

Summary
Secreted proteins mediate essential physiological processes. With conventional assays, it is challenging to map the spatial distribution of proteins secreted by single cells, to study cell-to-cell heterogeneity in secretion, or to detect proteins of low abundance or incipient secretion. Here, we introduce the “FluoroDOT assay,” which uses an ultrabright nanoparticle plasmonic-fluor that enables high-resolution imaging of protein secretion. We find that plasmonic-fluors are 16,000-fold brighter, with nearly 30-fold higher signal-to-noise compared with conventional fluorescence labels. We demonstrate high-resolution imaging of different secreted cytokines in the single-plexed and spectrally multiplexed FluoroDOT assay that revealed cellular heterogeneity in secretion of multiple proteins simultaneously. Using diverse biochemical stimuli, including Mycobacterium tuberculosis infection, and a variety of immune cells such as macrophages, dendritic cells (DCs), and DC-T cell co-culture, we demonstrate that the assay is versatile, facile, and widely adaptable for enhancing biological understanding of spatial and temporal dynamics of single-cell secretome.
Keywords: fluorescence imaging, plasmonic nanoparticles, single-cell secretion, tuberculosis, cytokine secretion, Interleukin-1 beta, Tumor Necrosis Factor-alpha
Graphical abstract
Highlights
-
•
FluoroDOT is an ultrasensitive assay for studying protein secretion dynamics of single cells
-
•
FluoroDOT provide a digital, quantifiable signal for measuring cell-to-cell heterogeneity
-
•
We demonstrate applicability on wide-ranging cell-types and stimuli
-
•
A multicolor plasmonic-fluor palette allows multiplexed detection of secreted proteins
Motivation
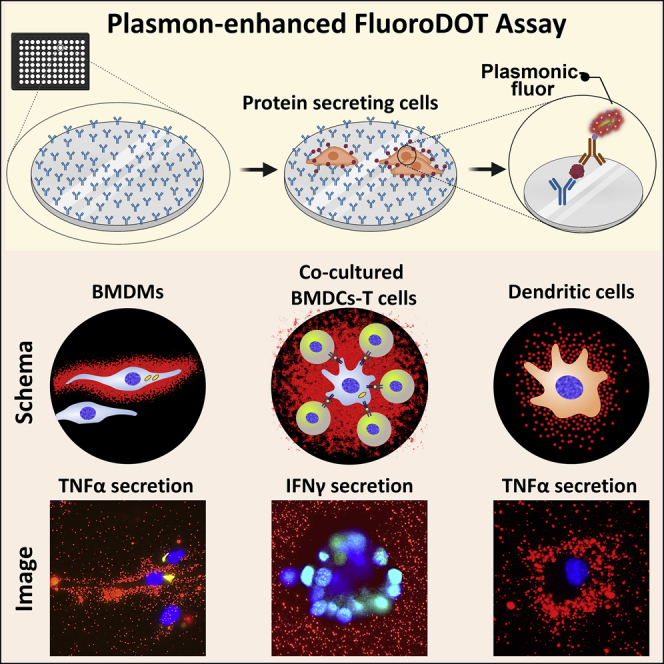
Understanding the spatial and temporal dynamics of protein secretion at a single-cell level is essential to understanding numerous biological systems relevant to health and disease. However, currently available methods for studying protein secretion at the single-cell level lack spatial resolution. High-resolution imaging of single-cell secretion typically requires expensive and specialized microscopy facilities that are not routinely available. We developed and validated a simple yet powerful method called the “FluoroDOT” assay to image the spatial distribution of secreted proteins around single cells, along with the capability to quantify cell-to-cell heterogeneity in secretion. The ultrabright nanolabels enable the quantification of the signals in a digital manner.
Understanding the spatial and temporal dynamics of protein secretion at a single-cell level is essential, but up until now remained challenging. To address this challenge, Seth et al. developed and validated a simple yet powerful method called "FluoroDOT assay" that enables the visualization of single-cell protein secretions with digital resolution.
Introduction
Proteins secreted by cells into the extracellular space constitute 13%–15% of the entire proteome and include growth factors, cytokines, chemokines, antibodies, extracellular matrix proteins, enzymes, hormones, and antimicrobial peptides (Uhlén et al., 2015, 2019). Secretory proteins facilitate essential physiological and pathological processes such as cell-to-cell communication, cell signaling, activation, inflammation, coagulation, hemostasis, differentiation, migration, toxicity, and defense (Uhlén et al., 2019). Understanding the cell secretome is essential in numerous life sciences disciplines, including immunology, oncology, neurobiology, microbiology, endocrinology, and stem cell biology. Researchers heavily rely on samples collected from cell culture supernatants and conventional immunoassays such as the enzyme-linked immunosorbent assay (ELISA) to assess the changes in proteins secreted by cells after various physicochemical or biological stimuli. However, for attaining a detectable signal in ELISA, one needs to collect supernatant from thousands of cells that have been incubated for an extended duration, typically ranging from 12 h to several days. Implicit averaging in these methods results in loss of information related to cell-to-cell heterogeneity, cell-cell interaction, and the spatial distribution of secreted proteins. This particularly confounds analysis when working with cells having subpopulations and multi-modal populations. Furthermore, due to the low sensitivity of these techniques, there is little information on the kinetics of protein secretion, particularly at early time points after stimulation and under low levels of stimulation. A recent technology feature highlights the imperative for detecting and measuring proteins secreted at single-cell resolution without having to rely on mRNA analysis and mass spectrometry (Marx, 2019). In more than 60% of cases, mRNA levels do not correlate with protein abundance (Maier et al., 2009; Vogel and Marcotte, 2012). On the other hand, mass spectrometry requires specialized strategies to isolate and handle single cells and expensive instrumentation and training, making it unsuitable for routine single-cell secretome analysis (Couvillion et al., 2019). Although ELISpot and FluoroSpot have emerged as powerful tools for studying protein secretions at the single-cell level, they result in low-resolution images. The assay readout is a colored or fluorescent “spot” on a white or dark background, either counted manually or with an ELISpot reader (Figure 1A). Each spot, which is often diffused, indicates a protein-secreting cell, and the size of the spot provides a qualitative and often vague estimate of the amount of protein secreted by the cell. In fact, several shortcomings, including large sample-to-sample and lab-to-lab variability in data acquisition and interpretation and poor accuracy in quantification of the secretion parameters, obligated extensive efforts to establish standardized and automated data acquisition and analysis procedures for evaluation of ELISpot assays (Janetzki et al., 2015). Further, the inherently low signal warrants the use of specialized membrane-coated plates to retain more protein and long incubation times for a discernable signal. The requirement for a dedicated reader further impedes its widespread applicability in laboratory settings.
Figure 1.

Comparison of conventional ELISpot/FluoroSpot assays with plasmon-enhanced FluoroDOT assay relying on plasmonic-fluors as ultrabright biolabels
(A and B) Schematic illustration depicting step-by-step method for (A) conventional ELISpot and FluoroSpot assays and (B) plasmon-enhanced FluoroDOT assay.
(C–E) Representative whole-well image (scale bar, 1000 μm) and zoomed-in image (scale bar, 100 μm) of PBMCs IFNγ assay. (C) ELISpot, (D) FluoroSPOT, and (E) FluoroDOT. The cells were incubated for 18 h for comparison of all three assays.
(F) JAWS II dendritic cells (DCs) were treated with 200 and 2,000 ng/mL lipopolysachharide (LPS), and bright-field images (left panel) and Cy5/DAPI merged images (middle panel) of a single cell secreting TNF-α, visualized using conventional fluor (Cy5), quantum dot 655, and Cy5-plasmonic-fluor.
(G) Representative line scans and signal-to-noise ratio (SNR) corresponding to conventional fluor (Cy5), quantum dot 655, and Cy5-plasmonic-fluor. To assess the signal of the secreted cytokine, the line scans for all fluors are placed just outside the boundary of the cell (using the corresponding bright-field image). High-power laser (Lumencor Aura III Light Engine) was used as light source. A standard Cy5 filter was used to image all three labels.
See also Figures S1 and S2.
Here, we introduce an ultrasensitive method for detecting and quantifying protein secretion at a single-cell level. The high sensitivity of the assay stems from an ultrabright plasmon-enhanced fluorescent nanolabel called plasmonic-fluor and is reported to sensitively detect biomarkers for biomedical research and clinical diagnostics (Kim et al., 2021; Liu et al., 2021; Luan et al., 2020; Wang et al., 2021). We and others have demonstrated that enhancement of the emission of fluorophores in proximity to plasmonic nanostructures such as gold and silver nanoparticles is largely attributed to (1) the enhanced electromagnetic field at the surface of the plasmonic nanoparticles and (2) the decrease in the fluorescence lifetime due to the coupling between the excited fluorophores and the surface plasmons of the nanoparticles (Abadeer et al., 2014; Bardhan et al., 2009; Gandra et al., 2014; Khatua and Orrit, 2014; Lu et al., 2020; Luan et al., 2018; Tam et al., 2007). The reported plasmon-enhanced method, which we call the “FluoroDOT assay,” is better than conventional approaches as (1) it provides a high-resolution digital signal in the form of dots (particles) per cluster (spot) as opposed to the analog signal in ELISpot and FluoroSPOT assays; (2) it provides spatial information (directional/polarized secretion), which is lost in the three-dimensional (3D) membrane of ELISpot and FluoroSPOT assays; (3) due to its high sensitivity, it can detect incipient protein secretions within 30 min to few hours compared with overnight or days of incubation for conventional assays; (4) the assay can be read using a standard epifluorescence microscope with a 20× objective as opposed to the requirement of a dedicated reader; and (5) the assay is performed on a glass-bottom plate, and the cells can be fixed, stained, and imaged. Retaining the cells and correlating their secretion provides significant advantage as it provides opportunity to reveal cellular and subcellular information (morphology of the cells, activation of fluorescent reporters, and/or correlation of functional significance of heterogeneity with protein secretion), which is difficult with conventional assays (Figure 1B; Table S1). For example, during innate immune responses, macrophages respond to a microbial challenge by inducing a variety of cytokines, including tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and interferon-α/β (IFN-α/β), among others. However, it has been challenging to assess whether secretion is driven by the majority of directly infected cells, a select subset of infected cells, or uninfected cells in the population. Most previous attempts to unravel single-cell protein secretion utilized microfluidics-based assays (micro-/nano-wells, micro-troughs, droplet platforms) or hyperspectral imaging (Armbrecht et al., 2020; Chen et al., 2019; Chiu et al., 2017; Juan-Colás et al., 2018; Murphy et al., 2018; Zhou et al., 2020). A major advantage of droplet-based microfluidic channels or small-volume microchamber systems are their ability to assess real-time single-cell secretion (Bounab et al., 2020; Li et al., 2018). Microfluidics-based single-cell secretion assays can achieve high-throughput measurements; however, they are prone to clogging by micron-size fragments and dust, affecting the reliability of the results (Jammes and Maerkl, 2020). Capillary-assisted microfluidics compartments can provide spatiotemporal dynamics of the single-cell secretion but fail to describe the secretion pattern around the cell (Hassanzadeh-Barforoushi et al., 2020). Some of these systems also incur high background due to the absence of washing steps (Choi, 2020). These methods have not been widely adopted as mainstream tools due to the compartmentalization of cells and sophisticated setup, which hinders or eliminates the interaction between cells for orchestrated protein secretion.
