

Abstract
Objective:
We investigated the duration of HIV transmission clusters.
Design:
54 subjects newly infected at enrollment in the ALIVE cohort were included, all of whom had sequences at an intake visit (T1) and from a 2nd (T2) and/or a 3rd (T3) follow-up visit, median 2.9 and 5.4 years later, respectively.
Methods:
Sequences were generated using the 454 DNA sequencing platform for portions of HIV pol and env (HXB2 positions 2717–3230; 7941–8264). Genetic distances were calculated using tn93 and sequences were clustered over a range of thresholds (1%−5%) using HIV-TRACE. Analyses were performed separately for subjects with pol sequences for T1 + T2 (n=40, “Set 1”) and T1 + T3 (n=25; “Set 2”), and env sequences for T1 + T2 (n=47, “Set 1”) and T1 + T3 (n=30; “Set 2”).
Results:
For pol, with one exception, a single cluster contained >75% of samples at all thresholds, and cluster composition was >=90% concordant between time points/thresholds. For env, two major clusters (A, B) were observed at T1 and T2/T3, although cluster composition concordance between time points/thresholds was low (<60%) at lower thresholds for both Sets 1 and 2. In addition, several subjects were included in clusters at T2/T3, although not at T1.
Conclusions:
Caution should be used in applying a single threshold in population studies where seroconversion dates are unknown. However, the retention of some clusters even after 5+ years is evidence for the robustness of the clustering approach in general.
INTRODUCTION
Identification of transmission clusters using HIV sequence data is a standard tool of molecular epidemiological studies (1–4) due in part to the computational efficiency and ease of use (5). Cluster analysis is particularly useful when analyzing large datasets, e.g. those generated from deep-sequencing technologies or country-wide HIV sequence databases (6). Cluster analysis can reveal epidemiologically important trends, help identify outbreaks, guide prevention strategies, and uncover significant risk factors for infection (7). Clustering methods are based on the underlying concept that genetically similar viruses likely shared a recent epidemiological history (e.g. direct transmission, derivation from the same source, or part of the same transmission chain)(5).
Interpretation of cluster dynamics is complicated by the observation that the same distance threshold, used with different methods (e.g. genetic distance (8) vs. tree-based (9, 10)), may result in different cluster compositions and capture widely different time spans (11). Furthermore, samples close to the time of seroconversion are more likely to cluster than samples from well after infection (5, 12). Since many cross-sectional and longitudinal population studies include subjects with unknown dates of seroconversion, clustering risk factors may therefore erroneously be identified (5) unless explicitly accounted for (1, 12, 13). A combination of the two approaches may provide additional epidemiological insight (5).
A threshold for genetic distance can be defined as the median, mean, or maximum genetic distance allowed for any two pairs of sequences within a cluster. Alternatively, “single linkage” methods can be used, where a sequence is included in a cluster if the distance between it and any other sequence is below the threshold (10). However, some of these methods (e.g. mean and single linkage) can result in large clusters where sequences are “chained” together (e.g. sequence A is related to sequence B, B is related to C, C is related to D) resulting in large distances above the threshold for many pairs (e.g. between A and D) (14).
The genetic distance threshold chosen to define clusters clearly has a strong impact on the resulting clusters (4, 15, 16). Objective methods to select the appropriate distance threshold include using a priori knowledge of either the distance between epidemiologically confirmed transmission pairs or the intra-host evolutionary rate of samples in the dataset under study (5). The optimal threshold is the value which most accurately separates known linked vs non-linked sequences, which will change among datasets depending on the pathogen, gene region, and sample composition (17). In cases lacking sufficient data for these approaches, conservative genetic distance thresholds for the pol gene of 1% - 2% are commonly used (2, 4, 5, 18), consistent with expected intra-host evolution of this region (4, 13, 19). While fewer studies have used the env gene to infer cluster dynamics, a threshold of up to 5.3% was found to correlate with known epidemiological linkage (17, 20), consistent with the higher evolutionary rate in this gene (21). Using a range of threshold values can provide insights that are lost when choosing only one value. For example, lower thresholds will capture the most closely related sequences (often interpreted as the leading edge of an epidemic)(18), while higher thresholds will capture mature epidemics and chronically infected individuals (10). Furthermore, the mode of transmission (i.e. intravenous drug use (IDU) vs. sexual) can impact transmission network patterns and may require different thresholds (22).
Here, we investigate another approach for determining appropriate thresholds by using sequence data from longitudinally sampled individuals who were all recently infected at enrollment. Individuals were identified as being recently infected by a multi-serology assay algorithm validated in an HIV-1 clade B setting that included members of the cohort analyzed in this paper (23). Our objective was to assess the persistence of transmission clusters detected soon after transmission at two follow-up time points (up to 7 years later). High throughput sequencing (HTS) was generated for pol and env, providing high resolution for determining linkage among individuals and the ability to compare the signal between genes.
METHODS
Subjects.
