

Abstract
There is exponential clinical and research interest in joint hypermobility due to recognition of the complexity of identification, assessment, and its appropriate referral pathways, ultimately impacting management. This state-of-the-science review provides an international, multidisciplinary perspective on the presentation, etiology, and assessment of joint hypermobility, as it presents in those with and without a systemic condition. We synthesize the literature, propose standardizing the use of terminology and outcome measures, and suggest potential management directions. The major topics covered are (i) historical perspectives; (ii) current definitions of hypermobility, laxity, and instability; (iii) inheritance and acquisition of hypermobility; (iv) traditional and novel assessments; (v) strengths and limitations of current assessment tools; (vi) age, sex, and racial considerations; (vii) phenotypic presentations; (viii) generalized hypermobility spectrum disorder and hypermobility Ehlers-Danlos syndrome; and (ix) clinical implications and research directions. A thorough understanding of these topics will equip the reader seeking to manage individuals presenting with joint hypermobility, while mindful of its etiology. Management of generalized joint hypermobility in the context of a complex, multisystem condition will differ from that of acquired hypermobility commonly seen in performing artists, specific athletic populations, posttrauma, and so on. In addition, people with symptomatic hypermobility present predominantly with musculoskeletal symptoms and sometimes systemic symptoms including fatigue, orthostatic intolerance, and gastrointestinal or genitourinary issues. Some also display skeletal deformities, tissue and skin fragility, and structural vascular or cardiac differences, and these warrant further medical follow-up. This comprehensive review on the full spectrum of joint hypermobility will assist clinicians, coaches/sports trainers, educators, and/or researchers in this area.
Key Words: Ehlers-Danlos syndromes, heritable disorders of connective tissue, hypermobility spectrum disorder, joint hypermobility, joint laxity
This article reviews various aspects pertaining to joint hypermobility. These include a historical perspective; current definitions; etiology; traditional and novel assessments and their utility; age, sex, and racial considerations; and phenotypic presentations. The primary aim is to provide a clinical overview about identification of hypermobility forms. The secondary aims are to highlight best available contemporary research on referral pathways and implications for clinical practice to highlight directions for future research. The resulting synthesis of the science article has been contributed to and reviewed by an internationally recognized multidisciplinary team of allied health professionals and medical specialists from the Ehlers-Danlos Syndrome International Consortium, who are tasked to develop evidence-informed management and care guidelines.
HISTORICAL PERSPECTIVES OF JOINT HYPERMOBILITY
Joint hypermobility was recognized as a clinical entity by Hippocrates in the fourth century bc when describing the use of medical interventions to improve capacity for javelin throwing or shooting arrows for family groups experiencing shoulder instability.1 The literature records similar descriptions from contortionists to musicians.2 Early reports of excessive joint mobility led to collections of signs and symptoms being ascribed diagnoses in the scientific literature of the late 1800s and early 1900s. In the 1960s, hypermobility syndrome was defined as “the occurrence of symptoms in otherwise healthy hypermobile individuals.”3 The term Ehlers-Danlos syndromes was ascribed in the genetic and dermatology fields,4 whereas the term joint hypermobility syndrome (JHS) was used in the rheumatology field.5 Specialists from these fields recognized the overlap of these conditions more recently,6 resulting in agreed terminology attributed to specific diagnoses of generalized hypermobility spectrum disorder (G-HSD) and hypermobile Ehlers-Danlos syndrome (hEDS).7
CURRENT DEFINITIONS OF HYPERMOBILITY, LAXITY, AND INSTABILITY
Although used interchangeably, the terms joint hypermobility, laxity, and instability are not synonymous. This is not a new phenomenon. In 1902, when reporting on a child with congenital hip dislocation, Ochsner8 described “hypermobility of the heads of the femora and of the thighs.” It is likely that he was referring to physiological motion of the hips (hypermobility) and accessory motion of the femoral heads (laxity).
Hypermobility describes an objective measure of a joint moving passively or actively beyond normal physiological limits around axes of motion.7 An example is hyperextension or recurvatum of the knee. The proposed multidimensional causes of joint hypermobility include bone morphology/shape seen in humeral and femoral torsion,9 increased surface area for articulation,10 and dysplastic or excessively compliant passive restraints to physiological joint motion.11
Laxity describes an objective measure of a joint to move passively beyond normal limits during accessory motion (commonly glide and spin).12 It is assessed using manual tests such as the Lachman test of the knee, anterior drawer of the ankle, or shoulder, or mechanically (e.g., KT-3000, a mechanical device that measures tibial displacement for an imposed load).13 The terms laxity and mechanical instability are often used interchangeably.
Functional Instability is a subjective/self-report by the patient as mistrust or insecurity that their joint will remain intact even under low force conditions. They might describe the joint subluxing, going out of place, giving way, or frankly dislocating. Measurement of functional instability or confidence in the joint of interest can be assessed by questionnaire.14
In the absence of functional instability (a symptom except where dislocation is evident), any signs of hypermobility and laxity may be clinically irrelevant and may even be an asset. Consider the gymnast who displays extreme physiological joint range of motion (hypermobility) and, if formally assessed, displays accessory motion in excess of what might be expected for his/her age and sex (laxity). Despite hypermobility and laxity, they do not report that their joints subluxate or dislocate during gymnastic routines or activities of daily life. That is, their joints are not unstable and do not need to be “managed” or medicalized. When joints are adequately stabilized by the active system (muscles and tendons) in the presence of adequate proprioception and kinesthesis, such that physiological and accessory motion is controlled, inadequacy of the passive stabilizers (fascia, ligaments, and joint capsules) is disguised. Affected joints become symptomatic (unstable) when the active system fails, which sometimes occurs following injury or deconditioning.
