

Abstract
An effective strategy to cure HIV will likely require a potent and sustained antiviral T cell response. Here we explored the utility of chimeric antigen receptor (CAR) T cells, expressing the CD4 ectodomain to confer specificity for the HIV envelope, to mitigate HIV-induced pathogenesis in bone marrow, liver, thymus (BLT) humanized mice. CAR T cells expressing the 4–1BB/CD3-ζ endodomain were insufficient to prevent viral rebound and CD4+ T cell loss after the discontinuation of antiretroviral therapy. Through iterative improvements to the CAR T cell product, we developed Dual-CAR T cells that simultaneously expressed both 4–1BB/CD3-ζ and CD28/CD3-ζ endodomains. Dual-CAR T cells exhibited expansion kinetics that exceeded 4–1BB-, CD28- and third-generation costimulated CAR T cells, elicited effector functions equivalent to CD28-costimulated CAR T cells and prevented HIV-induced CD4+ T cell loss despite persistent viremia. Moreover, when Dual-CAR T cells were protected from HIV infection through expression of the C34-CXCR4 fusion inhibitor, these cells significantly reduced acute-phase viremia, as well as accelerated HIV suppression in the presence of antiretroviral therapy and reduced tissue viral burden. Collectively, these studies demonstrate the enhanced therapeutic potency of a novel Dual-CAR T cell product with the potential to effectively treat HIV infection.
CAR T cell immunotherapies, in which engineered T cells are infused into patients, have induced durable remissions for treatment-refractory malignancies1. Although a potent and sustained T cell response of the kind that CAR T cells can afford is likely to be essential for the development of an effective HIV cure2, a successful CAR T cell therapy for HIV infection has remained elusive. CARs redirect T cell specificity by expressing an extracellular antigen recognition domain linked to an intracellular T cell costimulatory domain and the CD3-ζ chain3,4. The costimulatory domains for second-generation CARs are derived from the intracellular signaling domains of either CD28 or 4–1BB, which is one of the key differences between the two Food and Drug Administration-approved CD19-targeting CAR T cell therapies5,6. Preclinical cancer models demonstrate that CD28-costimulated CAR T cells exhibit profound effector function resulting in rapid tumor clearance, but have limited persistence in vivo7,8. In contrast, 4–1BB-costimulated CAR T cells have a slower antitumor response but sustained proliferation and survival9–12. The distinct signaling pathways used by CD28 and 4–1BB cause distinct metabolic, phenotypic and functional T cell profiles that can elicit optimal CAR T cell activity for specific diseases13–17, and there is great interest in tuning costimulatory signals to optimize CAR T cell function18,19.
The earliest clinical trials of CAR T cell therapy used first-generation, CD4-based CARs targeting the HIV Envelope glycoprotein via surface expression of the CD4 ectodomain and proved ineffective for treatment of either chronic or antiretroviral therapy (ART)-suppressed infection20–22. However, subsequent significant improvements in CAR technology by the cancer immunotherapy field have renewed interest in applying these advances to HIV treatment23–29. The continued investigation of the mechanistic underpinnings of successful and failed CAR T cell therapy, particularly in a model system that recapitulates HIV pathogenesis, will be critical for the development of a CAR T therapy for HIV cure initiatives.
Here we used the BLT humanized mouse model of HIV infection to iteratively test CD28 and 4–1BB costimulation in the context of optimizing HIV-specific CAR T cell therapy. We leveraged the BLT mouse model’s ability to support HIV infection, including high viral loads, rapid human CD4+ T cell depletion and ultimately T cell exhaustion30–33, to make stepwise improvements to the CAR T cell product (TCP). This effort culminated in the development of HIV-resistant (C34-CXCR4+), Dual-CAR T cells that express two CD4-based CARs independently encoding the CD28/CD3-ζ and 4–1BB/CD3-ζ endodomains. Collectively, these data provide important insight regarding the development of an engineered T cell–based therapy of HIV infection and highlight the novel function of a Dual-CAR TCP that mitigates HIV-induced disease.
Results
BLT mouse-derived CAR T cells are multifunctional and suppress HIV replication in vitro.
To determine whether T cells isolated from BLT mice generate potent CAR TCPs, we manufactured HIV-specific (CD4-based) CAR T cells expressing the CD3-ζ endodomain (CAR.ζ) from BLT mouse tissues and adult human peripheral blood mononuclear cells (PBMCs) (Supplementary Fig. 1a). BLT mouse- and human-derived CAR.ζ T cells exhibited comparable in vitro expansion kinetics and CAR surface expression levels (Supplementary Fig. 1b and Fig. 1a). Antigen-specific stimulation with K562 cells expressing HIVYU2 Envelope (K.Env) induced similar cytokine expression and polyfunctionality profiles between the CAR T cell sources (Fig. 1b,c and Supplementary Fig. 1c–e). Furthermore, CAR.ζ T cells from both donors suppressed viral outgrowth down to a 1:50 effector/target ratio in vitro (Fig. 1d,e), and induced similar levels of cleaved caspase-3 in HIV-infected CD4+ T cells (Supplementary Fig. 1f and Fig. 1f,g). The induction of caspase-3 combined with the coupregulation of granzyme B and perforin by CAR.ζ T cells (Supplementary Fig. 1g,h) indicates that elimination of virus-infected cells likely occurred via granule-mediated cytolysis. In total, the in vitro functional profile of BLT mouse-derived CAR.ζ T cells was indistinguishable from that of human-derived CAR.ζ T cells, demonstrating that highly functional CAR T cells can be manufactured from BLT mice.
Fig. 1 |. BLT mouse-derived HIV-specific CAR T cells are functionally indistinguishable from human-derived CAR T cells in vitro.
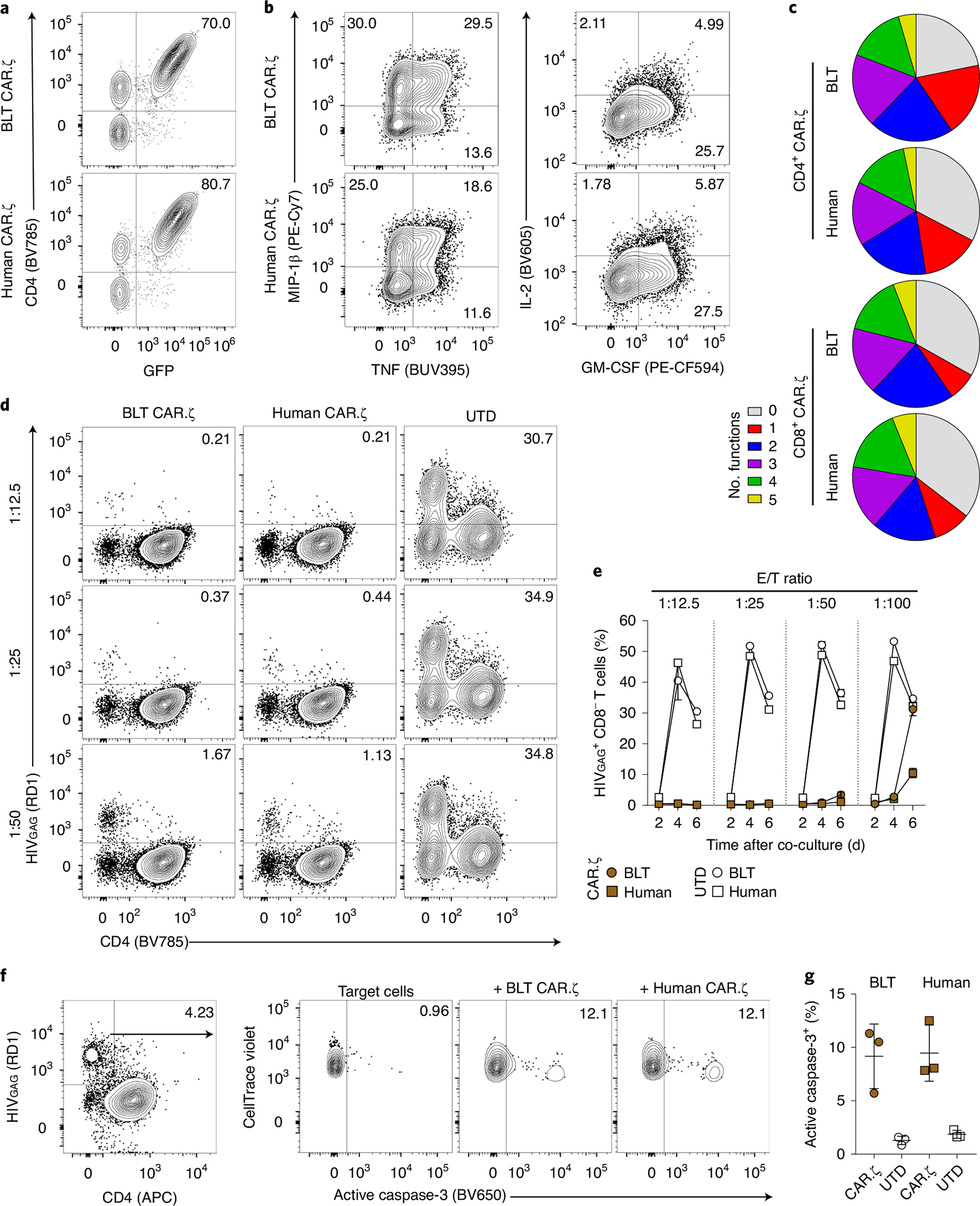
αCD3/CD28 Dynabeads were used to activate purified human T cells from a BLT mouse and healthy human donor, and then cells were transduced with the CD4-based CAR.ζ construct coexpressing GFP. a, FACS plots identify CAR.ζ T cells from each T cell source as GFP+ and CD4+. b, Following 10 d of culture, CD8+ CAR.ζ T cells were mixed with HIVYU2 Envelope+ K562 cells (K.Env) and upregulation of human cytokines was measured. c, Polyfunctionality profiles for combinatorial subsets of CD4+ and CD8+ CAR.ζ T cells producing 0 to 5 of the human cytokines GM-CSF, IFN-γ, IL-2, MIP-1β and TNF. Average of three unique donors per T cell source. d,e, HIV suppression assay as described in the Methods. d, FACS plots indicating the frequency of HIV-infected cells 6 d after coculturing with BLT mouse- or human-derived CAR.ζ T cells at denoted E/T ratios. e, Summary of frequency of HIV-infected cells (live CAR− CD8− cells) at 2, 4 and 6 d after coculture with BLT mouse- or human-derived CAR.ζ and untransduced (UTD) T cells at indicated E/T ratios. f,g, HIV elimination assay as described in the Methods. FACS plots (f) and summarized data (g) for frequency of active caspase-3 within HIV-infected cells (live CTV+ HIVGAG+ cells) 24 h after coculture with BLT mouse- or human-derived CAR.ζ and UTD T cells at 1:1 E/T ratio. Each symbol represents the average of technical duplicates per donor (n = 3 biologically independent donors). e,g, Symbols and lines reflect mean and error bars indicate ±s.e.m. RD1 is another name for phycoerythrin (PE).
Costimulation modulates CAR T cell persistence and function in vivo.
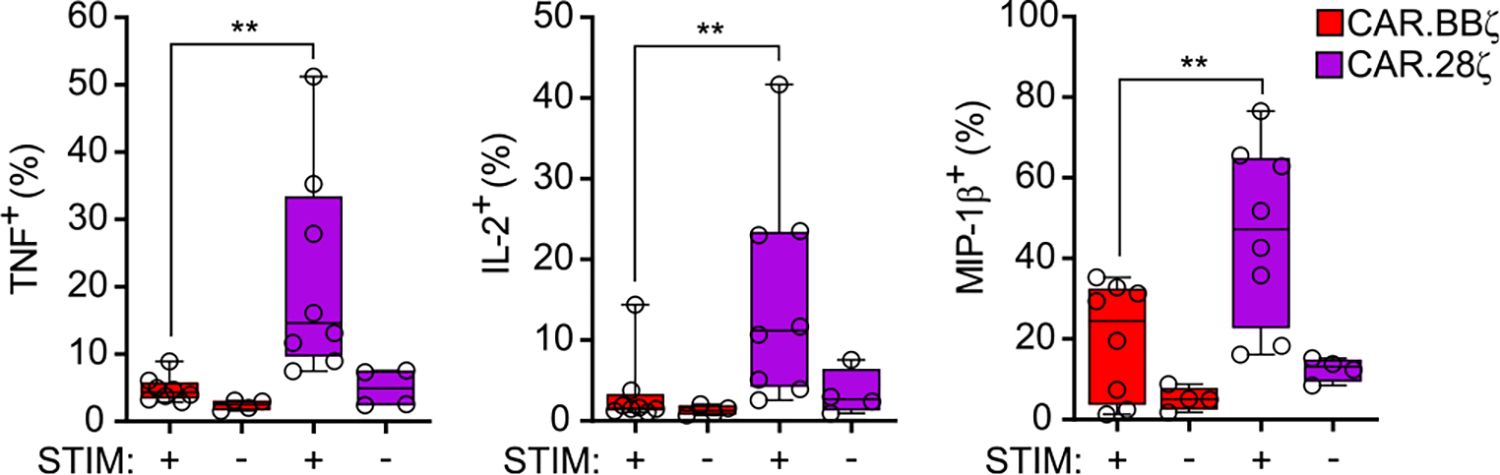
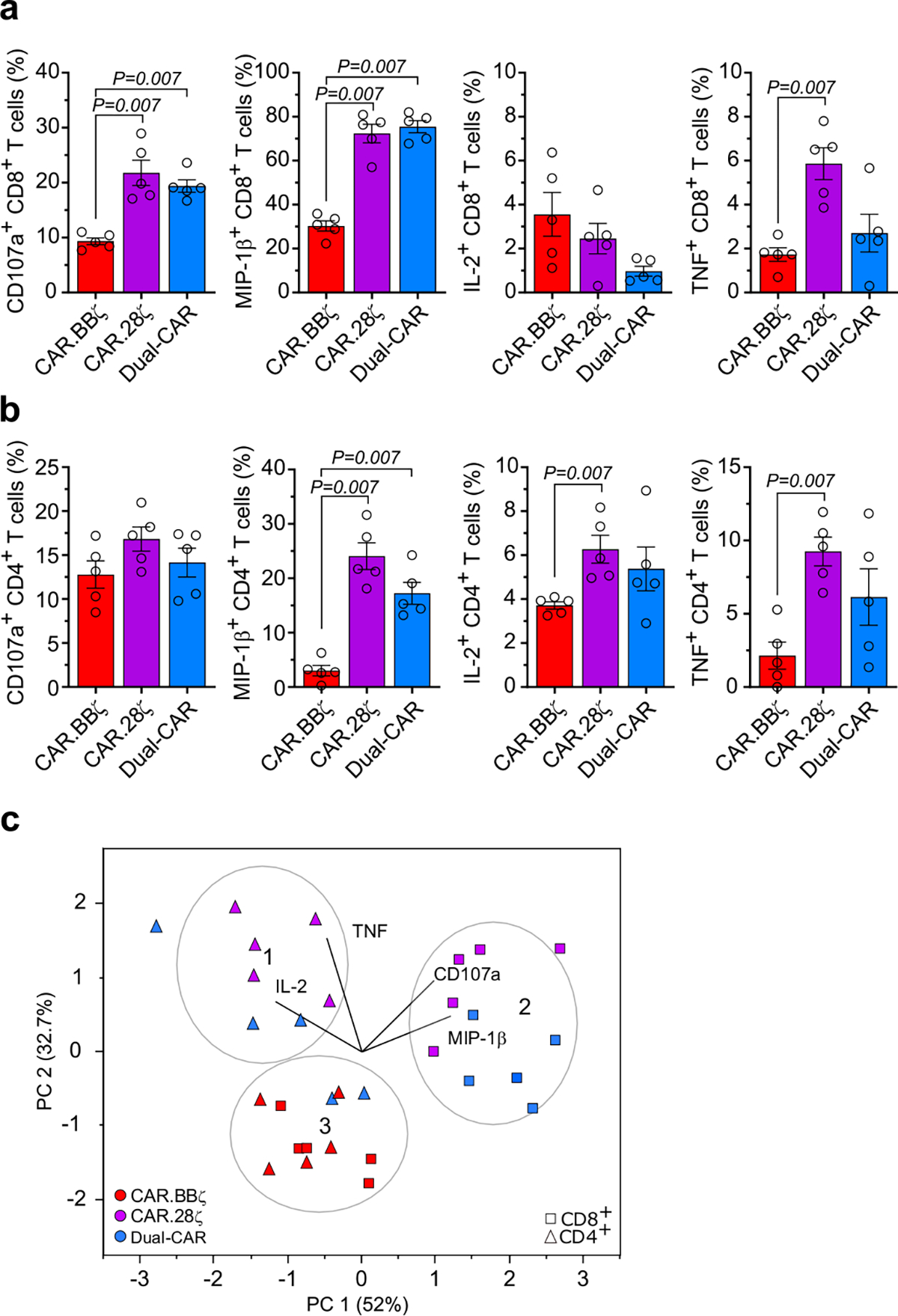
To identify a CAR TCP capable of long-term persistence against HIV, we compared the contribution of costimulatory domains with in vivo engraftment of T cells by creating an infusion product comprising equal frequencies of HIV-specific CAR.CD3-ζ (CAR.ζ), 4–1BB/CD3-ζ (CAR.BBζ) and CD28/CD3-ζ (CAR.28ζ) T cells, each of which was linked to a distinct fluorescent protein (Fig. 2a). After infusion, CAR.BBζ T cells exhibited significantly greater survival in the absence of HIV antigen (Fig. 2b–d), and constituted approximately 80% of total CAR T cells in tissues (Fig. 2e). Consistent with reports from the cancer field9–12, CAR.BBζ T cells also demonstrated superior in vivo antigen-driven proliferation upon infusion of K.Env cells (Fig. 2f). In contrast, CAR.28ζ T cells only exhibited a transient expansion followed by a progressive decline, and CAR.ζ T cells steadily declined demonstrating no evidence of expansion. Notably, however, CAR.28ζ T cells exhibited greater effector functions when stimulated ex vivo with K.Env cells, upregulating more MIP-1β, TNF and IL-2, and coexpressing greater levels of granzyme B and perforin than CAR.BBζ T cells from the same mice (Fig. 2g,h and Extended Data Fig. 1). Finally, we confirmed the in vivo cytotoxic potential of BLT mouse-derived CAR T cells by infusing CD19-specific CAR.BBζ T cells into recipient mice. We observed rapid and profound B cell aplasia in the blood (Fig. 2i), as well as in the spleen, lung, liver and bone marrow, consistent with a sustained cytotoxic CAR T cell response (Fig. 2j,k). Together, these data demonstrate the suitability of BLT mice for studying in vivo CAR T cell function, and the degree to which costimulation can modulate CAR T cell activity.