Using an ultrabright and specific fluorescent nanolabel developed in our lab, we can image low to high abundant proteins secreted by single cells (Luan et al., 2020). We extensively validated the assay by probing proteins secreted from macrophages, dendritic cells (DCs), and DC-T cell co-culture in response to diverse stimuli, including Toll-like receptor 4 (TLR4) stimulation, inflammasome activation, and Mycobacterium tuberculosis infection. Here we report high-resolution images of single-cell protein secretion, providing insight into cell-to-cell heterogeneity, directionality, and correlation with infectivity.
Results
Comparison of FluoroDOT assay with ELISpot/FluoroSPOT and with conventional fluorophores and quantum dots
First, we set out to compare the FluoroDOT assay with standard ELISpot and FluoroSPOT assays. We used healthy adult human peripheral blood mononuclear cells (PBMCs) and treated them with stimulants (phorbol 12-myristate 13-acetate [PMA] and ionomycin) to trigger secretion of IFN-γ. To compare the three assays, we used biotinylated detection antibody followed by streptavidin-fluor (FluoroSPOT), streptavidin-HRP (ELISpot), and streptavidin-plasmonic-fluor (FluoroDOT). We observed clear differences between unstimulated and stimulated wells in all three assays (Figures 1C–1E). Higher magnification images of cells in the FluoroDOT assay revealed a digitized “dot pattern,” corresponding to the proteins secreted by single cells. This was not apparent with the ELISpot or FluoroSPOT assay. The difference between the assays was not due to the distinct substrates the cells were grown on, as the amount of cytokine secreted on the glass substrate, used for the FluoroDOT assay, and the polyvinylidene fluoride (PVDF) membrane, used in the ELISpot/FluoroSPOT assay, was the same. The information deduced from the FluoroDOT assay is digitized (Figures S1A–S1C), which allowed us to quantify the signal for the FluoroDOT assay using the number of particles (dots) per cluster (spot) using a custom developed image-processing algorithm.
Next, we determined if the FluoroDOT assay is uniquely attuned for plasmonic-fluors or if it can also be achieved using conventional fluorophores or previously known bright nanolabels such as quantum dots. For this purpose, we used a DC line (JAWS II), which secretes the pro-inflammatory cytokine TNF-α after stimulation with the TLR4 agonist lipopolysaccharide (LPS) (Albrecht et al., 2008). We seeded the cells on a TNF-α capture-antibody-coated glass-bottom 96-well plate followed by LPS treatment at three different concentrations (20, 200, and 2,000 ng/mL). After 90 min of incubation, the cells were fixed and incubated with biotinylated TNF-α detection antibody. Different wells were then treated with streptavidin-Cy5, streptavidin-quantum dot 655 (Strep-Qdot 655), or Strep-Cy5-plasmonic-fluor, and the nuclei were stained with DAPI. Using an epifluorescence microscope, we could not discern a signal from the conventional fluorophores, and a very faint signal was observed with Strep-Qdot 655 (Figures S1D and S1E). In contrast, a robust signal was detected with the plasmonic-fluor, and, as expected, there was an increase in TNF-α secretion with increasing LPS concentration (Figure S1F). We further performed imaging using a 60× objective. We observed that the “secretion dot pattern” obtained using the plasmonic-fluor correlated well with the morphology of the cell observed in bright-field images. In contrast, no distinct secretion pattern was observed with the conventional fluor, and the signal-to-noise ratio for both the conventional fluor and Qdot was significantly lower compared with the plasmonic-fluors (Figure 1F). The signal-to-noise ratio (SNR) of the plasmonic-fluor was 680 ± 272, nearly 30-fold higher than the conventional fluor (23 ± 4) and 8-fold higher compared with Qdots (78 ± 50) (Figure 1G). For the conventional fluor, the small molecule diffused inside of the cells, leading to a small but finite level of non-specific signal seen in the absence of biotinylated antibody. We also assessed the performance of Strep-Qdot 655 using a Qdot-specific filter cube (Chroma qDOT655 filter cube). Again, we observed that the SNR of the Strep-Qdot 655 images were significantly lower than the Cy5-plasmonic-fluor (Figure S2). Due to compatibility issues of the Qdot-specific filter, we used a different excitation source for collecting the images shown in Figure S2 compared with those shown Figures 1F and S2. Thus, the absolute SNRs in Figures 1F and S2 are different, but in both cases, we observed an 8- to 15-fold higher SNR with the Cy5-plasmonic-fluor compared with the Strep-Qdot 655. In addition, as opposed to Qdots, individual Cy5-plasmonic fluors could be resolved using a 20× objective (0.75 numerical aperture [NA]). Finally, we observed that Qdots suffered from substantial blinking (fluorescence intermittency), as previously reported (Efros and Nesbitt, 2016; Yuan et al., 2018). To validate that the fluorescent dots observed are indeed single plasmonic-fluors, we correlated the fluorescence image and scanning electron microscopy (SEM) image obtained from the same location and found a one-to-one correspondence (Figure 2A). Transmission electron microscopy (TEM) images further revealed the size and shape of individual plasmonic-fluors (Figure 2B).
Figure 2.

Plasmonic-fluor brightness and single particle imaging
(A) Scanning electron microscopic (left) and epifluorescence microscopy (right) images from the same regions of substrate drop casted with plasmonic-fluors showing one-to-one correlation of individual plasmonic-fluors between the two images. Scale bar: 5 μm.
(B) Transmission electron microscopy images of streptavidin-Cy5-plasmonic-fluor and streptavidin-Cy3-plasmonic-fluor. Scale bar, 100 nm and scale bar, 200 nm.
(C) Fluorescence intensity of Cy5 conventional fluor and Cy5-plasmonic-fluor at their different molar concentrations. Data are represented as mean ± SD (n = 2 repeated tests). The data were fitted and graphically presented on a linear scale.
We further compared the fluorescence intensity of a defined molar concentration of plasmonic-fluors with that of the conventional fluorophore. The fluorescence intensity increased linearly with increasing concentrations of both the conventional fluorophore and the plasmonic-fluors (both Cy5 and Cy3) with at least three orders of magnitude difference in molar concentrations for similar fluorescence intensity. Notably, the slope of Cy5-plasmonic-fluor and Cy3-plasmonic-fluor were nearly 16,000- and 26,400-fold steeper than that of Cy5 and Cy3, respectively (Figures 2C and S4A). In our previous work, we have shown that a single plasmonic-fluor is comprised of ∼200 molecular fluorophores (Luan et al., 2020). The fold increase in brightness (16,000) is significantly higher than the number of fluorophores on a single plasmonic-fluor. This large enhancement in the brightness of the plasmonic-fluors compared with their conventional counterparts is critical for the high-resolution images in the FluoroDOT assay.
To confirm the analyte dose-dependent increase in the fluorescent dots, we performed a standard fluorescence-based sandwich immunoassay and correlated the particle count with that of concentration of the analyte (TNF-α, IL-6, and IFN-γ). We counted the particles using a custom-built algorithm. We observed excellent particle count dose dependence with a correlation coefficient of >0.99 for all three analytes (limits of detection [LODs] were TNF-α: 670 fg/mL, IL-6: 8 fg/mL, and IFN-γ: 3.17 pg/mL) (Figures S4B and S4C). To conclude, using JAWS II DCs, we established the assay’s sensitivity by demonstrating its ability to image cytokine secretion.
Interestingly, with certain cell types, we did not observe secreted TNF-α in the region overlapping the cell body. For tightly adherent cells such as macrophages and DCs, no signal was observed beneath the cell body, whereas for loosely adherent or suspension cells (PBMCs), we did detect a signal beneath the cell body. This suggested to us that either the adherent cells do not secrete in the region where they are adhered to the surface or that the capture antibody is not stable in that area. To distinguish these possibilities, we used JAWS II DCs and stimulated them with LPS (200 ng/mL), followed by washing with PBS after 1 h. We performed the experiment with and without fixing the cells (after the cells had secreted the cytokine and before adding the detection antibody). When the cells were not fixed, the cells were washed off during the subsequent processing and were not visible in the bright-field image. Irrespective of whether the cells were fixed or not fixed, the TNF-α secretion resembled a doughnut, with the empty space corresponding to where the cell body is present in the fixed samples, as shown in Figures S3A–S3C. We defined the boundary of the cell based on bright-field microscopy images. To determine whether the capture antibody beneath the cell is stable after the adhesion of cells, we used the unfixed samples, and we washed away the cells prior to adding 100 pg/mL of recombinant TNF-α to the well. We observed that the capture antibody in the empty space where the cell body had been is still able to bind recombinant TNF-α (Figure S3D). In contrast, if the cell was fixed onto the plate prior to the addition of TNF-α, the spiked TNF-α was not able to access the capture antibody beneath the cells, further confirming that the adhesion of the cell to the substrate prevents the access of secreted protein to capture antibody beneath the cell body (Figure S3E). Since the membrane staining was not used to define the boundaries of the cell, it is possible that the signal shown as secreted protein is on the cellular membrane at the extreme periphery of the cell. A membrane counter stain is required to study it in greater detail.
FluoroDOT assay detects protein secretions at an ultralow dose of stimulant and at very early time points
One of the fundamental questions in many cell biology studies is the degree of heterogeneity in cellular responses. When cells are subject to a stimulant, do all cells respond similarly, does a small subpopulation of cells exuberantly respond, or is it a combination of both, and to what extent does the heterogeneity depend upon the dose of stimulant? We used the plasmon-enhanced FluoroDOT assay to examine the dose response to LPS treatment, with a detailed assessment of the kinetics and cell-to-cell heterogeneity of the response. Using JAWS II DCs and LPS as a stimulant, the FluoroDOT assay revealed no significant secretion of TNF-α for an incubation duration of 90 min at 2 ng/mL of LPS. We observed substantial secretion at 20 ng/mL of LPS, which continued to increase with increasing LPS concentration (Figures 3A and S5A). Similarly, using an LPS concentration of 200 ng/mL, we found that TNF-α secretion was detectable after 40 min and increased at longer incubation time (60 and 90 min) (Figures 3C and S5B). Based on the images, we observed that both the number of cells secreting TNF-α and the amount of TNF-α secretion per cell increased with increasing dose and duration of LPS treatment (Figure 3B). This information cannot be derived from ELISA, which relies on the analysis of the cell culture medium, as the amount of secretion with low dose and short duration of stimulation was below its detection limit (Figure S6). We developed an algorithm to identify clusters of dots and count both the number of clusters in an image and the number of dots within each cluster. Using this algorithm, we quantified TNF-α-secreting cells, as indicated by the number of clusters (Figures 3D and 3F), and the amount of TNF-α secretion from each cell based upon the number of dots (i.e. particles) in each cluster (Figures 3E and 3G). The quantitative data corroborated our observations, as the difference between treated and untreated cells was only statistically significant when the LPS dose was higher than 20 ng/mL for 90 min incubation or the duration of treatment was more than 40 min with 200 ng/mL LPS. Interestingly, we observed that heterogeneity in cell secretion reflected not only the amount of secretion but also the directionality. The presence of capture antibody in the immediate vicinity of the secreting cell enables continuous capture of the secreted cytokine, thereby limiting diffusion of the analyte and retaining spatial information on the source of secretion. Interestingly, we found that at 20 ng/mL dose of LPS, approximately 55% of the cells exhibited isotropic secretion, whereas ∼45% of the cells secreted TNF-α preferentially on one side after 90 min (Figures S6C and S6D). There were no significant differences in the fraction of cells exhibiting isotropic secretion at 200 or 2,000 ng/mL LPS, suggesting that the phenomenon is not related to limiting ligand. The percentage of cells with anisotropic secretion was also relatively stable from 20 to 90 min. We also observed “doublet clusters” from cells that were either seeded close to each other or were undergoing cell division (Figure S7E). These intricate details of the secretion pattern revealed by the FluoroDOT assay are not achievable by conventional ELISpot and FluoroSPOT assays.
