

Abstract
In-vitro-transcribed messenger RNA (mRNA) has recently shown increasing significance in the development of vaccines and therapeutics. Immunogenic double-stranded RNA (dsRNA) is an undesired byproduct formed during in vitro transcription (IVT), and it is challenging to reduce dsRNA byproduct from mRNA due to their similar sizes and intrinsic characteristics. Removal of dsRNA relies heavily on post-IVT chromatography purifications, such as reverse-phase high-pressure liquid chromatography, which increase manufacturing costs, reduce yield, and often decrease integrity, especially for long mRNA. Thus, it would be ideal to reduce and control the level of dsRNA during IVT. We herein present a simple, scalable, and controllable method to reduce the formation of dsRNA byproducts during IVT. Selected chaotropic agents at optimized concentrations are included during IVT to create a mild denaturing environment to prevent the undesired intermolecular or intramolecular base-pairing that is thought to promote RNA-templated dsRNA formation by RNA polymerase. Compared with regular IVT, our improved method produces mRNA with significantly less dsRNA, much lower immuno-stimulation, and more efficient protein expression. Therefore, this method potentially eliminates dsRNA removal purification steps and does not require reduced magnesium concentration, elevated temperature, or custom reagents, enabling a straightforward, high-yield, and cost-effective scale-up approach for mRNA manufacturing.
Keywords: MT: RNA/DNA editing, messenger RNA, double-stranded RNA, in vitro transcription, mRNA purification, chaotropic agents
Graphical abstract
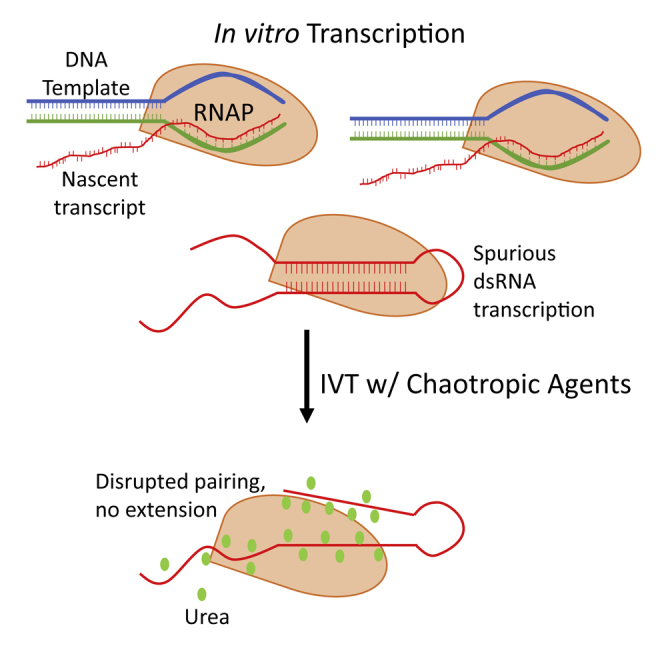
Addition of chaotropic agents to in vitro transcription reactions minimizes the formation of undesired double-stranded RNA byproducts while maintaining yield and transcript integrity, obviating the need for harsh or time-consuming HPLC purification techniques.
Introduction
Two mRNA vaccines were authorized for emergency use to combat the COVID-19 pandemic by multiple health authorities in the end of 2020,1,2 highlighting the value of in-vitro-transcribed mRNA for vaccines and the production of therapeutic proteins. For therapeutic applications that require chronic dosing, the immuno-stimulatory effect of the exogenous mRNA has to be reduced and controlled to a safe level,3 since an immuno-stimulatory response may lead to hypersensitivity reactions or loss of efficacy. Pioneering research has demonstrated that incorporation of modified nucleotides,4, 5, 6 depletion of uridine,7 improvement of 5’ capping structure,8 and removal of double-stranded RNA (dsRNA)9 all reduce mRNA immuno-stimulatory effect from different pathways. Compared with the first three optimizations that can be done prior to or during in vitro transcription (IVT), the removal of dsRNA remains a manufacturing challenge, as dsRNA is an IVT byproduct that shares similar size and intrinsic characteristics with the desired mRNA. Common methods for dsRNA removal rely heavily on post-synthesis purifications such as reverse-phase high-pressure liquid chromatography (HPLC)10 and hydroxyapatite chromatography.11 These two purification methods exhibit scalability issues due to limited column loading capacity. Furthermore, both methods have severe impact on mRNA integrity due to high temperature and sheer force from extensive processing. Cellulose powder as an improved approach has recently been demonstrated to be effective in removing dsRNA at ambient temperature.12 RNase III may be alternatively used to digest dsRNA in the presence of desired mRNA with additional steps to clear RNase III.13 Nevertheless, post-transcriptional purifications and digestions for dsRNA removal increase manufacturing costs, reduce yield, and often lower the percentage of intact mRNA, especially for long mRNA. Thus, it would be ideal to reduce the formation of dsRNA byproducts in the first place during in vitro transcription.
