

Abstract
Development of tyrosine kinase inhibitors (TKIs) targeting the BCR-ABL oncogene constitutes an effective approach for the treatment of chronic myeloid leukemia (CML) and/or acute lymphoblastic leukemia. However, currently available inhibitors are limited by drug resistance and toxicity. Ponatinib, a third-generation inhibitor, has demonstrated excellent efficacy against both wild type and mutant BCR-ABL kinase, including the “gatekeeper” T315I mutation that is resistant to all other currently available TKIs. However, it is one of the most cardiotoxic of the FDA-approved TKIs. Herein, we report the structure-guided design of a novel series of potent BCR-ABL inhibitors, particularly for the T315I mutation. Our drug design paradigm was coupled to iPSC–cardiomyocyte models. Systematic structure–activity relationship studies identified two compounds, 33a and 36a, that significantly inhibit the kinase activity of both native BCR-ABL and the T315I mutant. We have identified the most cardiac-safe TKIs reported to date, and they may be used to effectively treat CML patients with the T315I mutation.
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that accounts for approximately 15% of newly diagnosed leukemia cases in adults, and it is estimated that 61 090 new leukemia cases will be diagnosed in the United States in 2021.1 The fusion protein product of the Philadelphia chromosome (Ph), BCR-ABL,2−6 is associated with CML and a subset acute lymphoblastic leukemia (Ph+ ALL); therefore, the development of TKIs targeting the BCR-ABL oncogene constitutes an effective approach for the treatment of CML and/or ALL. Imatinib [Gleevec, ST1571 (Figure 1)], a first-line drug for patients diagnosed with CML, inhibits the activity of the BCR-ABL kinase protein. The clinical success of imatinib paved the way to consider kinases as druggable targets.7−10 However, despite its durable initial response in most of the CML patients, imatinib fails in ≤40% of patients due to the intolerance of the dose and drug resistance. Mutations within the kinase domain of BCR-ABL constitute the most frequent mechanism of drug resistance,11−14 as they cause ineffective binding of the inhibitor to the target.15 To date, more than 100 different point mutations have been identified in CML patients. Resistance to imatinib prompted the development of second-generation inhibitors, including nilotinib (Tasigna, AMN107), the multitarget kinase inhibitor dasatinib (Sprycel, BMS-354825), and bosutinib (Bosulif, SKI-606) (Figure 1), that were approved for second-line use.13,16,17 Although these second-generation inhibitors demonstrated superior potency over imatinib, they fail to inhibit many imatinib-resistant mutations,18−20 including the T315I “gatekeeper” mutation [replacement of threonine (Thr) with isoleucine (Ile) at position 315 in the ABL1 kinase domain] that occurs in >20% of CML patients.15,21,22 When Thr315 is mutated to Ile, its bulkier side chain protrudes into the enzyme active site and prevents imatinib and the second-generation inhibitors from entering and binding to the ATP-binding pocket. Consequently, the first- and second-generation inhibitors are ineffective against the tumors carrying T315I mutant BCR-ABL.17,23,24 Notably, several of these active site BCR-ABL inhibitors cause serious adverse side effects in patients. These include increased risks of vascular events for nilotinib,25 pulmonary hypertension and myelosuppression for dasatinib, and increased ALT and AST levels for bosutinib.13,26 There have been several attempts to develop inhibitors effective against the T315I mutant kinase; however, clinical development of these compounds has been halted due to toxicity concerns.21,27 In 2012, the FDA approved the third-generation multikinase inhibitor ponatinib (Figure 1) with a broad label as a second-line treatment option for the patients with CML and Ph+ ALL.28,29 It was shown to be most potent inhibitor among the TKIs that target BCR-ABL and has demonstrated excellent activity against T315I mutant clones.30−32 However, soon after its approval, it was found to cause serious vascular adverse events, and therefore, its use has been restricted for the treatment of tumors carrying the T315I mutant kinase. Ponatinib remains the only approved treatment option for the patients with the T315I mutation.33 Unfortunately, it is among the most cardiotoxic of all of the FDA-approved TKIs.34 Its cardiotoxicity is thought to be due to concurrent inhibition of kinases that are important for cardiovascular function,35 which are likely due to off-target effects caused by its binding to structurally similar ATP pockets.35−37 Therefore, the development of a new TKI that works against the T315I mutation with improved safety to meet clinical needs is warranted. Several approaches have been reported to address the challenges associated with ponatinib. In most of these studies, the new inhibitor showed efficacies similar to that of ponatinib on native protein kinase but lacked a desired effect on the T315I mutant protein kinase.38,39
Figure 1.

Chemical structures of the FDA-approved BCR-ABL inhibitors.
We envisioned that if a TKI were to be effective against both native and T315I mutant BCR-ABL, and highly cardiac-safe compared to ponatinib, it would gain a broader scope of utilization and provide much needed relief to the CML and Ph+ ALL patients with T315I mutations. Considering the cardiac-safe nature of imatinib and nilotinib as compared to ponatinib,26,34 we hypothesized that it should be possible to discover a cardiac-safe BCR-ABL inhibitor by modifying the structure of existing BCR-ABL inhibitors. We reasoned that H bond interactions between the TKIs and the Met318 residue in BCR-ABL are essential for the inhibitor to show efficacies against BCR-ABL. Therefore, re-engineering the core structure of each TKI responsible for H bond interaction with Met318 and studying the structure–activity relationship (SAR) for efficacies and cardiac safety would yield potential drugs that are more broadly applicable in the clinic.
We combined our drug design paradigm with iPSC-CM and vascular endothelial cell models to predict the cardiotoxicities of the new analogues. As expected, the newly designed inhibitors exhibited similar efficacies as benchmark FDA drugs against the K-562 cell line, a BCR-ABL positive CML tumor cell line. In addition, they also show excellent efficacies against K-562 cells engineered to express BCR-ABLT315I. Using the iPSC-CM cardiomyocyte contractility assays, we initially screened all new analogues and identified cardiotoxic cores. As a result, we finally identified cardiac-safe cores and determined the SAR around the core for efficacies against both native and T315I mutant cell lines, while maintaining cardiac safety. Subsequently, structural modifications led to the discovery of inhibitors 33a and 36a (Figure 2), which have significantly improved cardiac safety over ponatinib, inhibited the kinase activity of BCR-ABLT315I, and blocked the proliferation of K-562 CML cells carrying the mutated kinase.
Figure 2.
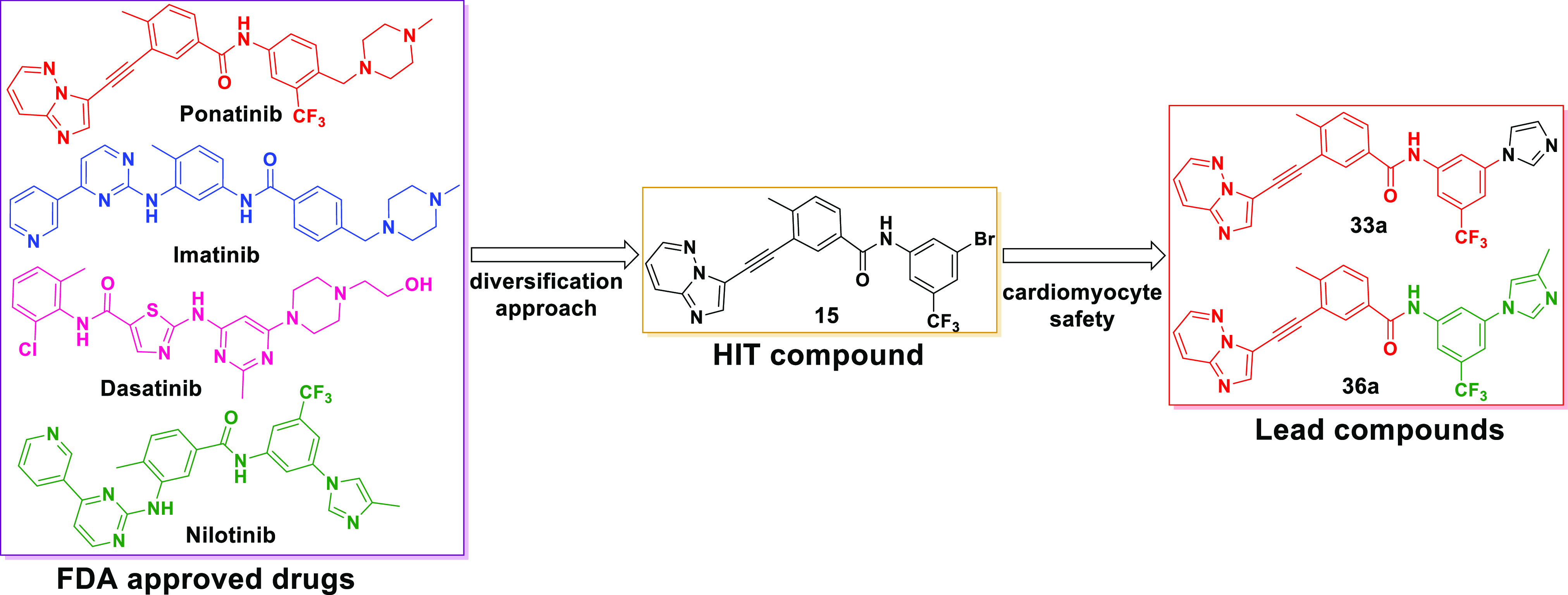
Design of the novel BCR-ABL inhibitors using a diversification strategy.
Results and Discussion
Molecular Design and Computational Studies
Figure 1 shows the FDA-approved BCR-ABL inhibitors. An important aspect of their activity is the interaction with the kinase through specific hydrogen bonding. These inhibitors form H bond interactions with the backbone of Met318 in the hinge region of native BCR-ABL. In addition, inhibitors such as imatinib, dasatinib, and nilotinib make a key hydrogen bond to the side chain of the gatekeeper residue Thr315.17,24,40 The formation of a H bond is critical for the activity of these inhibitors. Therefore, if the gatekeeper residue is mutated to Ile (T315I), this H bond is lost. Steric clashes of the bulkier Ile residue are posited to block entry of the inhibitor into the hydrophobic pocket, resulting in the loss of the H bond interaction with Met318 and efficacy against the T315I mutation. Similar steric hindrance of Ile was also observed for bosutinib. Consequently, the only H bond that bosutinib makes with Met318 in native BCR-ABL is inhibited when Thr at position 315 of BCR-ABL is mutated to Ile, and therefore, it is inactive.41 In contrast, ponatinib does not make H bond interactions with Thr315 in native BCR-ABL but makes H bond interactions with Met318 with both native and T315I mutant BCR-ABL kinase (Figure 3c,d), inhibiting both native BCR-ABL and BCR-ABLT315I kinases,30 explaining its unique efficacy against tumors carrying the T315I mutation.33
Figure 3.

Potential binding modes of ponatinib and lead inhibitors 33a and 36a with BCR-ABLWT (PDB entry 3OXZ) and BCR-ABLT315I (PDB entry 3IK3) proteins. (a and b) Binding interactions of inhibitors 33a and 36a, respectively. (c and d) Ponatinib binding interactions. The key residues, which will potentially make critical interactions with inhibitors, are shown in stick form and labeled. The distances between two atoms are indicated as yellow dashed lines and labeled in black.
On the basis of these observations, we reasoned that a H bond between an inhibitor and Met318 is crucial for activity against both native BCR-ABL and BCR-ABLT315I kinases. Therefore, we hypothesized that designing a hybrid molecule based on core structures of the existing BCR-ABL inhibitors that make H bond interactions with Met318 and studying their binding interactions with native BCR-ABL and BCR-ABLT315I protein would be an ideal first step. Using core structures of approved BCR-ABL inhibitors, several hybrid molecules were designed and computational studies were performed to investigate the potential binding modes of new compounds. These studies revealed that a majority of our designed molecules formed key H bond interactions with the backbone of Met318 in the hinge region of the native BCR-ABL. Moreover, some of the hybrids showed H bond interactions with Met318 in BCR-ABLT315I. Importantly, the hybrids that were designed using a core structure from ponatinib (structure similar to that of 9) occupied the ATP pocket of the BCR-ABLT315I and were predicted to form a H bond interaction with the backbone of Met318 (Figure 3 and Figure S1). Furthermore, as shown in Figure 3b, lead compounds 33a and 36a occupied the same binding region in BCR-ABLT315I as did ponatinib and formed the same distance between the N atom of the Met318 residue and the N atom of the imidazo[1,2-b]pyridazine moiety of inhibitors (Figure 3d). Moreover, the distances between the atoms of other key residues such as Glu286 and Asp381 and the atoms of our lead compounds [which could potentially interact with these residues by H bonding (Figure 3a,b)] were also similar to that observed for ponatinib with BCR-ABLT315I. The superimposition of both BCR-ABL and BCR-ABLT315I kinases (Figure 4) with the poses of lead compounds predicted that the ethyne linker in these inhibitors would skirt the mutated gatekeeper residue Ile315, similar to the case for ponatinib.42 Therefore, these compounds could possibly show efficacies similar to those of ponatinib in inhibiting BCR-ABLT315I.
Figure 4.

Potential binding interactions of (a) ponatinib and (b) 33a and 36a after alignment of protein structures of BCR-ABLWT (pale green, PDB entry 3OXZ) and BCR-ABLT315I (gray, PDB entry 3IK3).
Chemistry
Compound 3a was obtained from a commercial source (Ark Pharma). The synthesis of hit finder compounds 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide-based inhibitors 3b–d is shown in Scheme 1. N-(2-Chloro-6-methylphenyl)-2-[(2-methylpyrimidin-4-yl)amino]thiazole-5-carboxamide 3b was prepared according to the previously reported procedure for a similar analogue,43 by the SNAr displacement of 4-chloro-2-methylpyrimidine 2b with 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 1. Alternatively, 3c and 3d were obtained by amide coupling in the presence of EDC·HCl and HOBt.
Scheme 1. Synthesis of Hit Finder Compounds.
Reagents and conditions: (a) NaH (60% in mineral oil), DMF, 0 °C to rt, overnight; (b) EDC·HCl, HOBt, diisopropylethylamine, THF, rt, 18 h.
Inhibitors 11a–c were synthesized on the basis of the tandem Sonogashira strategy using a previously reported procedure for similar analogues.44 As illustrated in Scheme 2, two general methods (A and B) were explored using either 3-bromoimidazo[1,2-b]pyridazine 4 or methyl 3-iodo-4-methylbenzoate 6 as coupling agents in the first Sonogashira reaction. This was a straightforward reaction used for both reagents 4 and 6, and the corresponding products were isolated in good yields. However, the final Sonogashira reaction employed in method B resulted in very low yields of the desired product, with debromination of 4 being the major impurity. Therefore, method A was used to synthesize 8. Hydrolysis of 8 using a 1 M LiOH solution afforded 9, which upon reaction with appropriate amines 10a–c in standard amide coupling conditions, using EDC and HOBt, afforded the final compounds 11a–c, respectively. HIT compound 15 was synthesized using a convenient route outlined in Scheme 3. An amide coupling of the readily available 3-iodo-4-methylbenzoic acid 12 with 3-bromo-5-(trifluoromethyl)aniline 13 in the presence of SOCl2 and DIPEA yielded intermediate 14. Subsequent Sonogashira coupling of 14 with 5 provided inhibitor 15.
Scheme 2. Synthesis of HIT Finder SAR.
Reagents and conditions: (a) trimethylsilylacetylene, [PdCl2(Ph3P)2], CuI, K2CO3, acetonitrile, 100 °C, 24 h; (b) CuI, [Pd(Ph3P)4], diisopropylethylamine, DMF, sealed tube, 100 °C, 5 h; (c) 1 M LiOH solution in water, 1:1 THF/MeOH, rt, 24 h; (d) EDC·HCl, HOBt diisopropylethylamine, THF, rt, 18 h.
Scheme 3. Synthesis of the HIT Compound.
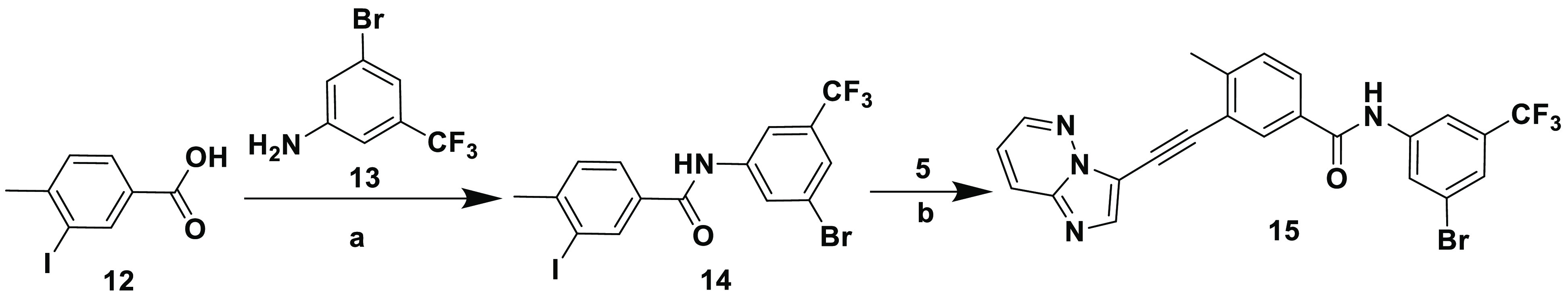
Reagents and conditions: (a) SOCl2, diisopropylethylamine, DMAP, THF, reflux, 5 h, THF; (b) CuI, [Pd(Ph3P)4], diisopropylethylamine, DMF, sealed tube, 100 °C, 5 h.
Inhibitors 19, 20, 21a, 21b, and 24 were synthesized according to the synthetic route outlined in Scheme 4. Starting material 3-ethynyl-4-methylbenzoic acid 16 was obtained from 3-iodo-4-methylbenzoate 6 via Sonogashira reaction followed by hydrolysis. Amide coupling of 17 with 16 resulted in 19, which underwent Sonogashira coupling with 4-iodo-1-methyl-1H-imidazole to yield 20. Inhibitors 21a and 21b were prepared like 19 using 18a and 18b instead of 16 as the carboxylic acid precursors. Inhibitor 24 was prepared from 17 via an amide coupling followed by a Sonogashira reaction, with the appropriate starting materials.
Scheme 4. Synthesis of Inhibitors 19–21 and 24.