Figure 3.

Studying the effect of stimulant dose and stimulation duration on single-cell protein secretions
(A and C) FluoroDOT assay performed on JAWS II dendritic cells (DCs) treated with (A) 0, 2, 20, 200, and 2,000 ng/mL LPS for 90 min and (C) 200 ng/mL of LPS treated for 20, 40, 60, and 90 min. Left panel: Cy5 epifluorescence microscopy images of the assay using Cy5-plasmonic-fluor. Scale bar: 100 μm. Right panel: Cy5/DAPI merged image of TNF-α-secreting cell visualized using Cy5-plasmonic-fluor at 60×. Scale bar: 50 μm.
(B) Illustration depicting the LPS treatment of JAWS II DCs for studying the secretion levels of TNF-α in a dose- and time-dependent manner.
(D–G) Quantification of the number of TNF-α-secreting cells and the amount of TNF-α secreted by individual cells with increasing dose of LPS (D and E) and increasing duration of LPS (F and G) treatment. Ten 20× images for each treatment condition were collected. 700–750 cells were analyzed. Each data point is the number of clusters in the field of a 20× image. The fields were randomly chosen. There were at least 10 cells in each field of view. ns, not significant; ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by one-way ANOVA and Tukey’s post test.
See also Figure S3.
FluoroDOT assay detects protein secreted through non-conventional pathway after treatment with two stimulants
Next, we set out to investigate if the FluoroDOT assay can reveal proteins secreted through a non-conventional pathway in primary cells. IL-1β is secreted in response to activation of the NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome (Nickel and Rabouille, 2009). IL-1β lacks a classical secretion signal and is secreted by a non-conventional transport pathway that involves caspase-dependent cleavage and Gasdermin-D (Evavold et al., 2018). IL-1β secretion depends upon a priming signal and inflammasome activation (Gritsenko et al., 2020). We treated alveolar macrophages with LPS, followed by nigericin, which leads to cleavage of pro-IL-1β and secretion of IL-1β in the extracellular space (Figure 4A) (Joshi et al., 2022). Using the FluoroDOT assay, we detected IL-1β secretion 30 min after NLRP3 activation by nigericin. In contrast, TNF-α secretion was detected in response to 4 h of LPS treatment (Figures 4B, S7A, and S7B). While most cells secreted TNF-α in response to LPS, less than half of the cells responded with IL-1β secretion. As we saw in the JAWS II cells, the alveolar macrophages secreted TNF-α in both an isotropic and anisotropic manner. In contrast, the majority of cells (82%) showed isotropic secretion of IL-1β. We confirmed the correlation between particle count and concentration of IL-1β by plotting a standard curve and found the LOD to be 3 fg/mL (Figures S7C and S7D). Using the algorithm, we determined the number of IL-1β-secreting cells along with the amount of IL-1β secreted by each cell as indicated by the number of clusters and the number of particles/clusters, respectively (Figures 4C and 4D). The larger amount of TNF-α compared with IL-1β secreted was confirmed by measuring the cytokine concentration in cell culture supernatant collected from 150,000 cells using ELISA (Figure S7E). These results suggest that the plasmon-enhanced FluoroDOT assay enables the quantification of the distinct and specific secretion of corresponding cytokines without cross-interference after treatment with two different stimulants.
Figure 4.

Detection of two secreted proteins in primary cells after multiple stimulants
(A) Illustration depicting the experiment performed using alveolar macrophages to study the effect of two stimulants (LPS and nigericin) on the secretion of TNF-α and IL-1β. Plasmon-enhanced FluoroDOT assay performed on mouse-derived alveolar macrophages treated with 500 ng/mL LPS with and without nigericin for inflammasome activation.
(B) TNF-α secretion and IL-1β secretion observed after 4 h of LPS treatment and 30 min of 20 μM nigericin treatment. Cy5 epifluorescence microscopy images of the assay using Cy5-plasmonic-fluor (red) overlaid with DAPI nuclei stain (blue). Each panel contains two representative 20× images stitched together. Scale bar: 100 μm.
(C and D) Quantification data showing (C) the number of cells secreting IL-1β and (D) the amount of IL-1β secreted by individual cells after inflammasome activation. Number of clusters and number of particles/cluster were calculated using twelve 20× images for each treatment condition. ∗∗∗∗p < 0.0001 by one-way ANOVA and Tukey’s post test.
See also Figure S6.
Multiplexed FluoroDOT assay for simultaneous detection of multiple secreted proteins from a single cell
As a proof of concept, we investigated the feasibility of a spectrally multiplexed analysis of two proteins at a single-cell level. For this purpose, we conjugated the detection antibody for TNF-α and IL-6 to Cy5- and Cy3-plasmonic-fluors, respectively. We first confirmed the specificity of individual plasmonic-flours to their respective analyte by spotting the capture antibody for analytes (i.e., IL-6 and TNF-α) within the same well of a microtiter plate (Figure S8A). As anticipated, both plasmonic-flours specifically recognized their respective analytes in single- and double-capture antibody-coated regions of the plate (Figure S8B). Further, we performed a multiplexed plasmon-enhanced fluorescent immunosorbent assay and observed a standard curve with a good correlation (R2 = 0.9942, LOD = 15.1 pg/mL for TNF-α and R2 = 0.9958, LOD = 2.5 pg/mL for IL-6) between the particle count and concentration of the protein analyte (Figures S8C and S8D). The multiplexed assay requires coating of both capture antibodies in a defined area, while the same area is available for coating a single-capture antibody for a singleplexed assay. Due to differences in the amount of available capture antibody, the sensitivity of the multiplexed assay is reduced compared with a singleplex assay. Using LPS-stimulated JAWS II DCs, we assessed the concurrent secretion of TNF-α and IL-6 at a single-cell level. We observed an increase in the overall secretion of both cytokines with the increasing duration of LPS incubation from 30 min to 3 h (Figure 5A). We noted significant heterogeneity in the secretion of both cytokines in terms of the amount of secretion (given by the number of particles) and temporal dynamics (Figure 5B). Interestingly, we observed significant cell-to-cell variation, even after 30 min of LPS treatment, such that some cells started secreting TNF-α, while no secretion for IL-6 was observed for the same cells (Figure 5C). At longer durations (2 and 3 h), we observed more predominant IL-6 secretion compared with TNF-α. Our observations align with previous findings in which mRNA quantification and the ELISA-based assay showed that TNF-α and IL-6 were produced with distinct kinetics in LPS-treated, activated DCs (Langenkamp et al., 2000).
Figure 5.

Multiplexed analysis of secretion of two proteins in the same cell using antibody-conjugated plasmonic-fluors
(A) Multiplexed analysis of secretion of TNF-α and IL-6 from a single JAWS II dendritic cell (DC) when subjected to 200 ng/mL LPS after 30 min, 1 h, 2 h, and 3 h of incubation. Epifluorescence microscopy images of the cells using TNF-α-detection antibody-conjugated Cy5-plasmonic-fluor (red) and IL-6-detection antibody-conjugated Cy3-plasmonic-fluor (yellow) overlaid with DAPI nuclei stain (blue). Each panel contains four representative 20× images stitched together. Scale bar: 100 μm.
(B) Quantification of both TNF-α and IL-6 from individual JAWS II DCs after LPS treatment for 30 min, 1 h, 2 h, and 3 h. Number of clusters and number of particles/cluster were calculated using seven 20× images for each treatment condition.
(C) Representative higher magnification images (60×) of JAWS II DCs showing the multiplexed detection of the TN-Fα and IL-6 secretion and the associated cell-to-cell heterogeneity indicated by white arrows. Scale bar: 50 μm.
See also Figure S8.
Thus, using the multiplexed FluoroDOT assay, we were able to ascertain population dynamics of two proteins secreted from single cells simultaneously. While as a proof of concept we have demonstrated cytokine secretion for two proteins, it is worth noting that the plasmonic-fluor can be generated with any molecular fluorophore, thus enabling higher multiplexing capability (up to 6 colors with minimal spectral overlap) (Chang and Murphy, 2018; Luan et al., 2020). The localized surface plasmon resonance (LSPR) wavelength of the plasmonic nanostructures is highly sensitive to its composition, size, and shape (Figure S4B) and can therefore be tuned over a wide range covering visible to near-infrared wavelengths.
FluoroDOT assay for assessment of cell-to-cell heterogeneity in infection-induced cytokine secretion
While the previous assays revealed heterogeneity when cells were exposed to a homogeneous stimulus, during actual infections, only a subpopulation of cells will be infected, and the resultant heterogeneity in secretion is likely to be more complex. One of the major advantages of the plasmon-enhanced FluoroDOT assay is its ability to retain the cells on the plate by fixing them, and later, imaging them. Taking advantage of this feature, we next set out to study the impact of infection by M. tuberculosis (Mtb) on TNF-α secretion by bone-marrow-derived macrophages (BMDMs) (Figure 6A). We infected BMDMs with DsRed-expressing Mtb and observed increased secretion of TNF-α by Mtb-infected cells compared with uninfected cells (Figure 6B). We could readily detect TNF-α secretion by the FluoroDOT assay at 6 h after infection, when TNF-α was undetectable by conventional ELISA, underscoring the sensitivity of the assay. Remarkably, the TNF-α-secretion pattern faithfully followed the elongated morphology of the BMDMs, and there was significant heterogeneity in terms of the direction in which secretion occurred.
Figure 6.

Cell-to-cell heterogeneity in cytokine secretion after Mtb infection
(A) Illustration depicting the heterogeneity in TNF-α secretion in terms of infection amount and direction of secretion by bone-marrow-derived macrophages (BMDMs) infected with M. tuberculosis (Mtb).
(B) Plasmon-enhanced FluoroDOT assay for TNF-α secretion from BMDMs in the absence and presence of DsRed-expressing Mtb. Epifluorescence microscopy images of the assay using Cy5-plasmonic-fluor (red) overlaid with Mtb (yellow) and DAPI nuclei stain (blue). Each panel contains three representative 20× images stitched together. Scale bar: 100 μm.
