

Abstract
Background and Objectives
We sought to characterize the natural history and standard-of-care practices between the radiologic appearance of brain lesions, the appearance of lesional enhancement, and treatment with hematopoietic stem-cell transplant or gene therapy among boys diagnosed with presymptomatic childhood-onset cerebral adrenoleukodystrophy (CCALD).
Methods
We analyzed a multicenter, mixed retrospective/prospective cohort of patients diagnosed with presymptomatic CCALD (Neurologic Function Score = 0, Loes Score [LS] = 0.5–9.0, and age <13 years). Two time-to-event survival analyses were conducted: (1) time from CCALD lesion onset-to-lesional enhancement and (2) time from enhancement-to-treatment. The analysis was repeated in the subset of patients with (1) the earliest evidence of CCALD, defined as an MRI LS ≤ 1, and (2) patients diagnosed between 2016 and 2021.
Results
Seventy-one boys were diagnosed with presymptomatic cerebral lesions at a median age of 6.4 years [2.4–12.1] with a LS of 1.5 [0.5–9.0]. Fifty percent of patients had lesional enhancement at diagnosis. In the remaining 50%, the median Kaplan-Meier (KM)-estimate of time from diagnosis-to-lesional enhancement was 6.0 months (95% CI 3.6–17.8). The median KM-estimate of time from enhancement-to-treatment is 3.8 months (95% CI 2.8–5.9); 2 patients (4.2%) developed symptoms before treatment. Patients with a diagnostic LS ≤ 1 were younger (5.8 years [2.4–11.5]), had a time-to-enhancement of 4.7 months (95% CI 2.7–9.30), and were treated in 3.8 months (95% CI 3.1–7.1); no patients developed symptoms before treatment. Time from CCALD diagnosis-to-treatment decreased over the course of the study (ρ = −0.401, p = 0.003).
Discussion
Our findings offer a more refined understanding of the timing of lesion formation, enhancement, and treatment among boys with presymptomatic CCALD. These data offer benchmarks for standardizing clinical care and designing future clinical trials.
X-linked adrenoleukodystrophy (ALD) is a genetic disorder that results from mutations in the ABCD1 gene. Pathogenic variants lead to the impairment of peroxisomal beta-oxidation resulting in the elevation of very long chain fatty acids which affect the nervous system and adrenal cortex.1 Approximately one-third of men with ALD will develop a cerebral demyelinating lesion during childhood (CCALD). CCALD lesions typically originate in the corpus callosum as small T2 hyperintense lesions which enlarge concentrically over many months before eventually manifesting clinical symptoms.2-5 Contrast enhancement is often absent in early stages of lesion development, but its appearance is considered an ominous prognostic marker of lesion progression.6 Lesional contrast enhancement is the indication for treatment with hematopoietic stem-cell transplant (HSCT),6,7 including hematopoietic stem-cell gene therapy currently under investigation.8-10 HSCT effectively halts progression but entails substantial risks.11,12 Clinical outcomes are superior among patients who receive HSCT although lesions are still small, before the onset of neurologic symptoms.11-17 Taken together, the clinical goal of treating actively progressing lesions that are still small and presymptomatic implies a narrow “window of opportunity” for optimal HSCT deployment. Newborn screening for ALD aims to maximize this presymptomatic window of opportunity18-20 by monitoring prospectively for early CCALD lesions,21 but the boundaries of this treatment window remain poorly defined.12,13,17
Our aim in this study was to clarify several key aspects of the CCALD treatment window. First, we set out to define the natural history of lesion-onset more precisely. Although past reports of CCALD onset have included patients manifesting symptomatic (i.e., large) and presymptomatic (i.e., small) lesions,21 we restricted our analysis to patients presenting only during the presymptomatic stage. Second, among presymptomatic lesions that were not yet enhancing, we sought to measure time-to-enhancement. Finally, we sought to measure time from lesion enhancement-to-HSCT infusion. These findings offer benchmarks for establishing clinical care standards and designing clinical trials for CCALD prevention and treatment.