Earlier work exploring the feasibility of reducing dsRNA during IVT used dramatically less divalent magnesium during IVT. As a result, a sharp yield loss was observed since magnesium is an essential cofactor for RNA polymerases.14,15 Three recent reports recruited competing oligonucleotides,16 co-tethered DNA promoter and RNA polymerase,17 and high temperature15 to reduce dsRNA formation during IVT, respectively. However, custom reagents or elevated temperature impose significant economic and technical burdens for scale-up mRNA manufacturing.
We herein present a simple, scalable, and cost-efficient method to reduce dsRNA formation during IVT. In the IVT reaction, when the 3’ end of a full-length RNA transcript hybridizes with itself (self-priming mode)18,19 or an abortive 5- to 11-nt RNA transcript anneals to another RNA transcript (antisense mode),20,21 bacteriophage-derived DNA-dependent RNA polymerases can use RNA as the template to extend the 3’ end of the full-length or abortive RNA transcript to form a duplex region. From the perspective of RNA thermodynamics, these two prevalent mechanisms of dsRNA formation reveal similar undesired intramolecular or intermolecular base-pairing as the root cause. We hypothesized that attenuating such uncontrolled base-pairing would reduce dsRNA formation during IVT. To test the hypothesis, we incorporated selected chaotropic agents such as urea and formamide at various concentrations in regular IVT reactions to create a mild chaotropic environment where enzymes remain functional, but weak undesired RNA hybridizations were disrupted. We evaluated the reduction and control of dsRNA byproducts in the desired mRNA, immuno-stimulatory effects, and efficiency of protein translation of the mRNA produced through this approach.
Results
Effective reduction of dsRNA formation by selected chaotropic agents
To explore the applicability of chaotropic agents to reduce dsRNA during IVT, a wide spectrum of common chaotropic agents were tested. In addition, mRNA molecules with different nucleoside triphosphate (NTP) chemistry were evaluated. To demonstrate that selected chaotropic agents effectively reduce dsRNA formation during IVT, we primarily used a 5000-nt N1-methylpseudouridine-modified mRNA encoding human amylo-1,6-glucosidase,4-alpha-glucanotransferase (hAGL) as the example since mRNA of this size is prone to degradation when the common HPLC methods are used for post-synthesis dsRNA removal. Ultraviolet (UV) spectrophotometry at 260 nm was used to evaluate mRNA yield after spin column purification. A dot blot method using the dsRNA-specific monoclonal antibody J2 was used to assess dsRNA content in the mRNA samples. Agarose gel electrophoresis was used to confirm the length of mRNA and assess integrity.
As shown in Figure 1, urea and formamide both displayed concentration-dependent dsRNA reduction, and the dsRNA content was close to that of HPLC-purified mRNA when higher concentrations of chaotropic agent were used, such as 0.8 M urea or 1.6 M formamide. This indicated that urea and formamide at higher concentrations were more effective in reducing the undesired nucleobase-pairing that caused dsRNA formation. More importantly, the mRNA yield from the urea or formamide-included IVT was only slightly impacted by the addition of chaotropic agents within the ranges tested in Figure 1, suggesting that these concentration ranges did not impact the efficiency of T7 RNA polymerase. Next, the urea concentration was increased up to 1.6 M at different temperatures. As seen in Figure S1, 1.2 M urea reduced dsRNA further compared with 0.8 M urea at 34°C and 37°C but almost shut down T7 RNA polymerase activity at 40°C. 1.6 M urea aborted the in vitro transcription at all temperatures that were tested (34°C, 37°C, and 40°C). The correlations between urea concentration, dsRNA reduction, and mRNA yield are summarized in Figures 1 and S2. Furthermore, the modified IVT conditions were effective for a variety of RNA molecules, independent of NTP chemistry or 5′ capping method, as significant dsRNA reduction was observed regardless of UTP base modifications (Figure 2).