(C) Bright-field and Cy5/TRITC/DAPI merged images at 60× with a zoom-in image on either side. Presence of Mtb is highlighted by white arrows. Scale bar: 50 μm.
We observed that some of the infected cells secreted TNF-α, while there were other infected cells that did not secrete the cytokine (Figure 6C). Thus, we verified the utility of the FluoroDOT assay for studying single-cell protein secretion within the context of infection and unraveling the underlying heterogeneity. Detailed investigation of protein secretions in single cells will help build enhanced models for understanding host-pathogen interactions in immune cells, emphasizing the value of extending the analysis beyond bulk measurements (Chattopadhyay et al., 2014; Toniolo et al., 2020).
FluoroDOT assay elucidates protein secretion in co-cultures
Lastly, we examined the FluoroDOT assay’s applicability in a multi-cellular system by co-culturing BMDCs infected with Mtb along with antigen-specific CD4+ T cells that expressed green fluorescent protein (GFP) under control of the Nur77 promoter (Ashouri and Weiss, 2017). We used CD4+ T helper type 1 (Th1) effector cells from transgenic mice (called P25 cells) that express a T cell receptor (TCR) specific for peptide 25 (amino acids 240–254) of Mtb Ag85B (Tamura et al., 2004). In response to co-culture with Mtb-infected BMDCs, the P25 cells secrete IFN-γ and induce GFP expression (Figure 7A). As a negative control, we infected major histocompatibility complex (MHC)-mismatched (MM) BMDCs (H-2k rather than H-2b). Before performing the experiment with Mtb, we first set out to estimate the minimum duration of co-culture required for detectable IFN-γ secretion using P25 peptide. We found that after 2–3 h of BMDC-T cell co-culture, clusters of IFN-γ secretions were evident (Figure S9). Next, we performed the FluoroDOT assay on Mtb-infected BMDCs co-cultured with T cells for 3 h, and we observed distinct clusters of IFN-γ secretion in wells with wild-type (WT) BMDCs, while almost no secretion was seen with the MM control BMDCs (Figures 7B, 7C; S10A, and S10B). Additionally, we did not observe IFN-γ secretion with BMDCs only, T cells only, or BMDCs and T cells without infection or peptide.
Figure 7.

Detection of IFN-γ secretion in a multi-cellular co-culture system
(A) Illustration depicting the experiment performed on wild-type (WT) and class II mismatched (MM) bone marrow-derived dendritic cells (BMDCs) infected with Mycobacterium tuberculosis (Mtb) and co-cultured with T cells (BMDCs: CD4+ T cells = 1:10). Antigen presentation leads to the activation of green fluorescent protein (GFP)-expressing CD4+ T cells and secretion of IFN-γ.
(B and C) IFN-γ secretion by Mtb MHCI-peptide-specific T cells when co-cultured with (B) wild-type (WT) BMDCs infected with Mtb and (C) mismatched (MM) BMDCs infected with Mtb. Epifluorescence microscopy images of the assay using Cy5-plasmonic-fluor as the label. Scale bar: 100 μm.
(D) IFN-γ secretion by Mtb MHCI-peptide (P25)-specific T cells when co-cultured with BMDCs for 1, 2, and 3 h. Cy5 (red) epifluorescence microscopy images of the assay using Cy5-plasmonic-fluor. Representative 60× image co-cultures of BMDCs and T cells (DAPI staining nuclei) along with GFP expression (green) triggered in activated CD4+ T cells. White arrows highlight dendritic cells (DCs) (bigger, blue nucleus stained) and T cells (smaller, green cells). Scale bar: 50 μm.
(E and F) Quantification of secreted IFN-γ represented as (E) number of particles in each cluster and (F) concentration in pg/mL of IFN-γ protein. Number of particles/clusters were calculated from 35 clusters using twenty 20× images for each treatment condition. ND, not detectable. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by one-way ANOVA and Tukey’s post test.
See also Figures S10 and S11.
We observed clusters of BMDCs and GFP-expressing CD4+ T cells with IFN-γ secretion surrounding them after 3 h of incubation, further confirming the specificity of the assay (Figure 7D). We detected IFN-γ secretion in the FluoroDOT assay after 3 h of co-culture, when it was undetectable in cell culture supernatants by ELISA. Finally, we compared the number of particles in each cluster for WT and MM and found that the number of particles/clusters in WT BMDCs was significantly higher than that of MM BMDCs (Figure 7E). We demonstrated the ability to correlate functional heterogeneity of cells in an antigen presentation assay for studying IFN-γ release. Using GFP-expressing cells, we further validated our findings that IFN-γ secretion was indeed a result of CD4+ T cell activation and that studying co-regulation of genes is achievable using this assay.
Discussion
ELISpot is a widely employed bioanalytical method that is used in both research and clinical settings for a wide range of applications such as screening antigen-specific immune cells and functional T cells in patients with cancer. It is used to assess response to immunotherapy, diagnose tuberculosis (T-SPOT.TB assay), map T cell responses against HIV, detect antibody-secreting cells in blood after vaccination, and for functional analysis of circulating tumor cells (CTCs) to understand drug susceptibility (Adetifa et al., 2013; Armbrecht et al., 2020; Malyguine et al., 2012; Pai et al., 2014; Saletti et al., 2013; Streeck et al., 2009; Zhou et al., 2020). Despite its enormous utility, this method has not undergone significant technological advances in spatial resolution or sensitivity over the past 3 decades. ELISpot assays thus remain severely limited by the inability to spatially resolve protein secretion, discrepancies in assay readout, and false negatives in clinical diagnosis (Janetzki et al., 2015; Pan et al., 2015). We introduce a simple yet powerful ultraresolved digital version of ELISpot and FluoroSpot assays called the FluoroDOT assay. The unmet need for an exceptionally bright fluorescent tag capable of detecting single-molecule fluorescence is served by plasmonic-fluors, which outperform conventional fluorophores and Qdots. Using plasmonic-fluors, we improved the LOD of the FluoroDOT assay by more than two orders of magnitude compared with conventional biolabels (Luan et al., 2020). We are able to digitalize the signal and count the fluorescent dots in contrast to integrating the signal as in the case of ELISpot and FluoroSpot, which can be considered analog versions of this assay. This method will provide deeper insights into how the mechanotransduction, polarization, and adhesion properties of cells correlate with single-cell protein secretion and application of this method for studying inflammasome-mediated protein secretion in diverse cell types including immune cells and endothelial cells (Bai et al., 2020; Joshi and Morley, 2019).
Since heterogeneity is a fundamental characteristic of cellular systems, single-cell technologies and measurements are of immense value for an in-depth understanding of individual cells, facilitating disease model development, drug discovery, and meaningful biological insights beyond ensemble population averages (Altschuler and Wu, 2010; Heath et al., 2016). We noted cell-to-cell heterogeneity in response to LPS treatment (Figures 3 and S5), inflammasome activation (Figure 4), and cytokine response to Mtb infection (Figure 6).
We found a difference in the secretion pattern of TNF-α and IL-1β. While many cells secreted TNF-α in an anisotropic manner, this was not the case with IL-1β secretion. These proteins are secreted in very different manners: TNF-α secretion depends upon cleavage of the membrane-bound precursor by the metalloprotease TNF-α-converting enzyme (TACE), whereas IL-1β secretion depends upon Gasdermin-D pores. The anisotropic nature of TNF-α secretion suggests that some required component is polarized in the cell, perhaps reflecting underlying polarity associated with DC migration (Mastrogiovanni et al., 2021). While the underlying reason behind the anisotropic secretion is a subject of future study, the ability of the FluoroDOT assay to faithfully capture these events further highlights its utility in biomedical research.
The FluoroDOT assay enables the visualization of heterogeneity, infectivity, and directionality, and it can be implemented for better understanding of host-pathogen interactions, neuronal secretions, and tumor cytotoxicity. As proof of principle, we demonstrate that it can be applied to study cytokine secretion during Mtb infection. There is an increasing appreciation that tuberculosis (TB) infection is characterized by bacterial and host heterogeneity (Avraham et al., 2015; Lin et al., 2014; Logsdon and Aldridge, 2018; Toniolo et al., 2020). However, the ability to assess protein secretion at a single-cell level has lagged behind single-cell technologies to assess mRNA or intracellular protein abundance of both the bacilli and host. The high resolution achieved here can be applied to determine the relationship between bacterial and host heterogeneity, mapping the spatial distribution of protein secretion in a multiplexed and multi-cellular situation to decode critical information on cellular behavior.
Overall, we developed an ultrasensitive digitized FluoroDOT immunoassay for the detection of protein secretions from cells. The assay can be employed for multiplexed detection of proteins. It can provide detailed spatial mapping of the secretion around the cells along with the subcellular information. When compared with any of the conventionally used approaches, we believe that the FluoroDOT assay will lead to transformative advances in single-cell secretion studies as it is versatile, low cost, and readily adaptable in any laboratory setting with regularly used supplies and reagents (glass-bottom 96-well plate, ELISA reagents), commercially available plasmonic-fluor (Auragent Bioscience), and a standard epifluorescence microscope. Considering the evolving landscape of cellular traits, this method holds the potential to provide a comprehensive understanding of protein secretion at a single cell level.