INHERITANCE AND ACQUISITION OF JOINT HYPERMOBILITY
The presentation and distribution of joint hypermobility vary widely between individuals. It may be present as monoarticular (found in a single joint), pauciarticular, or oligoarticular (a few joints), polyarticular (several joints often either central or peripheral, upper or lower limbs), or generalized joint hypermobility (GJH), present in all 4 limbs and the axial skeleton. These forms of hypermobility may be inherited as a normal trait with no identifiable genetic variant or as part of a heritable syndrome such as the Ehlers-Danlos syndromes (EDSs) and other heritable disorders of connective tissue. Joint hypermobility may also be acquired from trauma, joint disease, or training. For example, shoulder range has been shown to be greater in the dominant than nondominant arm of a baseball player.15 Joint hypermobility may also present bilaterally, for example, in both knees where extension range is reported to increase with training.16 There are also subpopulations for whom joint hypermobility is an asset such as dancers, musicians, gymnasts, and contortionists, and it is expected that these will have higher prevalence of GJH than the general population.2,17
The underlying determinants of GJH remain unknown. Given the trait of GJH running in families, it has been traditionally considered to be an autosomal dominant trait,18 most likely related to genes encoding collagen or a collagen-modifying enzyme.19 Candidate genes involved with GJH are, however, broader with joint hypermobility sometimes presenting as one sign of a heritable disorder of connective tissue. For example, one study reported that 5% to 10% of adults with hypermobile EDS demonstrate reduced tenascin-X serum levels.20 This has broadened the field of candidate genes responsible, as tenascin-X is a large extracellular matrix glycoprotein, the first noncollagenous protein identified with GJH in these conditions.19 Fragility of connective tissue, the most abundant tissue in the body, may be evident in tendons, septa, fascia, ligaments, and joint capsules due to deficient structural proteins including collagen, elastin, fibrillin, and tenascin.21 People with heritable disorders of connective tissue with known genetic causes, such as osteogenesis imperfecta,22 and neuromuscular conditions23 commonly present with GJH.
A more nuanced understanding of the spectrum of GJH and other copresenting signs, symptoms, and conditions is required. The complex interplay between an individual's genetic and functional physiological interactions and environmental influences contributes to their phenotypic presentation.24
TRADITIONAL AND NOVEL ASSESSMENTS OF JOINT HYPERMOBILITY
Interest in the clinical measurement of joint hypermobility has grown since the 1960s when Carter and Wilkinson25 published an assessment tool. Others from around the world adapted this assessment tool to include other joints relevant to the population of interest. The Nicholas Scale modified this tool to screen football players,26 and the Rotes-Querol Scale considered additional tests for shoulder, cervical, and lumbar spine mobility.27 Further development saw the Contompasis Score include a more complex grading system of mobility incorporating the ankle,28 whereas the Hospital del Mar Scale incorporated knee and shoulder rotation.29 Modified from the Carter and Wilkinson tool and originally designed to screen for GJH using a single cutoff score, the Beighton score is the most widely used tool30 (Supplemental Table, http://links.lww.com/RHU/A464). Recent research has demonstrated that extents of hypermobility vary and so recommends variable cutoffs for the age, sex, and cohort of interest based on race, occupation, and so on.31,32
More comprehensive peripheral joint hypermobility tools have been developed to provide greater detail and targeted management direction. The Upper Limb Hypermobility Assessment Tool (ULHAT)33 and Lower Limb Assessment Score34,35 are both 12-item tests covering the major joints of the upper and lower limbs, in multiple planes of movement (Supplemental Table, http://links.lww.com/RHU/A464). It should be noted that these tools incorporate a few tests of laxity also (described as passive accessory tests in Supplemental Table, http://links.lww.com/RHU/A464).
There are a few points to consider when using hypermobility assessments. Whereas age- and sex-specific cutoff scores have been determined for the Beighton score (Supplemental Table, http://links.lww.com/RHU/A464), these have not been established for the other tools, nor has any psychometric testing been undertaken to elucidate the key items to determine limb hypermobility. Despite a high interlimb mobility correlation,33–35 there are reports of the nondominant limb being more hypermobile than the dominant36,37 and side-to-side differences due to training effects.38
The 5-part questionnaire is a valid and reliable self-report questionnaire tool that can be used to measure GJH in adults. The tool is particularly useful in adults who have experienced age- and injury-related joint range reduction39 (Supplemental Table, http://links.lww.com/RHU/A464).
STRENGTHS AND LIMITATIONS OF CURRENT JOINT HYPERMOBILITY ASSESSMENT TOOLS
There are a number of tools designed to assess joint range of movement for which reliability has been evaluated.33–35,40 It should be noted that the Beighton score was designed for epidemiological screening purposes not for clinical use.30 An advantage of the Beighton score is that it is quick to perform and requires a goniometer only when joint range is equivocal. Limitations include that it is heavily upper-limb biased and that it incorporates a limited number of joints and assesses motion in only 1 plane of movement (sagittal). Perhaps its most significant limitation is that it does not include the joints that patients most commonly describe as unstable such as the shoulder, foot/ankle, and patellofemoral joints.41,42 While the 5-part Hypermobility Questionnaire has very good sensitivity and specificity for identifying GJH, it has been validated only in adults and does not provide clinicians with information about individual joints. The more recently developed and validated Lower Limb Assessment Score34,35 and the ULHAT33 have the potential to provide clinicians with richer insight into joint range and integrity to inform further functional assessments and interventions. These multidimensional assessments, however, require standardized procedures, skilled handling, and further psychometric testing. These insights into assessment strengths and limitations should prompt clinicians to apply the most appropriate assessment tool suited to the patient's presenting signs and symptoms.