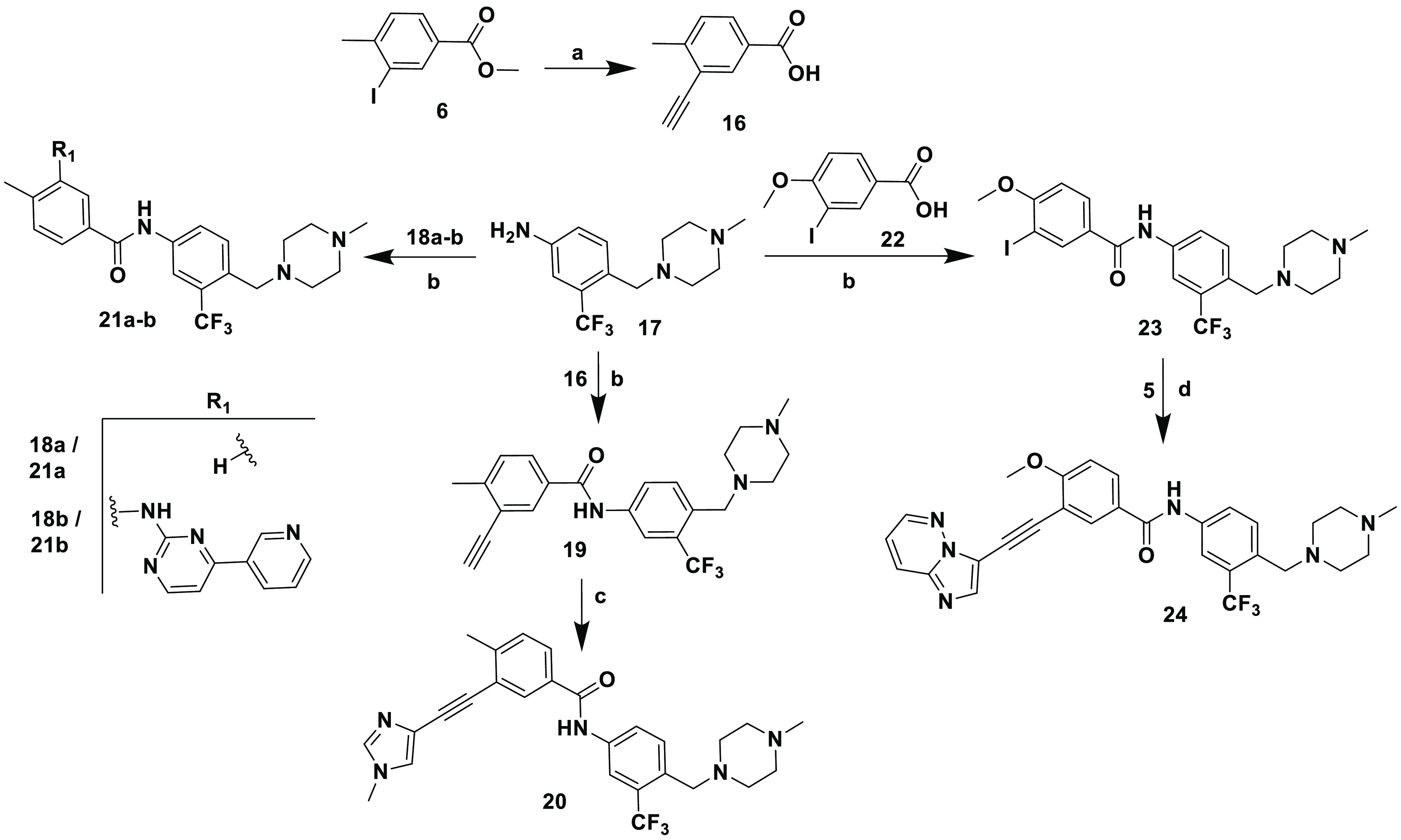
Reagents and conditions: (a) (i) trimethylsilylacetylene, [PdCl2(Ph3P)2Cl], CuI, triethylamine, THF, rt, 24 h; (ii) KOH, MeOH; (b) EDC·HCl, HOBt diisopropylethylamine, DMF, rt, 18 h; (c) 4-iodo-1-methyl-1H-imidazole, CuI, [PdCl2(Ph3P)2], diisopropylethylamine, DMF, sealed tube, 100 °C, 24 h; (d) CuI, [PdCl2(Ph3P)2], diisopropylethylamine, DMF, sealed tube, 100 °C, 24 h.
Synthesis of inhibitors 26, 29, and 32 is depicted in Scheme 5. Inhibitor 26 was obtained by reacting 3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline 25 with 16 using standard EDC-HOBt amide coupling conditions. Inhibitor 29 was prepared in a manner similar to that of 19, using the required starting materials for both Sonogashira reactions.45 The structure of inhibitor 32 resembled that of 11b; however, the sole difference is that the amide group in 32 was inverted between the two aryl groups. It was prepared in two steps. In the initial step, amide condensation was performed between 3-iodo-4-methylaniline 30 and 2d to afford intermediate 31, which was then reacted with 5 via Sonogashira reaction conditions to provide inhibitor 32.
Scheme 5. Synthesis of Inhibitors 26, 29, and 32.
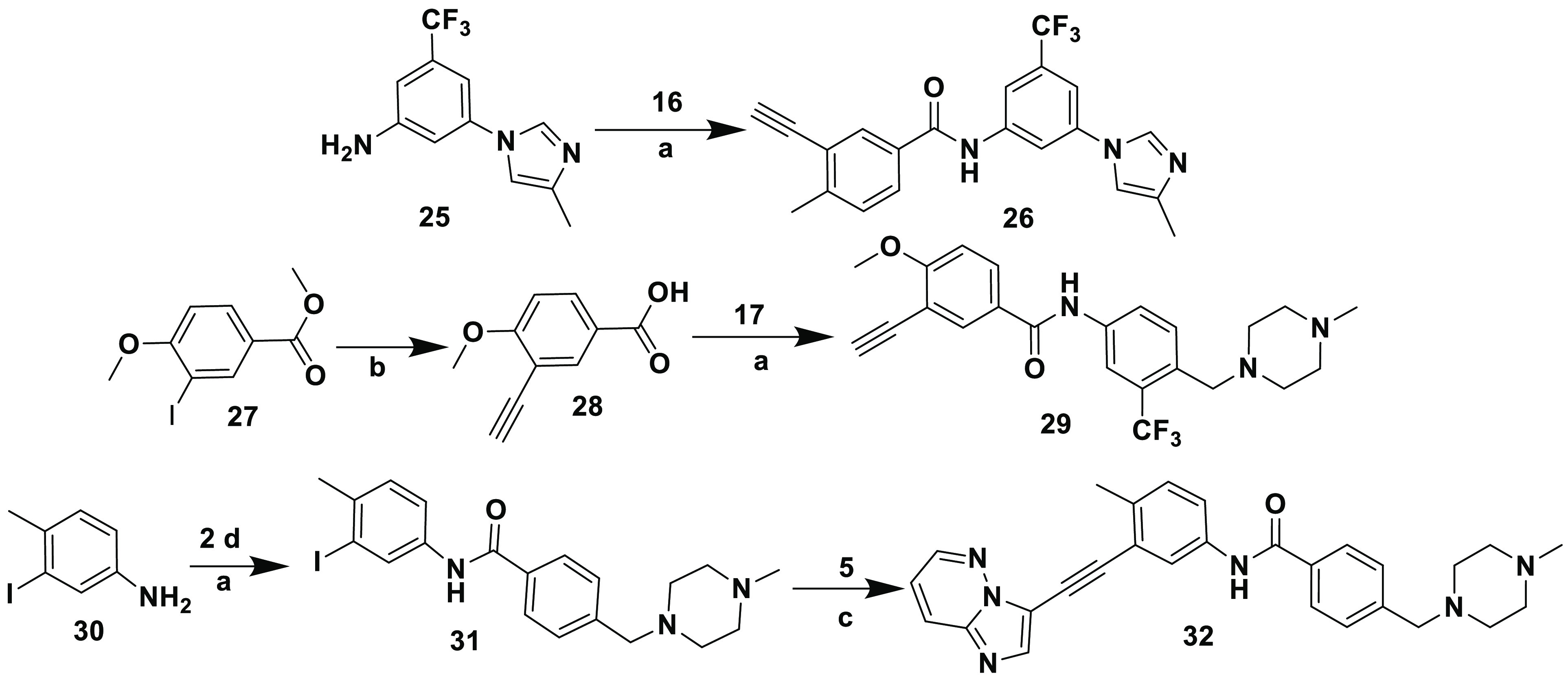
Reagents and conditions: (a) EDC·HCl, HOBt diisopropylethylamine, DMF, rt, 18 h; (b) (i) trimethylsilylacetylene, [PdCl2(Ph3P)2], CuI, triethylamine, THF, rt, 24 h; (ii) KOH, MeOH; (c) CuI, [Pd(Ph3P)4], diisopropylethylamine, DMF, sealed tube, 100 °C, 5 h.
Scheme 6 illustrates the synthesis of inhibitors 33a–h compiled in Table 5. Briefly, a copper-catalyzed N-arylation46−48 of imidazole or substituted imidazoles or methyl pyrrole or methyl piperazine with 15 yielded corresponding compounds 33a–h. Notably, the coupling reaction worked well for all of the substrates reported here; however, a slight decrease in yield was observed for inhibitors 33e and 33f, with pyrrole and methyl piperazine moieties, respectively. Notably, compound 33e was synthesized by microwave irradiation.
Scheme 6. Synthesis of Inhibitors 33a–h.
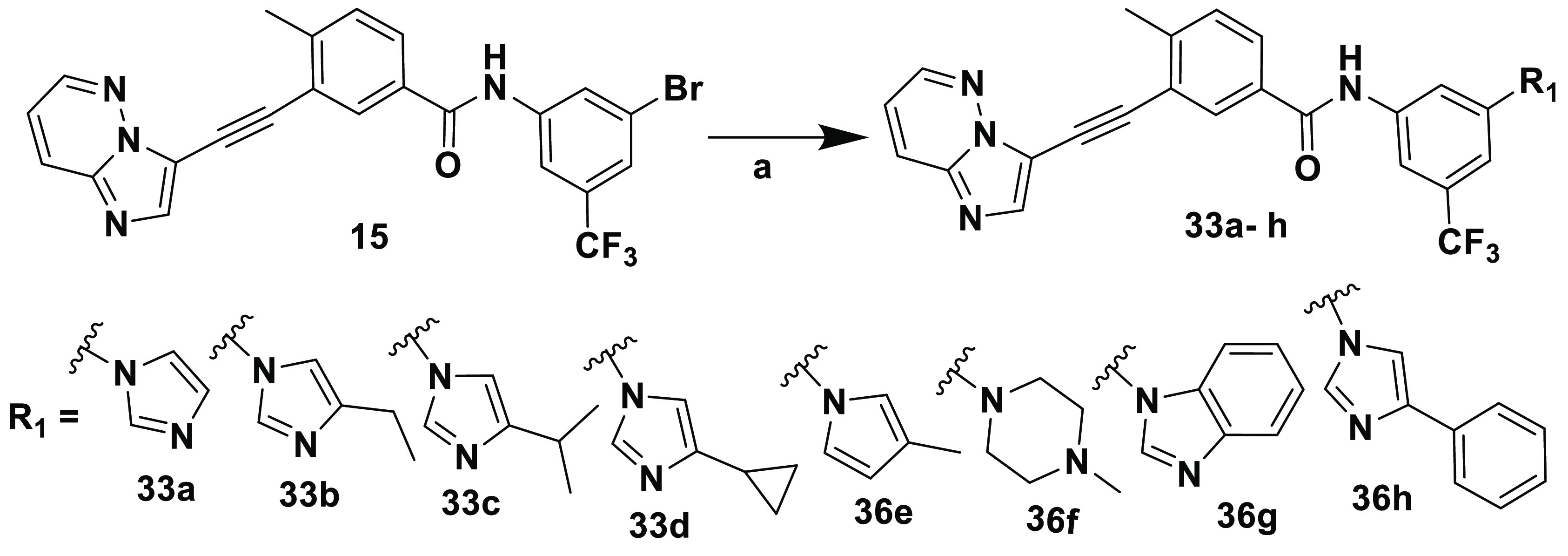
Reagents and conditions: (a) CuI, 8-Quinolinol, K2CO3, DMSO.
Table 5. SAR around 15e.

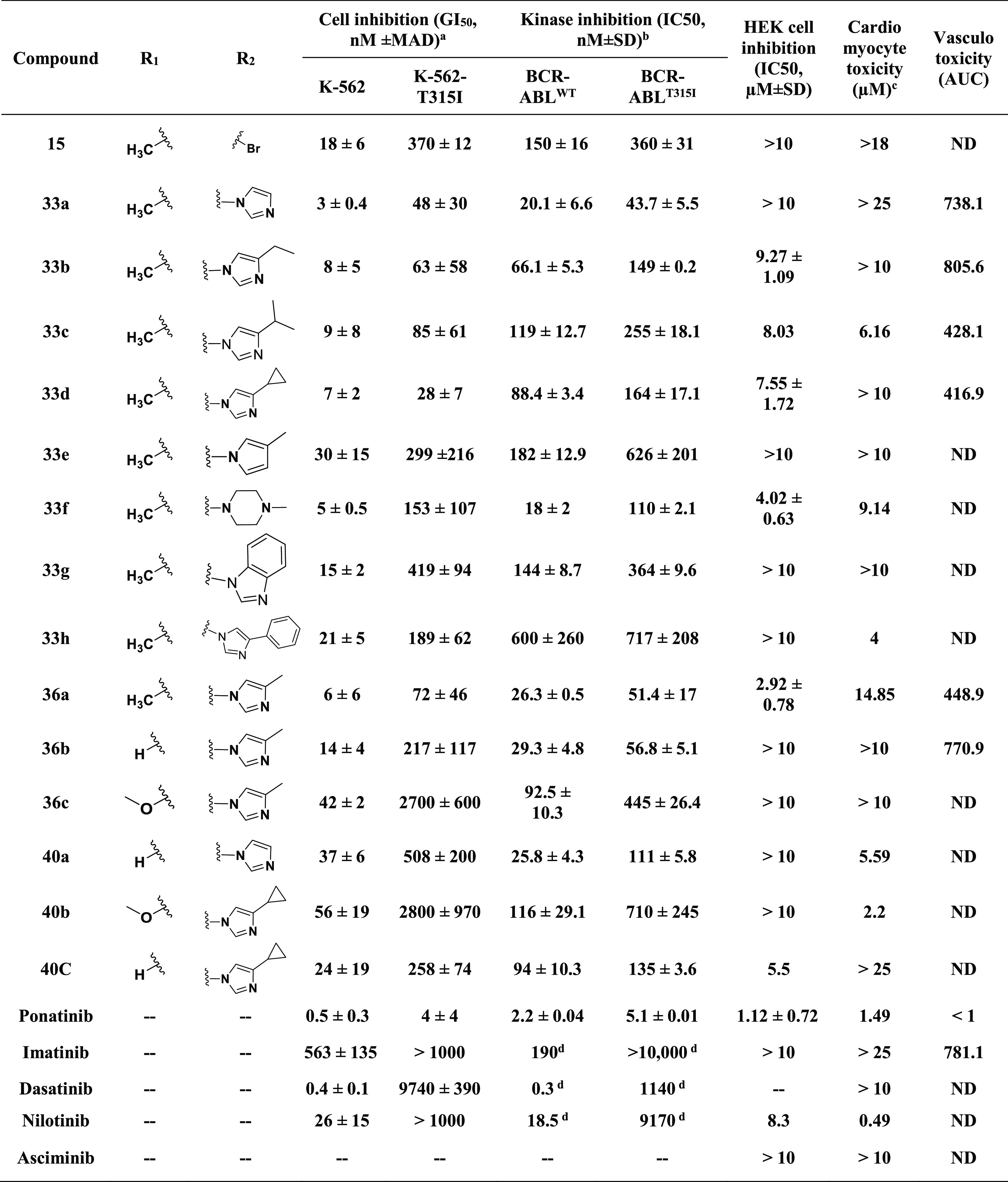
GI50 values are shown as the median and median absolute deviation (MAD) of at least two independent experiments performed in triplicate.
IC50 was determined by following the biochemical kinase assay protocol. The data represent the mean and standard deviation of at least two independent experiments performed in duplicate.
Overall smallest toxic dose.
Data taken from ref (52).
Cell viability in the presence of varying concentrations of compounds was measured by the AlamarBlue assay. HMVEC-Cs were treated with inhibitors, and GI50 values were measured to assess the vasculotoxicity as the AUC (area under the curve) as shown in Figure 6a. ND, no inhibition detected at concentrations of ≤10 μM.
The synthetic protocols for inhibitors 36a–c and 40a–c are outlined in Schemes 7 and 8, respectively. They were prepared using the amide coupling and Sonogashira procedures outlined in Scheme 5.
Scheme 7. Synthesis of Inhibitors 36a–c.
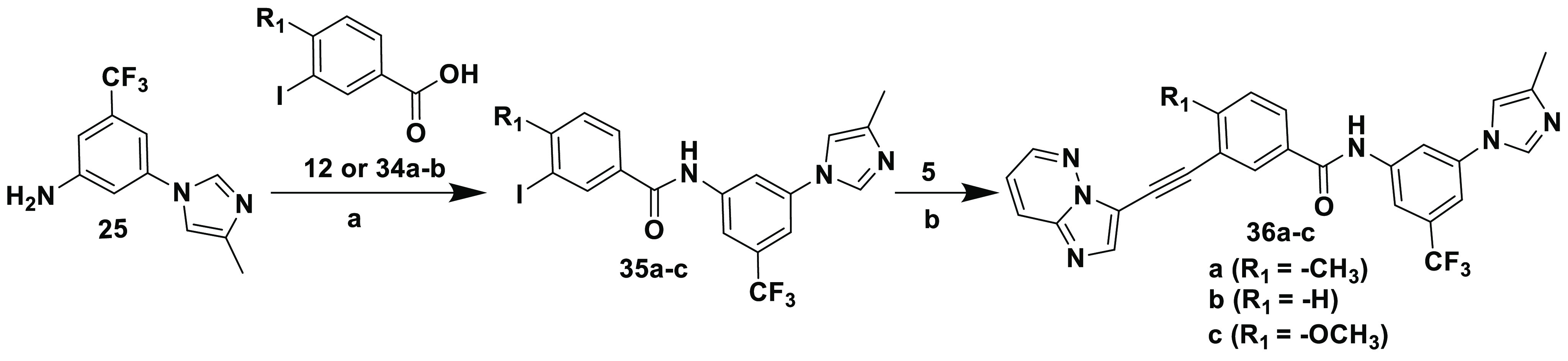
Reagents and conditions: (a) (i) SOCl2, reflux, 5 h; (ii) diisopropylethylamine, DMAP, THF, 18 h; (b) CuI, [Pd(Ph3P)4], diisopropylethylamine, DMF, sealed tube, 100 °C, 5 h.
Scheme 8. Synthesis of Inhibitors 40a–c.
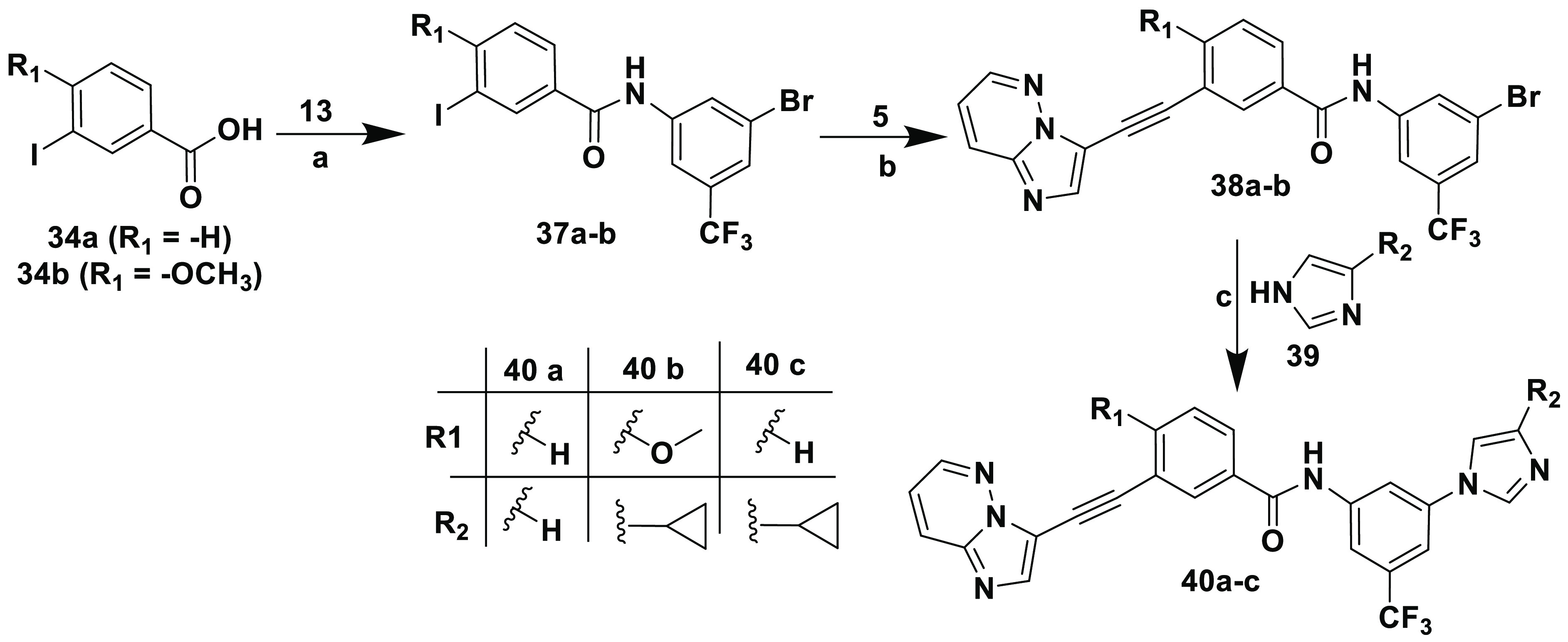
Reagents and conditions: (a) (i) SOCl2, reflux, 5 h; (ii) diisopropylethylamine, DMAP, THF, 18 h; (b) CuI, [Pd(Ph3P)4], diisopropylethylamine, DMF, sealed tube, 100 °C, 5 h; (c) CuI, 8-Quinolinol, K2CO3, DMSO.