Methods
Participants
We analyzed a mixed retrospective/prospective cohort of boys diagnosed with presymptomatic CCALD followed at Massachusetts General Hospital, Amsterdam University Medical Center, Weill Cornell Medicine, Stanford Health Care, and the Kennedy Krieger Institute between January 1, 1987, and July 1, 2021, by either extended family screening or newborn screening. Inclusion criteria for the study were (1) confirmed diagnosis of ALD defined by the presence of elevated very long chain fatty acids and/or a pathogenic mutation in ABCD1; (2) diagnosis of “early-stage disease”8,12 defined as the development of a characteristic demyelinating lesion on T2-weighted MRI5,7 with an MRI severity score, or Loes Score (LS, range 0–34),22 of 0.5–9.0; (3) childhood onset defined as age <13 years at cerebral adrenoleukodystrophy (CALD) diagnosis; and (4) neurologically presymptomatic at CALD diagnosis as defined by an Neurologic Function Scale (NFS, range 0–25) score of 0.3 Patients diagnosed with arrested CALD were excluded given the difference in natural history, and patients with self-halted disease do not qualify for HSCT or gene therapy.23-25 Excluded patients in this age range had radiographically stable lesions, with no change in LS for ≥6 months, no evidence of enhancement at any time point, no change in NFS, and remained untreated (although continuously monitored by MRI) at the time of this study. Participation in the Lorenzo's Oil trial did not preclude participation in this study.26
Clinical and Imaging Data
Three primary end points were collected per participant from routine clinical care: date of CCALD diagnosis, date of first contrast-enhancing lesion, and date of treatment (Figure 1). Patient age, NFS, LS, and lesion pattern were collected at each date. Lesions were subdivided into 5 patterns according to their primary neuroanatomic distribution7: (1) parieto-occipital lobe white matter or splenium of the corpus callosum, (2) frontal lobe white matter or genu of the corpus callosum, (3) frontopontine or corticospinal projection fibers, (4) cerebellar white matter, and (5) global involvement. The presence (+) or absence (−) of lesional gadolinium contrast enhancement on T1 postcontrast MR imaging at each scan date was recorded. MRI scans were classified as enhancement negative if contrast was administered and no lesional enhancement was present or a noncontrast MRI was performed at that time point. Treatment was defined as the date the patient underwent HSCT or gene therapy.
Figure 1. Visualization of Main Study Questions.
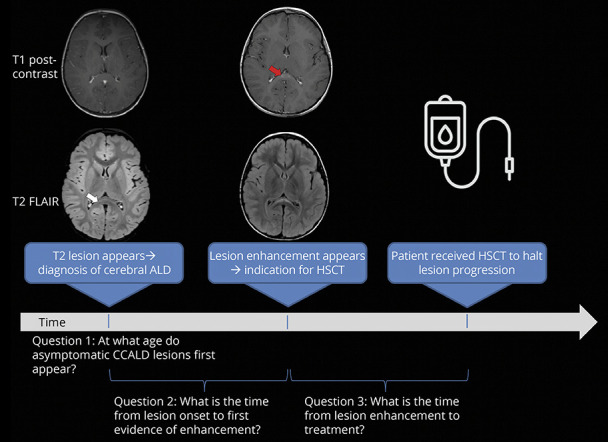
Illustration of the 3 primary analyses performed in this study: (1) age presymptomatic patients manifesting cerebral lesions, (2) measurement of time from diagnosis-to-enhancement, and (3) measurement of time from lesion enhancement-to-treatment.
Statistical Methods
Clinical and Imaging Characteristics
Continuous variables are reported as median and range. Categorical variables are reported as percentages. The unpaired Mann Whitney U test was used to compare the LS at diagnosis with the LS at first evidence of lesional enhancement, and LS between lesional enhancement and treatment. To quantify the potential relationships with age and LS at diagnosis and the emergence of cerebral inflammation, the Spearman rank correlation coefficient (ρ) was calculated between age of CCALD diagnosis and LS at diagnosis and time-to-lesional enhancement, respectively. Two-tailed tests with significance level of 0.5 were used to determine statistical significance. All data were annotated, analyzed, and visualized using RStudio (v.1.2.5033).
Primary Outcomes
Kaplan-Meier event-free survival was calculated from diagnosis of CCALD to the 2 major study end points: time from CCALD diagnosis-to-lesional enhancement and time from lesional enhancement-to-treatment (Figure 1). The results are plotted as cumulative incidence curves (1–Kaplan-Meier [KM] survival probability) and reported as median event-free survival and cumulative incidence ([1–KM survival probability] × 100%) of each outcome with 95% CIs.
Subgroup Analyses
To approximate the natural history of early lesions detected by newborn screening, the analysis was repeated in the subset of patients with a diagnostic LS ≤ 1.0.
Finally, we hypothesized that treatment performance has improved over the duration of the study, likely because of improved MR scanner strength and resolution (i.e., 1.5T vs 3.0T), disease awareness, and newborn screening. To test this hypothesis, the Spearman rank correlation coefficient (ρ) was calculated between year of CCALD diagnosis and the time interval from diagnosis to treatment. A time-to-treatment analysis was performed on the subset of patients diagnosed with CCALD within the past 5 years (January 1, 2016, to July 1, 2021).
Standard Protocol Approvals, Registrations, and Patient Consents
Patients were identified through each institution's respective institutional review board (IRB)-approved clinical research protocol (IRB Protocols: WCM 19-08020583, Stanford IRB 59014, JHMI IRB00105090, AUMC METC 2014_347, MGH 2012P000132). Participant data were reviewed, deidentified, and storage was encrypted and password protected at each site. Deidentified data were then transferred to Weill Cornell Medicine through interinstitutional data use agreements. Owing to anonymization, consent was waived.