Subjects were selected from the AIDS Linked to the IntraVenous Experience (ALIVE) cohort, one of the longest-running community-based cohorts of people who inject drugs (PWID) prospectively followed from 1988. At the conception of the ALIVE cohort, individuals enrolled were 89% Black and 81% male (24). All subjects were identified as recently infected (< 6 months) at enrollment using serological testing as follows: samples from subjects identified as recently infected had a BED-capture EIA value < 1.0, a BioRad avidity index < 80%, a viral load above 400 copies/ml and a CD4 count >200 cells/mm (25). A total of 54 subjects were included in the present study, all of whom had sequences at an intake visit (T1) and from a 2nd (T2) and/or a 3rd (T3) follow-up visit, which took place a median of 2.9 and 5.4 years later, respectively. Most of the 54 subjects (n=45) were enrolled in the period from February 1988 – April 1989; eight subjects were enrolled between December 1991 – October 1999; and two subjects were enrolled in 2006 (Table S1). Four subjects were treated with anti-retroviral therapy during the course of the study (Table S1).
Generation of Sequence Data.
Sample extraction and amplification was performed as previously described (26). Briefly, RNA was extracted from 140μL plasma. RT-PCR was used to generate portions of pol and env. PCR products were sequenced using the 454 DNA Sequencing platform (Roche, Branford, CT) as previously described (27). Sequence data were cleaned and analysis was performed on portions of HIV pol (HXB2 position 2717–3230) and env (HXB2 position 7941–8264). To determine the HIV subtype, env sequences from all subjects were grouped at a 95% similarity threshold using USEARCH (28), and the centroid sequence from each group was subtyped using the REGA HIV-1 Subtyping Tool (version 3.0) available at https://hivdb.stanford.edu/.
Genetic Distances.
Genetic distances were calculated using the tn93 program (https://github.com/veg/tn93). A receiver operating curve (ROC) analysis was used to determine the optimal threshold to differentiate self vs. non-self using R package pROC. Sequences were clustered over a range of thresholds (1%−5%) using HIV-TRACE (8), which uses the “single linkage method” described above. For the purposes of this study, clusters were defined as containing sequences from >1 subject.
RESULTS
Sequences generated.
A total of 28,427 (pol) and 9,548 (env) unique consensus sequences were generated representing 995,423 (pol) and 339,707 (env) sequence reads, which covered the entire region for both genes. Analyses were performed separately for pol and env. Because some subjects did not have sequences for all three visits, sequences for each gene were grouped into sets as follows: (1) subjects with pol sequences for T1 and T2 (“pol Set 1”), (2) subjects with env sequences for T1 and T2 (“env Set 1”), (3) subjects with pol sequences for T1 and T3 (“pol Set 2”), and (4) subjects with env sequences for T1 and T3 (“env Set 2”) (Table 1, Supplemental Table 1). All sequences were classified as HIV subtype B.
Table 1.
Number of subjects with sequences in each dataset.
| number subjects | pol Set 1 | pol Set 2 | env Set 1 | env Set 2 |
|---|---|---|---|---|
| 20 | x | x | x | x |
| 3 | x | x | x | |
| 4 | x | x | ||
| 2 | x | x | x | |
| 10 | x | x | ||
| 3 | x | x | ||
| 1 | x | x | ||
| 8 | x | |||
| 2 | x | |||
| 1 | x | |||
|
| ||||
| 54 | 40 | 25 | 47 | 30 |
X indicates that the subject has data for that dataset. Totals are given in bold at the bottom.
Optimized threshold based on intra-host and inter-host comparisons.
For pol, the ROC-determined optimal distance threshold for Set 1 subjects was 1% at T1 and 1.6% at T2. For pol Set 2 subjects, the optimal distance was also 1% at T1, and 2.2% at T3. For env, the distance threshold that optimally separated intra-host vs. inter-host comparisons for Set 1 subjects was 2.2% for T1 and 3.8% for T2. For env Set 2 subjects, the optimal distance for T1 was 1.3%, and 5.1% for T3.
Cluster composition for pol Set 1.
For the 40 subjects with pol sequence data at T1 and T2 (Set 1), for T1 a single cluster contained 31 subjects at 1% (Figure 1A, Supplemental Figure 1A). An additional four subjects were added at 2% (n=35) and another three at 3% (n=38). At 4% and 5%, all 40 subjects were in a single cluster. For T2, a single cluster was also observed at each threshold, with the composition being similar (although slightly smaller) than those at T1 (Figure 1A, Supplemental Figure 1B).
Figure 1. Cluster composition for composition for pol sequences for (A) Set 1 and (B) Set 2.