Anticancer Activity and Cardiac Safety
Because the H bond interactions between the inhibitor and Met318 in BCR-ABL were considered to play a crucial role in the activity of the desired compounds, we initially selected core fragments of the FDA-approved TKIs, which are important in making H bond interactions with Met318, and designed hybrid molecules to identify cardiac-safe “HIT” molecules. The new compounds were evaluated for their kinase and cellular activities in vitro, against both BCR-ABL and BCR-ABLT315I kinases and corresponding K-562 cell lines. Their cardiomyocyte toxicities were evaluated by probing the contractility and voltage transients in human iPSC-CMs to help guide template selection. The results from the in vitro human iPSC-CM assays have been shown previously to correlate well with clinical incidences of toxicity for small molecule kinase inhibitor oncology therapeutics.49−51 Briefly, the transients were quantified, and curve fitted, to extract several measures for voltage (action potential duration of 75%) and contractility (peak contraction amplitude). The minimal concentration at which the dose response curve of any metric or the variability of the given metric deviated beyond a threshold (25%) from the control was deemed the overall minimum toxic dose. Imatinib, dasatinib, and ponatinib were used as controls to validate the screening conditions.
As shown in Table 1, under the experimental conditions, the hybrids prepared from the dasatinib core (fragment) showed significant efficacies against native K-562 cells. In particular, 3d potently inhibited the growth of native K-562 cells with a GI50 value of 30 nM. Consistent with the cellular inhibition potency, it inhibited the activity of native BCR-ABL kinase (Table 2). However, similar to dasatinib, these hybrids were also ineffective against the T315I mutation and did not inhibit the activity of the BCR-ABLT315I kinase or the growth of the corresponding K-562 cell line.
Table 1. Cellular Activity of the Hit Finder Compoundsc.

GI50 values are shown as the median and median absolute deviation (MAD) of at least two independent experiments performed in triplicate.
Overall smallest toxic dose.
Cell viability in the presence of varying concentrations of compounds was measured by the AlamarBlue assay. ND, no inhibition detected at concentrations of ≤10 μM.
Table 2. Kinase Inhibition for the Selected Hit Finder Compounds.
| kinase
inhibition (IC50, nM ± SD)a |
||
|---|---|---|
| Compound | BCR-ABLWT | BCR-ABLT315I |
| 3c | 5.0 ± 0.3 | >10000 |
| 3d | 0.5 ± 0.3 | >10000 |
| 11b | 4.0 ± 0.3 | 547 ± 3.9 |
| 11c | 1000 | >10000 |
| 15 | 150 ± 16 | 360 ± 31 |
| 20 | 58.5 ± 1.0 | 120 ± 6.6 |
| 21b | 9.1 ± 2.8 | 670 ± 72 |
| 24 | 9.4 ± 2.0 | 16.1 ± 2.2 |
| 32 | 1.6 ± 0.3 | 390 ± 1.7 |
| imatinib | 190b | >10000b |
| dasatinib | 0.3b | 1140b |
| ponatinib | 2.2 ± 0.1 | 5.1 ± 0.1 |
IC50 was determined by following the biochemical kinase assay protocol. The data represent the mean and standard deviation of at least two independent experiments performed in duplicate.
Data taken from ref (52).
It is plausible that when the Thr at position 315 of BCR-ABL is mutated to the bulkier Ile residue, it could possibly block the entry of new compounds into the hydrophobic pocket. Therefore, the H bond interaction between new hybrids and the Met318 residue will be lost. Also, this could result in the loss of other key H bond interactions with Thr315 as observed in the case of dasatinib.24 Therefore, they are inefficient against BCR-ABLT315I. Moreover, our computational studies of 3d with BCR-ABLT315I revealed that none of the docking poses allowed H bond interactions with Met318. In the best pose, the position of 3d closest to Met318 (Figure S2a) was still too far away to form an H bond with Met318, whereas it was found to be within range to form H bond interactions with native BCR-ABL (Figure S2b).
The new compounds 3a–d appeared to be cardiac-safe (Table 1), as there was no decrease in the contractile amplitude or alterations in the iPSC-CM voltage waveforms at concentrations of ≤10 μM. Arrhythmia was assessed by analyzing cardiomyocyte voltage traces for evidence of action potential prolongation, after depolarizations, tachycardia and bradycardia, ectopy, etc. (see Experimental Section). Also, we did not observe a decrease in contractility up to this dose.
This initial findings with the dasatinib core encouraged us to explore SAR using core moieties of other inhibitors predicted to bind Met318 in the hinge region of BCR-ABL. Therefore, considering the similar approach of H bond interactions with Met318 in BCR-ABLT315I, we explored the SAR against a ponatinib fragment, 3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzoic acid (9) and designed new compounds (11a–c and 15). As shown in Table 3, significant improvement was observed in the growth inhibition of both native K-562 cells and K-562 cells expressing BCR-ABLT315I for these hybrids. 15 in particular showed remarkable growth inhibition against native and kinase-mutated K-562 cells, with GI50 values of 18 and 370 nM, respectively. Consistent with its cellular activity, 15 also strongly inhibited the native BCR-ABL and BCR-ABLT315I kinases in a biochemical kinase assay (Table 2), with IC50 values of 150 and 360 nM, respectively. These data suggest that 15 could access the hydrophobic pocket of BCR-ABLT315I. Docking studies predicted that 15 should interact with key residues, such as Met318, Glu286, and Asp381 of BCR-ABLT315I via H bond interactions (Figure S3), in a manner similar to that shown for ponatinib.30 On the contrary, 11a and 11c were found to be inactive and showed no efficacy against K-562 cells at concentrations of ≤10 μM. We found that 11c has dose-dependent cardiomyocyte toxicity, while other compounds derived from the ponatinib core moiety showed improved cardiac safety compared to that of ponatinib. We speculated that the cardiotoxicity of 11c could be due to its interaction with some other targets that would potentially cause cardiomyocyte toxicity. Therefore, the fragment (R group) used to prepare 11c was avoided in the subsequent studies.
Table 3. Cellular Activity of the Hit Finder Compoundsc.

GI50 values are shown as the median and median absolute deviation (MAD) of at least two independent experiments performed in triplicate.
Overall smallest toxic dose.
Cell viability in the presence of varying concentrations of compounds was measured by the AlamarBlue assay. ND, no inhibition detected at concentrations of ≤10 μM.
Despite being less potent than ponatinib, hybrids containing the fragment of ponatinib that interacts with Met318 exhibited significantly improved cardiac safety compared to that of ponatinib. We speculated, therefore, that other portions of ponatinib might be responsible for its cardiomyocyte toxicity. We explored this idea further in the next step by studying the SAR around the remaining core of ponatinib. Several structurally diverse hybrids were prepared using 4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)aniline 17 as a core. Consistent with our hypothesis, the majority of hybrids (Table 4, 19, 20, 21a, 21b, and 29) exhibited cardiomyocyte toxicity. Interestingly, some of the hybrids were inactive against K-562 cell lines yet exhibited cardiomyocyte toxicity significantly higher than that of the hybrids that were active in this series. For example, compounds 21a and 29 were ineffective against K-562 cells up to 10 μM but were found to be highly cardiomyocyte toxic at doses of 0.64 and 1.26 μM, respectively. Moreover, 21b, which is a hybrid molecule of imatinib and ponatinib, had significantly increased cardiomyocyte toxicity at 5.21 μM (Table 4). These findings suggest that the cardiomyocyte toxicity arises from fragment 17 because imatinib did not exhibit cardiomyocyte toxicity up to 25 μM, whereas appreciable cardiomyocyte toxicity was observed for 21b at a much lower concentration.
Table 4. Cellular Activity of the Hit Finder Compoundsc.

GI50 values are shown as the median and median absolute deviation (MAD) of at least two independent experiments performed in triplicate.
Overall smallest toxic dose.
Cell viability in the presence of varying concentrations of compounds was measured by the AlamarBlue assay. ND, no inhibition detected at concentrations of ≤10 μM.
Despite their cardiomyocyte toxicities, the hybrids that were generated from 17, such as 20, 21b, and 24, all were efficacious against K-562 cells, with GI50 values of 300, 3, and 3 nM, respectively. However, except for 24, none of these hybrids showed improved efficacies over 15 against BCR-ABLT315I kinase and the corresponding K-562 cell lines. Because compound 15 exhibited more favorable cardiomyocyte safety than 24, we decided to explore its SAR further.
SAR around 15
The SAR around the lead compound 15 was explored by investigating the influence of different R1 and R2 groups. Our computational investigation suggested that modifications at the R1 and R2 positions could preserve important elements of molecular recognition. The new analogues should access ATP-binding sites of both BCR-ABL and BCR-ABLT315I, and therefore, they would make key H bond interactions with Met318, Glu286, and Asp381 in both proteins (Figure S1). Hence, we expected either similar or enhanced potencies for the designed hybrids compared to that of 15. As shown in Table 5, most of the hybrids demonstrated improved efficacies in enzymatic and cellular assays relative to that of 15. We found that replacing the bromo group with imidazole or substituted imidazoles at the R2 position dramatically enhanced the activities for the inhibitors. For example, 33a–d and 36a have remarkably increased potencies compared to that of 15 (Table 5 and Figure 5a,b). Notably, 33a and 36a showed dramatically increased potencies (a 6–7-fold improvement compared to that of 15) in both enzymatic and cellular assays against BCR-ABLT315I. For these compounds, the bulkiness of the imidazole ring seemed to affect the potency of these hybrids. For example, compared to 33a, hybrids 36a, 33b–d, 33g, and 33h, which contain alkyl groups or bulky aromatic groups at position C-4 of the imidazole ring, were found to be less potent. Increasing the alkyl chain length at this position gradually decreased the activity of the hybrids. This phenomenon was generally observed for both BCR-ABL and BCR-ABLT315I protein inhibition. For example, 33a with no substitution on the imidazole showed superior activity among all of the hybrids, with IC50 values of 20.1 and 43.7 nM for BCR-ABL and BCR-ABLT315I, respectively, whereas 36a, with a methyl group at position C-4 of the imidazole ring, was slightly less potent than 33a (IC50 values of 26.3 and 51.4 nM for BCR-ABL and BCR-ABLT315I, respectively). Moreover, while the length of the alkyl chain of 33b and 33c was gradually increased upon incorporation of ethyl and isopropyl groups, respectively, their potencies also decreased. Finally, 33c with an isopropyl group was found to be the least potent among all of the n-alkyl-substituted analogues (IC50 values of 119 and 255 nM for BCR-ABL and BCR-ABLT315I, respectively). The sole exception to an increased alkyl chain length correlating with decreased activity was 33d. Unexpectedly, its cyclopropyl substitution corresponded to slightly improved activity (IC50 values of 88.4 nM for BCR-ABL and 164 nM for BCR-ABLT315I) compared to that seen with isopropyl analogue 33c.
Figure 5.
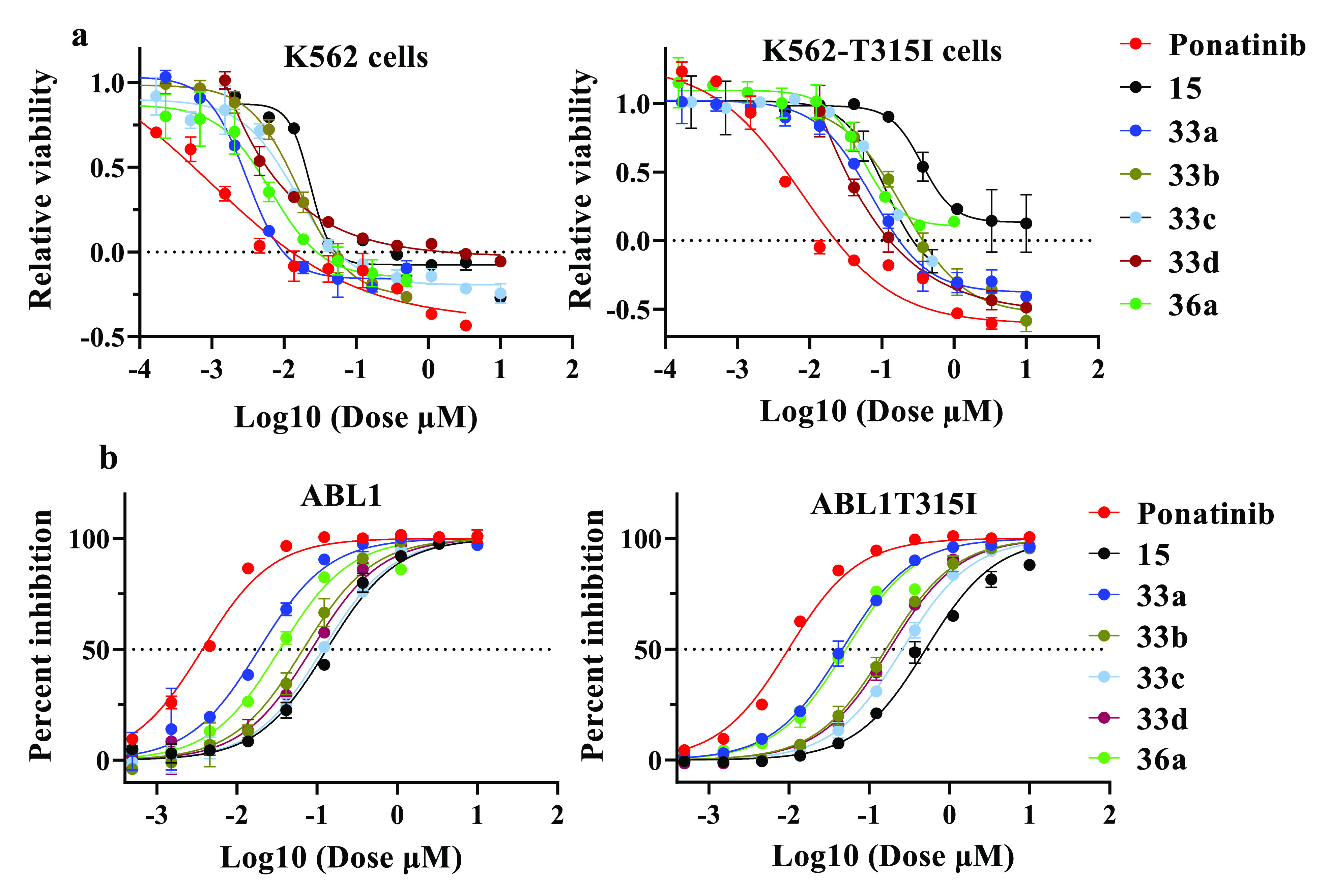
Antiproliferative activities of inhibitors. (a) Relative viability of K-562 cells and K-562-T315I cells. (b) Percent inhibition of ABL1 and ABL1T315I protein with drug treatment with ponatinib and hybrid compounds 15, 33a–d, and 36a.
As described in ref (53), the kinase profiles of 33a and 36a remain largely unchanged. Additional studies are needed to understand whether the altered potencies contribute to the improved cardiotoxicity profiles of 33a and 36a or to ascertain if they affect noncardiac toxicity. Overall, the BCR-ABLT315I kinase activity for these hybrids was reduced by 2–3-fold compared to the native BCR-ABL kinase activity, similar to the difference observed for ponatinib.42 A slight outward displacement of the Flag-methyl group containing the phenyl ring of the hybrids from the hydrophobic pocket of BCR-ABLT315I would account for the reduction in potency against BCR-ABLT315I. This is similar to the outward displacement observed for ponatinib in complex with BCR-ABLT315I that leads to reduced potencies against this mutated kinase and the corresponding cell lines.42
Next, we explored the effect of steric hindrance by using 1H-benzo[d]imidazole (33g) and 4-phenyl-1H-imidazole (33h) moieties. We found that hybrids 33g and 33h have markedly reduced kinase and cellular activities. Compared to 33a, the potencies of these compounds are decreased 7–16- and 4–9-fold in the BCR-ABLT315I enzymatic and cellular assays, respectively.
To further optimize the lead compound, we focused on improving solubility. We hypothesized that incorporation of the 1-methylpiperazine moiety would improve cell permeability and help reduce lipophilicity. However, we found that 33f did not show improved efficacies compared to 33a. Moreover, relative to 33a, compound 33f had 2-fold decreased activity against BCR-ABLT315I kinase. The cellular inhibition efficacy for 33f was consistent with that in the biochemical assay. Another hybrid, 33e, with 3-methyl-1H-pyrrole, was also less effective than 33a and showed significantly reduced BCR-ABLT315I kinase and cellular potencies of 14- and 6-fold, respectively. This suggests that the second nitrogen in the five-member ring is essential for improving the efficacies of the hybrids.
Next, a Flag-methyl54 group (R1) was briefly included to evaluate its impact on inhibitory activity. Hybrids 33a, 33d, and 36a were selected for the study, and the results are summarized in Table 5. When the methyl group in 33a was replaced with H, the resulting compound, 40a, displayed efficacies similar to those of 33a against the native BCR-ABL kinase. However, its activities against BCR-ABLT315I and the corresponding cell lines were dramatically decreased. Hybrids 40c and 36b, which were derived from 33d and 36a, respectively, maintained similar activities as seen with the corresponding analogues containing the methyl group against both native BCR-ABL and BCR-ABLT315I kinases. On the contrary, their cellular potencies decreased by 2–10-fold. We observed that large hydrophobic groups at the R1 position were detrimental to the activities on both kinase and cellular levels. For instance, relative to 33d and 36a, methoxy analogues 40b and 36c demonstrated 8–16- and 35–100-fold losses of potency against BCR-ABLT315I kinase and the corresponding K-562 cell lines, respectively. In line with previous findings,44,54,55 our results clearly demonstrated the importance of the Flag-methyl group in the selective inhibition of BCR-ABL. Furthermore, similar to ponatinib binding with BCR-ABL,42 the presence of the Flag-methyl group in hybrids favored desirable binding orientations within BCR-ABL. Accordingly, substituting the Flag-methyl group with either H or a large hydrophobic group resulted in a loss of selectivity,56 and the corresponding hybrids were found to be less potent than analogues containing the methyl group.