Limitations of the study
We recognize that fixing and retaining all non-adherent and loosely adherent cells (such as T cells and inflammasome-activated alveolar macrophages) on the plate is challenging in the FluoroDOT assay, as some of these cells are dislodged during the washing steps. We observed that this was not an issue with adherent cells such as DCs and BMDMs. This limitation can be addressed by employing more robust fixation conditions with gentler washing steps to ensure that we can retain all non-adherent cells seeded on the plate. Another shortcoming is that the algorithm used to quantify protein secretions is limited to analysis of isotropic secretions and is not suitable for quantitatively analyzing patterns of secretion that are unidirectional and an elongated shape. As a part of further investigation, we plan to improve the algorithm for identification of non-circular and directional protein-secretion patterns from elongated cells such as macrophages and neurons. Another limitation of this methodology is inherent to the difficulty in performing high-throughput experiments using conventional fluorescence microscopes. As with other microscopy-based assays, high throughput can be achieved but requires automated image acquisition and data analysis. Finally, further validation of the assay with a larger number of human PBMC samples is required before the assay can be translated to the real-world applications.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Mycobacterium tuberculosis | Dr. Philips lab | H37Rv |
| Biological samples | ||
| Human PBMC | Department of Anesthesiology, Washington University School of Medicine (WUSM) | N/A |
| Mouse alvealar macrophage | Division of Infectious Diseases, WUSM | N/A |
| Mouse bone marrow derived macrophages | Division of Infectious Diseases, WUSM | N/A |
| Mouse bone marrow derived dendritic cells | Division of Infectious Diseases, WUSM | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Streptavidin-conjugated Cy3-plasmonic-fluor | Auragent Bioscience | PF550 ultrabright fluor |
| Streptavidin-conjugated Cy5-plasmonic-fluor | Auragent Bioscience | PF650 ultrabright fluor |
| Streptavidin-Cy5 | Thermo Fisher Scientific | SA1011 |
| Streptavidin QD655™ | Thermo Fisher Scientific | Q10123MP |
| P25 Peptide | This paper | N/A |
| PBS | Thermo Fisher Scientific | 10010023 |
| RPMI 1640 | Thermo Fisher Scientific | 11875093 |
| DMEM | Thermo Fisher Scientific | 11966025 |
| Tween-20 | Millipore Sigma | P9416 |
| Lipopolysaccharides from Escherichia coli O111:B4 | Millipore Sigma | L4391 |
| Middlebrook 7H9 broth | Millipore Sigma | M0178-500G |
| Bovine Serum Albumin | Millipore Sigma | A9418 |
| GMCSF | Millipore Sigma | G0282-50UG |
| DAPI | Millipore Sigma | MBD0015 |
| Critical commercial assays | ||
| Mouse TNFα DuoSet ELISA kit | R&D systems | DY410 |
| Mouse IL-6 DuoSet ELISA kit | R&D systems | DY406 |
| Mouse IFNγ DuoSet ELISA kit | R&D systems | DY485-05 |
| Mouse IL-1β ELISA kit | Invitrogen | 88-7013-88 |
| Human IFNγ ELISpot development module | R&D systems | SEL285 |
| Experimental models: Cell lines | ||
| JAWS II dendritic cells | ATCC | ATCC® CRL-11904™ |
| Experimental models: Organisms/strains | ||
| Mice | Jackson Labs | C57BL/6 (H-2b) |
| Mice | Jackson Labs | C57BL/6 (MHCII mismatched) |
| Software and algorithms | ||
| Prism 6.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Image J 1.53a (64-bit) | NIH (open source) | https://imagej.nih.gov/ij/download.html |
| Source code | This paper |
https://github.com/singamaneni/Counting-single-plasmonic-nanoparticles. https://zenodo.org/badge/latestdoi/312896998 |
| Imunospot Professional 7.0 | CTL Immunospot | https://immunospot.com/products/analyzers/immunospot-s6-ultimate-m2.html |
| Image Studio Lite | Licor | https://www.licor.com/bio/image-studio-lite/ |
| Other | ||
| Nikon Eclipse epifluorescence microscope | Nikon | Ts2R-FL |
| Nikon Spinning Disk Confocal | Nikon | https://wucci.wustl.edu/facilities/light-2-2/ |
| Glass bottom plates | Cell-vis | P96-1.5 H-N |
| Cellular Technology series 6 ImmunoSpot Universal Analyzer | CTL Immunospot | S6 Univeral M2 |
| UV spectrophotometer | Shimadzu | UV-1800 |
| Field-emission scanning electron microscope | FEI | Nova 2300 |
| Biomolecular Imager | Azure Biosystems | Sapphire RGBNIR |
| Transmission electron microscope | JEOL | JEM-2100F |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Dr. Srikanth Singamaneni (singamaneni@wustl.edu).
Materials availability
All materials and constructs used in this study are maintained by Dr. Singamaneni’s laboratory and are available upon request.
Experimental model and subject details
Cell lines
Mouse JAWS II dendritic cells (immature, monocytes ATCC® CRL-11904™) were cultured in alpha minimum essential medium with ribonucleosides, deoxyribonucleosides, 4 mM L-glutamine, 1 mM sodium pyruvate and 5 ng/ml murine GM-CSF, 10% heat-inactivated fetal bovine serum, 50 IU/ml of penicillin, 50 μg/ml of streptomycin.
Primary cells
-
i)
Blood sampling and isolation of peripheral blood mononuclear cells (PBMCs) processing: Heathy individuals consented for blood samples. All samples were collected in accordance with the procedures approved by the Institutional Review Board at Washington University in St. Louis. Donor’s demographic data was deidentified. Samples were obtained in sodium heparin tubes. Fresh whole blood samples were processed within 90 min of collection as previously described.(Thampy et al., 2018) Briefly, blood was diluted in an equal volume of PBS and layered carefully on Ficoll Paque PLUS (GE Healthcare). The PBMC fraction was isolated following centrifugation at 500 × g for 30 minutes at room temperature. The number of total PBMCs was determined with a Vi-CELL Viability Analyzer (Beckman Coulter, Brea, CA).
-
ii)
Isolation of alveolar macrophages (AM): To obtain AMs, bronchoalveolar lavage (BAL) was performed in anesthetized mice lungs using 26G catheters via intra-tracheal route. DPBS containing 0.6 mM EDTA and 1% FBS was used as BAL buffer. Three washes were performed with 1 ml of BAL buffer. DMEM (Dulbecco’s Modified Eagle Medium) with 10% FBS, Glutamax (1:100) and freshly prepared HEPES (4-1-piperazineethanesulfonic acid) buffer (1:100) was used to culture the cells.
-
iii)
Isolation and culture of BMDMs and BMDCs: For BMDCs, the bone marrow cells were collected and cultured for seven days in 100 mm Petri dishes containing 10 ml of RPMI medium supplemented with 10% heat-inactivated fetal bovine serum, 50 IU/ml of penicillin, 50 μg/ml of streptomycin and 20 ng/ml of mouse recombinant granulocyte-macrophage colony-stimulating factor (R&D Systems). For differentiation into BMDMs, DMEM medium was supplemented with 10% FBS and 20% L929-conditioned medium for 7 days. The concentration of L929-conditioned medium was reduced to 10% before infections. The GMCSF was removed before infections. Murine hematopoietic stem cells were isolated from the tibia and femurs of 6- to 12-week-old C57BL/6 or MHC-II mismatch mice, obtained from the Jackson laboratory and processed as described above.
-
iv)
Bacterial Strains and Growth Conditions: Mtb strain H37Rv expressing DsRed (selected with 25ug/ml kanamycin) was grown at 37°C in 7H9 medium (Middlebrook 7H9 broth; Difco) supplemented with 0.05% Tyloxapol (Sigma), BBL Middlebrook OADC (oleic acid-albumin-dextrose-catalase) enrichment, and 0.2% glycerol (Sigma). For making single cells, Mtb were passed through a 5 μm filter before infection.
Mice
Female C57BL/6 (H-2b) mice that were 6- to 12-weeks of age were purchased from Jackson Labs. The mice were maintained under pathogen-free conditions. All experiments employing mice were performed in accordance with laboratory animal protocol approved by the School of Medicine Animal Studies Committee of Washington University in St. Louis. Mice were euthanized using CO2 asphyxiation and cervical dislocation. The euthanized mice were kept in 70% (v/v) ethanol for 1 min. Both the femurs and tibiae were isolated, and the muscle attachments were carefully removed using gauze pads. Both ends of the bones were cut with scissors and the marrow was centrifuged in an adapted centrifuge tube (0.65 ml tube with a hole inserted in 1.5 ml tube) at 1,000 rpm for 10 seconds. The pellet was resuspended by vigorous pipetting in RPMI 1640 medium. The cells were passed through a 70 μm cell strainer to prepare a single-cell suspension. After one wash (1,100 rpm, 5 min), red blood cells (RBC) were depleted with RBC lysis buffer (Millipore Sigma, St. Louis, MO, USA).
Method details
Plasmonic-fluor procurement and characterization
Streptavidin-conjugated Cy3-plasmonic-fluor (PF550™ ultrabright fluor) and streptavidin-conjugated Cy5-plasmonic-fluor (PF650™ ultrabright fluor) was purchased from Auragent Biosceince LLC (St. Louis, USA). The extinction was measured using a Shimadzu UV-1800 spectrophotometer. SEM images were obtained using a FEI Nova 2300 field-emission scanning electron microscope at an acceleration voltage of 10 kV. TEM images were obtained using a JEOL JEM-2100F field emission instrument. A drop of aqueous solution was dried on a carbon-coated grid, which had been made hydrophilic by glow discharge. Molar concentrations of plasmonic-fluors was calculated as described previously (Luan et al., 2020). Fluorescence intensity was recorded using Azure Biomolecular Imager: Sapphire RGBNIR (Azure Biosystems, Inc. Dublin, USA) and the images were analyzed using Licor Image Studio Lite.
Standard curve using plasmonic-fluors
Mouse TNFα DuoSet ELISA kit (R&D systems, catalog number DY410, lot number P189768), mouse IL-6 DuoSet ELISA kit (R&D systems, catalog number DY406, lot number P234212), mouse IFNγ DuoSet ELISA kits (R&D systems, catalog number DY485-05, lot number P234214) and mouse IL-1β (Invitrogen, catalog number 88-7013-88 and lot number 183204000) were used to perform the assays. Glass-bottom 96-well black plate (P96-1.5H-N, Cellvis, Mountain View, USA) was first coated with capture antibodies as per manufacturer’s instructions (100 μl/well) and incubated overnight at 4°C. The plate was washed three times with 1x PBST (1x PBS with 0.05% Tween-20) and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2μm filtered). After blocking the plates were washed three times with PBST, and serial dilutions of standard protein was added to different wells in duplicates and incubated for 2 hours at room temperature. The plates were washed three times with PBST and then incubated for 2 hours with 100 μl of biotinylated detection antibody as per the manufacturer’s instructions. The plates were washed three times with PBST and streptavidin Cy5-plasmonic-fluor (extinction 0.5) for 30 minutes at room temperature in dark. Finally, the plates were washed three times in PBST and imaged using Nikon TsR2 epifluorescence microscope.
FluoroDOT assay on JAWS II DC
Mouse TNFα DuoSet ELISA kits (R&D systems, catalog number DY410, lot number P189768) was used to perform the assays. Glass-bottom 96-well black plate (P96-1.5H-N, Cellvis, Mountain View, USA) was first coated with capture antibodies (0.8 μg/ml in PBS), 100 μl/well and incubated overnight at 4°C. The plate was washed three times with 1x PBS and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2μm filtered). After blocking, the plates were washed three times with PBS. JAWS II DCs were seeded on the capture coated plates, at the seeding density of 5,000 cells/well in 100 μl of medium followed by incubation at 37°C in 5% CO2 for 30 minutes. All the non-adherent cells were removed by taking out the medium and replacing it with 100 μl of fresh medium containing varying amounts of LPS ranging from (0 to 2000 ng/ml). The cells were incubated at 37°C in 5% CO2 for varying durations from 20 minutes to 90 minutes. After completion of the incubation duration, the medium was decanted, and the cells were fixed using 100 μl/well of 4% neutral buffered formalin (NBF) for 20 minutes at room temperature. The plates were then washed three times with PBS and incubated with biotinylated detection antibodies, 75 ng/ml in reagent diluent for 2 hours at room temperature. The plates were washed three times using PBS and then were incubated with 100 μl/well of streptavidin Cy5-plasmonic-fluors (extinction 0.5) for 30 minutes at room temperature in dark. For comparison, 100 μl of 1 μg/ml of streptavidin-Cy5 (Thermo Fisher Scientific, catalog number SA1011) as conventional fluorophore and 1 nM of streptavidin QD 655™ (Thermo Fisher Scientific, catalog number Q10123MP) was added to each well for 30 minutes at room temperature in dark. The plates were washed with PBS three times, and the nuclei of the cells were stained with 300 nM DAPI solution (Millipore Sigma, St. Louis, MO, USA) for 5 minutes at room temperature in dark. Finally, the plates were washed three times in PBS and imaged using Nikon TsR2 epifluorescence microscope. For ELISA, culture supernatants of JAWS II DC after treatment described above were collected and TNFα concentration was measured using mouse TNFα DuoSet ELISA kits (R&D systems, catalog number DY410, lot number P189768) as per manufacturer’s instructions.
