industrial activities such as fossil fuel combustion and natural events including wildfires and volcanic eruptions. Although agencies have attempted to reduce acceptable airborne particulate levels, with urbanization and population growth, these policies have been only moderately effective in mitigating disease progression. A growing area of research is focused on the role of PM exposure in the progression of Alzheimer’s disease. This review will summarize the knowns and unknowns of this expanding field.
Keywords: Alzheimer’s disease, air pollution, neurodegenerative disease, particulate matter
Can Particulate Matter Exposure Exacerbate Alzheimer’s Disease Development?
Particulate matter (PM) consists of a heterogeneous mixture of solid and liquid particles suspended in the air (Box 1)[1]. Current research has established compelling relationships between PM exposure and the development of ischemic heart disease, myocardial infarction, and lung diseases including aggravated asthma [1,2]. These connections are alarming due to the significant number of individuals routinely exposed to dangerous PM levels worldwide. Importantly, recent studies have also established connections between PM exposure and neurological diseases such as Alzheimer’s disease (AD)[3].
Box 1. What is PM?
PM is a heterogeneous mixture of particles derived from numerous anthropogenic and natural sources. These particles are classified by diameter as ultrafine (PM0.1, <0.1 μm), fine (PM2.5, <0.25 μm) and coarse (PM10, <10 μm). PM is generated by industrial activities, such as fossil fuel combustion, household activities, such as cooking or cleaning, and transportation, construction, and agriculture (Figure I). Particulates have been shown to enter the body through the lungs, gut, olfactory bulb, and various other points (Figure 1, Key Figure). Infiltration of PM into the body can cause a localized or systemic response, potentially affecting multiple organ systems following exposure. In 2021, the World Health Organization (WHO) stated that millions of deaths and over a hundred million years of healthy life are lost to air pollution exposure each year, ranking air pollution as a top ten risk factor for mortality worldwide [50,51].
In 2017, 92% of the world’s population resided in areas with PM2.5 levels above the WHO annual mean Air Quality Guideline (AQG) of 10 μg/m3; the annual mean AQG for PM2.5 was reduced further to 5 μg/m3 in 2021 [50,51]. PM exposure has been linked to respiratory, cardiovascular, and cognitive disease development as well as systemic inflammation. Furthermore, PM has been shown to directly cause neurotoxicity and oxidative stress in an in vitro cell model, but the effects are dependent on particulate size, seasonality of particles, and chemical composition [52–54].
Epidemiological data has shown that PM levels were significantly reduced during the latest COVID-19 pandemic, predominantly due to stay-at-home orders and shutdowns of businesses/work/etc. The WHO recently declared PM as a top ten health hazard, and its association with other main diseases including cardiovascular disease and cancer are alarming [55]. Although exposure to ambient/outdoor PM is assumed to be the main culprit of disease risk, the risk of indoor PM contributing to disease cannot be forgotten.
AD is a neurodegenerative (see Glossary) disorder that typically presents in individuals over 65 years old, has a wide variety of symptoms, and is the most common form of dementia [4–7] (Box 2). Recent studies have found positive correlations between PM exposure and dementia incidence in human cohorts after adjusting for community- and individual-level social and economic characteristics [8–10]. Furthermore, mouse models exposed to PM displayed elevated AD biomarkers compared to filtered air (FA) controls [10]. Studies have also shown PM exacerbates the aggregation and deposition of AD hallmarks amyloid beta plaques (Aβ) and neurofibrillary tangles (NFTs) in the brain through mechanisms related to microglia/astrocyte activation, oxidative stress, immune activation, and neuroinflammation [3,10–13]. This review presents evidence of neurologic changes induced by PM, with an emphasis on the mechanisms associated with AD pathogenesis.
Box 2. Alzheimer’s Disease Explained.
AD, a progressive, neurodegenerative disease, accounts for 50–75% of all dementia cases [7]. In 2021, approximately 6.2 million Americans ages 65+ lived with AD, while 55 million individuals globally lived with AD and other dementias; cases are expected to increase threefold by 2050 [56–58]. Symptoms of AD include reduced mental processing, working memory, creativity, mobility, attention, problem-solving, memory recall efficiency, odor identification, and language skills, and altered mood and sleep patterns [4,5]. Symptom severity ranges from mild to severe impairment, where daily living assistance is required. Unfortunately, a cure for AD does not exist; death due to AD and its associated complications is imminent and typically occurs 8–10 years after diagnosis [7,59].
The two main forms of AD are late onset AD (LOAD), occurring after age 65, and early onset AD (EOAD), occurring prior to age 65 [68]. LOAD accounts for 90–95% of cases and typically has sporadic origins, i.e., non-genetic etiologies [60,61]. EOAD accounts for the remaining 5–10% of cases and is commonly considered familial EOAD due to its heritability and established genetic mutations [60]. Of all EOAD patients, 35–60% have a first-degree relative suffering from AD. Mutations in the genes coding for amyloid precursor protein (APP), presenilin 1, and presenilin 2 are established causes of EOAD [70]. Meanwhile, LOAD develops from an amalgam of environmental and external risk factors [62]. Approximately two-thirds of all AD patients are female, leading researchers to believe that sex-driven developmental and physiological differences contribute to AD development [63]. Non-genetic risk factors associated with AD include, but are not limited to, high systolic blood pressure during midlife, Type 2 diabetes, obesity, and environmental exposures, such as PM [4,64–66].
AD is primarily diagnosed through psychiatric evaluations and memory and cognitive assessments as symptoms become more clinically apparent, though amyloid PET imaging may also be utilized [6,7]. Amyloid PET imaging detects the presence of β-amyloid in the brain antemortem and is available for clinical use, but low reimbursement rates currently minimize the accessibility of this diagnostic tool [67].
AD treatments remain limited due to an incomplete understanding of the mechanisms driving Aβ and NFTs formation. Current treatments focus on symptom alleviation, as therapeutics designed to slow or reverse AD progression are in the infancy stages of development. Acetyl-cholinesterase inhibitors are commonly used to treat AD symptoms, and memantine is another acceptable option for symptom relief [67].
PM Effects on the Brain
Environmental exposures, including PM, are thought to contribute to AD development and progression. Hallmarks of AD amplified by PM, such as Aβ deposition, NFT accumulation, and cognitive impairments, may manifest well before AD clinical diagnosis [14,15]. In addition to these disease characteristics, many other alterations that potentiate AD, i.e., microglial activation, systemic/neuro inflammation, oxidative stress, immune activation, and behavioral changes, have been found in PM-exposed animals [3,16–20]. The following section will discuss clinical hallmarks of AD and associated changes in the context of PM exposure through clinical and preclinical studies to better understand PM exposures and AD development.
