

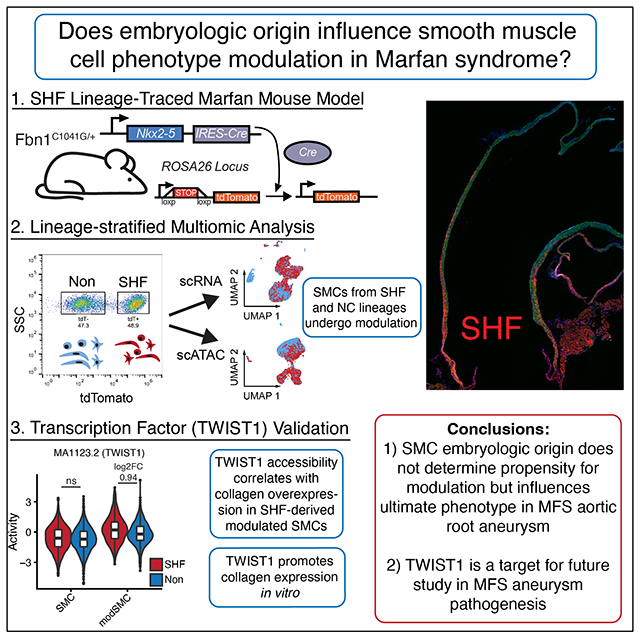
Abstract
Background:
Aortic root smooth muscle cells (SMC) develop from both the second heart field (SHF), and neural crest (NC). Disparate responses to disease-causing Fbn1 variants by these lineages are proposed to promote focal aortic root aneurysm formation in Marfan syndrome (MFS), but lineage-stratified SMC analysis in vivo is lacking.
Methods:
We generated SHF lineage-traced MFS mice and performed integrated multiomic (single-cell RNA and ATAC sequencing) analysis stratified by embryologic origin. SMC subtypes were spatially identified via RNA in situ hybridization. Response to TWIST1 overexpression was determined via lentiviral transduction in human aortic SMCs.
Results:
Lineage stratification enabled nuanced characterization of aortic root cells. We identified heightened SHF-derived SMC heterogeneity including a subset of Tnnt2-expressing cells distinguished by altered proteoglycan expression. MFS aneurysm-associated SMC phenotypic modulation was identified in both SHF-traced and non-traced (NC-derived) SMCs however transcriptomic responses were distinct between lineages. SHF-derived modulated SMCs overexpressed collagen synthetic genes and small leucine-rich proteoglycans while non-traced SMCs activated chondrogenic genes. These modulated SMCs clustered focally in the aneurysmal aortic root at the region of SHF/NC lineage overlap. Integrated RNA-ATAC analysis identified enriched Twist1 and Smad2/3/4 complex binding motifs in SHF-derived modulated SMCs. TWIST1 overexpression promoted collagen and SLRP gene expression in vitro, suggesting TWIST1 may drive SHF-enriched collagen synthesis in MFS aneurysm.
Conclusions:
SMCs derived from both SHF and NC lineages undergo phenotypic modulation in MFS aneurysm but are defined by subtly distinct transcriptional responses. Enhanced TWIST1 transcription factor activity may contribute to enriched collagen synthetic pathways SHF-derived SMCs in MFS.
Graphical Abstract
Introduction:
Thoracic aortic aneurysm (TAA) and the attendant risk of acute dissection remain significant clinical challenges without effective medical therapies. Monogenic hereditary thoracic aortic aneurysm syndromes such as Marfan syndrome (MFS) and Loeys-Dietz syndrome account for a minority of TAA patients but represent highly penetrant and clinically severe variants within a spectrum of heterogeneous clinical presentations. MFS is characterized by diffuse connective tissue pathology resulting from mutations in the extracellular matrix glycoprotein fibrillin-1 (FBN1)1. Intriguingly, although the biological effects of these genetic variants are systemic, MFS patients typically develop focal aortic root aneurysm while the adjacent ascending aorta and arch remain normal caliber. While the mechanisms behind this remain unclear, we hypothesize that disparate responses by aortic root SMCs arising from distinct embryologic origins (namely cardiac second heart field ‘SHF’ proximally and neural crest ‘NC’ distally)2–4 may represent a key feature in disease susceptibility.5 Previous studies using induced pluripotent stem cell models of aortic SMC development provide in vitro proof-of-concept for embryologic origin-specific pathology in cell lines with disease-causing FBN1 variants6,7 and SMAD3 loss-of-function8. Further, aortic SMCs demonstrated distinct, lineage-specific TGF-β responsiveness in a murine Loeys-Dietz syndrome model9. Despite this progress, the impact of embryologic origin on global SMC phenotype evolution during thoracic aortic aneurysm development in vivo remains incompletely characterized.
We recently reported dynamic SMC transcriptomic changes during aortic aneurysm formation in MFS at single-cell resolution, highlighting gradually reduced contractile gene expression and mixed synthetic/proteolytic pathway activation.10 Given the preponderance of SHF-derived SMCs within the aneurysmal aortic root, we hypothesized that the SMC subset destined for this modulated phenotype may derive specifically from SHF progenitors. Here, to specifically test this theory, we generated a novel SHF lineage-traced MFS mouse model and applied integrated single cell transcriptomics/epigenomics to rigorously parse the influence of embryologic origin on SMC phenotype modulation during MFS aortic root aneurysm development.
Methods:
Data Availability
All scRNAseq data (raw FASTQ and processed gene expression matrices) have been made publicly available at the gene expression omnibus (GEO) repository under accession GSE186845.
Statistical Analysis
For data with continuous variables from non-omics assays (echocardiography and RT-qPCR), normality of the distributions were assessed before testing. Data were first assessed for deviation from normality using the Shapiro-Wilk test in GraphPad. If no significant deviation from normal distribution was identified then parametric tests were used to improve statistical efficiency. Equal variance among groups was not assumed, therefore Brown-Forsythe and Welch ANOVA testing was performed using the Dunnett T3 multiple comparisons test. Adjusted p values <0.05 were considered statistically significant. RT-qPCR data from cultured cells was analyzed using four paired technical replicates from two separate human subjects (biological replicates). Relative expression values were determined by the ΔCT method using GAPDH as a housekeeping gene, where expression relative to GAPDH was calculated as 2-(CT(gene)-CT(GAPDH)). For experiments with sample size less than n=4, non-parametric (Kruskal-Wallis test followed by Dunn’s test for multiple comparisons) was used.
For single cell RNA and ATAC data, pairwise Wilcoxon rank-sum testing was applied between biologic groups wherever differential expression testing is described in the text. P values were adjusted for multiple comparisons using the Bonferroni method, adjusted p values <0.05 were considered significant.
Mouse Models
All animals were maintained according to protocols prospectively approved by the Institutional Animal Care and Use Committee. All models were maintained on the C57BL/6J background. Nkx2-5IRES-Cre (strain #024637) and fluorescent reporter RosatdTomato/tdTomato (strain #007909) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and successively crossed with Fbn1C1041G/+ Marfan mice (strain #012885) to generate experimental mice. Each allele was genotyped using sequence-specific PCR primers. Marfan (Fbn1C1041G/+) and littermate control (Fbn1+/+) mice from the same breeding pairs were utilized for all experiments.
Transthoracic Echocardiography
Mice were anesthetized with inhaled isoflurane and prepared for echocardiography as previously described.10 Briefly, replicate end-diastolic aortic root measurements were taken in long axis with an MS700 70 MHz MicroScan transducer on the Vevo 2100 system (VisualSonics, Toronto, Canada) by a blinded technician.
Aortic Tissue scRNAseq/scATACseq
Detailed methods for multiomic data acquisition and analysis are available in the Supplemental Materials.
Aortic histologic tissue preparation
Mice were anesthetized with inhaled isoflurane followed by cervical dislocation. Tissue fixation was achieved via whole-body perfusion with 10 mL of fresh 4% paraformaldehyde in PBS through left ventricular puncture. The aortic root was then resected en bloc with the left ventricular outflow tract and severed at the level of the transverse arch. Tissues were further fixed in 4% paraformaldehyde overnight then passed through a sucrose gradient and embedded in optimal cutting temperature (OCT) compound in either long or short axis orientation. Tissue blocks were then preserved at −80 degrees Celsius until cryosectioning at 10μM thickness.
RNA In Situ Hybridization
Aortic tissue sections were briefly dried at room temperature, rehydrated in PBS, and prepared for RNA hybridization via on-slide fixation with 4% paraformaldehyde, dehydration in ethanol, heat-activated target retrieval and protease treatment according to the RNAscope protocol (ACD Bio, Newark, CA). Sections were then incubated with sequence-specific probes for genes of interest (tdTomato, Igfbp2, Tnnt2) followed by serial amplification probes using the chromogenic red and duplex chromogenic RNAscope procedure kits according to supplier specifications. Following chromogenic detection, sections were counterstained with 50% hematoxylin and coverslipped with Vectamount media (Vector Laboratories, Burlingame, CA).