Epifluorescence microscopy
All images were acquired on a Nikon Eclipse Ts2R-FL epifluorescence illumination microscope with a 20x, 0.75–numerical aperture (NA) lens and 60x, 1.4-NA. The microscope is attached to Hamamatsu digital camera (ORCA-Flash 4.0) with aura light engine. We used NIS-Elements AR 5.11.01 64-bit software to acquire images. Bright field and fluorescence images were collected in four channels corresponding to DAPI, Cy5, TRITC and GFP. For Cy5, TRITC and GFP, 200 ms exposure time was used and for DAPI exposure time of 50 ms was used. All images were saved as .tif files and further processed.
Image processing and calculation of signal-to-noise ratio
Image J 1.53a (64-bit), was used for adjusting the brightness and contrast of .tif images. Pseudo-color was imparted to images collected from different channels and merged using Image J tool. Pseudo-line was drawn on the image and analyzed using “plot profile” feature to obtain intensity vs. distance (pixels) graph. The raw data of the graph was exported, and signal-to-noise ratio (SNR) was calculated using the following equation:
Average of 5 pixels with highest intensity was recorded as average signal. Standard deviation of first and last 50 pixels was recorded as standard deviation of noise. In case of ELISpot because the images had white background, “invert image” feature was used to create a dark background before the intensity vs. distance graph was obtained.
ELISpot/FluoroSPOT and FluoroDOT assay on PBMCs
Capture antibody pre-coated polyvinylidene difluoride–backed strip plates were used for ELISpot and FluoroSPOT assays as per the manufacturer’s instructions for detection of human IFNγ (ImmunoSPOT, Cellular Technology (CTL), Cleavland, OH). Samples were run in duplicate for each assay type. For ELISpot and FluoroSpot assays, 25,000 PBMCs/ well in 100 μl of media were seeded on the plate. Human IFNγ ELISpot development module (R&D systems, catalog number SEL285) was used to perform the FluoroDOT assays. Glass-bottom 96-well black plate (P96-1.5H-N, Cellvis, Mountain View, USA) was first coated with capture antibodies (1:60 dilution in PBS), 100 μl/well and incubated overnight at 4°C. The plate was washed three times with 1x PBS and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2μm filtered). After blocking, the plates were washed three times with PBS. For FluoroDOT assay 12,500 PBMCs/well in 100 μl of medium followed by incubation at 37°C in 5% CO2 for 30 minutes. All the non-adherent cells were removed by taking out the medium and replacing it with 100 μl of fresh medium containing PMA (1 ng/ml) and ionmycin (400 ng/ml). The cells were incubated at 37°C in 5% CO2 for 18 hours. After completion of the incubation duration, the medium was decanted, and the plates were then washed three times with PBS and incubated with biotinylated detection antibodies, 1:60 dilution in reagent diluent for 2 hours at room temperature. The plates were washed three times using PBS and then were incubated with 100 μl/well of streptavidin Cy5-plasmonic-fluors (extinction 0.5) for 30 minutes at room temperature in dark. Finally, the plates were washed three times in PBS and imaged using Nikon TsR2 epifluorescence microscope. For ELISpot assay, streptavidin-bound alkaline phosphatase and developer solution and for FluoroSPOT assay streptavidin-bound FITC were applied to samples as per manufacturer instructions before imaging and analysis using Cellular Technology series 6 ImmunoSpot Universal Analyzer with ImmunoSpot 7.0 professional software (Cellular Technology Analyzers, Shaker Heights, OH). Analysis parameters were optimized to obtain appropriate spot numbers (cytokine-secreting cells) and were maintained constant throughout each sample.
FluoroDOT assay using alveolar macrophages
Mouse TNFα DuoSet ELISA kits (R&D systems, catalog number DY410, lot number P189768) and mouse IL-1β (Invitrogen, catalog number 88-7013-88 and lot number 183204000) were used to perform the assays. Glass-bottom 96-well black plate (P96-1.5H-N, Cellvis, Mountain View, USA) was first coated with TNFα capture antibody (0.8 μg/ml in PBS) or 1L-1β capture antibody (1:250 dilution in PBS), 100 μl/well, and incubated overnight at 4°C. The plate was washed three times with 1x PBS and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2μm filtered). After blocking the plates were washed three times with PBS. Mouse alveolar macrophages were seeded on the capture antibody-coated plates, at the seeding density of 5,000 cells/well in 100 μl of medium followed by incubation at 37°C in 5% CO2 for 30 minutes. All the non-adherent cells were removed by taking out the medium and replacing it with 100 μl of fresh medium containing 500 ng/ml of LPS. The cells were incubated at 37°C in 5% CO2. After 4 hours, for some wells, 20 μM nigericin was added and the plate was incubated for another 30 minutes. Subsequently, the medium was decanted, and the cells were fixed using 100 μl/well of 4% neutral buffered formalin (NBF) for 20 minutes at room temperature. The plates were then washed three times with PBS and incubated with biotinylated detection antibodies (TNFα 75 ng/ml and IL-1β 1:250 dilution) in reagent diluent for 2 hours at room temperature. The plates were washed three times using PBS and then were incubated with 100 μl/well of streptavidin Cy5-plasmonic-fluors (extinction 0.5) for 30 minutes at room temperature in dark. The plates were washed with PBS three times and the nuclei of the cells were stained with 300 nM DAPI solution (Millipore Sigma, St. Louis, MO, USA) for 5 minutes at room temperature in dark. Finally, the plates were washed three times in PBS and imaged using Nikon TsR2 epifluorescence microscope.
TNFα and IL-1β ELISA of AM culture supernatant
AMs were seeded in 96-well plate (Corning® Costar®, Millipore Sigma, St. Louis, MO, USA) at a seeding density of 150,000 cell in 150 μl of media. Adherent AMs were selected by incubating for 1 hour in media and further, culture medium was replaced with DMEM containing 500 ng/ml LPS for priming. After 4 hours of LPS treatment, 20 μM nigericin was added to activate NLRP3 inflammasome for 30 minutes. Cell culture supernatant was collected and cytokine concentration was measured using mouse TNFα DuoSet ELISA kits (R&D systems, catalog number DY410, lot number P189768) and mouse IL-1β (Invitrogen, catalog number 88-7013-88 and lot number 183204000) as per manufacturer’s instructions.
Antibody conjugation on plasmonic-fluor for multiplexing and validation
Streptavidin-conjugated Cy3-plasmonic-fluor (40 μl, extinction 32) and streptavidin-conjugated Cy5-plasmonic-fluor (40 μl, extinction 30) was added to 50 μl of 4.5 μg/ml biotinylated IL-6 detection antibody and biotinylated TNF-α detection antibody, respectively. The mixture was incubated for 30 minutes at room temperature and then washed twice with pH 10 water. For washing, Cy3-plasmonic-fluor was centrifuged at 4,000 revolutions per minute (rpm) for 10 minutes, and Cy5-plasmonic-fluor was centrifuged at 6,000 rpm for 10 minutes. Finally, the pellet was resuspended in 1% BSA in 1x PBS and stored in 4°C until further use. In order to validate the successful conjugation of the antibody, 0.5 μl of TNFα capture antibody (0.8 μg/ml in 10% glycerol in 1x PBS) was deposited on top-left area of a 96-well glass-bottom plate. Similarly, 0.5 μl of IL-6 capture antibody (2 μg/ml in 10% glycerol in 1x PBS) was deposited on top-right area of the same 96-well glass-bottom plate. 0.5 μl of a mixture of TNFα capture antibody (0.8 μg/ml in 10% glycerol in 1x PBS) and IL-6 capture antibody (2 μg/ml in 10% glycerol in 1x PBS) was deposited on bottom-middle area of the same 96-well glass-bottom plate. The plate was sealed with plate sealant and incubated for 2 hours at room temperature. The plate was washed three times with 1x PBST and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2μm filtered). After blocking, the plates were washed three times with PBST and 5,000 pg/ml of both of standard proteins (TNFα and IL-6) in 100 μl of 1% BSA in 1x PBS was added to the well and incubated for 2 hours at room temperature. The plates were washed three times with PBST and then incubated for 2 hours with a suspension comprised of 50 μl of IL-6 detection antibody-conjugated Cy3-plasmonic-fluor and 50 μl of TNFα detection antibody-conjugated Cy5-plasmonic-fluor (extinction 1 each) in dark. Finally, the plates were washed three times in PBST and imaged using Nikon TsR2 epifluorescence microscope using a 4x objective.
Multiplexed FluoroDOT assay
Mouse TNFα DuoSet ELISA kits (R&D systems, catalog number DY410, lot number P189768) and mouse IL-6 DuoSet ELISA kit (R&D systems, catalog number DY406, lot number P234212) were used to perform the assays. Glass-bottom 96-well black plate (P96-1.5H-N, Cellvis, Mountain View, USA) was first coated with both TNFα capture antibody (0.8 μg/ml in PBS) and IL-6 capture antibody (2 μg/ml in PBS), 100 μl/well, and incubated overnight at 4°C. The plate was washed three times with 1x PBS and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2μm filtered). After blocking the plates were washed three times with PBS. JAWS II dendritic cells (ATCC® CRL-11904™) were cultured in alpha minimum essential medium with ribonucleosides, deoxyribonucleosides, 4 mM L-glutamine, 1 mM sodium pyruvate and 5 ng/ml murine GM-CSF, 10% heat-inactivated fetal bovine serum, 50 IU/ml of penicillin, 50 μg/ml of streptomycin. JAWS II DCs were seeded on the capture coated plates, at the seeding density of 5,000 cells/well in 100 μl of medium followed by incubation at 37°C in 5% CO2 for 30 minutes. All the non-adherent cells were removed by taking out the medium and replacing it with 100 μl of fresh medium containing 200 ng/ml of LPS. The cells were incubated at 37°C in 5% CO2 for 30 minutes, 1 hour, 2 hours, and 3 hours. The wells with medium and without LPS were incubated for 3 hours. Subsequently, the medium was decanted, and cells were fixed using 100 μl/well of 4% neutral buffered formalin (NBF) for 20 minutes at room temperature. The plates were then washed three times with PBS and incubated with biotinylated detection antibody conjugated plasmonic-fluors (TNFα-detection antibody conjugated Cy5-plasmonic-fluor and IL-6 detection antibody conjugated Cy3-plasmonic-fluor: extinction 0.5) in reagent diluent for 2 hours at room temperature. The plates were washed three times using PBS and the nuclei of the cells were stained with 300 nM DAPI solution (Millipore Sigma, St. Louis, MO, USA) for 5 minutes at room temperature in dark. Finally, the plates were washed three times in PBS and imaged using Nikon TsR2 epifluorescence microscope.