PM-Associated AD Markers in Human Subjects
Urban and rural locations experience varied levels of PM2.5 that periodically exceed quality recommendations, but areas, including Mexico City Metropolitan Area (MCMA) face lifetime PM2.5 exposures chronically higher than annual guidelines [21]. The Montreal Cognitive Assessment (MoCA) was validated in a study comparing MoCA to the MMSE, with a mild cognitive impairment (MCI) detection sensitivity rate of 90% compared to the MMSE rate of 18%; thus, the MoCA is considered a reliable screening tool for MCI by addressing several cognitive domains and offering insight into whether individuals require additional cognitive assessment/attention [21,22]. The MoCA was implemented in a study of young middle socioeconomic class urbanites (21.6 ± 5.8 years old) living in MCMA, Hermosillo (HER), and several other PM2.5-polluted cities exceeding United States Environmental Protection Agency (USEPA) annual standards of 12 μg/m3 to quantify individuals who performed abnormally on the assessment based on PM2.5 levels [21]. Findings revealed 55% of participants scored within the MCI score range, 30.3% of which were considered AD scores [21]. HER, with the lowest PM2.5 concentrations above USEPA standards, was a control city; MCMA was a megacity (~2.5x higher PM2.5 levels than USEPA standards); and all cities except the control were combined into a group (OTHERS) for comparison purposes [21]. MoCA scores were considered statistically equivalent across residencies, however, when considering specific tasks and tests, differences between HER, MCMA, and OTHERS became apparent in cube drawing, language fluency task, abstraction, and orientation, pointing to a stratification in cognitive ability based on PM2.5-exposure levels [21]. The cognitive domain index scores specifically showed participants with scores within the MCI range experienced significantly reduced Executive, Language, Visuospatial, and Composite Delay Memory [21]. Here, just 20 years of PM2.5 exposure above air quality standards appears to propel over half of the participants into the prodromal stage of AD, approximately 40 years earlier than symptoms are typically distinguishable in AD patients. A cross-sectional study of older adults (mean age of 75.8 years) suffering from cognitive impairment living in areas with air quality values ranging from 4.01 μg/m3 to 22.84 μg/m3 was conducted to determine whether air quality levels impact the likelihood of receiving a positive amyloid positron emission tomography (amyloid PET) scan, indicating the presence of neuronal Aβ [23]. Data from 18,178 individuals who experienced MCI (60.5%) or dementia (39.5%) from the Imaging Dementia—Evidence for Amyloid Scanning Study were used [23]. Those living in areas with higher PM2.5 concentrations had a greater likelihood of a positive amyloid PET scan result when factors including demographics, lifestyle, socioeconomics, and comorbidities were considered [23]. A similar study examined the association between AD hallmarks and PM2.5, PM10, and PM2.5abs, a marker of black/elemental carbon, in cognitively unimpaired 57-year-old Barcelona residents from the ALFA + study (Alzheimer and Families) [24]. AD hallmarks were measured by determining cerebrospinal fluid (CSF) biomarker concentrations (Aβ42, Aβ40, hyperphosphorylated tau (p-tau), total tau (t-tau), neurofilament light (NfL), and cerebral amyloid load (centiloid)) and Aβ deposition [24]. Family history and apolipoprotein E (APOE) ε4 status were considered, and land use regression models were implemented to estimate PM exposures for individuals based on residential addresses [24]. Positive associations were found between both PM2.5 and PM10 and CSF NfL levels. Upon adjusting for APOE-ε4 status, associations remained between individuals who were CSF Aβ-positive and PM2.5, PM10, and PM2.5abs and centiloid values, a scale used to compare neuronal Aβ deposits across positron emission tomography (PET) imaging tracers [24,25]. APOE-ε4 status did not alter associations, though a stronger effect of PM exposure on CSF NfL was found among carriers [24]. The APOE status of individuals must be considered when conducting research on AD and PM, as studies have found different rates of accelerated global cognitive decline and all-cause dementia based on APOE genotypes and 3-year average PM2.5 exposures. When individuals aged 65–79 years with the APOE-ε4 genotype resided in PM2.5 locations exceeding National Ambient Air Quality Standards, rates of cognitive decline and all-cause dementia were amplified [26]. Furthermore, studies by Calderón-Garcidueñas et al. have found increased p-tau and Aβ diffuse plaque levels in children and young adults exposed to PM2.5 and ozone concentrations exceeding USEPA standards in MCMA [27]. The Wechsler Intelligence Scale for Children-Revised was implemented and found MCMA children possessing the APOE-ε4 genotype performed significantly worse in Arithmetic, Information, Picture Completion, and Digit Span tests, indicating differences in short and working memory performance compared to non-carrier APOE-ε3 subjects [27]. These studies point to a reduction in cognitive performance and AD potentiation based on genotype and lifetime PM2.5 exposures exceeding US national guidelines. The changes in memory performance found among children demonstrate early consequences associated with PM and point to a compounding effect of PM exposures over time that may culminate in the development of AD.
Further evidence of long-term PM10 exposure on AD pathology is found by studying a population of cognitively normal and MCI older adults ages 65+ from the Korean Brain Aging Study for the Early Diagnosis and Prediction of AD via clinical assessments, PET scans, MRIs, and PM10mean (mean concentration of PM10 from the past 5 years) concentration matching with participants’ residences [28]. Individuals exposed to the highest tertile of PM10mean (48.0–67.0 μg/m3) experienced a greater Aβ positivity risk assessed via PET scan after controlling for confounding variables, but no significant association was found between tau accumulation nor AD-signature cortical thickness and PM exposure [28]. The human studies described suggest greater incidence of eventual AD diagnosis across locations and populations exposed to higher PM levels. These data should serve as an alarm for public health experts to examine healthy air quality levels and strategies to minimize air particulate levels to prevent lifetime exposures exceeding quality guidelines and mitigate disease [23,24,28]. Furthermore, these data point to the need to study animal and in vitro models to gain an increased understanding of the mechanisms driving AD development in the context of PM exposure.
PM-Associated AD Markers in Animal Models and Beyond
Behavioral Response.
Several animal studies offer insight into PM-induced behavioral changes in transgenic AD animal models. Memory and behavioral alterations were found after exposing 12.5–14-month male 3xTgAD and non-carrier control animals to PM0.1 (~58 μg/m3) or FA (4 h/day, 4 days/week) for 2-weeks; reduced spatial learning and impaired short-term memory were found in PM-exposed animals, independent of genotype [19]. Differences in depressive-like and anxiety-like responses in C57BL/6 male mice exposed to PM2.5 (16.85 μg/m3) versus FA over a 10-month (6 h/day, 5 days/week) period have also been detected [29]. Further, reduced spatial learning and memory in PM2.5-exposed animals compared to FA-exposed controls of the same C57BL/6 genotype were found [29]. These findings are especially important, as the animals exposed to PM2.5 were non-transgenic, and thus possessed no predisposition to AD symptomology, similar to most individuals diagnosed with AD. Additionally, the PM2.5 exposure concentrations are environmentally relevant to levels experienced by individuals living in metropolitan areas. Motor coordination deficits have been found in 5xFAD mice exposed to diesel engine exhaust for 13-weeks with a 0.95 mg/m3 dose (6 h/day, 5 days/week) compared to FA-exposed controls regardless of indistinguishable Aβ plaque loads between exposures [30]. Exposing 6-month-old 3xTgAD mice to PM2.5 at a mean concentration of 11.38 μg/m3 for three months failed to elicit spatial learning/memory and motor activity differences [31]. The failure of this PM2.5 exposure to elicit changes associated with clinical presentation of disease while promoting differences in p-tau and oxidative stress levels serves as an important reminder that hallmarks develop far before clinical diagnoses [31]. While findings vary in terms of the age at which behavioral changes present based on PM2.5 concentrations, exposure duration, and genotype, spatial learning and memory and motor coordination challenges are signs consistently found among PM-exposed animals, aligning with the clinical presentation of AD (Box 2) [19,29–31].
