

Abstract
Colorectal cancer (CRC) is the fourth most common cancer type and is the second leading cause of cancer deaths annually in the United States. Conventional treatment options include postoperative (adjuvant) and preoperative (neoadjuvant) chemotherapy and radiotherapy. Although these treatment modalities have shown to decrease tumor burden, a major limitation to chemothearpy/radiotherapy is the high recurrence rate in patients. Immune-modulation strategies have emerged as a promising new therapeutic avenue to reduce this recurrence rate while minimizing undesirable systemic side effects. This review will focus specifically on the mechanisms of monoclonal antibodies: immune checkpoint inhibitors and cytokines, as well as current drugs approved by the Food and Drug Administration (FDA) and new clinical/pre-clinical trials. Finally, this review will investigate emerging methods used to monitor tumor response post-treatment.
Keywords: Colorectal cancer, Checkpoint, Inhibition, Cytokine, Clinical, Immunotherapy
1. Introduction
Colorectal cancer (CRC) is the fourth most common cancer (by incidence) in the United States, accounting for 140,000 new cases and 50,000 deaths in 2018 [1]. Before advancements in treatment and cancer detection, patients with locally advanced CRC (high-risk stage II and stage III tumors) and metastatic CRC (mCRC) were treated via surgery followed by postoperative (adjuvant) chemotherapy.
In both the neoadjuvant and adjuvant settings, the current standard-of-care chemotherapy regimen is FOLFOX, a combination of 5-fluorouracil (5-FU), leucovorin and oxaliplatin [2]. FOLFOX, as an adjuvant therapy, is given in 12 cycles every two weeks through intravenous (IV) administration. Variations of this type of chemotherapy have been a fixture in CRC treatment since the 1960’s, and have been optimized since [3]. Although FOLFOX is the gold standard in treating mCRC, it is associated with myriad and sometimes debilitating systemic side effects (nausea, anemia, decrease in white blood cells, fatigue, etc.) [4].
In recent years, the addition of preoperative (neoadjuvant) chemotherapy for locally advanced CRC has become clinically accepted after success was demonstrated in esophageal [5] and gastric cancers [6], following a series of clinical studies by the Fluoropyrimidine, Oxaliplatin and Targeted-Receptor pre-Operative Therapy (FOxTROT) Collaborative Group [7]. The goals of neoadjuvant chemotherapy include achieving complete eradication of cancer cells or pathological complete response (pCR) prior to surgery, reducing intraoperative tumor cell shedding during surgery, and decreasing local recurrence rates [8]. In a feasibility phase trial by the FOxTROT Collaborative Group, 150 patients with locally advanced CRC were given a combination of chemotherapy drugs either in the neoadjuvant or adjuvant setting. Patients receiving neoadjuvant chemotherapy experienced significant tumor downstaging and regression [7].
Currently, in locally advanced colon cancer and mCRC, neoadjuvant chemotherapy is generally given to patients in 2–12 two-week cycles over 4–24 weeks [9]. After assessing tumor therapeutic response after 4–6 cycles (6–8 weeks after initiation) of neoadjuvant chemotherapy using techniques such as endorectal ultrasound [9], computed tomography (CT), positron emission tomography-computed tomography (PET-CT), or magnetic resonance imaging (MRI) (or a combination of these techniques), patients with locally advanced disease either receive additional neoadjuvant chemotherapy cycles or proceed to surgery [10].
Although there has been a steady reduction in CRC incidence and mortality since the 1970′s, primarily attributed to reduction in preventable risk factors, advances in early detection, [11] nationwide screening initiatives [1,12], and continued optimization of neoadjuvant and adjuvant chemotherapy regimens, current treatment standards and management of CRC remains problematic [8]. Although neoadjuvant therapy has shown significant tumor downstaging and regression, a low 5-year survival rate (∼10%) along with high recurrence rate (30–40%) is a concern to clinicans [13]. Therefore, researchers are exploring new therapeutic interventions to overcome these limitations. One broad-scale intervention that has gained clinical traction is immunotherapy.
Immunotherapy is an emerging technique to treat cancer by stimulating or enhancing a patient’s immune components to target and inhibit cancer cells, limiting the negative systemic effects associated with untargeted chemotherapy approaches [14]. Current clinically approved immunotherapy techniques for CRC treatment include monoclonal antibody therapy, adoptive cell transfer (ACT) therapy, cancer vaccines and cell therapy [13]. Among the many types of immunotherapy strategies, monoclonal antibody therapy has gained the most clinical traction for treating CRC in recent years [15]. This review discusses current CRC monoclonal antibody immunotherapy treatments, which can be divided into antibodies targeting either immune checkpoints or cytokines. Treatments discussed are either approved by the U.S. Food and Drug Administration (FDA), in clinical trials in humans, or in pre-clinical trials. Finally, this review discusses emerging methods (optical and non-optical) to monitor tumor response to immunotherapy treatments in CRC patients.
2. Immune checkpoints in colorectal cancer
Immune checkpoints are any set of ligand-mediated inhibitory pathways that maintain homeostasis of the immune system by regulating the duration and amplitude of immune responses [16]. Within these pathways, there are various types of cells that play a role in the regulation of the immune system (i.e. CD4, CD8, monocytes, natural killer (NK) cells and dendritic cells (DCs)). CD4 cells, a specific classification of T-cells, are responsible for immune system regulation, specifically the release of cytokines to increase the activity of other immune cells [17]. CD8 cells, also known as “killer cells”, suppress immune signaling during T cell activation [18]. Monocytes are types of white blood cells which can differentiate into macrophages and DCs [19]. When monocytes differentiate into macrophages, they can differentiate into one of two subtypes: M1 or M2. M1 macrophages produce pro-inflammatory cytokines (i.e. interleukin-1 (IL-1), IL-6, IL-12, tumor necrosis factor-alpha (TNF-α), etc.) that signals an immune response [19]. M2 macrophages produce polyamines (i.e. spermidine and spermine) that induce cell proliferation and extracellular matrix formation [19]. Natural killer cells (NK) are effector lymphocytes thatengages in interactions with other cell types (macrophages, T cells, etc.) to limit immune response [20]. DCs or antigen-presenting cells (APCs) are responsible for the activation of adaptive immune responses by presenting antigens to other cells such as T cells. Overall, the functions of these cells play a role in how the immune system is activated to decrease tumor burden.
Several immune checkpoints have been used as immunotherapeutic targets in various cancer types such as melanoma, kidney, bladder and non-small cell lung cancer as well as CRC, including cytotoxic T-lymphocyte antigen-4 (CTLA-4), programmed cell death protein-1 (PD-1), and programmed cell death ligand-1 (PD-L1).
2.1. CTLA4
CTLA4, and its homolog, CD28, are cell surface receptors found on CD4+ cells (helper T-cells) and CD8+ cells (cytotoxic T-cells). The ligands for CTLA4 and CD28 are the B7 proteins (B7–1 (CD80) and B7–2 (CD86)), which are produced by APCs. B7 ligands are upregulated and presented on the cell surface by APCs when the APCs encounter and acquire non-self-antigens [21]. When T-cells detect B7, along with major histocompatibility complex loaded with cognate peptide, competitive binding ensues between CD28/B7 and CTLA4/B7 to maintain T-cell homeostasis. CD28/B7 binding initiates immune stimulation by increasing T-cell proliferation whereas CTLA4/B7 binding initiates immunosuppression by competitively reducing signaling of the CD28/ B7 complex (Fig. 1) [22]. Then, CTLA4 reduces the probability of future CD28/B7 binding by removing B7 proteins from the APC surface via trans-endocytosis [23]. Thus, CTLA4/B7 interaction is involved in immune tolerance and immunosuppression, a hallmark of cancer [24]. Monoclonal antibodies which target and block the CTLA4 immune checkpoint pathway results in increased CD28/B7-dependent clonal expansion of T-cells [25].