Data Availability
After publication, any data not published within this article will be anonymized and shared by request from any qualified investigator.
Results
Diagnosis of Presymptomatic Childhood Cerebral Adrenoleukodystrophy
We identified 71 patients who were presymptomatic at the time of their first abnormal MRI. The median age was 6.4 years (2.4–12.1) (Figure 2) with an initial LS of 1.5 (0.5–9.0). There is a weak relationship between LS and age at diagnosis (ρ = 0.340, p = 0.004). Splenial lesions were most common at diagnosis (pattern 1, 69.0%), followed by internal capsule and brainstem involvement (pattern 3, 14.1%), frontal lesions (pattern 2, 9.9%), combined splenial-internal capsular lesions (pattern 1, 3, 5.6%), and one patient with combined genu, splenium, and internal capsular involvement (pattern 1, 2, 3, 1.4%).
Figure 2. Age Distribution of Presymptomatic CCALD Diagnosis.
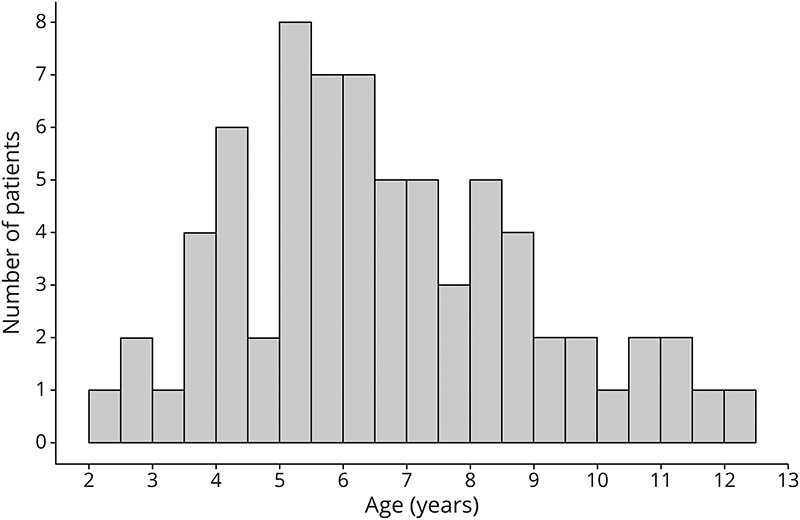
Median age of 71 patients diagnosed with CCALD before symptom onset (NFS = 0) was 6.4 years (2.4–12.1) with an initial LS of 1.5 (0.5–9.0). CCALD = childhood cerebral adrenoleukodystrophy; LS = Loes Score; NFS = Neurologic Function Scale.
Time From Presymptomatic CCALD Diagnosis-to-Lesion Enhancement
There is no relationship between age of diagnosis and time from diagnosis-to-lesional enhancement (ρ = 0.033, p = 0.822). There is no significant difference in LS between CCALD diagnosis and lesional enhancement. Imaging data were available for 44 patients, all of whom (100%) were presymptomatic at detection of enhancement (Table). Imaging data were not available for 27 patients at this time point.
Table.
Clinical and Imaging Scores per End Point

Two subgroups emerged in the analysis of time from CCALD diagnosis-to-lesional enhancement. Twenty-four patients had contrast-enhancing lesions at diagnosis (“immediate enhancement” subgroup). Twenty of 44 patients developed lesional enhancement after initial diagnosis, constituting the “delayed enhancement” subgroup. The median KM-estimate of time from diagnosis-to-lesional enhancement in the delayed enhancement cohort was 6.0 months (95% CI 3.6–17.8) (Figure 3). 81.5% (95% CI 53.0–92.7) of patients developed evidence of lesional inflammation by 18 months.
Figure 3. Time From CCALD Diagnosis-to-Enhancement.
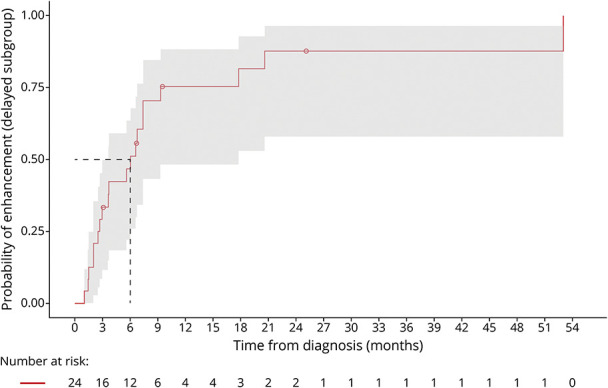
The median KM-estimate of time from diagnosis-to-lesional enhancement in the delayed enhancement cohort is 6.0 months (95% CI 3.6–17.8). 81.5% (95% CI 53.0–92.7) of patients developed enhancement by 18 months. All patients remained presymptomatic.