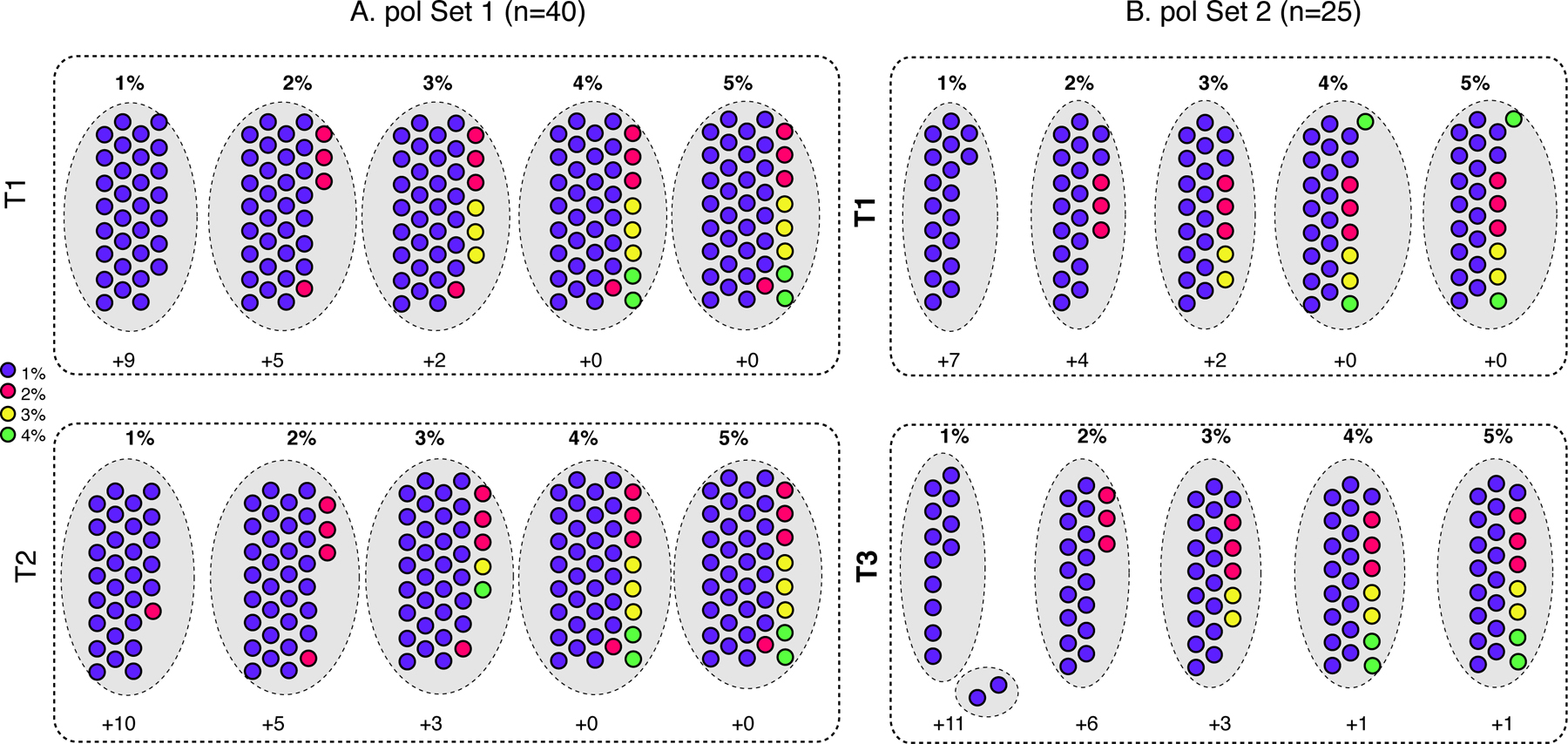
Filled circles represent samples, which are colored according to their cluster designation at T1 for both Sets, as specified in the legend, and retain the same color for the later time point. Larger grey dotted-line circles indicate clusters at the genetic distance thresholds (1% - 5%) listed across the top for each analysis. Time points for each set are denoted with dotted box: (A) T1, top; T2, bottom; (B) T1, top; T3, bottom. The number of samples that did not cluster is indicated at the bottom of each time point (+n).
To determine the cluster persistence between time points, we calculated, for each threshold, the number of subjects in the T2 cluster who were also in the cluster at T1, as a proportion of the total number of subjects in the cluster at T1. The results were as follows: 1%: 29/31 (0.94); 2%: 35/35 (1.0); 3%: 36/38 (0.95); 4% and 5%: 40/40 (1.0). Note that at 1% and 3%, the T2 cluster contained one subject who was not part of the T1 cluster at the corresponding threshold.
Cluster composition for pol Set 2.
For the 25 subjects with pol sequence data at T1 and T3 (pol Set 2), for T1 a single cluster contained 18 subjects at 1%, which increased by three subjects at 2% (n=21), another two subjects at 3% (n=23), and the remaining two subjects at 4% and 5% (n=25, Figure 1B, Supplemental Figure 1C). All subjects were in the cluster at the highest two thresholds.
For T3, two clusters were detected at 1%, containing 12 and two subjects, respectively (Figure 1B); however, one subject in the small cluster also had sequences in the larger cluster as well (Supplemental Figure 1D). At 2%, subjects in both clusters condensed into the single cluster, with seven additional subjects (n=19). At 3% an additional three subjects were included (n=22), and at 4% and 5%, two additional subjects were included (n=24). One subject did not cluster at all, even at the highest threshold.
For each threshold, the number of subjects in the T3 cluster who were also in the cluster at T1, as a proportion of the total number of subjects in the cluster at T1, were as follows: 1%: 12/18 (0.66); 2%: 19/21 (0.90); 3%: 22/23 (0.96); 4% and 5%: 24/25 (0.96).
