

Abstract
Bardet-Biedl syndrome (BBS) is a rare ciliopathy for which there are no current effective treatments. BBS is a genetically heterogeneous disease, though the M390R mutation in BBS1 is involved in approximately 25% of all genetic diagnoses of BBS. The principle features of BBS include retinal degeneration, obesity, male infertility, polydactyly, intellectual disability, and renal abnormalities. Patients with mutations in BBS genes often present with night blindness within the first decade of life, which progresses to complete blindness. This is due to progressive loss of photoreceptor cells. Male infertility is caused by a lack of spermatozoa flagella, rendering them immobile. In this study, we have crossed the wild-type human BBS1 gene, driven by the CAG promoter, onto the Bbs1M390R/M390R mouse model to determine if ectopic expression of BBS1 rescues male infertility and retinal degeneration. qRT-PCR indicates that the BBS1 transgene is expressed in multiple tissues throughout the mouse, with the highest expression seen in the testes, and much lower expression in the eye and hypothalamus. Immunohistochemistry of the transgene in the eye showed little if any expression in the photoreceptor outer nuclear layer. When male Bbs1M30R/M390R;BBS1TG+ mice are housed with WT females, they are able to sire offspring, indicating that the male infertility phenotype of BBS is rescued by the transgene. Using electroretinography (ERGs) to measure retinal function and optical coherence tomography to measure retinal thickness, we show that the transgene does not confer protection against retinal degeneration in Bbs1M300R/M390R;BBS1TG+ mice. The results of this study indicate the male infertility aspect of BBS is an attractive target for gene therapy.
Introduction:
Bardet-Biedl syndrome (BBS) is a rare, autosomal recessive ciliopathy for which there are no current treatment options (1). The prevalence of BBS in most populations is approximately 1:160,000, yet occurs as frequently as 1:13,500 in some isolated populations (2, 3). The clinical features of BBS include retinal degeneration, obesity, male infertility, renal abnormalities, intellectual disability, and polydactyly (1). BBS is a genetically heterogeneous disease with currently 22 genes known that can independently cause BBS (4). The most common cause of BBS is the M390R mutation in BBS1, which is found in approximately 25% of all BBS diagnoses (5). There can be varying severity of BBS symptoms, even in patients with the same molecular genotype (6, 7).
BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, and BBS18 (BBIP1) are known to form a complex called the BBSome, which is important for intraciliary trafficking, as well as trafficking of proteins to the cell membrane (8). This impaired trafficking of proteins is a driving force of several of the clinical features of BBS, including retinal degeneration and obesity. For instance, it has been shown that BBS1 directly binds to the leptin receptor (LepR) in the hypothalamus, and that the BBSome plays a key role in trafficking of LepR to the plasma membrane in hypothalamic neurons (9).
The role of protein trafficking by the BBSome also likely plays a major part in the pathology of retinal degeneration caused by mutations in BBS genes. The primary layer of the retina affected by BBS is the photoreceptor layer, a set of highly specialized neuroepithelial cells essential for phototransduction. The outer segment of photoreceptors, which consists of membranous discs harboring components of the visual signal transduction pathways, is constantly undergoing turnover, a process by which new discs are made and old discs are phagocytosed by the retinal pigment epithelium (10, 11). This turnover of discs requires the transport of proteins within a retinal structure known as the connecting cilium, which is a modified primary cilium (12). In mouse models of BBS, connecting cilia appear to be present, but retrograde trafficking of proteins has been shown to be abnormal, resulting in accumulation of proteins such as Stx3 and Stxbp1 in the outer segments of photoreceptors (13). It has been observed that BBS mutations cause morphological defects in rod outer segments even before notable photoreceptor degeneration has occurred (14).
One of the hallmark features of ciliopathies is male infertility. Male factor infertility is an important public health burden, where as many as 15% of couples struggle with fertility issues, and 50% of those issues are attributed to male factor infertility or sub-fertility (15). BBS patients and animal models exhibit a lack of flagella on sperm, resulting in complete immobility (2). While a small fraction of sperm do make flagella in BBS7 mutant mouse models, they are typically truncated and contain defects in microtubule or outer dense fiber structure (16). The exact mechanisms by which BBS mutations cause lack of flagella resulting in infertility are largely unknown.
Gene therapy has received considerable attention over the last several decades as a method for treating inherited diseases. The eye is a particularly attractive candidate for gene therapy trials due to its accessibility for direct delivery of gene therapy vectors. The first gene therapy to be approved to treat inherited targets a form of Leber congenital amaurosis (LCA), a progressive retinal dystrophy caused by mutations in RPE65, which affects approximately 1,000–3,000 people in the United States (17, 18). The treatment of LCA using viral vectors for gene therapy has set the stage for attempting to treat many other retinopathies using gene therapy, including BBS. Subretinal injection of an AAV2/4 expressing Bbs4 under the control of a mouse rod opsin (mOP) promotor delivered Bbs4-null mice resulted in protection against degeneration of the outer nuclear layer (ONL), improved electroretinograms (ERGs), and some improvement in optokinetic responses (19). Gene therapy for the visual phenotype in mice has not been uniformly straight forward, however. AAV2/5 expressing Bbs1 using a chicken β-actin promoter was delivered subretinally into Bbs1M390/M390R mice, a mouse model that recapitulates most aspects of human BBS1 (5). While some improvement in ERGs of treated mice was observed, overexpression toxicity seems to be an issue preventing full therapeutic value of the treatment (20).
There is a clear need for the development of viable treatments of BBS, and preferentially, one that targets many of the syndromic features. While little has been published on the prospects of treating multiple features of pleiotropic disease with viral vectors, systemic delivery of AAV vectors have been shown to successfully target several organ systems following delivery through the blood stream in mice (21). Whether or not systemic administration of gene therapy agents could ameliorate some of the features of BBS deserves attention. In this study, we tested the proof of principle of whether we can correct some of the features of BBS via ectopic expression of BBS1 in a mouse model of Bbs1M390R/M390R by expressing transgenic human BBS1 under the control of the CAG promoter, thus removing the potentially confounding effect of virus delivery and toxicity of viral vectors. We examined the therapeutic effects of human BBS1 transgene expression specifically on male infertility and retinal degeneration. Our results have implications for the future development of therapies for BBS and other related syndromic genetic diseases.
