

Abstract
Urothelial carcinoma (UC) comprises the majority of bladder cancers. Standard platinum-based chemotherapy has a response rate of approximately 50%, but drug resistance develops after short-term treatment. Deubiquitinating (DUB) enzyme inhibitors increase protein polyubiquitination and endoplasmic reticulum (ER) stress, which might further suppress cancer stemness and overcome cisplatin resistance. Therefore, we investigated the cytotoxic effect and potential mechanisms of b-AP15 on urothelial carcinoma. Our results revealed that b-AP15 induced ER stress and apoptosis in BFTC905, T24, T24/R (cisplatin-resistant), and RT4 urothelial carcinoma cell lines. Inhibition of the MYC signaling pathway and cancer stemness by b-AP15 was confirmed by RNA sequencing, RT-PCR, immunoblotting, and sphere-forming assays. In the mouse xenograft model, the combination of b-AP15 and cisplatin showed superior therapeutic effects compared with either monotherapy.
Keywords: bladder cancer, chemoresistance, cancer stem cell, deubiquitination, ER stress
Graphical abstract
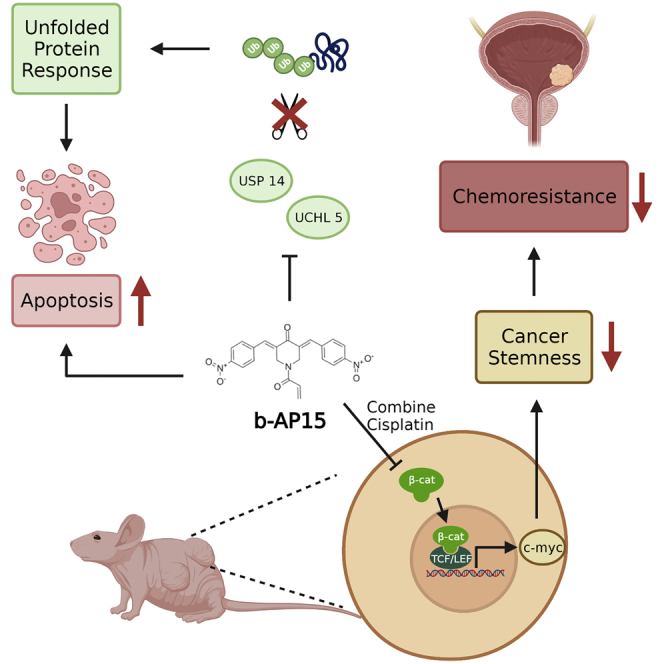
Standard platinum-based chemotherapy has a 50% drug resistance in urothelial carcinoma (UC). The DUB inhibitor b-AP15 induces ER stress and apoptosis, and it also inhibits the MYC signaling pathway and stemness-related chemoresistance in UC cell lines. The combination of b-AP15 and cisplatin is superior to either monotherapy in vivo.
Introduction
Urothelial carcinoma comprises more than 90% of bladder cancers. According to the 2021 Surveillance, Epidemiology, and End Results (SEER) database, bladder cancer accounts for 4.4% of all new cancer cases and 2.8% of all cancer deaths in the United States. The 5-year survival for in situ disease is 96%, but the 5-year survival for metastatic disease is only 6.4%.
The standard regimen for metastatic urothelial carcinoma is platinum-based chemotherapy, which is comprised of cisplatin1,2 for patients with intact renal functions and weaker carboplatin3 for patients with impaired renal functions. Chemotherapy was the only option until the invention of immune-checkpoint inhibitors, which exhibited overall response rates of approximately 20%–30% in cisplatin-ineligible patients.4, 5, 6 The first and only approved targeted therapy for urothelial carcinoma is erdafitinib, which only benefits patients who bear the uncommon FGFR3 mutation.7 Most recently, erfortumab vedolin, an antibody-drug conjugate directed against nectin-4, demonstrated positive results in third-line treatment for metastatic urothelial carcinoma (UC).8 Despite these advances in new treatment modalities, cisplatin is still the most important therapy for UC and has the highest response rate (approximately 50%). The study of drug resistance against cisplatin has been of upmost importance, and the underlying mechanisms include cancer stemness.9
Recently, proteasome inhibitors have gained attention in cancer treatments. The 26S proteasome degrades ubiquitin-tagged proteins and comprises a core catalytic 20S particle with two 19S regulatory caps. The deubiquitinating (DUB) inhibitor b-AP15 decreases the DUB activity of 19S particles, thus increasing protein ubiquitination and subsequent protein degradation. The actual mechanism involves the inhibition of two 19S deubiquitinases, ubiquitin-specific peptidase 14 (USP14) and ubiquitin C-terminal hydrolase 5 (UCHL5), resulting in accumulation the of polyubiquitin.10
Previously, b-AP15 was shown to increase the of the ubiquitination of the antiapoptotic protein BcL-2, inducing apoptosis in some cancer cells. The 19S regulatory particle and USP14 are therefore potential therapeutic targets.11,12 Other mechanisms have been proposed to be associated with the apoptotic effects, including endoplasmic reticulum (ER) stress, oxidative stress, and TRAIL-mediated apoptosis.13, 14, 15, 16
Most recently, Ding et al. reported suppression of Wnt/Notch1 signaling by b-AP15 in hepatocellular carcinoma cells, resulting in decreased cancer cell survival.17 In addition to its proapoptotic effect, this finding revealed the possible ability of b-AP15 to overcome stemness-related drug resistance in cancer cells. We herein present the first report regarding the therapeutic effect of b-AP15 against chemosensitive and chemoresistant UC and the synergistic effect of b-AP15 in combination with cisplatin.