Fig. 2 |. CAR T cells expressing the 4–1BB costimulatory domain exhibit a proliferative advantage and induce B cell aplasia in vivo.
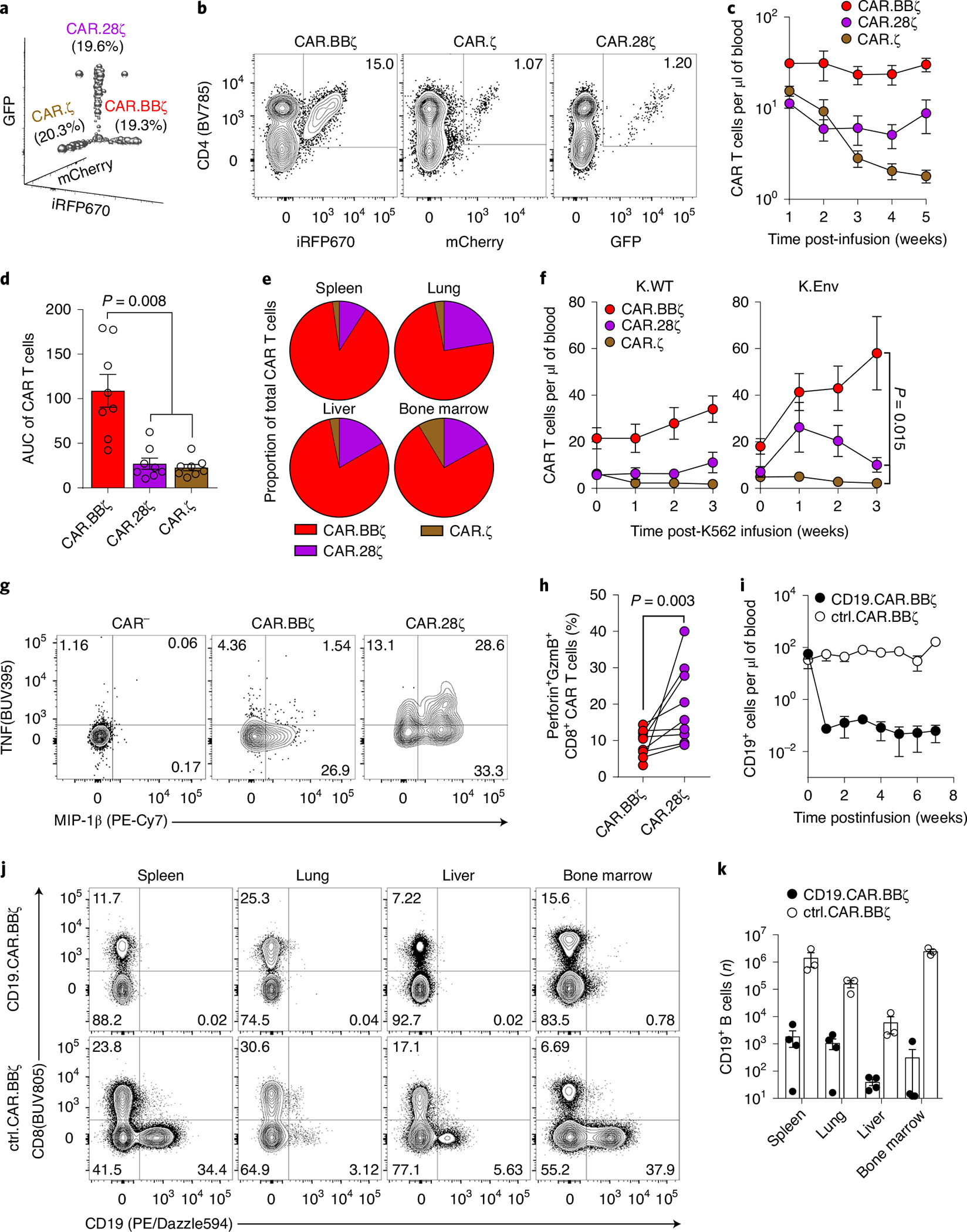
a–e, BLT mouse-derived T cells were transduced with mCherry.T2A.CAR.ζ, iRFP670.T2A.CAR.BBζ or GFP.T2A.CAR.28ζ. In total, 5 × 106 CAR-transduced T cells of each type were mixed before infusion into syngeneic mice (n = 8). a, Frequency of each CAR T cell type within the preinfusion TCP. b, Frequency of peripheral CAR T cells within the same mouse at 5 weeks postinfusion. c,d, Peripheral concentration (c) and cumulative persistence (d) of each CAR T cell type over 5 weeks of engraftment. e, Relative tissue frequency of each CAR T cell type at 7 weeks postinfusion. f, In a separate study, 2 weeks after infusion of the CAR T cell mixture described in a, BLT mice received 107 irradiated K.WT (n = 8) or HIVYU2 Envelope+ (K.Env; n = 8) K562 cells. Peripheral concentration of each CAR T cell type following K.WT or K.Env infusion. g, FACS plots indicating frequency of MIP-1β+ and TNF+ CAR.BBζ and CAR.28ζ T cells within the same mouse after ex vivo stimulation. CAR.ζ T cells were too infrequent for analysis. h, Frequency of granzyme B+ perforin+ CD8+ CAR T cells within the same mice ex vivo. i–k, Mice were infused with 5 × 106 CD19-specific CAR.BBζ (n = 4) or control CD4-based CAR.BBζ T cells (n = 3). i, Concentration of peripheral CD19+ cells following infusion. j,k, FACS plots showing frequency of CD19+ cells (j), and number of CD19+ cells (k) in tissues at 7 weeks postinfusion. For all data, symbols and bars reflect mean and error bars show ±s.e.m., except d and k where symbols represent individual mice. d, Friedman’s test with Dunn’s multiple corrections test. f,h, Two-sided Wilcoxon matched-pairs signed rank test was performed to calculate significance. Sample sizes for all mouse groups indicate biologically independent animals. AUC, area under the curve; ctrl, control.
CAR.BBζ T cells fail to control HIV rebound upon ART discontinuation.
After determining that the 4–1BB/CD3-ζ endodomain confers superior in vivo antigen-driven CAR T cell expansion and persistence, we sought to test the therapeutic potential of CAR. BBζ T cells in the context of ART-suppressed HIV infection. To do so, we infected BLT mice with CCR5-tropic HIVJRCSF and after 3 weeks initiated ART. At 2 weeks later, ART-suppressed mice were allocated into groups that received an infusion of either CAR.BBζ T cells (Group 1 (G1) and Group 3 (G3)), or inactive control CAR. BBΔζ T cells (Group 2 (G2) and Group 4 (G4)), which express a truncated CD3-ζ chain. In G1 and G2, ART was ceased immediately after infusion, whereas in G3 and G4 ART was continued for an additional 3 weeks to test whether the timing of ART interruption impacted the efficacy of CAR T cell therapy. In all groups, HIV rebounded by 2 weeks after treatment interruption, regardless of timing, and there were no observable differences in the kinetics or magnitude of viremia in CAR.BBζ-treated mice compared with matched control mice (Fig. 3a). Moreover, CAR.BBζ T cell therapy did not prevent memory CD4+ T cell loss in peripheral blood or tissues (Fig. 3b,c and Supplementary Fig. 2), which in BLT mice represents the CD4+ T cell subset preferentially infected and depleted by HIV due to high levels of CCR5 expression (Supplementary Fig. 3). Together, these data indicate that CAR.BBζ T cell therapy did not impact HIV progression.
Fig. 3 |. HIV-specific CAR.BBζ T cells display features of T cell exhaustion after failing to control viral rebound.
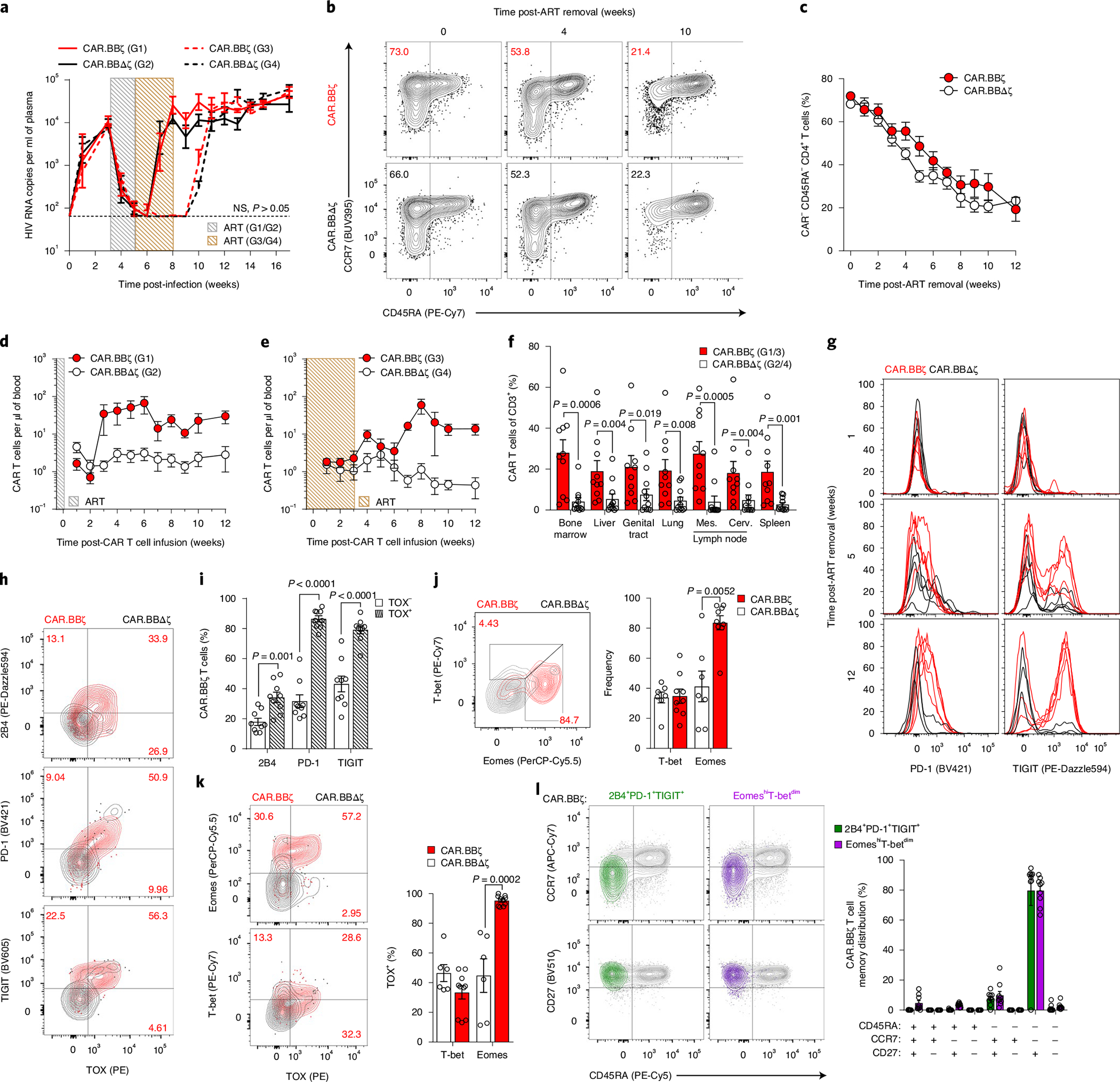
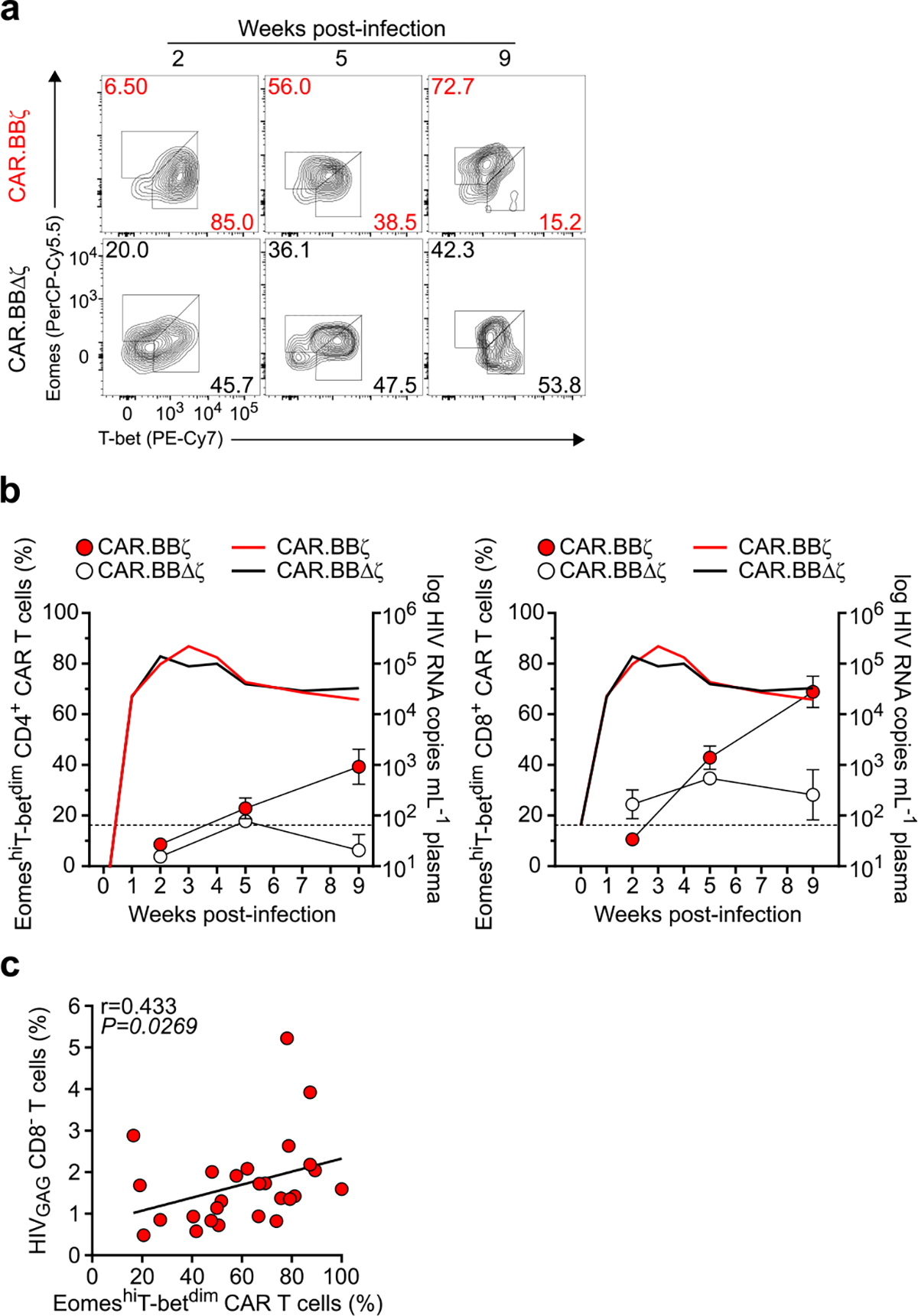
a, Mean log plasma viral RNA (copies per milliliter) in HIVJRCSF-infected mice treated with ART from week 3 to 5 (G1 and G2 mice; gray box) or from week 3 to 8 (G3 and G4 mice; brown box). At 5 weeks post-infection, mice in G1 (n = 6) and G3 (n = 10) received 107 CAR.BBζ T cells, and mice in G2 (n = 6) and G4 (n = 9) received 107 inactive control CAR.BBΔζ T cells. Thin, dotted line denotes limit of quantification. Sample sizes in these studies indicate biologically independent animals. b,c, FACS plots (b) and summary data (c) illustrate the frequency of total memory CD4+ T cells (CAR−) following ART cessation in CAR.BBζ and CAR.BBΔζ T cell–treated mice. d,e, Concentration of peripheral CAR T cells for G1/G2 (d) and G3/G4 (e). f, Frequency of CAR T cells in tissues at 12 weeks post-CAR T cell infusion for G1/G3 and G2/G4. g, PD-1 and TIGIT expression on peripheral CAR.BBζ or CAR.BBΔζ T cells from G1/G2 after ART discontinuation. h–l, FACS analysis of splenic tissue of BLT mice (G1 and G2) 12 weeks after ART cessation. h, Coexpression of TOX and 2B4, PD-1 or TIGIT on peripheral CAR.BBζ or CAR.BBΔζ T cells. i, Frequency of TOX− and TOX+ CAR.BBζ T cells positive for indicated inhibitory receptors. j, Frequency of T-bet- and Eomes-expressing CAR.BBζ and CAR.BBΔζ T cells. k, Frequency of TOX expression within T-bet+ and Eomes+ CAR.BBζ and CAR.BBΔζ T cells. l, Memory distribution of 2B4+PD-1+TIGIT+ and EomeshiT-betdim CAR.BBζ T cells. For all data, symbols and bars reflect mean and error bars indicate ±s.e.m., except f and i–l where symbols represent individual mice. In a, f and i–k, significance was calculated using a two-sided Wilcoxon rank-sum test. Cerv., cervical; Mes., mesenteric; NS, not significant.