Materials and Methods:
Mice
In this study, we used a knock-in mouse model of Bbs1M390R/M390R, which exhibits most of the common phenotypes of BBS, including male infertility, obesity, and retinal degeneration. This mouse model has been previously described (5). We also used a mouse model that expresses a human BBS1 transgene with a c-terminal flag tag under the control of a CAG promoter. We chose a CAG promoter due to its potential for ubiquitous expression among all tissue types in mice. Previous work has shown that the c-terminal flag tag does not interfere with BBSome assembly (20). The BBS1 transgenic mouse was produced using pronuclear injection at the Genome Editing Facility at the University of Iowa. The BBS1 transgenic mouse was produced on a B6 X SJL background, and was backcrossed seven times onto a 129/SvEv background to make a pure line. These mice were genotyped to ensure they are homozygous wild-type for the Crb1 gene (do not contain the Rd8 mutation, which is found in several strains of mice and can cause retinal lesions (22)). First, we crossed a female Bbs1M390R/WT mouse with a male BBS1TG+/TG+ mouse in order to obtain Bbs1M390R/WT;BBS1TG+/TG- mice. From this cross, we obtained three female mice with the desired genotype. These three females were bred with one of two male Bbs1M390R/WT mice in order to obtain all possible genotype combinations of Bbs1 and BBS1TG. To simplify nomenclature, we refer to Bbs1M390R/WT and Bbs1WT/WT mice as WT (as BBS is recessive and two copies of Bbs1M390R are required for BBS phenotypes). We refer to mice with or without a copy of the BBS1 transgene as TG+ or TG-, respectively. All procedures were performed in accordance with approved Institutional Animal Care and Use Committee (IACUC) protocol #8072147 of the University of Iowa.
Determine the expression level of the BBS1 transgene
To prepare a cDNA library from whole eye, hypothalamus, and testes, we first extracted RNA from three randomly chosen sacrificed animals that were positive for the transgene (by CO2 and cervical dislocation) using approximately 30 µg of each tissue using the NucleoSpinR RNA kit (Takara Bio USA, Inc.) according to manufacturer’s suggested protocol. RNA concentration was quantified using a Nanodrop 2000 spectrophotometer (Thermo Scientific). To reverse transcribe our RNA, we mixed 350 ng RNA, 4 µL SuperScript™ VILO™ MasterMix (Invitrogen), and nuclease free H2O to a volume of 20 µL, then incubated at 25°C for 10 minutes, 42°C for 60 minutes, and 85°C for 5 minutes.
Our primer sets were designed so that at least one per set overlapped exons, such that only cDNA should be amplified. Primer sets were chosen using the GenScript Real-time PCR (TaqMan) Primer Design tool:
BBS1-tg Forward: TTCTGCAGCTGGAGCTAAGT
BBS1-tg Reverse: GGGAAGGCTCATCTTGGCTA
Bbs1 Forward: TATCAAGCGCCAGACAGTCA
Bbs1 Reverse: TCAACCGGAACTCCACATCA
β-actin Forward: CCTGCTTCACCACCTTCTTG
β-actin Reverse: GTGTCCGTCGTGGATCTGA
The primers were tested with 10 ng cDNA template, 1x rTaq buffer, 1x betaine, 1 µM each forward and reverse primer, and rTaq DNA polymerase. The reactions were mixed thoroughly, and amplified by heating to 94°C for 3 minutes, then 35 cycles of 94° for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds, with a final extension of 72°C for 7 minutes. The PCR amplicons were run on a 1% agarose gel and examined to make sure there was only one product.
Each qRT-PCR reaction was performed in triplicate for each animal tissue. Our reactions were set up using 10 µL Powerup™ SYBR™ Green Master Mix (appliedbiosystems by Thermo Fisher Scientific), 10 ng cDNA, 1 µM each forward and reverse primer, and water to a total volume of 20 µL. The reaction was completed on a CFX96 Touch Real-Time PCR Detection System (BIO-RAD). We ran the CFX_3StepAmp+Melt protocol, using an initial melt temperature of 94°C for 3 minutes, 39 round of 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds, and ended with a melting curve with 95°C for 10 seconds, 65°C for 5 seconds, and 95°.
In order to calculate gene expression of the BBS1 transgene relative to endogenous mouse Bbs1, the measured CQ values were averaged for the technical triplicates within each animal. To calculate fold change of BBSTG mRNA expression relative to Bbs1WT mRNA expression, we used the following equations:
ΔCQ1 = CQ(BBS1TG) – CQ(β-actin)
ΔCQ2 = CQ(mBbs1) – CQ(β-actin)
ΔΔCQ = ΔCQ1 – ΔCQ2
2-ΔΔCQ = Fold change
The values for each animal and each tissue were calculated independently. The fold changes were then averaged among animals to give the relative expression of the transgene relative to endogenous Bbs1 for each tissue. In three cases, we only used two CQ values to calculate the average CQ for a gene in a tissue. There were two outliers that were removed from our analysis; a CQ value for transgene from one hypothalamus and a CQ value for endogenous Bbs1 from one eye. In the third case, there was a complete PCR failure for one technical replicate for endogenous Bbs1 in a testes sample.