Results
UCHL5 and USP14 are increased in bladder UC
First, we evaluated the presence of UCHL5 and USP14 in human bladder UC tissues. Immunohistochemistry (IHC) showed positive UCHL5 (Figures 1A and 1E) and USP14 (Figures 1B and 1F) staining in the tumor region. The IHC intensity score of UCHL5 and USP14 was higher in the tumor than in the non-tumor region (Figures 1A and 1B). The IHC intensity score of UCHL5 and USP14 was higher in chemoresistant patients than in chemosensitive patients (Figures 1C, 1D, 1G, and 1H). These findings suggested that UCHL5 and USP14 are expressed in bladder UC and are related to chemoresistance.
Figure 1.

The increase of UCHL5 in urothelial carcinoma is associated with chemo-resistance
(A and B) The expression of (A) UCHL5 or (B) USP14 in tumor or normal tissue was evaluated through the IHC staining. (C and D) Five chemoresistant and five chemosensitive specimens were immunohistochemically stained with (C) anti-UCHL5 or (D) anti-USP14 antibody. (A)–(D) were presented as Tukey’s range and analyzed using two-tailed Student’s t tests. Parafilm-embedded clinical tumor samples were sliced for immunochemistry analysis. (E and F) Tumor or normal tissue were stained with (E) anti-UCHL5 or (F) anti-USP14 antibody. (G and H) UCHL5 (G) or USP14 (H) expression of chemoresistant or chemosensitive samples was measured using IHC staining.
b-AP15 increases the accumulation of polyubiquitinated proteins and induces ER stress
To confirm the pharmacological action of b-AP15, the BFC905, T24, T24/R, and RT4 cell lines were treated with different concentrations of b-AP15 up to 2,000 nM for 24 h and then underwent immunoblotting using an anti-ubiquitin antibody. Immunoblotting indicated increased polyubiquitination in bladder UC cells as the drug concentration increased (Figure 2A). Immunoblotting with ER stress makers, including anti-Bip, anti-ATF4, and anti-CHOP, was increased in responses to b-AP15 treatment (Figure 2B). Comparing the RNA sequencing (RNA-seq) of T24 cells treated with 400 nM b-AP15 or normal saline for 6 h, the Gene Ontology (GO) terms of differentially expressed genes revealed enrichment in unfolded protein response, ER stress, and apoptosis (Figure 2C). These results are consistent with b-AP15, as a DUB inhibitor, enhancing the accumulation of polyubiquitinated proteins and subsequent ER stress.
Figure 2.

b-AP15 inhibits deubiquitination and leads to ER stress
The BFTC905, T24, T24/R, and RT4 cells were treated with b-AP15 at the indicated doses for 24 h. (A and B) The expression of (A) poly-ubiquitination level or (B) ER stress markers were measured by immunoblotting. (C) T24/R cells were treated with 1,500 nM b-AP15 for 6 h. Gene Ontology (GO) analysis demonstrates the top 30 terms upregulated within b-AP15 treatment. BP or MF displays Biological Process or Molecular Function groups of GO, respectively.
b-AP15 decreases cell viability and induces apoptosis in bladder UC
To evaluate the cytotoxicity of b-AP15 in human UC cells, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed after treating the cell lines with different concentrations of b-AP15. The decrease in cell viability was generally dose and time dependent, with half maximal inhibitory concentration (IC50) values ranging from 250 to 2,000 nM (Figure 3A). Flow cytometry revealed an approximately 3-fold increase in apoptotic cells after treatment with b-AP15 (Figures 3 and S1). Immunoblotting showed increased apoptotic markers including cleaved-PARP, cleaved caspase-3, and cleaved caspase-7 (Figure 3C). These findings confirm that the cytotoxicity of b-AP15 resulted from the induction of apoptosis.
Figure 3.

The treatment of b-AP15 induces apoptosis in human urothelial carcinoma cell lines
(A) The BFTC905, T24, T24/R, and RT4 cells were treated with b-AP15 at indicated concentrations for 24 or 48 h. MTT assay was performed to determine cell viability. (#p < 0.0001). (B) The cells were treated with b-AP15 at indicated concentrations for 24 h, followed by flow cytometry analysis. Apoptotic cells represent the cell population with higher Annexin-V expression (∗p < 0.05; ∗∗p < 0.01; #p < 0.0001). (C) The cells were treated with b-AP15 for 24 h, then immunoblotting was conducted to estimate the expression of apoptosis markers.