CAR.BBζ T cells display features of T cell exhaustion during uncontrolled HIV replication.
Despite the lack of efficacy following ART discontinuation, we observed profound CAR.BBζ T cell expansion during viral recrudescence with a median 75-fold increase in the blood (Fig. 3d,e). As expected, the control T cells did not expand in response to viral rebound (Fig. 3d,e), and the CAR. BBζ T cells were substantially more abundant throughout the body 12 weeks after infusion (Fig. 3f). These findings suggested that the inability of CAR.BBζ T cells to control viremia and HIV pathogenesis was not the result of poor proliferation, poor persistence or lack of migration to relevant anatomical compartments of virus replication34–36.
The proliferation of CAR.BBζ T cells was associated with upregulation of inhibitory receptors including PD-1, TIGIT and 2B4, which increased over time (Fig. 3g and Extended Data Fig. 2a–d). Importantly, these inhibitory receptors were not expressed to the same extent on endogenous CAR− T cells within the same mice, suggesting a CAR T cell–specific effect rather than generalized activation from inflammation or viral load (Extended Data Fig. 2e,f). Notably, elevated inhibitory receptor expression on CAR.BBζ T cells from chronically infected mice was associated with the expression of TOX (Fig. 3h,i), a transcription factor that regulates the T cell exhaustion program37–41. Further supporting the gradual emergence of a dysfunctional CAR T cell phenotype, T-bet expression in CAR. BBζ T cells waned as HIV infection progressed, culminating in a population of EomeshiT-betdim CAR T cells that were enriched in TOX expression and accumulated in tissues with higher viral burden (Fig. 3j,k and Extended Data Fig. 3). In addition, expression of multiple inhibitory receptors on CAR.BBζ T cells from chronic infection was linked to a transitional memory state that also displayed an EomeshiT-betdim phenotype (Fig. 3l), all of which is congruent with earlier studies identifying dysfunctional HIV-specific CD8+ T cells within this compartment in chronic human HIV infection42,43.
Finally, we compared the ex vivo functions of CAR.BBζ T cells isolated during chronic infection with the preinfusion CAR TCP. Although the CD8+ CAR.BBζ T cells from chronic infection retained the ability to upregulate MIP-1β and granzyme B, and degranulate based on CD107a expression, the degree of β-chemokine production and cytotoxic potential was attenuated (Supplementary Fig. 4). Taken together, these data indicate that CAR.BBζ T cells recognize HIV-infected cells, rapidly expand and upregulate markers of cellular activation, but that uncontrolled virus replication ultimately drives an exhaustion program that may diminish T cell function and subvert efficacy.
Augmented HIV-specific CAR TCP reduces CD4+ T cell loss during acute infection.
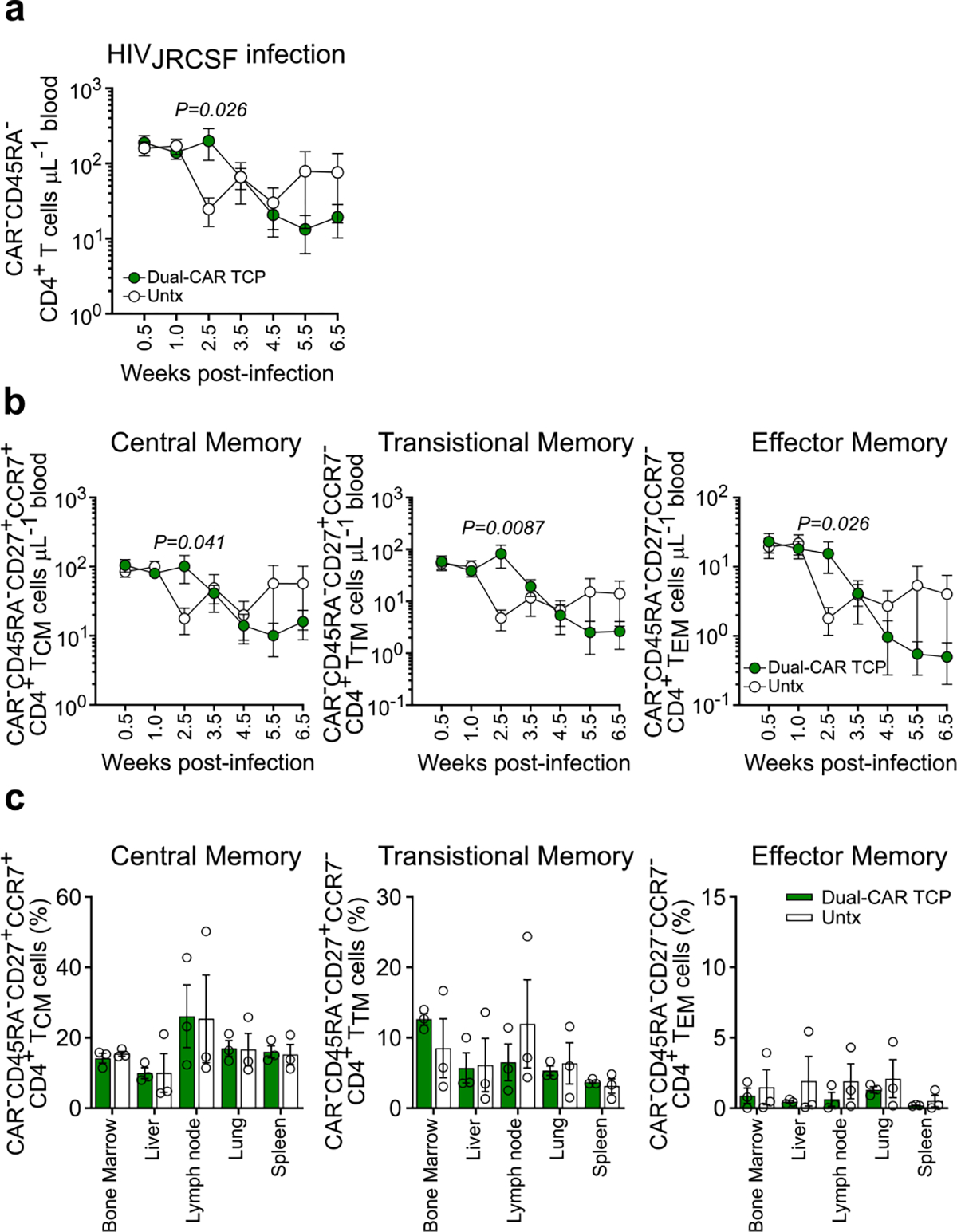
We hypothesized that combining the superior in vivo expansion and persistence of CAR.BBζ T cells with enhanced effector function could provide the necessary improvement to control HIV replication. To this end, we cotransduced CAR.BBζ T cells with the CD4-based, CD28-costimulated CAR that exhibited notable effector function (Extended Data Fig. 1) to create a novel Dual-CAR TCP. Due to cotransduction probabilities, the Dual-CAR TCP comprises three populations: CAR.BBζ, CAR.28ζ and Dual-CAR T cells, the last of which simultaneously expresses both CD4-based CARs (Fig. 4a). Inclusion of the CD28-costimulated CAR increased in vitro cytokine production of Dual-CAR T cells over CAR.BBζ T cells (Supplementary Fig. 5). To evaluate the Dual-CAR TCP in vivo, we used an acute infection model in which mice received CAR T cells 48 h after HIVJRCSF challenge to provide a more rapid model to test therapeutic efficacy. Although we observed no differences in acute viremia between the Dual-CAR TCP-treated and untreated groups (Fig. 4b), CAR T cell–treated mice exhibited a significant, albeit transient, delay in the loss of peripheral memory CD4+ T cells (CAR−), which coincided with peak expansion of total CAR T cells in peripheral blood (Fig. 4b,c and Extended Data Fig. 4a). Notably, this delay in CD4+ T cell loss was observed in central, transitional and effector memory subsets (Extended Data Fig. 4b), an effect that was not observed after ART discontinuation in the CAR.BBζ-treated mice in the previous study (Fig. 3b,c).
Fig. 4 |. Dual-CAR TCP mitigates CD4+ T cell loss and exhibits superior proliferative capacity.
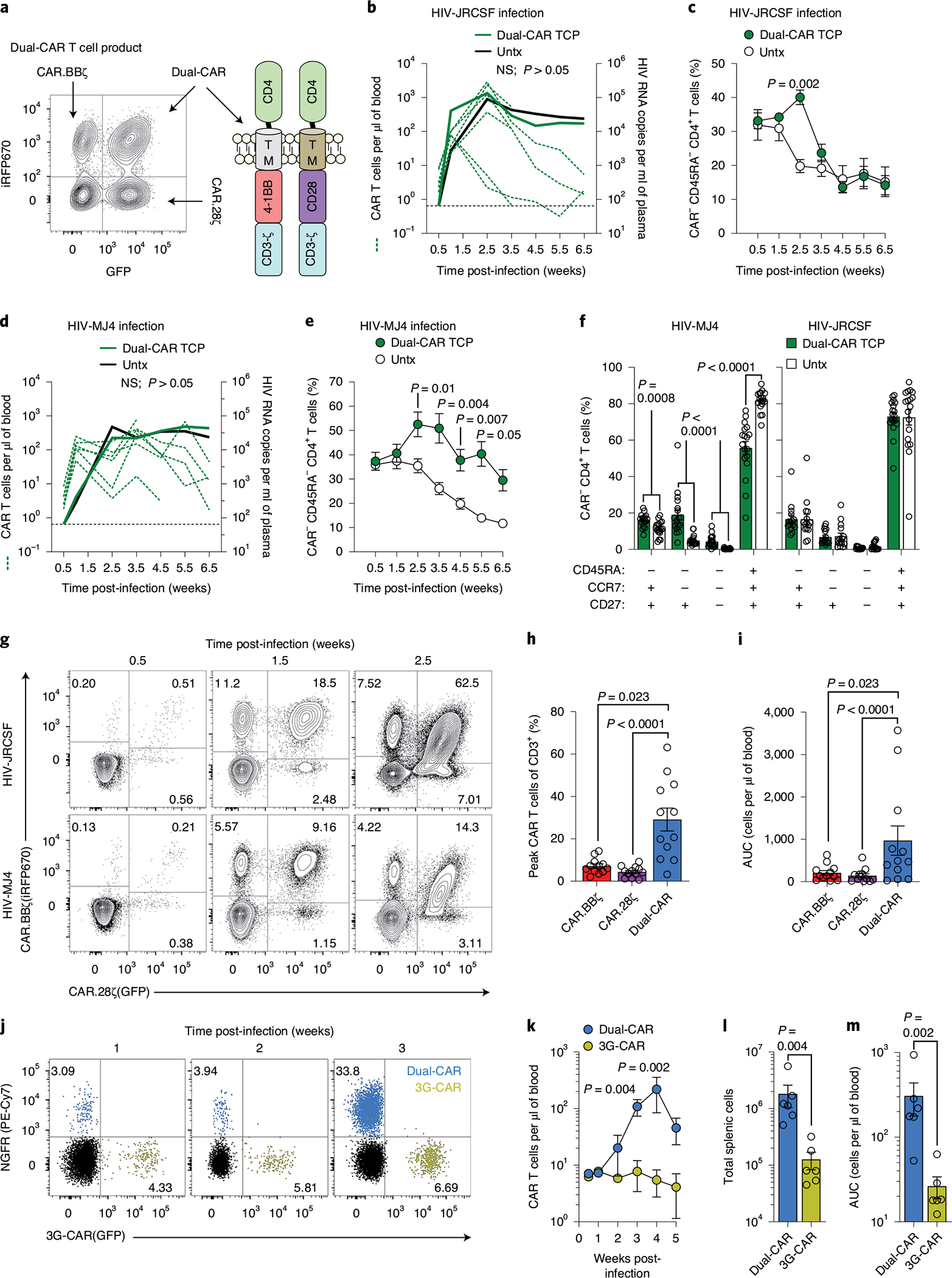
a–i, Mice were challenged with HIVJRCSF (n = 12) or HIVMJ4 (n = 12) and 48 h later six mice from each group were infused with Dual-CAR TCP (green lines) or were untreated (Untx, black lines). a, Dual-CAR TCP comprises CAR.BBζ, CAR.28ζ and Dual-CAR T cells. b, Concentration of total peripheral CAR T cells in individual mice (green dotted lines; left y axis) and mean log plasma viral RNA (copies per milliliter) (solid lines; right y axis) in HIVJRCSF-infected mice. Thin black dotted line denotes limit of quantification. c, Frequency of peripheral memory CD4+ T cells (CAR-) in HIVJRCSF-infected mice. d, Concentration of total peripheral CAR T cells in individual mice (green dotted lines; left y axis) and mean log plasma viral RNA (copies per milliliter) (solid lines; right y axis) in HIVMJ4-infected mice. Thin black dotted line denotes limit of quantification. e, Frequency of peripheral memory CD4+ T cells (CAR-) in HIVMJ4-infected mice. f, Frequency of CD4+ T cell (CAR−) memory subsets in tissue from HIVMJ4- (left) HIVJRCSF-infected mice (right) at 8 weeks post-CAR T cell infusion. Six distinct tissues were analyzed from three biologically independent animals per infection cohort. g, Peripheral longitudinal frequency of each CAR T cell type present in the Dual-CAR TCP. h,i, Peak peripheral frequency (h) and cumulative persistence (i) of CAR T cells. Data are aggregated from both infection cohorts (n = 12). j–m, Equal frequencies of Dual-CAR TCP and 3G CD4-based CAR T cells were combined (Extended Data Fig. 6c) before infusion into HIVMJ4-infected mice (n = 6). j, Overlaid FACS plots showing frequency of peripheral Dual-CAR (iRFP670+NGFR+) and 3G-CAR (GFP+) T cells within the same mouse. k, Concentration of peripheral CAR T cells. l,m, Total number of splenic CAR T cells (l) and cumulative CAR T cell persistence (m) at 5 weeks post-infection. For all data, bars and error bars show mean ± s.e.m., and symbols represent individual mice, except c and e where symbols represent mean. Two-sided Wilcoxon rank-sum test was used to calculate significance, except h and i where Friedman’s test with Dunn’s multiple corrections test was used. Sample sizes in these studies indicate biologically independent animals.
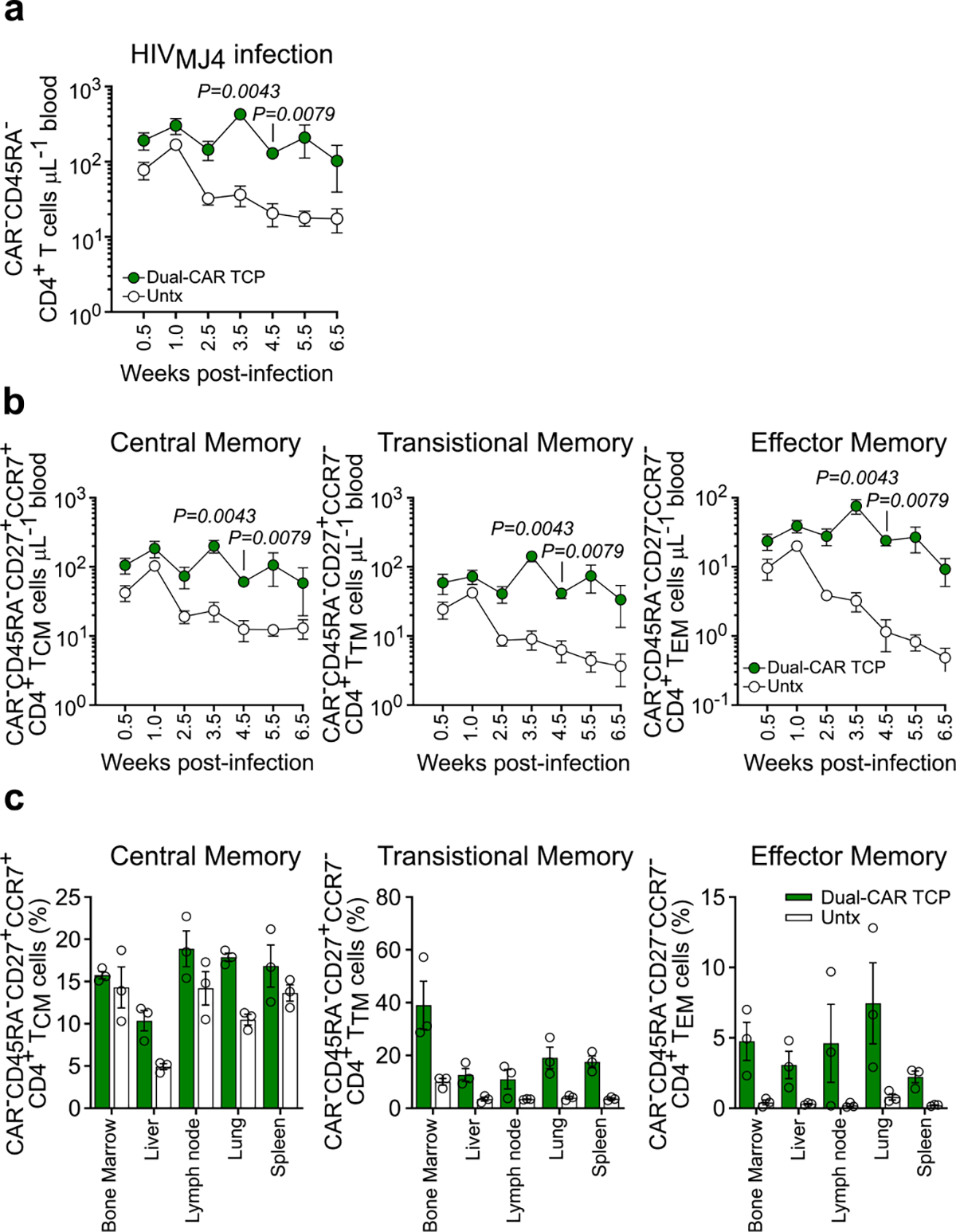
We next assessed the efficacy of the Dual-CAR TCP in the context of a more physiologically relevant strain of HIV. To do so, we infected additional mice from the same cohort as above with HIVMJ4, which exhibits slower acute-phase replication kinetics than HIVJRCSF, but ultimately achieves equivalent set-point viremia (Supplementary Fig. 6). Although the infusion of CAR T cells 48 h post-infection, again, did not alter viremia (Fig. 4d), we now observed more profound CD4+ T cell preservation and maintenance of the Dual-CAR TCP in peripheral blood as compared with the CAR T cell–treated mice infected with HIVJRCSF (Fig. 4e,f and Extended Data Fig. 5a). The preservation of CD4+ T cells was particularly accentuated in transitional and effector memory populations, which express greater levels of CCR5 (Extended Data Fig. 5b). Similarly, the percentages of all memory CD4+ T cell subsets in the tissues at necropsy were substantially preserved in Dual-CAR TCP-treated compared with untreated HIVMJ4-infected mice (Fig. 4f and Extended Data Fig. 5c), whereas there was no difference between CAR T cell–treated and control HIVJRCSF-infected mice (Fig. 4f and Extended Data Fig. 4c). Thus, treatment with the Dual-CAR TCP can effectively limit HIV-induced depletion of memory CD4+ T cells, an effect that is modulated by the pathogenicity of the infecting virus.