Histology
To look at the spatial expression of the BBS1 transgene, WT and Bbs1M390R/M390R mice with and without the BBS1 transgene were euthanized, and eyes were extracted and placed in 4% paraformaldehyde to fix the tissues. A hole was punctured in the cornea to allow for infiltration of the paraformaldehyde. The eyes were washed once with PBS, embedded in OCT and frozen using liquid nitrogen. 8–10 µM sections were cut from the frozen blocks using a cryostat, and the sections were stored at −80°C until histology was performed. To observe spatial expression of the BBS1 transgene, we used immunohistochemistry. The slides containing the sections were thawed at room temperature and blocked for 30 minutes using a buffer of 1% BSA and 0.3% Triton-X (FisherChemical, Fisher Scientific) in PBS. Monoclonal ANTI-FLAG M2 antibody, produced in mouse (SIGMA, cat. number F1804), was diluted in the blocking buffer at 1:250 and incubated on the sections for approximately one hour, and washed three times for five minutes each with PBS. A secondary antibody, Alexa Fluor 488 goat anti-mouse (Invitrogen, cat. number A11029), was diluted in the blocking buffer at 1:500 and incubated on the sections for an additional one hour. The secondary antibody was washed with PBS three times for five minutes each. A drop of Vectashield mounting medium for fluorescence with DAPI (Vector Laboratories) was placed on each of the sections, and slide covers were mounted on the sections using clear fingernail polish. The slides were imaged with using fluorescence microscopy using an Olympus IX71 microscope at 400x magnification.
To image seminiferous tubules of the testes, we extracted testes from mice euthanized with CO2 and cervical dislocation. Testes were fixed in 4% paraformaldehyde overnight, and then fixed for several hours in Richard-Allan Scientific™ Signature Series™ Pen-Fix™ (ThermoFisher Scientific) for several hours. The fixed testes were paraffin embedded using a Tissue-Tek® VIP® 6 AI Vacuum Infiltration Paraffin Processor, embedded in a paraffin block, and then sectioned at 8 µM thickness using a microtome. The sections were stained with hematoxylin and eosin (H&E) using a DRS-601 automated slide stainer (Sakura). The stained slides were imaged using an Olympus IX71 microscope at 400x and 600x magnification.
Optical coherence tomography
In order to observe whether there was any degeneration of retinas over time in WT or Bbs1M390R/M390R mice, with or without the BBS1 transgene, we used spectral-domain optical coherence tomography (SD-OCT). This non-invasive technique allows for in vivo longitudinal imaging and measuring of retinas over time. Mice were anesthetized with ketamine/xylazine, and eyes were hydrated with Balanced Salt Solution (BSS, Alcon Laboratories, Inc., Fort Worth, TX, USA) and then retinas, centered on the optic nerve, were imaged with the Mouse Retinal Bore using a Bioptigen SD-OCT (Bioptigen, Inc., USA). Both retinas of each mouse were imaged once a month for four months using a 1.4 mm x 1.4 mm rectangular scan with 1,000 A-scans/B-scan, 100 B-scans/volume, 1 frame/B-scan, and 1 volume. Following imaging, eyes were hydrated with Artificial Tears Ointment (AKORN, Inc., Lake Forest, IL, USA) and mice were provided indirect supplemental warmth for recovery. The retinal outer nuclear layer was measured using the Bioptigen InVivoVue Software by averaging 8 manually placed vertical angle-locked B-scan calipers for each mouse (4 calipers/eye). The caliper values and mouse genotypes were masked at the time of measurement in order to reduce any measurement bias.
Electroretinography
Photoreceptor function in WT and Bbs1M390R/M390R mice with and without the BBS1 transgene was assessed using electroretinography. Mice were dark adapted for at least 12 hours prior to examination, and anesthetized with a xylazine/ketamine solution. Phenylephrine Hydrochloride Ophthalmic Solution, USP 2.5% (Paragon Biotek, Inc.) was placed on the left eye of a mouse for five minutes, then dabbed off using a cotton swab. Gonosovic was put on the eye of the mouse for conductance, and a ground electrode was inserted subcutaneously on the snout. Electroretinography was performed using a Pheonix Ganfeld ERG IWX100 machine and LabScribe 2 software. Once the mouse was situated correctly and a baseline measurement showed a relatively flat ERG, measurements were taken at 0.01952, 0.1564, 1.252, and 40 cd sec/m2. Ten sweeps were taken and averaged for each intensity to obtain values for A and B waves. Following imaging, eyes were hydrated provided indirect warmth as described above. Each mouse was measured once a month over a period of four months.
Statistical analysis:
We used R to perform all statistical tests herein. GraphPad Prism 8.3.0 and Microsoft Excel software were used for all graphing. To determine the effect of the BBS1 transgene on WT and Bbs1M390R/M390R backgrounds with regards to retinal degeneration, we used mixed-effects models due to the repeat sampling of individual animals over four months, with a general form of the model: y = 1|(“Animal ID”) + AGE + GENDER + BBS1 + TG + BBS1 × AGE + BBS1 × TG. Terms that were not significant were removed to reach the final model. There were at least four animals per group in our analyses of retinal function and degeneration as determined by ERG and OCT, respectively. Statistical methods were not used to estimate sample size, as sample size was based on prior experience. The normality assumption on the random intercept and the residual was tested using the Shapiro-Wilk test for normality and the common variance assumption on the residuals was tested using Levene’s test for equality of variances. All data used for our analyses were deposited in Dryad (https://doi.org/10.5061/dryad.2rbnzs7jm).
Results:
Expression of BBS1 transgene
BBS1 transgene mRNA expression was assessed in whole eye, hypothalamus, and testes in three separate mice using technical triplicates. We used one animal with each of the following genotypes: Bbs1WT/WT;BBS1TG+/TG-, Bbs1M390R/WT;BBS1TG+/TG-, and Bbs1M390R/M390R;BBS1TG+/TG-. ΔCQ1 values were similar among the genotypes, indicating the M390R mutation in Bbs1 does not have an effect on mRNA levels. We found that BBS1 transgene expression level relative to endogenous WT Bbs1 varied among the tissue types tested (Figure 1). There was a much higher relative expression found in the testes than in the eye and hypothalamus. In the testes, the expression level of the transgene was approximately double the expression of endogenous Bbs1 mRNA (1.99 ± 0.50 fold change). In the whole eye and hypothalamus, the fold change of transgene relative to endogenous Bbs1 mRNA was 0.21 ± 0.07 and 0.19 ± 0.05, respectively. In the testes, the transgenic protein incorporated into the BBSome, as evidenced by our ability to pull down the BBSome via tandem affinity purification using the transgenic BBS1 (Figure S1).
