

Abstract
Objective:
Phosphomannomutase 2 deficiency (PMM2-CDG) is a disorder of protein N-glycosylation with a wide clinical spectrum. Hypoglycemia is rarely reported in PMM2-CDG. In this study, we evaluated cause, treatment options and outcomes in cases with hypoglycemia in the course of PMM2-CDG.
Methods:
Clinical records of patients followed with PMM2-CDG within the last two decades were reviewed. Medical data of patients with hypoglycemia were evaluated in more detail. Demographic and clinical findings, organ involvement and laboratory investigations at time of hypoglycemia were recorded. Time of first attack of hypoglycemia, cause, treatment modalities, duration of hypoglycemia (permanent/transient), and duration of treatment, as well as outcome were also recorded. Other published cases with PMM2-CDG and hypoglycemia are also reviewed in order to elucidate characteristics as well as pathophysiology of hypoglycemia.
Results:
Nine patients with PMM2-CDG were reviewed, and hypoglycemia was present in three cases. All three had hyperinsulinism as the cause of hypoglycemia. In the first two cases reported here, serum insulin level concurrent with hypoglycemic episodes was elevated, and glucose response was exaggerated during glucagon test, favoring hyperinsulinism. However, in the third case, the serum insulin level at time of hypoglycemia was not so high but hypoglycemia responded well to diazoxide. Hyperinsulinism was permanent in two of these three cases. No genotype-phenotype correlation was observed with respect to hyperinsulinism.
Conclusion:
The main cause of hypoglycemia in PMM2-CDG appears to be hyperinsulinism. Although insulin levels at the time of hypoglycemia may not be very high, hypoglycemia in patients with PMM2 responds well to diazoxide.
Keywords: Congenital disorders of glycosylation, diazoxide, hyperinsulinism, hypoglycemia, phosphomannomutase 2 deficiency
What is already known on this topic?
Phosphomannomutase 2 deficiency (PMM2-CDG) is an autosomal recessive disorder of protein N-glycosylation with a wide clinical spectrum ranging from life-threatening early-onset multisystem disease to milder phenotypes with isolated neurological involvement. PMM2-CDG is the most common form of congenital disorders of glycosylation.
What this study adds?
Hypoglycemia is a rare finding in PMM2-CDG, reported in 25 of 1064 patients in the literature (including the three new patients reported here). Half (13/25) of the cases have documented hyperinsulinism which was permanent in two thirds (9/13). Hyperinsulinemic hypoglycemia is usually a part of multisystemic disease with organ involvement. Although insulin levels taken at the time of hypoglycemia may not be very high, hypoglycemia seen in patients with PMM2 still responds well to diazoxide treatment.
Introduction
Phosphomannomutase 2 (PMM2) deficiency (PMM2-CDG, formerly named CDG-Ia, OMIM: #212065), which is by far the most common form of congenital disorders of glycosylation (CDG), is an autosomal recessive disorder of protein N-glycosylation with an estimated incidence of 1:20000 (1). It has a wide clinical spectrum, ranging from life-threatening early-onset multisystem disease to milder phenotypes with isolated neurological involvement. Clinical findings are quite variable, the most common findings being developmental delay, hypotonia and growth retardation (1). During infancy, neurological findings such as strabismus, abnormal eye movements, psychomotor retardation, cerebellar hypoplasia, and hypotonia and hyporeflexia, as well as multisystemic involvement including hepatic disease, chronic diarrhea, cystic renal dysplasia, nephrotic syndrome, hypertrophic cardiomyopathy and pericardial effusion may be observed (2,3,4,5,6). Approximately 20% of cases with visceral involvement die in the first year of life due to multiorgan failure (2,7,8,9,10,11). In addition, dysmorphic findings such as inverted nipples, abnormal distribution of adipose tissue in the supragluteal and suprapubic regions as well as facial dysmorphism, including large and dysplastic ears, are defined in PMM2-CDG (2,12,13,14,15,16,17). During childhood, neurological findings include developmental delay, ataxia, stroke-like episodes, seizures and peripheral neuropathy, as well as retinitis pigmentosa (5). Finally, adults with PMM2-CDG show stable intellectual impairment, stable or progressive ataxia, peripheral neuropathy, progressive kyphoscoliosis and osteoporosis. In addition, the risk of thrombosis is increased due to a decrease in serum levels of several anti-coagulation factors (13).
Most transport proteins and regulators involved in hormonal control are glycosylated so that the endocrine system is one of the main organ systems affected in PMM2-CDG (2). Four functions are especially affected: growth, thyroid function, sexual development and glucose regulation. Glycosylated proteins related to the endocrine system include insulin-like growth factor receptor-binding protein 3, thyroxin binding globulin (TBG), thyroid-stimulating hormone (TSH), prolactin (PRL), follicle-stimulating hormone, and luteinizing hormone. To date, endocrine disorders reported in PMM2-CDG include hyperprolactinemia, short stature despite increased growth hormone release, and hypo- or hyper-gonadotropic hypogonadism (18,19,20,21). Although clinical hypothyroidism is rare, a decrease in TBG and a temporary increase in TSH can be seen in cases with PMM2-CDG (22). Hypoglycemia is also rarely reported, since glycosylated proteins are also involved in glucose homeostasis. In a number of CDGs, including PMM2-CDG, hyperinsulinemic hypoglycemia has been reported. Other forms associated with hyperinsulinemic hypoglycemia are phosphomannose isomerase deficiency (MPI-CDG, CDG1b), phosphoglucomutase 1 (PGM1) deficiency (PGM1-CDG, CDG1t), and alpha-1,3-mannosyltransferase deficiency (ALG3-CDG, CDG1d) (23,24,25). However, data about hyperinsulinemic hypoglycemia in PMM2-CDG is quite sparse.
There is only one recently published systematic review that has collected PMM2-CDG cases with hypoglycemia (26). In this review, it is mentioned that only 23 (2.5%) of 933 cases had hypoglycemia and hyperinsulinemic hypoglycemia was reported as the underlying cause in about half of these cases (10/23). Among the patients with hyperinsulinemic hypoglycemia, clinical data of only seven cases who responded to diazoxide treatment were given. In the remaining 16 cases, neither the cause of hypoglycemia nor the results of the etiological evaluation were reported. There is lack of data in most of the papers mentioning hypoglycemia and little is known about the possible causes, treatment options and consequences of hypoglycemia in PMM2-CDG.
In this study, the cause, treatment options and outcomes in cases with hypoglycemia during the course of PMM2-CDG were evaluated. Other cases with PMM2-CDG and hypoglycemia from the literature are also reviewed in order to elucidate features and characteristics, as well as the pathophysiology of hypoglycemia in PMM2-CDG.