b-AP15 suppresses cancer stemness by inhibiting the β-catenin and c-myc signaling pathways
After treating T24 cells with 400 nM b-AP15 or normal saline for 6 h, gene set enrichment analysis (GSEA) of RNA-seq revealed downregulation of pathways related to β-catenin, MYC, cancer stem cell, and cisplatin resistance (Figures 4A–4D). More specifically, the treatment of b-AP15 downregulated genes related to proliferation and cell division, including MKI67, CCNB1/2, TOP2A, or CDK1 (Figure S2). Genes that are downregulated upon β-catenin depletion including EIF3A, EGFR, HMGA2, and SOX9 were suppressed within b-AP15 treatment; these genes are also involved in cancer progression (Table S6). Genes related to cancer stemness or drug resistance also declined, such as GJA1, VWDE, CDK6, MYB, and AKT3 (Figures S3 and S4). RT-PCR demonstrated decreased MYC mRNA transcription after b-AP15 treatment in a dose-dependent manner, except for RT4 cells (Figure 4E). Immunoblotting confirmed the decrease in β-catenin and c-myc protein levels (Figure 4F). The sphere-forming assays (Figure S5) showed a decrease in both sphere numbers (Figure 4G) and diameters (Figure 4H) after b-AP15 treatment. These results indicate that b-AP15 reduced cancer stemness by inhibiting the β-catenin and c-myc signaling pathways.
Figure 4.

b-AP15 inhibits cancer stemness via c-myc downregulation in UC cell lines
(A–D) RNA sequencing data from DMSO- or b-AP15-treated T24/R cells was analyzed via gene set enrichment analysis (GSEA). NES, normalized enrichment score. (E and F) The mRNA (E) expression or protein (F) expression of c-myc was determined via quantitative real-time PCR or immunoblotting (∗p < 0.05; ∗∗∗p < 0.001; #p < 0.0001). The cells were subjected to tumorsphere cultivation assay with or without a low-dose b-AP15 treatment (50 nM). (G and H) The results of this assay were presented as relative sphere numbers (G) and sphere diameters (H) (∗p < 0.05; ∗∗p < 0.01; #p < 0.0001).
b-AP15 and cisplatin exert synergistic effects in bladder UC by suppressing β-catenin and myc expression
By immunoblotting, the combination of b-AP15 and cisplatin induced higher levels of apoptotic proteins than either monotherapy (Figure 5A). The β-catenin level in RT4 and the c-myc level in T24 were induced by cisplatin. In other cell lines, the β-catenin and c-myc proteins were unchanged or only slightly suppressed by cisplatin. After combining with b-AP15, the β-catenin and c-myc proteins were markedly suppressed (Figure 5B). Under a fixed concentration ratio (Tables S2–S5) of b-AP15 and cisplatin, the combination index indicated a synergistic effect of combining b-AP15 and cisplatin in UC cell lines (Figure 5C). These results suggested that b-AP15 suppressed cisplatin-induced cancer stemness by inhibiting the β-catenin-c-myc signaling pathways and that the two drugs work synergistically against UC cells.
Figure 5.

The synergistic effect of b-AP15 and cisplatin on human UC cells
The BFTC905, T24, T24/R, and RT4 cells were treated with b-AP15 and/or cisplatin at the indicated doses for 24 h. (A and B) The expression of (A) apoptosis markers or (B) cancer stemness markers were determined by immunoblotting. (C) The MTT assay was performed to evaluate cell viability, and the cell viability data was subjected to calculate the combination index (CI) values. CI values less than 1 indicate the synergistic effects.
b-AP15 reduces tumor growth in mouse xenograft models
BFTC905 and T24 cells were injected into mice as subcutaneous xenografts. The mice were divided into 4 groups that received intraperitoneal injection with normal saline, b-AP15 (7.5 mg/kg thrice a week), cisplatin (5 mg/kg twice a week), or combined b-AP15 + cisplatin, respectively (Figure 6A). Tumor volume and weight were evaluated after 4 weeks of treatment, which revealed significant reductions in both tumor volume (Figures 6B–6E) and tumor weight (Figures 6F–6H) in the combination groups, compared with the control groups and the cisplatin groups. The body weights of the mice did not change significantly during the study period (Figures 6I–6K). These results indicated that the combination of b-AP15 and cisplatin is superior to cisplatin monotherapy against UC in vivo.
Figure 6.

b-AP15 sensitizes the therapeutic effects of cisplatin in mouse xenograft model
(A) The timeline and dosages of the treatment to the mouse xenografts. (B) Images of excised tumor from each group. (C–E) Tumor volume of each group was recorded during the treatment. (F–H) The weights of tumors were evaluated after the harvest. (I–K) The body weights of the mice were tracked at the indicated dates. Each set of data are presented as mean ± SEM (C–H) or ± SD value (I–K) (n = 6) (∗p < 0.05; ∗∗p < 0.01).
Discussion
To date, few attempts have been made to combine cisplatin with potential agents that target one or more of the resistance mechanisms. The once-promising immune-checkpoint inhibitor (ICI)-chemo combination turned out to be unbeneficial in a phase 3 clinical trial,18 presumably to have resulted from the inhibition of the immune system by chemotherapy. In recent years, b-AP15 has demonstrated in vitro and in vivo responses in multiple myeloma,12 breast cancer,19 oral squamous cell carcinoma,20 prostate cancer,21 leukemia,22 and mantle cell lymphoma.23 Specifically, Sooman et al. demonstrated synergistic effects of b-AP15 in combination with cisplatin, gefitinib, gemcitabine, or vinorelbine in lung cancer cells.24 Their results indicated that DUB inhibitors significantly potentiated the cytotoxic effects of all of these chemotherapies in at least one cell line.