Western Blotting
Tissue samples were snap frozen in liquid nitrogen immediately after dissection. Aortic segments were suspended in tissue protein extraction reagent (T-PER, ThermoFisher Scientific) and homogenized with a rotor-stator homogenizer. Lysates were centrifuged at 21,000xg to remove debris and supernatants were concentrated using an Amicon Ultra 10K 0.5mL spin column (EMD Millipore). Protein concentrations were determined via bicinchoninic acid assay (Thermo Scientific, Waltham, MA). 8ug of protein in Laemmli sample buffer with 5% 2-mercaptoethanol were prepared for each sample and were denatured at 95°C for 5 minutes. The proteins were loaded on 4-15% polyacrylamide gels (Bio-Rad Laboratories), separated onto gels by SDS-PAGE, and then transferred to nitrocellulose membranes (Thermo Scientific). After 30 minutes of blocking with 5% w/v bovine serum albumin in TBS, the membranes were incubated with primary antibodies overnight at 4°C to detect: Twist1 (1:500, Proteintech, Rosemont, IL), and β -Tubulin (1:500, Cell Signaling Technology, Boston, MA) as loading control. Secondary anti-rabbit HRP (1:3000, Cell Signaling Technology) was used to detect primary immuno-complexes. Detection was performed using an enhanced chemiluminescent (ECL) horseradish peroxidase (HRP) substrate (Thermo Scientific). Intensities of bands were measured by densitometry scanning using ImageJ software (National Institute of Health, Bethesda, MD).
Human aortic SMC tissue culture
All studies conducted on samples from human subjects were approved by the IRB and prospective informed consent was obtained. Human aortic tissue was processed for SMC culture as described previously.11 Briefly, aortic tissues were collected from the operating room immediately upon surgical resection and maintained in cold Hanks balanced salt solution (HBSS). Tissues were then dissected to remove the intimal and adventitial layers completely. The tunica media was then finely minced and placed in aortic smooth muscle cell media (Lonza, Basel Switzerland). Once a confluent monolayer of aortic SMCs was observed as outgrowth from the tissue, cells were trypsinized, filtered and passaged. Cells were then maintained at subconfluency and passaged at 70% confluence. Cells from passage 3-6 were used for in vitro experiments. Additional detailed methods for human cell line experiments is available in the Supplemental Materials.
Results:
Lineage-resolved single cell transcriptomics of the murine postnatal thoracic aorta
To enable aortic root cellular fate tracking and downstream isolation stratified by lineage, we bred a validated SHF lineage-tracing construct (Nkx2-5-cre knock-in12,13 combined with Cre-inducible tdTomato fluorescent reporter14, Figure 1A) into the Fbn1C1041G/+ MFS strain. Histologic assessment of 4-month-old lineage-traced (‘SHFlin’) mice demonstrated highest tdT fluorescent cell density (SHF-derived SMCs) in the aortic root tunica media with gradually reduced density encompassing only the outer layers more distally in the ascending aorta (Figure 1B). Lineage-traced cells were observed in tunica intima and adventitia layers throughout the proximal aorta and within the aortic valve annulus/interstitial cells, consistent with multiple prior transgenic SHF fate-mapping reports.3,4,12 As in prior studies10,15,16, MFS mice developed significant aortic root dilatation compared to littermate SHFlin controls regardless of sex, and male MFS mice had greater root diameter than age-matched MFS females (2.15±0.11mm vs. 1.96±0.08mm at age 16 weeks, n=6 p=0.041 Figure 1C). To encompass the overlapping regions of SHF- and NC-derived aortic SMCs, we performed aortic root/ascending tissue enzymatic digestion and fluorescence-activated cell sorting (FACS) to isolate SHF and non-traced cell suspensions from 4-month-old MFS and control SHFlin mice (Figure 1D). Lineage-segregated cell suspensions were then split into two partitions and either 1) processed fresh for single cell RNA sequencing (scRNAseq) library preparation or 2) subjected to cell lysis, nuclear extraction, and single cell assay for transposase-accessible chromatin (ATAC) sequencing. Separate cohorts of male and female mice (n=4 individual animals pooled in each cohort) were utilized for each genotype. Library preparation and sequencing were performed in parallel for all samples to negate batch effect.
Figure 1:
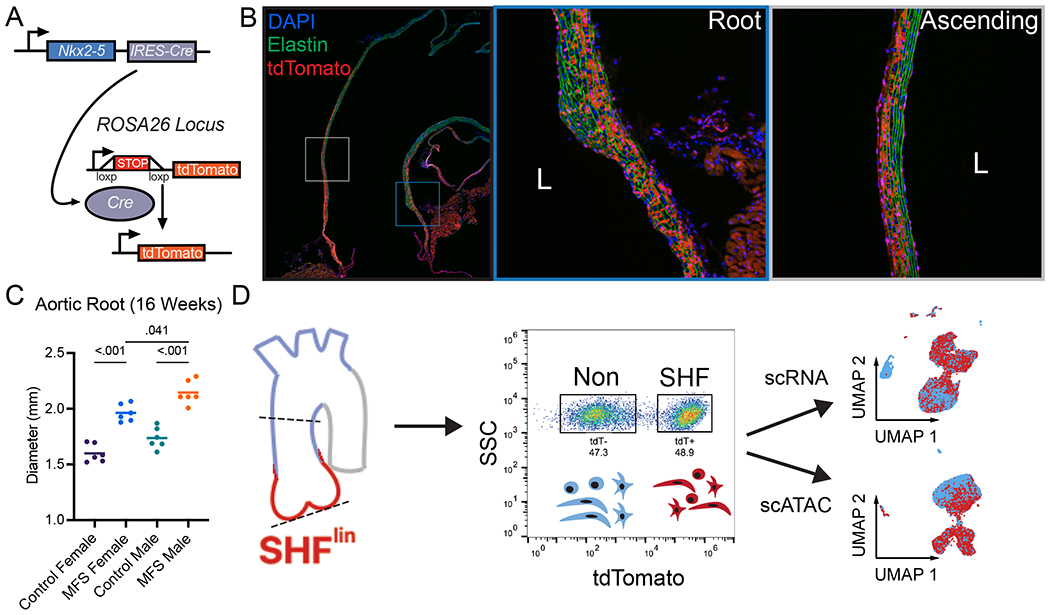
Second heart field (SHF) lineage-tracing Marfan mouse development and scRNAseq workflow. A SHF lineage-tracing strategy. IRES-Cre recombinase cassette inserted into the 3’ untranslated region of Nkx2-5 locus enables upstream stop signal deletion in tdTomato gene inserted into Rosa26 locus, leading to permanent fluorescence B Stitched serial long axis images and inset 20X magnification images of a representative 16-week-old healthy control male SHF-traced mouse thoracic aorta. DAPI = nuclear counterstain, Elastin = green autofluorescence, tdTomato = red fluorescent signal from SHF-derived cells C Transthoracic echocardiography data for aortic root diameter in 16-week-old Fbn1C1041G/+ (MFS) mice and littermate controls (WT). Line indicates mean diameter for n=6 animals of each sex. D Experimental workflow. Aortic tissue from the aortic valve to mid ascending aorta (dashed lines) digested to single cell suspension and sorted based on tdTomato fluorescent signal. Individual single cell (RNA) and nuclei (ATAC) libraries were generated and processed in parallel.
We first assessed the total dataset (comprising 39,031 cells from 8 libraries) to validate the separation of SHF and non-traced cells during sample processing. Unsupervised clustering of all scRNA samples identified eight major and five minor (each <1% of dataset) cell types. Major clusters broadly included four SMC subtypes, adventitial fibroblasts, aortic valve cells, macrophages, and endothelial cells (Figure 2A). Stratifying the dataset by lineage revealed largely similar distributions in uniform manifold approximation and projection (UMAP) space but distinct cell proportions between lineages including expected absence of SHF-derived macrophages (Figure 2B–C). Detection of tdTomato mRNA transcripts was restricted to SHF sorted cells, confirming successful lineage partitioning by FACS (Figure 2D). Having validated the SHF lineage-tracing model, we next examined the dataset stratified by genotype and sex. Consistent with our prior report comparing MFS to control animals with scRNAseq, we found that the principal distinctions were the presence of a disease-enriched SMC subset and heightened density of inflammatory cells in MFS (Figure 2E). These changes were identified in separate samples from both male and female mice (Supplemental Figure 1).
