

Abstract
Polychlorinated biphenyls (PCBs) are persistent organic pollutants that accumulate in adipose tissue and have been associated with cardiometabolic disease. We have previously demonstrated that exposure of human preadipocytes to the dioxin-like PCB126 disrupts adipogenesis via the aryl hydrocarbon receptor (AhR). To further understand how PCB126 disrupts adipose tissue cells, we performed RNAseq analysis of PCB126-treated human preadipocytes over a 3-day time course. The most significant predicted upstream regulator affected by PCB126 exposure at the early time point of 9 hours was the AhR. Progressive changes occurred in the number and magnitude of transcript levels of genes associated with inflammation, most closely fitting the pathways of cytokine-cytokine-receptor signaling and the AGE-RAGE diabetic complications pathway. Transcript levels of genes involved in the IL-17A, IL-1β, MAP kinase, and NF-κB signaling pathways were increasingly dysregulated by PCB126 over time. Our results illustrate the progressive time-dependent nature of transcriptional changes caused by toxicants such as PCB126, point to important pathways affected by PCB126 exposure, and provide a rich dataset for further studies to address how PCB126 and other AhR agonists disrupt preadipocyte function. These findings have implications for understanding how dioxin-like PCBs and other dioxin-like compounds are involved in the development of obesity and diabetes.
Keywords: Polychlorinated Biphenyls, PCB126, Adipose, AhR, RNAseq, Inflammation
GRAPHICAL ABSTRACT
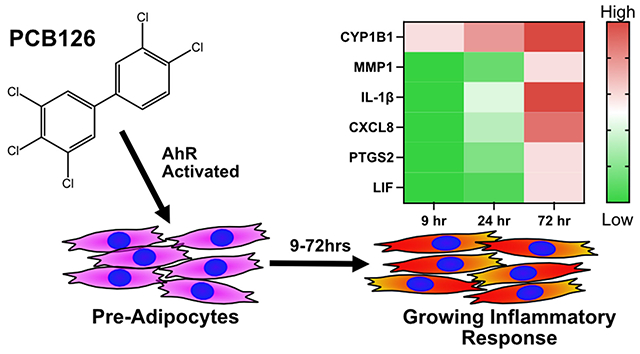
INTRODUCTION
Exposures to persistent organic pollutants (POPs) is associated with cardiometabolic disease, including obesity, diabetes, steatosis, and stroke (Dirinck et al. 2011; Dusanov et al. 2018; Grice et al. 2017; Henriquez-Hernandez et al. 2017; Lee et al. 2014; Ruzzin et al. 2010; Taylor et al. 2013). The mechanisms by which POPs cause cardiometabolic disease are unclear, but numerous studies have pointed to endocrine disruption that leads to adipose tissue dysfunction (Heindel et al. 2015). Adipose tissue, critical for regulating normal metabolism, stores and releases lipids, and secretes hormones and growth factors that act on other tissues to regulate energy homeostasis (Cohen and Spiegelman 2016; Rosen and Spiegelman 2014). Further, adipose tissue can become highly proinflammatory, particularly when existing adipocytes become hypertrophic during obesity (Gustafson et al. 2009). This condition is a precursor to insulin resistance and type II diabetes (Kohlgruber and Lynch 2015).
Adipocytes can also be thermogenic by upregulating mitochondrial uncoupling proteins (Cohen and Spiegelman 2015). For white adipocytes, this process is called “beiging”. The ability to beige indicates a healthy condition and is associated with exercise and weight loss (Stanford et al. 2013). Brown adipocytes, which decrease significantly with aging, perform this function naturally. Adipocytes are derived from preadipocytes which are themselves derived from adipocyte stem cells (Rosen and Spiegelman 2014). Disruption of the process of adipogenesis in both white and brown lineages has significant metabolic consequences. It can lead to diabetes and lipodystrophy, including steatosis, as lipids accumulate in other tissues besides fat (Cohen and Spiegelman 2016). POPs are well-known to disrupt adipogenesis (Gonzalez-Casanova et al.2020; Jackson et al. 2017).