Methods
Clinical records of patients followed with PMM2-CDG within the last two decades at one of the largest tertiary medical centers for inborn errors of metabolism in Turkey, were reviewed retrospectively for demographic, clinical, laboratory, imaging and molecular genetic findings. All patients were diagnosed by type 1 pattern on capillary zone electrophoresis of serum sialotransferrins (decreased tetrasialo- and increased disialo- and asialo-transferrin), followed by Sanger sequencing of the PMM2 gene. Medical data of patients with hypoglycemia were evaluated in more detail. The demographic and clinical findings including sex, age at onset of the disease, age at diagnosis, age at the last follow-up, organ involvements, and specific findings of the organ systems, as well as the results of the laboratory examinations at the time of hypoglycemia including serum glucose, insulin, cortisol, growth hormone levels, and urinary ketones were recorded. Time of the first attack of hypoglycemia, cause, treatment modalities, duration of hypoglycemia (permanent/transient), duration of treatment, and outcome were also recorded. If the patient died, the age and cause of death were documented. The NCBI reference sequence NM_000303.3 was used for reporting PMM2 gene variants.
We also performed a literature search in PubMed using the following search terms: carbohydrate-deficient glycoprotein syndrome OR CDG-Ia OR CDG type Ia OR PMM2 deficiency OR PMM2-CDG OR PMM2-CDG AND hypoglycemia OR hyperinsulinism OR hyperinsulinemia. We searched the literature from inception to the end of December 2020. Only the PMM2-CDG cases confirmed by enzymatic or molecular genetic analysis and who were reported to have hypoglycemia were included in the review.
Statistical Analysis
Data analyses were performed using Statistical Package for the Social Sciences (SPSS) for Windows, version 22.0 (SPSS Inc., Chicago, IL, USA). Normality was tested using the Shapiro-Wilk test. Continuous data are reported as mean±standard deviation and categorical data are reported as the number of cases (%). Statistical differences between two independent groups were compared by Student’s t-test. The differences among more than two independent groups were analyzed using one-way ANOVA.
Results
Nine patients who had been diagnosed with PMM2-CDG were included in the study, eight of whom had been previously reported by Yıldız et al. (27). Hypoglycemia was present in three cases.
Case 1
The male patient was referred at the age of 5 months because of chronic diarrhea, vomiting, seizures and failure to thrive. He was born at the 36th week of gestation as the second child of healthy, non-consanguineous parents with a birth weight of 2.8 kg [0.2 standard deviation score (SDS)]. He was hospitalized in the neonatal intensive care unit due to prematurity, respiratory distress syndrome and neonatal hypoglycemia. He had mild hypothyroxinemia and hyperthyrotropinemia [fT4: 11.2 pmol/L (N: 12-22), TSH: 8.7 µIU/mL (N: 0.27-4.2)], and was started on Na-L-thyroxine. At the time of referral, weight was 4 kg (-4.1 SDS, when corrected for gestastional age -3.5 SDS), height 59 cm (-2.6 SDS, -1.7 SDS when corrected for gestastional age) and head circumference 37 cm (-4.2 SDS, -3.5 SDS when corrected for gestastional age). He had dysmorphic facies, microcephaly, sensorineural hearing loss, inverted nipples, abnormal fat distribution, axial hypotonia and a hepatomegaly of 2 cm below the costal margin. In addition to feeding difficulties, he had vomiting, and chronic diarrhea. Investigations revealed hypoalbuminaemia, hypertransaminasemia, hypertriglyceridemia, ascites, increased renal echogenicity, pericardial effusion and cardiac hypertrophy. He had low blood glucose levels (<50 mg/dL) and an intravenous (iv) glucose infusion was given at a rate of 6-10 mg/kg/min. Hypoglycemia recurred when the glucose infusion rate was reduced. A critical sample during hypoglycemia (serum glucose 46 mg/dL) revealed elevated insulin and c peptide levels [6.0 µIU/mL (N: <1), and 9 ng/mL (N: <0.9) respectively] suggesting hyperinsulinemia. Simultaneous cortisol (35 µg/dL, N: >20) and growth hormone (19.6 ng/mL, N: >10) excluded adrenal insufficiency and growth hormone deficiency, respectively. Lactic acid (12 mg/dL; N: 4.5-19.8) and ammonia (46 µg/dL, N: 20-120) were normal, while urinary ketones were negative. Tandem mass spectrometry, plasma and urinary amino acid profiles, and urinary organic acid analyses were normal. A glucagon test during hypoglycemia showed an exaggerated glucose response (0, 15 and 30 min glucose levels: 35, 70, and 85 mg/dL, respectively) supporting hyperinsulinism. The patient died due to complications of epilepsia partialis continua before diazoxide could be initiated. Transferrin isoform electrophoresis for a preliminary diagnosis of CDG was abnormal with a type 1 pattern.
Case 2
The female patient was referred to our hospital at the age of 8 months with the complaints of poor feeding, failure to thrive and strabismus. She was born at term as the first child of healthy, non-consanguineous parents with a birth weight of 2.7 kg (-1.3 SDS). Her parents noticed failure to thrive and strabismus when she was 2 months old, however a specific diagnosis was not available. On admission to our hospital, she was underweight (6 kg, -2.5 SDS) with short stature (63 cm, -1.9 SDS) and microcephaly (41 cm, -2.0 SDS). She had abnormal eye movements, dysmorphic facies, inverted nipples, supragluteal fat pads, orange peel skin, axial hypotonia, and a hepatomegaly of 3 cm below the costal margin. Ophthalmological examination revealed internal strabismus and normal fundoscopy. There was no history of seizures, but she had hypotonia, hyporeflexia and developmental delay. She was fed by nasogastric feeding since she had swallowing difficulties. Feeding intolerance due to abdominal distention, and respiratory distress due to transient pleural effusion limited her oral and enteral intake. Biochemical investigations revealed hypoalbuminaemia, hypertransaminasemia, and hypertriglyceridemia, low high density lipoprotein (HDL) levels, and hypocholesterolemia. She had severe hypoglycemia (blood glucose <40 mg/dL) and iv glucose infusion was administered at a rate of 8-12 mg/kg/min. Again, a critical blood samples taken during an episode of hypoglycemia (serum glucose was 36 mg/dL) revealed an insulin of 3.1 µIU/mL (N: <1), c-peptide 1 ng/mL (N: <0.9), cortisol 6.19 µg/dL (N>20), adrenocorticotropin hormone (ACTH) 25.8 pg/mL (N: 0-46), growth hormone 8.01 ng/mL (N>10), lactic acid 14 mg/dL (N: 4.5-19.8), ammonia 33 µg/dL (N: 20-120), and urinary ketone was negative. Tandem mass spectrometry, plasma and urinary amino acid profiles, and urinary organic acid analyses were normal. Low dose ACTH stimulation was normal, ruling out central adrenal insufficiency as the cause of hypoglycemia. Hyperinsulinism was considered, since the levels of insulin and c-peptide were not suppressed appropriately during hypoglycemia, and an exaggerated glucose response was observed during glucagon test (0, 15 and 30 min glucose levels: 36, 84, and 95 mg/dL, respectively). The patient was started on diazoxide at a dose of 10 mg/kg/day, and experienced no new episodes of hypoglycemia after diazoxide.