Systemic Response.
Aβ plaques are considered hallmark characteristics of AD-brains, but these markers do not act independently. Convincing evidence that a compensatory response occurs in the brain to maintain homeostasis is supported by microglial activation which is often found in conjunction with Aβ plaques [32]. Microglia are activated and act protectively when the brain undergoes stress or injury [33]. Early activation allows for phagocytosis and antigen presentation; however, prolonged activation can have consequences including enhanced neuroinflammation. In AD, microglia appear to fail to clear Aβ and return to a resting state; though, it is also possible their presence may perpetuate Aβ accumulation [32]. Regardless, both scenarios result in an abundance of inflammatory mediators, and neuronal damage [33].
Exposing aged male 3xTgAD mice and non-carrier counterparts to PM0.1 or FA elicited significantly higher levels of total microglia in 3xTgAD mice than non-carrier counterparts, offering support for the complex nature of microglial activation and response in AD [16]. PM0.1 exposure significantly lowered overall microglia present in both 3xTgAD animals and non-carrier counterparts, as determined by ionized calcium-binding adapter molecule 1 (Iba1) and Sholl analysis [16]. Still, transgenic mice exposed to PM0.1 experienced significantly more microglia per plaque (9.3 microglia/plaque) compared to air-exposed controls (4.7 microglia/plaque) [16]. Another study further exemplifies the nuances between microglial activation and AD as sex-dependent differences in microglial responses were found based on air exposure [34]. After continuously exposing male and female transgenic (TgF344-AD) rats and non-carrier control littermates to traffic-related air pollution (TRAP) (15.6 ± 3.7 μg/m3), microglial activation differences were assessed by co-labeling brain sections with Iba1 and CD68, a marker of phagocytic microglia [34]. Among female rats, TRAP-exposure increased microglial activation in both genotypes by 3-months and in TgF344-AD at 6-months of age; however, microglial activation was reduced in both genotypes of TRAP-exposed 15-month-old animals (14-month exposure) [34]. Male TRAP-exposed rats experienced heightened microglial activation at 6 months of age, no change at 10 months, and greater activation among both genotypes at 15-months [34]. These findings demonstrate the complexity associated with microglial responses and AD; microglia can contribute as early responders to clear Aβ plaques, though as the plaques persist, their existence and ability to recruit additional inflammatory mediators may further contribute to plaque deposition [16,32,34].
Studies have shown enhanced circulating cytokines, such as IL-2, IL-6, IL-1β, TNF-α and TNF-β in AD patients, providing evidence of an elevated inflammatory response, but the relationship between PM exposures and systemic inflammation remains unclear [17,35]. A study exposing 11–12-month-old C57BL/6 mice to 1000 μg/m3 of PM0.1 for 3-weeks (8 h/day, 5 days/week) found nonsignificant differences in serum metabolite profiles post-PM0.1 exposure, representing the uncertainty associated with the ability of PM0.1 to induce systemic changes and impact neurological processes through this route [36]. Of note, transgenic animals were not utilized in this study, thus, the effect of PM alone on AD development is explored rather than its exacerbation of AD; it is unclear whether PM0.1 has comparable potential to PM2.5 to promote an AD phenotype.
Olfactory Route.
The olfactory route is a primary path by which PM reaches the brain, and olfactory dysfunction is an early clinical symptom of AD; thus, the olfactory route is a relevant region to study in the context of PM exposure and AD (Box 3) [37]. After PM is inhaled through the nose, it translocates from the olfactory nerve to the olfactory bulb. Continuously exposing C57BL/6 mice to PM2.5 (mean daily 140.18 μg/m3) for 4- or 8-weeks induced changes in the olfactory bulbs, including neuronal apoptosis, cell disorganization, and increased TNF-α signaling [38]. Transmission electron microscopy revealed particles accumulated in the olfactory bulbs of PM2.5-exposed animals [38]. Interestingly, several findings from this study, including the increased inflammatory response, were highest after 4-weeks, indicating a compensatory response or tolerance may have been achieved by 8-weeks of PM2.5 exposure [38]. Within the olfactory bulbs of PM2.5-exposed animals, significantly heightened p-tau levels have been found compared to FA-exposed counterparts [31]. Significant increases in malondialdehyde, indicative of increased free radicals/oxidative stress, have also been detected in the olfactory bulb and hippocampus of PM2.5-exposed animals [31]. Among PM0.1-exposed animals, inflammatory markers, i.e., TNF-α, IFN-y, and IL-12p70, were found in the olfactory bulb [36,39]. Heightened inflammatory markers and increased malondialdehyde suggest early changes seen in AD are associated with the olfactory route of entry, namely loss of smell [31,39]. Altered odor identification is considered a preclinical biomarker of AD; studies have found reduced olfactory performance in children exposed to unhealthy PM levels and an association between heightened PM2.5 levels and odds of developing anosmia (loss of smell), further supporting a clinical connection between the olfactory route changes found among PM-exposed animals [5,31,39–41].
Box 3. PM Modes of Entry.
Particulate size, chemical composition, and chemical solubility are critical in determining whether particles can access the brain [68–72]. There are two main proposed routes whereby PM reaches the brain via inhalation: 1) directly from nose to brain via the olfactory bulb or 2) indirectly by translocation from the respiratory tract into circulation by bypassing the blood-brain barrier (BBB) (Figure 1).
Direct delivery of particles or specific chemicals from the nose to the brain occurs via two modes of entry. The first involves transporting inhaled particles from the olfactory nerve to the olfactory bulb via olfactory receptor cells (Figure 1)[70,72–74]. Direct particle transport has been demonstrated in the olfactory nerves of squirrel monkeys, which showed translocation of gold particles anterogradely from olfactory epithelium to the olfactory bulb [75]. Similar studies of PM0.1 inhalation exemplify that particles can be translocated from nose to brain in rats [68,69]. Furthermore, studies have identified deposits of externally derived, ultra-fine magnetite particles in the frontal cells of human brains thought to have entered the brain via the olfactory unit due to the small particle size (<200 nm diameter) [76]. A second mechanism whereby chemicals or particles are transported from nose to brain is through intercellular clefts in the olfactory epithelium or along nerves [77]. However, this has only been demonstrated with intranasal administration and not inhalational exposure.
Indirect translocation of particles from the respiratory or intestinal tract into the circulation followed by BBB entry is not a simple process. While translocation of PM0.1 from the lungs to the circulation has been readily shown, the BBB makes it exceptionally difficult for particles, even PM0.1, to traverse [73,78]. When rats were exposed to radiolabeled iridium nanoparticles, only an exceptionally small fraction (0.001–0.01%) of particles were found in the brain; smaller nanoparticles (20 nm) were more commonly detected than larger counterparts (80 nm) [78]. Once the BBB is breached, PM0.1 has been shown to deposit in the striatum, frontal cortex, and cerebellum of rats exposed to ultra-fine manganese oxide particles [68]. Intubation and ventilation of exposed animals prevented particles from translocating directly from nose to brain [78]. PM can indirectly affect the brain via a systemic inflammation mechanism by infiltrating into the body and stimulating an increase in circulating pro-inflammatory cytokines in the pulmonary and cardiovascular systems, leading to a systemic inflammatory response, microglia activation, and neuroinflammation (Figure 1)[79,80].