Figure 1.

Impact of urea and formamide concentration on mRNA yield and dsRNA formation during in vitro transcription reaction
(A) J2 mAb dot blot image (top), agarose gel image (middle), and column comparison (bottom) for urea-included IVT to show reduced dsRNA byproducts and slight impact on mRNA yield. (B) J2 mAb dot blot image (top), agarose gel image (middle), and column comparison (bottom) for formamide-included IVT to show reduced dsRNA byproducts and slight impact on mRNA yield. Purple column indicates mRNA yield (left y axis) and green column indicates dsRNA content (right y axis). All experiments were repeated three times, and the error bars are standard deviation of the mean.
Figure 2.

Reduction of dsRNA by chaotropic agents for mRNA with different NTP chemistry
N1mΨ is N1-methylpseudo uridine triphosphate.
Impact on dsRNA formation by delayed addition of chaotropic agents
We observed that dsRNA formation could be controlled by adding different amounts of chaotropic agent (Figure 1) or through a delayed addition of chaotropic agent (Figure 3). In one example, multiple regular IVT reactions were set up and 8 M urea was added into the IVT mixture at various time points to reach a final concentration of 1 M. As seen in Figure 3, delayed addition of urea had less impact on dsRNA formation, presumably because urea was unable to eliminate dsRNA once it had already formed. More specifically, the five urea-containing samples all shared the same 1 M urea concentration at 180 min, yet the dsRNA content differed dramatically. These results favor a mechanism in which urea prevents undesired nucleobase hybridization in the IVT mixture to reduce the occurrence of RNA-templated RNA polymerase activity that leads to the formation of extended duplex regions. Once the dsRNA has already formed, urea at this concentration may be insufficient to break the extended duplex regions.
Figure 3.

J2 mAb dot blot to show increasing dsRNA byproduct with delayed addition of urea
Improved protein translation and reduced immuno-stimulatory effect
To assess the translatability of mRNA produced by inclusion of chaotropic agents in the IVT process, we prepared mRNA1 and mRNA2, which were synthesized through IVT in the absence of urea and in the presence of 1.2 M urea, respectively. Based on the testing results in Figure S2, 1.2 M urea was chosen to reduce dsRNA. HPLC mRNA was made from regular IVT, but the dsRNA was removed by HPLC. All three mRNA samples were fully buffer-exchanged into water to remove impurities. mRNA1, mRNA2, and HPLC mRNA were transfected in HepG2 cells, and protein expression was quantified after 24 h using In-Cell Western (ICW) blot technique. To our delight, both mRNA1 and mRNA2 had high protein expression (Figures 4C and S3). The results demonstrated that usage of chaotropic agents during IVT had no negative impact on the translation efficiency of the produced mRNA. Furthermore, mRNA2 that had less dsRNA also showed about 20% more protein expression. Compared with non-HPLC-purified mRNA samples mRNA1 and mRNA2, the translation efficiency of HPLC mRNA was surprisingly lower, possibly due to the reduced mRNA integrity as indicated by the more smeared appearance on an agarose gel (Figure 4A). Compared with smaller-size mRNA (<2000-nt),9 the 5000-nt mRNA seemed more prone to degradation caused by elevated temperatures and other HPLC conditions.
Figure 4.