Dual-CAR T cells exhibit vigorous in vivo proliferation in a competitive environment.
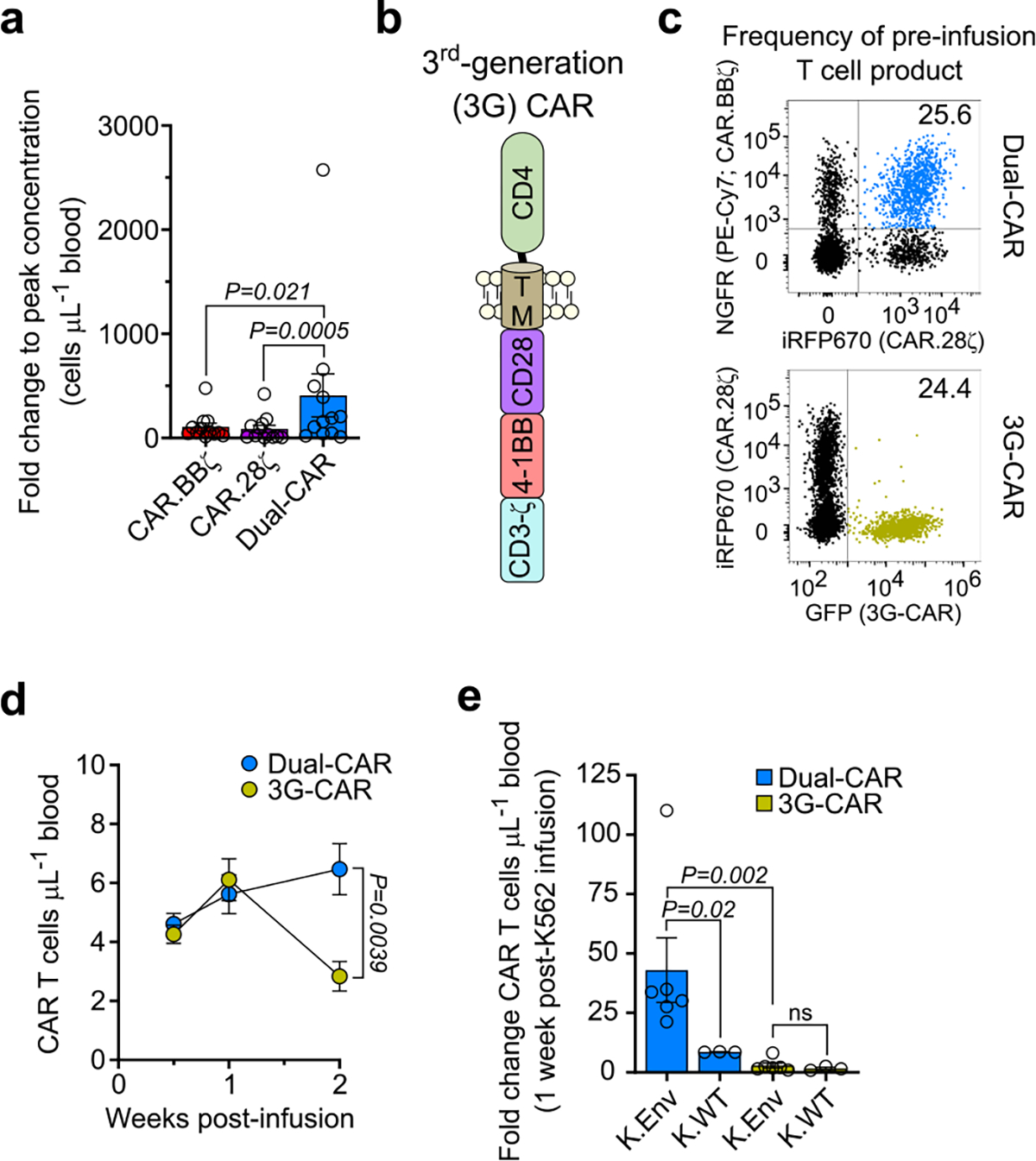
The linkage of each CAR to a unique fluorescent protein allowed for independent quantification of each CAR T cell type within the Dual-CAR TCP and revealed increased in vivo expansion of Dual-CAR T cells relative to either of the single costimulatory domain-expressing CAR T cells (Fig. 4g). Notably, significant differences were observed in the peak expansion and cumulative proliferation of Dual-CAR T cells (Fig. 4h,i), which remained significant after correcting for the baseline absolute count of each population (Extended Data Fig. 6a). In addition, we compared the proliferative capacity of Dual-CAR T cells with third-generation (3G)-CAR T cells, which express CD28 and 4–1BB linearly in the same construct (Extended Data Fig. 6b). Here we combined an equal amount of Dual-CAR and 3G-CAR T cells before adoptive transfer into recipient mice (Extended Data Fig. 6c). After infusion, Dual-CAR T cells showed significantly greater antigen-independent engraftment (Extended Data Fig. 6d), and also demonstrated superior antigen-driven proliferation after infusion of K.Env cells (Extended Data Fig. 6e). In contrast, 3G-CAR T cells marginally expanded and then progressively declined. Notably, during HIVMJ4 infection, Dual-CAR T cells exhibited profound proliferation (Fig. 4j,k) and long-term survival (Fig. 4l,m) relative to 3G-CAR T cells within the same mice. Together, these studies reveal the striking proliferative capacity exhibited by Dual-CAR T cells in a competitive setting under both antigen scarce and abundant in vivo environments.
Engineering HIV resistance augments CAR T cell persistence and function.
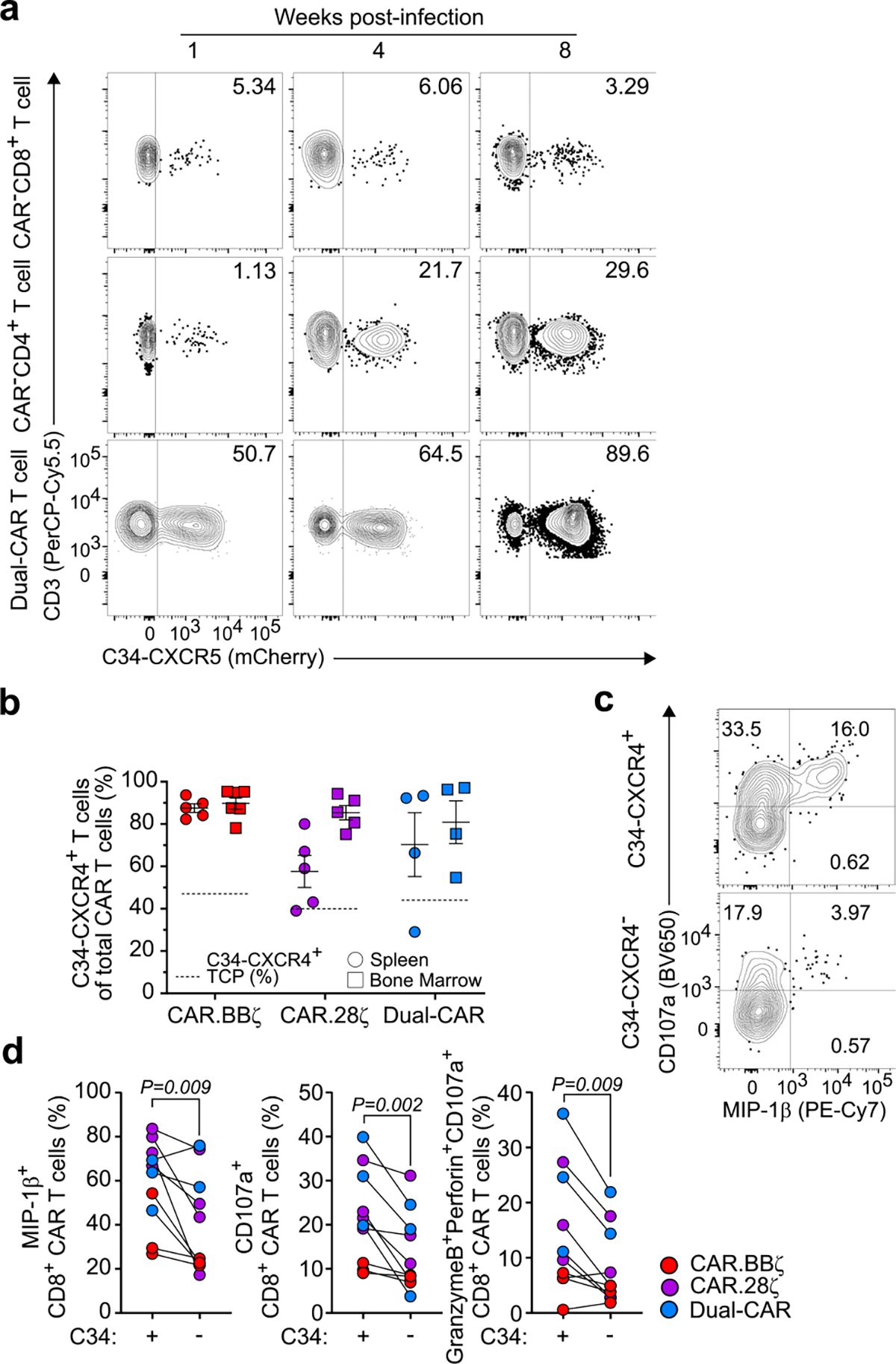
Despite the ability of CD4-based CARs to more efficiently suppress in vitro HIV replication versus several HIV-specific antibody-based CARs26, and the reduced likelihood for viral escape due to the requirement for HIV to bind CD4 for infection, this CAR results in the over-expression of the CD4 extracellular domain on the T cell surface, potentially increasing susceptibility to infection. Indeed, HIV-infected CAR T cells were detected in vivo, although the extent of total infection appeared to be indistinguishable from endogenous CAR− T cells (Supplementary Fig. 7a,b). More importantly, ex vivo stimulation revealed functional deficits in the capacity of HIV-infected CAR T cells to coupregulate granzyme B and perforin (Supplementary Fig. 7c,d). To confer HIV resistance, we cotransduced the Dual-CAR TCP with the surface-expressed HIV fusion inhibitor C34-CXCR4 (ref. 44) (Fig. 5a and Supplementary Fig. 8a). C34-CXCR4 was expressed on up to 50% of cells in the Dual-CAR TCP and provided protective benefit as the C34-CXCR4+ CAR T cells harbored significantly less HIV DNA than their unprotected counterparts (Fig. 5b), and were selected for over time in chronically infected mice (Extended Data Fig. 7a,b). Importantly, C34-CXCR4+ CAR T cells from chronic infection had markedly improved cytotoxic potential and MIP-1β expression relative to unprotected CAR T cells within the same mice (Extended Data Fig. 7c,d). Somewhat paradoxically, however, infusion of a Dual-CAR TCP where 50% of all cells were HIV-resistant was still insufficient to reduce acute virus replication (Supplementary Fig. 8b). These results demonstrate that CD4-based CAR T cells can be protected from HIV infection by the C34-CXCR4 fusion inhibitor and that such protection can preserve CAR T cell functionality during persistent exposure to HIV.
Fig. 5 |. HIV-resistant Dual-CAR T cells mediate superior virus-specific immune responses.
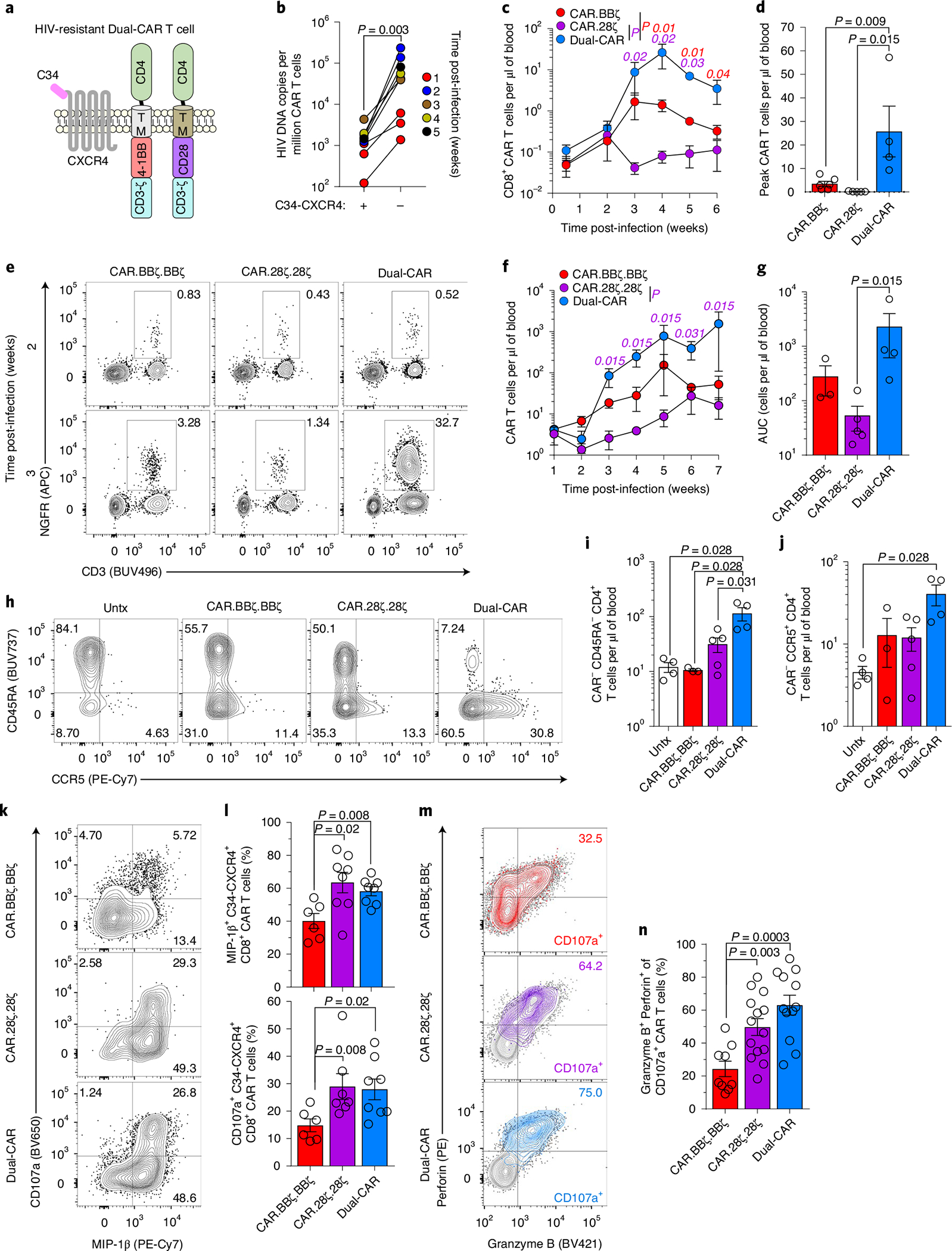
a, Schematic of HIV-resistant (C34-CXCR4+) Dual-CAR T cells. b, HIVJRCSF-infected BLT mice received 107 CAR T cells at 48 h postchallenge. HIV DNA load in sorted CAR T cells from individual mouse splenic tissue (n = 8). c,d, HIVMJ4-infected mice were infused at 48 h postchallenge with 106 C34-CXCR4+, CAR.BBζ (n = 6), CAR.28ζ (n = 5) or purified Dual-CAR (n = 4) T cells. Longitudinal peripheral concentration (c) and peak peripheral CAR T cell concentration (d). e–n, HIVMJ4-infected mice were infused at 48 h postchallenge with 106 C34-CXCR4+, purified CAR.BBζ.BBζ (n = 5), CAR.28ζ.28ζ (n = 5) or Dual-CAR (n = 5) T cells, or were untreated (n = 4). Purification strategy is described in Supplementary Fig. 10. e, Frequency of CAR T cell populations out of total human CD45+ cells 2 and 3 weeks post-infection. f,g, Longitudinal concentration (f) and cumulative peripheral CAR T cell persistence (g). h, FACS plots showing CCR5 expression within peripheral memory CD4+ T cells (CAR−). i,j, Concentration of total memory (i) and CCR5+ (j) CD4+ T cells (CAR−) at 6 weeks post-infection. k,l, FACS plots (k) and frequency (l) of MIP-1β+ and CD107a+ CD8+ CAR T cells from tissue at 8 weeks post-infection after ex vivo stimulation. m,n, Distribution (m) and frequency (n) of granzyme B+ perforin+ cells within CD107a+ CAR T cells from tissues after ex vivo stimulation. b, Two-sided Wilcoxon matched-pairs signed rank test was used to calculate significance. For remaining analyses, significance was calculated using a two-sided Wilcoxon rank-sum test. Bars and error bars indicate mean ± s.e.m., and symbols represent individual mice, except for c where symbols indicate mean. Sample sizes in these studies indicate biologically independent animals.