Figure 1.
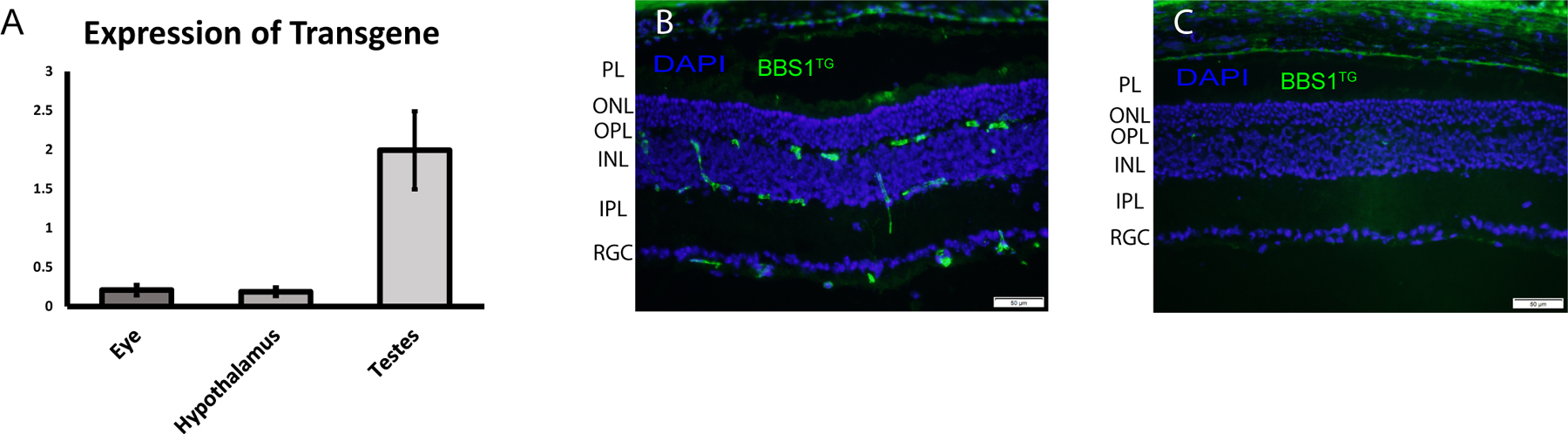
(A) BBS1 transgene mRNA expression, relative to endogenous mouse Bbs1 expression, as determined by qRT-PCR. The highest relative expression was observed in the testes, with relatively low expression in the eye and hypothalamus. Data displayed as mean ± SEM (n=3). Immunohistochemistry of BBS1 transgene in the retinas of (B) TG+ and (C) TG- mice. There are small islands of expression in the retinal ganglion cell layer (RGC) and inner nuclear layer (INL), with little to no expression observed in the outer nuclear layer (ONL) and photoreceptor layer (PL). The staining above the PL in the retinal pigmented epithelium is nonspecific. IPL: inner plexiform layer, OPL: outer plexiform layer. Scale bar = 50µM.
To examine the spatial expression of the BBS1 transgenic protein in the eye, we performed immunohistochemistry of retinal cross-sections from two Bbs1WT/WT;BBS1TG+/TG- mice (Figure 1). While the protein was observed in TG+ eyes and absent in negative controls, the expression of the transgene was diffuse, as expected considering the qRT-PCR data. Expression appeared to be limited to the ganglion cell layer and inner nuclear layer, with little to no expression observed in the outer nuclear layer or photoreceptor layer.
Male Fertility
Experimental breeder pairs were set up between Bbs1M390R/M390R;TG+ male mice and WT female mice to determine whether the transgene could rescue functional fertility in male Bbs1M390R/M390R mice. Two Bbs1M390R/M390R;TG+ male mice were housed with WT females for several months, and each of the males sired several litters of pups over the course of four months, indicating that functional fertility was rescued. Two male Bbs1M390R/M390R;TG- mice individually housed with multiple WT females did not sire any offspring.
Testes from euthanized mice were sectioned and stained with H&E as described above (n=1 for each representative genotype) (Figure 2). We noted complete lack of spermatozoa flagella in only the Bbs1M390R/M390R;TG- samples. The Bbs1M390R/M390R;TG+ mouse was indistinguishable from its WT counterpart. This result supports the functional rescue of fertility in the transgenic mice, as only spermatozoa with flagella will have functional mobility. We also evaluated sperm from excised epididymides of one Bbs1M390R/M390R;TG+ and several Bbs1M390R/M390R;TG- mice under light microscopy, and only observed active swimming spermatozoa in the Bbs1M390R/M390R;TG+ sample (data not shown).
Figure 2.
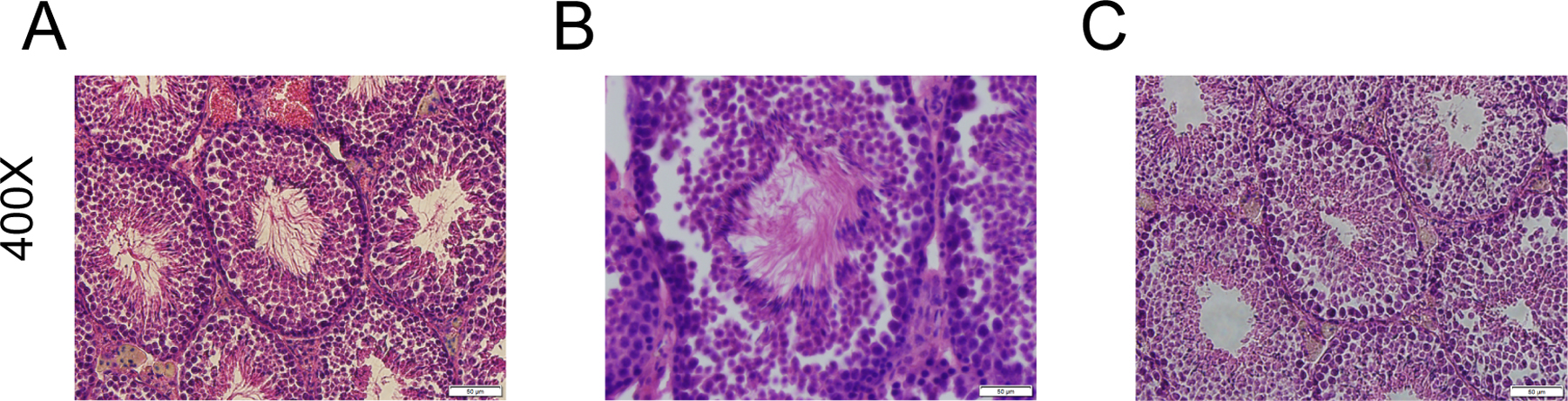
Seminiferous tubules from (A) WT;TG-, (B) Bbs1M390R/M390R;TG+, and (C) Bbs1M390R/M390R;TG- show flagella formation is rescued in the Bbs1M390R/M390R;TG+ mice, but is wholly absent in the Bbs1M390R/M390R;TG- mice. N=1 for each genotype. Scale bars: 50 µM.