Evaluating mRNA integrity, dsRNA content, protein expression, and immuno-stimulatory effect for mRNA1, mRNA2, and HPLC mRNA
(A) Agarose gel with RNA ladder. (B) J2 mAb dot blot. (C) Protein expression, the mRNA dose is 200 ng, and protein expression from mRNA2 was defined as 100% in this example. (D) immuno-stimulatory (IS) response; the mRNA dose is 1,250 ng; IS response triggered from HPLC mRNA was defined as 1 in this example. All experiments were repeated three times, and the error bars are standard deviation of the mean. mRNA1 is purified mRNA produced from regular IVT, mRNA2 is purified mRNA produced from IVT with 1.2 M urea, and HPLC mRNA is the mRNA produced from regular IVT and purified by RP-HPLC. Polyinosinic:polycytidylic acid [Poly(I:C)], a synthetic dsRNA was used as positive control in the assay.
The immuno-stimulatory effect of mRNA produced through chaotropic agent-included IVT was evaluated using a THP1-Dual Monocyte cell line. THP1-Dual cells express two stably transfected gene constructs: one of them is Lucia luciferase gene reporter, which is driven by an interferon (IFN) stimulated gene 54 (ISG54) promoter, and the other one is nuclear factor kappa B (NF-κB)-inducible secreted alkaline phosphatase gene. THP1-Dual cells are able to sense ssRNA- and dsRNA-triggered immune pathways. As expected, IFN-stimulated response was high in mRNA1 that contained the highest amount of dsRNA byproducts, whereas IFN response was dramatically lower in mRNA2 and HPLC mRNA (Figures 4B, 4D, and S4). The immuno-stimulatory response appeared to correlate with the amount of dsRNA byproduct in the mRNA samples.
Discussion
Chaotropic agents, like urea and formamide, have long been used in gel electrophoresis or other biological and analytical techniques to denature nucleic acids. To achieve a complete denaturation of nucleic acids, the concentrations of chaotropic agents are usually very high, such as 8 M urea and 95% formamide. Such a strong denaturing environment can also abolish enzyme function. Fortunately, the undesired intramolecular or intermolecular ribonucleobase-pairing that serves as the template for dsRNA formation during IVT seems relatively weak. For example, the 3’ polyA tail of a full RNA transcript may fold back and hybridize to a cluster of uridines in the intramolecular mode,22 or a short abortive RNA transcript may hybridize to another longer RNA transcript with only 5 to 11 nucleobase-pairing in the intermolecular mode.23,24 Accordingly, we hypothesized a mild chaotropic environment would be capable of attenuating the undesired nucleobase hybridization and reduce dsRNA formation while keeping all enzymes involved in IVT functionally active to produce mRNA with comparable yield and correct sequence. Within the working ranges, increasing chaotropic agent concentration reduced dsRNA further as the denaturing effect became stronger. Moderate yield loss was only observed when the chaotropic agent concentration approached the upper limit of the working range (1.2M urea, Figure S1). Meanwhile, little mRNA was yielded when the chaotropic agent concentration exceeded the limit (Figure S1), presumably due to the change of T7 RNA polymerase tertiary structure caused by chaotropic agent of high concentration. A feasible working concentration range of chaotropic agents may need to be tailored to different RNA molecules.
A mild chaotropic environment can be achieved by different means, and it seems promising for the concept of dsRNA reduction during IVT. Wu and colleagues reported the synthesis of low-immunogenicity RNA with high-temperature IVT at 52°C.15 The elevated temperature was a clean way without the addition of chemicals to create a mild denaturing environment and reduced the undesired intramolecular or intermolecular hybridizations that caused dsRNA formation. Undoubtedly, it was not possible without the use of an engineered RNA polymerase that can function at high temperatures. Gholamalipour and colleagues reported the use of a competing oligonucleotide strand to prevent the formation of a self-priming structure during IVT.16 Although this was not a common denaturing method, like heat, it prevented the 3’ end from folding back and acted as a blocker specific for the self-priming structure. A combination of two or three of the methods to create a mild denaturing environment may work well to reduce dsRNA formation with potential synergistic effects. We have tested the urea-containing IVT at higher temperature and found that lower amounts of urea are needed to achieve similar levels of dsRNA with higher mRNA yield (Figure S1). However, when it comes to industrial-scale mRNA manufacturing, significantly elevated temperatures or addition of custom reagents may impose extra economic and technical burdens. For example, a 50°C reaction temperature may require special equipment other than common bioreactors. High-quality custom reagents at scale may be costly.