Hippocampus.
The hippocampus is responsible for learning and memory processes and is implicated in AD development [34]. Studies have demonstrated PM’s ability to initiate neurological effects beyond the olfactory bulb and penetrate barriers within the body and brain [16,18,34]. Findings from TRAP-exposed animals support the hypothesis that PM nanoparticles can translocate to the brain, specifically the hippocampus, as exposed animals possessed visible refractive particles in the hippocampal region [34]. Hyperspectral brain imaging found a significant increase of hippocampal refractive particles in all TRAP-exposed animals after 3-, 6-, 10-, and 15-months of exposure compared to FA-controls [34]. These findings, paired with the increased microglia activation among TRAP-exposed rats discussed earlier, support the ability of PM to traverse barriers and initiate neuroinflammation [34]. Significant increases in tau pathology in 3xTgAD mice exposed to PM0.1 (~4%) compared to FA-exposed controls (~2%) was found in the model of aged 3xTgAD mice [16]. However, this study found no changes in soluble or insoluble Aβ42 concentrations from 3xTgAD hippocampal homogenates [16]. Microglia were detected via Iba1 staining, but no significant changes were found between exposure groups within the hippocampus [16]. Yet, when considering the subiculum, an area found to atrophy during early stages of AD, a significant interaction was found [16]. Failure to detect changes in Aβ42 concentrations may be explained by the acute PM0.1 exposure (2-weeks) or the genotype selected, though subiculum changes indicate that PM0.1 exposure effects 3xTgAD animals differently than non-carrier controls and may affect Aβ plaque aggregation patterns accordingly [16,42].
Park et al. found changed metabolic profiles and Aβ levels in 11–12-month-old C57BL/6 mice exposed to 1000 μg/m3 of PM0.1 compared to control air-exposed animals [36]. Thirty-one distinct metabolic changes between PM0.1-exposed and control air were visible in the hippocampus post-exposure, 25 of which were downregulated [36]. Of the downregulated metabolites, threonic acid and methionine sulfoxide experienced the greatest reductions [36]. These findings are of interest as studies indicate threonic acid combined with magnesium salt enhances learning and memory in APPswe/PS1sE9 mice, and AD brains exhibit reduced methionine sulfoxide reductase activity, which reverses methionine sulfoxide back to methionine to clear oxidants [36,43–45]. Additional findings include altered redox homeostasis through the methionine-glutathione pathway in PM0.1-exposed animals and significantly increased Aβ levels in the hippocampus of PM0.1-exposed animals [36]. Saveleva et al. conducted a shorter study (2-weeks) and found Aβ plaque aggregation was visible in hippocampal and cortical brain regions of transgenic mice, though there were no differences between transgenic animals exposed to PM0.1 (89.5 μg/m3) and control air after 2-weeks (4 h/day, 5 days/week) [39]. The discrepant findings between Park et al. and Saveleva et al. may be explained by differences in study duration (3- vs. 2-weeks), PM0.1 concentration (1000 vs. 89.5 μg/m3), mouse model (C57BL/6 vs. 5xFAD), and animal age (11–12 months vs. 6 months) [36,39]. While both studies provide evidence of Aβ plaque aggregation, the findings from Park et al. should be emphasized; with the lack of PM0.1 regulations at this time, the heightened exposure concentration and increased duration show the potential of PM0.1 to promote AD hallmarks in a non-transgenic animal model [36]. These results may have been achieved by Saveleva et al., had the study been longer, concentration higher, and animals older [39]. Still, evidence of early inflammatory changes associated with AD was found predominately in the hippocampal regions of PM0.1-exposed animals [39]. A PM0.1 inhalation effect was also detected, as non-carrier control and 5xFAD mice experienced significantly heightened IFNy and IL-12p70 levels compared to control air-exposed animals [39]. The increased cytokine levels and elevated mitochondrial superoxide dismutase found indicate significantly greater oxidative stress among animals exposed to PM0.1 in both studies [36,39]. Park et al. provide additional support for oxidative stress and inflammation within the hippocampus post PM0.1-exposure; 4-HNE (oxidative stress marker) was significantly increased in the hippocampus, as was TNF-α [36].
Entorhinal and Cerebral Cortices.
Continuous exposure of 6-month-old 3xTgAD mice to PM2.5 at a mean concentration of 11.38 μg/m3 for three months resulted in reduced cortical neurons in PM-exposed 3xTgAD mice, but visible differences related to neuron loss were not found elsewhere [31]. Significant reductions in Iba-1 were found in the cerebral cortex of exposed animals, though no changes in Aβ42 were found [31]. Continuous TRAP-exposure resulted in significantly greater neuronal cell loss in the entorhinal cortex of non-carrier control and TgF344-AD animals at 15 months compared to FA-exposed counterparts; TgF344-AD rats had significantly fewer neurons than non-carrier controls [34]. By 15-months, TRAP-exposed TgF344 animals had significantly higher anti-amyloid and Aβ levels compared to FA-controls [34]. Additionally, TRAP-exposed non-carrier control animals expressed significantly higher p-tau:t-tau levels; this change was not consistent among TRAP-exposed transgenic animals [34].
In Vitro Models.
An in vitro model of PM2.5-polluted human brains (PMBs) via a 3-D microfluidic platform was created to study the human brain immune response to PM-exposures [18]. After confirming the formation of tight junctions in the model, 0.1–10 μg/mL of PM2.5 was added to the lumen and ultimately precipitated into the brain side of the model when assessed 24-hours post-exposure [18]. This precipitation demonstrated the BBB integrity was compromised, and subsequent analysis revealed PM2.5 decreased zonula occludens-1 abundance, indicating tight junction downregulation was responsible for reduced BBB function [18]. PM2.5 treatment also induced astrogliosis and caused microglial infiltration [18]. Further investigation into the microglial response demonstrated an M1 phenotype as evidenced by enhanced CD11b and CD86 coupled with increased IL-6 and IL-8 by PM2.5-exposure [18,46]. Another study treated BV2 cells, a microglial cell line, with 100 μg/mL PM2.5 for 12 hours and neuroblastoma N2A cells with a conditioned medium from the BV2 cells for 24 hours[38]. PM2.5-treated BV2 cells exhibited significantly increased TNF-α levels, pointing to direct activation of microglia cells by PM2.5 [38]. The co-culture system resulted in apoptosis among N2A cells, indicating PM2.5 could cause neuronal cell death by inducing systemic inflammation and its associated downstream effects (microglia activation, TNF-α release) or via direct action [38]. Although several mouse models function to study the pathophysiological effects of PM on AD development, additional in vitro work is necessary to expand our understanding of mechanisms and modifiable factors related to AD and PM exposures, including further elucidating neuronal and microglia responses to PM [47].