Retina degeneration
We measured ERGs in mice once a month for four months using 1.252 cd sec/m2 stimulus and subsequently used a mixed effects model to test whether there was a significant effect of genotype on A and B waves. The data used for analysis of ERG values met all of the assumptions of the statistical tests performed. We observed that Bbs1M390R/M390R mice had significantly worse A and B waves than WT mice over the four months tested, regardless of transgene status (p=1.17×10−25 for A wave and p=1.22×10−21 for B wave; Figure 3). We found that the transgene did not have a significant impact on either A or B waves among Bbs1M390R/M390R or WT mice (p>0.05 for both A and B waves; Figure 3).
Figure 3.
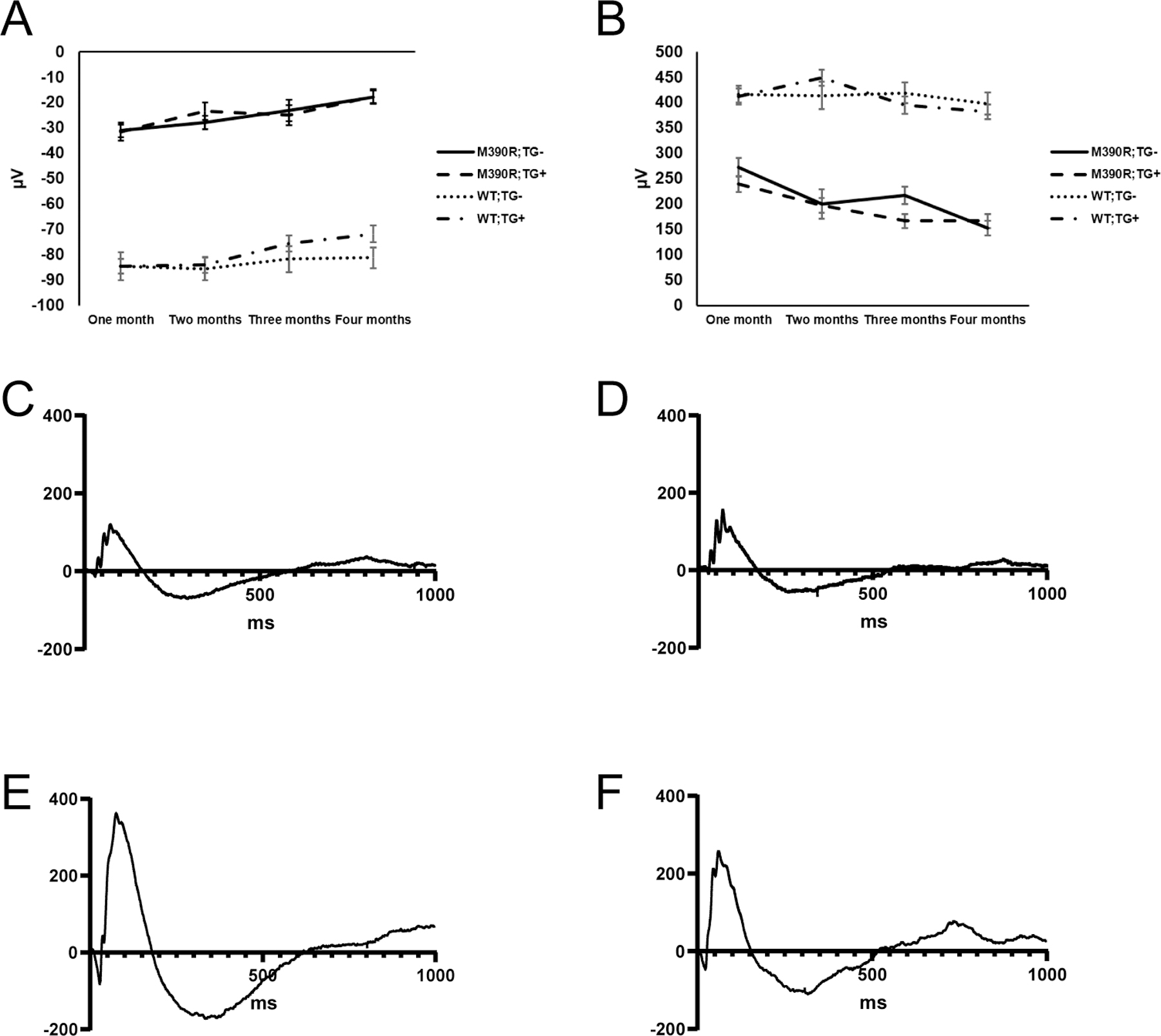
Comparison of ERG (A) A and (B) waves from each of the genotypes over four months indicates that the transgene does not have a significant effect on Bbs1M390R/M390R or WT mice. Error bars indicate standard error of the means. Displayed ERG values were captured using 1.252 cd sec/m2. N for each genotype group at each month (month 1, month 2, month 3, month 4): Bbs1M390R/M390R;TG- (4,4,4,4), Bbs1M390R/M390R;TG+ (8,8,7,7), WT;TG- (17,15,14,13), and WT;TG+ (26,22,21,21). Representative ERG traces from (C) Bbs1M390R/M390R;TG-, (D) Bbs1M390R/M390R;TG+, (E) WT;TG-, and (F) WT;TG+ mice at four months of age, captured using 1.252 cd sec/m2. In the figure M390R= Bbs1M390R/M390R.