Notably, lesional enhancement “flickered on” in 3 patients; an early MRI revealed a gadolinium-enhancing lesion, followed by an MRI without lesional enhancement, which on continued surveillance imaging again displayed positive enhancement 4.3–12.1 months from the initial enhancing scan.
Time From Lesional Enhancement-to-Treatment
The LS increased from detection of enhancement to treatment (median LS = 2.5 [0.5–11.0], p = 0.044). Forty-six of the 48 patients (95.8%) with available clinical data remained presymptomatic at treatment (Table). Two patients became symptomatic, each with an NFS = 2 at treatment: (1) a 3-year-old boy diagnosed with an initial LS = 4.0, mixed pattern 1 and 3 lesion distribution, with rapidly progressive disease who was transplanted 5 weeks after diagnosis and (2) a boy diagnosed at 7.2 years with a LS = 2.0 who was treated 26.1 months later after his parieto-occipital lesion expanded to a LS = 9.0.
Paired enhancement-to-treatment data were available for 43 patients. The median KM-estimate of time from enhancement-to-treatment is 3.8 months (95% CI 2.8–5.9). All patients were treated by 14.9 months (Figure 4).
Figure 4. Time From Enhancement-to-Treatment.
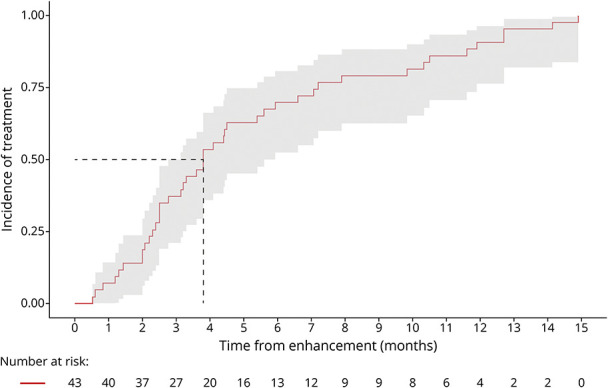
Paired enhancement-to-treatment data were available for 43 patients. The median KM-estimate of time from enhancement-to-treatment is 3.8 months (95% CI 2.8–5.9); all patients were treated by 14.9 months; and 95.8% of patients remained presymptomatic at treatment.
Overall treatment data were available for 54 patients. The median KM-estimate of time from initial CCALD diagnosis-to-treatment is 7.7 months (95% CI 5.8–11.9 months). One-year and 2-year treatment incidences from initial CCALD diagnosis were 66.7% (95% CI 51.4–77.1) and 90.7% (95% CI 78.7–96.0), respectively. Treatment data were not available for 17 patients at this time point.
Subgroup Analyses
Diagnostic LS Less Than or Equal to 1.0
Thirty-five patients had a diagnostic LS ≤ 1.0. The median age was 5.8 years (2.4–11.5), and all were presymptomatic at diagnosis.
Sixteen of 28 patients with available data were in the delayed enhancement subgroup. The median KM-estimate of time from diagnosis-to-lesional enhancement is 4.7 months (95% CI 2.7–9.3) (Figure 5). 93.7% (95% CI 58.3–99.1) of patients developed enhancement by 18 months. LS at lesional enhancement was 1.0 (0.5–4), and all patients remained presymptomatic.
Figure 5. Diagnostic LS < 1.0: Time From CCALD Diagnosis-to-Enhancement.
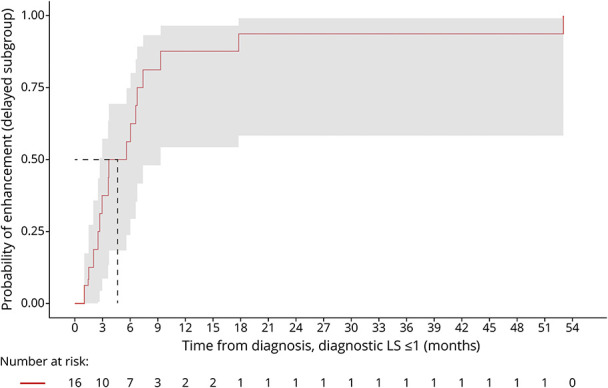
The median KM-estimate of time from diagnosis-to-lesional enhancement in the delayed enhancement cohort is 4.7 months (95% CI 2.7–9.3). 93.7% (95% CI 58.3–99.1) of patients developed enhancement by 18 months. All patients remained presymptomatic. CCALD = childhood cerebral adrenoleukodystrophy.