Interestingly, not all common chaotropic agents that we tested worked the same on dsRNA reduction. Dimethyl sulfoxide at tested concentrations did not reduce dsRNA, while sodium dodecyl sulfate at low concentrations abolished the IVT. An explanation could be that some chaotropic agents may have poor capabilities to dissociate the undesired nucleobase hybridization or a stronger tendency to denature enzymes than nucleic acid. Urea, formamide, and others that worked to reduce dsRNA formation also inactivated T7 RNA polymerase at high concentrations (Figure S1). These chaotropic agents weakened the undesired nucleobase hybridizations at lower concentrations than necessary to disrupt enzyme activity. Therefore, an optimum can be screened for selected chaotropic agents to reduce dsRNA formation during IVT while keeping mRNA yield relatively unchanged.
Like performing enzymatic reactions at higher temperatures, including selected chaotropic agents in the IVT introduces a potential risk of altering the mutation rate of the T7 RNA polymerase. The baseline nucleotide misincorporation rate of T7 RNA polymerase under regular reaction conditions is approximately 1 × 10−4.25 At this rate, occasional substitutions may occur, but they should be acceptable provided there is no demonstrable impact on therapeutic potency or toxicity. To assess whether our improved IVT conditions led to an elevation of the mutation rate, we reverse-transcribed the product mRNA into cDNA and performed Sanger sequencing with full coverage from the 5’ end to the start of the polyA tail. We did not observe any consistent errors or other evidence of gross loss of transcription fidelity in the sequencing chromatograms. While we cannot rule out a minor change in the error rate, there does not appear to be a major impact on overall mRNA quality.
In our study, the comparison of mRNA1 and mRNA2 provided an ideal insight for dsRNA-triggered immune response because the only difference that existed in the production (including mRNA synthesis and purification) of mRNA1 and mRNA2 was the use of urea during IVT. Clearly, the incorporation of urea during IVT caused about 60%–70% less dsRNA byproduct in mRNA as seen on J2 mAb dot blot, which in turn led to reduced immuno-stimulatory response as shown on THP1-Dual cell assay. Overall, mRNA produced through chaotropic agent-included IVT generates less immuno-stimulatory response. In addition, the reduction of immuno-stimulatory response may have positive feedback on protein expression. Both mRNA1 and mRNA2 displayed comparably high integrity (no smears seen in Figure 4A), yet mRNA1 that had higher dsRNA byproduct showed about 20% less protein expression by ICW assay than mRNA2. It was not unusual to observe that mRNA samples with reduced dsRNA byproducts triggered less immuno-stimulatory response and more protein expression based on previous reports.9,12 A possible explanation could be that dsRNA induced protein kinase R (PKR) activation, inhibiting translation of the mRNA by phosphorylating translation initiation factor eIF2α.26 However, the comparison of HPLC mRNA and mRNA2 may indicate that mRNA integrity played a more important role in protein translation, since dsRNA content (Figure 4B) and other critical quality attributes, including capping efficiency, residual plasmids, and proteins were comparable (data not shown) for the two mRNA samples, but the integrity of HPLC mRNA was noticeable lower than that of mRNA2 (Figure 4A).
In summary, incorporation of selected chaotropic agents at optimized concentrations during IVT dramatically reduces the formation of immunogenic dsRNA without comprising mRNA yield, and mRNA produced in this way showed higher translation efficiency and lower immuno-stimulatory effect. The level of dsRNA byproducts can be controlled by different amounts of chaotropic agents or additional time of chaotropic agents, and such a controllable approach may be desired for the production of mRNA vaccines. This innovative method represents a simple, practical, controllable, and cost-effective way for scale-up mRNA manufacturing. It potentially provides a promising solution for the challenging dsRNA removal problem and may benefit the development of mRNA vaccines and therapeutics.