ONL thickness of mice were measured once a month for four months using OCT to test the effect of the BBS1 transgene on Bbs1M390R/M390R and WT backgrounds (Figure 4). Shapiro-Wilk test for residual normality was significant (p=0.02), though recent research has indicated that the linear mixed effects model is robust to violation of the distributional assumptions (23, 24). All other assumptions of our analyses were met. Our mixed effects model indicated that ONL thickness was significantly lower in Bbs1M390R/M390R mice than WT mice (p=7.39×10−24). We also found that there was a significant interaction between age and Bbs1 genotype (p=1.62×10−32), where Bbs1M390R/M390R ONL thickness decreased dramatically over four months and WT ONL thickness remained relatively stable. The transgene did not have a significant effect on ONL thickness among Bbs1M390R/M390R or WT mice (p>0.05).
Figure 4.
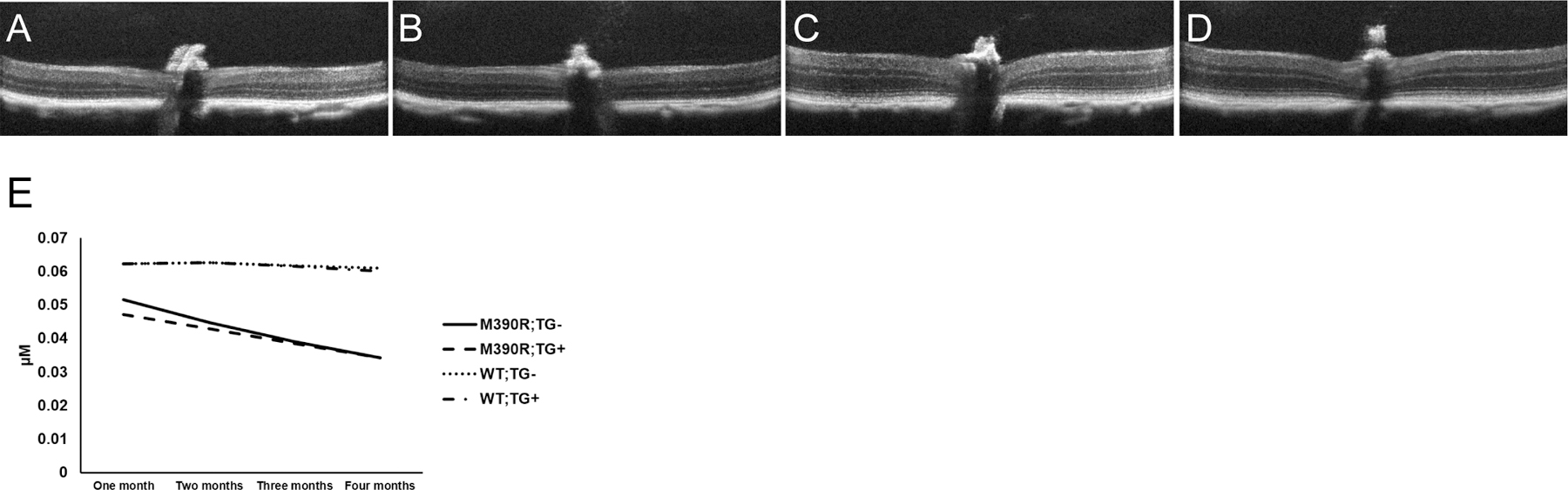
Representative OCT images of retinas at 4 months of age for (A) Bbs1M390R/M390R;TG-, (B) Bbs1M390R/M390R;TG+, (C) WT;TG-, and (D) WT;TG+ mice. Note the thin retinas in the Bbs1M390R/M390R mice with and without the transgene, while the WT mice with and without the transgene have significantly thicker retinas, especially at the outer nuclear layer. (E) Outer nuclear layer thickness of mice as measured by OCT over four months. Bbs1M390R/M390R mice exhibit significant retinal degeneration over time, and the transgene does not confer any protective effects against retinal degeneration. N for each genotype group at each month (month 1, month 2, month 3, month 4): Bbs1M390R/M390R;TG- (4,4,4,4), Bbs1M390R/M390R;TG+ (6,7,7,6), WT;TG- (17,14,14,15), and WT;TG+ (25,22,19,19). In the figure M390R= Bbs1M390R/M390R.
Discussion:
Syndromic ciliopathies such as BBS, Joubert syndrome, Senior-Loken syndrome, McKusick-Kaufman syndrome, and Meckel-Gruber syndrome display many similar disease features. The pathological phenotypes of these disorders can range from mild to severe or even lethal depending on the disease and mutations responsible, particularly for disorders such as Meckel-Gruber syndrome. While individually rare in most populations, together these diseases represent a significant public health burden, and can result in drastically reduced quality of life for affected individuals and their families. The development of efficacious therapeutic strategies would be a great step forward for patients with ciliopathies. The advances in gene therapy have come a long way over the last several decades, although diseases caused by mutations in some genes, like BBS1, have remained difficult targets due to issues such as the pleiotropic nature of the disorder, extensive heterogeneity and overexpression toxicity (20). In this report, we have shown that ectopic expression of BBS1 via transgene driven by a CAG promoter in a BBS1 mouse model (Bbs1M390R/M390R), harboring the most common mutation implicated in BBS, provides partial rescue of BBS phenotypes.