Concluding Remarks
Understanding how the environment can affect health and disease progression is important to inform policy as well as design novel mitigation strategies to combat the increase in PM-associated illness. It is apparent that exposure to consistently high levels of PM can have significant effects on AD progression; however, additional studies are needed to understand the mechanisms whereby PM exposure affects the brain and associated organs. Additionally, the majority of studies on PM-induced AD progression have examined PM2.5; therefore, further studies to define the relative contribution of PM0.1 to AD-associated effects are warranted. Determination of such mechanisms will promote prevention and illness alleviation and inform public health policy to enact stricter air quality guidelines. The role of systemic inflammation cannot be emphasized enough in beginning to understand AD progression and severity, and future studies focusing on PM may elucidate methods to delay the onset and/or severity of AD. For instance, it would be helpful to study “clean” versus “dirty” environments and the associated promotion of various neurodegenerative diseases. The Clinician’s Corner offers immediate strategies for short-term solutions to PM exposure, and the Outstanding Questions highlights areas of ongoing scientific inquiry in need of further exploration.
Clinician’s Corner
Mask wearing has been a long-time practice in many large urban areas in Asia and Europe and is now a common global practice due to the COVID-19 pandemic. Further messaging should be developed to encourage individuals to wear proper masks in outdoor spaces on high-pollution days to mitigate development of PM-associated diseases [48].
Indoor air purifier systems implementing high efficiency particulate air (HEPA) filters may be implemented throughout homes and businesses to reduce exposure to air particulates of different sizes and compositions [49].
Sensitive individuals with pre-existing medical diagnoses should be reminded that outdoor activities on days of high pollution are strongly discouraged.
Those that live in heavily polluted areas, or areas with improper ventilation, should be counseled on the potential risks of PM-associated disease development, including AD.
Patients who have the potential for occupational exposure to concentrated PM should be assessed for potential lung and brain effects, and this should be repeated routinely while remaining at risk.
Efforts should be focused on understanding the clinical effects of PM exposure to inform public health legislative bodies to enact stricter air quality control.
Figure I. Anthropogenic Sources of Particulate Matter.
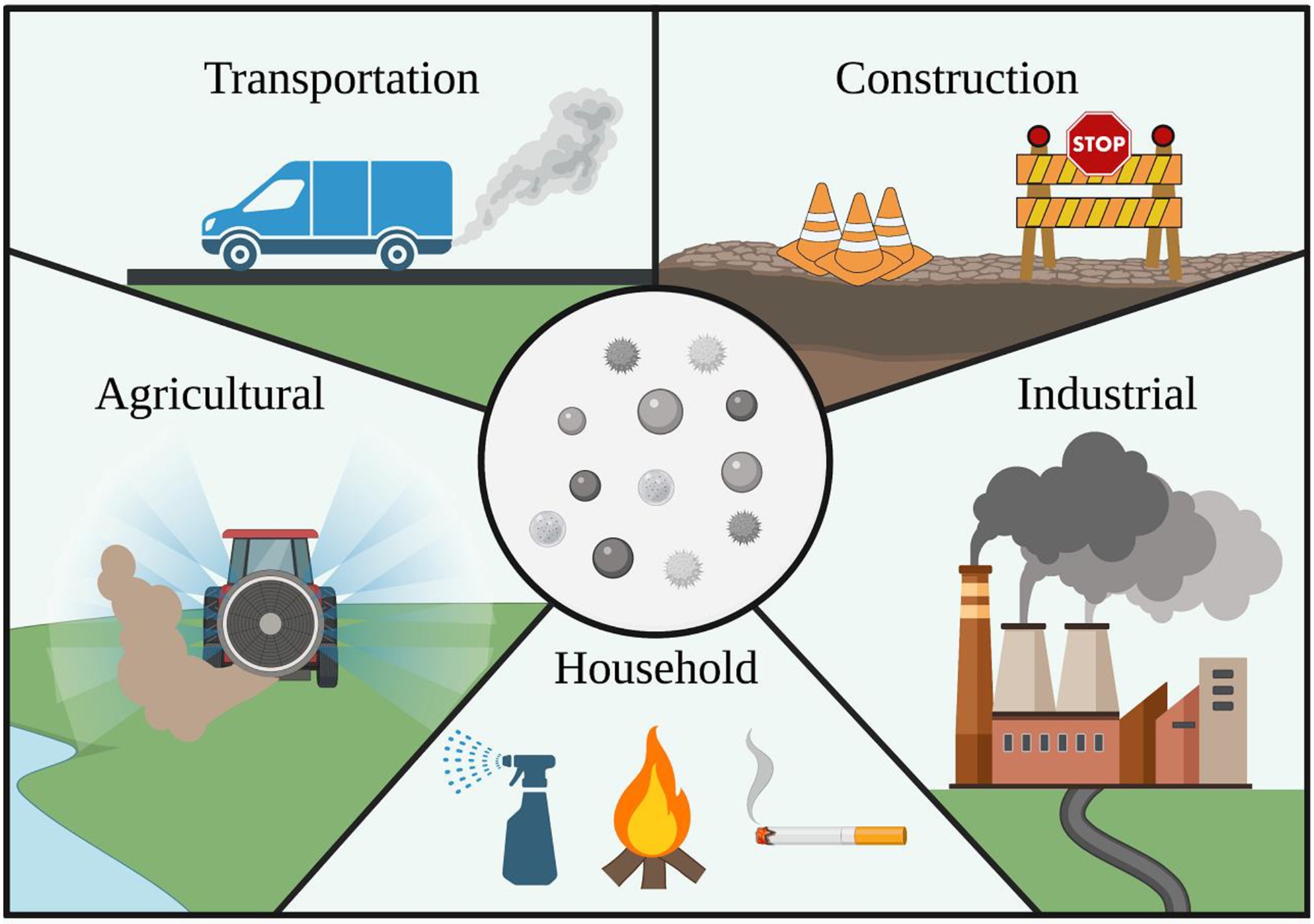
The common sources of human-derived particulate matter (PM) are transportation, construction, agricultural, industrial, and household pollutants. Agricultural sources of PM result from spraying crops with pesticides and fertilizers, combusting fossil fuels to power equipment, and aerosolizing dust during the movement of equipment and harvesting of crops. Industrial sources of PM originate mainly from the emissions of combusted fuels used to power manufacturing processes. Household sources of PM predominately affect the composition of interior air and are mainly derived from activities like cooking, using cleaning products, burning candles and fires, and smoking. Construction introduces various types of PM into the atmosphere through activities such as grinding and resurfacing roads, grinding metals, welding, cutting materials, excavating, moving materials, and emitting combustion fumes from construction equipment. Lastly, transportation sources of PM derive from brake dust, train and subway wheelsets interacting with rails, tires and pavement interacting, and fuel combustion associated powering the transporters.
Figure 1. Key Figure. PM Route to the Brain.