At treatment, all patients (100%) with available clinical data (n = 30) were presymptomatic, with a median LS of 1.5 (0.5–9.0). Paired enhancement-to-treatment data were available for 28 patients. The median KM-estimate of time from enhancement-to-treatment is 3.8 months (95% CI 3.1–7.1). All patients were treated by 12.7 months (Figure 6).
Figure 6. Diagnostic LS ≤ 1.0: Time From Enhancement-to-Treatment.
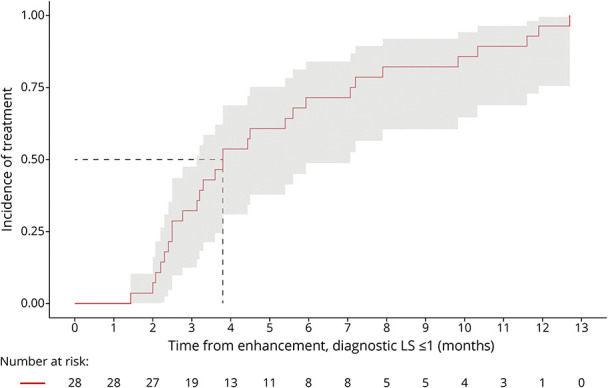
Paired enhancement-to-treatment data were available for 28 patients. The median KM-estimate of time from enhancement-to-treatment is 3.8 months (95% CI 3.1–7.1). All patients were treated by 12.7 months. All patients remained presymptomatic.
Treatment Performance Over Time
There is an inverse relationship between the year the patients were diagnosed with CCALD (1987–2021) and time from CCALD diagnosis-to-treatment (ρ = −0.401, p = 0.003). Overall, patients diagnosed with CCALD within the past 5 years were treated with HSCT or gene therapy in a median of 5.8 months (95% CI 3.5–9.7, n = 23) from initial diagnosis. The incidence of treatment at 12 months is 91.3% (95% CI 67.3–97.7). Twenty-two (95.7%) patients remained asymptomatic at treatment (NFS = 0 [0–2]).
Discussion
Childhood cerebral ALD lesions begin small but develop gadolinium enhancement as they enlarge and eventually manifest neurologic symptoms.5 Treatment with HSCT or gene therapy is reserved for actively enlarging lesions, which are demarcated by gadolinium enhancement. A delay in treatment initiation allows the lesion to progress further, yielding poorer clinical outcomes.8,12,17,27 Understanding the onset, progression, and time to treatment is necessary for optimizing care and designing clinical trials. In our cohort of 71 boys with CCALD, we find that lesions appeared at a median age of 6.4 years, but as early as 2 years of age. Half of the patients displayed lesional enhancement at diagnosis. The other half developed lesional enhancement on continued contrast-enhanced surveillance imaging in 6 months. The time from enhancement-to-treatment was 3.8 months for the whole cohort. Only 2 patients (4.2%) developed symptoms before treatment had begun.
Our previous analysis of mixed presymptomatic and early symptomatic patients suggested a median age of CCALD onset of 7.0.21 The younger median age in this study was expected because we restricted our analysis to presymptomatic lesions only. Therefore, the results likely offer a more accurate indication of disease onset, specifically 7 months earlier than the previously published estimate. Furthermore, we aimed to approximate the newborn screening experience by restricting the analysis to presymptomatic patients diagnosed with the earliest lesions (LS ≤ 1.0). In these cases, the median age of diagnosis was 5.8 years, 1.2 years earlier than the patient cohort in our previous work.
In patients who are diagnosed with an initial nonenhancing CCALD lesion, physicians and families should expect the emergence of lesional enhancement in 50% of patients by 4.7 months if the diagnostic LS was 1.0 or less and by 6.0 months overall. Lesional enhancement is to be expected in 81.5% of patients by 18 months. This supports the recommendation that patients with an identified cerebral lesion should, in parallel to continued contrast-enhanced MRI monitoring, be evaluated and prepared for HSCT or gene therapy to minimize treatment delivery time once lesional enhancement is present.
Importantly, physicians should be aware that enhancement may “flicker on” before developing sustained perilesional inflammation. This was a novel observation in 3 patients in this study captured by close interval follow-up imaging or pretransplant MRI. This radiographic phenomenon has been demonstrated in adults with arrested CALD who developed temporary lesional enhancement, step-wise disease progression, followed by spontaneous remission of contrast enhancement and disease restabilization.23,28 A similar observation has been noted in the residual enhancement present on MRI in some patients with CCALD who have received gene therapy.29 The phenomenon of “flickering” lesions bears further study because it may represent oscillations around immune and metabolic homeostasis that could shed new insights on disease pathogenesis and treatment.