HIV-resistant Dual-CAR T cells are responsible for mitigating HIV-induced CD4+ T cell loss.
To determine whether an infusion product of Dual-CAR T cells alone exhibits enhanced virus-specific responses during HIV infection, we infused a low dose of C34-CXCR4+, purified Dual-CAR T cells, CAR.BBζ or CAR.28ζ T cells into separate groups of HIVMJ4-infected mice. Dual-CAR T cells exhibited notable in vivo expansion kinetics that exceeded both single CAR-transduced T cell populations (Fig. 5c,d), and mitigated HIV-induced CCR5+ CD4+ T cell loss (Supplementary Fig. 9). However, to more stringently control for CAR surface expression we performed an additional study in another cohort of mice where HIV-resistant, purified Dual-CAR T cells were compared with HIV-resistant, purified CAR T cells transduced with two independent CAR.BBζ or CAR.28ζ constructs (Supplementary Fig. 10). Dual-CAR T cells again demonstrated remarkable sensitivity to acute virus replication, expanding 300-fold to represent 30% of total human cells in blood 3 weeks post-infection, whereas CAR.BBζ. BBζ and CAR.28ζ.28ζ T cells reached only 3% and 1%, respectively (Fig. 5e and Supplementary Fig. 11a). In addition, Dual-CAR T cells sustained greater long-term proliferation and maintenance in blood and tissues than CAR.28ζ.28ζ or CAR.BBζ.BBζ T cells (Fig. 5f,g and Supplementary Fig. 11b). Importantly, the infusion of purified Dual-CAR T cells resulted in the greatest protection against CD4+ T cell loss during HIVMJ4 infection (Fig. 5h–j), reflected in the preservation of total memory and CCR5+ CD4+ T cells especially late in the infection (Supplementary Fig. 11c,d). Furthermore, the magnitude of early CAR T cell expansion across all groups, but exemplified by Dual-CAR T cells, was positively correlated with CD4+ T cell preservation (Supplementary Fig. 11e). Together, these data indicate that after controlling for CAR surface expression, Dual-CAR T cells exhibit the greatest in vivo antiviral effect.
Ex vivo effector function of Dual-CAR T cells exceeds 4–1BB-costimulated CAR T cells.
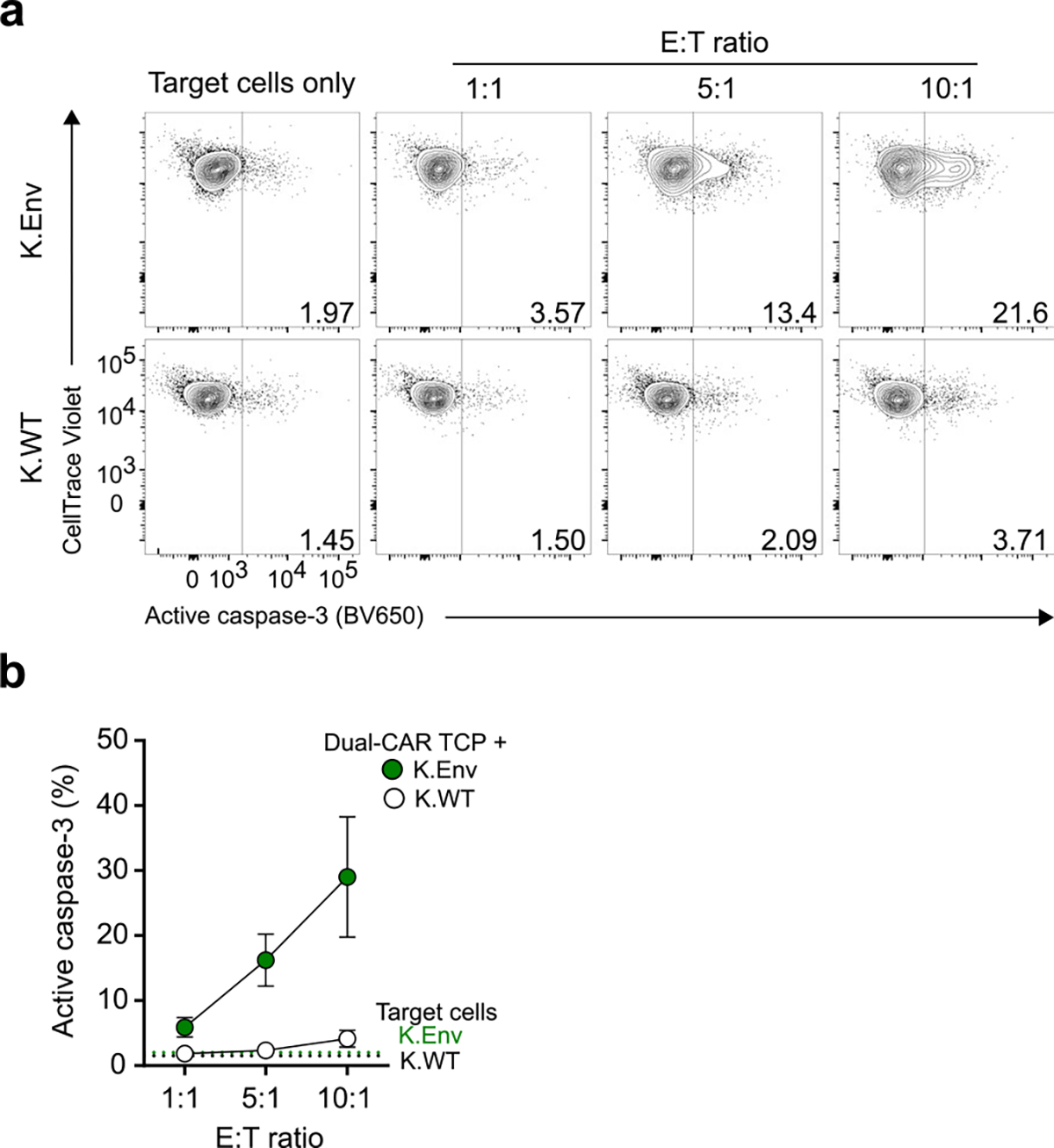
We next interrogated the ex vivo effector functions of CAR T cells from chronically infected mice. Dual-CAR T cells were superior to CAR.BBζ.BBζ T cells and equivalent to CAR.28ζ.28ζ T cells in their ability to produce MIP-1β and degranulate based on CD107a expression (Fig. 5k,l). Notably, a majority of CD107a+ Dual-CAR T cells coexpressed granzyme B and perforin compared with CAR.BBζ.BBζ T cells, indicating that these cells possess cytotoxic potential (Fig. 5m,n). In further support of cytolytic function, CAR T cells comprising the Dual-CAR TCP induced active caspase-3 expression in K.Env cells after ex vivo stimulation (Extended Data Fig. 8). Moreover, comparison of IL-2, TNF, MIP-1β and CD107a expression revealed distinct effector profiles between these CAR T cell populations (Extended Data Fig. 9a,b). Dual-CAR and CD28-costimulated CAR T cells clustered in a similar fashion, with CD4+ CAR T cells expressing more TNF and IL-2, and CD8+ CAR T cells upregulating more CD107a and MIP-1β. In contrast, CD4+ and CD8+ 4–1BB-costimulated CAR T cells clustered together and exhibited attenuated levels of effector molecules (Extended Data Fig. 9c). Together, these findings support our hypothesis that Dual-CAR T cells harness both the antigen-driven proliferation and effector functions mediated by 4–1BB and CD28 costimulation, respectively, for improved potency.
Protecting CAR T cells from HIV infection improves control over virus replication.
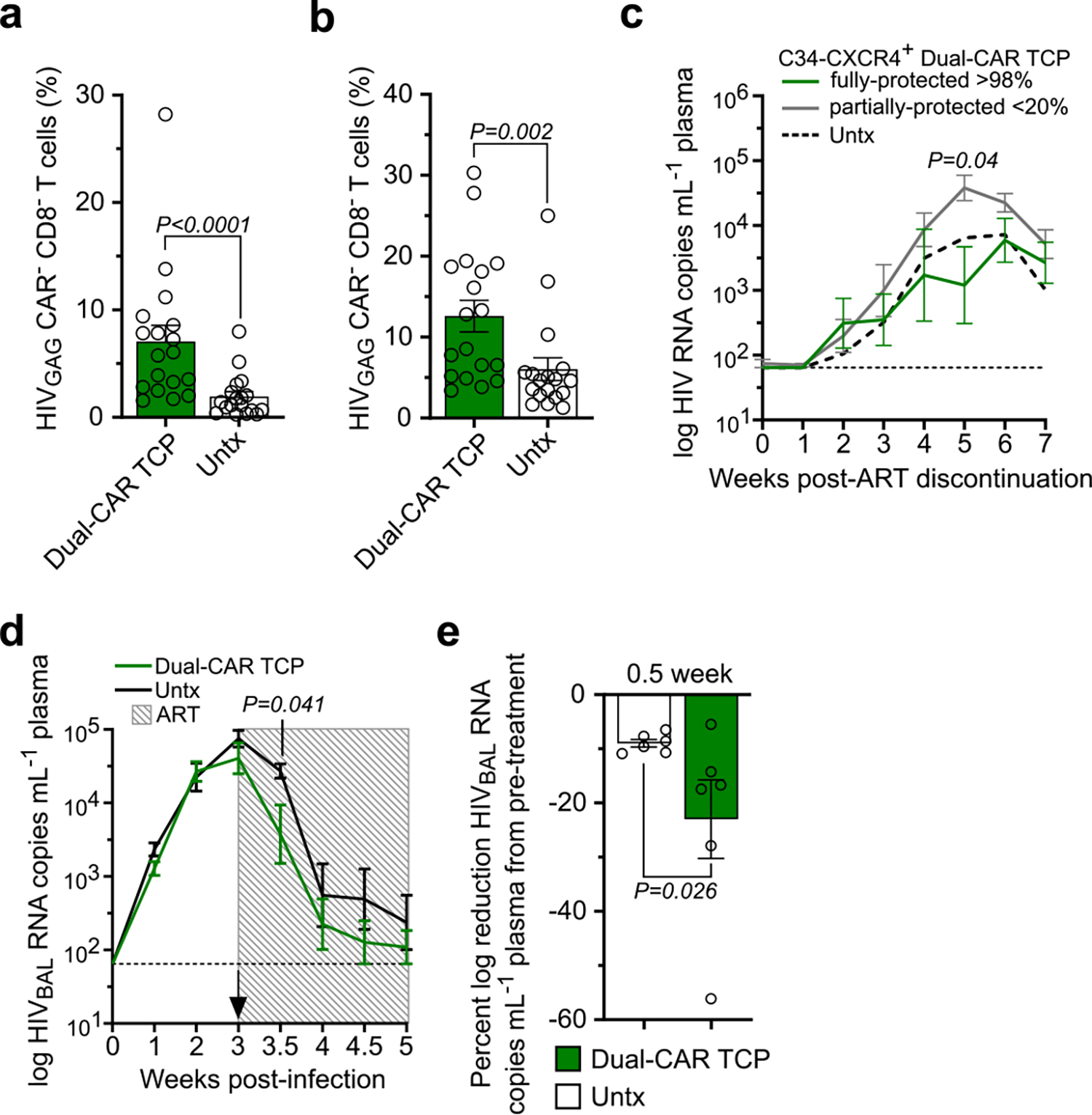
We hypothesized that the contribution of HIV-infected CAR T cells to viremia may be significant, in that virus secreted from infected CAR T cells could mask reductions in viral load caused by clearing infected CD4+ T cells. Indeed, after aggregating the data from all infection studies, we observed that infusion of HIV-susceptible CAR T cells significantly magnifies plasma viremia (Fig. 6a), as well as viral burden in tissues (Extended Data Fig. 10a,b). Thus, to test whether HIV infection of CD4-based CAR T cells negates CAR T cell–mediated reductions in viremia, we compared the outcomes of infusing a fully protected (>98% C34-CXCR4+) or a partially protected (<20% C34-CXCR4+) Dual-CAR TCP into HIVMJ4-infected, ART-suppressed mice followed by ART cessation. Strikingly, infusion of the partially protected Dual-CAR TCP increased rebound viremia over untreated mice to an average peak rebound of 4.6 log HIV RNA copies per ml versus 3.8 log copies per ml, whereas the fully protected Dual-CAR TCP significantly reduced viral load to 3.0 log copies per ml (Extended Data Fig. 10c). We confirmed this result by infusing the fully protected, Dual-CAR TCP into a larger cohort of BLT mice and observed significant reductions in acute viremia compared with untreated mice (Fig. 6b). Notably, treatment with the C34-CXCR4+ Dual-CAR TCP reduced the frequency of HIV-infected cells in tissues (Fig. 6c,d), contrasting the effect of unprotected CAR T cells on tissue viral burden in viremic mice (Extended Data Fig. 10a,b). Together, these data demonstrate the importance of safeguarding CAR T cells as HIV infection of unprotected CAR T cells can contribute to plasma viremia and potentially overwhelm CAR T cell–mediated control over virus replication.
Fig. 6 |. Mitigating CAR T cell infection improves control over HIV replication.
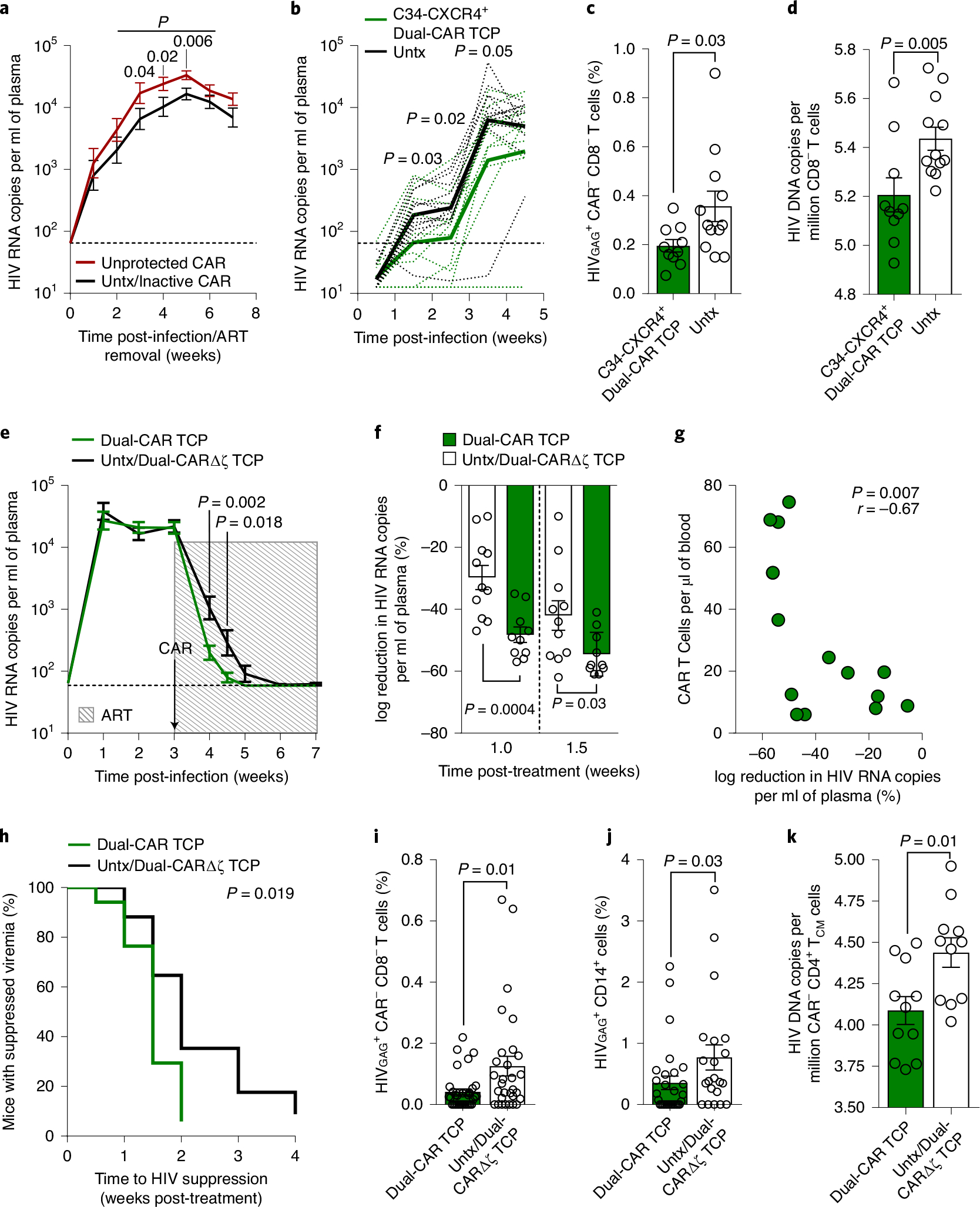
a, Mean log plasma viral RNA (copies per milliliter) of active, unprotected CAR T cell–treated mice (n = 38), and untreated/inactive CAR T cell–treated mice (n = 36). Data are aggregated across six independent studies. Thin dotted line denotes limit of quantification. b, Mean log plasma viral RNA (copies per milliliter) in mice infused at 48 h post-HIVMJ4 challenge with 107 protected (>98% C34-CXCR4+) Dual-CAR TCP (n = 12) or untreated mice (n = 12). c,d, Frequency of splenic HIV-infected CD8− T cells (CAR−) (c) and cell-associated HIV DNA load in lymph nodes (d) at 6–8 weeks post-infection. e–k, HIVJRCSF-infected mice were ART-treated and simultaneously infused with 107 HIV-resistant Dual-CAR TCP (n = 12) or inactive Dual-ΔCAR TCP (n = 5), or were untreated (n = 7). e, Mean log plasma HIV RNA (copies per milliliter). Shaded box indicates ART and arrow indicates CAR TCP infusion. f, Percentage log reduction in plasma HIV RNA from pre-ART (week 3) to 1 and 1.5 weeks post-ART. g,h, Data are aggregated from HIVJRCSF- and HIVBAL-infected cohorts. g, Correlation between percentage viral load reduction at first post-ART time-point and contemporaneous peripheral CAR T cell concentration. h, Kaplan–Meier curve of time to viral suppression after treatment initiation for Dual-CAR TCP versus control mice. i,j, Frequency of HIV-infected CD8− T cells (CAR−) (i) and HIV-infected CD14+ macrophages (j) aggregated from various tissues of plasma viremia suppressed mice. k, Cell-associated HIV DNA load in sorted central memory (CAR−CD45RA−CCR7+) CD4+ T cells. Statistical significance was calculated by one-sided unpaired Student’s t-test (a and b), two-sided Wilcoxon rank-sum test (c–f and i–k), Spearman correlation (g) and log-rank test (h). Bars and error bars indicate mean ± s.e.m. Symbols represent individual mice. Sample sizes in these studies indicate biologically independent animals. r, coefficient of correlation.