Cluster composition for env Set 1.
For the 47 subjects in Set 1, subjects were broadly grouped into two large clusters (Group A and Group B) at the lower thresholds (1% - 3%, Figure 2A, Supplemental Figure 2A). For T1 at 1%, two small Group A clusters (A1: n=5; A2: n=6) and two small Group B clusters (B1: n=2; B2: n=3) were observed. At 2%, clusters A1 and A2 merged with an additional 8 subjects, and B1 and B2 merged with an additional 5 subjects. At 3%, the single Group A cluster gained two subjects and the single Group B cluster remained the same. At 4%, the Group A and Group B clusters merged into a single cluster (with an additional three subjects), which then gained another three subjects at 5%. Nine subjects did not cluster at all, even at the highest threshold.
Figure 2. Cluster composition for composition for env sequences for (A) Set 1 and (B) Set 2.
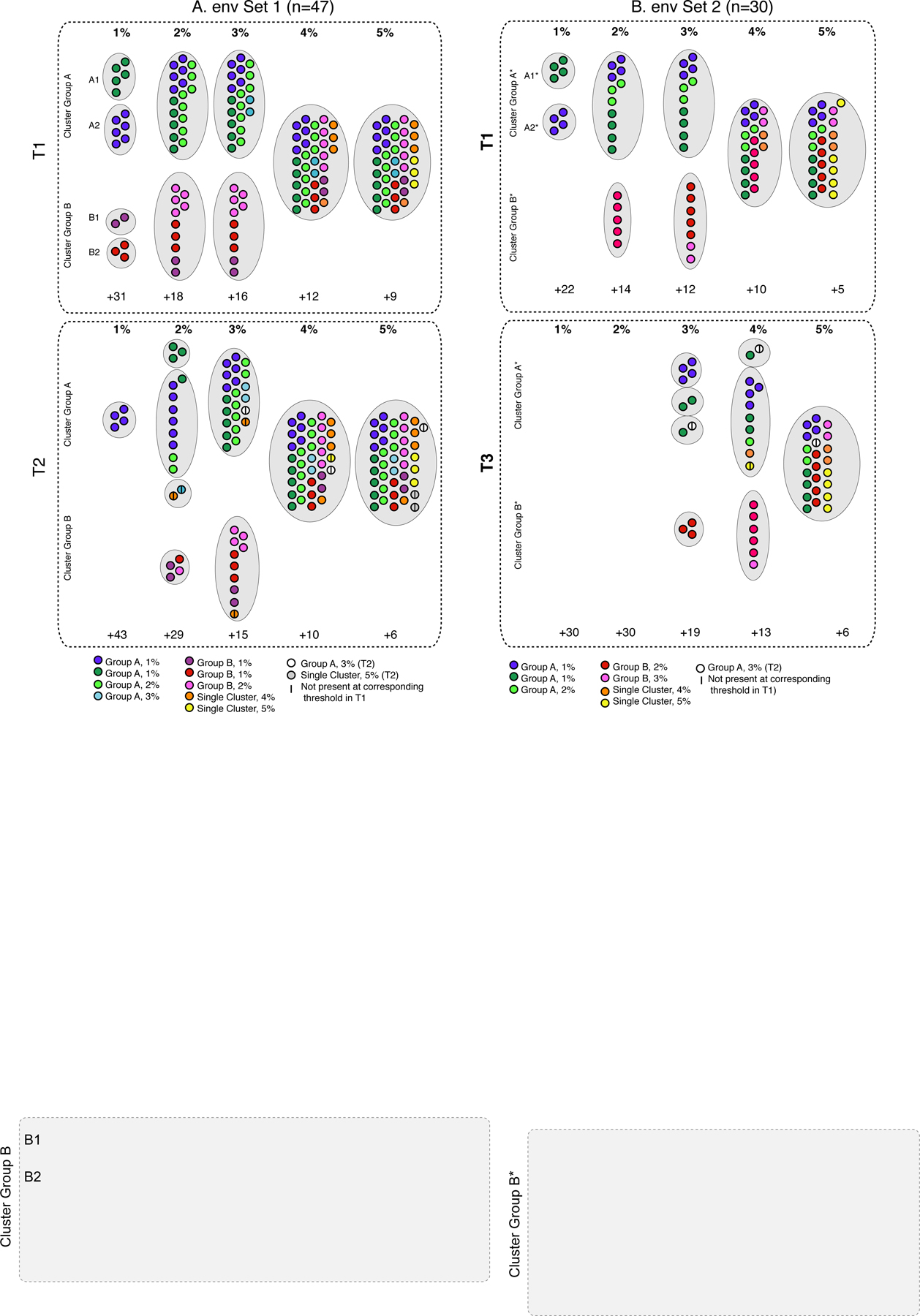
Filled circles represent samples; larger grey dotted-line circles indicate clusters at the genetic distance thresholds (1% - 5%) listed across the top for each analysis. Time points for each set are denoted with dotted box: (A) T1, top; T2, bottom; (B) T1, top; T3, bottom. Samples are colored according to their cluster designation at T1 for both Sets, as specified in the legend, and retain the same color for the later time point. Samples only appearing at the later time point are denoted in the legend. The line indicates samples that were not present at the earlier time point for the same threshold. Clusters are labeled (A, B, A*, B*) as described in the text. The number of samples that did not cluster is indicated at the bottom of each time point (+n).