Polychlorinated biphenyls (PCBs) were used extensively in industry for caulking, light ballast fluid, electrical transformers, and numerous other applications (Beyer and Biziuk 2009). While PCB production has been banned, they still exist at high levels in buildings and the environment. Further, PCBs are produced inadvertently in industrial processes. Humans are mainly exposed to PCBs through diet and air (Saktrakulkla et al. 2020; Wang et al. 2020). PCBs accumulate in fatty tissues (Beyer and Biziuk 2009; Jackson et al. 2017). Fish and other animals bioaccumulate PCBs within adipose, and these are passed to humans through diet. The highest levels of PCBs are found in adipose tissue, and weight loss is known to release PCBs into the serum, where they can affect other tissues (Grimm et al. 2015; Hu et al. 2010).
PCB mixtures comprise 209 different congeners, many of which have vastly different biological properties (Grimm et al. 2015). One of the most biologically active congeners is PCB 126, a dioxin-like molecule that potently activates the dioxin-responsive aryl hydrocarbon receptor (AhR) (Hestermann et al. 2000). We have demonstrated in previous studies that exposure of human preadipocytes to PCB126 for 3 days significantly disrupts adipogenesis and blocks the thermogenic response of adipocytes that are subsequently derived from the preadipocytes (Gadupudi et al. 2015; Gourronc et al. 2019; Gourronc et al. 2018). All these effects were found to be mediated through AhR. Our finding that preadipocytes are particularly sensitive to PCB126 motivated us to look further into how PCB126 affects preadipocytes. In this study, we have assessed the transcriptomic profile of preadipocytes that have been exposed to PCB126 over a 3-day time course. Our results demonstrate that PCB126 causes progressive changes in transcript levels of genes involved in AhR signaling, inflammation, and MAP-kinase signaling, among others. A key pathway found activated by PCB126 is the AGE-RAGE (Advanced Glycation End products-Receptor for Advanced Glycation End products) pathway of diabetic complications. Activation of this pathway is crucially important for the development of type II diabetes (Strieder-Barboza et al. 2019; Yamagishi et al. 2009). Overall, our studies provide a comprehensive view of transcriptomic changes caused by PCB126 exposure of human preadipocytes and point to important pathways that are likely to play a role in the disruption of adipogenesis, adipocyte tissue function, and subsequent development of diabetes caused by PCB126 and other dioxin-like compounds.
MATERIALS AND METHODS
Cells and Treatments
The human preadipocyte cell line, NPAD (Normal PreADipocyte), has been described previously (Gadupudi et al. 2015; Vu et al. 2013). It was derived from subcutaneous adipose of a non-diabetic female donor. The cell line is immortal and can differentiate into mature adipocytes that can be induced to beige (Gourronc et al. 2019; Vu et al. 2013). Early passage cells were cultured in PGM2 medium (Lonza). PCB126 (C12H5Cl5; InChI: 1S/C12H5Cl5/c13-8-2-1-6(3-9(8)14)7-4-10(15)12(17)11(16)5-7/h1-5H; InChI Key: REHONNLQRWTIFF-UHFFFAOYSA-N; Canonical SMILES: C1=CC(=C(C=C1C2=CC(=C(C(=C2)Cl)Cl)Cl)Cl)Cl) was synthesized in >99% purity from 3,4,5-trichlorobromobenzene and 3,4-dichlorophenylboronic acid using Pd(dba)2/DPDB as a catalyst, as described (Joshi et al. 2011). Details regarding the synthesis and authentication of this PCB126 batch are reported elsewhere (Gadupudi et al. 2018; Li 2019). PCB126 was dissolved in DMSO and used at a concentration of 10 μM in PGM2 medium for treatments. Cells that were approximately 90% confluent were treated with PCB126 or vehicle with complete medium change. Treatments were allowed to proceed for 9 hours, 1 day, or 3 days before the collection of RNA. Four biological replicates were prepared for each time point and treatment.