We show that our BBS1TG mouse model, when crossed with our strain that harbors the Bbs1M390R allele, expresses the transgene in all of the tissues we have tested, albeit at varying levels. The highest expressing tissue is the testes, which expresses BBS1TG approximately an order of magnitude higher than what we observed in the eye and the hypothalamus. In the retina, expression of BBSTG is very patchy, with small islands of expression, particularly in the ganglion cell layer and outer nuclear layer. Very little expression is observed in the inner nuclear layer or in the photoreceptor layer. Overall, qRT-PCR revealed that expression of BBS1TG in the eye and hypothalamus are only approximately 21% and 19% that of endogenous Bbs1 expression, respectively.
In light of our qRT-PCR data showing relatively high expression of the BBS1 transgene in the testes, it is perhaps not surprising that male infertility is rescued. H&E stained testes sections show that the lumens of seminiferous tubules in Bbs1M390R/M390R;TG- male mice are completely devoid of flagella, while the Bbs1M390R/M390R;TG+ seminiferous tubules were indistinguishable from their WT counterparts. Furthermore, excised epididymides of Bbs1M390R/M390R;TG+ mice examined under light microscopy reveal active swimming spermatozoa in the Bbs1M390R/M390R;TG+ epididymides, whereas swimming spermatozoa are not observed in Bbs1M390R/M390R samples. Finally, male Bbs1M390R/M390R;TG+ mice are able to sire multiple litters of offspring, whereas numerous Bbs1M390R/M390R;TG- male mice fail to sire any offspring when placed in breeder cages with WT females. While obesity can contribute to male fertility (25), the mechanisms of male infertility are not known to be related to abnormal flagella formation. Furthermore, obesity did not have an effect on male fertility in our BB1M390R/M390R;TG+ mice. The two Bbs1M390R/M390R;TG+ male mice that were tested for functional rescue of fertility were initially placed into breeding cages at approximately two months of age, and sired offspring before their weight significantly diverged from the average weight of age matched WT male mice. We continued to house one Bbs1M390R/M390R;TG+ male with a breeder female for several months until it became 37% heavier than the average of age matched WT mice (obesity was not rescued in 2/3 Bbs1M390R/M390R;TG+ male mice) and it continued to sire offspring when it was obese. As transgene mRNA is double that of endogenous Bbs1 mRNA in the testes, we have found that overexpression toxicity appears to be less of an issue in the testes than in the eye. This makes gene therapy to treat fertility in BBS1 theoretically easier than rescuing the eye phenotype.
Fertility issues affect up to 15% of couples, and male contribution to infertility is typically approximately 50% of cases (26). Male factors resulting in infertility include defects in motility (27), sperm head defects (28), and low sperm count (29). While environmental factors may cause some male infertility issues (30), a relatively large number of genes that, when mutated, result in male infertility. Our data here show proof of principle that BBS is a good disease model to test methods of correcting male infertility by using gene supplementation. There have been several studies aimed at treating male infertility using gene therapy and gene correction techniques. Azoospermia can be caused by several factors, including mutations in c-kit in Sertoli cells. Following injection of a lentivirus encoding c-kit into the seminiferous tubules of deficient mice, spermatozoa collected from injected mice were capable of fertilizing oocytes via intracytoplasmic injection (ICSI) (31). While ICSI is a common method of overcoming some forms of male infertility (32), a more attractive approach, perhaps, is to provide a therapeutic option that allows recipients to conceive naturally. Using CRIPSR technology, spermatogonial stem cells were harvested from one testis of a mouse with nonobstructive azoospermia, edited to WT using CRISPR mediated homologous recombination, enriched for corrected cells, and injected back into the remaining testis. The mouse was then able to naturally sire healthy offspring (33). This represents an important advance in the treatment of heritable disease and male infertility, and could be extended to treat male infertility caused by ciliopathies such as BBS. Due to the current moratorium on heritable genome editing in human patients (34), these techniques may not be available to treat humans in the foreseeable future.
The BBS1 transgene did not rescue retinal degeneration or retinal function on a Bbs1M390R/M390R background, as determined by OCT and ERG, respectively. This is likely due to the low expression of the transgene in the eye, and more importantly, the lack of expression in the ONL and photoreceptors. Our results indicate that preservation of visual function in BBS patients and mouse models will require expression of exogenous BBS specifically in the ONL and photoreceptors, which could be accomplished via subretinal injection of an AAV2/5 expressing the relevant functional BBS gene. It is possible that only 50% of endogenous levels of Bbs1 expression is necessary to preserve visual function in Bbs1M390R/M390R mouse models and humans with homozygous M390R mutations in the BBS1 gene, as we do not readily detect functional or phenotypic differences between homozygous WT individuals and heterozygous carriers of the M390R mutation. Considering the potential for overexpression toxicity by the BBS1 gene (20) and toxicity of ubiquitous promoters in the retina (35), care should be taken to choose a low expressing, retina specific promoter when designing a therapeutic vector to treat retinal degeneration caused by mutations in the BBS1 gene.
Supplementary Material
Funding:
This work was funded by the National Institutes of Health, grant numbers R01 EY011298 (V.C.S.) and R01 EY017168 (V.C.S.), as well as the Roy J. Carver Charitable Trust (V.C.S.). This work also benefitted from the University of Iowa Visual Science Core Facilities funded by NIH P30EY025580 (V.C.S.).
Footnotes
Conflict of interest:
None of the authors have any conflicts of interest to disclose.