Our data suggest that CCALD onset can occur at any time between age 2 and 12 years; capturing presymptomatic CCALD does not necessarily mean capturing patients at a younger age. The lack of a significant mathematical relationship between diagnostic age, LS at diagnosis, and time-to-detection of lesional enhancement further supports this notion. For lesion distribution, projection fiber involvement was observed at a higher rate than in previously published studies,5,7 appearing in 21.1% of all lesions in this cohort. The degree of internal capsular involvement is important to consider at diagnosis because the neurologic examination is sensitive to long-tract pathology, and it may frame clinical expectations given affected patients may be at higher risk for disease progression and posttransplant symptom development, as seen in the 3-year-old patient who became symptomatic in this study.30,31
Akin to the concept of “door-to-needle time,” patients with the earliest evidence of actively inflammatory CCALD should aim to be treated within 4 months from the onset of lesional enhancement, with the welcome expectation that nearly all patients will remain asymptomatic. Neurocognitive outcomes13,16 can be maximized if patients are treated with “very low severity” MRI scores (before the MRI exceeds a LS of 2.0). This was observed in the subanalysis of patients with a diagnostic LS ≤ 1.0, all of whom were treated presymptomatically with a median LS of 1.5.
There are features of CCALD that are patient-specific and not captured by group-level analyses. The rate of disease progression, onset of symptoms, and lesional distribution in the 2 patients who became symptomatic at treatment illustrate this interpatient variability. One boy developed enhancing cerebral disease at age 3 years, with splenial and internal capsular involvement, the latter of which was likely the cause of his hyperreflexia and running difficulty at the time of treatment 5 weeks later. Conversely, while the 26-month time interval from presymptomatic diagnosis to symptomatic treatment of the second patient affected by an isolated pattern 1 lesion indicates less overall disease burden and slower progression, it does highlight the danger in delayed treatment.
Ninety-one percent of patients diagnosed with progressive CCALD within the past 5 years were treated within 1 year. If follow-up contrast-enhanced MRI surveillance continues to reveal nonenhancing cerebral disease with no changes in the LS for more than a year to 18 months from the initial abnormal MRI, especially in an older child (older than 8 years), a diagnosis of arrested CCALD should be considered in the differential diagnosis, serial contrast-enhanced MRI monitoring should continue every 3 months, and treatment should be deferred.6,23 It is imperative that physicians do not prematurely diagnose the first signs of early, nonenhancing, demyelination as self-halted CCALD; patients in the delayed enhancement subgroup may initially meet arrested criteria by default. All early-stage lesions must be monitored every 3 months by MRI while the disease declares itself, and the differential diagnosis should only broaden to include spontaneous CCALD remission after a patient is outside of the expected 12–24 months expected treatment window. Novel, advanced imaging techniques, including MR perfusion and lesion volumetrics, may help to parse out actively progressing vs self-stabilizing lesions at these earliest time points.32-34
Important limitations apply to this study. Although the data are semilongitudinal, not all clinical and imaging variables were available at each time point, most often because of loss to follow-up or unavailability of historical medical records. Because of missing data, we opted for the more statistically conservative approach; we analyzed each of the end points as independent, nonpaired events. The incomplete availability of paired data limited our ability to calculate more robust survival estimates. Some boys early in the study (before 2005) participated in the Lorenzo's Oil (LO) trial. The results suggested that asymptomatic patients treated preventatively with LO may have altered the incidence of CCALD, possibly decreasing the number of patients available for this study. LO has not been shown to alter the course of disease once a cerebral lesion develops. In addition, the results have not been replicated, and the study was not placebo-controlled.26 The time-to-enhancement may be overestimated as not all initial scans were administered with gadolinium. The minimal amount of deidentified data required to perform the planned analyses were collected to promote feasibility of the study while protecting patient confidentiality. The data did not include annotation of which early MRI scans were contrast vs non–contrast-enhanced, thus leading to a potential overestimation to the time-to-enhancement estimate in the delayed enhancement cohort. The potential overestimation of this window may be favorable given all patients remained presymptomatic at this end point. It is important to note that MR sensitivity and overall treatment performance has improved over the course of this study, the effect of which we accounted for by analyzing the subset of patients diagnosed since 2016. Historically (before 2000), time-to-treatment was longer in presymptomatic patients because of the high mortality of HSCT, which introduced significant pause on the part of the families deciding to move forward with the procedure. Collectively, these limitations highlight the need for these results to be validated by analyzing other independent ALD cohorts. The presence of missing data also underscores the importance of a prospective ALD natural history study to more clearly define the longitudinal timing of early-stage CCALD onset, progression, divergence to arrested disease, clinical symptomatology, and treatment.
The results of this study provide insight into the presymptomatic "window of opportunity" by clarifying the timing of CCALD lesion development, and by quantifying the time frame from the detection of perilesional inflammation-to-treatment of boys diagnosed with presymptomatic CCALD. Our data will help optimize MRI surveillance, the timing and clinical expectations for treatment, and has the potential to frame preventative and early treatment trials for this devastating disease of childhood.