FluoroDOT assay using Mtb-infected BMDMs
Mouse TNFα DuoSet ELISA kit (R&D systems, catalog number DY410, lot number P189768) was used for this assay. Glass-bottom 96-well black plate (P96-1.5H-N, Cellvis, Mountain View, USA) was first coated with capture antibodies (0.8 μg/ml in PBS), 100 μl/well and incubated overnight at 4°C. The plate was washed 3 times with 1x PBS and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2 μm filtered). After blocking the plates were washed three times with PBS. BMDMs were seeded on the capture coated plates, at the seeding density of 5,000 cells/well in 100 μl of medium followed by incubation at 37°C in 5% CO2 for 30 minutes. All the non-adherent cells were removed by taking out the medium and replacing it with 100 μl of fresh medium containing DsRed-expressing Mtb strains at MOI of ∼5. The cells were incubated at 37°C in 5% CO2 for 3 hours, then the supernatant was replaced and incubated for another 3 hours. Subsequently, the medium is decanted, and the cells were fixed using 100 μl/well of 1% paraformaldehyde (PFA) in PBS, overnight at 4°C. The plates were then washed three times with PBS and incubated with biotinylated detection antibodies (75 ng/ml in reagent diluent for 2 hours at room temperature. The plates were washed three times using PBS and were incubated with 100 μl/well of streptavidin Cy5-plasmonic-fluors (extinction 0.5) for 30 minutes at room temperature in dark. The plates were washed with PBS 3 times and the nuclei of the cells were stained with 300 nM DAPI solution (Millipore Sigma, St. Louis, USA) for 5 minutes at room temperature in dark. Finally, the plates were washed three times in PBS and imaged using Nikon TsR2 epifluorescence microscope.
FluoroDOT assay using BMDC and T cell co-cultures
Mouse IFNγ DuoSet ELISA kit (R&D systems, catalog number DY485-05, lot number P234214) was used for this assay. Glass-bottom 96-well black plate (P96-1.5H-N, Cellvis, Mountain View, USA) was first coated with capture antibodies (4 μg/ml in PBS), 100 μl/well and incubated overnight at 4°C. The plate was washed three times with 1x PBS and then blocked with 200 μl of reagent diluent (1x PBS in 1% BSA, 0.2μm filtered). After blocking, the plates were washed three times with PBS. Wild-type and mismatch BMDCs were seeded on the capture coated plates, at the seeding density of 5,000 cells/well in 100 μl of medium followed by incubation at 37°C in 5% CO2 for 30 minutes. All the non-adherent cells were removed by taking out the medium and replacing it with 100 μl of fresh medium containing either DsRed-expressing Mtb strains at MOI of ∼5 or LPS (1,000 ng/ml). Incubate the cells at 37°C in 5% CO2 for 24 hours. Mycobacterium tuberculosis (Mtb) MHCII-peptide (P25)-specific GFP-expressing CD4+ T cells were prepared as reported previously.(Moran et al., 2011) LPS treated BMDCs were treated with 791 ng/ml of P25 peptide. GFP expressing CD4+ T cells (50,000 cells per well in 100 μl) were added to infected BMDCs, for 3 hours. For time-dependent study, the plates were incubated for 1 hour, 2 hours and 3 hours. Since T cells are non-adherent, after completion of the incubation duration, the plate was spun at 1100 rpm for 5 minutes. The medium was decanted, and the cells were fixed using 100 μl/well of 1% paraformaldehyde (PFA) in PBS, overnight at 4°C. The plates were then gently washed two times with PBS and incubated with biotinylated detection antibodies (200 ng/ml in reagent diluent for 2 hours at room temperature. The plates were gently washed two times using PBS and then were incubated with 100 μl/well of streptavidin Cy5-plasmonic-fluors (extinction 0.5) for 30 minutes at room temperature in dark. The plates were gently washed with PBS 2 times, and the nuclei of the cells were stained with 300 nM DAPI solution (Millipore Sigma, St. Louis, USA) for 5 minutes at room temperature in dark. Finally, the plates were gently washed two times in PBS and imaged using Nikon TsR2 epifluorescence microscope.
Quantification and statistical analysis
For analyzing the statistical difference between more than two groups, one-way analysis of variance (ANOVA) with a post-hoc Tukey’s honest significance test was used. Statistical significance of the data was calculated at 95% (p<0.05) confidence intervals. All values are expressed as mean ± standard deviation. GraphPad Prism 6 was used for all statistical analysis. Linear regression was used to calculate the equation and to derive the slope of fluorescence intensity vs. molar concentration graph. 4-parameter logistic (4-PL) was used to calculate the R2 values and LOD in the standard curves of immuno-assays. The LOD is defined as the analyte concentration corresponding to the mean fluorescence intensity of blank plus three times of its standard deviation (mean+3σ). For FluoroDOT assay, all measurements were taken from distinct samples as well as different regions of the same sample.
Acknowledgments
We thank Dr. Nathaniel Huebsch from the Department of Biomedical Engineering at Washington University in St. Louis for providing access to the epifluorescence microscope used in this study. The authors thank Nano Research Facility (NRF) and Institute of Materials Science and Engineering (IMSE) at Washington University in St. Louis for providing access to electron microscopy facilities. S.S. acknowledges support from the National Science Foundation (CBET-1900277) and the National Institutes of Health (R21CA236652, R21EB030171). J.A.P. acknowledges support from the National Institutes of Health (2R01 AI087682 and R01 304AI130454). S.C.M. acknowledges support from the National Institutes of Health (R56AI104732).
Author contributions
S.S. and A.S. conceived the project and designed the experiments. A.S. optimized and performed FluoroDOT assays and data analysis. J.A.P., E.M., and A.S. designed and performed the experiments for Mtb infection on BMDCs and BMDMs. J.L. helped with plasmonic-fluor optimization for the assay. S.K. developed the image-processing algorithm for particle counting and cluster identification. M.B.M. helped with acquiring PBMCs and performing and analyzing ELISpot and FluoroSPOT assays. S.C.M. and H.J. designed alveolar macrophage experiments and provided cells for the experiment. R.G. helped with antibody conjugation to plasmonic-fluor. P.R. acquired SEM images. Z.W. helped with BMDC isolation and calculation of molar concentration of plasmonic-fluor. J.J.M. helped with development of plasmonic-fluors. C.P.-C. and J.D.E. isolated and provided CD4+ Th1 effector cells. J.A.P. and S.S. supervised the project. A.S., J.A.P., and S.S. wrote the paper. All authors reviewed and commented on the manuscript.
Declaration of interests
The authors declare the following competing financial interest(s): J.L., J.J.M., and S.S. are inventors on pending patent related to plasmonic-fluor technology, and the technology has been licensed by the Office of Technology Management at Washington University in St. Louis to Auragent Bioscience, LLC. J.L., J.J.M., and S.S. are co-founders/shareholders of Auragent Bioscience, LLC. These potential conflicts of interest have been disclosed and are being managed by Washington University in St. Louis. A.S. is currently working with Auragent Bioscience.
Published: August 5, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100267.
Contributor Information
Jennifer A. Philips, Email: philips.j.a@wustl.edu.
Srikanth Singamaneni, Email: singamaneni@wustl.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
A Python code was used to count the number of particles and number of clusters. The latest version of the code is available at https://github.com/singamaneni/Counting-single-plasmonic-nanoparticles and https://zenodo.org/badge/latestdoi/312896998.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abadeer N.S., Brennan M.R., Wilson W.L., Murphy C.J. Distance and plasmon wavelength dependent fluorescence of molecules bound to silica-coated gold nanorods. ACS Nano. 2014;8:8392–8406. doi: 10.1021/nn502887j. [DOI] [PubMed] [Google Scholar]
- Adetifa I.M., Ota M.O.C., Jeffries D.J., Lugos M.D., Hammond A.S., Battersby N.J., Owiafe P.K., Donkor S.D., Antonio M., Ibanga H.B., et al. Interferon-γ ELISPOT as a biomarker of treatment efficacy in latent tuberculosis infection. Am. J. Respir. Crit. Care Med. 2013;187:439–445. doi: 10.1164/rccm.201208-1352OC. [DOI] [PubMed] [Google Scholar]
- Albrecht V., Hofer T.P.J., Foxwell B., Frankenberger M., Ziegler-Heitbrock L. Tolerance induced via TLR2 and TLR4 in human dendritic cells: role of IRAK-1. BMC Immunol. 2008;9:69. doi: 10.1186/1471-2172-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuler S.J., Wu L.F. Cellular heterogeneity: do differences make a difference? Cell. 2010;141:559–563. doi: 10.1016/j.cell.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrecht L., Rutschmann O., Szczerba B.M., Nikoloff J., Aceto N., Dittrich P.S. Quantification of protein secretion from circulating tumor cells in microfluidic chambers. Adv. Sci. 2020;7:1903237. doi: 10.1002/advs.201903237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashouri J.F., Weiss A. Endogenous Nur77 is a specific indicator of antigen receptor signaling in human T and B cells. J. Immunol. 2017;198:657–668. doi: 10.4049/jimmunol.1601301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham R., Haseley N., Brown D., Penaranda C., Jijon H.B., Trombetta J.J., Satija R., Shalek A.K., Xavier R.J., Regev A., Hung D.T. Pathogen cell-to-cell variability drives heterogeneity in host immune responses. Cell. 2015;162:1309–1321. doi: 10.1016/j.cell.2015.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B., Yang Y., Wang Q., Li M., Tian C., Liu Y., Aung L.H.H., Li P.-f., Yu T., Chu X.-m. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020;11:776. doi: 10.1038/s41419-020-02985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardhan R., Grady N.K., Cole J.R., Joshi A., Halas N.J. Fluorescence enhancement by Au nanostructures: nanoshells and nanorods. ACS Nano. 2009;3:744–752. doi: 10.1021/nn900001q. [DOI] [PubMed] [Google Scholar]
- Bounab Y., Eyer K., Dixneuf S., Rybczynska M., Chauvel C., Mistretta M., Tran T., Aymerich N., Chenon G., Llitjos J.-F., et al. Dynamic single-cell phenotyping of immune cells using the microfluidic platform DropMap. Nat. Protoc. 2020;15:2920–2955. doi: 10.1038/s41596-020-0354-0. [DOI] [PubMed] [Google Scholar]
- Chang H.-H., Murphy C.J. Mini gold nanorods with tunable plasmonic peaks beyond 1000 nm. Chem. Mater. 2018;30:1427–1435. doi: 10.1021/acs.chemmater.7b05310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay P.K., Gierahn T.M., Roederer M., Love J.C. Single-cell technologies for monitoring immune systems. Nat. Immunol. 2014;15:128–135. doi: 10.1038/ni.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Lu Y., Zhang K., Xiao Y., Lu J., Fan R. Multiplexed, sequential secretion analysis of the same single cells reveals distinct effector response dynamics dependent on the initial basal state. Adv. Sci. 2019;6:1801361. doi: 10.1002/advs.201801361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu D.T., deMello A.J., Di Carlo D., Doyle P.S., Hansen C., Maceiczyk R.M., Wootton R.C. Small but perfectly formed? Successes, challenges, and opportunities for microfluidics in the chemical and biological sciences. Chem. 2017;2:201–223. doi: 10.1016/j.chempr.2017.01.009. [DOI] [Google Scholar]