Further analyses that led to the diagnosis of PMM2-CDG were: prolonged prothrombin and activated prothrombin times; decreased antithrombin 3 and factor 11 levels; ascites; increased myocardial, hepatic and renal echogenicity; proteinuria; pleural and pericardial effusion; cardiac hypertrophy; cerebellar atrophy and diffuse volume loss in the brainstem. Minimally elevated serum TSH (5.6 µIU/mL) with normal free T4 level was detected in the examinations performed in terms of other endocrinological disorders that may accompany PMM2-CDG. Serum PRL level was normal. Serum IGF1 and IGBP3 levels were 5.6 ng/mL (-4.5 SDS) and 1530.8 ng/mL (-0.1 SDS), respectively. Transferrin isoform electrophoresis was abnormal with a type 1 pattern.
Around the time of discharge, her feeding improved and she was able to tolerate more enteral feeds, allowing the tapering and discontinuation of diazoxide after six months. At the age of four years she has global, but stable developmental delay. She requires intermittent transfusions for thrombocytopenia. Transaminase levels are elevated without clinical hepatic manifestations. Glucose levels are stable with frequent meals and continuous enteral feeding at night, without need for diazoxide.
Case 3
The male patient was referred to our hospital at the age of 2 months with the complaints of chronic diarrhea, vomiting, and failure to thrive. He was born at the 36th week of gestation as the second child of healthy, non-consanguineous parents with a birth weight of 2.1 kg (-2.3 SDS). He was hospitalized for 18 days due to prematurity, pneumonia and meconium aspiration syndrome. In follow-up he was diagnosed with primary congenital hypothyroidism and Na-L-thyroxine treatment was started [fT4: 6.1 pmol/L (N: 12-22), TSH: 100 µIU/mL (N: 0.27-4.2)]. He also had an afebrile seizure in the neonatal period but the etiology was not known. On admission at the age of 2 months, he was 3.1 kg (-3.3 SDS, when corrected for gestastional age -2.0 SDS) in weight, 50 cm (-3.4 SDS, when corrected for gestastional age -1.9 SDS) in height and had a head circumference of 35 cm (-2.8 SDS, when corrected for gestastional age -1.6 SDS). He had dysmorphic facies, microcephaly, sensorineural hearing loss, inverted nipples, generalized edema and hypotonia. The eye examination was normal. He had anemia with a normal thrombocyte count. He also had hypoglycemia, hypoalbuminaemia, proteinuria, hypertransaminasemia, hypocholesterolemia, decreased levels of coagulation and anti-coagulation factors, increased renal echogenicity, and mild right ventricular hypertrophy. In subsequent hospitalizations, he developed pericardial effusion necessitating pericardiocentesis, and deep venous thrombosis at sites of central venous catheters.
He had hypoglycemia and an iv glucose infusion was administered at a rate of 6-10 mg/kg/min. Hypoglycemia recurred when the iv glucose infusion rate was reduced. Serum insulin level was 1.7 µIU/mL (N: <1) and c-peptide was 1.5 ng/mL (N: <0.9) when serum glucose was 38 mg/dL. Concurrently measured cortisol level was 11.5 µg/dL (N>20), growth hormone 38 ng/mL (N>10), lactic acid 7.3 mg/dL (N: 4.5-19.8), ammonia 32.9 µg/dL (N: 20-120), and urinary ketones were negative. Tandem mass spectrometry, plasma and urinary amino acid profiles, and urinary organic acid analyses were normal. Growth hormone deficiency was excluded with high levels of growth hormone during hypoglycemia. Cortisol deficiency was excluded with a peak cortisol level of 20.6 µg/dL during low dose ACTH stimulation test. His other blood glucose levels and simultaneously taken insulin and c-peptide levels were: glucose 46 mg/dL; insulin 1.7 µIU/mL; c-peptide 1.2 ng/mL; and glucose 43 mg/dL; insulin 1.9 µIU/mL; c-peptide 1.0 ng/mL, respectively. Urinary ketones were negative during all hypoglycemic episodes. Hypoglycemia due to energy deficiency and/or storage deficiency could not be excluded since he had a low birth weight and thus his daily caloric, protein, carbohydrate and lipid intake was increased. The patient could not take the required calories orally so he was fed with nasogastric drip infusion. Insulin values obtained at the time of hypoglycemia were high and measurable (>1 µIU/mL) favoring hyperinsulinism, and since nonketotic hypoglycemia was persistent despite having a diet with enough calories, diazoxide was started at a dose of 10 mg/kg/day. His blood glucose levels normalized after the beginning of diazoxide treatment. Transferrin isoform electrophoresis, requested following a preliminary diagnosis of CDG was abnormal with a type 1 pattern.
Molecular Analyses
The PMM2 gene sequencing revealed compound heterozygous c.422G>A (p.Arg141His)/c.691G>A (p.Val231Met) variants in Case 1, homozygous c.385G>A (p.Val129Met) variant in Case 2, and compound heterozygous c.349G>T (p.Gly117Cys)/c.359T>C (p.Ile120Thr) variants in Case 3 (RefSeq NM_000303.3). The parents were heterozygous for one allele each, consistent with autosomal recessive inheritance. All of these variants, except for c.349G>T (p.Gly117Cys) have been previously reported in patients with PMM2-CDG (28). The c.349G>T (p.Gly117Cys) missense variant has a low allele frequency in the healthy population, is predicted to be pathogenic by multiple lines of computational evidence, and is located in a dense hot-spot where a different amino acid change (p.Gly117Arg) has been reported as pathogenic and is thus classified as pathogenic according to the American College of Medical Genetics and Genomics 2015 criteria (28,29).