Figure 2:
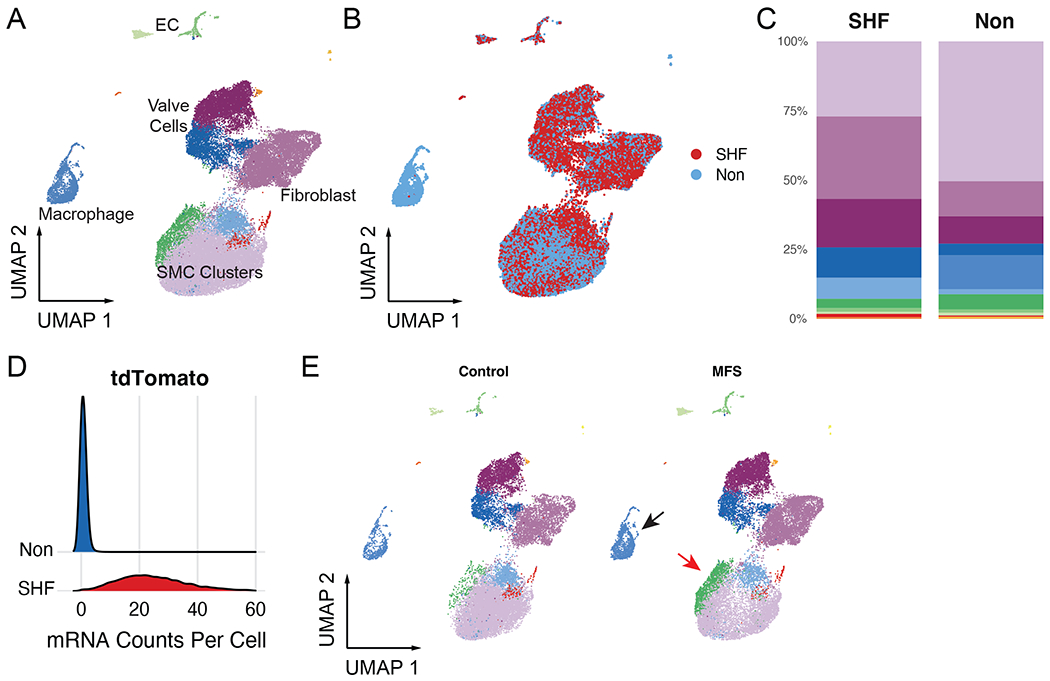
Complete aortic root scRNAseq dataset. A Unsupervised clustering of >39,000 aortic root cells from MFS and control mice. EC = endothelial cell B Overlaid UMAP distributions of second heart field (SHF) and non-traced cell populations C Cell cluster breakdown by lineage. Each lineage comprises aggregated separate male and female samples (n=3 each sex) of both Fbn1C1041G/+ and control genotype D Raw tdTomato mRNA transcript count per cell stratified by sorted lineage E Total scRNAseq dataset stratified by genotype demonstrates increased macrophage density (black arrow) and disease-associated smooth muscle cell cluster (red arrow) in MFS mice.
Lineage-stratified SMC profiling highlights aortic root phenotypic diversity
While the spatial distribution of SHF and NC lineage-derived aortic SMCs is well understood, the functional differences resulting from these distinct developmental pathways in the postnatal aorta in both homeostasis and disease are unknown. To better determine the broad differences between these SMC populations, we first assessed the four distinct SMC subtypes identified by unsupervised clustering (Figure 3B). The main sub-population consisted of quiescent SMCs, as distinguished by high mature contractile gene (e.g., Myh11) expression (‘SMC’ cluster, 79.1% of all SMCs). We also identified a distinct SMC subset primarily distinguished by highly specific expression of cardiac troponin T (‘Tnnt2+ SMC’, 10.2%), a modulated SMC cluster marked by reduced Myh11 and uniquely enriched Igfbp2 expression (‘modSMC’, 9.0%) and a rare Rgs5-expressing pericyte population (1.8%, Figure 3B). We next stratified the SMC dataset by genotype and lineage to determine the distribution of SMC types between samples. ‘Tnnt2+ SMC’ showed SHF enrichment in both MFS and control mice, comprising 16% of all SMCs in the SHF samples and only 2.5% of all non-traced SMCs (Figure 3C). To confirm these SMCs reside in the aortic tunica media (rather than epicardial or coronary arteries), we performed RNA in-situ hybridization with a Tnnt2 sequence-specific probe, confirming the restricted spatial distribution of Tnnt2+ SMC within outermost layers of the aortic root segment, consistent with aortic SHF derivatives (Figure 3D). We compared the Tnnt2+ subset to the main SMC cluster to determine its unique transcriptomic phenotype, identifying 253 differentially expressed genes (Figure 3E, Supplemental Table 1) including enriched the small leucine-rich proteoglycans (SLRP), lumican (Lum) and decorin (Dcn, Figure 3F). Conversely, Tnnt2+ SMCs expressed reduced elastin (Eln), periostin (Postn) and fibronectin (Fn1) compared to the major SMC cluster. Given the focal presence of Tnnt2+ SMCs within the site of aneurysm in the aortic root, we directly compared MFS and healthy control SMCs within this cluster. While we observed some transcriptomic differences between genotypes (104 DEGs) including modestly reduced Cnn1 and Myh11 expression, MFS Tnnt2+ SMCs did not show activation of typical genes associated with aneurysm (e.g., collagens, MMPs, or TGF-β ligands, Supplemental Figure 2). These findings suggest this SHF-derived SMC subset responds to perturbation related to MFS but may not directly be the pathogenic population for aneurysm.
Figure 3:
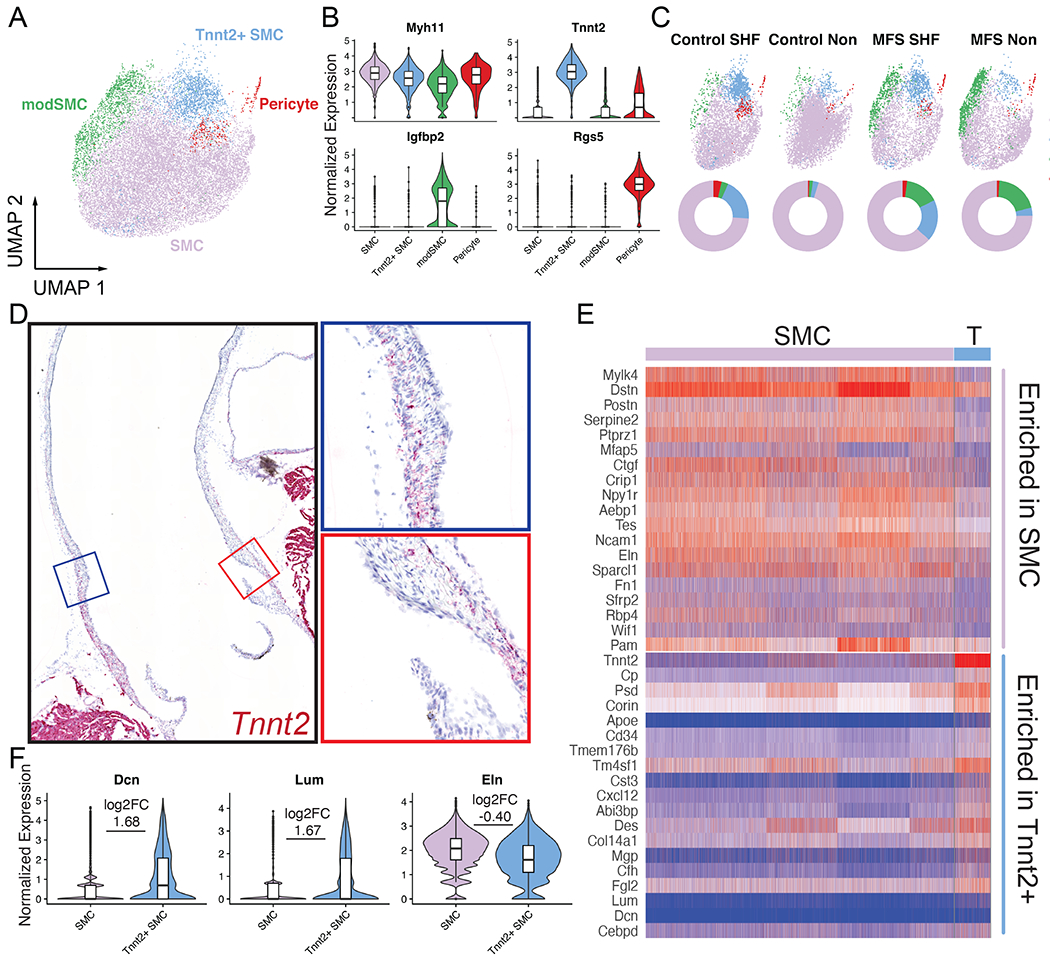
Aortic SMC subcluster analysis. A UMAP projection of four distinct SMC subtypes B Enriched markers for each subcluster. Major SMC group (purple) is characterized by peak expression of mature contractile markers such as Myh11 compared to other subsets C SMC population distribution in UMAP space split into original sample subtypes (top) and pie charts for fraction of each SMC cluster in sample (bottom). Tnnt2+ SMCs (light blue) are enriched in SHF lineage regardless of genotype, while modSMCs (green) are largely limited to MFS samples. D Long axis chromogenic RNA in situ hybridization for Tnnt2 localizes this SMC cluster within the outer layers of the aortic root segment. E Heatmap for top up- and down-regulated genes in Tnnt2+ cluster compared to major SMC subset. F Violin plots for markers of interest in Tnnt2+ SMCs. Boxes denote interquartile range (IQR), line denotes median, whiskers extend 1.5 IQR from box hinges. p value <0.001 for all comparisons, log2FC denotes Tnnt2+ subset over main SMC cluster.
We next examined the modSMC cluster to test our overall hypothesis that SHF-derived SMCs have altered propensity for phenotype shift during aneurysm progression. These modSMCs are remarkable for significantly reduced core SMC contractile gene expression (e.g., Cnn1, Myh11, Acta2) concomitantly with enriched Mmp2, Tgfb1, Col1a1, and were predominantly found in MFS samples (18.1% of all MFS SMCs vs. 2.2% of control, Supplemental Figure 3), consistent with prior studies of advanced MFS aneurysm. After stratifying the dataset by lineage and sex to examine individual scRNA libraries, we found that SMCs from both the SHF and non-traced (NC) lineages underwent transition to the modulated phenotype. Male MFS mice demonstrated higher modSMC density compared to females, particularly in the non-traced subset (18.3% of all SMCs vs. 11.5% for SHF lineage, 29.7% vs. 10.9% for NC, Figure 4A) though this analysis is limited by a single technical replicate of pooled mice for each sex. While this variable modSMC density between MFS male and female mice is consistent with the described sexual dimorphism in MFS, we found no significant difference in core modSMC markers between sexes (Supplemental Figure 4), confirming that more severe disease in males correlates with a greater fraction of modSMCs rather than a fundamentally distinct phenotypic modulation process. Accordingly, we compared SMC and modSMC clusters in downstream analyses in a sex-agnostic fashion.