Cancers can survive after conventional chemotherapy and radiotherapy due to the presence of cancer stem cells. Cancer stem cells exhibit dysregulated self-renewal capacity and can generate tumor cells in an unlimited fashion. Four transcription factors, MYC, OCT4, SOX2, and NANOG, are essential for maintaining pluripotency and self-renewal. Signaling pathways in normal stem cells, such as Wnt, Notch, and Shh, are dysregulated in cancer stem cells. While cancer stem cells are defined by their maintenance of long-term clonal growth in functional repopulation assays, stemness refers to the integrated functioning or molecular program that governs the stem cell state. Cancer stem cells are antiapoptotic, causing tumor recurrence, progression, and metastasis.25
Whether ER stress contributes or reduces stemness remains elusive. The chaperone GRP78 is overexpressed in many cancers.26 In colorectal cancer, ER stress has been shown to result in loss of stemness.27 In contrast, ER stress is also required to maintain stemness in cancer cells.28 Knockdown of GRP78, ATF6, and XBP1 has been shown to resensitize cancer to chemotherapy.29 Recently, many studies have investigated the role of DUB enzymes in the maintenance of cancer stemness. When ubiquitins are removed from substrates by DUB enzymes, these substrates escape proteasomal degradation. Many DUBs stabilize several cancer stem cell (CSC)-associated transcription factor and oncogenic proteins. For example, USP2a stabilizes MDM2 and thus suppresses TP53 function, USP21 deubiquitinates Nanog and increases stemness, and USP22 upregulates SOX2.30, 31, 32 Due to the paradoxical effect of ER stress on stemness, either activation or inhibition of ER stress and unfolded protein response may have therapeutic potential against CSCs.33
Our study confirmed the pharmacological actions of b-AP15, alone and in combination with cisplatin, in human UC cells. Induction of ER stress and apoptosis after b-AP15 treatment is evident. We also demonstrated the synergistic effect and the suppression of cancer stemness in the b-AP15-cisplatin combination. This combination was superior to either monotherapy in mouse xenograft model.
Materials and methods
Reagents and antibodies
b-AP15 (#HY-13989) was obtained from MedChemExpress (Monmouth Junction, NJ, USA). We purchased the UCHL5 (#11527-1-AP) antibody from Proteintech Group (Rosemont, IL, USA). Bip (#3183), ATF4 (#11815), CHOP (#2895S), cleaved caspase-3 (#9664), cleaved caspase-7 (#8438), caspase-3 (#14220), caspase-7 (#12827), c-myc (#18683), β-catenin (9582-P), cleaved PARP (#9541), and PARP (#9542) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The β-actin antibody (#sc-69879) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and an anti-ubiquitinylated protein antibody (#04–263) from Merck Millipore (Burlington, MA, USA).
IHC
We obtained bladder cancer tissue from patients who received systemic chemotherapy. The response evaluation criteria in solid tumors (RECIST) was applied to define the response to chemotherapy. Cases with partial or complete response to chemotherapy were considered chemosensitive, while patients with progressive disease were classified as chemoresistant. The specimens before chemotherapy were used for experiments in chemosensitive cases, whereas the specimens after chemotherapy were used in the chemoresistant cases.
The experiments involved in human material were approved and the informed consent for the use of existing tissues (from July 2009 to March 2019) was waivered by the 118th meeting of Research Ethics Committee B of the National Taiwan University Hospital on May 10, 2019 (approval no. 201901032RINB).
IHC experiments were performed as previously reported.34 In brief, the sections were incubated with UCHL5 (#11527-1-AP, Proteintech) antibody for 1.5 h. The immunoreactivity of UCHL5 and the IHC score were evaluated by W.C. Lin, a board-certificated uro-oncology specialist and pathologist who was blinded to the clinical data. The IHC score was calculated by multiplying the percentage of positive staining by the intensity score.
Cell culture and the chemo-resistant strain
The BFTC905 (#60068) cell line was obtained from the Bioresource Collection and Research Center (BCRC), Taiwan. We purchased the T24 (#HTB-4) cell line and RT4 (#HTB-2) cell line from the American Type Culture Collection (ATCC), USA. The T24/R cell line was derived from T24 cells with cisplatin resistance, which was established as previously described.35 Briefly, T24 cells were first treated with low-dose cisplatin for 1 month at first, followed by a gradual increase in cisplatin concentration until the acquisition of the T24/R resistant strain. The sensitivity to cisplatin of each cell line was tested using an MTT assay (Figure S6).