Fig. 1.
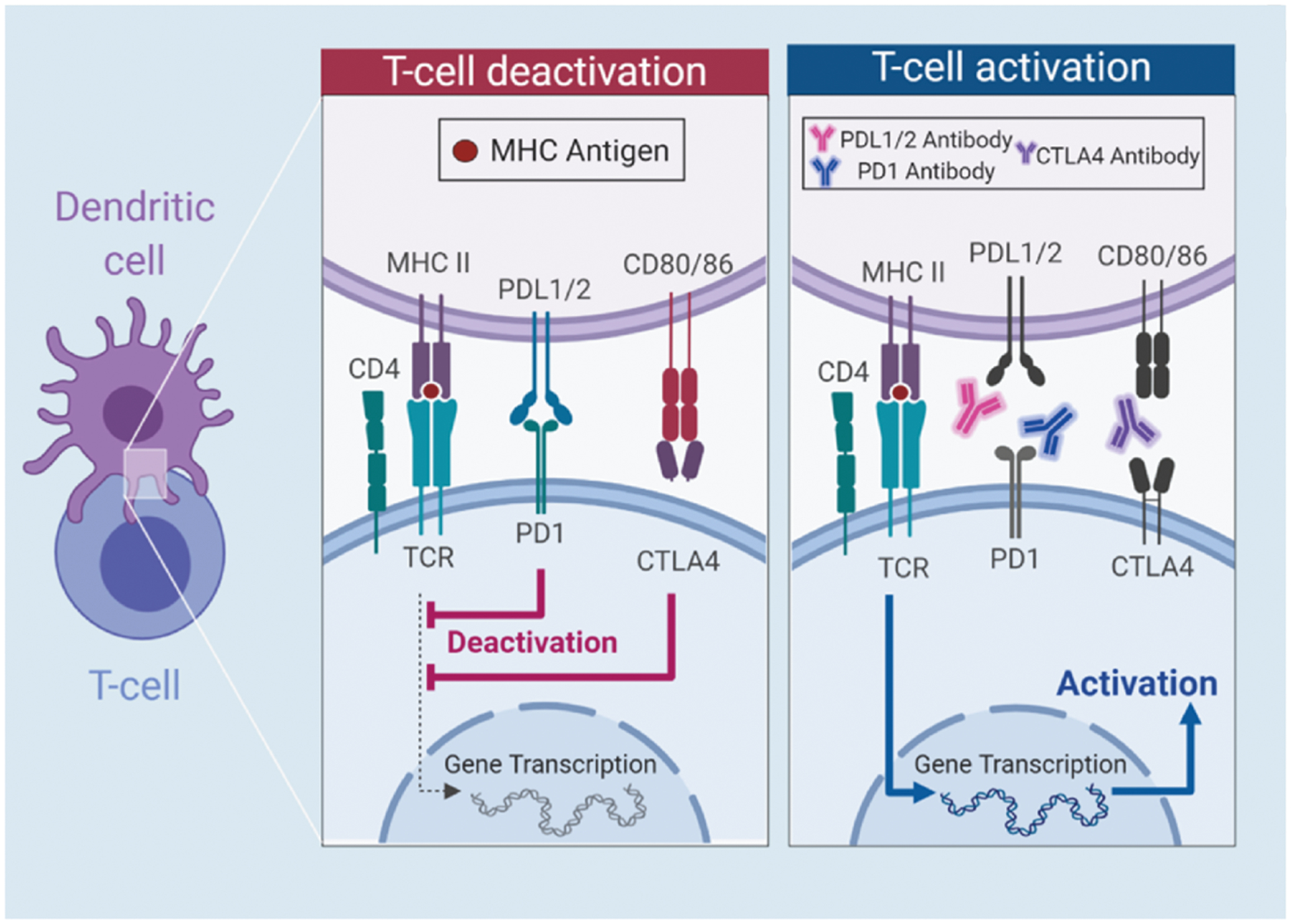
Schematic representation of CTLA-4 and PD-1/PD-L1 pathway through T-cell deactivation in CRC, along with T-cell activation through antibody binding. T-cell deactivation begins when the T-cell receptor (TCR) binds to the Major Histocompatibility Complex II (MHCII) and binding of PD1 and PDL1/2 and CTLA4 and CD80/86. When anti-CTLA4, anti-PDL1 and anti-PD1 are present, the antibodies bind to their respective ligands, inducing T cell activation.
2.2. PD-1
PD-1, a well-studied immune checkpoint, has a primary function to suppress the immune response to regulate tolerance and autoimmunity [26–28]. PD-1 is a cell surface receptor found on CD4+ cells, CD8+ cells, B-cells, NK cells, myeloid-derived cells, and macrophages [27]. The primary function of PD-1 is to suppress the immune response (Fig. 1) [26]. The ligands for PD-1 are the B7 proteins, B7-H1 (PD-L1) and B7-DC (PD-L2). PD-L2 is produced by APCs. PD-L1 is expressed by T-cells, B-cells, DCs, and macrophages and is upregulated by many pro-tumor cytokines such as IL-4, IL-10 VEGF, and TNF-α produced by infiltrating immune cells [26,29]. Additionally, PD-L1 is directly expressed by many types of cancer cell, including CRC and is associated with poor prognosis [30]. PD-1/PD-L1 binding results in T-cell apoptosis and reduced IL-2 (an anti-tumor cytokine) production [27]. Although the induction of T-cell apoptosis is problematic in tumors, it is essential for some T cells to survive apoptotic death in order to become memory T cells [31]. PD-1 and PD-L1 are active targets in CRC immunotherapy research with the goal of introducing monoclonal antibodies to block PD-1/PD-L1 binding and improve the anti-tumor immune response.
3. Immune checkpoint inhibition immunotherapy
3.1. FDA-approved drugs
Nivolumab (Opdivo®) is an immune checkpoint inhibitor that binds to PD-1 receptors, blocking PD-1 activation and resulting in T-cell activation and immune response. The first uses of Nivolumab was a first line treatment for metastatic melanoma as well as bladder cancer and brain metastases. Nivolumab was granted accelerated approval by the FDA in 2017 following an ongoing, multicenter Phase II trial (NCT02060188) [32], funded by Bristol-Myers Squibb, that indicated Nivolumab was effective for CRC patients with deficient DNA mismatch repair (dMMR)/microsatellite instability high (MSI-H) disease [33]. dMMR/MSI-H CRC makes up approximately 12–15% of cases and is phenotypically characterized by a high quantity of tumor infiltrating lymphocytes (TILs), prevalence in the right side of the colon (proximal colon), and poor differentiation [33]. The approval of Nivolumab was particularly important since standard FOLFOX-based chemotherapy has limited benefit for dMMR/MSI-H CRC patients as shown in five randomized clinical trials evaluating 5-FU vs surgical treatment [33]. There are currently 39 ongoing clinical trials further exploring Nivolumab as either stand alone or combinatorial treatment (Ipilimumab, Azacytidine, etc.) for CRC.