Literature Review
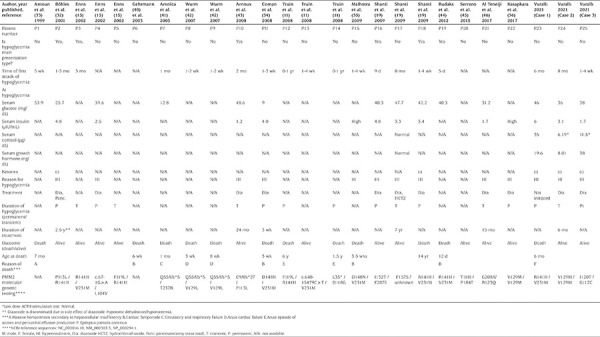
Hypoglycemia was reported in 37 cases (3.4%) among a total of 1.060 patients with PMM2-CDG in 171 articles in PubMed. However, in a few of these articles, only the number of hypoglycemic cases was reported without any further detail (30,31). Therefore, only data of 22 cases with hypoglycemia in whom clinical and laboratory features are published could be included in the current analysis. The clinical and laboratory findings related to hypoglycemia, as well as management and outcome of hypoglycemic patients with PMM2-CDG, are shown in Table 1. Hypoglycemia occurred in the first year of life in all cases, and in the early months of life in most cases. Hypoglycemia was one of the main presenting findings in six of the cases. Symptoms of hypoglycemia included seizures, decreased consciousness, and poor feeding. Blood glucose levels were between 9 and 50 mg/dL. Hyperinsulinism was the cause of hypoglycemia in approximately 45% (10/22) of the cases, and the cause of hypoglycemia was not specified in the remainder. Serum insulin levels ranged between 8.3 and 33 pmol/L (1.2 and 4.8 µIU/mL) at the time of hypoglycemia. Serum levels of ACTH, cortisol, growth hormone, and growth factors, which could clarify the cause of hypoglycemia were not specified in any of the cases, except one. Only two patients with hyperinsulinemic hypoglycemia had documentation of negative serum ketone levels, whereas ketone levels were not reported in others. A patient with hyperinsulinemic hypoglycemia had abdominal magnetic resonance imaging and no pathology was found in the pancreas (19). Abdominal ultrasonography was available in four of the cases. Normal pancreas anatomy was observed on ultrasonography in one patient in whom etiology of hypoglycemia was not specified, and in two hyperinsulinemic cases, while a cystic lesion on the head of the pancreas was detected in a hyperinsulinemic patient who had recurrent attacks of acute pancreatitis (19,32,33). Pancreatic biopsy was performed in a patient with hyperinsulinemic hypoglycemia who had normal abdominal ultrasonography, and histological examination revealed pancreatic islets cells of normal size and distribution with hypertrophic nuclei in a few beta cells (32). Hypertrophic nuclei in beta cells were evaluated as sign of functional activity.
A wide spectrum of multisystem involvement was present in cases with hypoglycemia. The specific organ involvements of all the cases with hypoglycemia are shown as supplementary material (Supplementary Table 1). Of all hypoglycemic cases, 73% had failure to thrive, 68% had inverted nipples, 64% had abnormal fat distribution, 55% had internal strabismus, 41% had dysmorphic facies, and 32% had microcephaly. Cardiac involvement (pericardial effusion and cardiac hypertrophy) was present in 59% of the cases, and gastrointestinal system involvement (feeding difficulties, vomiting, gastroesophageal reflux, gastrointestinal dysmotility, and chronic diarrhea) in 73%. Liver involvement, such as hypertransaminasemia and hepatomegaly, was present in 64% of all hypoglycemic patients, kidney involvement, such as proteinuria and tubulopathy, in 64%, coagulation problems, such as coagulation factor deficiencies, bleeding diathesis and thrombosis in 55%, and central nervous system involvement, such as hypotonia, developmental delay, cerebellar hypoplasia, hyporeflexia, seizures and ataxic gait, in 77%.
Some of the patients with hypoglycemia had severe progressive neurological, cardiovascular, hepatic, and gastrointestinal system involvement. Some had mild involvement of different organ systems. In some cases, skeletal findings, such as scoliosis, rhizomelia, generalized epiphyseal ossification delay, ovoid and anteriorly beaked vertebral bodies, bullet shaped short tubular bones of the hand, joint hyperlaxity, arachnodactyly, spinal canal stenosis, hypoplastic cervical vertebrae, thirteen pairs of costae, and shortening of long bones were also reported. Pseudocholinesterase deficiency and multiple episodes of sepsis were shown in one case. Other accompanying findings included thrombocytopenia, fetal hydrops, acute pancreatitis, stroke-like episodes, peripheral neuropathy, and athetosis. Hyperinsulinism was the main problem in one case and this case did not have other serious multisystem organ involvements (19).
When the cases are examined in terms of accompanying endocrinological disorders, thyroid dysfunction was reported in 10 (10/22, 45%) of the hypoglycemic cases, it was stated that thyroid function was normal in one case, and information about the thyroid function of the other cases was not provided. It was reported that TBG and serum T4 values were low in three cases, TSH was high in six cases and only T4 was low in one case. While three of the cases with high TSH also had accompanying low T4 (clinical hypothyroidism), T4 values were not given in the remaining three. One of the hypoglycemic cases also had hypocholesterolemia and another case had hypertriglyceridemia.
There was no difference in terms of demographic data, such as age at presentation and diagnosis, gender and accompanying organ involvement between hyperinsulinemic cases and cases with normal or unspecified insulin level (Supplementary Table 1). Nine of ten patients with hyperinsulinism were started on oral diazoxide treatment and all but one patient responded to diazoxide (Figure 1). The patient who did not respond to diazoxide had persistent hyponatremia as a side effect of diazoxide and diazoxide could not be continued. Glucose levels returned to normal in this case only after subtotal pancreatectomy (32). Treatment approach was not reported in one case with hyperinsulinemic hypoglycemia. Of the patients who needed diazoxide treatment to control hypoglycemia, hypoglycemia was transient in three patients and permanent in the others (Figure 1). One of the patients with transient hypoglycemia discontinued treatment after using diazoxide for 15 months, another for 24 months and another for 7 years. All cases with transient hyperinsulinism were alive at the time of reporting. One of the patients with permanent hyperinsulinemic hypoglycemia died at the age of 3 weeks, another at 3.5 weeks and another at the age of 14 years (19,34). There was severe biventricular hypertrophic cardiomyopathy and pericardial effusion in the prenatal period in cases who died at a very early age of 3 and 3.5 weeks, and the cause of death in both patients was severe pericardial tamponade (34,35). The cause of death in the child, who died at the age of 14, was not specified. Data on the etiology of hypoglycemia, treatment methods and outcome were lacking in patients who were not reported to be hyperinsulinemic. While two thirds (8/12) of the cases with undefined etiology of hypoglycemia died due to various causes, usually during the infancy period, this ratio was one third (4/13) in cases with hyperinsulinism (Figure 1).
Figure 1.