BFTC905 cells were cultured in high-glucose DMEM (#11995040, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 15% FBS (#10437028, Gibco). RT4 cells were cultured in modified McCoy’s 5A medium (#16600082, Gibco) containing 10% FBS (#10437028, Gibco) and 1 mM sodium pyruvate (#11360070, Gibco). T24 and T24/R cells were cultured in RPMI 1640 medium (#11875085, Gibco) containing 10% FBS. All media were supplemented with 2 mM L-glutamine (#A2916801, Gibco) and 100 μg/mL streptomycin-100 units/mL penicillin (#15140–122, Gibco). All cells were cultured at 37°C in a humidified 5% CO2 atmosphere, and the cells were harvested and subcultured using 0.05% trypsin-EDTA (#15400-054, Gibco) to reach approximately 90% confluence.
Cell viability assay
A total of 2∗103 cells per well were seeded into 96-well plates and grown overnight. Then, the cells were treated with a sequential concentration of b-AP15 and/or cisplatin for the indicated time or received no treatment. After treatment, cell viability was determined using an MTT (#21795, Cayman Chemical, Ann Arbor, MI, USA) assay by incubating the cells with 500 μg/mL MTT in culture medium at 37°C for 2 h. Next, we discarded the supernatant and dissolved the formazan crystals formed by metabolically viable cells in dimethyl sulfoxide (DMSO). Finally, the absorbance was measured at 570 nm using a Multiskan GO Microplate Spectrophotometer (Thermo Fisher Scientific).
Combination index
We analyzed the combined effects of b-AP15 and cisplatin using CalcuSyn software (v.1.1.1, Biosoft, Cambridge, UK) based on the method described by Chou and Talalay.36 Based on the various IC50 values of b-AP15 and cisplatin in each cell line, we treated the cells with different ratios of drugs as follows: 1:10 in BFTC905 cells, 1:50 in T24 cells, 1:100 in T24/R cells, and 1:10 in RT4 cells. A combination index (CI) analysis was performed following previous studies.37 CI effects were used to describe the combined dose effects, in which values less than 1 indicate synergistic effects. In addition, values greater than or equal to 1 indicated antagonistic or additive effects, respectively.36
Immunoblotting
Immunoblotting experiments were performed using standard protocols.38 Briefly, total cellular proteins were isolated in lysis buffer containing protease and phosphatase inhibitors. Cell lysates were quantified using a Pierce BCA Protein Assay Kit (#23225, Thermo Fisher Scientific) and denatured in 5× sodium dodecyl sulfate (SDS) sample buffer at 95°C for 10 min. Equal amounts of proteins were subjected to 8%–15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare Life-Sciences, Chicago, IL, USA). The membrane was blocked with 3% bovine serum albumin (BSA)/Tris-buffered saline with 0.1% Tween-20 (TBST) buffer and immunoblotted with selected primary antibodies at 4°C overnight. Membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (#GTX213110-01, #GTX213111-01, GeneTex, Alton Pkwy Irvine, CA, USA) after washing with TBST. The signals were developed using Immobilon Western Chemiluminescent HRP Substrate (ECL) (#WBKLS0500, Merck KGaA, Darmstadt, Germany) and captured with an ImageQuant LAS 4000 system (GE Healthcare Life-Sciences). We analyzed the images via ImageQuant TL8.1 and quantified them by Image J software.
Quantitative real-time PCR
Total RNA was extracted using NucleoZOL (#740404.200, Macherey-Nagel GmbH & Co. KG, Düren, Germany). The RevertAid First Strand cDNA Synthesis Kit (#K1622, Thermo Fisher Scientific) was used to reversetranscribe total RNA to complementary DNA (cDNA). Ten nanograms of cDNA from each sample were subjected to qPCR amplification using PowerUp SYBR Green Master Mix (#A25741, Thermo Fisher Scientific) using the QuantStudio 7 Flex Real-Time PCR system (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). All procedures were performed following the manufacturers’ instructions. Gene-specific primer sequences are shown in Table S1. The 2−ΔΔCt method was implemented to analyze relative gene expression, wherein target gene expression was normalized to GAPDH. The results of at least three independent experiments are displayed as the mean ± SD.
Flow cytometry
To detect the apoptotic cell population, b-AP15-treated cells were harvested using trypsin-EDTA followed by staining of the Muse Annexin V and Dead Cell Assay Kit (#MCH100105, Luminex, Austin, TX, USA) according to the manufacturer’s instructions. We quantified stained cells using a Guava Muse Cell Analyzer (Luminex) equipped with Muse Analysis software.
Tumorsphere cultivation
The tumorsphere cultivation method has been previously reported.39We seeded 1 × 104 cells/well into ultralow adherent 6-well plates (#3471, Corning, Corning, NY, USA) in serum-free DMEM/F-12 medium (#11330032, Gibco) containing N-2 supplement (#17502048, Gibco), 20 ng/mL FGF-basic (#AF-100-18C, PeproTech, Rocky Hill, NJ, USA), and 20 ng/mL EGF (#AF-100-15, PeproTech). We supplied freshly prepared medium every 3 days until day 14. The sizes of the tumorspheres were measured under an Eclipse Ti2-U inverted microscope (Nikon Instrument, Melville, NY, USA) equipped with NIS-Elements BR imaging software. Spheres with diameters greater than 50 μm were defined as tumorspheres.