Pembrolizumab (Keytruda®) is an IgG4-k monoclonal antibody that inhibits PD-1 binding with PD-L1 and PD-L2. This results in an upregulated immune response against CRC cells [30]. Pembrolizumab was granted accelerated approval by the FDA in 2017 as a second-line treatment for either unresectable, dMMR, or MSI-H CRC following multiple Phase II and III clinical trials [34,35]. There are currently 52 ongoing clinical trials further exploring Pembrolizumab as either stand alone or combinatorial treatment with standard chemotherapy for CRC.
3.2. Clinical studies
Table 1 shows a current list of ongoing clinical trials that use immune checkpoint inhibitors.
Table 1.
Current list of ongoing clinical trials using immune checkpoint inhibitors.
| Name | NCT.gov Identifier | Phase | Intervention | Results |
|---|---|---|---|---|
| Urelumab | NCT01471210 | I/II | Efficacy in patients with melanoma, B-cell non-Hodgkin’s lymphoma and colorectal cancer (CRC) [51] | Doses at 0.1 mg/kg every 3 weeks was proven effective |
| Varlilumab | NCT01460134 | II | Solid tumors in various cancer types including colorectal cancer (CRC) [52] | Dose of 10 mg/kg was well tolerated |
| Ipilimumab | NCT03271047 and NCT03350126 | I and II | Patients with metastatic CRC, microsatellite-stable (MSS) and dMMR/MSI-H CRC [53,54] | No published results at time of this writing |
3.3. Pre-clinical studies
Table 2 shows a list of ongoing pre-clinical trials that use immune checkpoint inhibitors.
Table 2.
Current list of ongoing pre-clinical trials using immune checkpoint inhibitors.
| Author | Study Title | Methods | Results |
|---|---|---|---|
| Jure-Kunkel et al. [55] | “Synergy between chemotherapeutic agents and CTLA-4 blockade in preclinical tumor models” | Subcutaneous CRC mouse model with CTLA4 | Reduced tumor growth in both treatment groups |
| Zhao et al. [56] | “Tumor location impacts immune response in mouse models of colon cancer” | Orthotopic CRC and subcutaneous allograft | Orthotopic models were more sensitive to checkpoint inhibition |
3.4. Conclusion
Immune checkpoint inhibition is a promising approach for CRC treatment [42], with several FDA-approved drugs already on the market and many more in clinical trials. Although immune checkpoint inhibition has shown success in treating CRC, the biggest challenge for investigators is identifying which patients may or not respond before treatment initiation [42] and overcoming tumor cell resistance to this immunotherapy [16]. Jenkins et al. provides a comprehensive review of tumor cell resistance to immune checkpoint inhibition [42]. This heterogeneous patient response to immune checkpoint inhibition is a strikingly similar problem to identifying responders vs. non-responders for standard first-line neoadjuvant chemotherapy in CRC [43]. The current state-of-the-art is to biopsy the tumor during colonoscopy and determine expression levels of markers such as a PD-L1 using immunohistochemistry (IHC). Patients overexpressing the target biomarker, such as PD-LI, are considered the best candidates for that immunotherapy [44]. In the future, investigators are looking into identifying other biomarkers and personalized gene-expression signatures to identify candidates most likely to respond to immune checkpoint inhibition [43,45].
4. Cytokines in colorectal cancer
Cytokines are small (∼5–20 kDa) cell-signaling proteins, produced by immune cells, that are involved in myriad pathways in CRC. [46] Chemokines, members of a family of cytokines able to induce cellular chemotaxis, are also involved in CRC pathways such as CCL2, IL-6, and other growth factor and their corresponding receptor pathways [47].
Interleukins (i.e. IL-6), are naturally occurring proteins that regulate communication between cells. Unlike most cytokines, interleukins are not stored within a cell, but it is secreted rapidly in response to stimuli. After being produced, interleukins travel to its target cells and binds to a receptor molecule on the cell surface, releasing a cascade of signals that controls a cell’s behavior.
Growth factors are signaling molecules between cells that are capable of stimulating cell growth, proliferation, healing and cell differentiation. Examples of growth factors include vascular endothelial growth factors (VEGFs) and epidermal growth factors (EGFs), which will be discussed in the following sections.
Thus, cytokines and chemokines, and their receptors, make attractive targets for CRC therapy, although pre-clinical and clinical research currently lags other discussed CRC immunotherapy techniques [48]. Development of cytokine-targeted immunotherapy can potentially be used as stand-alone treatment or, more likely, combinatorial treatment with either chemotherapy, radiotherapy, or other immunotherapy techniques to normalize the CRC tumor microenvironment (TME) [49].
5. Cytokine-targeted immunotherapy
5.1. FDA-approved drugs
All current FDA-approved cytokine-targeted immunotherapy drugs for melanoma, renal cell carcinoma, as well as CRC target either vascular endothelial growth factor receptors (VEGFRs) or epidermal growth factor receptors (EGFRs), depicted in Fig. 2. VEGFs are signaling proteins responsible for the simulation of blood vessel formation, while EGFRs are transmembrane proteins responsible for cell differentiation and proliferation upon activation. Cytokine-targeted immunotherapy drugs targeting VEGFRs include bevacizumab, aflibercept, and regorafenib. Drugs targeting EGFRs include cetuximab and panitumumab. All five FDA-approved drugs primarily benefit mCRC patients, although many clinical trials are ongoing for other CRC subtypes in both neoadjuvant and adjuvant settings.
Fig. 2.