AGE, SEX, AND RACIAL CONSIDERATIONS
Epidemiological studies report a large variation in hypermobility prevalence, depending on the clinical assessment method, cutoff score used, population, physical fitness, age, sex, and race.43 The prevalence of joint hypermobility has been reported as ranging from 5% to 40% in children and 10% to 20% in adults.44,45 Regardless, the prefix “hyper” signifies that the measure is not “normal,” where normal represents the mean and scores within 2 SDs for a population,46,47 factoring in age, sex, and race.
There is significant complexity in the assessment of joint range of motion of infants and children. Infant joint range of motion is dependent on a newborn's gestational age and is challenging to accurately assess because of a lack of bony landmarks and difficulty determining accurate planes of motion.48 Beyond the early years, children typically present with greater joint mobility than adults. No current assessments of GJH have been validated in children younger than 5 years and as such cannot be used to accurately identify GJH in this age range. Where available, age-, gender-, and race-specific norms of individual joints49 can be used to determine hypermobility of a child's individual joints only. Children exhibiting increased joint mobility in multiple individual joints can then be identified. Ongoing monitoring of joint range in children with suspected GJH and any potentially associated symptoms or concerns is recommended throughout the pediatric years before labeling a child with nongenetically confirmed conditions that can be associated with GJH. Although it is well-described that the prevalence of GJH reduces with age it remains unclear which children will continue to demonstrate GJH and which children will “tighten up postpuberty.”
Joint hypermobility is more prevalent in females than in males. Although similar age-related trends of joint hypermobility have been reported between sexes, the decrease in joint mobility after puberty is usually more pronounced among males.45,50 Proposed explanations for these sex-related differences include, but are not limited to, hormonal, anatomical, and neuromuscular differences.51,52
Concerning the racial variation of joint hypermobility, several studies suggest a higher prevalence in people of African, Asian, and Middle Eastern descent compared with White populations.44 Unfortunately, most studies of joint hypermobility do not report on the race of participants. Those that do incept mostly White participants. Consequently, subanalyses to determine the effect of race on joint hypermobility and related signs and symptoms are not possible.
PHENOTYPIC PRESENTATIONS OF JOINT HYPERMOBILITY
Most individuals with hypermobile joints remain asymptomatic throughout their life.7,53 This hypermobility is labeled as “asymptomatic joint hypermobility” if present in otherwise healthy individuals without any associated symptoms or complications. When the hypermobility is polyarticular or generalized, these individuals often describe themselves as “double jointed” and may undertake pursuits for which joint hypermobility is an asset, such as dancing, music, gymnastics, and contortion (Fig. 1, iceberg on the left).
FIGURE 1.
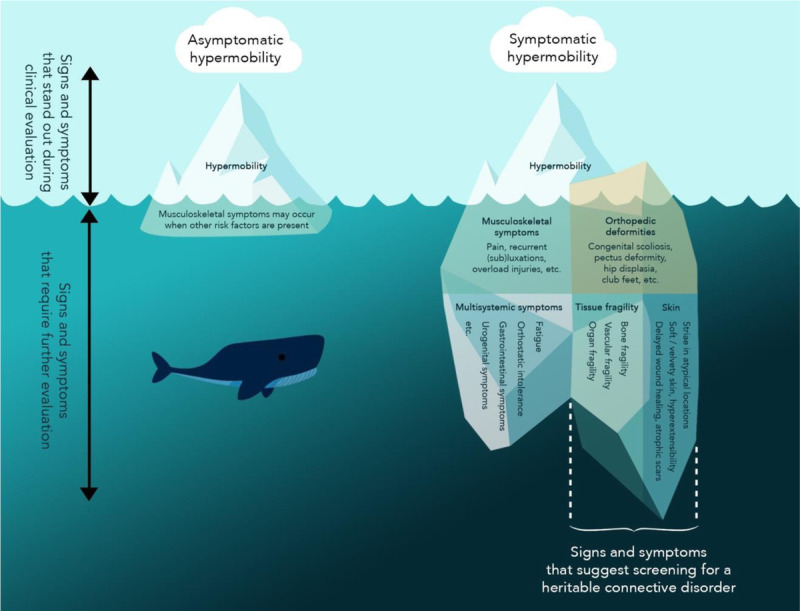
Phenotypic presentation of joint hypermobility. As a useful illustration, joint hypermobility can be compared with “the tip of an iceberg” that is visible above the water surface, as it can be a striking feature that stands out during clinical assessment. Whereas most individuals remain asymptomatic (iceberg on the left), some patients develop symptoms (iceberg on the right). If a hypermobile patient demonstrates orthopedic deformities and skin alterations and reports multiple signs of tissue fragility, ruling out a heritable connective tissue disorder is important (right part of the right iceberg). Color online-figure is available at http://www.jclinrheum.com.
The symptomatic threshold may be reached when multiple risk factors for pain onset are present in, or in proximity to, a hypermobile joint.54,55 These factors include muscle weakness,56 reduced mobility, insufficient musculotendinous length, muscle hypertonicity in proximity to a hypermobile joint, obesity,57 and altered movement patterns.42 If hypermobility is present in combination with pain, recurrent (sub)luxations, and musculoskeletal overload injuries,58 it can be categorized as “symptomatic joint hypermobility” (Fig. 1, left side of symptomatic hypermobility iceberg). A decrease in functional capacity,59 lower isometric strength,56 suboptimal muscle activation strategy, and quality of force control60 have been reported in people with symptomatic joint hypermobility. Despite growing attention in the past decade, researchers and clinicians are still exploring the determinants that cause joint hypermobility to become symptomatic.