Hybrids Decreased Adverse Cardiomyocyte and Vascular Toxicities
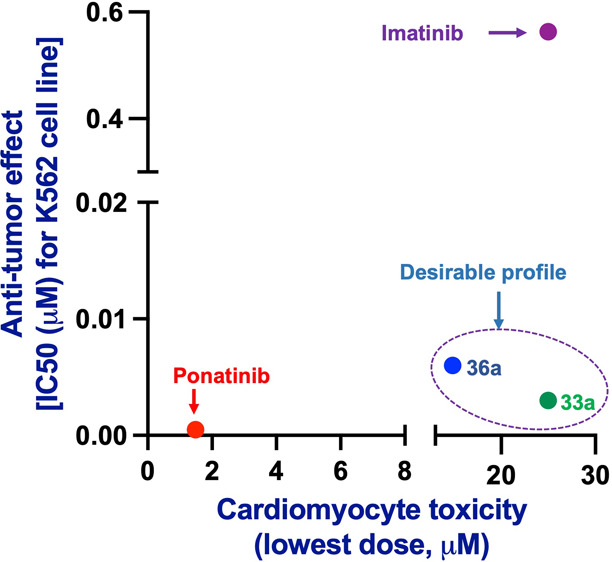
The TKIs currently used for the treatment of CML primarily target BCR-ABL kinase activity. However, most of them have shown distinctive off-target activities,30,57 which result in adverse effects.35 These cardiovascular complications have restricted the use of the most potent TKIs.34,58,59 Among these, ponatinib is currently the only FDA-approved drug that effectively inhibits the BCR-ABLT315I mutation and has been restricted due to cardiovascular adverse events to treating only patients carrying mutated tumors refractory to first- and second-line therapeutics.34,58 Ponatinib cardiomyocyte toxicity events were observed at a low dose of 1.49 μM in vitro (Table 5). Similarly, ponatinib inhibited the integrity of vascular structures formed by HMVEC-Cs in vitro (Figure 6) at <1 μM. In addition, it inhibited the growth of healthy HEK cells at 1.1 μM. These results demonstrated its toxicity in realistic assays and allowed us to develop improved hybrid compounds with excellent efficacies against both BCR-ABLT315I kinase and corresponding K-562 cells lines that were also safer in the vascular and cardiomyocyte assays (Figure 6). In addition, compounds 33a and 33b did not inhibit vascular integrity on HMVEC-Cs even at 10 μM, which suggests that these hybrids are cardiac-safe. In particular, highly potent hybrids 33a and 36a have shown superior cardiac safety up to 25 and ∼15 μM, respectively. Bulky substitutions at position C-4 of the imidazole ring, however, rendered hybrid molecules cardiotoxic. For example, whereas hybrids 33a, 36a, and 33b, with H, methyl, and ethyl groups, respectively, are cardiac-safe up to 25, ∼15, and 10 μM, respectively (Table 5 and Figure 6c–e), hybrid 33c with an isopropyl group demonstrated cardiomyocyte toxicity at 6.16 μM, suggesting that even a small modification on the imidazole ring could cause a significant change in cardiac safety. The cardiomyocyte toxicities caused by the bulkiness on the imidazole ring were clearly observed for hybrid 33h, which had a phenyl group on the imidazole moiety and was the most toxic hybrid among those examined during lead optimization. Notably, replacing the Flag-methyl group with H or a methoxy group also resulted in cardiotoxicity. These findings clearly suggest that a small change in the inhibitor structure could alter the balance of on-target versus off-target interactions56 to cause adverse effects. On the basis of the distinct SARs, we conclude that cardiotoxicity for ponatinib and the derivatives likely results from strong interactions with off-targets rather than BCR-ABL.
Figure 6.

Cardiac safety of inhibitors. (a) Vasculogenesis assay. HMVEC-Cs were treated for 6 h with a dose response 3-fold change with a maximum dose of 10 μM imatinib, ponatinib, 24, 33a–d, and 36a. DMSO was used as a control, and mean ± SEM values were used to plot the graph. (b) Images of the vasculogenesis assay for the control, imatinib, ponatinib, 24, 33a–d, and 36a at a dose of 1.1 μM (scale bar, 200 μM). Analogues preserved the ability to form a capillary-like network similar to control and imatinib, whereas loop formation was not observed for HMVEC-Cs treated with ponatinib. (c) Vasculotoxicity vs GI50 for imatinib, ponatinib, 33a–d, and 36a. AUC is area under the curves shown in panel a. (d) Cardiomyocyte toxicity vs GI50 for imatinib, ponatinib, 33a–d, and 36a. Cardiotoxicity is qualified as the smallest dose that caused cardiotoxicity. (e) Vasculotoxicity vs cardiomyocyte toxicity of all compounds tested. Note that 33a showed a safety profile similar to that of imatinib, whereas 36a showed an intermediate safety profile.
Considering the overall performance, including in vitro kinase and cellular potencies as well as cardiac safety, inhibitors 33a and 36a were selected to evaluate their pharmacokinetic profiles and antitumor activities in vivo. Our results suggest that these compounds have shown efficacies comparable to those of ponatinib in mouse models of CML driven by the T315I mutation. The complete details of these findings are reported in ref (53).
Conclusions
In summary, this paper describes a distinct SAR for the desirable anti-BCR-ABLT315I antitumor potency versus adverse cardiotoxicity for ponatinib. On the basis of this information, we successfully designed and synthesized a series of hybrid molecules that are more selective BCR-ABL inhibitors. The hybrids maintained significant inhibitory activities against K-562 human CML cells, including the most intractable gatekeeper T315I mutant associated with disease progression in CML. The most potent compounds, 33a and 36a, strongly inhibited the kinase activities of both native BCR-ABL and BCR-ABLT315I with mean IC50 values of 20.1 and 26.3 nM and 34.7 and 51.4 nM, respectively. Furthermore, they showed comparable efficacies and only slightly diminished potencies relative to those of ponatinib on K-562 cells expressing BCR-ABLT315I. Considering the ∼10-fold diminished potencies for in vitro inhibition of T315I mutant BCR-ABL, the net improved safety was nonetheless substantial. Compound 33a did not show cardiomyocyte or vascular toxicity at any dose (Table 5). We speculate that differential inhibition of kinases might contribute to the greater safety of 33a and 36a relative to ponatinib. As described in ref (53) and shown in the Supporting Information (page S54), several kinases are inhibited by ponatinib that are not significantly inhibited by 33a and 36a, at least in vitro. The in vivo biological consequences of different kinase targeting remain to be explored. Notably, hybrids 33a and 36a were found to be the most cardiac-safe TKIs reported to date. Moreover, 33a and 36a have shown excellent pharmacokinetics and achieved durable tumor regression in the K-562 xenograft model in mice upon oral administration. Therefore, they could serve as promising lead compounds for further development of a new class of BCR-ABL inhibitors overcoming the T315I mutation and cardiotoxicity.
Experimental Section
Chemical Synthesis
General Methods
All of the reagents and solvents were obtained at the highest commercial quality from sources such as Sigma-Aldrich, Fisher Scientific, TCI International, Acros organics, Alfa Aesar, Matrix Scientific, Chem-Implex, and Enamine and were used without further purification. Unless otherwise mentioned, all of the reactions were carried out under a nitrogen atmosphere with dry solvents. The reactions were monitored by TLC using precoated silica gel plates (Merck, silica gel 60 F254). Flash chromatography was carried out using a CombiFlash Rf+ Lumen chromatography system (Teledyne ISCO, Lincoln, NE). 1H (400 MHz) and 13C (101 MHz) NMR spectra were recorded either on an Agilent 400-MR NMR instrument or on a Bruker Avance 400 MHz spectrometer, using appropriate deuterated solvents, as needed. Chemical shifts (δ) were reported in parts per million upfield from tetramethylsilane (TMS) as an internal standard. Coupling constants (J) were reported in hertz, and s, br.s, d, t, and m denote singlet, broad singlet, doublet, triplet, and multiplet, respectively. LC-MS analysis was performed on an Agilent 6490 iFunnel Triple Quadrupole Mass Spectrometer from Agilent Technologies Inc. (Santa Clara, CA). An Agilent EclipsePlusC18 reverse phase column (1.8 μm, 2.1 mm × 50 mm) was used with solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) for LC-MS analysis. The solvent A:solvent B ratio was 1:9 at the beginning and gradually changed to 9:1 at the end. The flow rate was set to 0.4 mL/min. The detector wavelength was set to 254 nm. The mass spectrometer was operated under positive ionization mode. Purity testing was done by means of analytical HPLC on a Waters prep150 system with a Phenomenex Luna 3 μm PFP (2) column (150 mm × 3 mm) eluted at a rate of 0.6 mL/min with solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). The solvent A:solvent B ratio was 0.5:9.5 at the beginning and gradually changed to 9.5:0.5 after 5 min. The detector wavelength was set to 254–400 nm. All tested compounds were >95% pure.
N-(2-Chloro-6-methylphenyl)-2-[(2-methylpyrimidin-4-yl)amino]thiazole-5-carboxamide (3b)
Compound 3b was prepared on the basis of a literature procedure.43 Sodium hydride (60% in mineral oil, 0.186 g, 4.67 mmol) was added to a stirred solution of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 1 (0.5 g, 1.87 mmol) and 4-chloro-2-methylpyrimidine 2b (0.28 g, 2.24 mmol) in DMF (20 mL). The solution was heated at 100 °C overnight and cooled to room temperature (rt), and the reaction quenched by adding glacial acetic acid and water. The crude product was extracted into DCM (2 × 50 mL). The organic layers were combined and washed with water, followed by a saturated NaCl solution (25 mL). The organic phase was dried over Na2SO4, filtered, and then evaporated to dryness using a rotatory evaporator. The crude product was purified on a silica gel column with a 0% to 10% gradient of methanol in DCM to furnish the desired product as a pale yellow solid (0.07 g, 10% yield): HPLC purity 98.7% (tR = 1.75 min); 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 5.5 Hz, 1H), 8.12 (s, 1H), 7.58–7.48 (m, 1H), 7.47–7.37 (m, 2H), 6.90 (dd, J = 5.5, 0.7 Hz, 1H), 2.49 (s, 3H), 2.14 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 171.6, 167.1, 164.7, 159.6, 157.6, 156.4, 138.8, 132.0, 131.7, 131.3, 130.7, 128.6, 114.3, 98.6, 25.9, 17.8; LC-MS (ESI-QQQ) m/z 360.1 ([C16H14ClN5OS + H]+ calcd 360.06); purity 99% (tR = 3.287 min).
General Procedure for the Synthesis of 3c and 3d. Procedure for N-(2-Chloro-6-methylphenyl)-2-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}benzamido)thiazole-5-carboxamide (3c)
Under a nitrogen atmosphere, 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 1 (0.5 g, 1.87 mmol) and 4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}benoic acid 2c (0.57 g, 1.87 mmol) were added to dry THF (100 mL) at room temperature and the mixture was stirred for 10 min, which resulted in a clear solution. EDC·HCl (0.54 g, 2.80 mmol), HOBt (0.38 g, 2.80 mmol), and DIPEA (0.65 mL, 3.74 mmol) were added, and then the mixture was heated at 40 °C for 48 h. The progress of the reaction was monitored by TLC. Water (25 mL) was added, followed by EtOAc (25 mL). The organic phase was separated, and the aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic phase was washed with water (25 mL) followed by a brine solution (25 mL). The organic phase was dried over Na2SO4, filtered, and evaporated to dryness to afford the crude product that was purified on a silica gel column with a 0% to 10% gradient of methanol in DCM as an eluent to afford the desired compound as an off-white solid (0.08 g, 8% yield): HPLC purity 95.3% (tR = 2.71 min); 1H NMR (400 MHz, DMSO-d6) δ 12.90 (bs, 1H), 9.84 (s, 1H), 9.28 (d, J = 2.3 Hz, 1H), 9.08 (s, 1H), 8.69 (dd, J = 4.8, 1.6 Hz, 1H), 8.55 (d, J = 5.1 Hz, 1H), 8.49 (dt, J = 8.1, 2.0 Hz, 1H), 8.47–8.41 (m, 1H), 8.29 (s, 1H), 7.89 (dd, J = 7.9, 1.8 Hz, 1H), 7.54 (dd, J = 8.0, 4.8 Hz, 1H), 7.47 (d, J = 5.2 Hz, 1H), 7.43–7.33 (m, 2H), 7.32–7.20 (m, 2H), 2.34 (s, 3H), 2.25 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 167.5, 166.4, 162.1, 161.9 161.6, 160.6, 160.0, 151.9, 148.6, 141.7, 139.3, 138.4, 136.7, 134.9, 134.2, 132.9, 132.6, 130.6, 129.5, 128.5, 127.4, 125.4, 125.0, 124.6, 124.4, 108.3, 18.8, 18.7; LC-MS (ESI-QQQ) m/z 556.20 ([C28H22ClN7O2S + H]+ calcd 556.12); purity 96.3% (tR = 4.853 min).
N-(2-Chloro-6-methylphenyl)-2-{4-[(4-methylpiperazin-1-yl)methyl]benzamido}thiazole-5-carboxamide (3d)
The title compound was synthesized from 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 1 (0.62 g, 2.34 mmol) and 4-[(4-methylpiperazin-1-yl)methyl]benzoic acid 2d (0.5 g, 2.13 mmol), as described for the synthesis of 3c. The crude product was purified on a silica gel column using a 0% to 10% gradient of methanol in DCM as an eluent to yield the desired compound as an off-white solid (0.2 g, 19% yield): HPLC purity 96.8% (tR = 1.87 min); 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.39 (s, 1H), 8.13–8.03 (m, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.41 (dd, J = 7.5, 2.0 Hz, 1H), 7.35–7.20 (m, 3H), 3.55 (s, 2H), 2.41 (bs, 8H), 2.25 (s, 3H), 2.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.8, 162.4, 160.0, 144.1, 141.0, 139.2, 133.8, 132.8, 130.8, 129.5, 129.3, 128.8, 127.5, 127.3, 123.8, 61.9, 54.9, 52.7, 45.8, 18.7; LC-MS (ESI-QQQ) m/z 484.10 ([C24H26ClN5O2S + H]+ calcd 484.15); purity 97.9% (tR = 3.520 min).
3-Ethynylimidazo[1,2-b]pyridazine (5)
Compound 5 was prepared according to the previously reported method,44 with several modifications. To a solution of 3-bromoimidazo[1,2-b]pyridazine 4 (10.0 g, 50.5 mmol) in acetonitrile were added CuI (0.5 g, 2.63 mmol), Pd(PPh3)2Cl2 (1.8 g, 2.63 mmol), and TEA (21.0 mL, 150.6 mmol). The solution was purged with a nitrogen flow for 10 min, and then ethynyltrimethylsilane (21.0 mL, 151.8 mmol) was added. The mixture was heated to reflux overnight. After being cooled to rt, the reaction mixture was filtered to remove the undissolved solid. The solid was washed with copious amounts of acetonitrile. The filtrate was evaporated to dryness and then placed in methanol (300 mL). To this mixture was added K2CO3 (14.3 g, 103.5 mmol) at room temperature, and then the mixture was stirred for 4 h. The progress of the reaction was monitored by TLC. The reaction mixture was filtered to remove excess K2CO3. The solid was washed with a minimal amount of methanol. The filtrate was concentrated to dryness, dissolved in excess EtOAc, and then washed with water followed by a brine solution. The organic phase was dried over Na2SO4, filtered, and evaporated to dryness to afford the crude product, which was purified on a silica gel column using a 0% to 50% gradient of EtOAc in hexane to afford the desired product as a pale-brown solid (5.0 g, 69%): 1H NMR (400 MHz, CDCl3) δ 8.47 (dd, J = 4.4, 1.7 Hz, 1H), 8.03–7.96 (m, 2H), 7.12 (dd, J = 9.1, 4.5 Hz, 1H), 3.80 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 143.9, 139.0, 132.0, 128.4, 126.0, 117.9, 87.3, 70.6; LC-MS (ESI-QQQ) m/z 144.10 ([C8H5N3 + H]+ calcd 144.05); purity 99% (tR = 2.680 min).
Methyl 3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzoate (8)
Compound 8 was prepared according to the literature procedure,60 with few modifications. Methyl 3-iodo-4-methylbenzoate 6 (1.85 g, 6.71 mmol) was added to a stirred solution of 3-ethynylimidazo[1,2-b]pyridazine 5 (0.8 g, 5.59 mmol) in DMF (10 mL). The mixture underwent three cycles of vacuum/filling with nitrogen, and then CuI (0.21 g, 1.11 mmol), Pd(PPh3)4 (0.64 g, 0.55 mmol), and diisopropylethylamine (1.94 mL, 11.17 mmol) were added. The reaction mixture was stirred at 80 °C for 2 h before it was cooled to rt. Water (25 mL) was added, and the product extracted into EtOAc (3 × 25 mL). The organic layers were combined and washed with water (20 mL) followed by a brine solution (20 mL). The organic phase was dried over Na2SO4, filtered, and then evaporated to dryness to afford a gummy solid, which was then triturated with minimal acetonitrile to yield a solid. The solid was collected by filtration, washed with a minimal amount of acetonitrile, and dried under vacuum for 2 h to furnish the desired compound as an off-white solid (0.82 g, 50% yield): 1H NMR (400 MHz, CDCl3) δ 8.53 (dd, J = 4.4, 1.0 Hz, 1H), 8.25 (d, J = 1.9 Hz, 1H), 8.17–8.02 (m, 2H), 7.92 (dd, J = 8.0, 1.9 Hz, 1H), 7.33 (dt, J = 8.0, 0.7 Hz, 1H), 7.20 (dd, J = 9.1, 4.4 Hz, 1H), 3.91 (s, 3H), 2.62 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.4, 145.6, 144.4, 136.7, 133.2, 130.7, 129.9, 129.8, 128.0, 125.6, 122.5, 118.6, 113.7, 97.1, 79.9, 52.2, 21.1; LC-MS (ESI-QQQ) m/z 292.00 ([C17H13N3O2 + H]+ calcd 292.10); purity 99% (tR = 5.027 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzoic Acid (9)
Compound 9 was prepared on the basis of a literature procedure,60 with few modifications. Methyl 3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzoate 8 (0.81 g, 2.78 mmol) was placed in a 1:1 MeOH/THF mixture (120 mL). To this mixture was added a freshly prepared 1.0 M LiOH solution in water (15.0 mL), and the mixture was stirred at rt for 24 h. The pH was adjusted to 2 before the volume was reduced to 15% on a rotatory evaporator. The off-white solid that had appeared was collected by filtration, washed with copious amounts of ether, and dried under vacuum for 4 h to give the title compound (0.7 g, 91% yield): 1H NMR (400 MHz, DMSO-d6) δ 13.09 (bs, 1H), 8.70 (dd, J = 4.4, 1.6 Hz, 1H), 8.28–8.15 (m, 2H), 8.03 (d, J = 1.8 Hz, 1H), 7.87 (dd, J = 7.9, 1.9 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.37 (dd, J = 9.2, 4.4 Hz, 1H), 2.57 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.9, 145.5, 144.8, 140.1, 138.7, 132.4, 130.7, 130.1, 129.3, 126.5, 122.5, 119.5, 112.1, 96.6, 81.5, 20.9; LC-MS (ESI-QQQ) m/z 277.9 ([C16H11N3O2 + H]+ calcd 278.09); purity 99% (tR = 4.16 min).
Compounds 11a–c were prepared from compound 9 and the corresponding reactants 10a–c, respectively, using a similar method that was described for the synthesis of 3d.