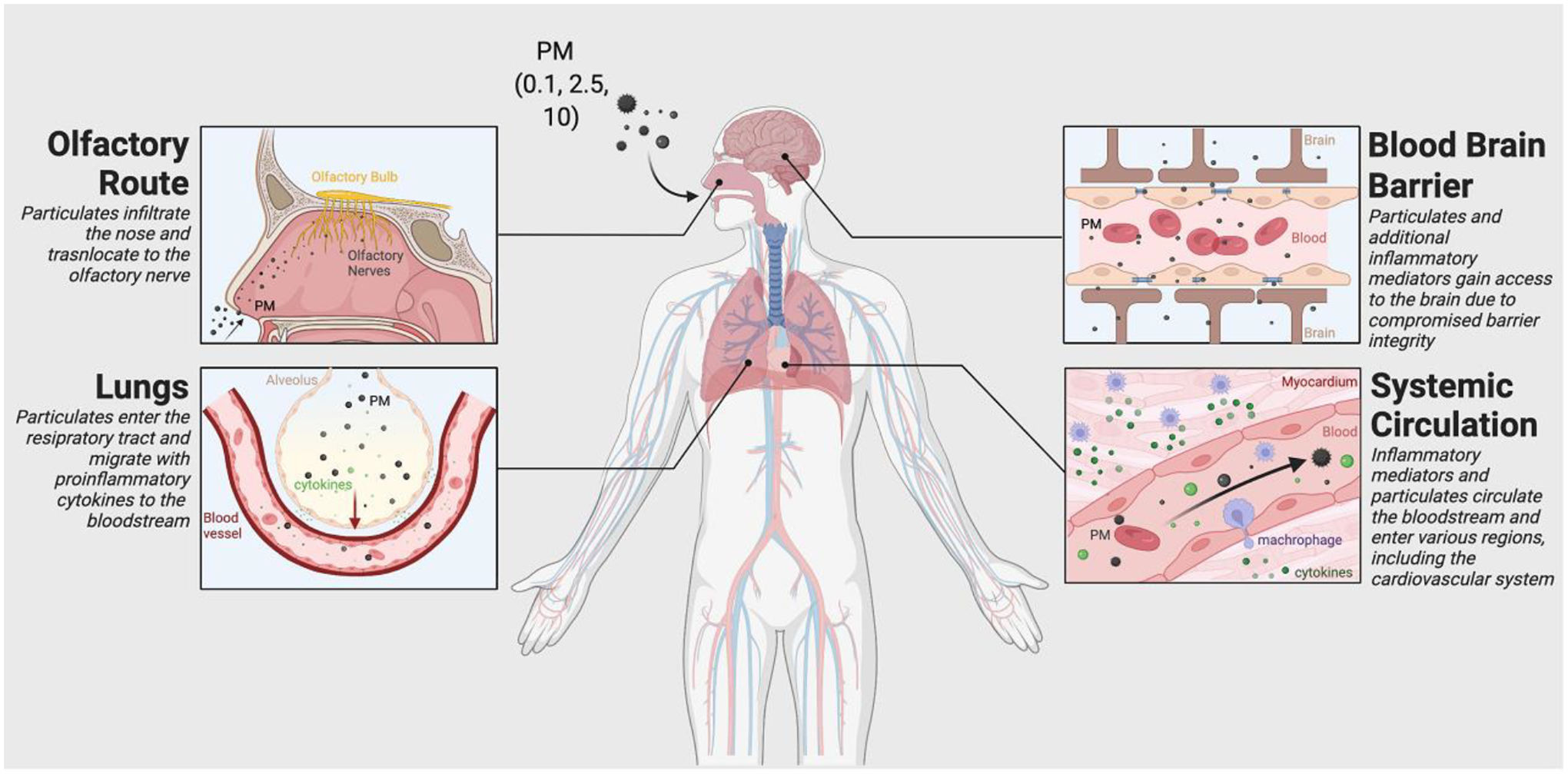
Particulate matter of various sizes (PM2.5, PM10, PM0.1) is inhaled through the nose and can directly affect the olfactory bulb or follow a peripheral route through the lungs. Upon entering the olfactory system, particulates may infiltrate the olfactory nerve and translocate to the brain. When PM follows the peripheral route, it navigates the respiratory tract and eventually reaches the lungs. Here, PM elicits an inflammatory response; proinflammatory cytokines and particulates subsequently migrate to the bloodstream and into the systemic circulation. After entering the bloodstream, PM and other inflammatory mediators can travel throughout the body and cross the BBB due to compromised barrier integrity. PM may also reach the cardiovascular system, where a localized inflammatory response may occur, thus releasing additional inflammatory mediators into the circulation. This reaction may amplify systemic inflammation and ultimately contribute to chronic inflammation in AD patients.
Outstanding Questions:
We know that the environment influences AD progression; however, is PM-associated AD reversible?
Will the recent COVID-19 pandemic cause the general public to view mask wearing as a common measure to prevent disease or a nuisance?
While post-natal exposure to environmental toxicants, including PM, are understood to cause systemic inflammation and enhance reactive oxygen species, does in utero, or even preconception exposure to PM potentiate adulthood development of AD?
Since the data on PM-associated AD have been variable, particularly with regards to the PM source and type, what are the roles of each particulate in AD development? This information will promote effective study designs to combat such associations.
Highlights:
Worldwide, individuals are routinely exposed to levels of particle matter (PM) above the air quality guideline levels set by the WHO, placing them at risk for developing PM-induced diseases.
PM enters the body and affects the progression of numerous diseases, many of which impact vital organs such as the heart, lungs, brain, and gut.
PM may infiltrate the brain and engender the development of numerous cognitive diseases by crossing the blood-brain barrier or traveling through the olfactory bulb.
PM exposure and AD development are strongly correlated, and PM is believed to exacerbate AD development via a complex interplay of mechanisms.
Treatments for AD remain limited, but a deeper understanding of the risk factors and AD pathogenesis will enable researchers to design studies aimed to mitigate AD symptoms and development.
Acknowledgments
This work was supported by salary support for Dr. Yael-Natalie H. Escobar (T32HL149637) and grants from the National Institutes of Health (R01 HL139348, R01 AG057046) and the American Heart Association (20YVNR35490079) to LEW. All figures were created using BioRender.com.
Glossary
- Aβ40
40-reside peptide derived from the cleavage of the protein amyloid precursor protein and is major component of Aβ plaques
- Aβ42
42-residue peptide derived from the cleavage of the protein amyloid precursor protein and is major component of Aβ plaques
- amyloid beta plaques (Aβ)
insoluble extracellular aggregations produced from the processing of amyloid precursor protein (APP) by β and γ secretases
- amyloid positron emission tomography scan (amyloid PET)
a functional imaging technique that detects the presence of amyloid in brain tissue, and brain blood vessels by detecting positrons emitted during the decay of a radioisotope-labeled ligand that binds to amyloid
- anthropogenic
derived from human activity – not naturally occurring
- apolipoprotein E (APOE)
a multifunctional protein that is mainly involved in the transportation of lipids through the cerebrospinal fluid and plasma while simultaneously being a risk factor for AD pathology. The APOE-ε4 allele is the most risk-inducing allele for the development of AD
- blood-brain barrier (BBB)
the specialized microvasculature that surrounds the brain. The microvasculature consists of non-fenestrated blood vessels with specialized endothelial cells which tightly regulate what can and cannot pass from the circulating blood system to the brain
- cerebral cortex
outer brain layer responsible for higher level processes, including emotion, thought, language, memory, reasoning, and consciousness
- C57BL/6
an inbred mouse strain widely used in research as a control
- entorhinal cortex
located within the medial temporal lobe and responsible for shuttling information to and from the hippocampus, this structure plays a critical role in memory and navigation
- hippocampus
brain structure found within the temporal lobe with major learning and memory responsibilities
- hyperphosphorylated tau (p-tau)
an abnormal, pathological form of tau protein found to aggregate and contribute to neurofibrillary tangles and the associated physiological degradation and neuronal death seen in AD patients
- mild cognitive impairment (MCI)
cognitive and/or memory decline that is above normal aging, but below diagnostic standards for dementia. Affected persons are able to lead independent lives. The condition may return to normal, remain stable, or progress to dementia
- neurodegenerative
involving the death or dysfunction of cells in the central nervous system
- neurofibrillary tangles (NFTs)
dense intracellular accumulations of the hyperphosphorylated protein tau that contribute to neuronal death
- olfactory route
the route by which inhaled particles are translocated from the nasal cavity to olfactory bulb
- TgF344-AD
a transgenic AD rat model which has two mutant alleles associated with AD: APPSwe, PSEN1dE9
- 3xTgAD
a transgenic AD mouse model which has three mutant alleles associated with AD: APPSwe, tauP301L, PS1M146V
- 5xFAD
a transgenic AD mouse model which has five mutant alleles associated with AD: APPSwe, APPI716V, APPV717I, PSEN1M146L, PSEN1L286V
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors have no interests to declare.