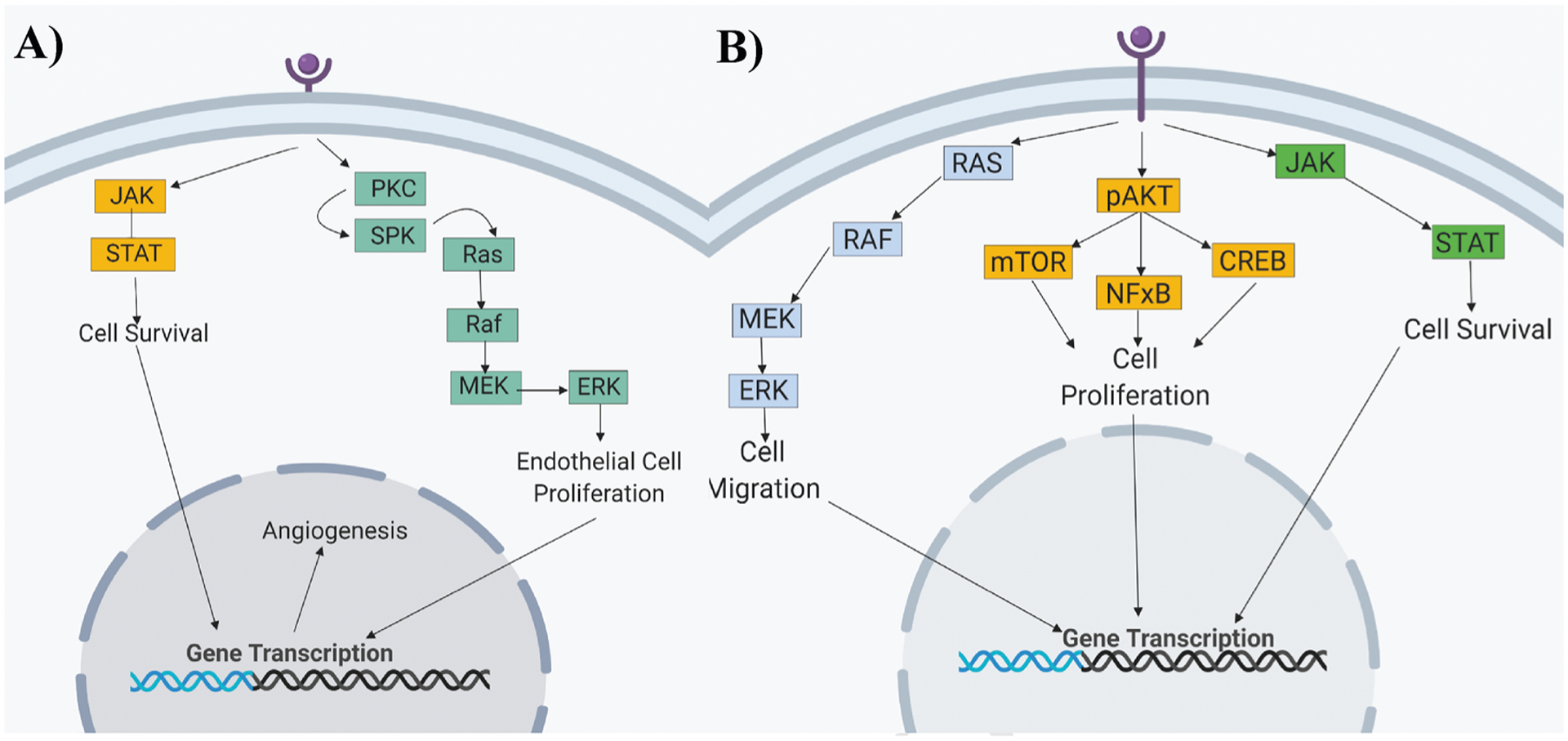
A). Through the activation of the A) VEGF and B) EGF pathway, intercellular pathways are also activated. These intercellular signaling pathways control cell survival, migration and proliferation, affecting the production of blood vessels (angiogenesis). Binding of an anti-VEGF/EGF to a VEGF/EGF receptor, inhibits receptor dimerization, preventing activation.
5.1.1. Anti-vascular endothelial growth factors (anti-VEGFs)
The FDA approved bevacizumab as first line treatment for mCRC in 2004 [50] and in 2006 for second-line treatment of mCRC in combination with FOLFOX4 [51], making it the first anti-VEGF drug for CRC. A phase III clinical trial by Eastern Cooperative Oncology Group (ECOG) tested bevacizumab’s efficacy and safety in combination with FOLFOX4 [52]. Patients treated with the combination therapy saw a longer median overall survival of 12.9 months with a 22.2% response rate compared to an overall survival of 10.8 months and an 8.6% response rate for patients receiving standalone FOLFOX4 chemotherapy [53]. Additional studies have confirmed the benefits of bevacizumab in treating mCRC [54,55]. In three phase III clinical trials, the addition of bevacizumab to a chemotherapy regime was well-tolerated and improved progression-free survival [54]. However, even though the findings from these clinical studies have been supported by a large clinical practice-based study (ATHENA), the efficacy of bevacizumab in terms of overall survival showed no significant benefit [54]. At an average cost of $100,000 a year for treatment and an average increase in overall survival of an average of two-four months, many clinicians have restricted the use bevacizumab. The FDA reversed the fast-track approval for bevacizumab in 2010, leaving the future of this agent in limbo.
Six years later in 2012, aflibercept, an antiangiogenic VEGF inhibitor, was approved by the FDA as a second-line treatment for mCRC in combination with the FOLFIRI chemotherapy regimen (leucovorin calcium, fluorouracil, and irinotecan hydrochloride) [56]. Aflibercept (Zaltrap®/Eylea®) is meant to be used for mCRC patients who failed to respond to previous FOLFOX-based chemotherapy [57]. In a phase III clinical trial (NCT00561470), the addition of aflibercept to FOLFIRI improved overall median survival from 12.1 to 13.5 months and progression-free survival from 4.7 to 6.9 months for stage IV mCRC patients [58]. In an update to this same phase III clinical trial, published in 2014, investigators found that overall survival increased by 0.8 months for mCRC patients with no prior treatment and 1.5 months for patients with no prior treatment [59].
5.1.2. Anti-epidermal growth factor receptors (anti-EGFRs)
EGFRs are cellular receptor located on a cell’s surface that activates tyrosine kinase that phosphorylates intracellular substrates responsible for the genetic transcription for cell proliferation, angiogenesis, and invasion (Fig. 2).
In 2004, the FDA approved cetuximab to treat advanced CRC patients who have failed standard chemotherapy [62–65]. Cetuximab, also approved for use in breast cancer, targets the ligand-binding domain of EGFR, as a mutation (i.e. Kristen rat sarcoma (KRAS) gene mutation) in this pathway results in an increase in uncontrolled cell growth. A clinical trial conducted by the North Central Cancer Treatment Group (NCCTG) N0147 compared the use of FOLFIRI with and without cetuximab in stage III CRC with both wild-type KRAS and mutant KRAS. In the clinical trial, a combination treatment with cetuximab plus FOLFIRI showed that 5-year disease-free survival, overall survival and time to recurrence in patients with wild-type KRAS improved from 64% to 83% (p = 0.10), 76% to 87% (p-0.21), and 67% to 86% (p = 0.09), respectively after 10 to 11 months [66]. Based in part on this study, as well as the CEGOG trial, the FDA approved cetuximab in 2012 as a first-line treatment in KRAS−/EGFR+ mCRC in combination with FOLFIRI.
In 2017, panitumumab, another EGFR inhibitor, was granted FDA approval to treat mCRC patients with wild-type KRAS as a first-line treatment in combination with FOLFOX [69,70]. A study by Leone et al. used panitumumab in combination with capecitabine plus oxaliplatin (XELOX) to study its efficacy in patients with liver only mCRC. Out of the forty-six patients, the objective response rate was 54% with two patients with complete responses and 23 with a partial response. Overall, the combination of panitumumab with XELOX (P-XELOX) yields a high response for patients with liver only mCRC [71].
Like many other clinically approved drugs, anti-VEGFs and anti-EGFRs also have their limitations in mCRC and other cancer types. Many clinicians and researchers have listed a number of explanations for the generally limited efficacy for these inhibitors. One explanation is that the formation of tumor blood vessels and how they are maintained is not fully understood113. After treatment with an anti-VEGF therapy, it has been shown that residual hypoxic tumor cells are simulated to increase production in VEGF-A which can inhibit the anti-VEGF therapy [72]. Also, in many pre-clinical studies, many protocols induce mice with tumor xenografts and immediately start treatment after implantation [72]. This treatment pattern does not follow the clinical treatment pathway in tumors that have already established tumor vasculature [72]. In conclusion, the limitations of anti-VEGF and anti-EGFR therapies have pushed researchers to create new clinical trials and pre-clinical trials to use other cytokine pathways for mCRC treatment.
5.2. Clinical studies
Table 3 shows a list of ongoing clinical trials using cytokine inhibitors.
Table 3.