Figure 4:
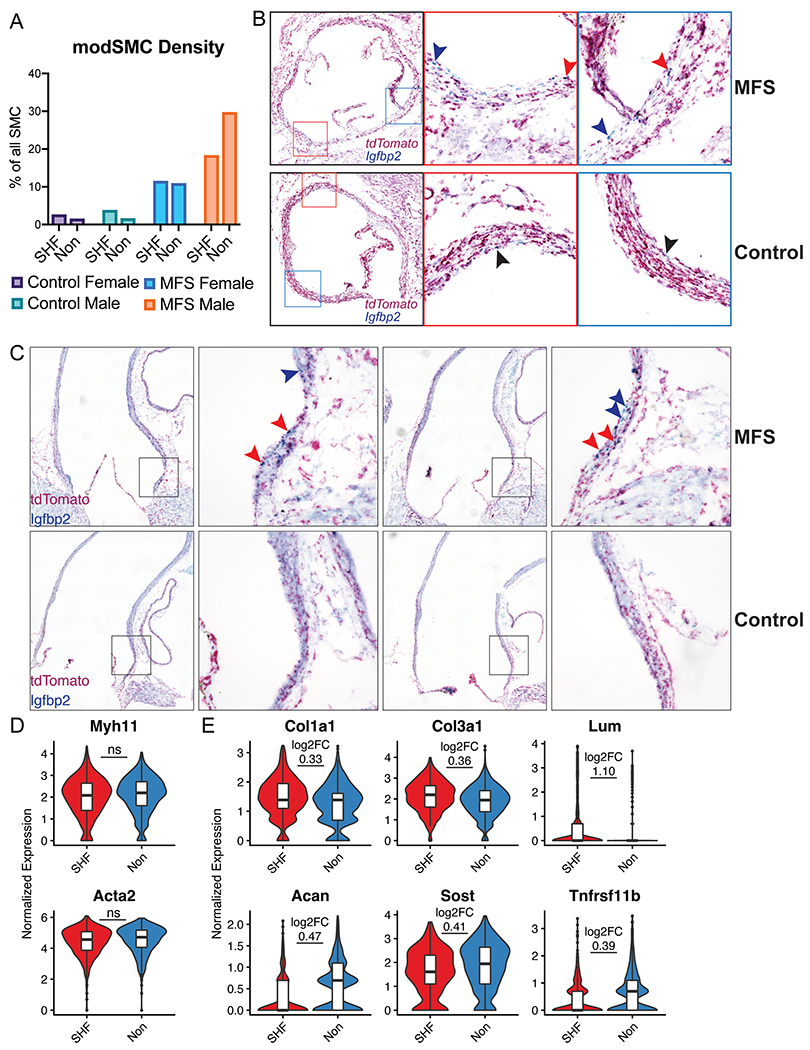
Modulated SMC localization and phenotype stratified by lineage. A Proportion of all SMCs belonging to modSMC cluster by sample. SHF=lineage traced second heart field-derived cells, Non=untraced cells. B Short axis chromogenic RNA in-situ hybridization for SHF lineage marker (tdTomato) and insulin-like growth factor binding protein-2 (Igfbp2), a specific marker for the modSMC subset. 16-week-old MFS animals show modSMCs from both SHF (double stain, red arrowheads) and non-traced (blue stain only, blue arrowheads) while control animals demonstrate rare Igfbp2-positive cells (black arrowheads). Left image is stitched image of entire root circumference with boxes marking inset 20X magnification images. C Long-axis RNA localization shows modSMCs are concentrated in the MFS aortic root segment regardless of lineage (SHF-derived = red arrowheads, non-traced = blue arrowheads) while control animals show trivial Igfbp2 expression D Myh11 and Acta2 expression levels are similar between MFS modSMCs from SHF and non-traced lineages E Multiple collagen isoforms and proteoglycans are enriched in SHF-derived modSMCs (top panel) while Acan, Sost, and Tnfrsf11b are enriched in non-traced modSMCs (bottom). Boxes denote interquartile range (IQR), line denotes median, whiskers extend 1.5 IQR from box hinges. ‘ns’ indicates fold change not calculated between lineages due to non-significant difference, otherwise p < 0.001 for all genes with log2FC displayed.
To validate the finding of lineage-agnostic phenotype shift, we performed duplexed RNA in situ hybridization for tdTomato (SHF lineage) and Igfbp2, a robust and highly specific marker for early activated MFS modulated SMCs.10 We found enriched Igfbp2 transcripts in the MFS aortic root tunica media layer corresponding to both tdTomato+ (SHF-traced) and non-traced SMCs (Figure 4B). We also identified small Igfbp2-expressing cell populations in control mouse aortic root sinuses consistent with rare modSMCs in the scRNAseq dataset. To spatially assess whether modSMCs arise diffusely in the diseased MFS aorta or focally within the aneurysmal segment we studied Igfbp2 expression in long-axis aortic sections. MFS aortas showed grouped Igfbp2+ cells from both SHF and non-traced SMCs at the site of aneurysm formation with minimal expression distal to the sinotubular junction (Figure 4C).
After discovering MFS modSMCs develop from both lineages, we hypothesized embryologic origin may promote distinctly abnormal phenotypes in MFS. Because modSMCs from control mice represented a tiny fraction of total SMCs (2.2%), the small absolute number of cells from each lineage precluded robust statistical comparisons and suggested limited biologic relevance of these cells in healthy aortas. We therefore compared only MFS modSMCs stratified by lineage, identifying 79 genes with lineage-specific expression patterns (complete list in Supplemental Table 2). While SMC contractile genes were similarly reduced compared to the quiescent SMC cluster between lineages (Figure 4D), SHF-derived modSMCs showed significantly increased expression of multiple fibrillar collagen isoforms: Col1a1 (average log2FC=0.33, p=3.8x10−5), Col3a1 (average log2FC=0.36, p=4.7x10−6)), and SLRPs: Dcn (average log2FC=1.27, p=2.2x10−18) and Lum (average log2FC=1.10, p=1.8x10−12) suggesting enhanced collagen biosynthetic pathway activation. Conversely, NC-derived modSMCs expressed greater aggrecan (Acan, average log2FC=0.47, p=1.3x10−22)), sclerostin (Sost, average log2FC=0.41, p=9.7x10−4) and osteoprotegerin (Tnfrsf11b, average log2FC=0.39, p=1.2x10−3) consistent with a more chondrogenic phenotype (Figure 4E). These data suggest both SHF and NC SMC undergo phenotypic modulation in the setting of MFS, however, their distinct embryologic origins trigger varying transcriptomic responses to the aneurysmal trigger.
Single-cell epigenomics identifies lineage-specific regulatory motif enrichment
Given the finding of differential gene expression in modSMCs from distinct lineages, we hypothesized that altered epigenetic landscapes may account for distinct responses; and that activation of unique transcription factors may influence the SMC modulatory process in each embryologic origin. To study this, we assessed the concurrently generated scATAC dataset to enable integrated gene regulatory network analysis of distinct SMC phenotypic subtypes in vivo. The scATAC dataset was initially subjected to independent unsupervised dimensional reduction and clustering using the Signac package in R17 (Figure 5A) and SMC clusters were selected based on enriched DNA accessibility peaks within typical SMC genes (e.g., Myh11, Figure 5B). SMCs from SHF and non-traced lineages occupied distinct projections in UMAP space (Figure 5C) suggesting unique epigenetic profiles. To broadly establish the unique epigenomic distinctions between SMC lineages, we first performed differential peak accessibility testing between all SMCs in the dataset based exclusively on lineage. Using the FindMarkers function, we identified 33 peaks enriched in SHF SMCs and a single enriched peak within the upstream regulatory region of the Nr2f1 gene, a critical regulator of NC cell gene expression,18 in non-lineage traced cells. Using GREAT19, we found that the 33 SHF marker peaks are enriched for regulatory regions relevant to multiple cardiac developmental processes including endocardial cushion development, regulation of heart morphogenesis, and outflow tract morphogenesis (Figure 5D). Enrichment of these pathways was primarily driven by peaks within the regulatory regions for critical cardiac related transcription factors including Isl1, Gata4, Twist1, and Hand2 (Supplemental Table 3).