The symptoms of hypermobile patients can extend far beyond the confines of the musculoskeletal system,61 as depicted in Figure 1 (right side of symptomatic hypermobility iceberg). Approximately two-thirds of patients with symptomatic hypermobility also report various functional multisystemic symptoms,61 including, but not limited to, gastrointestinal dysfunction, orthostatic intolerance,62 postural orthostatic tachycardia syndrome, urogynecological problems, symptoms of mast cell activation, physical and cognitive fatigue, and anxiety and depression,63–68 constituting a syndromic presentation. At present, there is an association between GJH and many of these conditions, but it remains unknown if the same underlying pathology drives some or all the reported multisystemic features, or if they result from other associated factors such as deconditioning or chronic widespread pain and chronic fatigue, which are nonspecific symptoms.7
Some proposed pathogeneses link joint hypermobility to other multisystemic conditions. For example, orthostatic intolerance may be attributed to an increased vascular distensibility based on altered connective tissue or to peripheral small fiber neuropathy,69 whereas gastrointestional disorders are linked to autonomic dysfunction/dysmotility, interoceptive sensitivity, and/or altered stretch and mechanoreceptor function of the intestines.63 The pathogenesis of an association between mast cell activation and joint hypermobility is yet to be determined.70
The mechanisms behind the complex pain states that can be observed in this latter subgroup of hypermobile individuals are poorly understood.21,71 Subsequent lesions of capsuloligamentous and other soft tissues may occur in conjunction with irritation of peripheral nerves due to compression, elongation, or entrapment,54,57 which are some of the mechanisms proposed. The symptom profile may vary widely between patients as illustrated by the Spider chart (Fig. 2),72 with symptoms presenting on a broad spectrum of severity.73
FIGURE 2.
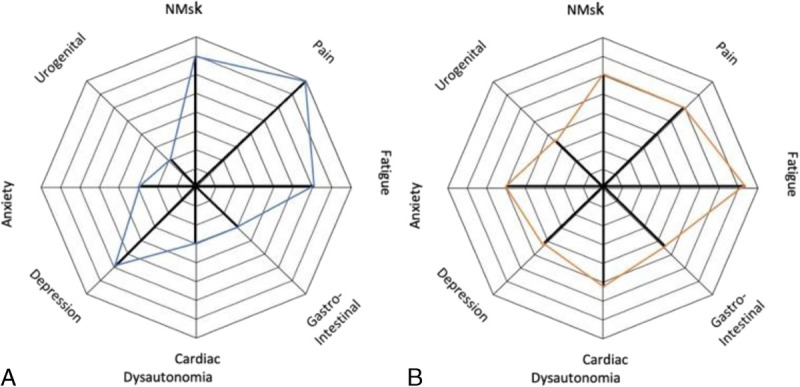
Example of heterogeneous multisystem symptom presentations in 2 patients diagnosed with hEDS. The distance from the center of the web denotes the severity of the symptom. Patient A predominantly experiences neuromuscular symptoms and pain. The symptoms of patient B suggest a more global presentation of symptoms. Color online-figure is available at http://www.jclinrheum.com.
If a hypermobile patient also demonstrates several orthopedic deformities and skin abnormalities and vascular fragility and reports multiple signs of tissue fragility, screening for an underlying heritable condition is warranted to ensure proper medical follow-up and avoid more severe complications. Examples of such pathologies are the heritable disorders of connective tissue such as Marfan syndrome, EDSs, osteogenesis imperfecta, and Stickler syndrome. Several other chromosomal disorders such as Down syndrome and metabolic disorders such as homocystinuria and hyperlysinemia may also need to be excluded as part of the differential diagnosis. Features that alert the clinician to attribute signs and symptoms to a heritable syndrome are evidence of:
Tissue fragility: bone fragility (recurrent fractures), and/or soft tissue fragility (abdominal hernia, bladder, uterine, or bowel prolapse) or organ fragility (rupture of hollow organs). The most important to recognize is vascular fragility (easy bruising, bleeding tendency, vascular aneurysms, dilatations, or dissections) associated with Marfan syndrome, vascular EDS, familial thoracic aortic aneurysm, and so on.
Skin fragility and altered skin structure: soft and velvety skin surface, stretchy skin, skin hyperextensibility, atrophic scarring, the presence of multiple scars, delayed wound healing, wound dehiscence, and so on.
Orthopedic deformities: pectus deformity, congenital club feet, short or tall stature, congenital scoliosis, bilateral congenital hip dysplasia and/or hip dislocations, increased arm span to length ratio, arachnodactyly, and so on.
G-HSD and hEDS
Consistent diagnostic terminology and standardized, valid assessments are important enablers for research, evidence-informed treatments, and communication between health providers and patients. We have presented hypermobility terminology, variable presentations, sign and symptoms of GJH, and how it is assessed. Through this exploration, we have highlighted the diagnostic labels currently adopted.