Current list of ongoing clinical trials using cytokine inhibitors.
| Name | NCT.gov Identifier | Phase | Intervention | Results |
|---|---|---|---|---|
| Siltuximab113 | — | I/II | CRC patients with advanced solid tumors using interleukin-6 (IL-6) | Increased tumor hemoglobin; low response rate |
| AM0100 | — | I | Solid tumors in CRC using IL-10) | No results posted |
| Nimotuzumab | NCT00972465 | II | Advanced CRC by binding to an EGFR | No results posted |
| Imalumab | NCT02448810 | II | Patients with metastatic colorectal cancer through binding to a migration inhibitory factor (MIF) | No patients completed treatment. Two deaths reported. |
5.3. Pre-clinical studies
The effect of modulating cytokines and chemokines in the human CRC TME is mostly hypothesized and has not yet been rigorously tested in clinical trials. Most CRC cytokine modulation research, besides the aforementioned interleukins, exists in the pre-clinical and basic biology realms.
Two chemokine receptors, C-C chemokine receptor type 1 (CCR1) and chemokine C-C motif receptor-like 2 (CCRL2), have been recently implicated in aiding in liver metastasis [48], the primary cause of death for CRC patients [73]. Ligands for CCR1 and CCRL2 are the chemokines CCL3, CCL5, CCL7, and CCL23, and are suggested as potential targets for cytokine-targeted immunotherapy [48]. CCL2 and CCL24 were also found to be highly elevated (> 100-fold) in CRC liver metastases compared to healthy adjacent liver tissue, implying that these chemokines could also be targets for cytokine-targeted immunotherapy [74].
Chemokine neutralization, especially of CCL2, has gained traction in both CRC and non-CRC studies of mice [75]. CRC, independent of subtype [76,77], recruits circulating monocytes via chemotaxis to the TME primarily through the release of CCL2, also known as monocyte chemoattractant protein-1 (MCP1), a highly elevated chemokine in CRC [68–71]. In the TME, monocytes differentiate into TAMs, partially as a result of CCL2. TAMs, the most abundant immune cell in the TME, also have the most substantial and pervasive effect of any immune cell in the TME [72–,73,74,75]. In CRC, TAMs have been shown to have both anti-tumor and pro-tumor functions, depending on whether they are polarized more towards an M1 (classical) or M2 (alternative) phenotype and their physical location within the tumor [76]. Pro-tumor functions of alternatively activated M2-polarized TAMs include tumor growth, angiogenesis, immunosuppression, and matrix remodeling [77]. Additionally, CCL2 binding to its receptor, CCR2, on endothelial cells increases vascular permeability and metastatic risk [78]. Thus, targeting CCL2 to reduce M2-polarized, pro-tumor TAMs is an attractive ongoing cytokine-targeted immunotherapy strategy in pre-clinical settings. In mouse models, CCL2 blockade has resulted in reduced neovascularization and tumor size of orthotopic colon tumors in Balb/c mice, suggesting that CCL2 may be a promising target for treating colitis-associated colon cancer [79]. Additionally, anti-CCL2 immunotherapy prolonged survival in C57BL/6 mice with GL261 glioma [80], and reduced TAM infiltration in FVB/N mice with MCF-7 breast cancer [81]. However, few cytokine-targeted immunotherapy techniques have been tested for efficacy in human CRC, although oral N-acetyl-L-cysteine (NAC) co-administered with mesalamine, an anti-inflammatory, has benefitted ulcerative colitis patients, attributed in part to the down-regulation of CCL2 and IL-8 [82]. In summary, many investigators now believe that CCL2-neutralizing immunotherapy will play an important role in early-stage CRC treatment in future clinical studies [83].
Besides CCL2, other cytokines and chemokines have been explored. For example, blocking the pro-angiogenic and pro-tumor chemokine ligand 1 (CXCL1), whose gene is also known as growth-regulated oncogene-α, using an anti-CXCL1 neutralizing antibody inhibited tumor growth and angiogenesis in a mouse xenograft model of human CRC [84]. Blockade of IL-1β reduced tumor formation in a mouse model of colitis-associated CRC [85]. TNF blockade reduced CRC carcinogenesis in an AOM/DSS (colitis-induced) mouse model [86]. On the other hand, the addition of IL-15, which has anti-tumor effects in CRC, was shown to increase the therapeutic effects of anti-PD-L1 and anti-CTLA4 treatment in a CT26 colon carcinoma mouse model [87]. The overarching current hypothesis is that cytokine-targeted immunotherapy, especially the blockade of pro-tumor cytokines in CRC, may enhance tumor therapeutic response in CRC tumors treated with chemotherapy, radiation, or approved checkpoint inhibitors.
5.4. Conclusion
Cytokine-targeted immunotherapy research lags other discussed CRC immunotherapy methods, although further investigation is justified. The biggest challenge facing this type of therapy is determining which pharmacokinetic and pharmacodynamic variables are important navigating cytokine pathways while decreasing systemic toxicity in CRC patients. Additionally, the FDA approved drugs, cetuximab and panitumumab are ineffective in patients with RAS mutations (∼23% of stage IV CRC patients). Overall, cytokine therapies will likely be most effective in combination with other immunotherapies or chemo- and/or radiotherapy.
6. Assessing tumor therapeutic response
In addition to new CRC therapies being investigated, there is significant interest in the development of clinically-translatable methods to rapidly assess whether a therapy regimen is effective on a per patient basis [87–90]. Rapid assessment of therapy can prevent unnecessary chemotherapy in both responders and non-responders [91]. Currently, tumors are assessed based on the widely accepted Response Evaluation Criteria in Solid Tumors (RECIST) criteria, which grades tumors as, from most desirable to least desirable, complete responders, partial responders, stable disease, or progressive disease [92–94]. The overall goal of assessing tumor therapeutic response is adjusting treatment if necessary, avoiding surgery and reducing morbidity [95]. The standards for monitoring tumor therapeutic response to neoadjuvant therapy (chemotherapy, radiation, and/or immunotherapy) using RECIST are digital rectal examination (DRE), rigid proctoscopy, biopsy, carcinoembryonic antigen (CEA) level, and a radiological technique such as CT [96], PET-CT, MRI, or Diffusion-Weighted (DW)-MRI [97]. However, following neoadjuvant treatment initiation, assessing tumor response does not occur for approximately two months [10]. Additionally, for patients showing evidence of partial or complete response after these two months of neoadjuvant treatment, they must wait an additional 1–2 months for follow-up as part of the “Wait and Watch Protocol.” Finally, studies have shown that current radiological techniques are insufficient to identify responders with positive predictive values less than 50% [93]. Several research groups are investigating emerging optical and imaging methods to rapidly assess therapeutic response on a scale of days or weeks, rather than months.
Some advantages to using optical methods to monitor tumor response include non-ionizing radiation, better spatial resolution, sensitivity to biological molecules, etc. Since the CRC screening, diagnostic, and, in some cases, therapeutic standard (in early CRC stages only) is colonoscopy, investigators are aiming to create minimally-invasive endoscopy-compatible techniques. Techniques currently being evaluated, mostly in pre-clinical laboratory settings, for use in CRC include nonlinear optical imaging, fluorescence-based endoscopy, and diffuse reflectance spectroscopy.
6.1. Fluorescence-based endoscopy
Fluorescence-based endoscopy integrates colonoscopy with optical imaging. This technique is a “robust method for early detection of CRC owing to its intrinsic coupling of detection with the underlying molecular-level pathology of the disease”. With the use of molecular imaging, this type of optical system can detect variations in tissues unlike other system that only detect changes in structure [94].