Figure 5:
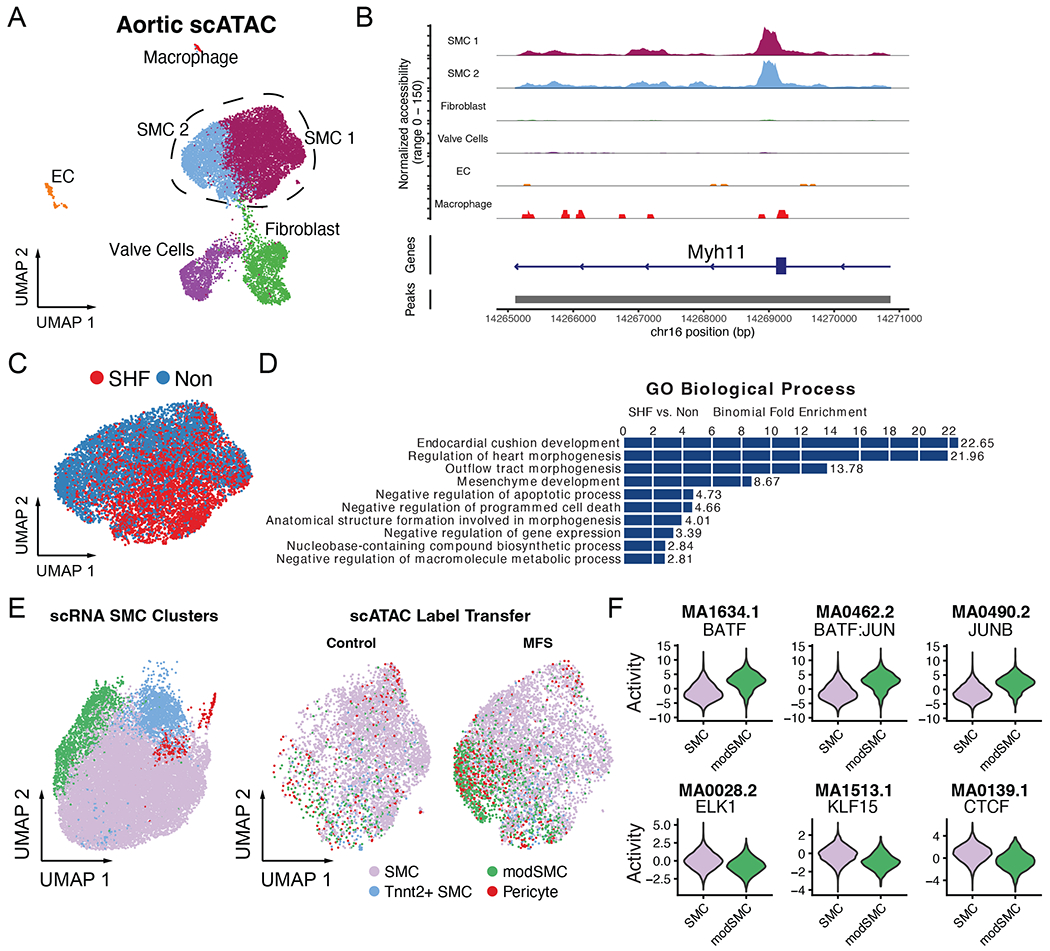
Single-cell ATAC dataset. A Unsupervised clustering of total scATAC data comprising MFS and control samples from SHF and non-traced lineages. Dashed lines indicate cells sub-selected for analysis B Enriched DNA accessibility peaks within the Myh11 gene confirm SMC identity of sub-selected clusters C UMAP projection of MFS SMCs reveals distinct profiles for SHF (red) and non-traced (blue) SMCs D GREAT output depicting top enriched biological processes for genes with regulatory regions corresponding to SHF-derived SMC marker peaks from the scATAC dataset E Integrated label transfer from scRNA dataset (left) onto scATAC dataset confirms modSMC enrichment in MFS samples. F ChromVAR output depicting top transcription factor DNA binding motifs in genome-wide assessment of enriched peak regions for modSMC subset (top) and quiescent SMC (bottom) from MFS mice only, all results statistically significant (p < 1x10−50).
Combining scRNA and scATAC data enables investigation of the epigenetic changes associated with transcriptomic phenotype modulation. To fully integrate the scRNA and scATAC datasets, we next performed integrative label transfer for these SMCs using the SHFlin scRNA dataset as a reference, identifying enriched modSMCs presence in MFS (Figure 5E). To specifically parse the epigenetic signals associated with SMC modulation in MFS and interrogate the influence of embryologic origin on this process, we selected the SMC/modSMC clusters from MFS mice only for downstream analyses. Direct comparison of the modSMC/SMC clusters identified 336 peaks enriched in modSMCs compared to only 29 enriched peaks in SMCs, suggesting generally heightened chromatin accessibility and phenotypic heterogeneity associated with this transition. We assessed for potential upstream regulatory transcription factors associated with global SMC modulation by employing chromVAR20 to quantify the genome-wide signature of accessible transcription factor motifs within the SMC/modSMC subsets. We identified 242 transcription factor motifs with enriched activity and 103 with diminished activity associated when comparing modSMCs to SMC (top three up- and down-regulated in Figure 5F, complete list in Supplemental Table 4). To identify regulatory signals with lineage-specific activity profiles, we directly compared modSMCs from SHF and non-traced lineages, identifying 36 transcription factors with enriched activity in SHF cells, while non-traced cells had no specific motif enrichment. Among these 36 SHF-enriched transcription factor motifs, 22 overlapped with the modSMC-enriched set, including NFIC (2.1-fold enriched, p=6.6x10−7), TWIST1 (1.9-fold, p=7.7X10−7), ZBTB18 (1.9-fold, p=3.3x10−6), OSR2 (1.8-fold, p=2.6x10−7), and the SMAD2:SMAD3:SMAD4 complex (1.8-fold p=1.3x10-8). These findings identify candidate regulatory signals that may contribute to distinct epigenetic and transcriptomic responses to upstream signals in aortic aneurysm progression on the basis of embryologic origin.
We examined this set of 22 overlapping transcription factor sets to identify possible novel regulators of SMC modulation in SHF derivatives. Critically, enhanced SMAD complex motif accessibility in SHF modSMCs suggests variable responses to transforming growth factor-beta (TGF-β) signaling, a central pathway dysregulated in MFS aneurysm.21–23 We identified TWIST1 as a transcription factor of particular interest due to the combination of heightened motif accessibility and enriched promoter accessibility at the Twist1 locus in SHF cells (Supplemental Table 3), suggesting this ‘epigenetic memory’ in SHF derivatives may impact their transcriptome. Furthermore, given the known roles of Twist1 as a promoter of fibrotic gene activation24 and a critical regulator of ECM organization during cardiac outflow tract development25, we hypothesized that this transcription factor may play an active role in the evolution of MFS aneurysm. The parallel enrichment of TWIST1 motif activity and canonical TGF-β signaling mediators (SMADs) in the SHF modSMC subset (Figure 6A) suggested a potential synergistic effect of these transcription factors. Interrogating individual cell activity levels for both TWIST1 and SMAD2:SMAD3:SMAD4 motifs identified a moderate linear correlation for these values (Pearson coefficient 0.57, Figure 6B). Because unsupervised genome-wide transcription factor motif accessibility (chromVAR) analysis suggested a functional role for Twist1 in MFS, particularly within SHF-derived modSMCs, we next assessed Twist1 mRNA and protein expression in MFS. Within the scRNA dataset, we identified a greater fraction of SMC/modSMCs with detectable Twist1 mRNA expression (at least one transcript) in SHF cells compared to non-traced for both control samples and MFS, with a heightened fraction of Twist1-expressing cells for each lineage in MFS vs. control (Figure 6C). This coincided with significantly greater Twist1 protein expression in 16-week-old MFS aneurysm tissue (aortic root and ascending aorta) compared to controls (3.3-fold, p=0.009, Figure 6D). Conversely, we found no significant difference in Twist1 protein level in the non-dilated descending thoracic aorta between genotypes (p=0.62). While these data confirm Twist1 overexpression correlates with aneurysm in MFS and suggest a heightened functional role in SHF-derived SMCs during modulation, they do not provide a direct association between with the SHF-enriched gene signature.
Figure 6:
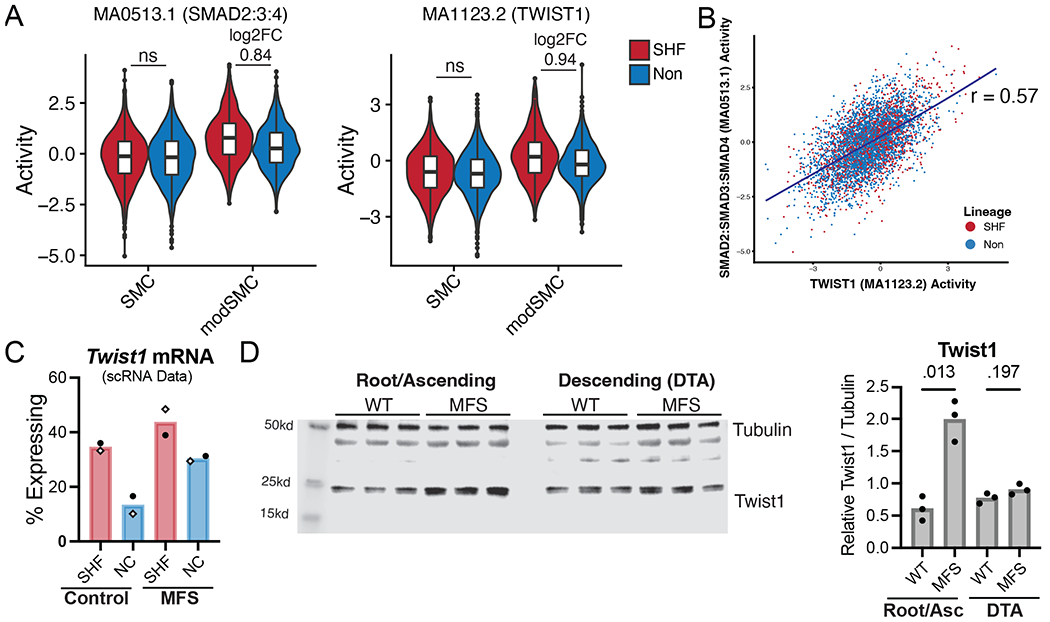
Integrated scRNA/scATAC reveals Twist1 as a marker of SHF-SMC modulation. A Transcription factor binding motif accessibility signal for SHF-enriched motifs with enhanced signal in modSMC cluster compared to quiescent subset in MFS SMC/modSMC clusters. ‘ns’ indicates fold change not calculated between lineages due to non-significant difference, otherwise p < 0.001 where log2FC displayed. B Scatter plot for SMAD2:SMAD3:SMAD4 and TWIST1 motif activity for individual cells within the scATAC dataset. C Twist1 mRNA expression for SMC/modSMCs from individual samples within scRNA dataset (white diamonds indicate males, circles indicate females). Cells with at least one Twist1 transcripts considered positive. D Aortic tissue lysate western blots for aneurysmal segment (root/ascending) and non-dilated region (descending) from n=3 male 16-week-old mice with band density quantification relative to loading control (tubulin).