For T2, at the 1% threshold, a single cluster contained a subset (n=4) of the A2 cluster (Figure 2A, Supplemental Figure 2B). At 2%, a total of four clusters contained: (1) a subset of A1 (n=3); (2) all of A2 (n=6), plus one subject from A1 and two subjects from the larger Group A; (3) one subject from Group A and one non-A/B subject; and (4) all of B1 (n=2), a subset of B2 (n=1), and one additional subject from Group B. At 3%, the single Group A cluster now comprised all of A1, all of A2, nine additional Group A subjects, one non-A/B subject, plus another subject who was not detected in any cluster at the first time point. The single Group B cluster contained all of B1, all of B2, plus four additional Group B subjects and one non-A/B subject. At 4%, all subjects merged into a single cluster, plus three additional subjects. At 5%, two additional subjects were added who were present at in the large single cluster at T1, plus another two subjects who were not.
Comparing the cluster composition between time points at the same distance threshold was not as straightforward for env as with pol as there were multiple clusters at each threshold. We therefore calculated, for each threshold, the number of subjects in any cluster at T2 who were also in any cluster at T1, as a proportion of the total number of subjects in a cluster at T1. The proportions were as follows: 1%: 4/16 (0.25); 2: 16/29 (0.55); 3%: 29/31 (0.94); 4%: 35/35 (1.0); 5%: 38/38 (1.0). (Note that some subjects in T2 clusters were not included as they did not appear in T1 clusters at the corresponding threshold.)
For the ROC-defined thresholds (rounded to the nearest whole number), two clusters were found for T1 at 2%, and only one cluster for T2 at 4%.
Cluster composition for env Set 2.
For the 30 subjects in Set 2, subjects also clustered into two major groups (denoted A* and B*; Figure 2B, Supplemental Figure 2C). Subjects in the Group A* and Group B* clusters were largely a subset of those in the Group A and B clusters from the Set 1 analysis. For T1 at 1%, two clusters were observed: (1) A1* (n=4) and (2) A2* (n=4). At 2%, two clusters contained: (1) all of A1*, all of A2*, plus three other Group A* subjects, and (2) five B* subjects. At 3%, the Group A* cluster remained unchanged, while the Group B* cluster gained two subjects. At 4%, both clusters merged into a single cluster, plus an additional two subjects. At 5%, the single cluster added another five subjects. Five subjects remained unclustered at the highest threshold.
For T3, no clusters were observed at 1% or 2% (Figure 2B, Supplemental Figure 2D). At 3%, four clusters were observed: (1) all of A2*; (2) a subset of A1* (n=2); (3) one subject from A1*, plus an additional subject not observed in any cluster at T1; and (4) three Cluster Group B* subjects. At 4%, three clusters were observed: (1) one subject from A1* plus an additional subject not observed in any cluster at T1; (2) all of A2* (n=4), a subset of A1* (n=2), one additional Group A* subject, and two non-A*/B* subjects; and (4) six Group B* subjects. At 5%, the Group A* and B* cluster merged, and included all of A1* (n=4); all of A2* (n=4), two Group A* subjects, one subject not observed at T1, seven Cluster B* subjects, and an additional four non-A*/B* subjects. Six subjects remained unclustered at the highest threshold.
For each threshold, the number of subjects in any cluster at T2 who were also in any cluster at T1, as a proportion of the total number of subjects in a cluster at T1, were as follows: 1%: 0/8 (0); 2%: 0/16 (0); 3%: 10/19 (0.53); 4%: 15/20 (0.75); 5%: 23/25 (0.92).
For the ROC-defined thresholds (rounded to the nearest whole number), two clusters were found for T1 at 1%, and one cluster for T3 at 5%.
Time of samples.
We then sought to determine if the time between visits was associated with clustering patterns in env (Supplemental Figure 3). In general, the times between T1 and T2 and T1 and T3 were similar for the subjects in small clusters (A1, A2, B1, B2) compared to the rest of the subjects. The exceptions were for env Set 1, where the time between T1 and T2 was well below the interquartile range for two A1 and one A2 subjects. These three samples did not have a T3 sample. Finally, we considered whether sampling year was associated with clustering patterns in env (Supplemental Figure 4). Of the 11 subjects who did not cluster in at least one env analysis, five were initially sampled in 1994 or later (out of a total of seven), while six were initially sampled from 1998–1998 (of a total of 41 subjects).
DISCUSSION
Here, we investigated the duration of cluster composition among 54 subjects who were newly infected at study enrollment of the ALIVE cohort using HTS for the pol and env genes. Our goal was to determine whether transmission clusters present at the first visit (T1) were detectable at the second (T2) and third (T3) visits.