In a study by Mitsunaga et al., they developed a “rapid fluorescent detection method” using a “topically applied enzymatically activatable probe (gGlu-HMRG)” to detect the γ-glutamyltranspeptidase (GGT) enzyme during a colonoscopy. Expression of GGT was higher in mouse models with CRC than those without. Five minutes after topical administration, gGlu-HMRG fluorescent lesions were detected using fluorescent microscopy. Based on these results, the use of gGlu-HMRG can improve detection of colitis-associated colon cancer (CAC) with a “higher target to background ratio” compared to conventional white light colonoscopy [98].
In a human study by Watanabe et al., used the PINPOINT® Endoscopic Fluorescence Imaging System intraoperatively to identify tumor sites using indocyanine green during laparoscopic surgery. Using this system, surgeons saw a tumor visibility rate of 93.8%. No adverse effects were observed during these procedures. As a result, this study provided evidence that the PINPOINT® system was able to identify colorectal tumors without adverse effects [99].
6.2. Diffuse reflectance spectroscopy
Diffuse reflectance spectroscopy (DRS) is a non-invasive or minimally-invasive technique that uses a small probe to deliver broadband light to tissue and collect the diffusely reflected light with a spectrometer [97]. DRS can provide relevant clinical information such as total hemoglobin content, tissue oxygen saturation, oxy- and deoxyhemoglobin, lipid and water content, and tissue scattering properties, and can thus be applied to monitoring tumor response to therapy [97,99].
DRS has recently been used in an ex vivo study of resected human colon tissue to differentiate tissue type with an overall accuracy of 95%. The investigators hope to eventually apply this technology in an in vivo setting for real-time guidance during CRC surgery. DRS has also been integrated into a fiber-optic biopsy needle to assess functional tissue properties in an in vivo study of lung cancer patients. Greening et al. used their DRS system to monitor tumor response to chemotherapy in a murine subcutaneous colonic tumor model. The investigators are currently studying short-term vs long-term data points after various treatment modalities and believe this technology can someday help optimize personalized cancer treatments.146
One of the primary limitations with optical methods, such as DRS, is relatively poor sampling depth into highly scattering tissues, especially when compared to methods such as MRI. However, DRS sampling depth is greater than 0.5 mm at 630 nm at source-detector separations (< 1 mm) compatible with the biopsy port of standard colonoscop (1.5 mm) [97]. This indicates that data obtained via DRS endoscopy could yield data from a similar region of the tumor as can be obtained via endoscopic biopsy. Although the entire tumor volume may not be accessible, this may provide enough functional information to guide clinical decision-making with respect to therapeutic monitoring.
As of yet, DRS applied to CRC is in its infancy; it has only been applied to monitor tumor therapeutic response to chemotherapy in mouse models, although investigators believe DRS technology can be used to quantify volumetric tumor perfusion in response to immunotherapies, which can eventually help guide clinicians in identifying potential responders and non-responders during early therapy [97].
7. Conclusion
Colorectal cancer is still one of the most prominent cancer types within the United States. Although current treatment standards (neoadjuvant therapy, surgery, and adjuvant therapy) treat a wide spectrum of cancer patients, recurrence, patient heterogeneity, toxicity, and poor survival rate remain problematic. Therefore, research into antibody-based immunotherapies in both clinical and pre-clinical settings is highly active. Clinical research into immune checkpoint inhibitors is more mature than cytokine-targeted immunotherapy. At present, cytokine-targeted immunotherapy is limited to anti-VEGF, anti-VEGFR, and anti-EGFR therapies for mCRC patients, although there is a growing interest in interleukin and chemokine therapies in both pre-clinical and early clinical trials. Additionally, monitoring CRC tumor response is a major problem, and investigators are continuing to engineer optical methods to improve the state-of-the-art. One of the biggest emerging challenges for immunotherapy in CRC is elucidating the genomic biomarkers for identifying patients likely to be responders or non-responders for certain immunotherapy regimens and monitoring response in real-time.
Acknowledgements
This material is based on work supported by the National Science Foundation (CBET 1751554) National Institutes of Health (R15-CA202662-01), the National Science Foundation Graduate Research Fellowship Program (G.G., DGE-1450079), and the Arkansas Biosciences Institute. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the acknowledged funding agencies.
Biographies

Shelby N. Bess received her bachelor’s degree in Biomedical Engineering from the University of Arkansas in Fayetteville, Arkansas. She is currently a Graduate Researcher under Dr. Timothy Muldoon at the University of Arkansas, studying the long-term effects of 5-FU and anti-CCL2 treatment response in murine models using diffuse reflectance spectroscopy.

Gage J. Greening received his Ph.D. in Biomedical Engineering from the University of Arkansas in Fayetteville, AR. He did his doctoral work on using diffuse reflectance spectroscopy to quantify in vivo tissue optical properties in human epithelium and subcutaneous murine colon cancer. He is currently working for NanoMatronix using computer aided design on various government contracts.

Timothy J. Muldoon received his Ph.D. from Rice University in Houston, Texas. He is currently an Associate Professor at the University of Arkansas where his research encompasses designing multimodal microendoscopy and spectroscopy technologies used to monitor colorecta cancer and other gastric diseases such as ulcerative colitis.
Footnotes
Declaration of Competing Interest
The authors declare no conflicts of interest or comp shows a list of ongoing pre-clinical trials that use immune checkpoeting interests.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2018, Cancer J. Clin 68 (2018) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Jeon HJ, Woo JH, Lee HY, et al. , Adjuvant chemotherapy using the FOLFOX regimen in colon cancer, J. Korean Soc. Coloproctol 27 (2011) 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sharp GS, Benefiel WW, 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum, Cancer Chemother. Rep 20 (1962) 97–101. [PubMed] [Google Scholar]
- [4].General Cancer Information, FOLFOX | Cancer Information | Cancer Research UK, Cancer Research UK, 17 May (2018) www.cancerresearchuk.org/about-cancer/cancer-in-general/treatment/cancer-drugs/drugs/folfox.