TWIST1 promotes SHF-enriched gene signature in vitro
To determine whether TWIST1 augments the fibrillar collagen and SLRP overexpression identified in vivo in SHFlin mice, we sought to translate our findings to a clinically relevant model by manipulating TWIST1 expression in primary aortic root smooth muscle cell lines derived from two young MFS patients. We performed lentiviral-mediated transduction of 1) TWIST1 shRNA, 2) scrambled (Scr) shRNA, or 3) TWIST1 overexpression (OE) constructs. Stably transfected lines were established with puromycin treatment and TWIST1 overexpression was confirmed in both cell lines by RT-PCR and western blot (Supplemental Figure 5). We then performed RT-PCR for SHF-enriched markers from the mouse scRNAseq dataset, demonstrating significantly enriched expression for COL1A1, COL3A1, LUM, and DCN following TWIST1 overexpression compared to scrambled control. Furthermore, COL1A1 expression was significantly reduced following TWIST1 silencing via shRNA in both patient lines (Figure 7A). Conversely, expression of multiple non-lineage-traced SMC markers (TNFRSF11B and SOST) increased with TWIST1 silencing in both cell lines, with trivial (mean 1.35-fold) enrichment of TNFRSF11B in one cell line with TWIST1 overexpression (Figure 7B). Finally, to determine whether TWIST1 overexpression induces altered chromatic accessibility for SHF-enriched genes, we performed bulk ATAC sequencing on stably transduced aortic SMCs (shRNA, Scr, and OE). We observed enriched chromatin accessibility in the promoter regions for both COL1A1 and LUM as well as and in multiple enhancer peaks upstream of COL1A1 for TWIST1-OE cells compared to Scr and shRNA treated cells (Figure 7C). Collectively, these data suggest a critical role for Twist1 activity as a contributor to the SHF-specific modSMC transcriptome in MFS.
Figure 7:
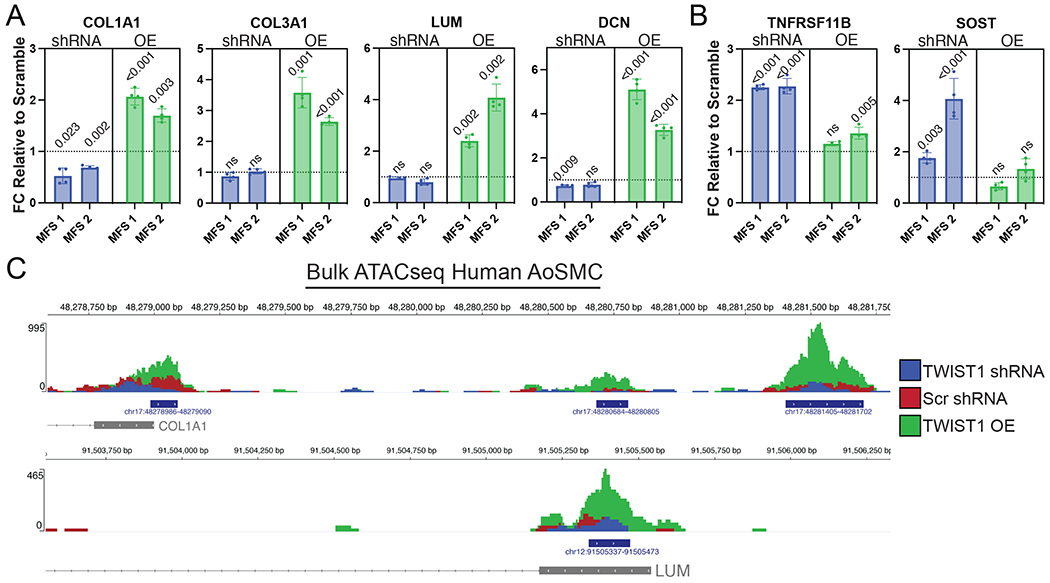
TWIST1 overexpression induces fibrillar collagen and small leucine-rich proteoglycan expression in aortic SMCs. A RT-qPCR data for SHF-enriched modSMC markers from MFS mouse model (n=4 paired technical replicate values in n=2 distinct MFS patient aortic root SMC lines) following TWIST1 silencing (shRNA) or overexpression (OE). All values plotted as fold change (FC) relative to scrambled shRNA control. Dashed lines indicate FC=1 B RT-qPCR data for chondrogenic markers of non-SHF traced modSMCs from MFS mouse model C Chromatic accessibility tracks for single MFS aortic SMC line following stable lentiviral transduction of denoted construct. Blue bars indicate peaks called by MACS2 for total sample pool, grey boxes indicate first exon for denoted genes.
Discussion
Vascular SMC phenotype modulation is a core marker of thoracic aortic aneurysm in MFS and may participate in aortic wall degeneration and ECM remodeling.10,11,26 The mechanisms underlying focal dilation of the aortic root segment in MFS remain elusive. By utilizing a SHF lineage-tracing strategy in a well-studied MFS mouse model and applying integrated, lineage-stratified single cell transcriptomics/epigenomics, we have identified critical gene regulatory pathways involved in MFS SMC phenotype modulation and parsed the influence of embryologic origin on this process.
Conceptually, the presence of SMCs derived from two distinct developmental pathways in this region has presented an attractive model whereby one lineage (presumably SHF-derived, given the predominance of these cells in the aortic root) either demonstrates heightened propensity for modulation in response to disease-causing FBN1 mutations or undergoes a uniquely pathologic process during disease development. In MFS, applying single-cell multiomics enabled rigorous, parallel investigation of SMC phenotype in distinct SHF- and NC-derived SMC subsets. We demonstrate that SMCs from both lineages exhibit similar propensity for de-differentiation in MFS, but that the SHF lineage retains DNA accessibility surrounding critical cardiac-related developmental TFs which may ultimately impact the ECM-modulatory transcriptome adopted during aneurysm development. While SMC modulation was globally similar between lineages, SHF-derived SMCs exhibited enriched expression of multiple fibrillar collagen isoforms (e.g., Col1a1, Col3a1) and activation of the SLRPs Lum and Dcn. This is consistent with our previous report identifying heightened type I collagen expression in induced pluripotent stem cell-derived SMCs from MFS patients directed through the SHF developmental pathway compared to NC.7 Furthermore, while the precise effect of collagen deposition in MFS remains controversial, anatomically specific type 1 collagen overexpression in the root segment is a reproducible clinical finding in MFS patient tissues.11,26 Our general hypothesis based on clinical studies27–29 is that this process promotes enhanced tissue stiffness, reduced distensibility and ultimately increases propensity for aneurysm growth and acute dissection; data from this study suggests that SHF-specific overactivation of the collagen synthetic pathway mechanistically may contribute to the anatomically focal nature of this phenomenon.
The role played by the SLRPs Lum and Dcn during aneurysm development is comparatively less well understood. Clinically, increased circulating Lum may be a useful biomarker for thoracic aortic aneurysm and dissection.30,31 SLRPs are known ECM structural components of semilunar valves and the aortoventricular junction expressed spatiotemporally with TGF-β signaling activation during development.32 Physiologically, SLRPs participate in collagen fibrillogenesis and matrix organization33 and regulate diverse cell signaling pathways.34 Notably, Lum and Dcn expression also marked the distinct Tnnt2+ SMC cluster. The physiologic significance of this subset of SMCs, which predominantly arose from the SHF, is unclear. While Tnnt2+ SMCs represented a distinguishing feature of the SHF and the anatomic aortic root, these cells appeared resistant to phenotype modulation in MFS and are therefore of uncertain significance to aneurysm progression. Nevertheless, we consider the unique transcriptome of these cells (including reduced Eln and increased SLRP expression) a defining feature of the normal aortic root microenvironment. Taken together, we propose that SHF lineage-specific Dcn/Lum overexpression, which distinguishes the aortic root segment at baseline and is further activated during aneurysm development, may participate in enhanced collagen synthesis33,35 and global ECM disarray, ultimately contributing to distinct regional aortic wall biomechanical properties.