3-[(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylphenyl](4-methylpiperazin-1-yl)methanone (11a)
Compound 11a was prepared using 3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzoic acid 9 (0.1 g, 0.36 mmol) and 1-methylpiperazine 10a (0.04 g, 0.54 mmol) as shown in Scheme 2. The desired product was obtained as an off-white solid (0.05 g, 39% yield): HPLC purity 96.6% (tR = 1.61 min); 1H NMR (400 MHz, CDCl3) δ 8.46 (dd, J = 4.4, 1.7 Hz, 1H), 8.03 (s, 1H), 7.99 (dd, J = 9.1, 1.6 Hz, 1H), 7.62 (d, J = 1.2 Hz, 1H), 7.31 (s, 1H), 7.30 (s, 1H), 7.12 (dd, J = 9.1, 4.5 Hz, 1H), 3.84–3.63 (m, 4H), 2.59–2.55 (m, 7H), 2.42 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 169.5, 143.9, 142.3, 139.7, 138.3, 133.0, 130.4, 129.9, 127.5, 125.9, 122.8, 117.7, 113.1, 96.8, 80.7, 54.7, 54.7, 45.5, 20.8; LC-MS (ESI-QQQ) m/z 519.3 ([C28H25F3N6O + H]+ calcd 519.2); purity 93.6% (tR = 4.24 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]phenyl}benzamide (11b)
Compound 11b was prepared using 3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzoic acid 9 (0.1 g, 0.36 mmol) and 4-[(4-methylpiperazin-1-yl)methyl]aniline 10b (0.07 g, 0.36 mmol) as shown in Scheme 2. The desired product was obtained as an off-white solid (0.02 g, 12% yield): HPLC purity 98.5% (tR = 2.00 min); 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 8.73 (dd, J = 4.5, 1.6 Hz, 1H), 8.26 (dd, J = 9.2, 1.6 Hz, 1H), 8.24 (s, 1H), 8.18 (d, J = 2.0 Hz, 1H), 7.93 (dd, J = 8.0, 2.0 Hz, 1H), 7.78–7.68 (m, 2H), 7.53 (d, J = 8.1 Hz, 1H), 7.40 (dd, J = 9.2, 4.5 Hz, 1H), 7.32–7.21 (m, 2H), 3.42 (s, 2H), 2.60 (s, 3H), 2.36 (bs, 8H), 2.17 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.7, 145.5, 143.6, 140.1, 138.7, 138.3, 134.0, 133.2, 130.6, 130.5, 129.6, 128.9, 126.6, 122.2, 120.7, 119.6, 112.2, 97.0, 81.5, 62.1, 55.1, 52.8, 46.1, 20.8; LC-MS (ESI-QQQ) m/z 465.0 ([C28H28N6O + H]+ calcd 465.2); purity 99% (tR = 3.557 min).
N-{6-[4-(2-Hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4-yl}-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzamide (11c)
Compound 11c was prepared from 3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzoic acid 9 (0.1 g, 0.36 mmol) and 2-[4-(6-amino-2-methylpyrimidin-4-yl)piperazin-1-yl]ethan-1-ol 10c (0.09 g, 0.36 mmol) as shown in Scheme 2. The desired product was obtained as an off-white solid (0.04 g, 22% yield): HPLC purity 96.7% (tR = 1.90 min); 1H NMR (400 MHz, DMSO-d6) δ 8.72 (dd, J = 4.5, 1.6 Hz, 1H), 8.30–8.21 (m, 2H), 8.07 (d, J = 1.8 Hz, 1H), 7.91 (dd, J = 8.0, 1.9 Hz, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.39 (dd, J = 9.2, 4.4 Hz, 1H), 6.09 (s, 2H), 5.44 (s, 1H), 4.43 (t, J = 5.8 Hz, 2H), 3.41 (t, J = 4.9 Hz, 4H), 2.75 (t, J = 5.8 Hz, 2H), 2.60 (s, 3H), 2.54 (d, J = 5.1 Hz, 4H), 2.15 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.9, 165.4, 164.9, 163.2, 145.5, 145.4, 140.2, 138.8, 132.1, 130.9, 130.0, 128.4, 126.6, 122.7, 119.6, 112.1, 96.4, 81.8, 80.0, 62.8, 56.5, 53.0, 44.1, 26.1, 21.0; LC-MS (ESI-QQQ) m/z 497.40 ([C27H28N8O2 + H]+ calcd 497.23); purity 99% (tR = 3.230 min).
N-[3-Bromo-5-(trifluoromethyl)phenyl]-3-iodo-4-methylbenzamide (14)
Under a nitrogen atmosphere, 3-iodo-4-methylbenzoic acid 12 (5.0 g, 19.08 mmol) was taken in SOCl2 (6.5 mL, 89.6 mmol) and then two drops of DMF was added at rt. The reaction mixture was stirred at reflux for 5 h before it was cooled to rt, and the excess SOCl2 was carefully removed. The crude material was co-evaporated with benzene and dried under vacuum to afford the desired acid chloride. The acid chloride was dissolved in anhydrous THF (20 mL) and then added dropwise to a stirred mixture of 3-bromo-5-(trifluoromethyl)aniline 13 (4.57 g, 19.08 mmol), diisopropylethylamine (3.97 mL, 22.8 mmol), and DMAP (0.23 g, 1.88 mmol) in THF at 0 °C. Upon completion of the addition, the reaction mixture was warmed to rt and stirred overnight. The reaction was quenched with water, and the product was extracted into EtOAc (3 × 50 mL). The combined organic extracts were washed with a brine solution (25 mL), dried over Na2SO4, filtered, and evaporated to dryness to afford a crude material that was purified on a silica gel column using a 0% to 50% gradient of EtOAc in hexane as the eluent to obtain the desired product as an off-white solid (7.6 g, 82% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 8.49–8.29 (m, 2H), 8.19 (s, 1H), 7.91 (dd, J = 7.9, 1.9 Hz, 1H), 7.67 (s, 1H), 7.50 (d, J = 7.9 Hz, 1H), 2.44 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.6, 145.9, 141.7, 137.9, 133.5, 131.6 (q, J = 32.4 Hz), 130.4, 128.3, 126.5, 123.6 (q, J = 274.7 Hz), 123.0 (q, J = 4.1 Hz), 122.7, 115.9 (q, J = 4.0 Hz), 101.6, 28.1; LC-MS (ESI-QQQ) m/z 483.90 ([C15H10BrF3INO + H]+ calcd 483.89); purity 99% (tR = 6.410 min).
N-[3-Bromo-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzamide (15)
This was prepared using N-[3-bromo-5-(trifluoromethyl)phenyl]-3-iodo-4-methylbenzamide 14 (2.0 g, 4.13 mmol) and 3-ethynylimidazo[1,2-b]pyridazine 5 (0.62 g, 4.33 mmol) as shown in Scheme 3 using a similar method that was described for the synthesis of 8. The desired product was obtained as an off-white solid (1.42 g, 69% yield): HPLC purity 95.6% (tR = 5.95 min); 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 8.72 (dd, J = 4.5, 1.5 Hz, 1H), 8.39 (s, 1H), 8.26 (dd, J = 9.3, 1.6 Hz, 1H), 8.24–8.19 (m, 2H), 7.94 (dd, J = 8.0, 2.0 Hz, 1H), 7.68 (d, J = 0.7 Hz, 1H), 7.65–7.58 (m, 1H), 7.58–7.54 (m, 1H), 7.39 (dd, J = 9.2, 4.4 Hz, 1H), 2.61 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.5, 144.3, 141.8, 138.7, 132.2, 131.9, 131.5 (q, J = 32.5 Hz), 130.6, 129.3, 129.1, 129.0, 126.5, 126.4, 123.6 (q, J = 274.7 Hz), 123.0 (q, J = 4.0 Hz), 122.6, 122.3, 119.5, 115.8 (q, J = 4.0 Hz), 96.8, 81.7, 20.9; LC-MS (ESI-QQQ) m/z 499.1 ([C23H14BrF3N4O + H]+ calcd 499.03); purity 95.8% (tR = 6.040 min).
3-Ethynyl-4-methylbenzoic Acid (16)
Methyl 3-iodo-4-methylbenzoate 6 (3.0 g, 10.86 mmol) was placed in anhydrous THF (30 mL). The solution underwent three cycles of vacuum/filling with nitrogen, and then CuI (0.17 g, 0.89 mmol), [Pd(PPh3)2Cl2] (0.4 g, 0.56 mmol), and ethynyltrimethylsilane (5.0 mL, 36.14 mmol) were added. The mixture was stirred overnight at rt. EtOAc (50 mL) was added followed by a 0.5 M aqueous NH4OH solution (100.0 mL). Aqueous and organic phases were separated. The organic phase was washed with 0.5 N HCl (50 mL) followed by a brine solution (25 mL), dried over Na2SO4, filtered, evaporated to dryness to afford a brown oil that was dissolved in a freshly prepared methanolic KOH solution (13 g of KOH flakes dissolved in 50 mL of MeOH), and stirred at rt for 2 h. EtOAc (100 mL) was added, and the undissolved solid was removed by filtration. The solid was washed with copious amounts of methanol. The filtrate was evaporated to dryness and placed in water (50 mL). The pH was adjusted to 5 using 0.5 N HCl, during which time an off-white solid was observed. The solid obtained was collected by filtration and washed with cold water followed by hexane. The solid was dried under vacuum for 4 h to obtain the desired compound as an off-white solid (1.5 g, 86%): 1H NMR (400 MHz, DMSO-d6) δ 13.10 (bs, 1H), 7.92 (d, J = 1.8 Hz, 1H), 7.84 (dd, J = 8.0, 1.8 Hz, 1H), 7.42 (d, J = 8.0 Hz, 1H), 4.47 (d, J = 0.9 Hz, 1H), 2.44 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 167.0, 145.4, 133.2, 130.4, 130.0, 129.5, 122.3, 85.8, 81.8, 20.8.
3-Ethynyl-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (19)
The title compound was prepared using the general procedure that was described for the synthesis of 3c, except for using 4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)aniline 17 (1.0 g, 3.66 mmol) and 3-ethynyl-4-methylbenzoic acid 16 (0.58 g, 3.66 mmol) as the starting materials, as depicted in Scheme 4. The desired compound was obtained as an off-white solid (0.9 g, 59% yield): HPLC purity 99.0% (tR = 3.49 min); 1H NMR (400 MHz, DMSO-d6) δ 11.94 (s, 1H), 10.80 (s, 1H), 8.37 (s, 1H), 8.19 (s, 1H), 8.11 (d, J = 2.0 Hz, 1H), 7.96 (dd, J = 8.0, 2.0 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 4.53 (s, 1H), 4.35 (s, 2H), 3.75–3.28 (m, 8H), 2.81 (s, 3H), 2.45 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.4, 144.7, 140.7, 134.2, 132.3, 131.7, 130.3, 129.2 (q, J = 29.8 Hz), 128.9, 128.3 124.3 (q, J = 274.7 Hz), 123.9, 122.2, 117.8 (q, J = 6.0 Hz), 86.0, 82.0, 55.6, 50.2, 48.6, 42.2, 20.7; LC-MS (ESI-QQQ) m/z 416.2 ([C23H24F3N3O + H]+ calcd 416.19); purity 99% (tR = 4.273 min).
4-Methyl-3-[(1-methyl-1H-imidazol-4-yl)ethynyl]-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (20)
The title compound was prepared following the general Sonogashira coupling, as described for the synthesis of 8, except for using 19 (0.25 g, 0.60 mmol) and 4-iodo-1-methyl-1H-imidazole (0.14 g, 0.66 mmol) as starting materials. Instead of Pd(PPh3)4, [Pd(PPh3)2Cl2] was used as a catalyst. The desired compound was obtained as an off-white solid (0.05 g, 18% yield): HPLC purity 99.9% (tR = 2.29 min); 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H), 8.22 (d, J = 2.2 Hz, 1H), 8.12 (d, J = 2.0 Hz, 1H), 8.06 (dd, J = 8.5, 2.2 Hz, 1H), 7.88 (dd, J = 8.0, 2.0 Hz, 1H), 7.74–7.66 (m, 2H), 7.59 (d, J = 1.2 Hz, 1H), 7.48 (d, J = 8.1 Hz, 1H), 3.69 (s, 3H), 3.56 (s, 2H), 2.50 (s, 3H), 2.47–2.22 (m, 8H), 2.16 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.2, 143.6, 139.4, 138.7, 132.5, 131.7, 130.6, 130.3, 128.9, 128.1, 127.9 (q, J = 30.3 Hz), 126.0, 124.8 (q, J = 275.7 Hz), 123.9, 123.2, 122.8, 117.7 (q, J = 6.0 Hz), 89.8, 86.5, 57.9, 55.2, 53.1, 46.2, 33.7, 20.8; LC-MS (ESI-QQQ) m/z 496.20 ([C27H28F3N5O + H]+ calcd 496.22); purity 99% (tR = 3.610 min).
Compounds 21a and 21b were prepared from 17 and the corresponding reactant 18 by a similar method that was described for the synthesis of 3c.
4-Methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (21a)
The title compound was obtained as an off-white solid (59%): HPLC purity 98.5% (tR = 2.85 min); 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.19 (d, J = 2.2 Hz, 1H), 8.02 (dd, J = 8.5, 2.2 Hz, 1H), 7.92–7.83 (m, 2H), 7.67 (d, J = 8.5 Hz, 1H), 7.39–7.28 (m, 2H), 3.54 (s, 2H), 2.37 (s, 3H), 2.33 (s, 8H), 2.14 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.0, 142.4, 138.8, 132.3, 132.0, 131.6, 129.4, 128.2, 127.8 (q, J = 29.3 Hz), 124.8 (q, J = 275.7 Hz), 123.9, 117.6 (q, J = 7.0 Hz), 57.9, 55.2, 53.1, 46.1, 21.5; LC-MS (ESI-QQQ) m/z 392.2 ([C21H24F3N3O + H]+ calcd 392.19); purity 99% (tR = 4.020 min).
4-Methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}-3-[{4-(pyridin-3-yl)pyrimidin-2-yl]amino}benzamide (21b)
The title compound was obtained as an off-white solid (10%): HPLC purity 96.2% (tR = 2.48 min); 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 9.31–9.23 (m, 1H), 9.14 (s, 1H), 8.68 (dt, J = 4.8, 1.3 Hz, 1H), 8.54 (dd, J = 5.2, 0.9 Hz, 1H), 8.44 (dt, J = 7.9, 1.4 Hz, 1H), 8.28 (s, 1H), 8.19 (d, J = 2.2 Hz, 1H), 8.06 (dd, J = 8.5, 2.2 Hz, 1H), 7.74 (dd, J = 8.4, 1.6 Hz, 1H), 7.69 (d, J = 8.5 Hz, 1H), 7.54–7.45 (m, 2H), 7.42 (d, J = 8.0 Hz, 1H), 3.56 (s, 2H), 2.35 (bs, 8H), 2.33 (s, 3H), 2.16 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.9, 162.1, 161.5, 160.0, 151.9, 150.3, 148.6, 146.9, 138.8, 138.6, 137.0, 134.7, 132.6 (q, J = 4.0 Hz), 132.4, 131.7, 130.8, 127.8 (q, J = 30.3 Hz), 124.8 (q, J = 275.7 Hz), 124.8, 124.3, 124.0, 117.7 (q, J = 4.0 Hz), 108.4, 57.9, 55.2, 53.1, 46.2, 18.7; LC-MS (ESI-QQQ) m/z 562.30 ([C30H30F3N7O + H]+ calcd 562.25); purity 97.7% (tR = 3.863 min).
3-Iodo-4-methoxy-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (23)
The title compound was prepared using a similar method that was described for the synthesis of 3c, except for using 17 (0.5 g, 1.83 mmol) and 3-iodo-4-methoxybenzoic acid 22 (0.53 g, 1.92 mmol) as the starting materials as shown in Scheme 4. The desired product was obtained as an off-white solid (0.86 g, 89% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.42 (d, J = 2.2 Hz, 1H), 8.17 (d, J = 2.2 Hz, 1H), 8.03 (dt, J = 8.6, 2.5 Hz, 2H), 7.69 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 8.7 Hz, 1H), 3.92 (s, 3H), 3.55 (s, 2H), 2.36 (d, J = 20.7 Hz, 8H), 2.15 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.2, 160.9, 138.7, 138.7, 132.4, 131.6, 130.5, 128.6, 127.8 (q, J = 30.3 Hz), 124.8 (q, J = 275.7 Hz), 123.9, 117.7 (q, J = 6.0 Hz), 111.5, 86.3, 57.9, 57.2, 55.2, 53.1, 46.2; LC-MS (ESI-QQQ) m/z 534.1 ([C21H23F3IN3O2 + H]+ calcd 534.08); purity 94.2% (tR = 4.237 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methoxy-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (24)
The title compound was prepared following the general Sonogashira coupling, as described for the synthesis of 8, except for using 23 (0.2 g, 0.37 mmol) and 5 (0.05 g, 0.37 mmol) as the starting material as shown in Scheme 4. The desired product was obtained as an off-white solid (0.04 g, 20% yield): HPLC purity 96.2% (tR = 2.55 min); 1H NMR (400 MHz, DMSO-d6) δ 10.49 (s, 1H), 8.71 (dd, J = 4.7, 1.6 Hz, 1H), 8.31–8.22 (m, 2H), 8.20 (dd, J = 4.6, 1.4 Hz, 2H), 8.08 (ddd, J = 10.7, 8.5, 2.3 Hz, 2H), 7.70 (d, J = 8.6 Hz, 1H), 7.42–7.34 (m, 1H), 7.31 (d, J = 8.9 Hz, 1H), 3.98 (s, 3H), 3.57 (s, 2H), 2.41 (bs, 8H), 2.21 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.7, 162.6, 145.4, 140.0, 138.8, 138.7, 132.9, 132.3, 131.7, 131.6, 127.9 (q, J = 29.3 Hz), 127.0, 126.5, 124.8 (q, J = 275.7 Hz), 124.0, 119.5, 117.7 (q, J = 6.0 Hz), 112.4, 112.0, 111.0, 94.6, 81.1, 57.8, 56.8, 55.0, 52.8, 45.8; LC-MS (ESI-QQQ) m/z 549.30 ([C29H27F3N6O2 + H]+ calcd 549.21); purity 99% (tR = 3.943 min).
3-Ethynyl-4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (26)
The title compound was prepared using a similar method that was described for the synthesis of 3c, except for using 3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline 25 (1.0 g, 4.14 mmol) and 3-ethynyl-4-methylbenzoic acid 16 (0.67 g, 4.14 mmol) as the starting materials as depicted in Scheme 5. The desired compound was obtained as an off-white solid (0.4 g, 25% yield): HPLC purity 94.6% (tR = 3.39 min); 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 8.26 (s, 1H), 8.18 (s, 1H), 8.15–8.07 (m, 2H), 7.91 (dd, J = 8.1, 1.8 Hz, 1H), 7.71 (s, 1H), 7.63–7.53 (m, 1H), 7.48 (t, J = 5.8 Hz, 1H), 4.53 (s, 1H), 2.45 (s, 3H), 2.16 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 144.9, 141.6, 138.4, 132.1, 132.0, 131.9, 131.5, 131.4, 131.1, 130.5, 129.3, 129.1, 128.8, 122.3, 115.4, 114.7, 86.0, 81.9, 20.7, 14.0; LC-MS (ESI-QQQ) m/z 384.10 ([C21H16F3N3O + H]+ calcd 384.12); purity 89.6% (tR = 4.510 min).
3-Ethynyl-4-methoxybenzoic Acid (28)
Compound 28 was prepared according to the general procedure described for the synthesis of 16 except for using methyl 3-iodo-4-methoxybenzoate 27 (1.0 g, 3.42 mmol) as the starting material. The desired product was obtained as an off-white solid (0.19 g, 32% yield): 1H NMR (400 MHz, DMSO-d6) δ 12.83 (bs, 1H), 7.95 (dd, J = 8.7, 2.3 Hz, 1H), 7.90 (d, J = 2.1 Hz, 1H), 7.17 (d, J = 8.8 Hz, 1H), 4.31 (d, J = 0.8 Hz, 1H), 3.90 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.7, 163.9, 135.0, 132.5, 123.4, 111.8, 111.3, 85.6, 79.5, 56.6.