- [5].Surgical resection with or without preoperative chemotherapy in oesophageal cancer: a randomised controlled trial, Lancet 359 (2002) 1727–1733. [DOI] [PubMed] [Google Scholar]
- [6].Cunningham D, Allum WH, Stenning SP, et al. , Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer, N. Engl. J. Med 355 (2006) 11–20. [DOI] [PubMed] [Google Scholar]
- [7].Group FC, Feasibility of preoperative chemotherapy for locally advanced, operable colon cancer: the pilot phase of a randomised controlled trial, Lancet Oncol. 13 (2012) 1152–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Van Cutsem E, Borràs JM, Castells A, et al. , Improving outcomes in colorectal cancer: where do we go from here? Eur. J. Cancer 49 (2013) 2476–2485. [DOI] [PubMed] [Google Scholar]
- [9].Cercek A, Goodman KA, Hajj C, et al. , Neoadjuvant chemotherapy first, followed by chemoradiation and then surgery, in the management of locally advanced rectal cancer, J. Compr. Canc. Netw 12 (2014) 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Habr-Gama A, Perez RO, Wynn G, et al. , Complete clinical response after neoadjuvant chemoradiation therapy for distal rectal cancer: characterization of clinical and endoscopic findings for standardization, Dis. Colon Rectum 53 (2010) 1692–1698. [DOI] [PubMed] [Google Scholar]
- [11].Miles A, van Duijnhoven F, McQueen A, et al. , Colorectal cancer: advances in prevention and early detection, Biomed Res. Int 2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bénard F, Barkun AN, Martel M, et al. , Systematic review of colorectal cancer screening guidelines for average-risk adults: summarizing the current global recommendations, World J. Gastroenterol 24 (2018) 124–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lynch D, Murphy A, The emerging role of immunotherapy in colorectal cancer, Ann. Transl. Med 4 (2016) 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boland PM, Ma WW, Immunotherapy for colorectal cancer, Cancers 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Noguchi T, Ritter G, Nishikawa H, Antibody-based therapy in colorectal cancer, Immunotherapy 5 (2013) 533–545. [DOI] [PubMed] [Google Scholar]
- [16].Lee L, Gupta M, Sahasranaman S, Immune Checkpoint inhibitors: an introduction to the next-generation cancer immunotherapy, J. Clin. Pharmacol 56 (2016) 157–169. [DOI] [PubMed] [Google Scholar]
- [17].S, et al. , Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent B cell differentiation (help), J. Exp. Med 175 (4) (1992) 1091–1101 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2119166/Lederman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gao Gynix, Jakobsen K, Bent, Molecular interactions of coreceptor CD8 and MHC class I: the molecular basis for functional coordination with the T-cell receptor, Immunol. Today 21 (2001) 630–636, https://doi.org/10.1016/S0167-5699(00)01750-3 https://www.cell.com/trends/immunology/fulltext/S0167-5699(00)01750-3?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0167569900017503%3Fshowall%3Dtrue. [DOI] [PubMed] [Google Scholar]
- [19].Nichols BA, et al. , Differentiation of monocytes. Origin, nature, and fate of their azurophil granules, J. Cell Biol 50 (2) (1971) 498–515 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2108281/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vivier Eric, et al. , Functions of natural killer cells, Nature Immunology, U.S. National Library of Medicine, 2008. May www.ncbi.nlm.nih.gov/pubmed/18425107. [DOI] [PubMed] [Google Scholar]
- [21].Buchbinder EI, Desai A, CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition, Am. J. Clin. Oncol 39 (2016) 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Seidel JA, Otsuka A, Kabashima K, Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations, Front. Oncol. 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Qureshi OS, Zheng Y, Nakamura K, et al. , Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4, Science 332 (2011) 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Passardi A, Canale M, Valgiusti M, et al. , Immune checkpoints as a target for colorectal cancer treatment, Int. J. Mol. Sci 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sun X, Suo J, Yan J, Immunotherapy in human colorectal cancer: challenges and prospective, World J. Gastroenterol 22 (2016) 6362–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Riley JL, PD-1 signaling in primary T cells, Immunol. Rev 229 (2009) 114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Valentini AM, Di Pinto F, Cariola F, et al. , PD-L1 expression in colorectal cancer defines three subsets of tumor immune microenvironments, Oncotarget 9 (2018) 8584–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Keir ME, Liang SC, Guleria I, et al. , Tissue expression of PD-L1 mediates peripheral T cell tolerance, J. Exp. Med 203 (2006) 883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang X, Yang L, Huang F, et al. , Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells, Immunol. Lett 184 (2017) 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].O’Neil BH, Wallmark JM, Lorente D, et al. , Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma, PLoS One 12 (2017) e0189848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hildeman A. David, et al. , T cell apoptosis and reactive oxygen species, J. Clin. Invest 111 (5) (2003) 575–581, https://doi.org/10.1172/JCI18007 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC151907/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Overman MJ, McDermott R, Leach JL, et al. , Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study, Lancet Oncol. 18 (2017) 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Smith KM, Desai J, Nivolumab for the treatment of colorectal cancer, Expert Rev. Anticancer Ther. 18 (2018) 611–618. [DOI] [PubMed] [Google Scholar]
- [34].Diaz LA, Le DT, Yoshino T, et al. , KEYNOTE-177: randomized phase III study of pembrolizumab versus investigator-choice chemotherapy for mismatch repair-deficient or microsatellite instability-high metastatic colorectal carcinoma, J. Clin. Oncol 35 (2017) TPS815–TPS815. [Google Scholar]
- [35].Andre T Le DT, Kim TW, et al. , KEYNOTE-164: phase 2 study of pembrolizumab for patients with previously treated, microsatellite instability-high advanced colorectal carcinoma, J. Clin. Oncol 34 (2016) TPS3631–TPS3631. [Google Scholar]
- [42].Jenkins RW, Barbie DA, Flaherty KT, Mechanisms of resistance to immune checkpoint inhibitors, Br. J. Cancer 118 (2018) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tsuji S, Midorikawa Y, Takahashi T, et al. , Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis, Br. J. Cancer 106 (2012) 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Patel SP, Kurzrock R, PD-L1 expression as a predictive biomarker in cancer immunotherapy, Mol. Cancer Ther 14 (2015) 847–856. [DOI] [PubMed] [Google Scholar]
- [45].Li B, Chan HL, Chen P, Immune checkpoint inhibitors: basics and challenges, Curr. Med. Chem (2017). [DOI] [PubMed] [Google Scholar]
- [46].West NR, McCuaig S, Franchini F, et al. , Emerging cytokine networks in colorectal cancer, Nat. Rev. Immunol 15 (2015) 615–629. [DOI] [PubMed] [Google Scholar]
- [47].Itatani Y, Kawada K, Inamoto S, et al. , The role of chemokines in promoting colorectal cancer invasion/metastasis, Int. J. Mol. Sci 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Akram IG, Georges R, Hielscher T, et al. , The chemokines CCR1 and CCRL2 have a role in colorectal cancer liver metastasis, Tumour Biol. 37 (2016) 2461–2471. [DOI] [PubMed] [Google Scholar]
- [49].Klampfer L, Cytokines, inflammation and colon cancer, Curr. Cancer Drug Targets 11 (2011) 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Strickler JH, Hurwitz HI, Bevacizumab-based therapies in the first-line treatment of metastatic colorectal cancer, Oncologist 17 (2012) 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cohen MH, Gootenberg J, Keegan P, et al. , FDA drug approval summary: bevacizumab plus FOLFOX4 as second-line treatment of colorectal cancer, Oncologist 12 (2007) 356–361. [DOI] [PubMed] [Google Scholar]
- [52].Giantonio BJ, Catalano PJ, Meropol NJ, et al. , Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200, J. Clin. Oncol 25 (2007) 1539–1544. [DOI] [PubMed] [Google Scholar]
- [53].Hurwitz H, Fehrenbacher L, Novotny W, et al. , Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer, N. Engl. J. Med 350 (2004) 2335–2342. [DOI] [PubMed] [Google Scholar]