We also report the activation of several chondrogenic ECM-modulatory genes (e.g., Acan, Sost, Tnfrsf11b) by untraced (ostensibly NC-derived) SMCs in this model. Acan accumulation is a recognized marker of thoracic aortic aneurysm which is proposed to induce tissue swelling and ECM disarray.36,37 By contrast, enriched sclerostin (Sost) expression has not been previously described in thoracic aortic aneurysm, but forced overexpression was shown to reduce atherosclerosis and abdominal aortic aneurysm in Apoe−/− mice.38 Notably, these cells did not progress fully to a characteristic chondromyocyte phenotype (characterized by Sox9 and Col2a1 expression39) though our analysis was limited to a fairly early time point in the disease spectrum. The functional significance of this NC-specific ECM modulatory signature is unclear but given that modSMCs were anatomically restricted to the aortic root regardless of lineage, it is likely that both lineages contribute to the characteristic ECM disarray found in MFS aortic root aneurysm in distinct ways. The finding of greater modSMC density (particularly in the non-traced SMC lineage) in MFS males compared to females (Figure 4A) is noteworthy given the propensity of males in the C1041G/+ MFS strain to develop greater aneurysmal dilation of the distal ascending aorta.16 As in our previous report, direct comparison of modSMCs between male and female MFS mice did not identify sex-specific pathways, instead disclosing distinctions only in the ratio of quiescent to modulated SMCs. We interpret this data as a further indication that SMC modulation is a reliable marker of aneurysm disease severity, though it remains possible that sex-specific mechanisms promote this process disproportionately in males. Ultimately a devoted investigation with multiple technical replicates for each sex will be needed to further explore these mechanisms.
Uniquely, we performed concurrent scATACseq to simultaneously profile genome-wide chromatin accessibility toward the goal of identifying lineage-specific enriched transcription factor motifs. To our knowledge, this is the first application of integrated single cell epigenomic techniques to complement gene expression analyses in thoracic aortic aneurysm. In assessing the SMC-modSMC transition we identified over 200 transcription factor motifs with differential activity across this phenotypic spectrum, generating a new pool of potential regulatory targets governing patterned SMC transcriptomic changes in MFS. We further identified enrichment of the SMAD2/3/4 and TWIST1 motifs in modSMCs from the SHF lineage, suggesting an amplified role for these regulatory signals within the SHF-enriched aortic root. Activation of the TGF-β-Smad2/3/4 signaling axis is known to participate in later stages of aneurysm progression in murine MFS models22,23,40 and drives excess collagen I expression in MFS patient-derived aortic SMCs in vitro.11,26 We therefore propose that SHF-specific enriched TGF-β-SMAD mediated gene regulation may participate in this anatomically focal collagen synthetic pathway gene activation. Conversely, little is known about the role of Twist1 in aortic aneurysm development. While we note that heightened Twist1 mRNA expression and DNA motif accessibility were not specific to the SHF lineage, enrichment of these signals suggested a potential role for this regulatory signal in SHF-derived SMCs. Confirmation that forced overexpression of TWIST1 in human aortic SMCs could effectively induce the SHF-enriched gene signature (fibrillar collagen and SLRP overexpression) without activating markers of non-traced SMCs (e.g., TNFRSF11B and SOST) and of a TWIST1-responsive enhancer for COL1A1 further supports this concept. Given the recognized role of TWIST1 as a mediator of pathologic fibrosis24 and SMC contractile gene silencing41 as well as its SMC phenotype-modulatory role in coronary artery disease42, we conclude that this transcription factor represents an important target for further mechanistic investigation via targeted knockdown and potential future therapeutic design.
Given that aortic SMCs from both SHF and NC lineages showed propensity for SMC modulation but that this process was anatomically confined to the root segment, a critical question remains what factors influence this regional specificity in MFS. We found that a rare subset (approximately 2%) of SMCs in control aortas demonstrated the modulated phenotype and that these cells were also restricted to the aortic root sinuses (Figure 4C), suggesting that physiologic conditions alone may be sufficient to drive focal but macroscopically inconsequential SMC pathology in this region. Indeed, supraphysiologic pressure overload is sufficient to induce a remarkably similar SMC phenotypic signature to MFS in the proximal thoracic aorta.43 We therefore propose that a unique combination of region-specific ECM properties and mechanical strain distribution enhance SMC propensity for modulation in the root, which is further exacerbated by underlying loss-of-function mutations in the elastic ECM apparatus (e.g., Fbn1). Furthermore, the embryologic origin-specific, ECM-modulatory transcriptome adopted by resident SMCs may further exacerbate this pathology, in particular the activation of collagen deposition by SHF-derived SMCs. Clinically, epidemiologic data highlights the more virulent clinical course of dilatation within the aortic root segment compared to the ascending aorta in non-syndromic TAA, suggesting heightened propensity for aortic degeneration in the root segment in general.44
This study has several limitations. We chose to use a single lineage-tracing marker for SHF-derived cells, leading to potential uncertainty about the origins of non-traced SMCs which we consider as NC-derived. While this allows direct comparison of SMCs from the same pool of mice, adding an NC lineage-tracing strain (e.g., Wnt1-Cre) would further augment the study of NC-specific changes. Our analysis is also limited by the inclusion of a single time point for this study. We elected to study mice at 16 weeks to augment our previous report of SMC phenotype at 4 and 24 weeks and predictably found an intermediate level of de-differentiation in the MFS-specific modSMC cluster. It remains possible that the SMC phenotypes of may diverge further or converge upon a more cohesive identity with aneurysm progression over time. Ultimately, longitudinal analyses of these mechanisms will further augment our understanding of the functional consequences of lineage-specific pathology. While we separated male and female data with respect to aneurysm severity and ratios of modSMCs to quiescent SMCs, analyzing scRNA/scATAC data from males and females together limits our ability to comment on the interplay of sex dimorphism and embryologic origin. Nevertheless, this approach may provide a more heterogeneous sampling of SMC phenotype within the spectrum of MFS biology enabling more robust statistical comparisons. Our analysis is also limited by the application of several, discrete pairwise comparisons within the complete scRNAseq dataset comprising multiple experimental variables (genotype, sex, embryologic origin). While this methodology lends itself to implementation of well-validated computational pipelines employing pairwise non-parametric tests, performing select comparisons based on pre-determined experimental questions may induce bias and increase error compared to a more comprehensive multiple group comparison. Finally, while we have identified graded Twist1 activation based on SMC origin as a likely regulator of spatially augmented collagen synthetic pathways in the aortic root, we cannot definitively determine whether this represents a deleterious response. Future targeted inhibition of this gene regulatory axis in vivo will ultimately be necessary to make this determination.
Supplementary Material
Highlights:
Aortic root smooth muscle cells derived from both second heart field and neural crest lineages undergo aneurysm-associated phenotypic modulation in murine Marfan syndrome
The smooth muscle cell modulation signature is distinguished by enriched fibrillar collagen and small leucine-rich proteoglycan expression.
Integrated single-cell transcriptomic and epigenomic analysis enables the identification of novel aneurysm-related gene regulatory signals in the second heart field lineage, including Twist1 and TGF-β-Smad2/3/4
Acknowledgments:
The authors thank the staff scientists at the Stanford Genome Sequencing Service Center for critical assistance in single-cell RNA library preparation and sequencing.
Sources of Funding:
This work was supported by grants from National Institute of Health F32HL154681 (AJP), F32HL160058 (ARD), R01HL157949 (MPF), K08HL153798 (PC), K08HL152308 (RW), R01HL139478 (TQ), R01HL145708 (TQ), R01HL134817 (TQ), R01HL151535 (TQ), R01HL156846 (TQ), a Human Cell Atlas grant from the Chan Zuckerberg Foundation (TQ), and from the American Heart Association #834986 (RS), 20CDA35310303 (PC) The NovaSeq 6000 device used in this study was purchased using NIH grant S10OD025212 funds.
Non-standard Abbreviations and Acronyms:
- ECM
Extracellular matrix
- MFS
Marfan syndrome
- NC
Neural crest
- scRNAseq
Single-cell RNA sequencing
- scATACseq
Single-cell assay for transposase-accessible chromatic by sequencing
- SHF
Second heart field
- SMC
Smooth muscle cell
- TAA
Thoracic aortic aneurysm
Footnotes
Disclosures: None.