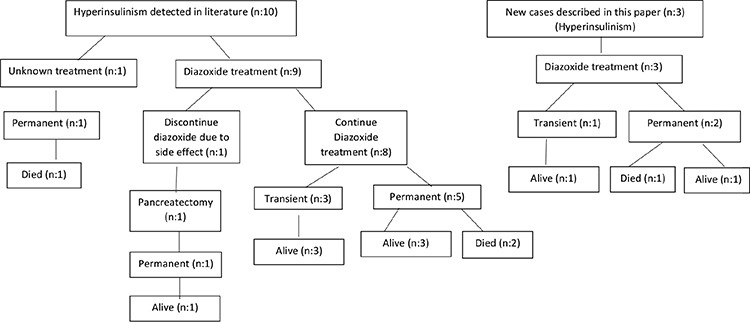
Etiology and outcome of hypoglycemic PMM2-CDG patients
PMM2-CDG: Phosphomannomutase 2 deficiency
Genotype analysis of the patients with hypoglycemia did not reveal any correlation to the phenotype with respect to hyperinsulinism. Missense mutations have been reported in affected individuals, causing amino acid alterations in conserved residues. Most cases with hypoglycemia were compound heterozygous. The most common mutant alleles in the hypoglycemic patients were p.Arg141His and p.Val231Met; and their compound heterozygosity was the most common genotype, associated with severe disease and mortality similar to the general PMM2-CDG population. Other relatively common mutant alleles were p.Phe119Leu, p.Val129Met, and p.Pro113Leu (Table 1). Homozygous known pathogenic variants p.Val129Met/p.Val129Met were present in two Turkish cases, including one reported in this paper. As neither of these patients had consanguineous parents, increased allele frequency of this variant in the Turkish population has been proposed, but not demonstrated (27,36). One of the cases with this genotype had transient and the other had permanent hyperinsulinism (Patient 22 and 24).
Table 1. Clinical and laboratory findings related to hypoglycemia as well as management and outcome of hypoglycemic patients with PMM2-CDG.
Discussion
The literature review indicated that hypoglycemia is a rare finding seen in PMM2-CDG. Including the three new cases described in this article, hypoglycemia was detected in only 25 of 1064 patients in total. In about one fourth of the PMM2-CDG cases with hypoglycemia, hypoglycemia was the main presenting finding and symptoms, such as seizures and decreased consciousness associated with hypoglycemia, have been reported in these cases. However, in the remainder, no major symptom or sign related to hypoglycemia was observed, and hypoglycemia was discovered during routine investigations. This situation suggests that some cases with hypoglycemia may remain undetected since there are no symptoms specific to hypoglycemia, especially in cases with severe neurological deficits. Therefore, we suggest close monitoring of blood glucose in cases with CDG, even if there is no clinical finding related to hypoglycemia.
Half of the cases with hypoglycemia had documented hyperinsulinism. However, in the other half, the cause of hypoglycemia could not be determined, since there is not sufficient data. Hormone levels, including cortisol and GH whose deficiencies could cause hypoglycemia, and other laboratory investigations, such as serum or urine ketone levels, were not reported in most cases. A complete etiological evaluation is essential to determine the cause of hypoglycemia and to plan the appropriate treatment. Although it is difficult to interpret the etiology due to the lack of data, hyperinsulinemic hypoglycemia may be the main cause of hypoglycemia in cases with PMM2-CDG. Besides, no case of hypoglycemia due to cortisol or GH deficiency is reported in the literature in patients with PMM2-CDG. Probably because the cases have not been investigated in detail in this respect, hyperinsulinemic hypoglycemia has been rarely reported in cases with PMM2-CDG. It is possible that at least some of the cases of hypoglycemia of unknown cause may be attributable to hyperinsulinism.
Cases with PMM2-CDG should be evaluated for possible hypoglycemia, and if it is documented further analysis for etiology, especially hyperinsulinism, should be carried out. We believe that if a detailed etiological evaluation of each hypoglycemia case is made properly, the majority of PMM2-CDG cases will have hyperinsulinism as the etiology and, with the appropriate treatment, hypoglycemia, which has a high probability of severe sequelae if left untreated, can be prevented.
One of the cases in the current series (case 3) did not have a high level of insulin at the time of hypoglycemia although it was measurable, but this patient responded well to diazoxide. Therefore, patients with PMM2-CDG can benefit from diazoxide treatment even if insulin levels were not so high. It should also be noted that poor oral feeding, feeding intolerance, vomiting and diarrhea, and hepatic dysfunction may contribute to hypoglycemia. Optimization of feeding may contribute to better glycemic control, as witnessed in cases 2 and 3.
Hypoglycemia seems to appear during early infancy in cases with PMM2-CDG but the age of diagnosis may be delayed. Failure to thrive is a common finding (approximately 75%) in cases with hypoglycemia. All organ system involvement, but especially central nervous system and gastrointestinal system involvement, can be seen in these cases with variable frequencies. Besides, CDG specific findings, such as inverted nipples and abnormal fat distribution, liver, kidney, and cardiac involvement are observed in 2/3 of the cases with hypoglycemia, and coagulopathies are observed in approximately half of the cases. More rarely, skeletal findings, thrombocytopenia, fetal hydrops, acute pancreatitis, stroke-like episodes, peripheral neuropathy, and athetosis have been described in these cases. Hyperinsulinemic hypoglycemia is usually a part of a multisystemic disease with different organ involvement. The only exception is a case who had no serious organ involvement other than hyperinsulinism (19). While this case was followed up due to failure to thrive, she had afebrile seizures due to hypoglycemia at the age of 8 months and hyperinsulinism was determined as the etiology of her hypoglycemia. She did not have signs more specific to CDG, such as strabismus, inverted nipples or an abnormal fat pad distribution. Her hypoglycemia responded well to diazoxide and she was being followed up as a case with normal psychomotor development and no cerebellar abnormality. Since the age of this case was young when published, it is also possible that other organ involvements might become more pronounced during follow-up.
There was no difference in terms of demographic and clinical findings between cases with hyperinsulinism and cases with unspecified insulin level. In hyperinsulinemic cases, no pancreatic pathology was detected to explain hyperinsulinism. All cases in whom oral diazoxide treatment was initiated responded well to this treatment. In only one case, diazoxide treatment was discontinued due to severe hyponatraemia with fluid retention, which is a side effect of diazoxide. It was not stated whether other medical treatment options were evaluated in that case, but it was reported that he had subtotal pancreatectomy due to hyperinsulinism. Including the three new patients presented in this article, hyperinsulinism was permanent in two thirds (9/13) of the cases, while it was transient in the remaining one third (4/13). In cases where hyperinsulinism was transient, diazoxide treatment was usually discontinued one or two years after the initiation of treatment. In one case, it was discontinued after using diazoxide for a longer period of 7 years. Half of the patients with permanent, patients died mostly due to cardiac abnormalities, while no patient with transient hyperinsulinism died. No genotype-phenotype correlation was observed in the cases with respect to hyperinsulinism. It is interesting to note that while the most common p.Arg141His allele is reported to be present in approximately 60% of reported cases of PMM2-CDG (30), this allele is slightly underrepresented (30%) in those with hypoglycemia (7 of 23 patients with known genotypes; Table 1).