- [54].Saltz LB, Clarke S, Diaz-Rubio E, et al. , Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study, J. Clin. Oncol 26 (2008) 2013–2019. [DOI] [PubMed] [Google Scholar]
- [55].Ilic I, Jankovic S, Ilic M, Bevacizumab combined with chemotherapy improves survival for patients with metastatic colorectal cancer: evidence from meta analysis, PLoS One 11 (2016) e0161912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Van Cutsem E, Tabernero J, Lakomy R, et al. , Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen, J. Clin. Oncol 30 (2012) 3499–3506. [DOI] [PubMed] [Google Scholar]
- [57].Tabernero J, Van Cutsem E, Lakomy R, et al. , Aflibercept versus placebo in combination with fluorouracil, leucovorin and irinotecan in the treatment of previously treated metastatic colorectal cancer: prespecified subgroup analyses from the VELOUR trial, Eur. J. Cancer 50 (2014) 320–331. [DOI] [PubMed] [Google Scholar]
- [58].Dhillon S, Regorafenib: a review in metastatic colorectal cancer, Drugs 78 (2018) 1133–1144. [DOI] [PubMed] [Google Scholar]
- [59].Grothey A, Van Cutsem E, Sobrero A, et al. , Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial, Lancet 381 (2013) 303–312. [DOI] [PubMed] [Google Scholar]
- [62].Hubbard JM, Alberts SR, Alternate dosing of cetuximab for patients with metastatic colorectal cancer, Gastrointest. Cancer Res 6 (2013) 47–55. [PMC free article] [PubMed] [Google Scholar]
- [63].Cunningham D, Humblet Y, Siena S, et al. , Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer, N. Engl. J. Med 351 (2004) 337–345. [DOI] [PubMed] [Google Scholar]
- [64].Lenz HJ, Cetuximab in the management of colorectal cancer, Biologics 1 (2007) 77–91. [PMC free article] [PubMed] [Google Scholar]
- [65].Huang J, Nair SG, Mahoney MR, et al. , Comparison of FOLFIRI with or without cetuximab in patients with resected stage III colon cancer; NCCTG (Alliance) intergroup trial N0147, Clin. Colorectal Cancer 13 (2014) 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ocvirk J, Brodowicz T, Wrba F, et al. , Cetuximab plus FOLFOX6 or FOLFIRI in metastatic colorectal cancer: CECOG trial, World J. Gastroenterol 16 (2010) 3133–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Price TJ, Peeters M, Kim TW, et al. , Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study, Lancet Oncol. 15 (2014) 569–579. [DOI] [PubMed] [Google Scholar]
- [69].Leone F, Artale S, Marino D, et al. , Panitumumab in combination with infusional oxaliplatin and oral capecitabine for conversion therapy in patients with colon cancer and advanced liver metastases. The MetaPan study, Cancer 119 (2013) 3429–3435. [DOI] [PubMed] [Google Scholar]
- [70].“A Safety, Efficacy and Pharmacokinetic Study of Siltuximab (CNTO 328) in Participants With Solid Tumors - Full Text View.” Full Text View - ClinicalTrials.gOv, US National Library of Medicine, 2014 May clinicaltrials.gov/ct2/show/NCT00841191. [Google Scholar]
- [71].Valderrama-Trevino AI, Barrera-Mera B, Ceballos-Villalva JC, et al. , Hepatic metastasis from colorectal Cancer, Euroasian J. Hepatogastroenterol 7 (2017) 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cheadle EJ, Riyad K, Subar D, et al. , Eotaxin-2 and colorectal cancer: a potential target for immune therapy, Clin. Cancer Res 13 (2007) 5719–5728. [DOI] [PubMed] [Google Scholar]
- [73].Chun E, Lavoie S, Michaud M, et al. , CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function, Cell Rep. 12 (2015) 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lim SY, Yuzhalin AE, Gordon-Weeks AN, et al. , Targeting the CCL2-CCR2 signaling axis in cancer metastasis, Oncotarget 7 (2016) 18697–18710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Becht E, De Reyniès A, Giraldo NA, et al. , Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy, Clin. Cancer Res 22 (2016) 4057–4066. [DOI] [PubMed] [Google Scholar]
- [76].Marech I, Ammendola M, Sacco R, et al. , Tumour‐associated macrophages correlate with microvascular bed extension in colorectal cancer patients, J. Cell. Mol. Med 20 (2016) 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Erreni M, Mantovani A, Allavena P, Tumor-associated macrophages (TAM) and inflammation in colorectal cancer, Cancer Microenviorn. 4 (2011) 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Allavena P, Sica A, Solinas G, et al. , The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages, Crit. Rev. Oncol. Hematol 66 (2008) 1–9. [DOI] [PubMed] [Google Scholar]
- [79].Chen JJ, Lin YC, Yao PL, et al. , Tumor-associated macrophages: the double-edged sword in cancer progression, J. Clin. Oncol 23 (2005) 953–964. [DOI] [PubMed] [Google Scholar]
- [80].Liu Y, Cao X, The origin and function of tumor-associated macrophages, Cell. Mol. Immunol 12 (2015) 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Popivanova BK, Kostadinova FI, Furuichi K, et al. , Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice, Cancer Res. 69 (2009) 7884–7892. [DOI] [PubMed] [Google Scholar]
- [82].Zhu X, Fujita M, Snyder LA, et al. , Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy, J. Neurooncol 104 (2011) 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Svensson S, Abrahamsson A, Vazquez Rodriguez G, et al. , CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer, Clin. Cancer Res 21 (2015) 3794–3805. [DOI] [PubMed] [Google Scholar]
- [84].Guijarro LG, Mate J, Gisbert JP, et al. , N-acetyl-L-cysteine combined with mesalamine in the treatment of ulcerative colitis: randomized, placebo-controlled pilot study, World J. Gastroenterol 14 (2008) 2851–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang D, Wang H, Brown J, et al. , CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer, J. Exp. Med 203 (2006) 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang Y, Wang K, Han GC, et al. , Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis, Mucosal Immunol. 7 (2014) 1106–1115. [DOI] [PubMed] [Google Scholar]
- [87].Popivanova BK, Kitamura K, Wu Y, et al. , Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis, J. Clin. Invest 118 (2008) 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Roblyer D, Ueda S, Cerussi A, et al. , Optical imaging of breast cancer oxyhemoglobin flare correlates with neoadjuvant chemotherapy response one day after starting treatment, PNAS 108 (2011) 14626–14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Chen CJ, Sung WW, Chen HC, et al. , Early assessment of colorectal Cancer by quantifying circulating tumor cells in peripheral blood: ECT2 in diagnosis of colorectal cancer, Int. J. Mol. Sci 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Granata V, Fusco R, Catalano O, et al. , Early assessment of colorectal cancer patients with liver metastases treated with antiangiogenic drugs: the role of intravoxel incoherent motion in diffusion-weighted imaging, PLoS One 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Eisenhauer EA, Therasse P, Bogaerts J, et al. , New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1), Eur. J. Cancer 45 (2009) 228–247. [DOI] [PubMed] [Google Scholar]
- [94].Wahl RL, Jacene H, Kasamon Y, et al. , From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors, J. Nucl. Med 50 (2009) 122s–150s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chung WS, Park MS, Shin SJ, et al. , Response evaluation in patients with colorectal liver metastases: RECIST version 1.1 versus modified CT criteria, AJR Am. J. Roentgenol 199 (2012) 809–815. [DOI] [PubMed] [Google Scholar]
- [96].Walker AS, Zwintscher NP, Johnson EK, et al. , Future directions for monitoring treatment response in colorectal cancer, J. Cancer 5 (2014) 44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kekelidze M, D’Errico L, Pansini M, et al. , Colorectal cancer: current imaging methods and future perspectives for the diagnosis, staging and therapeutic response evaluation, World J. Gastroenterol 19 (2013) 8502–8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Mitsunaga M, Kosaka N, Choyke PL, et al. , Fluorescence endoscopic detection of murine colitis-associated colon cancer by topically applied enzymatically rapid-activatable probe, Gut 62 (2013) 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Watanabe M, Murakami M, Ozawa Y, et al. , Intraoperative identification of colonic tumor sites using a near-infrared fluorescence endoscopic imaging system and indocyanine green, Dig. Surg 34 (2017) 495–501. [DOI] [PubMed] [Google Scholar]