RNA-seq analysis
T24/R cells were seeded in a 6 cm dish and treated with b-AP15 or DMSO. RNA was isolated from UC cells using NucleoZOL reagent (Macherey-Nagel). Quality control and sequencing of RNA samples were conducted by Biotools Biotech. The original data were acquired from the NovaSeq 6000 Sequencing System (Illumina, San Diego, CA, USA). We performed GO pathway enrichment analysis using clusterProfiler (v.3.10.1).40 Gene expression profiling and GSEA were conducted with 1,000 permutations to identify the enrichment of biological functions and the molecular signatures database (MSigDB) was applied for activated pathways.41 Raw sequencing data is available under BioProject accession number PRJNA780349.
UC xenograft model
The IACUC of National Taiwan University approved all in vivo experiment protocols (IACUC approval no. 20210201). T24 cells (5 × 106) or BFTC905 cells (5 × 106) were suspended in 100 μL medium without serum supplementation. After adding an equal volume of Matrigel Matrix (#354234, Corning), the mixture was subcutaneously injected into 8-week-old male BALB/cAnN mice weighted 20 ± 2 g. Cg-Foxnlnu/CrlNarl mice were obtained from the National Laboratory Animal Center (Taipei, Taiwan). The cages of mice were located in the same rack to minimize the potential confounders. The mice were irregularly divided into four groups 2 weeks after injection and treated with b-AP15 and/or cisplatin or normal saline. The treatment solution is prepared in the following proportion: 45% normal saline, 40% polyethylene glycol 300, 10% b-AP15 and/or cisplatin dissolved in DMSO, and 5% Tween 80. We measured the tumor volumes using the following formula: V = (π/6) × [(A + B)/2]3, where A and B are the shortest and longest tumor diameters, respectively. The mice with symptoms, including losing over 20% of their body weight, severe infection, skin ulcer, depraved appetite, over 2 cm in tumor diameter, or dying, would be sacrificed. No blinding has been done during in vivo experiments. All animal experiments complied with the ARRIVE guidelines and followed the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Statistical analysis
Quantitative data are presented as the mean ± SD or mean ± standard error of the mean (SEM). Comparisons between groups were analyzed using two-tailed Student’s t tests, one-way analysis of variance (ANOVA), or Mann-Whitney U tests. A p value of less than 0.05 was considered statistically significant.
Data availability statement
All data are available in the main text. The datasets analyzed during this study are available in the clusterProfiler (v3.10.1) repository, https://www.liebertpub.com/doi/10.1089/omi.2011.0118 and the gene expression profiling and GSEA repository, https://www.pnas.org/content/102/43/15545.
Acknowledgments
We thank the assistance provided from the RCF6 and RCF3 Laboratories, Department of Medical Research, National Taiwan University Hospital. We thank Dr. Yu-Lun Kuo at Biotools, Co., Ltd., Taiwan, for assisting with the analysis of next-generation sequencing (NGS) data. English editing was provided by Elsevier Language Editing Services. Graphical abstract was created with BioRender.com. This work was supported by the Ministry of Science and Technology (MOST), Taiwan (grant number 109-2314-B-002-173-MY3) and by the National Taiwan University Hospital, Taiwan (grant numbers 108-M4137, 109-O05, 109-M4544, and 110-O04).
Author contributions
P.-M.C., Y.-W.C., K.-L.K., S.-H.L., and K.-H.H designed the study. P.-M.C., J.-R.D., and Y.-W.C. administered the experiments. J.-R.D. and Y.-W.C. investigated the data. The IHC data were evaluated by W.-C.L. P.-M.C. and J.-R.D. wrote the paper. The data and the manuscript have been discussed and approved by all authors.
Declaration of interests
The authors declare no conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.08.004.