It is unclear why hyperinsulinism is seen in cases with PMM2-CDG, and the pathophysiology is not fully explained. Hypoglycosylation of sulfonylurea receptor 1 (SUR1) and other proteins involved in insulin secretion has been suggested as the responsible mechanism. It is known that all PMM2-CDG patients with hyperinsulinemic hypoglycemia respond well to diazoxide. Diazoxide directly acts on beta cells in the pancreas, opening the KATP channel and preventing insulin release from beta cells (37). The good response of the cases with PMM2-CDG to diazoxide suggests that the hyperinsulinism seen in these cases may be due to the impaired function of KATP channels. KATP channels contain four ion channels (Kir6.2) and four regulatory SUR1 receptors and control glucose-stimulated release of insulin. Glycosylation of SUR1 is required for the expression of KATP channels on the surface (38). In addition, the dysfunction of the insulin receptor may be the cause of the hyperinsulinism seen in these cases. The insulin receptor consists of two extracellular α subunits and two transmembrane β subunits, and both subunits are glycosylated. Each of the α monomers contains 13 N-glycans, while the β-monomers contain four N-glycans and six O-glycans. It is not known whether glycosylation of insulin receptors is affected in CDGs (39). The insulin level in one case described in this article was not markedly elevated at the time of hypoglycemia. A possible hypothesis for this case may be hypoglycolization at the postreceptor level, and even if insulin levels are not very high, hyperinsulinism findings and hypoketotic hypoglycemia may be observed in these cases, similar to AKT2 activating mutation.
Study Limitations
Although hypoglycemia was detected in 37 cases with PMM2-CDG during the literature review, only 22 cases could be evaluated in detail due to the lack of sufficient clinical and laboratory data.
Conclusion
The main cause of hypoglycemia in PMM2-CDG appears to be hyperinsulinism. Abnormal counter-regulatory hormone response has not been identified in cases with PMM2-CDG, suggesting hyperinsulinism may still be the underlying cause of hypoglycemia in those with undetermined cause. Other possible causes of hypoglycemia, such as inadequate feeding, malnutrition, chronic diarrhea, and hepatic disease may also contribute to low levels of blood glucose. In cases with PMM2-CDG, even if there are no symptoms specific to hypoglycemia, blood glucose should be closely monitored and a detailed etiological evaluation should be performed when hypoglycemia is detected. Although insulin levels taken at the time of hypoglycemia may not be very high, hypoglycemia seen in patients with PMM2-CDG may respond well to diazoxide treatment.
Demographic characteristics and organ involvements of the hypoglycemic patients with PMM2-CDG
Footnotes
Ethics
Ethics Committee Approval: This study protocol was reviewed and approved by Hacettepe University Ethics Committee (approval number GO 17/141-14).
Informed Consent: Retrospective study.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Doğuş Vurallı, Yılmaz Yıldız, Alev Ozon, Ali Dursun, Nazlı Gönç, Ayşegül Tokatlı, H. Serap Sivri, Ayfer Alikaşifoğlu, Concept: Doğuş Vurallı, Yılmaz Yıldız, H. Serap Sivri, Design: Doğuş Vurallı, Yılmaz Yıldız, H. Serap Sivri, Data Collection or Processing: Doğuş Vurallı, Alev Ozon, Ayşegül Tokatlı, Analysis or Interpretation: Doğuş Vurallı, Yılmaz Yıldız, Alev Ozon, Ali Dursun, Nazlı Gönç, Ayşegül Tokatlı, H. Serap Sivri, Ayfer Alikaşifoğlu, Literature Search: Doğuş Vurallı, Alev Ozon, Writing: Doğuş Vurallı, Yılmaz Yıldız, Alev Ozon, H. Serap Sivri, Ayfer Alikaşifoğlu.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Péanne R, de Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, Pérez B, Seta N, Thiel C, Van Schaftingen E, Matthijs G, Jaeken J. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. 2018;61:643–663. doi: 10.1016/j.ejmg.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 2.de Lonlay P, Seta N, Barrot S, Chabrol B, Drouin V, Gabriel BM, Journel H, Kretz M, Laurent J, Le Merrer M, Leroy A, Pedespan D, Sarda P, Villeneuve N, Schmitz J, van Schaftingen E, Matthijs G, Jaeken J, Korner C, Munnich A, Saudubray JM, Cormier-Daire V. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet. 2001;38:14–19. doi: 10.1136/jmg.38.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grünewald S, Imbach T, Huijben K, Rubio-Gozalbo ME, Verrips A, de Klerk JB, Stroink H, de Rijk-van Andel JF, Van Hove JL, Wendel U, Matthijs G, Hennet T, Jaeken J, Wevers RA. Clinical and biochemical characteristics of congenital disorder of glycosylation type Ic, the first recognized endoplasmic reticulum defect in N-glycan synthesis. Ann Neurol. 2000;47:776–781. [PubMed] [Google Scholar]
- 4.Erlandson A, Bjursell C, Stibler H, Kristiansson B, Wahlström J, Martinsson T. Scandinavian CDG-Ia patients: genotype/phenotype correlation and geographic origin of founder mutations. Hum Genet. 2001;108:359–367. doi: 10.1007/s004390100489. [DOI] [PubMed] [Google Scholar]
- 5.Monin ML, Mignot C, De Lonlay P, Héron B, Masurel A, Mathieu- Dramard M, Lenaerts C, Thauvin C, Gérard M, Roze E, Jacquette A, Charles P, de Baracé C, Drouin-Garraud V, Khau Van Kien P, Cormier- Daire V, Mayer M, Ogier H, Brice A, Seta N, Héron D. 29 French adult patients with PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J Rare Dis. 2014;9:207. doi: 10.1186/s13023-014-0207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson DA, Lyons RJ, Russell-Eggitt I, Liasis A, Jagle H, Grünewald S. Retinal characteristics of the congenital disorder of glycosylation PMM2-CDG. J Inherit Metab Dis. 2013;36:1039–1047. doi: 10.1007/s10545-013-9594-2. [DOI] [PubMed] [Google Scholar]