We found a single cluster for pol at nearly all time points/thresholds, which likely reflects the shared geographic location, risk group, and early sampling dates of the subjects. However, it is somewhat surprising that 5+ years later, the cluster composition was basically unchanged. This may be explained in part by the single linkage clustering algorithm itself, which adds a sequence to a cluster if the distance between it and at least one other sequence in the cluster is less than the distance threshold (as opposed to the distance between the query sequences and all other sequences being less than the threshold). This can result in clusters containing pairs of sequences separated by distances higher than the threshold. HTS data may exaggerate this behavior, since each subject has multiple sequences, rather than just a single consensus. Further investigation of this point may clarify the issue (18). In this study, the majority of subjects (n=47) provided all samples prior to the introduction of HAART in 1996, and therefore the impact of drug-resistant mutations in pol is not expected to impact the results.
For env, the ROC-defined thresholds of self vs. non-self also increased with time, although possibly more rapidly than expected given the intra-host env evolutionary rate of ~ 0.005 substitutions/site/year (29, 30). Interestingly, at the ROC-defined thresholds at T2 and T3, only a single large cluster was observed. When comparing cluster composition at the same thresholds, the number of subjects in any cluster at T2 who were also in a cluster at T1, as a proportion of the total number of subjects in a cluster at T1, were highest at the 4% - 5% thresholds. This behavior is somewhat expected: low thresholds are expected to capture more recent transmission events, and the accumulated divergence among subjects over time will eventually exceed lower diversity thresholds. On the other hand, the clusters at 4% - 5% also contained the majority of subjects. These results suggest that the use of a single threshold may obscure epidemiologically relevant information.
The composition of the two large clusters (A and B) at T1 in env was generally maintained at T2 and T3. However, an entirely new cluster of two subjects was observed at both T2 and T3 that was not present at T1, and subjects who did not cluster at all at T1 were found in clusters at T2. These results are somewhat surprising: while the breakdown of clusters would be expected over time as more lineage-specific mutations accumulate in each subject, the addition of subjects into clusters (and the generation of new clusters entirely) at later time points is unexpected, since this implies that related sequences accumulate more diversity over time than non-related sequences over time. Again, this may have resulted from the single linkage clustering method and/or the use of HTS data. Alternatively, the observed “new” clusters at later time points may actually represent true transmission clusters. Intravenous drug use allows more virus to be transmitted than sexual transmission and the possibility of multiple founder populations (31). While HTS is expected to capture greater diversity than older Sanger-sequencing method it is possible that unsampled lineages at the first time point obscured true transmission events (32).
The difference in clustering patterns between pol and env are not unexpected, as different regions of the HIV genome are subject to different selective pressures. For example, env diversification resulting from host immune pressure and changes in tropism is expected to contribute to a higher substitution rate than pol, and recombination can further unlink transmission histories among HIV genes (33). The use of multiple regions may therefore provide enhanced ability to recover epidemiolocal linkages (34).
These results are consistent with those reported by Redd et al (35), which analyzed a subset (n=23) of the subjects in the present study. Redd and co-authors used HIV-TRACE at a single distance threshold (2%) to cluster HTS sequences from the first time point. They found three distinct clusters, which corresponded to the clusters defined here as Group A2, Group A1/larger Group A, and larger Group B, respectively. They confirmed these results using single genome amplification of the same samples. The inexact congruence of cluster assignment between the two studies is very likely due to the inclusion of additional subjects in the present analysis. Here, a lower distance threshold (1%) was necessary to obtain similar resolution as found in the earlier study (35), suggesting that that the choice of distance threshold might be further conditioned on the number of subjects included in the analysis.
This study has a several limitations. Epidemiological confirmation of transmission pairs was not available, so the genetic clusters could not be confirmed. Because the methods used here did not account for unsampled subjects, and the subjects included in this study were only a small subsample of the PWID epidemic in Baltimore at the time, it is likely that clustered subjects were not involved in direct transmission events. A strength of the study was the use of high-throughput sequencing, which provided much greater depth of diversity than bulk Sanger sequencing. Follow-up studies using phylogenetic methods would provide additional insight into the maintenance of clustering signal over time. Overall, our results suggest caution in equating clusters with transmission events, particularly at higher thresholds. However, the fact that some clustering patterns were retained after 5+ years of divergence, provides confidence for the use of these methods in studies where seroconversion times are unknown.
Supplementary Material
Subject IDs are shown, with one row per subject across all analyses. Columns contain the IDs for all subjects who clustered at the indicated distance threshold (top, 1% - 5%) for each time point/analysis: (A) T1; (B) T2; (C) T1; (D) T3. Black boxes indicate clustered subjects. Colored dots next to the sample names correspond with those in Figure 1.
Subject IDs are shown, with one row per subject across all analyses. Columns contain the IDs for all subjects who clustered at the indicated distance threshold (top, 1% - 5%) for each time point/analysis: (A) T1; (B) T2; (C) T1; (D) T3. Black boxes indicate clustered subjects. The light grey box indicates the Group A/A* and Group B/B* subjects. Subjects appearing in the white space between grey boxes did not cluster until all other clusters were merged at the higher thresholds. Colored dots next to the sample names correspond with those in Figure 2.