Although C34-CXCR4 reduces HIV infection of CAR T cells, we have shown that the protection is not sterilizing in the presence of persistent viremia (Fig. 5b). To test whether providing ART to prevent new rounds of infection at the time of CAR T cell infusion could further reveal CAR T cell–mediated viral load reduction, we challenged mice with HIVJRCSF and initiated combination therapy (ART and Dual-CAR TCP) at peak viremia. After 1 week of combination therapy, the Dual-CAR TCP-treated mice achieved approximately a 1-log greater reduction in viral load relative to the ART-only control group, which corresponded to a 50% reduction in viremia from pretreatment levels (Fig. 6e,f). We confirmed the suppressive effect of the Dual-CAR TCP in a separate cohort of mice infected with a different HIV strain (HIVBAL) (Extended Data Fig. 10d,e). Aggregation of the data from the two studies showed that the magnitude of early viral load reduction was associated with the contemporaneous concentration of CAR T cells in peripheral blood (Fig. 6g), and that CAR T cell treatment significantly accelerated HIV suppression, with nearly all combination therapy-treated mice reaching full suppression by 2 weeks after treatment initiation versus 4 weeks for ART-treated control mice (Fig. 6h). Furthermore, the Dual-CAR TCP reduced tissue viral burden in mice with suppressed plasma viremia, evidenced by fewer HIV-infected CD8− T cells (CAR−) and CD14+ macrophages in the tissues (Fig. 6i,j). Notably, central memory CD4+ T cells (CAR−) sorted from mice treated with the Dual-CAR TCP exhibited a significant, albeit modest, reduction in cell-associated HIV DNA load compared with the control group (Fig. 6k), suggesting that CAR T cell therapy is capable of reducing the size of the virus reservoir that forms during ART. Together, these findings highlight the potential for the HIV-resistant Dual-CAR TCP to mediate direct antiviral activity to clear infected cells in vivo.
Discussion
Here we use the BLT humanized mouse model to iteratively develop a CD4-based CAR T cell treatment of HIV infection. Although, initially, 4–1BB-costimulated CAR T cells demonstrated marked in vivo antigen-driven proliferation and survival, they failed to control viremia after ART cessation. To increase effector function, we created a Dual-CAR T cell that simultaneously expresses two CD4-based CARs independently encoding 4–1BB/CD3-ζ and CD28/CD3-ζ endodomains. These Dual-CAR T cells exhibited both the cytotoxic potential and cytokine expression of CD28-costimulated CAR T cells and the proliferative capacity of 4–1BB-costimulated CAR T cells, suggesting concurrent contribution by both costimulatory signals. In contrast, 3G CD4-based CAR T cells exhibited the more limited expansion of CAR.28ζ T cells despite containing a 4–1BB costimulatory domain within the same construct, likely the result of a dominant effect of the membrane-proximal CD28 domain as described for 3G cancer-specific CAR T cells45–48. This Dual-CAR approach introduces the potential to produce a CAR T cell with the benefits of both 4–1BB and CD28 costimulation.
The immunodeficiency caused by HIV infection is characterized by infection of CD4 T cells and concomitant immune activation and dysfunction49. Importantly, the rapid antigen-driven proliferation of CAR T cells makes them susceptible to these same pathogenic mechanisms. In particular, the extracellular domain of CD4-based CARs renders both CD4+ and CD8+ CAR T cells infectable, resulting in a substantial contribution to plasma viremia, as well as functional deficits. To combat this, we coexpressed a C34-CXCR4 (ref. 44) fusion inhibitor, which significantly improved CAR T cell survival and effector function during early HIV infection. Notably, the HIV-resistant Dual-CAR T cell treatment also resulted in a reduction in HIV burden in a variety of tissues and cell types, including long-lived memory CD4+ T cells, which suggests for the first time that a CAR T cell therapy targeted the HIV reservoir. However, the benefit of C34-CXCR4 waned over time, which highlights the need to confer complete and permanent HIV resistance, perhaps through the deletion of CCR5 (refs. 50–53).
The HIV-resistant Dual-CAR T cell treatment resulted in substantial protection of memory and CCR5+ CD4+ T cells from HIV-induced depletion despite failing to durably control viremia. Interestingly, the degree of protection was associated with the viral replication capacity (vRC) of the infecting strain. This is consistent with earlier findings that vRC affects many aspects of HIV-associated pathogenesis, including the kinetics of CD4+ T cell loss in acute infection54. Since the vRC of transmitted/founder viruses can vary by orders of magnitude across infected individuals55–57, vRC may become an important clinical consideration for the efficacy of CAR T cell treatment of HIV.
The HIV/BLT mouse model recapitulates key characteristics of HIV infection that are necessary to explore the efficacy of HIV-specific CAR T cell therapy. This small animal model may actually provide an overtly stringent test of CAR T cell efficacy given the general inability of BLT mice to develop affinity-matured antibodies58–60. We have used this model to develop a Dual-CAR T cell that successfully exhibits the functionalities mediated by both CD28 and 4–1BB costimulation, and was capable of reducing viremia, limiting HIV-induced CD4+ T cell loss and diminishing viral tissue burden in the context of a fully disseminated infection. Collectively, these findings provide critical insight into the iterative development of an engineered T cell–based therapy for HIV and describe a product that not only mitigates HIV pathogenesis but also has broad utility for viral infections and malignancies.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-020-1039-5.
Methods
Ethics.
Anonymized human fetal tissue (gestational age of 17–19 weeks) was acquired with informed consent from healthy donors by Advanced Bioscience Resources, and used under Partners Healthcare Institutional Review Board-approved protocols. Humanized mouse experiments performed at the Ragon Institute and at the University of Pennsylvania were approved by the Massachusetts General Hospital and the University of Pennsylvania Institutional Animal Care and Use Committee under the approved protocols 2016N000483 and 805606, respectively. Animal studies were performed following the instructions detailed in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Purified CD3+ and CD4+ T cells from anonymous healthy human donors were obtained by the University of Pennsylvania Human Immunology Core/Centers for AIDS Research (CFAR) Immunology Core.
Humanized mice.
Male and female NOD/SCID/IL2Rγ−/− (NSG) mice (Jackson Laboratory) were maintained in pathogen-free facilities at the Ragon Institute of MGH, MIT and Harvard or the University of Pennsylvania. Microisolator cages were used to house mice, and mice were fed autoclaved food and water. Animal rooms were maintained at 72 ± 2 °F (22.2 ± 1.1 °C) and 30–70% relative humidity and were on a 12:12-h light/dark cycle. BLT humanized mice were generated at the Ragon Institute as previously described32,61,62. Briefly, 6–8-week-old NSG mice were anesthetized and whole-body irradiated (2 Gy), followed by implantation with 1-mm3 fragments of human fetal liver and thymus tissue under the murine kidney capsule. Then, 105 autologous fetal liver-derived CD34+ hematopoietic stem cells (HSCs) were injected intravenously within 6 h of transplantation. BLT humanized mice were also generated at the University of Pennsylvania as previously described63. Briefly, 1–1.5 × 105 human fetal liver-derived CD34+ HSCs were administered intravenously into 7–10-week-old NSG mice 24 h after busulfan (30 mg kg−1) conditioning. At 3–6 d following stem cell transplant, mice were surgically implanted with 3–5 fragments of autologous human fetal thymus tissue measuring 3–5 mm3 under the murine kidney capsule. For all BLT humanized mice, human immune reconstitution was assessed from 12 to 17 weeks after transplantation. Mice were included in experiments when over 50% of cells in the lymphocyte gate were positive for human CD45 and, of those human cells, greater than 40% were CD3+ T cells.
Flow cytometry and cell sorting.
Washed cells were resuspended in 50 μl of PBS containing 2 mM EDTA and 2% FCS, and then surface stained with anti-human antibodies from BioLegend: CD45 (HI30 and 2D1), CD19 (HIB19), CD3 (OKT3), CD45RA (HI100), CD27 (LG.3A10), CD8 (RPA-T8), CCR7 (G043H7), CCR5 (J418F1), CD4 (OKT4), CD271 (ME20.4), PD-1 (EH12.2H7), TIGIT (VSTM3), 2B4 (C1.7), CD107a (H4A3); BD Biosciences: CD45 (HI30), CD3 (UCHT1), CD8 (SK1), CD45RA (HI100), CCR7 (3D12); and R&D: Human EGFR (Cetuximab Biosimilar, Hu1). Live cells were identified through staining negative for Fixable Viability Dye eFlour 780 (eBioscience) or LIVE/DEAD Fixable Blue (Invitrogen). For the detection of intracellular proteins, the cells were treated with Cell Fixation & Cell Permeabilization Kit (Invitrogen) or True-Nuclear Transcription Factor Buffer Set (BioLegend) in accordance with the manufacture’s protocols with antibodies from: BioLegend: IL-2 (MQH-17H12), Perforin (B-D48); BD Biosciences: TNF (Mab11), IFN-γ (4 S.B3), Granzyme B (GB11), MIP-1β (D21–1351), GM-CSF (BVD2–21C11), Active Caspase-3 (C92605); Beckman Coulter: HIV-1 Core Antigen (KC57); and eBioscience: T-bet (4B10), EOMES (WD1928), TOX (TXRX10). Flow cytometry data were acquired on BD LSR II, BD LSRFortessa and BD FACS Symphony instruments using BD FACSDiva Software v.8.0.1 (BD Biosciences). Data were analyzed using FlowJo software v.10 (TreeStar). Sorting of C34-CXCR+ and C34-CXCR4− CAR T cells for quantitation of viral burden by digital-droplet PCR was performed by sorting live C34-CXCR4+ and C34-CXCR4− CAR T cells from splenocytes after surface staining with the following Biolegend antibodies: CD45 (2D1), CD3 (OKT3), CD8 (RPA-T8), CD4 (OKT4). Living CAR T cells were identified by staining negative for Fixable Viability Dye eFlour 780 (Supplementary Fig. 12a). Sorting of endogenous central memory CD4+ T cell populations for quantitation of viral burden by droplet-digital PCR (ddPCR) was performed by staining splenocytes with the following antibodies from BioLegend: anti-mouse CD45 (30-F11), anti-human CD45 (HI30), CD20 (2H7), CD14 (HCD14), CD56 (HCD56), CD3 (OKT3), CD4 (RPA-T4), CD8 (SK1), CCR7 (G043H7), CD45RA (HI100). Live cells were discriminated using LIVE/DEAD Fixable Blue (Invitrogen). FACSAria II (BD Biosciences) was used for all cell sorting (Supplementary Fig. 12b). A complete list of antibodies used including additional technical information for flow cytometry experiments can be found in the Nature Research Reporting Summary.
HIV inoculum preparation.
Viral stocks of the molecular clones, HIVJRCSF and HIVMJ4, were generated through transfections of HEK293T cells (ATCC: CRL-3216) and titered as previously described64. HIVBAL virus stocks were generated by passage in anti-CD3/CD28 Dynabead stimulated human CD4+ T cells as previously described26.
Viral (HIV) load quantification.
Peripheral blood plasma-derived HIV RNA was isolated using the QiaAmp Viral RNA Mini Kit (Qiagen). Quantitative RT–PCR using the QuantiFast Syber Green RT–PCR kit (Qiagen) was used as previously described to measure viral loads64. Data were collected on a LightCycler 480 System (Roche) or a ViiA 7 Real-Time PCR system (Applied Biosystems) using Lightcycler 480 software release 1.5.1.62 SP3 or Viia 7 QuantStudio Real-Time PCR software v.1.3, respectively.
Plasmid construction.
Amino acid sequences specific to the CD4-based CAR constructs containing the intracellular signaling domains: CD3-ζ, 4–1BB/ CD3- ζ and CD28/CD3- ζ, are described elsewhere26. Each CAR was amplified from its parent plasmid with 5′-CACGTCCTAGGATGGCCTTACCAGTG and 5′-GTGGTCGACTTATGCGCTCCTGCTGAAC and inserted into the AvrII and SalI restriction enzyme sites of the pTRPE transfer plasmid downstream of GFP, mCherry or iRFP670, and an intervening T2A linker permits expression of both proteins. To construct the plasmids for CAR T cell selection, double-stranded DNA fragments (IDT) encoding NGFR (CD271)65 and truncated EGFR66 were custom synthesized, flanked with suitable restriction enzyme sites and cloned into the second position of the pTRPE plasmid preceded by the CAR-BBζ and CAR-28ζ gene and T2A linker. The C34-CXCR4 construct’s sequence is denoted elsewhere44. A previously described67 Asp mutation (D97N) in CXCR4 was introduced to impair SDF-1 binding and limit receptor internalization.
Lentivirus production and transfection.
Lentiviral particles were generated using packaging expression vectors that encode VSV or Cocal glycoprotein, HIV Rev and HIV Gag/Pol (pTRPE pVSV-g, pCocal-g, pTRPE.Rev and pTRPE g/p, respectively), and were synthesized using DNA 2.0 or ATUM. The packaging plasmids along with the appropriate pTRPE transfer vector were transfected into HEK293T cells using Lipofectamine 2000 (Life Technologies)26,68. At 24 and 48 h after transfection, the HEK293T cell supernatant was collected, filtered through a 0.45-μm syringe-driven filter and then concentrated by ultracentrifugation for 2.5 h at 25,000 r.p.m. at 4 °C. The supernatant was aspirated and the virus pellet was resuspended in 800 μl of complete RPMI and stored at −80 °C.
Cell culture.
For preparation of CAR T cells: de-identified human donor T cells were isolated by negative selection with RosetteSep Human CD3+ Enrichment Cocktails (Stem-Cell Technologies) following the manufacturer protocol. T cells from BLT humanized mice were purified by processing spleen, bone marrow and liver tissues into a single-cell suspension. Density gradient centrifugation using Lymphoprep (Stem-Cell Technologies) was used to isolate Mononuclear cells. Human CD2+ cells were purified by CD2 Microbeads (Miltenyi Biotec) per the manufacturer’s protocol. T cells were placed in culture at 106 cells per ml in either complete RPMI: RPMI 1640, 1% Penicillin-Streptomycin, 2 mM GlutaMax and 25 mM HEPES buffer from Life Technologies, and 10% FCS (Seradigm), or CTS OpTmizer T-Cell Expansion SFM (Gibco) with 1% Penicillin-Streptomycin, 2 mM GlutaMax and 25 mM HEPES buffer. T cell expansion medium was complemented with human IL-15 (5 ng ml−1, Biolegend) and IL-7 (10 ng ml−1, R&D). T cells were stimulated at a 3:1 (bead/cell) ratio with anti-CD3/CD28 Dynabeads (Life Technologies), and incubated at 37 °C, 5% CO2 and 95% humidity. At 18 h after stimulation the medium was reduced by and replaced with 300 μl of the appropriate lentivirus supernatant for CAR transduction. At 5 d after T cell activation, magnetic separation was used to remove Dynabeads. Throughout CAR T cell manufacturing, medium was changed every second day, which spanned 8–10 d, or as needed to adjust cell counts to 0.5 × 106 cells per ml.
Two-step immunomagnetic selection of CAR T cells during manufacturing: on day 4 after initial T cell activation, anti-CD3/CD28 Dynabeads were removed by magnetic bead separation. T cells were counted and then incubated at a 1:2 cell/bead ratio with CELLection Biotin Binder Dynabeads (Life Technologies) conjugated to anti-EGFR (Cetuximab) antibody. Truncated EGFR+ T cells were isolated according to the manufacturer’s protocol. The cell concentration was adjusted to 0.5 × 106 cells per ml with medium and expanded as described in the preceding paragraph. At 7 d after initial activation, EGFR+ T cells were counted and incubated with CD271 Microbeads (Miltenyi Biotec) to positively select for NGFR+ T cells according to the manufacturer’s instructions. The eluted fraction of T cells contained 85–95% EGFR+NGFR+ T cells. Before infusion into BLT mice, the T cells were cultured for an additional day at the adjusted cell concentration.
HIV treatment and ART discontinuation mouse model.