- Choi J.R. Advances in single cell technologies in immunology. Biotechniques. 2020;69:226–236. doi: 10.2144/btn-2020-0047. [DOI] [PubMed] [Google Scholar]
- Couvillion S.P., Zhu Y., Nagy G., Adkins J.N., Ansong C., Renslow R.S., Piehowski P.D., Ibrahim Y.M., Kelly R.T., Metz T.O. New mass spectrometry technologies contributing towards comprehensive and high throughput omics analyses of single cells. Analyst. 2019;144:794–807. doi: 10.1039/C8AN01574K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efros A.L., Nesbitt D.J. Origin and control of blinking in quantum dots. Nat. Nanotechnol. 2016;11:661–671. doi: 10.1038/nnano.2016.140. [DOI] [PubMed] [Google Scholar]
- Evavold C.L., Ruan J., Tan Y., Xia S., Wu H., Kagan J.C. The pore-forming protein Gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandra N., Portz C., Tian L., Tang R., Xu B., Achilefu S., Singamaneni S. Probing distance-dependent plasmon-enhanced near-infrared fluorescence using polyelectrolyte multilayers as dielectric spacers. Angew. Chem. Int. Ed. Engl. 2014;53:866–870. doi: 10.1002/anie.201308516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsenko A., Yu S., Martin-Sanchez F., Diaz-del-Olmo I., Nichols E.-M., Davis D.M., Brough D., Lopez-Castejon G. Priming is dispensable for NLRP3 inflammasome activation in human monocytes in vitro. Front. Immunol. 2020;11:565924. doi: 10.3389/fimmu.2020.565924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanzadeh-Barforoushi A., Warkiani M.E., Gallego-Ortega D., Liu G., Barber T. Capillary-assisted microfluidic biosensing platform captures single cell secretion dynamics in nanoliter compartments. Biosens. Bioelectron. 2020;155:112113. doi: 10.1016/j.bios.2020.112113. [DOI] [PubMed] [Google Scholar]
- Heath J.R., Ribas A., Mischel P.S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 2016;15:204–216. doi: 10.1038/nrd.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jammes F.C., Maerkl S.J. How single-cell immunology is benefiting from microfluidic technologies. Microsyst. Nanoeng. 2020;6:45. doi: 10.1038/s41378-020-0140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetzki S., Price L., Schroeder H., Britten C.M., Welters M.J.P., Hoos A. Guidelines for the automated evaluation of Elispot assays. Nat. Protoc. 2015;10:1098–1115. doi: 10.1038/nprot.2015.068. [DOI] [PubMed] [Google Scholar]
- Joshi H., Almgren-Bell A., Anaya E.P., Todd E.M., Van Dyken S.J., Seth A., McIntire K.M., Singamaneni S., Sutterwala F., Morley S.C. L-plastin enhances NLRP3 inflammasome assembly and bleomycin-induced lung fibrosis. Cell Rep. 2022;38:110507. doi: 10.1016/j.celrep.2022.110507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi H., Morley S.C. Cells under stress: the mechanical environment shapes inflammasome responses to danger signals. J. Leukoc. Biol. 2019;106:119–125. doi: 10.1002/jlb.3mir1118-417r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan-Colás J., Hitchcock I.S., Coles M., Johnson S., Krauss T.F. Quantifying single-cell secretion in real time using resonant hyperspectral imaging. Proc. Natl. Acad. Sci. USA. 2018;115:13204–13209. doi: 10.1073/pnas.1814977115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatua S., Orrit M. Probing, sensing, and fluorescence enhancement with single gold nanorods. J. Phys. Chem. Lett. 2014;5:3000–3006. doi: 10.1021/jz501253j. [DOI] [PubMed] [Google Scholar]
- Kim Y., Wang Z., Li C., Kidd K., Wang Y., Johnson B.G., Kmoch S., Morrissey J.J., Bleyer A.J., Duffield J.S., et al. Ultrabright plasmonic fluor nanolabel-enabled detection of a urinary ER stress biomarker in autosomal dominant tubulointerstitial kidney disease. Am. J. Physiol. Renal Physiol. 2021;321:F236–F244. doi: 10.1152/ajprenal.00231.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenkamp A., Messi M., Lanzavecchia A., Sallusto F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 2000;1:311–316. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- Li X., Soler M., Szydzik C., Khoshmanesh K., Schmidt J., Coukos G., Mitchell A., Altug H. Label-free optofluidic nanobiosensor enables real-time analysis of single-cell cytokine secretion. Small. 2018;14:1800698. doi: 10.1002/smll.201800698. [DOI] [PubMed] [Google Scholar]
- Lin P.L., Ford C.B., Coleman M.T., Myers A.J., Gawande R., Ioerger T., Sacchettini J., Fortune S.M., Flynn J.L. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat. Med. 2014;20:75–79. doi: 10.1038/nm.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Wang Z., Wang Y., Luan J., Morrissey J.J., Naik R.R., Singamaneni S. Plasmonically enhanced CRISPR/Cas13a-based bioassay for amplification-free detection of cancer-associated RNA. Adv. Healthcare Mater. 2021:2100956. doi: 10.1002/adhm.202100956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon M.M., Aldridge B.B. Stable regulation of cell cycle events in mycobacteria: insights from inherently heterogeneous bacterial populations. Front. Microbiol. 2018;9:514. doi: 10.3389/fmicb.2018.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Ye G., Punj D., Chiechi R.C., Orrit M. Quantum yield limits for the detection of single-molecule fluorescence enhancement by a gold nanorod. ACS Photonics. 2020;7:2498–2505. doi: 10.1021/acsphotonics.0c00803. [DOI] [Google Scholar]
- Luan J., Morrissey J.J., Wang Z., Derami H.G., Liu K.-K., Cao S., Jiang Q., Wang C., Kharasch E.D., Naik R.R., Singamaneni S. Add-on plasmonic patch as a universal fluorescence enhancer. Light Sci. Appl. 2018;7:29. doi: 10.1038/s41377-018-0027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan J., Seth A., Gupta R., Wang Z., Rathi P., Cao S., Gholami Derami H., Tang R., Xu B., Achilefu S., et al. Ultrabright fluorescent nanoscale labels for the femtomolar detection of analytes with standard bioassays. Nat. Biomed. Eng. 2020;4:518–530. doi: 10.1038/s41551-020-0547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T., Güell M., Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Malyguine A.M., Strobl S., Dunham K., Shurin M.R., Sayers T.J. ELISPOT assay for monitoring cytotoxic T lymphocytes (CTL) activity in cancer vaccine clinical trials. Cells. 2012;1:111–126. doi: 10.3390/cells1020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V. A dream of single-cell proteomics. Nat. Methods. 2019;16:809–812. doi: 10.1038/s41592-019-0540-6. [DOI] [PubMed] [Google Scholar]
- Mastrogiovanni M., Di Bartolo V., Alcover A. Cell polarity regulators, multifunctional organizers of lymphocyte activation and function. Biomed. J. 2022;45:299–309. doi: 10.1016/j.bj.2021.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran A.E., Holzapfel K.L., Xing Y., Cunningham N.R., Maltzman J.S., Punt J., Hogquist K.A. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy T.W., Zhang Q., Naler L.B., Ma S., Lu C. Recent advances in the use of microfluidic technologies for single cell analysis. Analyst. 2017;143:60–80. doi: 10.1039/C7AN01346A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W., Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009;10:148–155. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- Pai M., Denkinger C.M., Kik S.V., Rangaka M.X., Zwerling A., Oxlade O., Metcalfe J.Z., Cattamanchi A., Dowdy D.W., Dheda K., Banaei N. Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin. Microbiol. Rev. 2014;27:3–20. doi: 10.1128/CMR.00034-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L., Jia H., Liu F., Sun H., Gao M., Du F., Xing A., Du B., Sun Q., Wei R., et al. Risk factors for false-negative T-SPOT.TB assay results in patients with pulmonary and extra-pulmonary TB. J. Infect. 2015;70:367–380. doi: 10.1016/j.jinf.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Saletti G., Çuburu N., Yang J.S., Dey A., Czerkinsky C. Enzyme-linked immunospot assays for direct ex vivo measurement of vaccine-induced human humoral immune responses in blood. Nat. Protoc. 2013;8:1073–1087. doi: 10.1038/nprot.2013.058. [DOI] [PubMed] [Google Scholar]
- Streeck H., Frahm N., Walker B.D. The role of IFN-γ Elispot assay in HIV vaccine research. Nat. Protoc. 2009;4:461–469. doi: 10.1038/nprot.2009.7. [DOI] [PubMed] [Google Scholar]
- Tam F., Goodrich G.P., Johnson B.R., Halas N.J. Plasmonic enhancement of molecular fluorescence. Nano Lett. 2007;7:496–501. doi: 10.1021/nl062901x. [DOI] [PubMed] [Google Scholar]
- Tamura T., Ariga H., Kinashi T., Uehara S., Kikuchi T., Nakada M., Tokunaga T., Xu W., Kariyone A., Saito T., et al. The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int. Immunol. 2004;16:1691–1699. doi: 10.1093/intimm/dxh170. [DOI] [PubMed] [Google Scholar]
- Thampy L.K., Remy K.E., Walton A.H., Hong Z., Liu K., Liu R., Yi V., Burnham C.-A.D., Hotchkiss R.S. Restoration of T Cell function in multi-drug resistant bacterial sepsis after interleukin-7, anti-PD-L1, and OX-40 administration. PLoS One. 2018;13:e0199497. doi: 10.1371/journal.pone.0199497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toniolo C., Rutschmann O., McKinney J.D. Do chance encounters between heterogeneous cells shape the outcome of tuberculosis infections? Curr. Opin. Microbiol. 2021;59:72–78. doi: 10.1016/j.mib.2020.08.008. [DOI] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Uhlén M., Karlsson M.J., Hober A., Svensson A.-S., Scheffel J., Kotol D., Zhong W., Tebani A., Strandberg L., Edfors F., et al. The human secretome. Sci. Signal. 2019;12:eaaz0274. doi: 10.1126/scisignal.aaz0274. [DOI] [PubMed] [Google Scholar]
- Vogel C., Marcotte E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Luan J., Seth A., Liu L., You M., Gupta P., Rathi P., Wang Y., Cao S., Jiang Q., et al. Microneedle patch for the ultrasensitive quantification of protein biomarkers in interstitial fluid. Nat. Biomed. Eng. 2021;5:64–76. doi: 10.1038/s41551-020-00672-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G., Gómez D.E., Kirkwood N., Boldt K., Mulvaney P. Two mechanisms determine quantum dot blinking. ACS Nano. 2018;12:3397–3405. doi: 10.1021/acsnano.7b09052. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Shao N., Bessa de Castro R., Zhang P., Ma Y., Liu X., Huang F., Wang R.-F., Qin L. Evaluation of single-cell cytokine secretion and cell-cell interactions with a hierarchical loading microwell chip. Cell Rep. 2020;31:107574. doi: 10.1016/j.celrep.2020.107574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
A Python code was used to count the number of particles and number of clusters. The latest version of the code is available at https://github.com/singamaneni/Counting-single-plasmonic-nanoparticles and https://zenodo.org/badge/latestdoi/312896998.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.