3-Ethynyl-4-methoxy-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (29)
The title compound was prepared using a similar method that was described for the synthesis of 3c, except for using 28 (0.19 g, 1.10 mmol) and 17 (0.3 g, 1.10 mmol) as the starting materials as shown in Scheme 5. The title compound was obtained as an off-white solid (0.15 g, 32% yield): HPLC purity 97.4% (tR = 2.66 min); 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.19 (d, J = 2.2 Hz, 1H), 8.11 (d, J = 2.3 Hz, 1H), 8.03 (dt, J = 8.9, 2.6 Hz, 2H), 7.69 (d, J = 8.5 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 4.36 (s, 1H), 3.91 (s, 3H), 3.56 (s, 2H), 2.40 (bs, 8H), 2.20 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.7, 163.2, 138.8, 133.3, 132.3, 131.7, 131.1, 127.8 (q, J = 29.3 Hz), 126.7, 124.8 (q, J = 274.7 Hz), 123.9, 117.7 (q, J = 6.0 Hz), 111.7, 111.0, 85.7, 79.9, 57.9, 56.6, 55.1, 52.9, 45.9; LC-MS (ESI-QQQ) m/z 432.2 ([C23H24F3N3O2 + H]+ calcd 432.18); purity 99% (tR = 3.950 min).
N-(3-Iodo-4-methylphenyl)-4-[(4-methylpiperazin-1-yl)methyl]benzamide (31)
The title compound was prepared using a similar method that was described for the synthesis of 3c, except for using 4-[(4-methylpiperazin-1-yl)methyl]benzoic acid 2d (0.5 g, 2.14 mmol) and 3-iodo-4-methylaniline 30 (0.6 g, 2.56 mmol) as the starting materials as shown in Scheme 5. The title compound was obtained as an off-white solid (0.72 g, 75% yield): 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 2.2 Hz, 1H), 7.81 (dd, J = 8.1, 6.4 Hz, 3H), 7.57 (dd, J = 8.2, 2.3 Hz, 1H), 7.48–7.37 (m, 2H), 7.20 (dd, J = 8.2, 0.8 Hz, 1H), 3.57 (s, 2H), 2.54 (s, 8H), 2.41 (s, 3H), 2.35 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.4, 142.6, 137.5, 136.5, 133.4, 130.2, 129.6, 129.4, 127.1, 120.1, 100.6, 62.3, 54.9, 52.6, 45.7, 27.4; LC-MS (ESI-QQQ) m/z 450.1 ([C20H24IN3O + H]+ calcd 450.19); purity 99% (tR = 3.843 min).
N-[3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylphenyl]-4-[(4-methylpiperazin-1-yl)methyl]benzamide (32)
The title compound was prepared following the general Sonogashira coupling, as described for the synthesis of 8, except for using 31 (0.34 g, 0.768 mmol) and 5 (0.1 g, 0.70 mmol) as the starting materials as shown in Scheme 5. The title compound was obtained as an off-white solid (0.062 g, 19% yield): HPLC purity 97.9% (tR = 2.10 min); 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.71 (dd, J = 4.4, 1.6 Hz, 1H), 8.25 (dd, J = 9.2, 1.6 Hz, 1H), 8.22 (s, 1H), 8.08 (d, J = 2.2 Hz, 1H), 7.93 (d, J = 8.3 Hz, 2H), 7.72 (dd, J = 8.3, 2.3 Hz, 1H), 7.45 (d, J = 8.3 Hz, 2H), 7.38 (dd, J = 9.2, 4.5 Hz, 1H), 7.36–7.31 (m, 1H), 3.54 (s, 2H), 2.50 (s, 3H), 2.39 (s, 8H), 2.19 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.4, 145.0, 142.3, 139.5, 138.0, 137.2, 134.4, 133.4, 130.0, 128.7, 127.6, 126.0, 122.5, 121.5, 121.1, 118.9, 111.9, 97.1, 80.1, 61.5, 54.6, 52.4, 45.5, 19.7; LC-MS (ESI-QQQ) m/z 465.40 ([C28H28N6O + H]+ calcd 465.23); purity 99% (tR = 3.673 min).
General Procedure for the Synthesis of 33a–h. Procedure for N-[3-(1H-Imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzamide (33a)
Compound 33a was prepared according to the previously reported methods for similar compounds,46,47 with several modifications. N-[3-Bromo-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzamide 15 (3.0 g, 6.00 mmol) and 1H-imidazole (0.45 g, 6.61 mmol) were placed in dry DMSO (50 mL) in a pressure tube. The solution was purged with a nitrogen flow for 10 min; CuI (0.17 g, 0.90 mmol), K2CO3 (2.5 g, 18.0 mmol), and 8-hydroxyquinoline (0.13 g, 0.90 mmol) were added, and purging was continued for an additional 10 min. The pressure tube was then sealed tightly and stirred at 100 °C for 18 h. Upon being cooled to rt, the reaction mixture was poured into ice-cold water (∼50 mL) and allowed to stir for 30 min, during which time a pale yellow solid was observed. The solid was collected by filtration and then dissolved in 10% MeOH in DCM (100 mL). The undissolved solid was removed by filtration. The filtrate was evaporated to dryness to afford the crude product, which was purified on a silica gel column using a 0% to 10% gradient of methanol in DCM as an eluent to afford the desired product as a pale yellow solid (1.67 g, 57% yield): HPLC purity 98.4% (tR = 3.16 min); 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.73 (dt, J = 4.5, 1.4 Hz, 1H), 8.34 (s, 2H), 8.30–8.19 (m, 4H), 7.98 (dd, J = 8.1, 1.9 Hz, 1H), 7.81 (d, J = 9.6 Hz, 2H), 7.59 (d, J = 8.1 Hz, 1H), 7.40 (ddd, J = 9.2, 4.5, 1.1 Hz, 1H), 7.18 (s, 1H), 2.63 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.5, 144.3, 141.7, 140.2, 138.8, 138.5, 132.3, 131.3 (q, J = 32.2 Hz), 130.9, 130.7, 130.6, 129.0, 126.6, 124.1 (q, J = 273.7 Hz), 122.4, 119.6, 116.0, 115.2 (q, J = 4.0 Hz), 112.8, 112.7, 112.2, 100.3, 96.8, 81.7, 20.9; LC-MS (ESI-QQQ) m/z 487.20 ([C26H17F3N6O + H]+ calcd 487.14); purity 99% (tR = 4.510 min).
N-[3-(4-Ethyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzamide (33b)
Compound 33b was synthesized from 15 (0.1 g, 0.20 mmol) and 4-ethyl-1H-imidazole (0.03 g, 0.30 mmol) according to the general procedure for the synthesis of 33a–h. After the reaction had reached completion, the reaction mixture was cooled to rt and a 10% NH4OH solution was added. The product was extracted into EtOAc (3 × 25 mL). The combined organic layers were washed with water followed by a 10% NH4OH solution, dried over Na2SO4, filtered, and evaporated to dryness. The crude product was purified on a silica gel column using a 0% to 10% gradient of methanol in DCM as an eluent to yield the title compound as a pale yellow solid (3.0 mg, 3% yield): HPLC purity 97.5% (tR = 4.22 min); 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.82–8.67 (m, 1H), 8.38–8.12 (m, 6H), 7.98 (dd, J = 8.0, 2.0 Hz, 1H), 7.77 (s, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.52 (s, 1H), 7.40 (dd, J = 9.2, 4.4 Hz, 1H), 2.62 (s, 3H), 2.56 (q, J = 7.4 Hz, 2H), 1.22 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.6, 144.3, 141.7, 138.8, 138.5, 132.3, 131.3 (q, J = 32.3 Hz), 131.1, 131.0, 130.7, 130.6, 129.0, 126.6, 124.1, 122.4, 119.6, 115.4, 114.7 (q, J = 4.0 Hz), 114.7, 113.6, 112.2, 112.2, 96.8, 81.7, 21.7, 20.9, 13.8; LC-MS (ESI-QQQ) m/z 515.30 ([C28H21F3N6O + H]+ calcd 515.17); purity 87.5% (tR = 4.663 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-N-[3-(4-isopropyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-4-methylbenzamide (33c)
Compound 33c was prepared from 15 (0.1 g, 0.20 mmol) and 4-isopropyl-1H-imidazole (0.03 g, 2.40 mmol) using a similar method that was described for the synthesis of 33b. The desired product was obtained as a pale yellow solid (0.03 g, 29% yield): HPLC purity 98.4% (tR = 3.99 min); 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 8.73 (dd, J = 4.5, 1.4 Hz, 1H), 8.37–8.13 (m, 6H), 7.97 (dd, J = 8.0, 1.9 Hz, 1H), 7.78 (d, J = 2.1 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.49 (s, 1H), 7.40 (ddd, J = 9.3, 4.4, 1.2 Hz, 1H), 2.84 (septet, J = 6.9 Hz, 1H), 2.62 (s, 3H), 1.24 (d, J = 6.8, Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.6, 144.3, 141.6, 138.8, 138.5, 135.3, 132.3, 131.3 (q, J = 32.2 Hz), 130.8, 130.7, 130.6, 129.0, 128.2, 126.6, 124.2 (q, J = 272.6 Hz), 122.4, 119.6, 115.4, 114.7 (q, J = 4.0 Hz), 112.6, 112.3, 112.2, 96.8, 81.7, 27.8, 22.7, 20.9; LC-MS (ESI-QQQ) m/z 529.2 ([C29H23F3N6O + H]+ calcd 529.19); purity 99% (tR = 4.847 min).
N-[3-(4-Cyclopropyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzamide (33d)
Compound 33d was prepared from 15 (0.03 g, 0.06 mmol) and 4-cyclopropyl-1H-imidazole (8.0 mg, 0.07 mmol) using a similar method that was described for the synthesis of 33b. The desired product was obtained as a pale yellow solid (20.0 mg, 29% yield): HPLC purity 98.1% (tR = 5.97 min); 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.73 (dd, J = 4.5, 1.5 Hz, 1H), 8.27 (dd, J = 16.4, 2.2 Hz, 4H), 8.17 (s, 2H), 7.97 (dd, J = 8.0, 2.0 Hz, 1H), 7.74 (s, 1H), 7.56 (dd, J = 13.1, 4.8 Hz, 2H), 7.40 (dd, J = 9.2, 4.5 Hz, 1H), 2.62 (s, 3H), 1.87 (s, 1H), 0.90–0.62 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.6, 144.3, 141.7, 138.8, 138.4, 135.3, 132.3, 131.5, 131.3 (q, J = 32.3 Hz), 130.7, 130.6, 129.0, 126.6, 125.5, 124.1 (q, J = 272.7 Hz), 122.4, 119.6, 115.4, 114.8, 113.3, 113.0, 112.2, 96.8, 81.7, 20.9, 9.4, 7.5; LC-MS (ESI-QQQ) m/z 527.30 ([C29H21F3N6O + H]+ calcd 527.17); purity 99% (tR = 4.910 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-[3-(3-methyl-1H-pyrrol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (33e)
Compound 33e was prepared from 15 (0.1 g, 0.20 mmol) and 3-methyl-1H-pyrrole (0.02 g, 0.30 mmol) using a similar method that was described for the synthesis of 33b. Instead of conventional heating, the reaction was carried out in a microwave at 100 °C for 1 h. The desired product was obtained as a pale yellow solid (4.0 mg, 4% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.73 (d, J = 4.8 Hz, 1H), 8.29–8.26 (m, 1H), 8.25 (s, 3H), 8.08 (s, 1H), 7.98 (d, J = 10.1 Hz, 1H), 7.62–7.53 (m, 2H), 7.42–7.38 (m, 1H), 7.33 (d, J = 2.6 Hz, 1H), 7.21 (s, 1H), 6.17 (s, 1H), 2.62 (s, 3H), 2.10 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.6, 144.3, 141.6, 141.3, 138.8, 134.3, 132.5, 131.5, 131.4, 130.7, 130.6, 129.0, 126.6, 124.3 (q, J = 273.7 Hz), 122.3, 121.6, 119.6, 119.4, 117.3, 114.1, 113.5, 112.2, 101.9, 96.8, 81.7, 20.9, 12.3; LC-MS (ESI-QQQ) m/z 500.30 ([C28H20F3N5O + H]+ calcd 500.16); purity 76.6% (tR = 6.180 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-[3-(4-methylpiperazin-1-yl)-5-(trifluoromethyl)phenyl]benzamide (33f)
Compound 33f was prepared from 15 (0.1 g, 0.20 mmol) and 1-methylpiperazine (0.03 g, 0.30 mmol) using a similar method that was described for the synthesis of 33b. The desired product was obtained as a pale yellow solid (3.0 mg, 4% yield): HPLC purity 97.5% (tR = 2.97 min); 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.73 (d, J = 7.9 Hz, 1H), 8.31–8.22 (m, 2H), 8.21 (t, J = 1.9 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.70 (d, J = 1.6 Hz, 1H), 7.65 (d, J = 2.0 Hz, 1H), 7.55 (d, J = 8.3 Hz, 1H), 7.40 (dd, J = 9.2, 4.4 Hz, 1H), 6.96 (s, 1H), 3.23 (s, 4H), 2.61 (s, 3H), 2.51 (s, 4H), 2.25 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.6, 151.6, 145.1, 143.5, 140.6, 139.7, 138.3, 132.3, 130.2, 130.1, 130.1, 129.9, 128.5, 126.1, 124.4 (q, J = 273.7 Hz), 121.8, 119.1, 111.7, 109.6, 106.7, 96.4, 81.1, 54.3, 47.5, 45.6, 20.4; LC-MS (ESI-QQQ) m/z 519.30 ([C28H25F3N6O + H]+ calcd 519.20); purity 93.5% (tR = 4.247 min).
N-[3-(1H-Benzo[d]imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methylbenzamide (33g)
Compound 33g was prepared from 15 (0.1 g, 0.20 mmol) and 1H-benzo[d]imidazole (0.03 g, 0.24 mmol) using a similar method that was described for the synthesis of 33b. The desired product was obtained as a pale yellow solid (15.0 mg, 14% yield): HPLC purity 97.6% (tR = 4.77 min); 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 8.73 (dd, J = 4.5, 1.5 Hz, 2H), 8.49 (d, J = 2.0 Hz, 1H), 8.36 (d, J = 1.8 Hz, 1H), 8.26 (td, J = 5.9, 2.4 Hz, 3H), 7.99 (dd, J = 8.0, 2.0 Hz, 1H), 7.90–7.74 (m, 3H), 7.59 (d, J = 8.2 Hz, 1H), 7.40 (ddd, J = 14.4, 10.3, 7.2 Hz, 3H), 2.62 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.5, 145.6, 144.5, 144.4, 143.8, 141.8, 138.8, 137.6, 132.4, 131.6 (q, J = 32.3 Hz), 131.4, 130.7, 130.7, 129.1, 126.6, 125.5, 124.3, 124.1 (q, J = 273.7 Hz), 122.8, 122.4, 120.7, 119.6, 118.5, 115.6, 115.5 (q, J = 4.0 Hz), 112.2, 111.2, 96.8, 81.8, 20.9; LC-MS (ESI-QQQ) m/z 537.20 ([C30H19F3N6O + H]+ calcd 537.16); purity 92% (tR = 5.613 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-[3-(4-phenyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (33h)
Compound 33h was prepared from 15 (0.1 g, 0.20 mmol) and 4-phenyl-1H-imidazole (0.03 g, 0.24 mmol) using a similar method that was described for the synthesis of 33b. The desired product was obtained as a pale yellow solid (0.04 g, 36% yield): HPLC purity 97.5% (tR = 5.05 min); 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 8.73 (dd, J = 4.4, 1.4 Hz, 1H), 8.51–8.34 (m, 3H), 8.31–8.16 (m, 4H), 7.98 (dd, J = 8.0, 1.9 Hz, 1H), 7.93–7.82 (m, 3H), 7.58 (d, J = 8.1 Hz, 1H), 7.47–7.34 (m, 3H), 7.26 (t, J = 7.3 Hz, 1H), 2.62 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.5, 144.4, 142.7, 141.7, 140.2, 138.8, 138.2, 136.6, 134.2, 132.3, 131.4 (q, J = 32.3 Hz), 130.7, 130.6, 129.1, 129.0, 127.4, 126.6, 125.0, 124.1 (q, J = 273.7 Hz), 122.4, 119.6, 115.8, 115.3, 114.6, 112.6, 112.1, 96.8, 81.7, 20.9; LC-MS (ESI-QQQ) m/z 563.2 ([C32H21F3N6O + H]+ calcd 563.17); purity 99% (tR = 5.793 min).
3-Iodo-4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (35a)
The title compound was synthesized following the procedure described for the synthesis of 14 except for using 3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline 25 (2.0 g, 8.3 mmol) and 3-iodo-4-methylbenzoic acid 12 (2.5 g, 9.12 mmol) as the starting materials as depicted in Scheme 7. The desired product was obtained as an off-white solid (3.83 g, 95% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.45 (s, 1H), 8.27 (s, 1H), 8.20 (s, 1H), 8.13 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.72 (s, 1H), 7.55–7.40 (m, 2H), 2.44 (s, 3H), 2.18 (d, J = 2.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.6, 145.8, 141.6, 139.4, 138.4, 137.9, 135.4, 133.6, 131.3 (q, J = 32.2 Hz), 130.3, 128.3, 124.1 (q, J = 272.7 Hz), 115.4, 114.7 (d, J = 4.1 Hz), 114.6, 112.1 (d, J = 4.0 Hz), 101.6, 28.0, 14.0; LC-MS (ESI-QQQ) m/z 486.00 ([C19H15F3IN3O + H]+ calcd 486.02); purity 97.5% (tR = 4.773 min).
3-Iodo-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (35b)
The title compound was synthesized following the procedure described for the synthesis of 14 except for using 3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline 25 (2.43 g, 10.07 mmol) and 3-iodobenzoic acid 34a (2.5 g, 10.07 mmol) as the starting materials as depicted in Scheme 7. The desired product was obtained as an off-white solid (4.06 g, 86% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 8.35 (q, J = 1.8 Hz, 1H), 8.28–8.07 (m, 3H), 8.00 (dq, J = 7.8, 1.3 Hz, 2H), 7.74 (d, J = 1.9 Hz, 1H), 7.48 (q, J = 1.2 Hz, 1H), 7.38 (td, J = 7.8, 1.5 Hz, 1H), 2.22–2.13 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.9, 141.5, 141.1, 139.4, 138.4, 136.5, 136.4, 135.4, 131.3 (q, J = 32.2 Hz), 131.18, 127.75, 124.0 (d, J = 273.7 Hz), 115.4, 114.8 (d, J = 4.0 Hz), 114.6, 112.3, 95.3, 14.0; LC-MS (ESI-QQQ) m/z 472.00 ([C18H13F3IN3O + H]+ calcd 472.01); purity 99% (tR = 4.457 min).
3-Iodo-4-methoxy-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (35c)
The title compound was synthesized following the procedure described for the synthesis of 14 except for using 3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline 25 (2.0 g, 7.2 mmol) and 3-iodo-4-methoxybenzoic acid 34b (1.73 g, 7.2 mmol) as the starting materials as depicted in Scheme 7. The desired product was obtained as an off-white solid (2.63 g, 73% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H), 8.46 (dd, J = 2.3, 1.2 Hz, 1H), 8.27 (t, J = 1.9 Hz, 1H), 8.20 (t, J = 1.4 Hz, 1H), 8.13 (d, J = 1.8 Hz, 1H), 8.06 (ddd, J = 8.6, 2.3, 1.2 Hz, 1H), 7.74–7.67 (m, 1H), 7.47 (d, J = 1.5 Hz, 1H), 7.17 (dd, J = 8.8, 1.3 Hz, 1H), 3.93 (s, 3H), 2.18 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.4, 161.2, 141.7, 139.4, 138.7, 138.3, 135.4, 131.2 (q, J = 32.3 Hz), 130.6, 128.2, 124.1 (q, J = 273.7 Hz), 115.3, 114.7 (q, J = 5.0 Hz), 114.6, 112.0, 111.6, 86.3, 57.3, 14.0; LC-MS (ESI-QQQ) m/z 502.00 ([C19H15F3IN3O2 + H]+ calcd 502.02); purity 99% (tR = 4.450 min).