Materials and methods
In vitro transcription
Regular IVT mixture included 40 mM HEPES, 10 mM DTT, 2 mM spermidine, 5 mM ribonucleotide triphosphate (N1-methylpseudouridine triphosphate is used unless otherwise specified), 50 ng/μL linear DNA plasmid, 4 KU/mL T7 RNA polymerase, 1 KU/mL RNase inhibitor, and 2 U/mL inorganic pyrophosphatase. All enzymes used in IVT were purchased from New England BioLabs (Ipswich, MA). 15–25 mM Mg2+ was also included in the IVT mixture depending on RNA sequences and capping methods. IVT mRNA was capped co-transcriptionally by using 4 mM CleanCap AG (TriLink BioTechnologies, San Diego, CA) or otherwise specified. DNA template was a linearized plasmid encoding human amylo-1,6-glucosidase,4-alpha-glucanotransferase. The plasmid contained T7 promoter and about 100 nt polyA tail. IVT mixture was incubated at 37°C for 2–3 h. For chaotropic agent-included IVT, the selected chaotropic agent was added into the regular IVT mixture to achieve the target concentration prior to incubation. After IVT, DNase I was added to remove template plasmid DNA followed by proteinase K treatment and spin column purification (Macherey-Nagel NUCLEOSPIN RNA) for enzyme and other impurities removal. All mRNA samples used in cell-based assays were buffer-exchanged into water through tangential flow filtration. mRNA yield was measured by the absorbance at 260 nm.
Gel electrophoresis
The same volume of crude IVT mRNA (after DNase I, but prior to spin column purification) was applied on 0.7% agarose gel to show the difference of mRNA yield visually. The gel was run in 1X TAE buffer at 90 V for 1 h. Gel was pre-stained with GelRed (Biotium) and imaged under green fluorescence or UV on GE Amersham Imager 680.
dsRNA dot blot
J2 mAb (SCICONS) was used for dsRNA dot blot, and 1,000 ng mRNA (500 ng/μL) was applied onto each dot. dsRNA dot blot was performed according to a previous publication.10 Dot blot was visualized on GE Amersham Imager 680.
Delayed addition of urea
A master mix of regular IVT components was prepared and aliquoted into multiple vials. 8 M urea was added into a vial at specific time points to achieve a target concentration of 1 M. At 120 min, 8 M urea and water of the same volume were added into the last two vials. All samples were incubated for a total of 180 min.
Reverse-phase HPLC
Ion-pair RP-HPLC was done according to a previous publication10 using a linear gradient of 30%–70% buffer B (0.1 M triethylammonium acetate, pH 7.0, 25% [v/v] acetonitrile) in buffer A (0.1 M TEAA, pH 7.0) at 55°C. Fractions were combined based on J2 mAb dot blot results and buffer-exchanged into water by tangential flow filtration.
Sanger sequencing
Transcription accuracy of in-vitro-transcribed RNAs was confirmed by Sanger sequencing of cDNA synthesized from the RNA by reverse transcription (RT). Reverse transcription was performed using the SuperScript IV Reverse Transcriptase enzyme (Invitrogen) according to the manufacturer recommended protocol using 2 μg of RNA template and an oligo-dT18 primer. The template RNA was digested away using RNase H (NEB), and the resulting first strand cDNA was purified using RNACleanXP (Beckman-Coulter) SPRI beads. The first strand cDNA was sequenced directly using 12 complementary sequencing primers providing full overlapping coverage from roughly position 147 through to the start of the polyA sequence.
For coverage of the 5’ end, a separate reverse transcription reaction was prepared using the Template Switching RT Enzyme Mix from NEB with a template switching oligo and separate reverse transcription primer closer to the 5’ end of the RNA. Following the RT step, which was performed according to the manufacturer’s recommended protocol, the template RNA was digested with RNase H, and second strand cDNA was synthesized using Q5 Hot Start High-Fidelity 2X Master Mix (NEB). The double-stranded cDNA was purified using SPRI beads, and a sequencing primer complementary to the second strand cDNA was used to determine the sequence of the template RNA through the 5’ end. All primer sequences are provided Table S1.
HepG2 and THP1-Dual cell lines
Hep G2 cells were purchased from American Type Culture Collection and maintained at 37°C in a humidified incubator containing 5% CO2 using Eagle’s minimal essential medium with 10% HI-FBS.
THP1-Dual cells were purchased from InvivoGen and routinely cultured in RPM1 1640 growth medium with 10% HI-FBS at 37°C in a humidified incubator containing 5% CO2. Selective antibiotics, i.e., Blasticidin (10 μg/mL) and Zeocin (100 μg/mL), were added alternatively for routine culture.