Supplemental information
Target genes enrichment scores of the GSEA FEVR_CTNNB1_TARGETS_DN were listed in this heatmap
References
- 1.von der Maase H., Hansen S.W., Roberts J.T., Dogliotti L., Oliver T., Moore M.J., Bodrogi I., Albers P., Knuth A., Lippert C.M., et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J. Clin. Oncol. 2000;18:3068–3077. doi: 10.1200/JCO.2000.18.17.3068. [DOI] [PubMed] [Google Scholar]
- 2.von der Maase H., Sengelov L., Roberts J.T., Ricci S., Dogliotti L., Oliver T., Moore M.J., Zimmermann A., Arning M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005;23:4602–4608. doi: 10.1200/JCO.2005.07.757. [DOI] [PubMed] [Google Scholar]
- 3.De Santis M., Bellmunt J., Mead G., Kerst J.M., Leahy M., Maroto P., Gil T., Marreaud S., Daugaard G., Skoneczna I., et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 30986. J. Clin. Oncol. 2012;30:191–199. doi: 10.1200/JCO.2011.37.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balar A.V., Castellano D., O'Donnell P.H., Grivas P., Vuky J., Powles T., Plimack E.R., Hahn N.M., de Wit R., Pang L., et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18:1483–1492. doi: 10.1016/S1470-2045(17)30616-2. [DOI] [PubMed] [Google Scholar]
- 5.Balar A.V., Galsky M.D., Rosenberg J.E., Powles T., Petrylak D.P., Bellmunt J., Loriot Y., Necchi A., Hoffman-Censits J., Perez-Gracia J.L., et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389:67–76. doi: 10.1016/S0140-6736(16)32455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellmunt J., de Wit R., Vaughn D.J., Fradet Y., Lee J.L., Fong L., Vogelzang N.J., Climent M.A., Petrylak D.P., Choueiri T.K., et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 2017;376:1015–1026. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loriot Y., Necchi A., Park S.H., Garcia-Donas J., Huddart R., Burgess E., Fleming M., Rezazadeh A., Mellado B., Varlamov S., et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 2019;381:338–348. doi: 10.1056/NEJMoa1817323. [DOI] [PubMed] [Google Scholar]
- 8.Powles T., Rosenberg J.E., Sonpavde G.P., Loriot Y., Durán I., Lee J.L., Matsubara N., Vulsteke C., Castellano D., Wu C., et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N. Engl. J. Med. 2021;384:1125–1135. doi: 10.1056/NEJMoa2035807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 10.D'Arcy P., Brnjic S., Olofsson M.H., Fryknäs M., Lindsten K., De Cesare M., Perego P., Sadeghi B., Hassan M., Larsson R., Linder S. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011;17:1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 11.D'Arcy P., Linder S. Proteasome deubiquitinases as novel targets for cancer therapy. Int. J. Biochem. Cell Biol. 2012;44:1729–1738. doi: 10.1016/j.biocel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Feng X., Holmlund T., Zheng C., Fadeel B. Proapoptotic effects of the novel proteasome inhibitor b-AP15 on multiple myeloma cells and natural killer cells. Exp. Hematol. 2014;42:172–182. doi: 10.1016/j.exphem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Sarhan D., Wennerberg E., D'Arcy P., Gurajada D., Linder S., Lundqvist A. A novel inhibitor of proteasome deubiquitinating activity renders tumor cells sensitive to TRAIL-mediated apoptosis by natural killer cells and T cells. Cancer Immunol. Immunother. 2013;62:1359–1368. doi: 10.1007/s00262-013-1439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brnjic S., Mazurkiewicz M., Fryknäs M., Sun C., Zhang X., Larsson R., D'Arcy P., Linder S. Induction of tumor cell apoptosis by a proteasome deubiquitinase inhibitor is associated with oxidative stress. Antioxid. Redox Signal. 2014;21:2271–2285. doi: 10.1089/ars.2013.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarhan D., D'Arcy P., Lundqvist A. Regulation of TRAIL-receptor expression by the ubiquitin-proteasome system. Int. J. Mol. Sci. 2014;15:18557–18573. doi: 10.3390/ijms151018557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chitta K., Paulus A., Akhtar S., Blake M.K.K., Caulfield T.R., Novak A.J., Ansell S.M., Advani P., Ailawadhi S., Sher T., et al. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenstrom macroglobulinaemia tumour cells. Br. J. Haematol. 2015;169:377–390. doi: 10.1111/bjh.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding Y., Chen X., Wang B., Yu B., Ge J. Deubiquitinase inhibitor b-AP15 activates endoplasmic reticulum (ER) stress and inhibits Wnt/Notch1 signaling pathway leading to the reduction of cell survival in hepatocellular carcinoma cells. Eur. J. Pharmacol. 2018;825:10–18. doi: 10.1016/j.ejphar.2018.02.020. [DOI] [PubMed] [Google Scholar]
- 18.Powles T., Csőszi T., Özgüroğlu M., Matsubara N., Géczi L., Cheng S.Y.S., Fradet Y., Oudard S., Vulsteke C., Morales Barrera R., et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:931–945. doi: 10.1016/S1470-2045(21)00152-2. [DOI] [PubMed] [Google Scholar]
- 19.Vogel R.I., Coughlin K., Scotti A., Iizuka Y., Anchoori R., Roden R.B.S., Marastoni M., Bazzaro M. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget. 2015;6:4159–4170. doi: 10.18632/oncotarget.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X., Wu J., Chen Y., Ye D., Lei H., Xu H., Yang L., Wu Y., Gu W. Ubiquitin-specific protease 14 regulates cell proliferation and apoptosis in oral squamous cell carcinoma. Int. J. Biochem. Cell Biol. 2016;79:350–359. doi: 10.1016/j.biocel.2016.08.038. [DOI] [PubMed] [Google Scholar]