- 7.Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr. 2003;162:359–379. doi: 10.1007/s00431-002-1136-0. [DOI] [PubMed] [Google Scholar]
- 8.Kristiansson B, Stibler H, Hagberg B, Wahlström J. [CDGS-1--a recently discovered hereditary metabolic disease. Multiple organ manifestations, incidence 1/80,000, difficult to treat] Lakartidningen. 1998;95:5742–5748. [PubMed] [Google Scholar]
- 9.Marquardt T, Hülskamp G, Gehrmann J, Debus V, Harms E, Kehl HG. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur J Pediatr. 2002;161:524–527. doi: 10.1007/s00431-002-1029-2. [DOI] [PubMed] [Google Scholar]
- 10.Romano S, Bajolle F, Valayannopoulos V, Lyonnet S, Colomb V, de Baracé C, Vouhe P, Pouard P, Vuillaumier-Barrot S, Dupré T, de Keyzer Y, Sidi D, Seta N, Bonnet D, de Lonlay P. Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia) J Med Genet. 2009;46:287–288. doi: 10.1136/jmg.2008.057620. [DOI] [PubMed] [Google Scholar]
- 11.Truin G, Guillard M, Lefeber DJ, Sykut-Cegielska J, Adamowicz M, Hoppenreijs E, Sengers RCA, Wevers RA, Morava E. Pericardial and abdominal fluid accumulation in congenital disorder of glycosylation type Ia. Mol Genet Metab. 2008;94:481–484. doi: 10.1016/j.ymgme.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Barone R, Carrozzi M, Parini R, Battini R, Martinelli D, Elia M, Spada M, Lilliu F, Ciana G, Burlina A, Leuzzi V, Leoni M, Sturiale L, Matthijs G, Jaeken J, Di Rocco M, Garozzo D, Fiumara A. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol. 2015;262:154–164. doi: 10.1007/s00415-014-7549-7. [DOI] [PubMed] [Google Scholar]
- 13.Krasnewich D, O’Brien K, Sparks S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG-Ia) Am J Med Genet C Semin Med Genet. 2007;145:302–306. doi: 10.1002/ajmg.c.30143. [DOI] [PubMed] [Google Scholar]
- 14.Krasnewich D, Gahl WA. Carbohydrate-deficient glycoprotein syndrome. Adv Pediatr. 1997;44:109–140. [PubMed] [Google Scholar]
- 15.Enns GM, Steiner RD, Buist N, Cowan C, Leppig KA, McCracken MF, Westphal V, Freeze HH, O’brien JF, Jaeken J, Matthijs G, Behera S, Hudgins L. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr. 2002;141:695–700. doi: 10.1067/mpd.2002.128658. [DOI] [PubMed] [Google Scholar]
- 16.Pérez Jde J, Udeshi ND, Shabanowitz J, Ciordia S, Juárez S, Scott CL, Olszewski NE, Hunt DF, García JA. O-GlcNAc modification of the coat protein of the potyvirus Plum pox virus enhances viral infection. Virology. 2013;442:122–131. doi: 10.1016/j.virol.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child. 2001;85:236–239. doi: 10.1136/adc.85.3.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller BS, Freeze HH. New disorders in carbohydrate metabolism: congenital disorders of glycosylation and their impact on the endocrine system. Rev Endocr Metab Disord. 2003;4:103–113. doi: 10.1023/a:1021883605280. [DOI] [PubMed] [Google Scholar]
- 19.Shanti B, Silink M, Bhattacharya K, Howard NJ, Carpenter K, Fietz M, Clayton P, Christodoulou J. Congenital disorder of glycosylation type Ia: heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis. 2009;32(Suppl 1):241–251. doi: 10.1007/s10545-009-1180-2. [DOI] [PubMed] [Google Scholar]
- 20.de Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res. 1995;37:395–401. doi: 10.1203/00006450-199504000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Kristiansson B, Stibler H, Wide L. Gonadal function and glycoprotein hormones in the carbohydrate-deficient glycoprotein (CDG) syndrome. Acta Paediatr. 1995;84:655–659. doi: 10.1111/j.1651-2227.1995.tb13720.x. [DOI] [PubMed] [Google Scholar]
- 22.Mohamed M, Theodore M, Claahsen-van der Grinten H, van Herwaarden AE, Huijben K, van Dongen L, Kouwenberg D, Lefeber DJ, Wevers RA, Morava E. Thyroid function in PMM2-CDG: diagnostic approach and proposed management. Mol Genet Metab. 2012;105:681–683. doi: 10.1016/j.ymgme.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Kranz C, Basinger AA, Güçsavaş-Calikoğlu M, Sun L, Powell CM, Henderson FW, Aylsworth AS, Freeze HH. Expanding spectrum of congenital disorder of glycosylation Ig (CDG-Ig): sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am J Med Genet A. 2007;143:1371–1378. doi: 10.1002/ajmg.a.31791. [DOI] [PubMed] [Google Scholar]
- 24.Miller BS, Freeze HH, Hoffmann GF, Sarafoglou K. Pubertal development in ALG6 deficiency (congenital disorder of glycosylation type Ic) Mol Genet Metab. 2011;103:101–103. doi: 10.1016/j.ymgme.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HH. Congenital disorder of glycosylation id presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J Clin Endocrinol Metab. 2005;90:4371–4375. doi: 10.1210/jc.2005-0250. [DOI] [PubMed] [Google Scholar]
- 26.Moravej H, Altassan R, Jaeken J, Enns GM, Ellaway C, Balasubramaniam S, De Lonlay P, Coman D, Mercimek-Andrews S, Witters P, Morava E. Hypoglycemia in CDG patients due to PMM2 mutations: Follow up on hyperinsulinemic patients. JIMD Rep. 2020;51:76–81. doi: 10.1002/jmd2.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yıldız Y, Arslan M, Çelik G, Kasapkara ÇS, Ceylaner S, Dursun A, Sivri HS, Coşkun T, Tokatlı A. Genotypes and estimated prevalence of phosphomannomutase 2 deficiency in Turkey differ significantly from those in Europe. Am J Med Genet A. 2020;182:705–712. doi: 10.1002/ajmg.a.61488. [DOI] [PubMed] [Google Scholar]