Protein expression In-Cell Western assay
HepG2 cells were seeded in 96-well black plate for 24 h before transfection. Cells were transfected with different concentrations of mRNA1, mRNA2, and HPLC mRNA (starting conc. 200 ng) using Lipofectamine Messenger Max (Invitrogen) according to manufacturer instructions. After 24 h, media was aspirated off, and cells were immuno-stained for antibody using the LI-COR’s ICW procedure. Cell Tag dye (provided by LI-COR) was used to stain cells. Protein expression was visualized by LI-COR Odyssey and normalized with Cell Tag signal. Data were analyzed by PLA 3.0 software and Relative Potency was measured using 4 PL analysis.
THP1-Dual reporter assay
THP1-Dual reporter assay was performed according to a previous publication27 with minor modification. Briefly, THP1-Dual cells were seeded in 96-well flat bottom culture plates at 80,000 cells per well in RPMI 1640 growth media with 10% HI-FBS and PMA (625 ng/ml) for 24 h. Cells were transfected with Mock, mRNA (starting conc 5,000 ng per well) and Poly (I:C) (1,250 ng per well). After 24 h of transfection, Luciferase activity was measured in the supernatant by QUANTI-Luc assay using Spectramax M3 plate reader.
Acknowledgments
The authors would like to thank Ultragenyx Pharmaceutical Inc for support and Drs. Jin Zhou and Zheng Meng for insightful discussions.
Author contributions
X.P. and S.W. developed the concept. X.P., V.Y., E.W., W.C., and B.E.L. designed experiments. X.P., W.C., and L.T. prepared all mRNA samples used in cell-based experiments. X.P. performed IVT, gel electrophoresis, and J2 mAb dot blot. V.Y. conducted cell experiments. E.W. performed Sanger sequencing. X.P., V.Y., and E.W. wrote the manuscript, and all authors reviewed and approved the manuscript.
Declaration of interests
All authors are employees and shareholders of Ultragenyx Pharmaceutical Inc. Part of the manuscript contents are included in a patent application.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.08.001.
Supplemental information
References
- 1.Fortner A., Schumacher D. First COVID-19 vaccines receiving the US FDA and EMA emergency use authorization. Discoveries. 2021;9:e122. doi: 10.15190/d.2021.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jhaveri R. The COVID-19 mRNA vaccines and the pandemic: do they represent the beginning of the end or the end of the beginning? Clin. Ther. 2021;43:549–556. doi: 10.1016/j.clinthera.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines — a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karikó K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Karikó K., Muramatsu H., Welsh F.A., Ludwig J., Kato H., Akira S., Weissman D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008;16:1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson B.R., Muramatsu H., Nallagatla S.R., Bevilacqua P.C., Sansing L.H., Weissman D., Karikó K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010;38:5884–5892. doi: 10.1093/nar/gkq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaidyanathan S., Azizian K.T., Haque A.K.M.A., Henderson J.M., Hendel A., Shore S., Antony J.S., Hogrefe R.I., Kormann M.S.D., Porteus M.H., McCaffrey A.P. Uridine depletion and chemical modification increase Cas9 mRNA activity and reduce immunogenicity without HPLC purification. Mol. Ther. Nucleic Acids. 2018;12:530–542. doi: 10.1016/j.omtn.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sikorski P.J., Warminski M., Kubacka D., Ratajczak T., Nowis D., Kowalska J., Jemielity J. The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5′ cap modulates protein expression in living cells. Nucleic Acids Res. 2020;48:1607–1626. doi: 10.1093/nar/gkaa032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karikó K., Muramatsu H., Ludwig J., Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011;39:e142. doi: 10.1093/nar/gkr695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weissman D., Pardi N., Muramatsu H., Karikó K. HPLC purification of in vitro transcribed long RNA. Methods Mol. Biol. 2013;969:43–54. doi: 10.1007/978-1-62703-260-5_3. [DOI] [PubMed] [Google Scholar]