References
- 1.Kim K-H et al. (2015) A review on the human health impact of airborne particulate matter. Environment International 74, 136–143 [DOI] [PubMed] [Google Scholar]
- 2.Hadei M and Naddafi K (2020) Cardiovascular effects of airborne particulate matter: A review of rodent model studies. Chemosphere 242, 125204. [DOI] [PubMed] [Google Scholar]
- 3.Block ML and Calderón-Garcidueña. (2009) L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends in Neurosciences 32, 506–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kioumourtzoglou MA et al. (2016) Long-term PM2.5 exposure and neurological hospital admissions in the northeastern United States. Environmental Health Perspectives 124, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lafaille-Magnan M-E et al. (2017) Odor identification as a biomarker of preclinical AD in older adults at risk. Neurology 89, 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atri A (2019) The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Medical Clinics of North America 103, 263–293 [DOI] [PubMed] [Google Scholar]
- 7.Lane CA et al. (2018) Alzheimer’s disease. European Journal of Neurology 25, 59–70 [DOI] [PubMed] [Google Scholar]
- 8.Chen H et al. (2017) Exposure to ambient air pollution and the incidence of dementia: A population-based cohort study. Environment International 108, 271–277 [DOI] [PubMed] [Google Scholar]
- 9.Ailshire JA and Crimmins EM (2014) Fine particulate matter air pollution and cognitive function among older US adults. American Journal of Epidemiology 180, 359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahu B et al. (2021) Particulate Matter Exposure Exacerbates Amyloid-β Plaque Deposition and Gliosis in APP/PS1 Mice. Journal of Alzheimer’s Disease 80, 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozben T and Ozben S (2019) Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clinical Biochemistry 72, 87–89 [DOI] [PubMed] [Google Scholar]
- 12.Calsolaro V and Edison P (2016) Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s and Dementia 12, 719–732 [DOI] [PubMed] [Google Scholar]
- 13.Cacciottolo M et al. (2020) Traffic-related air pollutants (TRAP-PM) promote neuronal amyloidogenesis through oxidative damage to lipid rafts. Free Radical Biology and Medicine 147, 242–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKay NS et al. (2022) Beta-amyloid moderates the relationship between cortical thickness and attentional control in middle- and older-aged adults. Neurobiology of Aging 112, 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sperling RA et al. (2020) Association of Factors with Elevated Amyloid Burden in Clinically Normal Older Individuals. JAMA Neurology 77, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herr D et al. (2021) Effects of concentrated ambient ultrafine particulate matter on hallmarks of Alzheimer’s disease in the 3xTgAD mouse model. NeuroToxicology 84, 172–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng C et al. (2016) The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Translational Neurodegeneration 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang YJ et al. (2021) An Air Particulate Pollutant Induces Neuroinflammation and Neurodegeneration in Human Brain Models. Advanced Science 8, 2101251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jew K et al. (2019) Selective memory and behavioral alterations after ambient ultrafine particulate matter exposure in aged 3xTgAD Alzheimer’s disease mice. Particle and Fibre Toxicology 16, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu P et al. (2020) Fine particulate matter aggravates intestinal and brain injury and affects bacterial community structure of intestine and feces in Alzheimer’s disease transgenic mice. Ecotoxicology and Environmental Safety 192, 110325. [DOI] [PubMed] [Google Scholar]
- 21.Calderón-Garcidueñas L et al. (2019) Mild Cognitive Impairment and Dementia Involving Multiple Cognitive Domains in Mexican Urbanites. J Alzheimers Dis 68, 1113–1123 [DOI] [PubMed] [Google Scholar]
- 22.Nasreddine ZS et al. (2005) The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool for Mild Cognitive Impairment. J Am Geriatr Soc 53, 695–699 [DOI] [PubMed] [Google Scholar]
- 23.Iaccarino L et al. (2021) Association Between Ambient Air Pollution and Amyloid Positron Emission Tomography Positivity in Older Adults with Cognitive Impairment. JAMA Neurol 78, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alemany S et al. (2021) Associations between air pollution and biomarkers of Alzheimer’s disease in cognitively unimpaired individuals. Environment International 157, 106864. [DOI] [PubMed] [Google Scholar]
- 25.Klunk WE et al. (2015) The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s & Dementia 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cacciottolo M et al. (2017) Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Translational Psychiatry 7, e1022–e1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calderón-Garcidueñas L et al. (2016) Interactive and additive influences of Gender, BMI and Apolipoprotein 4 on cognition in children chronically exposed to high concentrations of PM2.5 and ozone. APOE 4 females are at highest risk in Mexico City. Environmental Research 150, 411–422 [DOI] [PubMed] [Google Scholar]
- 28.Lee JH et al. (2020) Long-Term Exposure to PM10 and in vivo Alzheimer’s Disease Pathologies. Journal of Alzheimer’s Disease 78, 745–756 [DOI] [PubMed] [Google Scholar]
- 29.Fonken LK et al. (2011) Air pollution impairs cognition, provokes depressive-like behaviors and alters hippocampal cytokine expression and morphology. Molecular Psychiatry 16, 987–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hullmann M et al. (2017) Diesel engine exhaust accelerates plaque formation in a mouse model of Alzheimer’s disease. Particle and Fibre Toxicology 14, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S-H et al. (2021) Three month inhalation exposure to low-level PM2.5 induced brain toxicity in an Alzheimer’s disease mouse model. PLOS ONE 16, e0254587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen D. v et al. (2018) Microglia in Alzheimer’s disease. Journal of Cell Biology 217, 459–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fakhoury M (2018) Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr Neuropharmacol 16, 508–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patten KT et al. (2021) The Effects of Chronic Exposure to Ambient Traffic-Related Air Pollution on Alzheimer’s Disease Phenotypes in Wildtype and Genetically Predisposed Male and Female Rats. Environmental Health Perspectives 129, EHP8905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huberman M et al. (1995) IL-2 and IL-6 secretion in dementia: correlation with type and severity of disease. Journal of the Neurological Sciences 130, 161–164 [DOI] [PubMed] [Google Scholar]
- 36.Park SJ et al. (2020) Exposure of ultrafine particulate matter causes glutathione redox imbalance in the hippocampus: A neurometabolic susceptibility to Alzheimer’s pathology. Science of The Total Environment 718, 137267. [DOI] [PubMed] [Google Scholar]
- 37.Zou Y et al. (2016) Olfactory dysfunction in Alzheimer’s disease. Neuropsychiatric Disease and Treatment 2016, 869–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji X et al. (2022) Olfactory bulb microglia activation mediated neuronal death in real-ambient particulate matter exposure mice with depression-like behaviors. Science of The Total Environment 821, 153456. [DOI] [PubMed] [Google Scholar]