Acknowledgment
We thank Jordan Goodman and Kerry Gao for providing additional help collecting participant data.
Glossary
- ALD
X-linked adrenoleukodystrophy
- CALD
cerebral adrenoleukodystrophy
- CCALD
childhood cerebral adrenoleukodystrophy
- HSCT
hematopoietic stem-cell transplant
- IRB
institutional review board
- KM
Kaplan-Meier
- LO
Lorenzo's Oil
- LS
Loes Score
- NFS
Neurologic Function Scale
Appendix. Authors
Study Funding
Funding was received from the NIH for activities related to this project (E.J.M. K12NS066274, K23NS118044; A.F. P50HD103538, U54NS115052; F.S.E. U54NS115052). A portion of the subject monitoring was funded in part by the Johns Hopkins University School of Medicine General Clinical Research Center Grant M01-RR00052 from the National Center for Research Resources/NIH; Grants HD10981 and HD39276 from the NIH; Grant 685–008 from the Office of Orphan Drug Products of the Food and Drug Administration.
Disclosure
E.J. Mallack receives research support from the National Institute of Neurological Disorders and Stroke (K12NS066274, K23NS118044), serves on a DSMB for Lysogene, has institutional contracts with Passage Bio and Viking Therapeutics, and received support for leukodystrophy education from Prime, PCORI, Orchard Therapeutics, and Neurology Alert. K. Van Haren receives research support from Bluebird Bio, Minoryx, and is a scientific consultant for Poxel and Viking Therapeutics. A. Torrey reports no disclosures relevant to the manuscript. S. van de Stadt reports no disclosures relevant to the manuscript. M. Engelen receives research support from the Dutch Research Council (NWO; Vidi grant 016.196.310), Minoryx, Vertex, Autobahn Therapeutics, and SwanBio. G. Raymond is a consultant for Minoryx (Barcelona), Viking, and Bluebird Bio (Cambridge, MA). He serves on the DSMBs for Ionis and Retrophin. A. Fatemi receives research support from the NICHD (P50HD103538), National Institute of Neurological Disorders and Stroke/NCATS (U54NS115052), NCATS (UL1TR003098), NCATS (U01NS103882), Institutional Contract with Minoryx, Viking Therapeutics, Autobahn Therapeutics, Poxel, Personal Contract with Bluebird Bio (DSMB member). F.S. Eichler receives research support from FDA Orphan Disease Group (R01FD004127), National Institute of Neurological Disorders and Stroke (R01NS072446, R01NS082331,U54NS115052), Retrophin, Minoryx, Bluebird Bio, AGTC, and SwanBio. Go to Neurology.org/N for full disclosures.
References
- 1.Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120(pt 8):1485-1508. [DOI] [PubMed] [Google Scholar]
- 2.Huffnagel IC, Laheji FK, Aziz-Bose R, et al. The natural history of adrenal insufficiency in X-linked adrenoleukodystrophy: an international collaboration. J Clin Endocrinol Metab. 2019;104:118-126. [DOI] [PubMed] [Google Scholar]
- 3.Moser HW, Loes DJ, Melhem ER, et al. X-linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality. A study involving 372 patients. Neuropediatrics. 2000;31(5):227-239. [DOI] [PubMed] [Google Scholar]
- 4.Van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol. 2001;49:186-194. [DOI] [PubMed] [Google Scholar]
- 5.Liberato AP, Mallack EJ, Aziz-Bose R, et al. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy. Neurology. 2019;92(15):e1698–e1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melhem ER, Loes DJ, Georgiades CS, Raymond GV, Moser HW. X-linked adrenoleukodystrophy: the role of contrast-enhanced MR imaging in predicting disease progression. AJNR Am J Neuroradiol. 2000;21(5):839-844. [PMC free article] [PubMed] [Google Scholar]
- 7.Loes DJ, Fatemi A, Melhem ER, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology. 2003;61(3):369-374. [DOI] [PubMed] [Google Scholar]
- 8.Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377(17):1630-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mallack EJ, Turk B, Yan H, Eichler FS. The landscape of hematopoietic stem cell transplant and gene therapy for X-linked adrenoleukodystrophy. Curr Treat Options Neurol. 2019;21(12):61. [DOI] [PubMed] [Google Scholar]
- 10.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326(5954):818-823. [DOI] [PubMed] [Google Scholar]
- 11.Moser HW. Adrenoleukodystrophy–phenotypic variability and implications for therapy. J Inherit Metab Dis. 1992;15(4):918. [DOI] [PubMed] [Google Scholar]
- 12.Raymond GV, Aubourg P, Paker A, et al. Survival and functional outcomes in boys with cerebral adrenoleukodystrophy with and without hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2019;25:538-548. [DOI] [PubMed] [Google Scholar]