References:
- 1.Sheffield V, Zhang Q, Heon E, Drack A, Stone E, Carmi R. The Bardet-Biedl Syndrome. Third ed. Erickson R, Wynshaw-Boris A, editors: Oxford University Press; 2016. [Google Scholar]
- 2.Forsythe E, Beales P. Bardet-Biedl syndrome. European Journal of Human Genetics. 2013;21:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farag T, Teebi A. High incidence of Bardet Biedl syndrome among the Bedouin. Clinical Genetics. 1989;36:463–5. [DOI] [PubMed] [Google Scholar]
- 4.Schaefer E, Delvalee C, Mary L, Stoetzel C, Geoffroy V, Marks-Delesalle C, et al. Identification and characterization of known biallelic mutations in the IFT27 (BBS19) gene in a novel family with Bardet-Biedl syndrome. Frontiers in Genetics. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis R, Swiderski R, Rahmouni K, Nishimura D, Mullins R, Agassandian K, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. PNAS 2007;4(104):19422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azari A, Aleman T, Cideciyan A, Schwartz S, Windsor E, Sumaroka A, et al. Retinal disease expression in Bardet-Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina-wide degeneration. Investigative Ophthalmology and Visual Science. 2006;47(11):5004–10. [DOI] [PubMed] [Google Scholar]
- 7.Cox K, Kerr N, Kedrov M, Nishimura D, Jennings B, Stone E, et al. Phenotypic expression of Bardet-Biedl syndrome in patients homozygous for the common M390R mutation in the BBS1 gene. Vision Research. 2012;75(15):77–87. [DOI] [PubMed] [Google Scholar]
- 8.Jin H, Nachury M. The BBSome. Current Biology. 2009;19(12):482–73. [DOI] [PubMed] [Google Scholar]
- 9.Seo S, Guo D, Bugge K, Morgan D, Rahmouni K, Sheffield V. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Human molecular genetics. 2009;18(7):1323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young R The renewal of photoreceptor cell outer segments. J Cell Biol 1967;33(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen J, Hicks D. Renewal of photoreceptor outer segments and their phagocytosis by the retinal pigment epithelium. International Review of Cytology. 2000;196:245–313. [DOI] [PubMed] [Google Scholar]
- 12.Tian G, Lodowski K, Lee R, Imanishi Y. Retrograde intraciliary trafficking of opsin during the maintenance of cone‐shaped photoreceptor outer segments of Xenopus laevis. Journal of Comparative Neurology. 2014;522(16):3577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datta P, Allamargot C, Hudson J, Anderson E, Bhattarai S, Drack A, et al. Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet-Biedl syndrome. PNAS 2015;112(32):E4400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu Y, Garrison J, Kim G, Schmitz A, Searby C, Zhang Q, et al. BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLOS Genetics. 2017;13(10):e1007057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal A, Mulgund A, Hamada A, Chyatte M. A unique view on male infertility around the globe. Reproductive Biology and Endocrinology. 2015;13(37). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q, Nishimura D, Vogel T, Shao J, Swiderski R, Yin T, et al. BBS7 is required for BBSome formation and its absence in mice results in Bardet-Biedl syndrome phenotypes and selective abnormalities in membrane protein trafficking. Journal of Cell Science. 2013;126(11):2372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smalley E First AAV gene therapy poised for landmark approval. Nature Biotechnology. 2017;35:998–9. [DOI] [PubMed] [Google Scholar]
- 18.Cai X, Conley S, Naash M. RPE65: role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic Genetics. 2009;30(2):57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simons D, Boye S, Hauswirth W, Wu S. Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet-Biedl syndrome mouse model. PNAS 2011;108(15):6276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seo S, Mullins R, Dumitrescu A, Bhattarai S, Gratie D, Wang K, et al. Subretinal gene therapy of mice with Bardet-Biedl syndrome type 1. Investigative Ophthalmology and Visual Science. 2013;11(54):6118–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zincarelli C, Soltys S, Rengo G, Rabinowitz J. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 2008;16(6):1073–80. [DOI] [PubMed] [Google Scholar]
- 22.Mattapallil J, Wawrousek E, Chan C, Zhao H, Roychoudhury J, Ferguson T, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Immunology and Microbiology. 2012;53(6):2921–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schielzeth H, Dingemanse N, Nakagawa S, Westneat D, Allegue H, Teplitsky C, et al. Robustness of linear mixed‐effects models to violations of distributional assumptions. Methods in Ecology and Evolution. 2020;11(9). [Google Scholar]
- 24.Knief U, Forstmeier W. Violating the normality assumption may be the lesser of two evils. bioRxiv 2018. [DOI] [PMC free article] [PubMed]
- 25.Du Plessis S, Cabler S, McAlister D, Sabanegh E, Agarwal A. The effect of obesity on sperm disorders and male infertility. Nature Reviews Urology. 2010;7:153–61. [DOI] [PubMed] [Google Scholar]
- 26.Jodar M, Soler-Ventura A, Oliva R. Semen proteomics and male infertility. Journal of Proteomics. 2017;162:125–34. [DOI] [PubMed] [Google Scholar]
- 27.Pereira R, R S, Barros A, Sousa M. Major regulatory mechanisms involved in sperm motility. Asian Journal of Andrology. 2017;19:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goyal R, Kotru M, Gogia A, Sharma S. Qualitative defects with normal sperm counts in a patient attending infertility clinic. Indian Journal of Pathology and Microbiology. 2018;61(2):233–5. [DOI] [PubMed] [Google Scholar]
- 29.Levine H, Jorgensen N, Martino-Andrade A, Mendiola J, Weksler0Derri D, Mindlis I, et al. Temporal trends in sperm count: a systematic review and meta-regression analysis. Human Reproduction Update. 2017;23(6):646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skakkebaek N Sperm counts, testicular cancers, and the environment. BMJ 2017;359:j4517. [DOI] [PubMed] [Google Scholar]
- 31.Ikawa M, Tergaonkar V, Ogura A, Ogonuki N, Inoue K, Verma I. Restoration of spermatogenesis by lentiviral gene transfer: offspring from infertile mice. PNAS 2002;99(11):7524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jain T, Grainger D, Ball G, Biggons W, Rebar R, Robins J, et al. 30 years of data: impact of the United States in vitro fertilization data registry on advancing fertility care. Fertility and Sterility. 2019;111(3):477–88. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Sun T, Wang X, Tang J, Liu Y. Restore natural fertility of Kitw/Kitwv mouse with nonobstructive azoospermia through gene editing on SSCs mediated by CRISPR-Cas9. Stem Cell Research and Therapy. 2019;10(271). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dyer O Scientists call for moratorium on editing heritable genes. BMJ 2019;364. [DOI] [PubMed]
- 35.Xiong W, Wu D, Xue U, Wang S, Chung M, Ji X, et al. AAV cis-regulatory sequences are correlated with ocular toxicity. PNAS 2019;116(12):5785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.