Compounds 36a–c were prepared from 5 and the corresponding reactant 35 by a similar method that was described for the synthesis of 8.
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (36a)
The title compound was obtained as a pale yellow solid (49% yield): HPLC purity 98.7% (tR = 3.25 min); 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.73 (dd, J = 4.5, 1.6 Hz, 1H), 8.32–8.29 (m, 1H), 8.27 (dd, J = 9.2, 1.6 Hz, 1H), 8.25 (d, J = 2.1 Hz, 2H), 8.22 (s, 1H), 8.18 (d, J = 1.7 Hz, 1H), 7.97 (dd, J = 8.0, 2.0 Hz, 1H), 7.74 (d, J = 1.5 Hz, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.50 (s, 1H), 7.40 (dd, J = 9.2, 4.5 Hz, 1H), 2.62 (s, 3H), 2.18 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 145.6, 144.3, 141.7, 140.2, 140.1, 139.4, 138.8, 138.4, 135.5, 132.3, 131.3 (q, J = 33.3 Hz), 130.7, 130.6, 129.0, 126.6, 124.1 (q, J = 273.7 Hz), 122.4, 119.6, 115.5, 114.8, 114.8, 112.1 (q, J = 4. 0 Hz), 96.8, 81.7, 20.9, 14.1; LC-MS (ESI-QQQ) m/z 501.30 ([C27H19F3N6O + H]+ calcd 501.16); purity 99% (tR = 4.427 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (36b)
The title compound was obtained as a pale yellow solid (61% yield): HPLC purity 99.5% (tR = 2.88 min); 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 8.72 (dd, J = 4.4, 1.6 Hz, 1H), 8.33–8.20 (m, 5H), 8.17 (d, J = 1.7 Hz, 1H), 8.05 (dt, J = 7.9, 1.5 Hz, 1H), 7.86 (dt, J = 7.7, 1.4 Hz, 1H), 7.75 (t, J = 1.8 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.49 (s, 1H), 7.40 (dd, J = 9.2, 4.4 Hz, 1H), 2.18 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.4, 145.5, 141.6, 140.1, 139.4, 139.0, 138.4, 135.4, 135.1, 134.9, 131.3 (q, J = 32.3 Hz), 130.5, 129.9, 129.1, 126.6, 124.1 (q, J = 273.7 Hz), 122.5, 119.6, 115.5, 114.8, 114.7 (q, J = 6.0 Hz), 112.3, 112.0, 97.8, 78.1, 14.0; LC-MS (ESI-QQQ) m/z 487.20 ([C26H17F3N6O + H]+ calcd 487.14); purity 99% (tR = 4.197 min).
3-(Imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methoxy-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]benzamide (36c)
The title compound was obtained as a pale yellow solid (55% yield): HPLC purity 98.2% (tR = 2.80 min); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.72 (dd, J = 4.4, 1.5 Hz, 1H), 8.34–8.19 (m, 5H), 8.17 (d, J = 1.7 Hz, 1H), 8.11 (dd, J = 8.8, 2.4 Hz, 1H), 7.73 (s, 1H), 7.51 (s, 1H), 7.40 (dd, J = 9.2, 4.5 Hz, 1H), 7.34 (d, J = 8.9 Hz, 1H), 4.00 (s, 3H), 2.19 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.9, 162.8, 145.4, 141.8, 140.1, 139.5, 138.7, 138.4, 135.5, 132.8, 131.7, 131.2 (q, J = 32.2 Hz), 126.6, 124.1 (q, J = 273.7 Hz), 119.5, 115.4, 114.7 (q, J = 4.04 Hz), 112.4, 112.3, 112.1, 112.0, 112.0, 111.1, 94.5, 81.2, 56.9, 14.1; LC-MS (ESI-QQQ) m/z 517.20 ([C27H19F3N6O2 + H]+ calcd 517.17); purity 99% (tR = 4.137 min).
Compounds 37a and 37b were prepared from 13 and the corresponding reactant 34 by a similar method that was described for the synthesis of 14.
N-[3-Bromo-5-(trifluoromethyl)phenyl]-3-iodobenzamide (37a)
The title compound was obtained as an off-white solid (76% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.34 (dd, J = 6.4, 1.7 Hz, 2H), 8.19 (d, J = 1.9 Hz, 1H), 8.02–7.92 (m, 2H), 7.70–7.64 (m, 1H), 7.36 (td, J = 7.9, 1.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 164.9, 141.6, 141.1, 136.4, 136.4, 131.6 (q, J = 32.4 Hz), 131.1, 127.8, 126.5, 123.5 (q, J = 273.7 Hz), 123.1 (q, J = 4.0 Hz), 122.7, 115.9 (q, J = 4.2 Hz), 95.2; LC-MS (ESI-QQQ) m/z 469.8 ([C14H8BrF3INO + H]+ calcd 469.88); purity 99% (tR = 6.240 min).
N-[3-Bromo-5-(trifluoromethyl)phenyl]-3-iodo-4-methoxybenzamide (37b)
The title compound was obtained as an off-white solid (78% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.42 (dd, J = 2.5, 1.1 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 8.19 (s, 1H), 8.06–7.95 (m, 1H), 7.63 (d, J = 2.1 Hz, 1H), 7.14 (d, J = 8.8 Hz, 1H), 3.92 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.4, 161.2, 141.9, 138.8, 131.5 (q, J = 32.4 Hz), 130.6, 128.1, 126.3, 123.6 (q, J = 273.1 Hz), 122.7 (q, J = 3.8 Hz), 122.6, 115.8 (q, J = 4.1 Hz), 111.5, 86.3, 57.2; LC-MS (ESI-QQQ) m/z 499.9 ([C15H10BrF3INO2 + H]+ calcd 499.89); purity 98.8% (tR = 6.197 min).
Compounds 38a and 38b were prepared from 5 and the corresponding reactant 37 by a similar method that was described for the synthesis of 8.
N-[3-Bromo-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)benzamide (38a)
The title compound was obtained as a rust-colored solid (63% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H), 8.73 (dd, J = 4.5, 1.6 Hz, 1H), 8.39 (s, 1H), 8.34–8.19 (m, 4H), 8.10–7.99 (m, 1H), 7.91–7.83 (m, 1H), 7.74–7.63 (m, 2H), 7.41 (dd, J = 9.2, 4.4 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 165.5, 145.5, 141.7, 140.1, 139.0, 135.1, 135.0, 131.6 (q, J = 32.4 Hz), 130.5, 129.9, 129.1, 126.6, 126.5, 123.6 (q, J = 273.7 Hz), 123.1, 122.7, 122.5, 119.7, 116.0, 112.0, 97.8, 78.1; LC-MS (ESI-QQQ) m/z 485.00 ([C22H12BrF3N4O + H]+ calcd 485.01); purity 89.2% (tR = 5.850 min).
N-[3-Bromo-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methoxybenzamide (38b)
The title compound was obtained as an off-white solid (39% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 8.71 (dd, J = 4.4, 1.3 Hz, 1H), 8.38 (d, J = 1.8 Hz, 1H), 8.31–8.18 (m, 4H), 8.12–8.04 (m, 1H), 7.68 (s, 1H), 7.39 (dd, J = 8.6, 4.4 Hz, 1H), 7.33 (dd, J = 8.9, 1.3 Hz, 1H), 3.99 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.0, 162.8, 145.4, 142.0, 139.0, 132.9, 131.7, 131.7, 131.6 (q, J = 33.3 Hz) 126.7, 126.5, 126.4, 125.3 (q, J = 274.7 Hz), 122.9, 122.8 (q, J = 4.0 Hz), 122.7, 119.5, 115.9 (q, J = 4.0 Hz), 112.1, 111.1, 94.5, 81.2, 56.9; LC-MS (ESI-QQQ) m/z 515.00 ([C23H14BrF3N4O2 + H]+ calcd 515.03); purity 90% (tR = 5.763 min).
Compounds 40a–c were prepared following the general procedure described for 33a–h.
N-[3-(1H-Imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)benzamide (40a)
Compound 40a was prepared from 38a (0.1 g, 0.20 mmol) and 1H-imidazole (15 mg, 0.22 mmol) as depicted in Scheme 8. The desired product was obtained as a pale yellow solid (14 mg, 14% yield): HPLC purity 97.5% (tR = 2.80 min); 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 8.72 (dd, J = 4.4, 1.4 Hz, 1H), 8.33 (s, 1H), 8.33–8.13 (m, 5H), 8.06 (d, J = 8.1 Hz, 1H), 7.96–7.83 (m, 2H), 7.81 (s, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.40 (dd, J = 9.3, 4.4 Hz, 1H), 7.27 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 165.5, 145.5, 141.6, 139.0, 135.1, 135.1, 135.0, 131.6, 131.5 (q, J = 32.3 Hz), 130.5, 130.1, 130.0, 129.1, 126.6, 124.1 (q, J = 273.7 Hz), 122.7, 122.5, 119.6, 116.1, 115.2 (q, J = 4.0 Hz), 112.9, 112.9, 112.0, 97.8, 78.1; LC-MS (ESI-QQQ) m/z 473.20 ([C25H15F3N6O + H]+ calcd 473.13); purity 99% (tR = 4.250 min).
N-[3-(4-Cyclopropyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methoxybenzamide (40b)
Compound 40b was prepared from 38b (85 mg, 0.16 mmol) and 4-cyclopropyl-1H-imidazole (21 mg, 0.20 mmol) as depicted in Scheme 8. The desired product was obtained as a pale yellow solid (20 mg, 22% yield): HPLC purity 95.6% (tR = 3.19 min); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.71 (dd, J = 4.4, 1.5 Hz, 1H), 8.31–8.23 (m, 3H), 8.21 (s, 1H), 8.16 (s, 2H), 8.11 (dd, J = 8.8, 2.4 Hz, 1H), 7.72 (s, 1H), 7.54 (s, 1H), 7.39 (dd, J = 9.2, 4.4 Hz, 1H), 7.34 (d, J = 8.9 Hz, 1H), 3.99 (s, 3H), 1.86 (td, J = 8.3, 4.3 Hz, 1H), 0.82 (dd, J = 8.3, 2.4 Hz, 2H), 0.71 (dd, J = 5.0, 2.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 164.9, 162.8, 145.7, 145.4, 141.8, 140.0, 138.7, 138.4, 135.4, 135.4, 132.8, 131.7, 131.3 (q, J = 32.3 Hz), 126.6, 124.1 (q, J = 273.7 Hz), 119.5, 115.3, 114.7 (q, J = 4.0 Hz), 113.2, 112.3, 112.1, 112.0, 111.1, 94.5, 81.2, 56.9, 9.4, 7.5; LC-MS (ESI-QQQ) m/z 543.3 ([C29H21F3N6O2 + H]+ calcd 543.17); purity 95.9% (tR = 4.590 min).
N-[3-(4-Cyclopropyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-(imidazo[1,2-b]pyridazin-3-ylethynyl)benzamide (40c)
Compound 40c was prepared from 38a (0.1 g, 0.20 mmol) and 4-cyclopropyl-1H-imidazole (26 mg, 0.24 mmol) as depicted in Scheme 8. The desired product was obtained as a pale yellow solid (20 mg, 19% yield): HPLC purity 95.0% (tR = 3.4 min); 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.73 (dd, J = 4.5, 1.6 Hz, 1H), 8.32–8.21 (m, 4H), 8.17 (s, 2H), 8.06 (dd, J = 7.9, 1.5 Hz, 1H), 7.87 (d, J = 7.7 Hz, 1H), 7.76 (s, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.55 (s, 1H), 7.41 (dd, J = 9.2, 4.4 Hz, 1H), 1.88 (ddd, J = 8.2, 5.9, 3.3 Hz, 1H), 0.91–0.66 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 165.4, 145.7, 145.5, 141.6, 140.1, 139.0, 138.4, 135.3, 135.1, 135.0, 131.3 (q, J = 32.3 Hz), 130.5, 130.0, 129.1, 126.6, 124.1 (q, J = 273.7 Hz), 122.5, 119.7, 115.4, 114.8 (q, J = 4.0 Hz), 113.2, 112.3, 112.0, 97.8, 78.1, 9.4, 7.5; LC-MS (ESI-QQQ) m/z 513.2 ([C28H19F3N6O + H]+ calcd 513.16); purity 92.5% (tR = 4.670 min).
Docking Studies
Molecular docking simulations were performed using AutoDock Vina 1.1.2. Pymol 2.3.1 was employed to analyze the docking results.61 The crystal structures of wild type BCR-ABL and BCR-ABLT315I were taken from PDB entries 3OXZ and 3IK3, respectively. The protein structure was prepared by adding polar hydrogens, deleting water molecules, and adding charges. The grid box was prepared on the basis of the ligand sites that were defined in the crystal structure. The coordinate center of the search space for 3OXZ was set to 12.110, −5.407, 15.591 (x, y, z). The x, y, and z dimensions were set to 22, 24, and 34, respectively. The coordination center of the search space for 3IK3 was set to 6.487, 1.061, 17.621 (x, y, z), and the x, y, and z dimensions were set to 22, 30, and 26, respectively. For both structures, a grid-point spacing of 0.375 Å was applied. The exhaustiveness was set to 48, and the maximum number of binding modes was set to 100. Other docking parameters were kept to the default values. Docking calculations were performed with full flexibility of the ligand inside the search space.
Biological Characterization of Compounds
K-562 and K-562-T315I
Leukemia cell line K-562 was purchased from ATCC and maintained as recommended by ATCC (Manassas, VA). Briefly, K-562 cells were cultured in suspension in RPMI1640 (ThermoFisher Scientific) supplemented with 10% fetal bovine serum and Pen/Strep/l-glutamine. The K-562-T315I cell line was derived from the K-562 line by CRISPR. Briefly, 1 million K-562 cells were seeded in six-well plates and transfected with Lipofectamine 2000 and 1 μg of CRISPR/Cas9 vector [pSpCas9(BB)-2A-GFP] (Addgene 48138) incorporating the guide sequence (CTCAGTGATGATATAGAACG), and Lipofectamine RNAiMax (Invitrogen 13778500, Thermo Fisher Scientific) and 4 μg of ssDNA donors (1 μg of each donor, HDR template 1, CGTGTTGAAGTCCTCGTTGTCTTGTTGGCAGGGGTCTGCACCCGGGAGCCCCCGTTCTATATCATCATTGAGTTCATGACCTACGGGAACCTCCTGGACT; HDR template 2, TTCAGTTGGGAGCGGAGCCACGTGTTGAAGTCCTCGTTGTCTTGTTGGCAGGGGTCTGCACCCGGGAGCCCCCGTTCTATATCATCATTGAGTTCATGAC; HDR template 3, CGTGTTGAAGTCCTCGTTGTCTTGTTGGCAGGGGTCTGCACCCGGGAGCCACCGTTCTATATCATCATTGAGTTCATGACCTACGGGAACCTCCTGGACT; and HDR template 4, TTCAGTTGGGAGCGGAGCCACGTGTTGAAGTCCTCGTTGTCTTGTTGGCAGGGGTCTGCACCCGGGAGCCACCGTTCTATATCATCATTGAGTTCATGAC) for each well of a six-well plate. The cells were left to recover and proliferate before being selected using 1 μM imatinib in RPMI supplemented with 10% FBS. When an enriched T315I polyclonal line was achieved, imatinib selection was stopped.
HEK 293 Cells
Human embryonic kidney cells (HEK293) were cultured in high-glucose DMEM medium (Gibco 11965092) supplemented with 10% FBS (Gibco 26140079) and Pen/Strep/l-glutamine (Gibco 10378016).
Human iPSC-CMs
iPSC lines were as described (SCVI-15S1 from the Stanford Cardiovascular Institute Biobank). The derivation of the line was approved by the Stanford University Institutional Review Board and Stem Cell Research Oversite board. hiPSC lines were cultured in E8 cell culture medium (Life Technologies) in plates coated with growth factor-reduced Matrigel (Corning) until at least passage 20 before differentiation. hiPSC cells were differentiated into cardiomyocytes (CMs) utilizing a chemically defined cardiomyocyte differentiation protocol62 and fatty acid rich maturation protocol.63
HMVEC-Cs Cells
Human microvascular cardiac endothelial cells (HMVEC-Cs) were purchased from LONZA (CC-7030 batch 0000550176). The HMVEC cells were cultured in EBMTM-2 Basal Medium (Lonza, CC-3156) and EGMTM-2 MV supplemented with microvascular endothelial Cell Growth Medium SingleQuots (Lonza, CC-4147). HMVEC-Cs were expanded and used at passage 7 for the vasculogenesis assay.
Cell Viability and Growth Inhibition Assay
Growth inhibitory activities were evaluated on K-562 CML cancer cell lines. The effects of the compounds on cell viability were evaluated using the AlamarBlue assay using the NCI60 methodology.64 Cells were harvested and plated in 384-well plates (Greiner μClear) at a concentration of 1250 cells/well in 40 μL and incubated for 24 h at 37 °C. The next day, test compounds were added to the cells as a 2× 40 μL solution and incubated for 48 h at 37 °C. Then, the cells were treated with Resazurin (final concentration of 10%) and incubated for 2 h before the fluorescence was measured on a plate reader (excitation at 544 nm, emission at 590 nm) to quantify the antiproliferative effects of the compounds. The 50% growth inhibition (GI50) values are reported as medians ± median absolute differences (MADs).
Kinase Activity Assays
The kinase activity for ABL1 and ABL1T315I was performed using the SelectScreen Biochemical Kinase Profiling service of ThermoFisher Scientific (Madison, WI). For each kinase, an IC50 was calculated on the basis of a 10-point concentration curve of the test article and converted into Ki values.
Vasculogenesis Assay
HMVEC-Cs were plated on a Geltrex surface in 96-well plates at a density of 25 000 cells/well (Greiner μClear) supplemented with EBMTM-2 Basal Medium. The cells were incubated with the compounds for 6 h at 37 °C at three doses per compound: 10, 3.33, and 1.11 μM for all of the compounds. DMSO was used as a vehicle for the compounds and also tested. After the treatment, the HMVEC cells were stained with calcein AM fluorescent dye (Corning 354216) at a 1/200 dilution in 1× HBSS medium (ThermoFisher, 14185052) for 25 min and imaged without washing the dye on the IC200 Kinetic Imaging Platform (Vala Sciences) with a 4× objective. The vasculogenesis assay was generally performed for compounds that had cell inhibition GI50 values of <100 nM for K-562-T315I cells. Vessel formation was quantified using ImageJ version 1.53e analyzer, quantifying the number of complete formed loops in a capillary-like web and AUC for the overall dose response curve.
Cardiomyocyte Toxicity Assays
Human iPSC-CMs were plated on Matrigel-coated 384-well plates at a density of 20 000 cells/well (Greiner μClear) in 50 μL cardiomyocyte media (fatty acid rich maturation medium63) supplemented with 10% knockout replacement serum. The subsequent day, an additional 50 μL of medium was added and cells were grown for a minimum of 5 days prior to analysis. Action potential kinetics and contractility were measured sequentially on the same cells. First, action potential kinetics were recorded using the protocol as established by McKeithan et al.50 Briefly, the cells were washed five times with FluoroBrite, loaded with VF2.1.Cl dye for 50 min at 37 °C, and washed again five times with FluoroBrite. Voltage time series were acquired at a frequency of 33 Hz for a duration of 10 s on the IC200 Kinetic Imaging Platform (Vala Sciences). Then, the cells were loaded with 1/50000 tetramethylrhodamine and a methyl ester perchlorate (TMRM) wheat germ agglutinin–Alexa Fluor 555 conjugate (10 nM, T668 ThermoFisher) for 50 min at 37 °C and then washed four times with FluoroBrite.49 Contractile time series were acquired at a frequency of 50 Hz for a duration of 6.5 s. The resulting recordings were subsequently analyzed using Vala Sciences and custom software, and action potential kinetics, action potential durations and rates,50 or contractile activity parameters (peak divergence, area under the curve) were used as measures of cardiotoxicity.49 To determine human iPSC-CM toxicity, the transients were quantified, and curves fitted, to extract several measures for voltage (action potential duration of 75%) and contractility (peak contraction amplitude). The minimal concentration at which the dose response curve of any metric or the variability of the given metric deviated beyond a threshold (25% or 4.5×, respectively) from control was deemed the overall minimum toxic dose.