- 21.Cai J., Xia X., Liao Y., Liu N., Guo Z., Chen J., Yang L., Long H., Yang Q., Zhang X., et al. A novel deubiquitinase inhibitor b-AP15 triggers apoptosis in both androgen receptor-dependent and -independent prostate cancers. Oncotarget. 2017;8:63232–63246. doi: 10.18632/oncotarget.18774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song C., Ma R., Yang X., Pang S. The deubiquitinating enzyme USP14 regulates leukemic chemotherapy drugs-induced cell apoptosis by suppressing ubiquitination of aurora kinase B. Cell. Physiol. Biochem. 2017;42:965–973. doi: 10.1159/000478679. [DOI] [PubMed] [Google Scholar]
- 23.Kropp K.N., Maurer S., Rothfelder K., Schmied B.J., Clar K.L., Schmidt M., Strunz B., Kopp H.G., Steinle A., Grünebach F., et al. The novel deubiquitinase inhibitor b-AP15 induces direct and NK cell-mediated antitumor effects in human mantle cell lymphoma. Cancer Immunol. Immunother. 2018;67:935–947. doi: 10.1007/s00262-018-2151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sooman L., Gullbo J., Bergqvist M., Bergström S., Lennartsson J., Ekman S. Synergistic effects of combining proteasome inhibitors with chemotherapeutic drugs in lung cancer cells. BMC Res. Notes. 2017;10:544. doi: 10.1186/s13104-017-2842-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kreso A., Dick J.E. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Avril T., Vauléon E., Chevet E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis. 2017;6:e373. doi: 10.1038/oncsis.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermeulen L., Snippert H.J. Stem cell dynamics in homeostasis and cancer of the intestine. Nat. Rev. Cancer. 2014;14:468–480. doi: 10.1038/nrc3744. [DOI] [PubMed] [Google Scholar]
- 28.Dauer P., Sharma N.S., Gupta V.K., Durden B., Hadad R., Banerjee S., Dudeja V., Saluja A., Banerjee S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining "stemness". Cell Death Dis. 2019;10:132. doi: 10.1038/s41419-019-1408-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madden E., Logue S.E., Healy S.J., Manie S., Samali A. The role of the unfolded protein response in cancer progression: from oncogenesis to chemoresistance. Biol. Cell. 2019;111:1–17. doi: 10.1111/boc.201800050. [DOI] [PubMed] [Google Scholar]
- 30.Lei H., Shan H., Wu Y. Targeting deubiquitinating enzymes in cancer stem cells. Cancer Cell Int. 2017;17:101. doi: 10.1186/s12935-017-0472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haq S., Suresh B., Ramakrishna S. Deubiquitylating enzymes as cancer stem cell therapeutics. Biochim. Biophys. Acta Rev. Cancer. 2018;1869:1–10. doi: 10.1016/j.bbcan.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Kaushal K., Antao A.M., Kim K.S., Ramakrishna S. Deubiquitinating enzymes in cancer stem cells: functions and targeted inhibition for cancer therapy. Drug Discov. Today. 2018;23:1974–1982. doi: 10.1016/j.drudis.2018.05.035. [DOI] [PubMed] [Google Scholar]
- 33.Chua B.A., Van Der Werf I., Jamieson C., Signer R.A.J. Post-transcriptional regulation of homeostatic, stressed, and malignant stem cells. Cell Stem Cell. 2020;26:138–159. doi: 10.1016/j.stem.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow P.-M., Liu S.-H., Chang Y.-W., Kuo K.-L., Lin W.-C., Huang K.-H. The covalent CDK7 inhibitor THZ1 enhances temsirolimus-induced cytotoxicity via autophagy suppression in human renal cell carcinoma. Cancer Lett. 2020;471:27–37. doi: 10.1016/j.canlet.2019.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Kuo K.-L., Liu S.-H., Lin W.-C., Hsu F.-S., Chow P.-M., Chang Y.-W., Yang S.-P., Shi C.-S., Hsu C.-H., Liao S.-M., et al. Trifluoperazine, an antipsychotic drug, effectively reduces drug resistance in cisplatin-resistant urothelial carcinoma cells via suppressing Bcl-xL: an in vitro and in vivo study. Int. J. Mol. Sci. 2019;20:3218. doi: 10.3390/ijms20133218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chou T.-C., Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 37.Vogus D.R., Pusuluri A., Chen R., Mitragotri S. Schedule dependent synergy of gemcitabine and doxorubicin: improvement of in vitro efficacy and lack of in vitro-in vivo correlation. Bioeng. Transl. Med. 2018;3:49–57. doi: 10.1002/btm2.10082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuo K.-L., Ho I.L., Shi C.-S., Wu J.-T., Lin W.-C., Tsai Y.-C., Chang H.-C., Chou C.-T., Hsu C.-H., Hsieh J.-T., et al. MLN4924, a novel protein neddylation inhibitor, suppresses proliferation and migration of human urothelial carcinoma: in vitro and in vivo studies. Cancer Lett. 2015;363:127–136. doi: 10.1016/j.canlet.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 39.Chow P.-M., Chang Y.-W., Kuo K.-L., Lin W.-C., Liu S.-H., Huang K.-H. CDK7 inhibition by THZ1 suppresses cancer stemness in both chemonaïve and chemoresistant urothelial carcinoma via the hedgehog signaling pathway. Cancer Lett. 2021;507:70–79. doi: 10.1016/j.canlet.2021.03.012. [DOI] [PubMed] [Google Scholar]
- 40.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Target genes enrichment scores of the GSEA FEVR_CTNNB1_TARGETS_DN were listed in this heatmap
Data Availability Statement
All data are available in the main text. The datasets analyzed during this study are available in the clusterProfiler (v3.10.1) repository, https://www.liebertpub.com/doi/10.1089/omi.2011.0118 and the gene expression profiling and GSEA repository, https://www.pnas.org/content/102/43/15545.