The previous diagnoses of JHS or EDS–hypermobility type, essentially considered the same condition,6 were revised in 2017. The revised 2017 EDS classification reports that for individuals with otherwise unclassified joint hypermobility, the term hEDS should be restricted to those with features suggestive of a systemic and/or Mendelian connective tissue disorder.74 Based on a population study of health care records in Wales,75 the prevalence of JHS, likely a combination of those currently diagnosed with hEDS and G-HSD, is 1 in 500 to 600.76 Of the 13 types of EDSs, hEDS is the most common accounting for greater than 95% of all EDS presentations to specialist clinics.77 The conditions G-HSD and hEDS share many clinical characteristics, including GJH, joint instability, and widespread chronic pain, all of which are included in the criteria for both diagnoses (https://www.ehlers-danlos.com/heds-diagnostic-checklist). The new diagnostic labels of hEDS and G-HSD have caused some confusion between researchers, clinicians, and those with a lived experience of these conditions, and from a symptom-management perspective, the 2 conditions can be managed identically. The heterogeneous presentation of multisystem signs and symptoms depicted in Figure 2 means that management must be individualized. Most pertinent to management is that thorough history-taking and clinical assessment must combine with equally thorough clinical reasoning, informed by patient expectations and goals.
Currently, the hEDS diagnostic criteria are not valid for use in children and adolescents as multiple multisystemic features that comprise criterion 2 of the 2017 classification are not appropriate in the assessment of children. For example, atrophic scarring cannot be seen in a young child who has not had a skin tear, and dental crowding cannot be determined in a child who has not yet had all adult teeth erupted. In light of these difficulties applying the current criteria to children, the development of pediatric specific criteria is underway led by the Pediatric Working Group of the International Consortium on EDS and HSD. Until that time, children should be monitored to document changes in GJH and other potentially associated multisystemic features until skeletal maturity is reached, at which time a diagnostic label such as hEDS can be reevaluated using diagnostic adult criteria.
Depending on the geographical location, the medical professionals best placed to oversee the multidisciplinary management of patients based on their individual presentation are clinical geneticists, rheumatologists, rehabilitation physicians, or pediatricians (for children/adolescents). These specialists will then confer with the patients' general practitioner and refer on to appropriate allied health or other specialists, and clinics with specific expertise (e.g., chronic pain).
CLINICAL IMPLICATIONS AND RESEARCH DIRECTIONS
The heterogeneity of measurement outcomes reported in the literature has hindered attempts to produce clinical assessment and management guidelines. The EDS Society is currently collating self-report and objective outcome measures (common data elements) including those appropriate for hypermobility assessment to standardize their use in this population globally.
While joint hypermobility is most commonly asymptomatic warranting identification only for risk surveillance and management, task-specific injury prevention, or performance enhancement,78 this form of hypermobility does not need to be medicalized. In these cases, the Beighton score functions well to screen. However, in those who experience symptoms associated with their joint hypermobility, more extensive assessment to determine the joint(s) and plane(s) of movement affected together with patient-specific and objective functional assessments will permit targeted management. For example, a patient who describes shoulder instability and tests positive to all 3 ULHAT shoulder tests (Supplemental Table, http://links.lww.com/RHU/A464) and demonstrates functional movement impairments may best be managed with exercises that improve strength and control in more than 1 anatomical plane.
Although focusing on a diagnosis is important, from a clinical/management perspective, the focus should be on the patient's individual symptom presentation. This review highlights the strengths and limitations of many of the contemporary assessment methods and prompts clinicians to consider how these assessments add value or align with the presenting complaints or treatment goals of the patient. It is possible that those diagnosed with G-HSD are undermanaged because of the perception that their condition is less severe than hEDS. Large, cross-sectional and prospective studies are needed to determine whether there are phenotypic differences between the 2 cohorts if they exist and whether they should be managed similarly.
Finally, future research is warranted to validate diagnostic criteria, self-report, and physical measures in the pediatric population. Such research will need to incorporate standardized outcome measures on homogenous patient cohorts to enable the development of best-practice guidelines for children with symptomatic hypermobility.
CONCLUSIONS
Joint hypermobility presents on a spectrum from an asymptomatic physiological phenomenon through to 1 component of a complex myriad of multisystem presentations. This article has presented the current state of the science and research relating to the terminology, etiology, presentation, assessment, diagnosis, and future directions for research.
KEY POINTS
Using standardized terminology about joint hypermobility will assist researchers to compare findings related to prevalence, diagnostics, and management.
Joint hypermobility presents on a spectrum from an asymptomatic physiological phenomenon through to 1 component of multisystem presentations.
Whereas joint hypermobility is often asymptomatic, symptomatic forms require specialized assessment, management, and follow-up.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the entire EDS Society and the International Consortium on EDS and HSD for their support of this review project and specifically Dr Alan Hakim, consultant rheumatologist, London, United Kingdom; Dr Fransiska Malfait, geneticist, Ghent University Hospital, Belgium; and Dr Louise Tofts, pediatric rehabilitation specialist, The Children's Hospital at Westmead, Sydney, Australia; for their valuable contribution to the review of this article. This review was written on behalf of the Allied Health Professionals Working Group of the International Consortium on EDS and HSD (https://www.ehlers-danlos.com/international-consortium-working-groups/#allied-health-working-group). They also acknowledge all the clinicians and researchers who have invested their energies to improve knowledge in this field and to improve diagnosis and management for people with symptomatic joint hypermobility and whose work is included and reflected in this review article.
Footnotes
Open access funding of this article is supported by the EDS International Consortium.
The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s Web site (www.jclinrheum.com).
Contributor Information
Jane Simmonds, Email: jane.simmonds@ucl.ac.uk.
Verity Pacey, Email: verity.pacey@mq.edu.au.
Inge De Wandele, Email: inge.dewandele@ugent.be.
Lies Rombaut, Email: lies.rombaut@ugent.be.
Cylie M. Williams, Email: cylie.williams@monash.edu.