For the study in Fig. 3, BLT humanized mice were administered 2 mg of medroxyprogesterone (McKesson) subcutaneously 1 week before intravaginal challenge with 20,000 tissue culture infectious dose 50 (TCID50) HIVJRCSF in 20-μl total volume. Then, 75–100 μl of blood was obtained through puncture of the retro-orbital sinus weekly to quantify viral load and immunophenotype circulating blood cells. At 3 weeks post-HIV challenge, all infected mice were administered daily intraperitoneal injections of ART consisting of 10 mg kg−1 EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine, LeadGen Labs) and 50 mg kg−1 Dolutegravir (Sigma) for 1 week and then every second day thereafter. Following 2 weeks of ART, four treatment groups were defined based on normalization of plasma viral load, body weight and human reconstitution percentages. G1 (n = 6) and G3 (n = 10) are treatment groups that were infused with 107 CAR-BBζ T cells, while G2 (n = 6) and G4 (n = 9) are control groups that were infused with 107 CAR-BBΔζ T cells that express a defective CD3-ζ endodomain. T cells were administered in a 300-μl volume via tail vein injection. ART was interrupted immediately after adoptive T cell transfer for G1 and G2, while ART discontinuation was delayed for 3 weeks in G3 and G4. At necropsy, 17 weeks after HIV challenge, various tissues were collected to analyze the CAR T cells.
For the study described in Extended Data Fig. 10c, BLT humanized mice were infected via the intraperitoneal route with 20,000 TCID50 HIVMJ4. At 3 weeks post-infection, all mice received ART and an HIV-resistant (<20% C34-CXCR4+) Dual-CAR TCP (n = 7), an HIV-resistant Dual-CAR TCP with further magnetic bead selection to obtain a >98% C34-CXCR4+ transfer product (n = 5) or no CAR T cells (n = 9). After plasma viremia was fully suppressed in all three groups, ART was discontinued and virus rebound was monitored via weekly blood draws from the retro-orbital sinus.
Acute HIV infection treatment model.
BLT humanized mice were challenged with 20,000 TCID50 HIVJRCSF or HIVMJ4 via intraperitoneal injection. For the study comparing the replication capacity of HIVJRCSF and HIVMJ4 (Fig. 4a–i): HIVJRCSF-infected mice (n = 6) and HIVMJ4-infected mice (n = 6) were infused with the Dual-CAR TCP consisting of 2 × 107 total CAR T cells. T cells were administered via tail vein injection 48 h after HIV challenge. Control mice that were infected with HIVJRCSF (n = 5) or HIVMJ4 (n = 6) received no T cells. For the study comparing Dual-CAR T cells and 3G CD4-based CAR T cells: Dual-CAR TCP was combined with 3G-CAR T cells, normalizing the frequency of Dual-CAR and 3G-CAR T cells before infusion into mice. Nine HIV-uninfected mice were infused with this mixture, where each mouse received 2.5 × 106 Dual-CAR T cells and 2.5 × 106 3G-CAR T cells via tail vein injection. After 2 weeks, six mice were infused via intravenous injection with 107 irradiated K.Env cells and three mice received 107 irradiated wild-type K562 (K.WT) cells (Extended Data Fig. 6). Additional mice (n = 6) were challenged with 20,000 TCID50 HIVMJ4 and infused 48 h later with the same Dual-CAR TCP/3G-CAR T cell mixture described above (Fig. 4j–m). For the study in Fig. 5c, HIVMJ4-infected mice were allocated into four groups. The groups were infused with 106 C34-CXCR4+, CAR.BBζ (n = 6), CAR.28ζ (n = 5) or purified Dual-CAR (n = 4) T cells, while the remaining mice were untreated (n = 6). For the study comparing purified Dual-CAR and double CAR-transduced BBζ.BBζ and 28ζ.28ζ T cell populations (Fig. 5f): HIVMJ4-infected mice were distributed into four groups and normalized based upon body weight and the absolute count of CD4+ T cells in blood. The groups were infused with 106 C34-CXCR4+, purified CAR.BBζ.BBζ (n = 5), CAR.28ζ.28ζ (n = 5) or Dual-CAR (n = 5) T cells via tail vein injection 48 h after HIV challenge. Control mice did not receive T cells (n = 5). For the study evaluating efficacy of enriched C34-CXCR4+ (>98%) Dual-CAR T cells (Fig. 6b–d), HIVMJ4-infected mice were divided into two groups that received 107 C34-CXCR4-enriched Dual-CAR TCP (n = 12) or were untreated (n = 12). In all studies mice were bled by retro-orbital puncture 1 d after adoptive T cell transfer, and then weekly thereafter until their respective endpoint and tissue collection.
CAR T cell therapy and ART model.
In the study described in Fig. 6e, BLT humanized mice were challenged with 20,000 TCID50 HIVJRCSF via intraperitoneal injection. At 3 weeks post-HIV challenge, all infected mice were given low-dose ART consisting of 1 mg kg−1 EFdA and 25 mg kg−1 Dolutegravir every other day by intraperitoneal injection for 4 weeks. At the time of ART initiation, HIV-infected mice were allocated into three groups. Treated mice (n = 11) were infused with an HIV-resistant (C34-CXCR4+) Dual-CAR TCP consisting of 107 total CAR T cells. Control mice were infused with either 107 total CAR T cells from the HIV-resistant Dual-ΔCAR TCP which expresses defective CD3-ζ signaling domains (n = 5) or were untreated (n = 7). The mice were euthanized and tissues were collected for analysis 7 weeks post-infection. For the study described in Extended Data Fig. 10d, BLT humanized mice were challenged with 20,000 TCID50 HIVBAL via intraperitoneal injection. At 3 weeks post-HIV challenge, all infected mice were administered ART consisting of 10 mg kg−1 EFdA and 25 mg kg−1 Dolutegravir every other day by intraperitoneal injection for 2 weeks. At the time of ART initiation, HIV-infected mice were distributed into two groups. Treated mice (n = 6) were infused with an HIV-resistant (C34-CXCR4+) Dual-CAR TCP comprising 107 total CAR T cells, or were untreated (n = 6).
C34-CXCR4 protection of CAR T cells in vivo.
In Fig. 5b, BLT humanized mice were challenged with 20,000 TCID50 HIVJRCSF via intraperitoneal injection. At 48 h after challenge, mice received the HIV-resistant (C34-CXCR4+) Dual-CAR TCP consisting of 2 × 107 total CAR T cells. In 1-week intervals after challenge, mice were euthanized and tissues were collected at necropsy. Splenocytes were prepared and freshly sorted; isolated cells were used to quantify the amount of cell-associated HIV DNA harbored within C34-CXCR4+ and C34-CXCR4− CAR T cells.
HIV suppression assay.
At 7 d following initial activation with anti-CD3/CD28 Dynabeads, we infected primary CD4+ T cells with CCR5-tropic HIVJRCSF at a multiplicity of infection (MOI) of 1. After 1 d, we washed HIV-challenged CD4+ T cells with complete RPMI supplemented with 100 U ml−1 IL-2 and mixed with CAR.ζ or control untransduced (UTD) T cells at effector-to-target (E/T) ratios of 1:12.5, 1:25, 1:50, 1:100 and 1:200. The E/T ratios reflect the number of CAR.ζ T cells to HIV-challenged, CD4+ T cells. Mixtures of cells were plated in triplicate and HIV replication was measured by sampling 100 μl per well for flow cytometric detection and intracellular cytokine staining for HIV-1 Core Antigen at 2, 4 and 6 d postcoculture. We added fresh media to each well following staining.
HIV-infected cell elimination assay.
A similar target cell elimination assay was performed as described69. Here we prepared HIV-infected CD4+ T cells as described above. Once 30% of total cells stained positively for HIV-1 Core Antigen, cells were membrane labeled using CellTrace Violet (CTV) (ThermoFisher) to identify HIV-infected cells from CAR or UTD T cells. For characterizing the cytotoxic function of the preinfusion TCP, CAR-ζ and UTD T cells were cultured with CTV-labeled HIV-infected cells at 0.25:1, 1:1 and 4:1 E/T ratios. For ex vivo stimulation, single-cell suspensions of bone marrow from HIV-infected mice treated with the Dual-CAR TCP were cultured with CTV-labeled HIV-infected target cells at 1:1, 5:1 and 10:1 E/T ratios. HIV-infected cells were assessed for the induction of active caspase-3 24 h later by flow cytometry. Active caspase-3 was identified in living CTV+ HIVGAG+ T cells. The gating strategy is outlined in Supplementary Fig. 1f.
Cytotoxicity, CD107a degranulation and cytokine assays.
In vitro CAR T cell functionality was assessed following stimulation of 2 × 105 CAR-ζ or UTD T cells with 2 × 105 K.WT or K562 cells engineered to express the extracellular and transmembrane regions of HIVYU2GP160 (K.Env). Anti-CD107a antibody was included at the beginning of the stimulation followed by 1X Brefeldin A and Monensin Solution (BioLegend) an hour later. In total, cells were incubated for 6 h at 37 °C. Cytokine production was measured by flow cytometry and intracellular staining with anti-human antibodies specific for IL-2, IFN-γ, MIP-1β, TNF and GM-CSF, while cytotoxic potential was measured by staining with antibodies specific for granzyme B and perforin. Cytokine-positive CAR T cells were reported as the difference in the production of cytokines after stimulation with K.Env and K.WT cells.
Measurement of CAR T cell responses ex vivo.
Functionality of CAR T cells from HIV-infected BLT humanized mice was measured after ex vivo stimulation with K562 target cells. Density gradient centrifugation was used to isolate mononuclear cells after preparing a single-cell suspension from tissues. Between 0.5 and 1 × 106 mononuclear cells were cultured with 2 × 105 K562.WT or K562.Env cells. The assessment of cytotoxic potential, degranulation and cytokine production was performed using the same protocol described above.
Cell-associated HIV DNA quantitation.
Mononuclear cell suspensions obtained from spleens were stained and sorted as described above. After sorting, samples were frozen as cell pellets and stored at −80 °C. To obtain genomic DNA, cell pellets were thawed and total DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN) per the manufacturer’s protocol. Total HIV DNA was measured in each sample using a multiplexed ddPCR assay specific for HIV gag and the human RPP30 gene. Gag forward and reverse amplification primer sequences were 5′- AGTGGGGGGACATCAAGCAGCCATGCAAAT and 5′-TGCTATGTCAGTTCCCCTTGGTTCTCT, respectively. Gag sequence was detected using a 5′ HEX-labeled hydrolysis probe (HEX-CCATCAATGAGGAAGCTGCAGAATGGGA). RPP30 forward and reverse amplification primer sequences were 5′- GATTTGGACCTGCGAGCG and 5′-GCGGCTGTCTCCACAAGT, respectively. Human RPP30 sequence was detected with a 5′ 6-FAM-labeled hydrolysis probe (6-FAM-CTGACCTGAAGGCTCT). The RPP30 primer/probe set has been described previously70. ddPCR reactions were performed using the manufacturer-recommended consumables and the ddPCR supermix for probes (No UTP) (Bio-Rad). Thermal cycling conditions are as follows: 1 cycle of 95 °C for 10 min, 45 cycles of 94 °C for 30 s followed by 60 °C for 1 min, 1 cycle of 98 °C for 10 min. Droplets were generated using a QX100 droplet generator and subsequently analyzed on a QX200 droplet reader (Bio-Rad) using the QuantaSoft v.1.7.4.0917 software (Bio-Rad). All samples were run in duplicate.
vRC assay.
In vitro replication assays were performed essentially as previously described57. Human PBMCs were isolated from whole blood by density gradient centrifugation using Histopaque-1077 (Sigma). PBMCs were stimulated with 3 μg ml−1 phytohemagglutinin (PHA) in complete RPMI (1% Penicillin-Streptomycin, 2 mM L-Glutamine, 25 mM HEPES buffer and 10% FCS) supplemented with 20 U ml−1 recombinant human IL-2 at a concentration of 1 × 106 cells ml–1. After 72 h of stimulation, PBMCs were washed twice with complete RPMI, and resuspended in complete RPMI supplemented with 50 U ml−1 IL-2 at a concentration of 5 × 106 cells ml–1. Cells were infected by combining 1,000 TCID50 HIVJRCSF or HIVMJ4 with 5 × 105 cells and a final concentration of 5 μg ml−1 polybrene in a 200-μl total volume. Cells were infected by spinoculuation at 1,200 r.p.m. and 25 °C for 2 h. Cells were then washed five times to remove excess virus, and plated in 500 μl of complete RPMI including 50 U ml−1 IL-2 in a 48-well plate. Infections were incubated at 37 °C and 5% CO2, and 50 μl of media was removed every 2 d and frozen. Supernatant Gag p24 levels were measured using the Alliance HIV-1 p24 antigen ELISA kit per the manufacturer’s instructions (PerkinElmer). Plates were read on a Tecan Infinite 200 Pro plate reader (Tecan Life Sciences) using the Magellan software v.7.2. All infections were carried out in triplicate.
Statistical analysis.
Statistical analyses were performed using JMP Pro v.14.2.0 (SAS Institute) and Prism v.7 for PC v.7.05 and Prism v.8 for macOS v.8.2.1 (GraphPad). Spearman rank correlation was used to test for all bivariate continuous correlations. Wilcoxon rank-sum test was conducted to test for all one-way comparisons of means from unpaired samples, and Wilcoxon matched-pairs signed rank test was used for comparing means of paired samples. Kaplan–Meier survival curves were performed using an endpoint defined as the limit of detection of the viral load quantification assay (1.81 log RNA copies per ml), and statistics are generated from the log-rank test. K-means clustering was performed using the JMP Pro v.12 statistical package to generate principal component plots with circles denoting where 90% of the observations would fall. We performed area-under-the-curve calculations in GraphPad Prism v.7 using cell concentration data normalized to 1 μl of blood. Additional information regarding how sample sizes were determined and randomized for mouse experiments can be found in the Nature Research Reporting Summary.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All of the construct sequences described in this article are published (see ref. 26). All materials described in this manuscript are available via a material transfer agreement with the University of Pennsylvania or the Ragon Institute. All data are available from the corresponding authors upon request. Source data are provided with this paper.
Extended Data
Extended Data Fig. 1 |. CD28 costimulation enhances the ex vivo effector function of CAR T cells.
HIV-uninfected mice were infused with an equal mixture of CD4-based CAR T cells expressing either CD3-ζ, 4–1BB/CD3-ζ and CD28/CD3-ζ costimulatory domains linked to unique fluorescent proteins to facilitate identification in vivo as described in Fig. 2 legend. Cumulative data indicating the frequency of TNF+, IL-2+ and MIP-1β+ CAR.BBζ and CAR.28ζ T cells within the same mice after ex vivo stimulation with K.Env (+) or K.WT (−) cells. Data represents the aggregate of cytokine producing cells from liver and terminal blood (n = 8). CAR.ζ T cells were too infrequent for analysis. Data shows box and whisker plots where the middle line indicates median, bounds of the box show 25th to 75th percentiles, and bars extend to min and max values. Symbols represent biologically independent animals. Significance was calculated using two-sided Wilcoxon matched-pairs signed rank test.
Extended Data Fig. 2 |. CAR.BBζ T cells accumulate multiple inhibitory receptors as disease progresses.
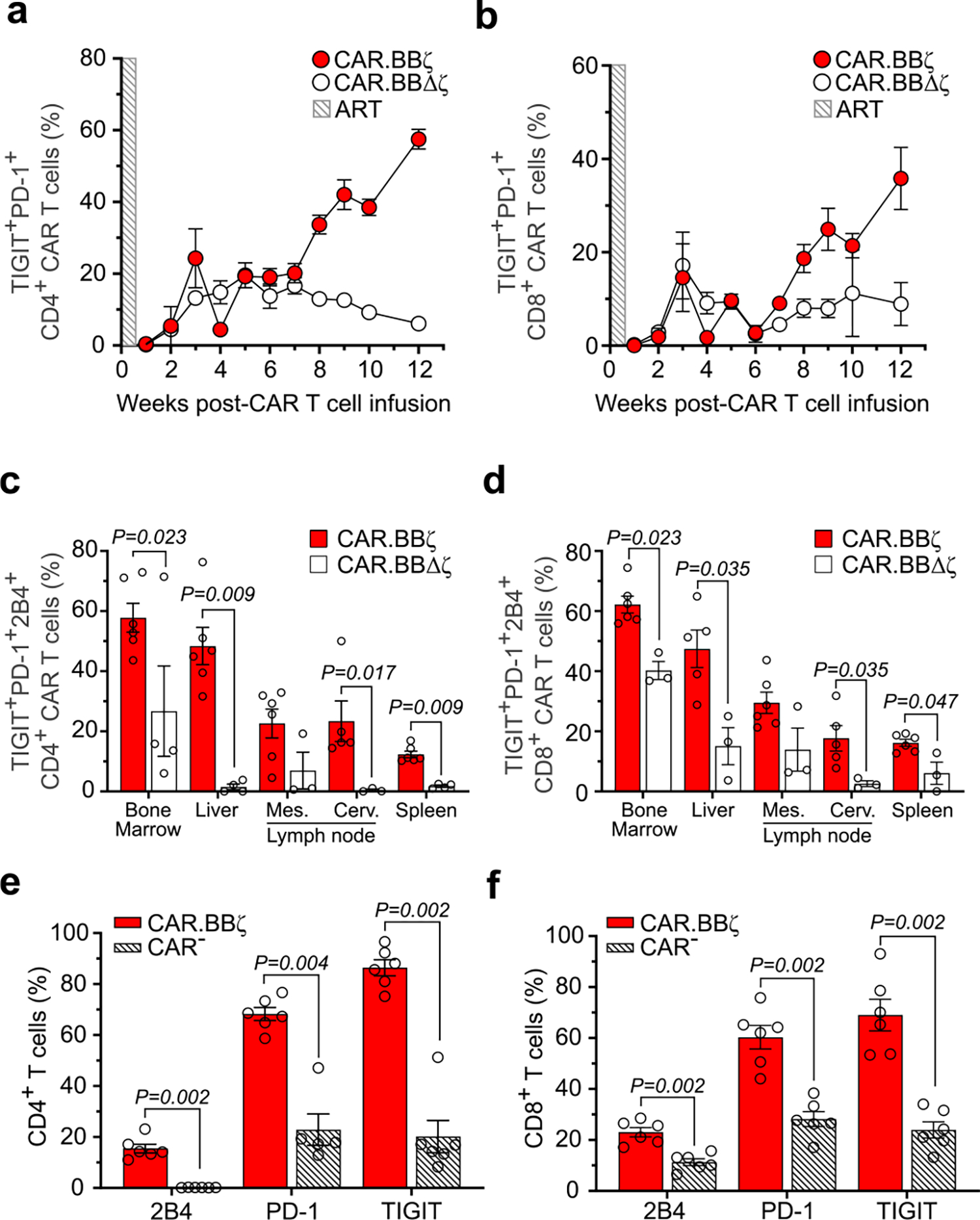
a, Frequency of CD4+ and (b) CD8+ CAR.BBζ T cells (G1; n = 6) and CAR.BBΔζ T cells (G2; n = 6) co-expressing TIGIT and PD-1 after infusion. Shaded box indicates the window of ART. Symbols and error bars indicate mean ± SEM. c, Frequency of CD4+ and (d) CD8+ CAR.BBζ T cells (G1) and CAR.BBΔζ T cells (G2) co-expressing TIGIT, PD-1 and 2B4 in tissues 12 weeks post-infusion. e, Cumulative data indicating the frequency of 2B4+, PD-1+ and TIGIT+ CD4+ CAR.BBζ T cells (G1) compared to CAR− CD4+ T cells (G1) within the spleens of the same mice, and (f) CD8+ CAR.BBζ T cells (G1) compared to CAR− CD8+ T cells (G1) within the spleens of the same mice. c–f, Bars indicate mean, error bars show ± SEM and symbols represent individual mice. Significance was calculated using two-sided Wilcoxon rank-sum test. Sample sizes in these studies represent biologically independent animals.