- 13.Pierpont EI, Eisengart JB, Shanley R, et al. Neurocognitive trajectory of boys who received a hematopoietic stem cell transplant at an early stage of childhood cerebral adrenoleukodystrophy. JAMA Neurol. 2017;74(6):710-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118(7):1971-1978. [DOI] [PubMed] [Google Scholar]
- 15.Miller W, Askin G, vad de Stadt S, et al. Hematopoietic cell transplantation (HCT) for adrenoleukodystrophy: ten year experience with 62 patients. Ann Neurol. 2010;68:S121. [Google Scholar]
- 16.Pierpont EI, Nascene DR, Shanley R, et al. Neurocognitive benchmarks following transplant for emerging cerebral adrenoleukodystrophy. Neurology. 2020;95(5):e591–e600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. 2007;6(8):687-692. [DOI] [PubMed] [Google Scholar]
- 18.Raymond GV, Jones RO, Moser AB. Newborn screening for adrenoleukodystrophy: implications for therapy. Mol Diagn Ther. 2007;11(6):381-384. [DOI] [PubMed] [Google Scholar]
- 19.Moser AB, Fatemi A. Newborn screening and emerging therapies for X-linked adrenoleukodystrophy. JAMA Neurol. 2018;75(10):1175-1176. [DOI] [PubMed] [Google Scholar]
- 20.Moser AB, Jones RO, Hubbard WC, et al. Newborn screening for X-Linked adrenoleukodystrophy. Int J Neonatal Screen. 2016;2(4):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallack EJ, Turk BR, Yan H, et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: meta-analysis and consensus guidelines. J Inherit Metab Dis. 2021;44(3):728-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15(9):1761-1766. [PMC free article] [PubMed] [Google Scholar]
- 23.Mallack EJ, van de Stadt S, Caruso PA, et al. Clinical and radiographic course of arrested cerebral adrenoleukodystrophy. Neurology. 2020;94(24):e2499–e2507. doi: 10.1212/WNL.0000000000009626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin JE, Armour EA, Heshmati A, et al. Pearls & Oy-sters: adolescent-onset adrenomyeloneuropathy and arrested cerebral adrenoleukodystrophy. Neurology. 2019;93(2):81-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korenke GC, Pouwels PJ, Frahm J, et al. Arrested cerebral adrenoleukodystrophy: a clinical and proton magnetic resonance spectroscopy study in three patients. Pediatr Neurol. 1996;15(2):103-107. [DOI] [PubMed] [Google Scholar]
- 26.Moser HW, Raymond GV, Lu SE, et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo's oil. Arch Neurol. 2005;62(7):1073-1080. [DOI] [PubMed] [Google Scholar]
- 27.Miller WP, Srinivasan S, Panoskaltsis-Mortari A, et al. GVHD after haploidentical transplantation: a novel, MHC-defined rhesus macaque model identifies CD28- CD8+ T cells as a reservoir of breakthrough T-cell proliferation during costimulation blockade and sirolimus-based immunosuppression. Blood. 2010;116(24):5403-5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlson AM, Huffnagel IC, Verrips A, van der Knaap MS, Engelen M, Van Haren K. Five men with arresting and relapsing cerebral adrenoleukodystrophy. J Neurol. 2021;268(3):936-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eichler F, Duncan C, Orchard P, et al. Disease stabilization following treatment with elivaldogene autotemcel (eli-cel, lenti-D) gene therapy for the treatment of cerebral adrenoleukodystrophy: interim results from phase 2/3 (ALD-102) and phase 3 (ALD-104) studies (2064). Neurology. 2021;96. [Google Scholar]
- 30.Kühl JS, Suarez F, Gillett GT, et al. Long-term outcomes of allogeneic haematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Brain. 2017;140(4):953-966. [DOI] [PubMed] [Google Scholar]
- 31.Eichler F, Mahmood A, Loes D, et al. Magnetic resonance imaging detection of lesion progression in adult patients with X-linked adrenoleukodystrophy. Arch Neurol. 2007;64(5):659-664. [DOI] [PubMed] [Google Scholar]
- 32.Musolino PL, Rapalino O, Caruso P, Caviness VS, Eichler FS. Hypoperfusion predicts lesion progression in cerebral X-linked adrenoleukodystrophy. Brain. 2012;135(pt 9):2676-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lauer A, et al. DSC MR Perfusion detects white matter microvascular perfusion abnormalities in X-linked adrenoleukodystrophy. J Cereb Blood Flow Metab. 2017;37:47-48. [Google Scholar]
- 34.Mallack EJ, Askin G, van de Stadt S, et al. A longitudinal analysis of early lesion growth in presymptomatic patients with cerebral adrenoleukodystrophy. AJNR Am J Neuroradiol. 2021;42(10):1904-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
After publication, any data not published within this article will be anonymized and shared by request from any qualified investigator.