Cliffton Chan, Email: cliffton.chan@sydney.edu.au.
REFERENCES
- 1.Adams F. On the Articulations. Glasgow Scotland UK: Good Press; 2021. [Google Scholar]
- 2.Van Dongen PW. Hypermobility in reproduction. S Afr J Obstet Gynaecol. 2006;12:38–43. [Google Scholar]
- 3.Kirk JA, Ansell BM, Bywaters EG. The hypermobility syndrome. Musculoskeletal complaints associated with generalized joint hypermobility. Ann Rheum Dis. 1967;26:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denko C. Chernogubov's syndrome: a translation of the first modern case report of the Ehlers-Danlos syndrome. J Rheumatol. 1978;5:347–352. [PubMed] [Google Scholar]
- 5.Finkelstein H. Joint hypotonia. NY Med J. 1916;104:942–943. [Google Scholar]
- 6.Tinkle BT Bird HA Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A:2368–2370. [DOI] [PubMed] [Google Scholar]
- 7.Castori M Tinkle B Levy H, et al. A framework for the classification of joint hypermobility and related conditions. Am J Med Genet C Semin Med Genet. 2017;175:148–157. [DOI] [PubMed] [Google Scholar]
- 8.Ochsner EH. IV. Congenital dislocation of hips with report of cases and description of a pelvis obtained three years after successful reduction by the Lorenz method. Ann Surg. 1902;36:198–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whiteley R Ginn K Nicholson L, et al. Indirect ultrasound measurement of humeral torsion in adolescent baseball players and non-athletic adults: reliability and significance. J Sci Med Sport. 2006;9:310–318. [DOI] [PubMed] [Google Scholar]
- 10.Molleson T. The case for signs of joint hypermobility on disarticulated human bones. Bioarchaeol Near East. 2018;12:33–45. [Google Scholar]
- 11.Kanazawa K Hagiwara Y Kawai N, et al. Correlations of coracohumeral ligament and range of motion restriction in patients with recurrent anterior glenohumeral instability evaluated by magnetic resonance arthrography. J Shoulder Elbow Surg. 2017;26:233–240. [DOI] [PubMed] [Google Scholar]
- 12.MacConaill MA. The movements of bones and joints. V. The significance of shape. J Bone Joint Surg Br. 1953;35-B:290–297. [DOI] [PubMed] [Google Scholar]
- 13.Chen K Zhu W Zheng Y, et al. A retrospective study to compare the clinical effects of individualized anatomic single- and double-bundle anterior cruciate ligament reconstruction surgery. Sci Rep. 2020;10:14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerometta A Klouche S Herman S, et al. The Shoulder Instability—Return to Sport After Injury (SIRSI): a valid and reproducible scale to quantify psychological readiness to return to sport after traumatic shoulder instability. Knee Surg Sports Traumatol Arthrosc. 2018;26:203–211. [DOI] [PubMed] [Google Scholar]
- 15.Greenberg EM Lawrence JT Fernandez-Fernandez A, et al. Humeral retrotorsion and glenohumeral motion in youth baseball players compared with age-matched nonthrowing athletes. Am J Sports Med. 2017;45:454–461. [DOI] [PubMed] [Google Scholar]
- 16.Hahn T Foldspang A Vestergaard E, et al. Active knee joint flexibility and sports activity. Scand J Med Sci Sports. 1999;9:74–80. [DOI] [PubMed] [Google Scholar]
- 17.Vera AM Peterson LE Dong D, et al. High prevalence of connective tissue gene variants in professional ballet. Am J Sports Med. 2020;48:222–228. [DOI] [PubMed] [Google Scholar]
- 18.Hakim AJ Cherkas LF Grahame R, et al. The genetic epidemiology of joint hypermobility: a population study of female twins. Arthritis Rheum. 2004;50:2640–2644. [DOI] [PubMed] [Google Scholar]
- 19.Malfait F Hakim AJ De Paepe A, et al. The genetic basis of the joint hypermobility syndromes. Rheumatology (Oxford). 2006;45:502–507. [DOI] [PubMed] [Google Scholar]
- 20.Zweers MC, Kucharekova M, Schalkwijk J. Tenascin-X: a candidate gene for benign joint hypermobility syndrome and hypermobility type Ehlers-Danlos syndrome? Ann Rheum Dis. 2005;64:504–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Syx D De Wandele I Rombaut L, et al. Hypermobility, the Ehlers-Danlos syndromes and chronic pain. Clin Exp Rheumatol. 2017;35 Suppl 107:116–122. [PubMed] [Google Scholar]
- 22.Paterson CR, McAllion S, Miller R. Heterogeneity of osteogenesis imperfecta type I. J Med Genet. 1983;20:203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voermans NC Bonnemann CG Hamel BC, et al. Joint hypermobility as a distinctive feature in the differential diagnosis of myopathies. J Neurol. 2009;256:13–27. [DOI] [PubMed] [Google Scholar]
- 24.Castori M. Deconstructing and reconstructing joint hypermobility on an evo-devo perspective. Rheumatology. 2021;60:2537–2544. [DOI] [PubMed] [Google Scholar]
- 25.Carter C, Wilkinson J. Persistent joint laxity and congenital dislocation of the hip. J Bone Joint Surg Br. 1964;46:40–45. [PubMed] [Google Scholar]
- 26.Nicholas JA. Injuries to knee ligaments. Relationship to looseness and tightness in football players. JAMA. 1970;212:2236–2239. [DOI] [PubMed] [Google Scholar]
- 27.Rotes-Quérol J Duran J Subiros R, et al. La laxité articulaire comme facteur d'alterations de l'appareil locomoteur (Nouvelle étude 1971). Rhumatol Mai. 1972;24:179–191. [PubMed] [Google Scholar]