- 11.Kalmakoff J., Payne C.C. A simple method for the separation of single-stranded and double-stranded RNA on hydroxyapatite. Anal. Biochem. 1973;55:26–33. doi: 10.1016/0003-2697(73)90287-x. [DOI] [PubMed] [Google Scholar]
- 12.Baiersdörfer M., Boros G., Muramatsu H., Mahiny A., Vlatkovic I., Sahin U., Karikó K. A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids. 2019;15:26–35. doi: 10.1016/j.omtn.2019.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foster J.B., Choudhari N., Perazzelli J., Storm J., Hofmann T.J., Jain P., Storm P.B., Pardi N., Weissman D., Waanders A.J., et al. Purification of mRNA encoding chimeric antigen receptor is critical for generation of a robust T-cell response. Hum. Gene Ther. 2019;30:168–178. doi: 10.1089/hum.2018.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanwal F., Chen T., Zhang Y., Zhang Y., Simair A., Rujie C., Sadaf Zaidi N.U.S., Guo X., Wei X., Siegel G., Lu C. Large-scale in vitro transcription, RNA purification and chemical probing analysis. Cell. Physiol. Biochem. 2018;48:1915–1927. doi: 10.1159/000492512. [DOI] [PubMed] [Google Scholar]
- 15.Wu M.Z., Asahara H., Tzertzinis G., Roy B. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA. 2020;26:345–360. doi: 10.1261/rna.073858.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gholamalipour Y., Johnson W.C., Martin C.T. Efficient inhibition of RNA self-primed extension by addition of competing 3’-capture DNA-improved RNA synthesis by T7 RNA polymerase. Nucleic Acids Res. 2019;47:e118. doi: 10.1093/nar/gkz700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavac E., Ramírez-Tapia L.E., Martin C.T. High-salt transcription of DNA cotethered with T7 RNA polymerase to beads generates increased yields of highly pure RNA. J. Biol. Chem. 2021;297:100999. doi: 10.1016/j.jbc.2021.100999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Triana-Alonso F.J., Dabrowski M., Wadzack J., Nierhaus K.H. Self-coded 3′-extension of run-off transcripts produces aberrant products during in vitro transcription with T7 RNA polymerase. J. Biol. Chem. 1995;270:6298–6307. doi: 10.1074/jbc.270.11.6298. [DOI] [PubMed] [Google Scholar]
- 19.Gholamalipour Y., Karunanayake Mudiyanselage A., Martin C.T. 3’ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character-RNA-Seq analyses. Nucleic Acids Res. 2018;46:9253–9263. doi: 10.1093/nar/gky796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mu X., Greenwald E., Ahmad S., Hur S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018;46:5239–5249. doi: 10.1093/nar/gky177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nacheva G.A., Berzal-Herranz A. Preventing nondesired RNA-primed RNA extension catalyzed by T7 RNA polymerase. Eur. J. Biochem. 2003;270:1458–1465. doi: 10.1046/j.1432-1033.2003.03510.x. [DOI] [PubMed] [Google Scholar]
- 22.Moqtaderi Z., Geisberg J.V., Struhl K. Secondary structures involving the poly(A) tail and other 3’ sequences are major determinants of mRNA isoform stability in yeast. Microb. Cell. 2014;1:137–139. doi: 10.15698/mic2014.04.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin C.T., Muller D.K., Coleman J.E. Processivity in early stages of transcription by T7 RNA polymerase. Biochemistry. 1988;27:3966–3974. doi: 10.1021/bi00411a012. [DOI] [PubMed] [Google Scholar]
- 25.Huang J., Brieba L.G., Sousa R. Misincorporation by wild-type and mutant T7 RNA polymerases: identification of interactions that reduce misincorporation rates by stabilizing the catalytically incompetent open conformation. Biochemistry. 2000;39:11571–11580. doi: 10.1021/bi000579d. [DOI] [PubMed] [Google Scholar]
- 26.Balachandran S., Roberts P.C., Brown L.E., Truong H., Pattnaik A.K., Archer D.R., Barber G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13:129–141. doi: 10.1016/s1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- 27.Nelson J., Sorensen E.W., Mintri S., Rabideau A.E., Zheng W., Besin G., Khatwani N., Su S.V., Miracco E.J., Issa W.J., et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020;6:eaaz6893. doi: 10.1126/sciadv.aaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.