Boxes represent the interquartile range (IQR); vertical lines represent the full range of the data. Grey circles represent the time (y-axis) between T1 and T2 (left) and T1 and T3 (right). Colored circles indicate subjects in clusters A1/A1* (blue), A2/A2* (green), B1/B1* (red) and B2/B2* (pink). Labeled circles indicate subjects with time intervals below the IQR.
Dots indicate the sampling date (year on y-axis) for samples from T1 (blue), T2 (red) and T3 (green). Subject identifiers are given on the x-axis. Clustering patterns for env are noted below the subject ID as described in the legend. Subjects with any history of drug treatment during the study period are denoted in bold.
FUNDING SOURCES
Support was provided by the Division of Intramural Research, NIAID.
References
- 1.Leigh Brown AJ, Lycett SJ, Weinert L, Hughes GJ, Fearnhill E, Dunn DT, et al. Transmission network parameters estimated from HIV sequences for a nationwide epidemic. J Infect Dis 2011;204(9):1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wertheim JO, Oster AM, Hernandez AL, Saduvala N, Bañez Ocfemia MC, Hall HI. The International Dimension of the U.S. HIV Transmission Network and Onward Transmission of HIV Recently Imported into the United States. AIDS Res Hum Retroviruses 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wertheim JO, Leigh Brown AJ, Hepler NL, Mehta SR, Richman DD, Smith DM, et al. The global transmission network of HIV-1. J Infect Dis 2014;209(2):304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wertheim JO, Kosakovsky Pond SL, Forgione LA, Mehta SR, Murrell B, Shah S, et al. Social and Genetic Networks of HIV-1 Transmission in New York City. PLoS Pathog 2017;13(1):e1006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Vu S, Ratmann O, Delpech V, Brown A, Gill O, Tostevin A, et al. Comparison of cluster-based and source-attribution methods for estimating transmission risk using large HIV sequence databases. Epidemics 2018;23:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dennis AM, Herbeck JT, Brown AL, Kellam P, de Oliveira T, Pillay D, et al. Phylogenetic studies of transmission dynamics in generalized HIV epidemics: an essential tool where the burden is greatest? J Acquir Immune Defic Syndr 2014;67(2):181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grabowski MK, Herbeck JT, Poon AFY. Genetic Cluster Analysis for HIV Prevention. Curr HIV/AIDS Rep 2018;15(2):182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kosakovsky Pond SL, Weaver S, Leigh Brown AJ, Wertheim JO. HIV-TRACE (Transmission Cluster Engine): a tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol Biol Evol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norström MM, Prosperi MC, Gray RR, Karlsson AC, Salemi M. PhyloTempo: A Set of R Scripts for Assessing and Visualizing Temporal Clustering in Genealogies Inferred from Serially Sampled Viral Sequences. Evol Bioinform Online 2012;8:261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ragonnet-Cronin M, Hodcroft E, Hué S, Fearnhill E, Delpech V, Brown AJ, et al. Automated analysis of phylogenetic clusters. BMC Bioinformatics 2013;14:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibson KM, Steiner MC, Kassaye S, Maldarelli F, Grossman Z, Pérez-Losada M, et al. A 28-Year History of HIV-1 Drug Resistance and Transmission in Washington, DC. Front Microbiol 2019;10:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Impacts Poon A. and shortcomings of genetic clustering methods for infectious disease outbreaks. Virus Evol 2016;2(2):vew031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poon AF, Joy JB, Woods CK, Shurgold S, Colley G, Brumme CJ, et al. The impact of clinical, demographic and risk factors on rates of HIV transmission: a population-based phylogenetic analysis in British Columbia, Canada. J Infect Dis 2015;211(6):926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aldous JL, Pond SK, Poon A, Jain S, Qin H, Kahn JS, et al. Characterizing HIV transmission networks across the United States. Clin Infect Dis 2012;55(8):1135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ragonnet-Cronin M, Ofner-Agostini M, Merks H, Pilon R, Rekart M, Archibald CP, et al. Longitudinal phylogenetic surveillance identifies distinct patterns of cluster dynamics. J Acquir Immune Defic Syndr 2010;55(1):102–8. [DOI] [PubMed] [Google Scholar]
- 16.Lewis F, Hughes GJ, Rambaut A, Pozniak A, Leigh Brown AJ. Episodic sexual transmission of HIV revealed by molecular phylodynamics. PLoS Med 2008;5(3):e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rose R, Lamers SL, Dollar JJ, Grabowski MK, Hodcroft EB, Ragonnet-Cronin M, et al. Identifying Transmission Clusters with Cluster Picker and HIV-TRACE. AIDS Res Hum Retroviruses 2017;33(3):211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassan AS, Pybus OG, Sanders EJ, Albert J, Esbjörnsson J. Defining HIV-1 transmission clusters based on sequence data. AIDS 2017;31(9):1211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hightower GK, May SJ, Pérez-Santiago J, Pacold ME, Wagner GA, Little SJ, et al. HIV-1 clade B pol evolution following primary infection. PLoS One 2013;8(6):e68188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eshleman SH, Hudelson SE, Redd AD, Wang