- 28.McNerney JE, Johnston WB. Generalized ligamentous laxity, hallux abducto valgus and the first metatarsocuneiform joint. J Am Podiatry Assoc. 1979;69:69–82. [DOI] [PubMed] [Google Scholar]
- 29.Bulbena A Duro J Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19:115–122. [PubMed] [Google Scholar]
- 30.Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis. 1973;32:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh H McKay M Baldwin J, et al. Beighton scores and cut-offs across the lifespan: cross-sectional study of an Australian population. Rheumatology. 2017;56:1857–1864. [DOI] [PubMed] [Google Scholar]
- 32.Chan C Hopper L Zhang F, et al. The prevalence of generalized and syndromic hypermobility in elite Australian dancers. Phys Ther Sport. 2018;32:15–21. [DOI] [PubMed] [Google Scholar]
- 33.Nicholson LL, Chan C. The upper limb hypermobility assessment tool: a novel validated measure of adult joint mobility. Musculoskelet Sci Pract. 2018;35:38–45. [DOI] [PubMed] [Google Scholar]
- 34.Ferrari J Parslow C Lim EJ, et al. Joint hypermobility: the use of a new assessment tool to measure lower limb hypermobility. Clin Exp Rheumatol. 2005;23:413–420. [PubMed] [Google Scholar]
- 35.Meyer KJ Chan C Hopper L, et al. Identifying lower limb specific and generalised joint hypermobility in adults: validation of the lower limb assessment score. BMC Musculoskelet Disord. 2017;18:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El-Garf AK, Mahmoud GA, Mahgoub EH. Hypermobility among Egyptian children: prevalence and features. J Rheumatol. 1998;25:1003–1005. [PubMed] [Google Scholar]
- 37.Verhoeven JJ, Tuinman M, Van Dongen PW. Joint hypermobility in African non-pregnant nulliparous women. Eur J Obstet Gynecol Reprod Biol. 1999;82:69–72. [DOI] [PubMed] [Google Scholar]
- 38.Phan K Nicholson LL Hiller CE, et al. Prevalence and unique patterns of lower limb hypermobility in elite ballet dancers. Phys Ther Sport. 2020;41:55–63. [DOI] [PubMed] [Google Scholar]
- 39.Hakim A, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57:163–166. [PubMed] [Google Scholar]
- 40.Juul-Kristensen B Schmedling K Rombaut L, et al. Measurement properties of clinical assessment methods for classifying generalized joint hypermobility—a systematic review. Am J Med Genet C Semin Med Genet. 2017;175:116–147. [DOI] [PubMed] [Google Scholar]
- 41.Pacey V Tofts L Adams RD, et al. Quality of life prediction in children with joint hypermobility syndrome. J Paediatr Child Health. 2015;51:689–695. [DOI] [PubMed] [Google Scholar]
- 42.Spanhove V Van Daele M Van den Abeele A, et al. Muscle activity and scapular kinematics in individuals with multidirectional shoulder instability: a systematic review. Ann Phys Rehabil Med. 2020;107:11–18. [DOI] [PubMed] [Google Scholar]
- 43.Remvig L, Jensen DV, Ward RC. Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: review of the literature. J Rheumatol. 2007;34:804–809. [PubMed] [Google Scholar]
- 44.Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17:989–1004. [DOI] [PubMed] [Google Scholar]
- 45.Quatman CE Ford KR Myer GD, et al. The effects of gender and pubertal status on generalized joint laxity in young athletes. J Sci Med Sport. 2008;11:257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Surgeons AAoO . Joint Motion: Method of Measuring and Recording. Edinburgh and London UK: Churchill Livingstone; 1965. [Google Scholar]
- 47.Fairbank JC, Pynsent PB, Phillips H. Quantitative measurements of joint mobility in adolescents. Ann Rheum Dis. 1984;43:288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mondal R Nandy A Datta D, et al. Newborn joint mechanics. J Matern Fetal Neonatal Med. 2021;1–8. [DOI] [PubMed] [Google Scholar]
- 49.Cahill SV, Sharkey MS, Carter CW. Revisiting the Beighton criteria: does ligamentous laxity testing correlate with shoulder range of motion norms in a North American, pediatric population? J Pediatr Orthop. 2020;40:536–542. [DOI] [PubMed] [Google Scholar]
- 50.Nicholson LL McKay MJ Baldwin JN, et al. Is there a relationship between sagittal cervical spine mobility and generalised joint hypermobility? A cross-sectional study of 1000 healthy Australians. Physiotherapy. 2021;112:150–157. [DOI] [PubMed] [Google Scholar]
- 51.Graf C Schierz O Steinke H, et al. Sex hormones in association with general joint laxity and hypermobility in the temporomandibular joint in adolescents—results of the epidemiologic LIFE child study. J Oral Rehabil. 2019;46:1023–1030. [DOI] [PubMed] [Google Scholar]
- 52.Jansson A Saartok T Werner S, et al. General joint laxity in 1845 Swedish school children of different ages: age- and gender-specific distributions. Acta Paediatr. 2004;93:1202–1206. [DOI] [PubMed] [Google Scholar]
- 53.Malfait F Francomano C Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26. [DOI] [PubMed] [Google Scholar]
- 54.Mulvey MR Macfarlane GJ Beasley M, et al. Modest association of joint hypermobility with disabling and limiting musculoskeletal pain: results from a large-scale general population–based survey. Arthritis Care Res. 2013;65:1325–1333. [DOI] [PubMed] [Google Scholar]