Acknowledgments
This research was supported by grants from the National Institutes of Health (R21GM137151, R01HL152055, R01HL130840, and P01HL141084 to M.M. and R01DK114174 and R01EY032159 to S.V.M.) and the Joan and Sanford I. Weill Scholars Endowment (to M.M.). M.P. and S.V.M. acknowledge the OCTRI Biomedical Innovation Program (BIP) Drug Discovery for partial support of this work. Also, S.V.M. would like to acknowledge the partial support from the Knight Cancer Institute for this project.
Glossary
Abbreviations Used
- ABL
Abelson tyrosine-protein kinase
- BCR
breakpoint cluster region protein
- CM
cardiomyocyte
- CML
chronic myeloid leukemia
- PDB
Protein Data Bank
- iPSCs
induced pluripotent stem cells
- Ph+ ALL
Philadelphia positive acute lymphoblastic leukemia
- SAR
structure–activity relationship
- SNAr
nucleophilic aromatic substitution
- TKI
tyrosine kinase inhibitor.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01853.
Author Contributions
M.P.: conceptualization, designing molecules, molecular docking studies, chemical synthesis, analysis, and manuscript preparation. A.A.N.B.: in vitro analysis and review and editing. A.P.H.: in vitro analysis and review and editing. S.V.M.: conceptualization, resources, project supervision, project administration, funding acquisition, and manuscript preparation and editing. M.M.: conceptualization, resources, supervision, funding acquisition, and manuscript editing.
The authors declare the following competing financial interest(s): M.P., A.A.N.B., A.P.H., M.M., and S.V.M. have a pending patent application on the refined analogues.
This paper was originally published ASAP on August 9, 2022, with an error in the TOC graphic. The corrected version was reposted on August 12, 2022.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Fuchs H. E.; Jemal A. Cancer Statistics, 2021. Ca-Cancer J. Clin. 2021, 71 (1), 7–33. 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- Rowley J. D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243 (5405), 290–293. 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- Daley G. Q.; Van Etten R. A.; Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990, 247 (4944), 824–830. 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- Faderl S.; Garcia-Manero G.; Thomas D. A.; Kantarjian H. M. Philadelphia chromosome-positive acute lymphoblastic leukemia-current concepts and future perspectives. Reviews in clinical and experimental hematology 2002, 6 (2), 142–160. 10.1046/j.1468-0734.2002.00066.x. [DOI] [PubMed] [Google Scholar]
- Ben-Neriah Y.; Daley G. Q.; Mes-Masson A.-M.; Witte O. N.; Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986, 233 (4760), 212–214. 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- Shtivelman E.; Lifshitz B.; Gale R. P.; Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315 (6020), 550–554. 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- Druker B. J.; Guilhot F.; O’Brien S. G.; Gathmann I.; Kantarjian H.; Gattermann N.; Deininger M. W.; Silver R. T.; Goldman J. M.; Stone R. M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355 (23), 2408–2417. 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Druker B. J.; Tamura S.; Buchdunger E.; Ohno S.; Segal G. M.; Fanning S.; Zimmermann J.; Lydon N. B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine 1996, 2 (5), 561–566. 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Capdeville R.; Buchdunger E.; Zimmermann J.; Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discovery 2002, 1 (7), 493–502. 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein kinases—the major drug targets of the twenty-first century?. Nat. Rev. Drug Discovery 2002, 1 (4), 309–315. 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Deininger M.; O’Brien S. G.; Guilhot F.; Goldman J. M.; Hochhaus A.; Hughes T. P.; Radich J. P.; Hatfield A. K.; Mone M.; Filian J.; et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood 2009, 114 (22), 1126. 10.1182/blood.V114.22.1126.1126. [DOI] [Google Scholar]
- Hochhaus A.; O’brien S.; Guilhot F.; Druker B.; Branford S.; Foroni L.; Goldman J.; Müller M.; Radich J.; Rudoltz M.; et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009, 23 (6), 1054–1061. 10.1038/leu.2009.38. [DOI] [PubMed] [Google Scholar]
- Pophali P. A.; Patnaik M. M. The role of new tyrosine kinase inhibitors in chronic myeloid leukemia. Cancer journal (Sudbury, Mass.) 2016, 22 (1), 40–50. 10.1097/PPO.0000000000000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T.; Deininger M.; Hochhaus A.; Branford S.; Radich J.; Kaeda J.; Baccarani M.; Cortes J.; Cross N. C.; Druker B. J. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood 2006, 108 (1), 28–37. 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare T.; Eide C. A.; Deininger M. W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110 (7), 2242–2249. 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- Shah N. P.; Tran C.; Lee F. Y.; Chen P.; Norris D.; Sawyers C. L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305 (5682), 399–401. 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- Weisberg E.; Manley P. W.; Breitenstein W.; Brüggen J.; Cowan-Jacob S. W.; Ray A.; Huntly B.; Fabbro D.; Fendrich G.; Hall-Meyers E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7 (2), 129–141. 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Saglio G.; Kim D.-W.; Issaragrisil S.; Le Coutre P.; Etienne G.; Lobo C.; Pasquini R.; Clark R. E.; Hochhaus A.; Hughes T. P.; et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2010, 362 (24), 2251–2259. 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- Kantarjian H.; Shah N. P.; Hochhaus A.; Cortes J.; Shah S.; Ayala M.; Moiraghi B.; Shen Z.; Mayer J.; Pasquini R.; et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2010, 362 (24), 2260–2270. 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- Vener C.; Banzi R.; Ambrogi F.; Ferrero A.; Saglio G.; Pravettoni G.; Sant M. First-line imatinib vs second-and third-generation TKIs for chronic-phase CML: a systematic review and meta-analysis. Blood advances 2020, 4 (12), 2723–2735. 10.1182/bloodadvances.2019001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintás-Cardama A.; Cortes J. Therapeutic options against BCR-ABL1 T315I-positive chronic myelogenous leukemia. Clin. Cancer Res. 2008, 14 (14), 4392–4399. 10.1158/1078-0432.CCR-08-0117. [DOI] [PubMed] [Google Scholar]
- O’Hare T.; Walters D. K.; Stoffregen E. P.; Jia T.; Manley P. W.; Mestan J.; Cowan-Jacob S. W.; Lee F. Y.; Heinrich M. C.; Deininger M. W.; Druker B. J. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005, 65 (11), 4500–4505. 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- Miller G. D.; Bruno B. J.; Lim C. S. Resistant mutations in CML and Ph(+)ALL - role of ponatinib. Biologics 2014, 8, 243–254. 10.2147/BTT.S50734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarski J. S.; Newitt J. A.; Chang C. Y. J.; Cheng J. D.; Wittekind M.; Kiefer S. E.; Kish K.; Lee F. Y.; Borzillerri R.; Lombardo L. J.; et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006, 66 (11), 5790–5797. 10.1158/0008-5472.CAN-05-4187. [DOI] [PubMed] [Google Scholar]
- Hughes T. P.; Saglio G.; Larson R. A.; Kantarjian H. M.; Kim D.-W.; Issaragrisil S.; Le Coutre P.; Etienne G.; Boquimpani C.; Clark R. E.. Long-term outcomes in patients with chronic myeloid leukemia in chronic phase receiving frontline nilotinib versus imatinib: ENESTnd 10-year analysis; American Society of Hematology: Washington, DC, 2019; pp 2924–2924. [Google Scholar]
- Jabbour E.; Kantarjian H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol 2020, 95 (6), 691–709. 10.1002/ajh.25792. [DOI] [PubMed] [Google Scholar]
- Giles F. J.; Cortes J.; Jones D.; Bergstrom D.; Kantarjian H.; Freedman S. J. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood 2007, 109 (2), 500–502. 10.1182/blood-2006-05-025049. [DOI] [PubMed] [Google Scholar]
- FDA approves Ponatinib. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm381452.htm.
- Attwood M. M.; Fabbro D.; Sokolov A. V.; Knapp S.; Schiöth H. B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discovery 2021, 20, 839–861. 10.1038/s41573-021-00252-y. [DOI] [PubMed] [Google Scholar]
- O’Hare T.; Shakespeare W. C.; Zhu X.; Eide C. A.; Rivera V. M.; Wang F.; Adrian L. T.; Zhou T.; Huang W. S.; Xu Q.; Metcalf C. A. 3rd; Tyner J. W.; Loriaux M. M.; Corbin A. S.; Wardwell S.; Ning Y.; Keats J. A.; Wang Y.; Sundaramoorthi R.; Thomas M.; Zhou D.; Snodgrass J.; Commodore L.; Sawyer T. K.; Dalgarno D. C.; Deininger M. W.; Druker B. J.; Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16 (5), 401–412. 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes J. E.; Kim D. W.; Pinilla-Ibarz J.; le Coutre P.; Paquette R.; Chuah C.; Nicolini F. E.; Apperley J. F.; Khoury H. J.; Talpaz M.; DiPersio J.; DeAngelo D. J.; Abruzzese E.; Rea D.; Baccarani M.; Muller M. C.; Gambacorti-Passerini C.; Wong S.; Lustgarten S.; Rivera V. M.; Clackson T.; Turner C. D.; Haluska F. G.; Guilhot F.; Deininger M. W.; Hochhaus A.; Hughes T.; Goldman J. M.; Shah N. P.; Kantarjian H.; Investigators P. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369 (19), 1783–1796. 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes J. E.; Kim D.-W.; Pinilla-Ibarz J.; Le Coutre P.; Paquette R.; Chuah C.; Nicolini F. E.; Apperley J. F.; Khoury H. J.; Talpaz M.. Long-term follow-up of ponatinib efficacy and safety in the phase 2 PACE trial; American Society of Hematology: Washington, DC, 2014. [Google Scholar]
- Gainor J. F.; Chabner B. A. Ponatinib: accelerated disapproval. oncologist 2015, 20 (8), 847–848. 10.1634/theoncologist.2015-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. P.; Umbarkar P.; Tousif S.; Lal H. Cardiotoxicity of the BCR-ABL1 tyrosine kinase inhibitors: Emphasis on ponatinib. Int. J. Cardiol 2020, 316, 214–221. 10.1016/j.ijcard.2020.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moslehi J. J.; Deininger M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J. Clin Oncol 2015, 33 (35), 4210–4218. 10.1200/JCO.2015.62.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifi Y.; Moccetti F.; Wu M.; Xie A.; Packwood W.; Qi Y.; Ozawa K.; Shentu W.; Brown E.; Shirai T.; et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood 2019, 133 (14), 1597–1606. 10.1182/blood-2018-10-881557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishoey M.; Chorn S.; Singh N.; Jaeger M. G.; Brand M.; Paulk J.; Bauer S.; Erb M. A.; Parapatics K.; Müller A. C.; et al. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol. 2018, 13 (3), 553–560. 10.1021/acschembio.7b00969. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Gao H.; Sun X.; Sun Y.; Qiu Y.; Weng Q.; Rao Y. Global PROTAC Toolbox for Degrading BCR-ABL Overcomes Drug-Resistant Mutants and Adverse Effects. J. Med. Chem. 2020, 63 (63), 8567–8585. 10.1021/acs.jmedchem.0c00967. [DOI] [PubMed] [Google Scholar]
- Larocque E.; Chu E. F. Y.; Naganna N.; Sintim H. O. Nicotinamide-Ponatinib Analogues as Potent Anti-CML and Anti-AML Compounds. ACS Omega 2020, 5 (6), 2690–2698. 10.1021/acsomega.9b03223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B.; Bornmann W. G.; Pellicena P.; Schindler T.; Veach D. R.; Miller W. T.; Clarkson B.; Kuriyan J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62 (15), 4236–4243. [PubMed] [Google Scholar]
- Levinson N. M.; Boxer S. G. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PloS one 2012, 7 (4), e29828 10.1371/journal.pone.0029828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Commodore L.; Huang W. S.; Wang Y.; Thomas M.; Keats J.; Xu Q.; Rivera V. M.; Shakespeare W. C.; Clackson T.; Dalgarno D. C.; Zhu X. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem. Biol. Drug Des 2011, 77 (1), 1–11. 10.1111/j.1747-0285.2010.01054.x. [DOI] [PubMed] [Google Scholar]
- Chen P.; Norris D.; Das J.; Spergel S. H.; Wityak J.; Leith L.; Zhao R.; Chen B.-C.; Pitt S.; Pang S.; et al. Discovery of novel 2-(aminoheteroaryl)-thiazole-5-carboxamides as potent and orally active Src-family kinase p56Lck inhibitors. Bioorg. Med. Chem. Lett. 2004, 14 (24), 6061–6066. 10.1016/j.bmcl.2004.09.093. [DOI] [PubMed] [Google Scholar]
- Huang W. S.; Metcalf C. A.; Sundaramoorthi R.; Wang Y.; Zou D.; Thomas R. M.; Zhu X.; Cai L.; Wen D.; Liu S.; Romero J.; Qi J.; Chen I.; Banda G.; Lentini S. P.; Das S.; Xu Q.; Keats J.; Wang F.; Wardwell S.; Ning Y.; Snodgrass J. T.; Broudy M. I.; Russian K.; Zhou T.; Commodore L.; Narasimhan N. I.; Mohemmad Q. K.; Iuliucci J.; Rivera V. M.; Dalgarno D. C.; Sawyer T. K.; Clackson T.; Shakespeare W. C. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-y l)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J. Med. Chem. 2010, 53 (12), 4701–4719. 10.1021/jm100395q. [DOI] [PubMed] [Google Scholar]
- Chinchilla R.; Nájera C. The Sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107 (3), 874–922. 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- Huang W.-S.; Shakespeare W. C. An efficient synthesis of nilotinib (AMN107). Synthesis 2007, 2007 (14), 2121–2124. 10.1055/s-2007-983754. [DOI] [Google Scholar]
- Wen M.; Shen C.; Wang L.; Zhang P.; Jin J. An efficient D-glucosamine-based copper catalyst for C-X couplings and its application in the synthesis of nilotinib intermediate. RSC Adv. 2015, 5 (2), 1522–1528. 10.1039/C4RA11183D. [DOI] [Google Scholar]
- Bhunia S.; Pawar G. G.; Kumar S. V.; Jiang Y.; Ma D. Selected Copper-Based Reactions for C- N, C- O, C- S, and C- C Bond Formation. Angew. Chem., Int. Ed. 2017, 56 (51), 16136–16179. 10.1002/anie.201701690. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Burridge P. W.; McKeithan W. L.; Serrano R.; Shukla P.; Sayed N.; Churko J. M.; Kitani T.; Wu H.; Holmström A.; et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med. 2017, 9 (377), eaaf2584 10.1126/scitranslmed.aaf2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeithan W. L.; Savchenko A.; Yu M. S.; Cerignoli F.; Bruyneel A. A.; Price J. H.; Colas A. R.; Miller E. W.; Cashman J. R.; Mercola M. An automated platform for assessment of congenital and drug-induced arrhythmia with hiPSC-derived cardiomyocytes. Front. Physiol. 2017, 8, 766. 10.3389/fphys.2017.00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer E. R.; Vega R.; McDonough P. M.; Price J. H.; Whittaker R. Specific prediction of clinical QT prolongation by kinetic image cytometry in human stem cell derived cardiomyocytes. Journal of Pharmacological and Toxicological Methods 2016, 81, 263–273. 10.1016/j.vascn.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Uitdehaag J. C.; de Roos J. A.; van Doornmalen A. M.; Prinsen M. B.; de Man J.; Tanizawa Y.; Kawase Y.; Yoshino K.; Buijsman R. C.; Zaman G. J. Comparison of the cancer gene targeting and biochemical selectivities of all targeted kinase inhibitors approved for clinical use. PloS one 2014, 9 (3), e92146 10.1371/journal.pone.0092146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnatiuk A. P.; Bruyneel A. A. N.; Tailor D.; Pandrala M.; Dheeraj A.; Li W.; Fernandez R. S.; Feyen D. A.; Vu M.; Amatya P.; Gupta S.; Morgado I.; Wiebking V. A.; Porteus M. H.; Malhotra S. V.; Mercola M. Reengineering Ponatinib to Minimize Cardiovascular Toxicity. Cancer Res. 2022, 10.1158/0008-5472.CAN-21-3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J.; Buchdunger E.; Mett H.; Meyer T.; Lydon N. B. Potent and selective inhibitors of the Abl-kinase: phenylamino-pyrimidine (PAP) derivatives. Bioorg. Med. Chem. Lett. 1997, 7 (2), 187–192. 10.1016/S0960-894X(96)00601-4. [DOI] [Google Scholar]
- Li Y.; Shen M.; Zhang Z.; Luo J.; Pan X.; Lu X.; Long H.; Wen D.; Zhang F.; Leng F.; et al. Design, synthesis, and biological evaluation of 3-(1 H-1, 2, 3-triazol-1-yl) benzamide derivatives as potent pan Bcr-Abl inhibitors including the threonine315→ isoleucine315 mutant. J. Med. Chem. 2012, 55 (22), 10033–10046. 10.1021/jm301188x. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Zhang Y.; Pinkas D. M.; Fox A. E.; Luo J.; Huang H.; Cui S.; Xiang Q.; Xu T.; Xun Q.; et al. Design, Synthesis, and Biological Evaluation of 3-(Imidazo [1, 2-a] pyrazin-3-ylethynyl)-4-isopropyl-N-(3-((4-methylpiperazin-1-yl) methyl)-5-(trifluoromethyl) phenyl) benzamide as a Dual Inhibitor of Discoidin Domain Receptors 1 and 2. J. Med. Chem. 2018, 61 (17), 7977–7990. 10.1021/acs.jmedchem.8b01045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix U.; Hantschel O.; Dürnberger G.; Remsing Rix L. L.; Planyavsky M.; Fernbach N. V.; Kaupe I.; Bennett K. L.; Valent P.; Colinge J.; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110 (12), 4055–4063. 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- Singh A. P.; Glennon M. S.; Umbarkar P.; Gupte M.; Galindo C. L.; Zhang Q.; Force T.; Becker J. R.; Lal H. Ponatinib-induced cardiotoxicity: delineating the signalling mechanisms and potential rescue strategies. Cardiovascular research 2019, 115 (5), 966–977. 10.1093/cvr/cvz006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Xu Z.; Yan H.; He Q.; Yang X.; Luo P. A comprehensive review of clinical cardiotoxicity incidence of FDA-approved small-molecule kinase inhibitors. Front. Pharmacol. 2020, 11, 891. 10.3389/fphar.2020.00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar M.; Suebsuwong C.; Ray S. S.; Thapa R. J.; Maki J. L.; Nogusa S.; Shah S.; Saleh D.; Gough P. J.; Bertin J.; et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 2015, 10 (11), 1850–1860. 10.1016/j.celrep.2015.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X.; Hsiao C.; Wilson G.; Zhu K.; Hazeltine L. B.; Azarin S. M.; Raval K. K.; Zhang J.; Kamp T. J.; Palecek S. P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (27), E1848-E1857 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyen D. A.; McKeithan W. L.; Bruyneel A. A.; Spiering S.; Hörmann L.; Ulmer B.; Zhang H.; Briganti F.; Schweizer M.; Hegyi B.; et al. Metabolic maturation media improve physiological function of human iPSC-derived cardiomyocytes. Cell Rep. 2020, 32 (3), 107925. 10.1016/j.celrep.2020.107925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R. H. The NCI60 human tumour cell line anticancer drug screen. Nature Reviews Cancer 2006, 6 (10), 813–823. 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.