L, Debes R, Chen YQ, et al. Analysis of genetic linkage of HIV from couples enrolled in the HIV Prevention Trials Network 052 trial. J Infect Dis 2011;204(12):1918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alizon S, Fraser C. Within-host and between-host evolutionary rates across the HIV-1 genome. Retrovirology 2013;10:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maljkovic Berry I, Ribeiro R, Kothari M, Athreya G, Daniels M, Lee HY, et al. Unequal evolutionary rates in the human immunodeficiency virus type 1 (HIV-1) pandemic: the evolutionary rate of HIV-1 slows down when the epidemic rate increases. J Virol 2007;81(19):10625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konikoff J, Brookmeyer R, Longosz AF, Cousins MM, Celum C, Buchbinder SP, et al. Performance of a limiting-antigen avidity enzyme immunoassay for cross-sectional estimation of HIV incidence in the United States. PLoS One 2013;8(12):e82772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vlahov D, Anthony JC, Munoz A, Margolick J, Nelson KE, Celentano DD, et al. The ALIVE study, a longitudinal study of HIV-1 infection in intravenous drug users: description of methods and characteristics of participants. NIDA Res Monogr 1991;109:75–100. [PubMed] [Google Scholar]
- 25.Laeyendecker O, Brookmeyer R, Cousins MM, Mullis CE, Konikoff J, Donnell D, et al. HIV incidence determination in the United States: a multiassay approach. J Infect Dis 2013;207(2):232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Redd AD, Wendel SK, Longosz AF, Fogel JM, Dadabhai S, Kumwenda N, et al. Evaluation of postpartum HIV superinfection and mother-to-child transmission. AIDS 2015;29(12):1567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redd AD, Collinson-Streng A, Martens C, Ricklefs S, Mullis CE, Manucci J, et al. Identification of HIV superinfection in seroconcordant couples in Rakai, Uganda, by use of next-generation deep sequencing. J Clin Microbiol 2011;49(8):2859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26(19):2460–1. [DOI] [PubMed] [Google Scholar]
- 29.Lemey P, Rambaut A, Pybus O. HIV evolutionary dynamics within and among hosts. AIDS Rev 2006;8(3):125–40. [PubMed] [Google Scholar]
- 30.Raghwani J, Redd AD, Longosz AF, Wu CH, Serwadda D, Martens C, et al. Evolution of HIV-1 within untreated individuals and at the population scale in Uganda. PLoS Pathog 2018;14(7):e1007167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grabowski MK, Redd AD. Molecular tools for studying HIV transmission in sexual networks. Curr Opin HIV AIDS 2014;9(2):126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ratmann O, Grabowski MK, Hall M, Golubchik T, Wymant C, Abeler-Dörner L, et al. Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis. Nat Commun 2019;10(1):1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vrancken B, Rambaut A, Suchard MA, Drummond A, Baele G, Derdelinckx I, et al. The genealogical population dynamics of HIV-1 in a large transmission chain: bridging within and among host evolutionary rates. PLoS Comput Biol 2014;10(4):e1003505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson KM, Jair K, Castel AD, Bendall ML, Wilbourn B, Jordan JA, et al. A cross-sectional study to characterize local HIV-1 dynamics in Washington, DC using next-generation sequencing. Sci Rep 2020;10(1):1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redd AD, Doria-Rose NA, Weiner JA, Nason M, Seivers M, Schmidt SD, et al. Longitudinal Antibody Responses in People Who Inject Drugs Infected With Similar Human Immunodeficiency Virus Strains. J Infect Dis 2020;221(5):756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Subject IDs are shown, with one row per subject across all analyses. Columns contain the IDs for all subjects who clustered at the indicated distance threshold (top, 1% - 5%) for each time point/analysis: (A) T1; (B) T2; (C) T1; (D) T3. Black boxes indicate clustered subjects. Colored dots next to the sample names correspond with those in Figure 1.
Subject IDs are shown, with one row per subject across all analyses. Columns contain the IDs for all subjects who clustered at the indicated distance threshold (top, 1% - 5%) for each time point/analysis: (A) T1; (B) T2; (C) T1; (D) T3. Black boxes indicate clustered subjects. The light grey box indicates the Group A/A* and Group B/B* subjects. Subjects appearing in the white space between grey boxes did not cluster until all other clusters were merged at the higher thresholds. Colored dots next to the sample names correspond with those in Figure 2.
Boxes represent the interquartile range (IQR); vertical lines represent the full range of the data. Grey circles represent the time (y-axis) between T1 and T2 (left) and T1 and T3 (right). Colored circles indicate subjects in clusters A1/A1* (blue), A2/A2* (green), B1/B1* (red) and B2/B2* (pink). Labeled circles indicate subjects with time intervals below the IQR.
Dots indicate the sampling date (year on y-axis) for samples from T1 (blue), T2 (red) and T3 (green). Subject identifiers are given on the x-axis. Clustering patterns for env are noted below the subject ID as described in the legend. Subjects with any history of drug treatment during the study period are denoted in bold.