- 55.Cattalini M, Khubchandani R, Cimaz R. When flexibility is not necessarily a virtue: a review of hypermobility syndromes and chronic or recurrent musculoskeletal pain in children. Pediatr Rheumatol Online J. 2015;13:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jindal P Narayan A Ganesan S, et al. Muscle strength differences in healthy young adults with and without generalized joint hypermobility: a cross-sectional study. BMC Sports Sci Med Rehabil. 2016;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tobias JH Deere K Palmer S, et al. Joint hypermobility is a risk factor for musculoskeletal pain during adolescence: findings of a prospective cohort study. Arthritis Rheum. 2013;65:1107–1115. [DOI] [PubMed] [Google Scholar]
- 58.Bin Abd Razak HR, Bin Ali N, Howe TS. Generalized ligamentous laxity may be a predisposing factor for musculoskeletal injuries. J Sci Med Sport. 2014;17:474–478. [DOI] [PubMed] [Google Scholar]
- 59.Scheper MC de Vries JE Juul-Kristensen B, et al. The functional consequences of generalized joint hypermobility: a cross-sectional study. BMC Musculoskelet Disord. 2014;15:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jensen BR Olesen AT Pedersen MT, et al. Effect of generalized joint hypermobility on knee function and muscle activation in children and adults. Muscle Nerve. 2013;48:762–769. [DOI] [PubMed] [Google Scholar]
- 61.Hakim A, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology. 2004;43:1194–1195. [DOI] [PubMed] [Google Scholar]
- 62.Peebles KC Tan I Butlin M, et al. The prevalence and impact of orthostatic intolerance in young women across the hypermobility spectrum [published online February 27, 2022]. Am J Med Genet A. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beckers AB Keszthelyi D Fikree A, et al. Gastrointestinal disorders in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: a review for the gastroenterologist. Neurogastroenterol Motil. 2017;29:e13013. [DOI] [PubMed] [Google Scholar]
- 64.Celletti C Camerota F Castori M, et al. Orthostatic intolerance and postural orthostatic tachycardia syndrome in joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type: neurovegetative dysregulation or autonomic failure? Biomed Res Int. 2017;2017:9161865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gilliam E, Hoffman JD, Yeh G. Urogenital and pelvic complications in the Ehlers-Danlos syndromes and associated hypermobility spectrum disorders: a scoping review. Clin Genet. 2020;97:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krahe AM, Adams RD, Nicholson LL. Features that exacerbate fatigue severity in joint hypermobility syndrome/Ehlers-Danlos syndrome–hypermobility type. Disabil Rehabil. 2018;40:1989–1996. [DOI] [PubMed] [Google Scholar]
- 67.Sinibaldi L, Ursini G, Castori M. Psychopathological manifestations of joint hypermobility and joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type: the link between connective tissue and psychological distress revised. Am J Med Genet C Semin Med Genet. 2015;169:97–106. [DOI] [PubMed] [Google Scholar]
- 68.Chang AR, Vadas P. Prevalence of symptoms of mast cell activation in patients with postural orthostatic tachycardia syndrome and hypermobile Ehlers-Danlos syndrome. J Allergy Clin Immunol. 2019;143:AB182. [Google Scholar]
- 69.De Wandele I Rombaut L Leybaert L, et al. Dysautonomia and its underlying mechanisms in the hypermobility type of Ehlers-Danlos syndrome. Semin Arthritis Rheum. 2014;44:93–100. [DOI] [PubMed] [Google Scholar]
- 70.Kohn A, Chang C. The relationship between hypermobile Ehlers-Danlos syndrome (hEDS), postural orthostatic tachycardia syndrome (POTS), and mast cell activation syndrome (MCAS). Clin Rev Allergy Immunol. 2020;58:273–297. [DOI] [PubMed] [Google Scholar]
- 71.Malfait F Colman M Vroman R, et al. Pain in the Ehlers-Danlos syndromes: mechanisms, models, and challenges. Am J Med Genet C Semin Med Genet. 2021;187:429–445. [DOI] [PubMed] [Google Scholar]
- 72.Simmonds JV. Advances in assessment of hypermobility-related disorders. Am J Med Genet C Semin Med Genet. 2021;187:453–457. [DOI] [PubMed] [Google Scholar]
- 73.Russek LN, Stott P, Simmonds J. Recognizing and effectively managing hypermobility-related conditions. Phys Ther. 2019;99:1189–1200. [DOI] [PubMed] [Google Scholar]
- 74.Malfait F Castori M Francomano CA, et al. The Ehlers-Danlos syndromes. Nat Rev Dis Primers. 2020;6:64. [DOI] [PubMed] [Google Scholar]
- 75.Demmler JC Atkinson MD Reinhold EJ, et al. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: a national electronic cohort study and case-control comparison. BMJ Open. 2019;9:e031365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hakim AJ, Tinkle BT, Francomano CA. Ehlers-Danlos syndromes, hypermobility spectrum disorders, and associated co-morbidities: reports from EDS ECHO. Am J Med Genet C Semin Med Genet. 2021;187:413–415. [DOI] [PubMed] [Google Scholar]
- 77.Conaghan P Denton C Foster H, et al. Oxford Textbook of Rheumatology. Edinburgh and London UK: Oxford University Press; 2013. [Google Scholar]
- 78.Pacey V Nicholson LL Adams RD, et al. Generalized joint hypermobility and risk of lower limb joint injury during sport: a systematic review with meta-analysis. Am J Sports Med. 2010;38:1487–1497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.