Extended Data Fig. 3 |. eomeshiT-betdim CAR.BBζ T cells accumulate from acute to chronic phases of infection.
BLT mice were infected with HIVJRCSF and infused 48 h later with either 2×107 CAR.BBζ T cells (n = 5) or inactive control CAR.BBΔζ T cells (n = 3). a, FACS plots show the change in Eomes and T-bet expression within the different CAR T cell types over time. b, Summary data indicating the longitudinal frequency of EomeshiT-betdim CD8+ (left panel) and CD4+ (right panel) CAR T cells (left y-axis), and mean log plasma HIV RNA (copies mL−1) (right y-axis). Thin dotted line denotes limit of viral load quantification. Symbols and error bars indicate mean ± SEM. c, Spearman correlation analysis of frequency of EomeshiT-betdim CD8+ CAR.BBζ T cells compared with viral burden measured as the frequency of HIVGAG+ CD8− T cells in various tissues 10 weeks post-infection. Sample sizes in these studies indicate biologically independent animals.
Extended Data Fig. 4 |. Dual-CAR T cell product transiently delays CD4+ T cell loss despite persistent HIVJRCSF infection.
BLT mice received Dual-CAR T cell product (TCP) (n = 6) 48 h after HIVJRCSF challenge, while control mice were untreated (Untx) (n = 5). a, Concentration of peripheral total memory CD4+ T cells (CAR−). b, Concentration of peripheral central memory (CD45RA−CD27+CCR7+; left panel), transitional memory (CD45RA−CD27+CCR7−; middle panel), and effector memory (CD45RA−CD27−CCR7−; right panel) CD4+ T cells (CAR−). c, Frequency of memory CD4+ T cell (CAR−) subsets in tissues 8 weeks post-infection. a, b, Significance was calculated using a two-sided Wilcoxon rank-sum test. Symbols and bars indicate mean, and error bars show ± SEM. Sample sizes indicate biologically independent animals.
Extended Data Fig. 5 |. Dual-CAR T cell product prevents CD4+ T cell loss despite persistent HIVMJ4 infection.
BLT mice were infused with Dual-CAR T cell product (TCP) (n = 6) 48 h post-HIVMJ4 challenge, while control mice were untreated (Untx) (n = 6). a, Concentration of peripheral total memory CD4+ T cells (CAR−). b, Concentration of peripheral central memory (CD45RA−CD27+CCR7+; right panel), transitional memory (CD45RA−CD27+CCR7−; middle panel), and effector memory (CD45RA−CD27−CCR7−; left panel) CD4+ T cells (CAR−). c, Frequency of memory CD4+ T cell (CAR−) subsets in tissues 8 weeks post-infection. a, b, Significance was calculated using a two-sided Wilcoxon rank-sum test. Symbols and bars indicate mean, while error bars show ± SEM. Sample sizes indicate biologically independent animals.
Extended Data Fig. 6 |. Dual-CAR T cells exhibit superior in vivo expansion compared to 4–1BB-costimulated, CD28-costimulated, and 3rd-generation CAR T cells.
a, BLT mice were challenged with either HIVJRCSF (n = 6) or HIVMJ4 (n = 6) and infused with 2×107 Dual-CAR T cell product (TCP). Fold-change in CAR T cell concentration from baseline to peak levels in peripheral blood. Data is the aggregate of both infection cohorts. b, Schematic shows the structural components of the 3rd-generation (3 G) CD4-based CAR construct. c–e, Dual-CAR T cell product and 3G-CAR T cells were combined at an equal frequency prior to infusion into uninfected mice (n = 9). c, FACS plots indicate the frequency of Dual-CAR and 3G-CAR T cells present within the pre-infusion T cell product. d, Longitudinal concentration of peripheral CAR T cells following adoptive transfer into HIV-negative mice. Symbols and error bars indicate mean ± SEM. e, At 2 weeks post-infusion, mice received either 107 irradiated K.Env cells (n = 6) or 107 irradiated K.WT cells (n = 3). Fold change in the concentration of peripheral CAR T cells 1-week post-K562 infusion from baseline concentration prior to K562 infusion. a, e, Bar and error bars indicate mean ± SEM, and symbols represent individual mice. a, d, e, Two-sided Wilcoxon rank-sum test was used to calculate significance. Sample sizes in these studies indicate biologically independent animals.
Extended Data Fig. 7 |. C34-CXCR4+ CAR T cells are selected for during chronic infection and exhibit superior ex vivo effector functions.
a, BLT mice were infected with HIVJRCSF and 48 h later infused with 107 C34-CXCR4+ Dual-CAR T cell product (TCP). FACS plots indicate the frequency of C34-CXCR4+ throughout infection. b, Mice were infected with HIVMJ4 and 48 h later were infused with 106 C34-CXCR4+ CAR.BBζ (n = 5), CAR.28ζ (n = 5), or purified Dual-CAR (n = 4) T cells. Frequency of C34-CXCR4+ CAR T cells in tissue 8 weeks post-infection. Thin dotted line indicates the frequency of C34-CXCR4+ CAR T cells in the pre-infusion TCP for the indicated CAR T cell type. Line and error bars indicate mean ± SEM. c, d, Mice were infected with HIVMJ4 and 48 h later received 106 C34-CXCR4+, purified CAR.BBζ.BBζ (n = 3), CAR.28ζ.28ζ (n = 4), or Dual-CAR (n = 3) T cells. c, FACS plots and (d) cumulative data show the frequency of each CD8+ CAR T cell population expressing MIP-1β and CD107a, and the frequency of CAR T cells with cytotoxic potential (granzyme B+ perforin+ CD107a+). CAR T cells were isolated from the spleen and bone marrow of mice 8 weeks post-infection and ex vivo stimulated. Significance was calculated using two-sided Wilcoxon matched-pairs signed rank test. For all data, symbols represent individual mice. Sample sizes in these studies indicate biologically independent animals.
Extended Data Fig. 8 |. CAR T cells from HIV-infected mice exhibit ex vivo cytotoxic function.
HIVJRCSF-infected mice (n = 3) treated with the Dual-CAR TCP were euthanized and the bone marrow cells were ex vivo stimulated with K.Env or K.WT cells for 24 h at the indicated E:T ratios. a, Representative FACS plots and (b) cumulative data shows the induction of active caspase-3 within the different target cell populations. Symbols and error bars indicate mean ± SEM. Sample size indicates biologically independent animals.
Extended Data Fig. 9 |. Dual-CAR and CAR.28ζ T cells exhibit similar ex vivo functional profiles.
BLT mice were challenged with HIVJRCSF (n = 5) and infused with 2×107 Dual-CAR T cell product (TCP) 48-hours post infection. a, Frequency of CD8+ and (b) CD4+ CAR T cell populations from tissue at necropsy (8-weeks post-infection) within the same mice expressing CD107a, MIP-1β, IL-2 and TNF after ex vivo stimulation. Bars and error bars indicate mean ± SEM, and symbols represent individual mice. Significance was calculated using two-sided Wilcoxon rank-sum test. c, Principle Components Analysis (PCA) of IL-2, TNF, MIP-1β, and CD107a expression in ex vivo stimulated CD8+ and CD4+ CAR T cells from PBMCs of HIVJRCSF-infected mice (n = 5). Sample sizes in these studies represent biologically independent animals.
Extended Data Fig. 10 |. HIV-resistant Dual-CAR TCP reduces virus replication in vivo.
a, Frequency of HIVGAG+ CD8− T cells (CAR−) within the bone marrow and spleen of HIVJRCSF-infected mice (n = 6) and (b) HIVMJ4-infected mice (n = 6) that were treated 48 h post-challenge with the Dual-CAR T cell product (TCP) or were untreated (Untx). c, Mean log plasma HIVMJ4 RNA (copies mL−1) after ART discontinuation of mice infused at ART initiation with 107 protected >98% C34-CXCR4+ (n = 5) or partially-protected <20% C34-CXCR4+ (n = 7) Dual-CAR TCP, or were untreated (n = 9). d, e, HIVBAL-infected mice were ART-treated and simultaneously infused with 107 HIV-resistant Dual-CAR TCP (n = 6) or were untreated (n = 6). d, Mean log plasma HIV RNA (copies mL−1). Shaded box indicates ART and arrow indicates CAR TCP infusion. e, Percent log reduction in plasma HIV RNA from pre-ART (week 3) and 0.5 and 1 week post-ART. For all data, bars and error bars indicate mean ± SEM, and symbols represent individual mice. Significance was calculated for (a–d) by two-sided Wilcoxon rank-sum test and (e) two-sided Kolmogorov-Smirnov test. Sample sizes in these studies indicate biologically independent animals.
Supplementary Material
Acknowledgements
We thank C. Ellebrecht and A. Payne (University of Pennsylvania) for the HIVYU2 Envelope-transduced K562 cell line. We thank W. Garcia-Beltran for artistic contributions. This study was supported by NIH grants P01HL129903 (T.M.A.), U19AI117950 (J.L.R.) and UM1AI126620 (J.L.R.) funded by NIAID, NIDA, NIMH and NINDS; T32 grant AI007632 (C.R.M.); and an NIAID F32 grant AI136750 (D.T.C.). We also acknowledge support from the Ragon Institute of MGH, MIT and Harvard and the MGH Scholars program (T.M.A.). We thank the Human Immune System Mouse Core at the Ragon Institute of MGH, MIT and Harvard and the Stem Cell and Xenograft Core at the University of Pennsylvania for the generation of BLT humanized mice, the Penn Center for AIDS Research (P30-AI045008) for purified human T cells, and the Allen and Riley laboratories for helpful comments.
Footnotes
Competing interests
C.R.M. and J.L.R. have filed an institution-owned patent (20180265565: Method of Redirecting T Cells to Treat HIV Infection) describing the construction of these HIV-specific CARs specific to Figs. 1 and 2. This patent application has been published but has not yet been granted. C.R.M. and J.L.R. have also filed an institution-owned patent (Dual CAR Expressing T Cells Individually Linked to CD28 and 4–1BB) specific to Figs. 4–6. G.J.L., J.A.H. and J.L.R. have also filed an institution-owned patent (Non-Signaling HIV Fusion Inhibitors And Methods Of Use Thereof) specific to Figs. 5 and 6. J.L.R. cofounded a company called Tmunity Therapeutics that has the rights to license the technology described in this paper. J.L.R. holds an equity interest in Tmunity. C.R.M. and J.L.R. declare no other competing financial interests. No other authors declare any competing financial interests. No authors declare any nonfinancial interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41591-020-1039-5.
Supplementary information is available for this paper at https://doi.org/10.1038/s41591-020-1039-5.
Reprints and permissions information is available at www.nature.com/reprints.
Peer review information Alison Farrell was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
References
- 1.June CH & Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med. 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker B & McMichael A The T-cell response to HIV. Cold Spring Harb. Perspect. Med. 2, 1–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross G, Waks T & Eshhar Z Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl Acad. Sci. USA 86, 10024–10028 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Stegen SJ, Hamieh M & Sadelain M The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Disco. 14, 499–509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porter DL et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJ et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 13, 5426–5435 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Song DG et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4–1BB). Cancer Res. 71, 4617–4627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carpenito C et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl Acad. Sci. USA 106, 3360–3365 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milone MC et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 17, 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Z et al. In vivo expansion and antitumor activity of coinfused CD28- and 4–1BB-engineered CAR-T cells in patients with B cell leukemia. Mol. Ther. 26, 976–985 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quintarelli C et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology 7, e1433518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawalekar OU et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Neelapu SS et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park JH et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salter AI et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 11, eaat6753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuster SJ et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380, 45–56 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Abate-Daga D & Davila ML CAR models: next-generation CAR modifications for enhanced T-cell function. Mol. Ther. Oncolytics 3, 16014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinkove R, George P, Dasyam N & McLellan AD Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin. Transl. Immunol. 8, e1049 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deeks SG et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 5, 788–797 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Colovos C, Villena-Vargas J & Adusumilli PS Safety and stability of retrovirally transduced chimeric antigen receptor T cells. Immunotherapy 4, 899–902 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Scholler J et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 4, 132ra153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim GB, Hege K & Riley JL CAR talk: how cancer-specific CAR T cells can instruct how to build CAR T cells to cure HIV. Front. Immunol. 10, 2310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali A et al. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J. Virol. 90, 6999–7006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hale M et al. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol. Ther. 25, 570–579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leibman RS et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 13, e1006613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhen A et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. 13, e1006753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anthony-Gonda K et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci. Transl. Med. 11, eaav5685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herzig E et al. Attacking latent HIV with convertibleCAR-T cells, a highly adaptable killing platform. Cell 179, e810 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Z et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J. Exp. Med. 204, 705–714 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denton PW et al. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med. 5, e16 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brainard DM et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J. Virol. 83, 7305–7321 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhen A et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J. Clin. Invest. 127, 260–268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boritz EA et al. Multiple origins of virus persistence during natural control of HIV infection. Cell 166, 1004–1015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Estes JD et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 23, 1271–1276 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ventura JD et al. Longitudinal bioluminescent imaging of HIV-1 infection during antiretroviral therapy and treatment interruption in humanized mice. PLoS Pathog. 15, e1008161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Khan O et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott AC et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seo H et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl Acad. Sci. USA 116, 12410–12415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao C et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 20, 890–901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Appay V et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8, 379–385 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Buggert M et al. T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog. 10, e1004251 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leslie GJ et al. Potent and broad inhibition of HIV-1 by a peptide from the gp41 heptad repeat-2 domain conjugated to the CXCR4 amino terminus. PLoS Pathog. 12, e1005983 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Z et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guedan S et al. Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI Insight 3, e96976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pule MA et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 12, 933–941 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Hombach AA, Heiders J, Foppe M, Chmielewski M & Abken H OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. Oncoimmunology 1, 458–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moir S, Chun TW & Fauci AS Pathogenic mechanisms of HIV disease. Annu. Rev. Pathol. 6, 223–248 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Perez EE et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26, 808–816 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.. Holt N et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 28, 839–847 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tebas P et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peterson CW et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: implications for HIV gene therapy. PLoS Pathog. 14, e1006956 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Claiborne DT et al. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc. Natl Acad. Sci. USA 112, E1480–E1489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wright JK et al. Influence of Gag-protease-mediated replication capacity on disease progression in individuals recently infected with HIV-1 subtype C. J. Virol. 85, 3996–4006 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prince JL et al. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog. 8, e1003041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deymier MJ et al. Heterosexual transmission of subtype C HIV-1 selects consensus-like variants without increased replicative capacity or interferon-ɑ resistance. PLoS Pathog. 11, e1005154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seung E & Tager AM Humoral immunity in humanized mice: a work in progress. J. Infect. Dis. 208(Suppl 2), S155–S159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez-Torres F, Nochi T, Wahl A, Garcia JV & Denton PW Hypogammaglobulinemia in BLT humanized mice—an animal model of primary antibody deficiency. PLoS ONE 9, e108663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karpel ME, Boutwell CL & Allen TM BLT humanized mice as a small animal model of HIV infection. Curr. Opin. Virol. 13, 75–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dudek TE et al. Rapid evolution of HIV-1 to functional CD8+ T cell responses in humanized BLT mice. Sci. Transl. Med. 4, 143ra198 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Claiborne DT et al. Immunization of BLT humanized mice redirects T cell responses to Gag and reduces acute HIV-1 viremia. J. Virol. 93, e00814–e00819 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pardi N et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 8, 14630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boutwell CL, Rowley CF & Essex M Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 83, 2460–2468 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson D et al. Expression and structure of the human NGF receptor. Cell 47, 545–554 (1986). [DOI] [PubMed] [Google Scholar]
- 66.Wang X et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118, 1255–1263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brelot A, Heveker N, Montes M & Alizon M Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 275, 23736–23744 (2000). [DOI] [PubMed] [Google Scholar]
- 68.Richardson MW et al. Mode of transmission affects the sensitivity of human immunodeficiency virus type 1 to restriction by rhesus TRIM5ɑ. J. Virol. 82, 11117–11128 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clayton KL et al. Resistance of HIV-infected macrophages to CD8+ T lymphocyte-mediated killing drives activation of the immune system. Nat. Immunol. 19, 475–486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hindson BJ et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 83, 8604–8610 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the construct sequences described in this article are published (see ref. 26). All materials described in this manuscript are available via a material transfer agreement with the University of Pennsylvania or the Ragon Institute. All data are available from the corresponding authors upon request. Source data are provided with this paper.