- 39.Saveleva L et al. (2022) Subacute inhalation of ultrafine particulate matter triggers inflammation without altering amyloid beta load in 5xFAD mice. NeuroToxicology 89, 55–66 [DOI] [PubMed] [Google Scholar]
- 40.Guarneros M et al. (2020) Metal-containing Particulate Matter and Associated Reduced Olfactory Identification Ability in Children from an Area of High Atmospheric Exposure in Mexico City. Chemical Senses 45, 45–58 [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z et al. (2021) Exposure to Particulate Matter Air Pollution and Anosmia. JAMA Network Open 4, e2111606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlesimo GA et al. (2015) Atrophy of presubiculum and subiculum is the earliest hippocampal anatomical marker of Alzheimer’s disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring 1, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gabbita SP et al. (1999) Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. J Neurochem 73, 1660–6 [DOI] [PubMed] [Google Scholar]
- 44.Sun Q et al. (2016) Regulation of structural and functional synapse density by L-threonate through modulation of intraneuronal magnesium concentration. Neuropharmacology 108, 426–39 [DOI] [PubMed] [Google Scholar]
- 45.Li W et al. (2014) Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer’s disease mouse model. Molecular Brain 7, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jurga AM et al. (2020) Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Frontiers in Cellular Neuroscience 14, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costa LG et al. (2014) Neurotoxicants are in the air: convergence of human, animal, and in vitro studies on the effects of air pollution on the brain. Biomed Res Int 2014, 736385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ravindra K et al. (2022) COVID-19 pandemic: What can we learn for better air quality and human health? Journal of Infection and Public Health 15, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dubey S et al. (2021) Assessing effectiveness of air purifiers (HEPA) for controlling indoor particulate pollution. Heliyon 7, e07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.World Health Organization (2021) WHO global air quality guidelines: particulate matter (PM2.5 and PM10), ozone, nitrogen dioxide, sulfur dioxide and carbon monoxide: executive summary. World Health Organization; [PubMed] [Google Scholar]
- 51.Health Effects Institute (2019) State of Global Air 2019, Health Effects Institute. [Google Scholar]
- 52.Chen M et al. (2017) Particulate matter (PM2.5) exposure season-dependently induces neuronal apoptosis and synaptic injuries. J Environ Sci (China) 54, 336–345 [DOI] [PubMed] [Google Scholar]
- 53.Wei H et al. (2017) Role of oxidative stress and DNA hydroxymethylation in the neurotoxicity of fine particulate matter. Toxicology 380, 94–103 [DOI] [PubMed] [Google Scholar]
- 54.Gillespie P et al. (2013) Particulate matter neurotoxicity in culture is size-dependent. Neurotoxicology 36, 112–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prüss-Ustün A et al. (2016) Preventing disease through healthy environments: A global assessment of the burden of disease from environmental risks, World Health Organization. [Google Scholar]
- 56.(2021) 2021 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 17, 327–406 [DOI] [PubMed] [Google Scholar]
- 57.Gauthier S et al. (2021) World Alzheimer Report 2021 Abridged version,
- 58.Tiwari S et al. (2019) Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. International Journal of Nanomedicine Volume 14, 5541–5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eratne D et al. (2018) Alzheimer’s disease: clinical update on epidemiology, pathophysiology and diagnosis. Australasian Psychiatry 26, 347–357 [DOI] [PubMed] [Google Scholar]
- 60.Ayodele T et al. (2021) Early-Onset Alzheimer’s Disease: What Is Missing in Research? Current Neurology and Neuroscience Reports 21, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bird TD (1993) Alzheimer Disease Overview,
- 62.Cacace R et al. (2016) Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s & Dementia 12, 733–748 [DOI] [PubMed] [Google Scholar]
- 63.Pike CJ (2017) Sex and the development of Alzheimer’s disease. Journal of Neuroscience Research 95, 671–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kivipelto M (2001) Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ 322, 1447–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehla J et al. (2014) Experimental induction of type 2 diabetes in aging-accelerated mice triggered alzheimer-like pathology and memory deficits. Journal of Alzheimer’s Disease 39, 145–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Picone P et al. (2020) Obesity and Alzheimer’s disease: Molecular bases. European Journal of Neuroscience 52, 3944–3950 [DOI] [PubMed] [Google Scholar]
- 67.Briggs R et al. (2016) Drug treatments in Alzheimer’s disease. Clinical Medicine 16, 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elder A et al. (2006) Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environmental Health Perspectives 114, 1172–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oberdörster G et al. (2004) Translocation of inhaled ultrafine particles to the brain. Inhalation Toxicology 16, 437–445 [DOI] [PubMed] [Google Scholar]
- 70.Brenneman KA et al. (2000) Direct olfactory transport of inhaled manganese (54MnCl2) to the rat brain: Toxicokinetic investigations in a unilateral nasal occlusion model. Toxicology and Applied Pharmacology 169, 238–248 [DOI] [PubMed] [Google Scholar]
- 71.Rao DB et al. (2003) Inhaled iron, unlike manganese, is not transported to the rat brain via the olfactory pathway. Toxicology and Applied Pharmacology 193, 116–126 [DOI] [PubMed] [Google Scholar]
- 72.Dorman DC et al. (2002) Olfactory transport: A direct route of delivery of inhaled manganese phosphate to the rat brain. Journal of Toxicology and Environmental Health - Part A 65, 1493–1511 [DOI] [PubMed] [Google Scholar]
- 73.Oberdörster G et al. (2002) Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. Journal of Toxicology and Environmental Health - Part A 65, 1531–1543 [DOI] [PubMed] [Google Scholar]
- 74.Illum L (2000) Transport of drugs from the nasal cavity to the central nervous system, 11 [DOI] [PubMed] [Google Scholar]
- 75.de Lorenzo AJD (2008) The Olfactory Neuron and the Blood-Brain Barrier. In Taste and Smell in Vertebrates: A Ciba Foundation symposium (Wolstenholme GEW and Knight J, eds) pp. 151–176, Churchill Livingstone [Google Scholar]
- 76.Maher BA et al. (2016) Magnetite pollution nanoparticles in the human brain. Proc Natl Acad Sci U S A 113, 10797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frey WH et al. (1997) Delivery of 125I-NGF to the brain via the olfactory route. Drug Delivery: Journal of Delivery and Targeting of Therapeutic Agents 4, 87–92 [Google Scholar]
- 78.Kreyling WG et al. (2009) Size dependence of the translocation of inhaled iridium and carbon nanoparticle aggregates fürom the lung of rats to the blood and secondary target organs. Inhalation Toxicology 21, 55–60 [DOI] [PubMed] [Google Scholar]
- 79.van Berlo D et al. (2012) Toxicology of Ambient Particulate Matter. In Molecular, Clinical and Environmental Toxicology: Volume 3: Environmental Toxicology (Luch A, ed), pp. 165–217, Springer Basel; [DOI] [PubMed] [Google Scholar]
- 80.Qin L et al. (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. GLIA 55, 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]