References
- 1.Judge DP, Dietz HC. Marfan’s syndrome. Lancet Lond Engl 2005;366:1965–1976. doi: 10.1016/S0140-6736(05)67789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Dev Camb Engl 2000;127:1607–1616. [DOI] [PubMed] [Google Scholar]
- 3.Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol 2005;281:78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Sawada H, Rateri DL, Moorleghen JJ, Majesky MW, Daugherty A. Smooth Muscle Cells Derived From Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report. Arterioscler Thromb Vasc Biol 2017;37:1722–1726. doi: 10.1161/ATVBAHA.117.309599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruddy JM, Jones JA, Ikonomidis JS. Pathophysiology of thoracic aortic aneurysm (TAA): is it not one uniform aorta? Role of embryologic origin. Prog Cardiovasc Dis 2013;56:68–73. doi: 10.1016/j.pcad.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet 2017;49:97–109. doi: 10.1038/ng.3723. [DOI] [PubMed] [Google Scholar]
- 7.Iosef C, Pedroza AJ, Cui JZ, Dalal AR, Arakawa M, Tashima Y, Koyano TK, Burdon G, Churovich SMP, Orrick JO, Pariani M, Fischbein MP. Quantitative proteomics reveal lineage-specific protein profiles in iPSC-derived Marfan syndrome smooth muscle cells. Sci Rep 2020;10:20392. doi: 10.1038/s41598-020-77274-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jian Gong, Dong Zhou, Longtan Jiang, Ping Qiu, Milewicz Dianna M, Eugene Chen Y., Bo Yang. In Vitro Lineage-Specific Differentiation of Vascular Smooth Muscle Cells in Response to SMAD3 Deficiency. Arterioscler Thromb Vasc Biol 2020;40:1651–1663. doi: 10.1161/ATVBAHA.120.313033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacFarlane EG, Parker SJ, Shin JY, et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J Clin Invest n.d;129:659–675. doi: 10.1172/JCI123547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedroza AJ, Tashima Y, Shad R, Cheng P, Wirka R, Churovich S, Nakamura K, Yokoyama N, Cui JZ, Iosef C, Hiesinger W, Quertermous T, Fischbein MP. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler Thromb Vasc Biol 2020;40:2195–2211. doi: 10.1161/ATVBAHA.120.314670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedroza AJ, Koyano T, Trojan J, Rubin A, Palmon I, Jaatinen K, Burdon G, Chang P, Tashima Y, Cui JZ, Berry G, Iosef C, Fischbein MP. Divergent effects of canonical and non-canonical TGF-β signalling on mixed contractile-synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J Cell Mol Med 2020;24:2369–2383. doi: 10.1111/jcmm.14921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harmon AW, Nakano A. Nkx2-5 lineage tracing visualizes the distribution of second heart field-derived aortic smooth muscle. Genes N Y N 2000 2013;51:862–869. doi: 10.1002/dvg.22721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L, Harvey RP. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3’UTR-ires-Cre allele of the homeobox gene Nkx2–5. Int J Dev Biol 2002;46:431–439. [PubMed] [Google Scholar]
- 14.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tashima Y, He H, Cui JZ, Pedroza AJ, Nakamura K, Yokoyama N, Iosef C, Burdon G, Koyano T, Yamaguchi A, Fischbein MP. Androgens Accentuate TGF-β Dependent Erk/Smad Activation During Thoracic Aortic Aneurysm Formation in Marfan Syndrome Male Mice. J Am Heart Assoc 2020;9:e015773. doi: 10.1161/JAHA.119.015773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen JZ, Sawada H, Ye D, Katsumata Y, Kukida M, Ohno-Urabe S, Moorleghen JJ, Franklin MK, Howatt DA, Sheppard MB, Mullick AE, Lu HS, Daugherty A. Deletion of AT1a (Angiotensin II Type 1a) Receptor or Inhibition of Angiotensinogen Synthesis Attenuates Thoracic Aortopathies in Fibrillin1C1041G/+ Mice. Arterioscler Thromb Vasc Biol 2021;41:2538–2550. doi: 10.1161/ATVBAHA.121.315715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuart T, Srivastava A, Lareau C, Satija R. Multimodal single-cell chromatin analysis with Signac. BioRxiv 2020:2020.11.09.373613. doi: 10.1101/2020.11.09.373613. [DOI] [Google Scholar]
- 18.Rada-Iglesias A, Bajpai R, Prescott S, Brugmann SA, Swigut T, Wysocka J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 2012;11:633–648. doi: 10.1016/j.stem.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 22.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol 2015;35:911–917. doi: 10.1161/ATVBAHA.114.305150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ning X, Zhang K, Wu Q, Liu M, Sun S. Emerging role of Twist1 in fibrotic diseases. J Cell Mol Med 2018;22:1383–1391. doi: 10.1111/jcmm.13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vrljicak P, Cullum R, Xu E, Chang ACY, Wederell ED, Bilenky M, Jones SJM, Marra MA, Karsan A, Hoodless PA. Twist1 Transcriptional Targets in the Developing Atrio-Ventricular Canal of the Mouse. PLoS ONE 2012;7:e40815. doi: 10.1371/journal.pone.0040815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crosas-Molist E, Meirelles T, López-Luque J, et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler Thromb Vasc Biol 2015;35:960–972. doi: 10.1161/ATVBAHA.114.304412. [DOI] [PubMed] [Google Scholar]
- 27.Baumgartner C, Mátyás G, Steinmann B, Baumgartner D. Marfan syndrome--a diagnostic challenge caused by phenotypic and genetic heterogeneity. Methods Inf Med 2005;44:487–497. [PubMed] [Google Scholar]
- 28.Baumgartner D, Baumgartner C, Schermer E, Engl G, Schweigmann U, Mátyás G, Steinmann B, Stein JI. Different patterns of aortic wall elasticity in patients with Marfan syndrome: a noninvasive follow-up study. J Thorac Cardiovasc Surg 2006;132:811–819. doi: 10.1016/j.jtcvs.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, LeMaire SA, Chen L, Shen YH, Gan Y, Bartsch H, Carter SA, Utama B, Ou H, Coselli JS, Wang XL. Increased collagen deposition and elevated expression of connective tissue growth factor in human thoracic aortic dissection. Circulation 2006;114:I200–205. doi: 10.1161/CIRCULATIONAHA.105.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu G, Cheng W, Yao C, Yin J, Tong C, Rao A, Yen L, Ku M, Rao J. Quantitative proteomics analysis by isobaric tags for relative and absolute quantitation identified Lumican as a potential marker for acute aortic dissection. J Biomed Biotechnol 2011;2011:920763. doi: 10.1155/2011/920763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.GU G, WAN F, XUE Y, CHENG W, ZHENG H, ZHAO Y, FAN F, HAN Y, TONG C, YAO C. Lumican as a novel potential clinical indicator for acute aortic dissection: A comparative study, based on multi-slice computed tomography angiography. Exp Ther Med 2016;11:923–928. doi: 10.3892/etm.2016.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dupuis LE, Kern CB. Small leucine-rich proteoglycans exhibit unique spatiotemporal expression profiles during cardiac valve development. Dev Dyn Off Publ Am Assoc Anat 2014;243:601–611. doi: 10.1002/dvdy.24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S, Birk DE. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. FEBS J 2013;280:2120–2137. doi: 10.1111/febs.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaefer L, Iozzo RV. Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem 2008;283:21305–21309. doi: 10.1074/jbc.R800020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalamajski S, Oldberg A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol J Int Soc Matrix Biol 2010;29:248–253. doi: 10.1016/j.matbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Cikach FS, Koch CD, Mead TJ, Galatioto J, Willard BB, Emerton KB, Eagleton MJ, Blackstone EH, Ramirez F, Roselli EE, Apte SS. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight n.d;3. doi: 10.1172/jci.insight.97167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dupuis LE, Nelson EL, Hozik B, Porto SC, Rogers-DeCotes A, Fosang A, Kern CB. Adamts5−/− Mice Exhibit Altered Aggrecan Proteolytic Profiles That Correlate with Ascending Aortic Anomalies. Arterioscler Thromb Vasc Biol 2019;39:2067–2081. doi: 10.1161/ATVBAHA.119.313077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krishna SM, Seto S-W, Jose RJ, Li J, Morton SK, Biros E, Wang Y, Nsengiyumva V, Lindeman JHN, Loots GG, Rush CM, Craig JM, Golledge J. Wnt Signaling Pathway Inhibitor Sclerostin Inhibits Angiotensin II-Induced Aortic Aneurysm and Atherosclerosis. Arterioscler Thromb Vasc Biol 2017;37:553–566. doi: 10.1161/ATVBAHA.116.308723. [DOI] [PubMed] [Google Scholar]
- 39.Alencar GF, Owsiany KM, Karnewar S, et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020;142:2045–2059. doi: 10.1161/CIRCULATIONAHA.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA. Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor β Signaling. J Am Heart Assoc 2017;6. doi: 10.1161/JAHA.116.004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Guo J-R, Wu X-L, Wang X, Zhu Z-M, Wang Y, Gu X, Fan Y. TWIST1 induces phenotypic switching of vascular smooth muscle cells by downregulating p68 and microRNA-143/145. FEBS Open Bio 2021;11:932–943. doi: 10.1002/2211-5463.13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nurnberg ST, Guerraty MA, Wirka RC, et al. Genomic profiling of human vascular cells identifies TWIST1 as a causal gene for common vascular diseases. PLoS Genet 2020;16:e1008538. doi: 10.1371/journal.pgen.1008538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pedroza AJ, Shad R, Dalal AR, Yokoyama N, Nakamura K, Hiesinger W, Fischbein MP. Acute Induced Pressure Overload Rapidly Incites Thoracic Aortic Aneurysmal Smooth Muscle Cell Phenotype. Hypertens Dallas Tex 1979 2022;79:e86–e89. doi: 10.1161/HYPERTENSIONAHA.121.18640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalogerakos PD, Zafar MA, Li Y, Mukherjee SK, Ziganshin BA, Rizzo JA, Elefteriades JA. Root Dilatation Is More Malignant Than Ascending Aortic Dilation. J Am Heart Assoc 2021:e020645. doi: 10.1161/JAHA.120.020645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scRNAseq data (raw FASTQ and processed gene expression matrices) have been made publicly available at the gene expression omnibus (GEO) repository under accession GSE186845.