- 28.Matthijs G, Schollen E, Bjursell C, Erlandson A, Freeze H, Imtiaz F, Kjaergaard S, Martinsson T, Schwartz M, Seta N, Vuillaumier-Barrot S, Westphal V, Winchester B. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia) Hum Mutat. 2000;16:386–394. doi: 10.1002/1098-1004(200011)16:5<386::AID-HUMU2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 29.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altassan R, Péanne R, Jaeken J, Barone R, Bidet M, Borgel D, Brasil S, Cassiman D, Cechova A, Coman D, Corral J, Correia J, de la Morena-Barrio ME, de Lonlay P, Dos Reis V, Ferreira CR, Fiumara A, Francisco R, Freeze H, Funke S, Gardeitchik T, Gert M, Girad M, Giros M, Grünewald S, Hernández-Caselles T, Honzik T, Hutter M, Krasnewich D, Lam C, Lee J, Lefeber D, Marques-de-Silva D, Martinez AF, Moravej H, Õunap K, Pascoal C, Pascreau T, Patterson M, Quelhas D, Raymond K, Sarkhail P, Schiff M, Seroczyńska M, Serrano M, Seta N, Sykut-Cegielska J, Thiel C, Tort F, Vals MA, Videira P, Witters P, Zeevaert R, Morava E. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up. J Inherit Metab Dis. 2019;42:5–28. doi: 10.1002/jimd.12024. [DOI] [PubMed] [Google Scholar]
- 31.Witters P, Honzik T, Bauchart E, Altassan R, Pascreau T, Bruneel A, Vuillaumier S, Seta N, Borgel D, Matthijs G, Jaeken J, Meersseman W, Cassiman D, Pascale de L, Morava E. Long-term follow-up in PMM2-CDG: are we ready to start treatment trials? Genet Med. 2019;21:1181–1188. doi: 10.1038/s41436-018-0301-4. [DOI] [PubMed] [Google Scholar]
- 32.Böhles H, Sewell AA, Gebhardt B, Reinecke-Lüthge A, Klöppel G, Marquardt T. Hyperinsulinaemic hypoglycaemia--leading symptom in a patient with congenital disorder of glycosylation Ia (phosphomannomutase deficiency) J Inherit Metab Dis. 2001;24:858–862. doi: 10.1023/a:1013944308881. [DOI] [PubMed] [Google Scholar]
- 33.Antoun H, Villeneuve N, Gelot A, Panisset S, Adamsbaum C. Cerebellar atrophy: an important feature of carbohydrate deficient glycoprotein syndrome type 1. Pediatr Radiol. 1999;29:194–198. doi: 10.1007/s002470050571. [DOI] [PubMed] [Google Scholar]
- 34.Coman D, Bostock D, Hunter M, Kannu P, Irving M, Mayne V, Fietz M, Jaeken J, Savarirayan R. Primary skeletal dysplasia as a major manifesting feature in an infant with congenital disorder of glycosylation type Ia. Am J Med Genet A. 2008;146:389–392. doi: 10.1002/ajmg.a.32119. [DOI] [PubMed] [Google Scholar]
- 35.Malhotra A, Pateman A, Chalmers R, Coman D, Menahem S. Prenatal cardiac ultrasound finding in congenital disorder of glycosylation type 1a. Fetal Diagn Ther. 2009;25:54–57. doi: 10.1159/000196816. [DOI] [PubMed] [Google Scholar]
- 36.Kasapkara ÇS, Barış Z, Kılıç M, Yüksel D, Keldermans L, Matthijs G, Jaeken J. PMM2-CDG and sensorineural hearing loss. J Inherit Metab Dis. 2017;40:629–630. doi: 10.1007/s10545-017-0073-z. [DOI] [PubMed] [Google Scholar]
- 37.Virgili N, Mancera P, Wappenhans B, Sorrosal G, Biber K, Pugliese M, Espinosa-Parrilla JF. K(ATP) channel opener diazoxide prevents neurodegeneration: a new mechanism of action via antioxidative pathway activation. PLoS One. 2013;8:e75189. doi: 10.1371/journal.pone.0075189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conti LR, Radeke CM, Vandenberg CA. Membrane targeting of ATP-sensitive potassium channel. Effects of glycosylation on surface expression. J Biol Chem. 2002;277:25416–25422. doi: 10.1074/jbc.M203109200. [DOI] [PubMed] [Google Scholar]
- 39.Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, Morrione A, Vigneri R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev. 2017;38:379–431. doi: 10.1210/er.2017-00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gehrmann J, Sohlbach K, Linnebank M, Böhles HJ, Buderus S, Kehl HG, Vogt J, Harms E, Marquardt T. Cardiomyopathy in congenital disorders of glycosylation. Cardiol Young. 2003;13:345–351. [PubMed] [Google Scholar]
- 41.Aronica E, van Kempen AA, van der Heide M, Poll-The BT, van Slooten HJ, Troost D, Rozemuller-Kwakkel JM. Congenital disorder of glycosylation type Ia: a clinicopathological report of a newborn infant with cerebellar pathology. Acta Neuropathol. 2005;109:433–442. doi: 10.1007/s00401-004-0975-3. [DOI] [PubMed] [Google Scholar]
- 42.Wurm D, Hänsgen A, Kim YJ, Lindinger A, Baghai A, Gortner L. Early fatal course in siblings with CDG-Ia (caused by two novel mutations in the PMM2 gene): clinical, molecular and autopsy findings. Eur J Pediatr. 2007;166:377–378. doi: 10.1007/s00431-006-0240-y. [DOI] [PubMed] [Google Scholar]
- 43.Arnoux JB, Boddaert N, Valayannopoulos V, Romano S, Bahi-Buisson N, Desguerre I, de Keyzer Y, Munnich A, Brunelle F, Seta N, Dautzenberg MD, de Lonlay P. Risk assessment of acute vascular events in congenital disorder of glycosylation type Ia. Mol Genet Metab. 2008;93:444–449. doi: 10.1016/j.ymgme.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 44.Rudaks LI, Andersen C, Khong TY, Kelly A, Fietz M, Barnett CP. Hypertrophic cardiomyopathy with cardiac rupture and tamponade caused by congenital disorder of glycosylation type Ia. Pediatr Cardiol. 2012;33:827–830. doi: 10.1007/s00246-012-0214-y. [DOI] [PubMed] [Google Scholar]
- 45.Serrano M, de Diego V, Muchart J, Cuadras D, Felipe A, Macaya A, Velázquez R, Poo MP, Fons C, O’Callaghan MM, García-Cazorla A, Boix C, Robles B, Carratalá F, Girós M, Briones P, Gort L, Artuch R, Pérez- Cerdá C, Jaeken J, Pérez B, Pérez-Dueñas B. Phosphomannomutase deficiency (PMM2-CDG): ataxia and cerebellar assessment. Orphanet J Rare Dis. 2015;10:138. doi: 10.1186/s13023-015-0358-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al Teneiji A, Bruun TU, Sidky S, Cordeiro D, Cohn RD, Mendoza-Londono R, Moharir M, Raiman J, Siriwardena K, Kyriakopoulou L, Mercimek-Mahmutoglu S. Phenotypic and genotypic spectrum of congenital disorders of glycosylation type I and type II. Mol Genet Metab. 2017;120:235–242. doi: 10.1016/j.ymgme.2016.12.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Demographic characteristics and organ involvements of the hypoglycemic patients with PMM2-CDG