RNA isolation and processing for RNAseq
Cells were homogenized in 1 ml of TRIzol Reagent (Invitrogen). Total RNA from the aqueous phase was further purified using RNeasy Columns (Qiagen). Transcription profiling using RNAseq was performed by the University of Iowa Genomics Division using manufacturer-recommended protocols. Briefly, 500 ng of DNase I-treated total RNA was used to enrich for polyA-containing transcripts using beads coated with oligo(dT) primers. The enriched RNA pool was then fragmented, converted to cDNA and ligated to sequencing adaptors containing indexes using the Illumina TruSeq stranded mRNA sample preparation kit (Cat. #RS-122-2101, Illumina, Inc., San Diego, CA). The molar concentrations of the indexed libraries were measured using the 2100 Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA) and combined equally into pools for sequencing. The concentration of the pools was measured using the Illumina Library Quantification Kit (KAPA Biosystems, Wilmington, MA) and sequenced on the Illumina HiSeq 4000 genome sequencer using 50 bp paired-end SBS chemistry.
RNAseq bioinformatic analysis
Four biological replicates were prepared per treatment and time point. Barcoded samples were pooled and sequenced, using an Illumina NovaSeq6000 in the Iowa Institute of Human Genetics (IIHG) Genomics Division, to obtain a minimum of 25 million paired-end 50 bp reads per sample. Reads were converted from the Illumina BCL format to fastq format and were processed using the ‘bcbio-nextgen’ pipeline (https://github.com/chapmanb/bcbio-nextgen). This pipeline includes ‘best practices’ approaches for quality control (QC), alignment, and read quantitation. Reads were aligned against the hg38 reference genome using the hisat2 aligner and stored as indexed BAM files (Kim et al. 2015; Kim et al. 2019). In parallel, rapid quantification of reads to the human transcriptome (GENCODE 39) was performed using the salmon aligner (Patro et al. 2017). Analysis of BAM files showed that for all samples, ~95% of RNA-seq reads were uniquely mapped to the reference, with ~90% of mapped reads originating in exonic regions. The ‘bcbio-nextgen’ pipeline runs MultiQC, a computational tool that provides an overview of the data to detect common QC problems (Ewels et al. 2016). No significant QC problems were detected for any samples reported here. All four replicates for each time point and treatment passed QC parameters and were not excluded as outliers by Principal Component Analysis (Figure S1). RNAseq data (fastq and processed data) was deposited on Gene Expression Omnibus (GEO) and is available for review (Record GSE193578, reviewer token: axgxwimqfvcjrad). Following alignment, salmon transcript expression values were summarized to the gene level using tximport (Soneson et al. 2015). Estimated, non-normalized gene-level counts from this procedure were used for differential gene expression analysis using DESeq2 (Love et al. 2014). The sets of differentially expressed genes (DEGs) between PCB126-treated and DMSO-treated cells at the same time point are provided in Supplementary Data and with the GEO deposit. Code to recreate this data analysis can be found online (https://github.com/mchimenti/klingelhutz_rnaseq_july2020_pcb126). The sets of DEGs were imported into iPathwayGuide commercial software (https://advaitabio.com/ipathwayguide) to perform “impact analysis” to generate reports of statistically enriched pathways, upstream regulatory genes, biological processes, diseases and functions, and interaction with metabolites and chemicals. These analyses consider the direction and type of all signals on a pathway along with the position, role and type of each gene (Draghici et al. 2007; Khatri and Draghici 2005; Nguyen et al. 2019).
RESULTS
Our previous studies found that preadipocytes treated with PCB126 become refractory to adipogenesis even if the PCB126 is removed before adding differentiation medium (Gadupudi et al. 2015; Gourronc et al. 2019; Gourronc et al. 2018). Thus, PCB126 exposure causes stable changes in preadipocytes that prevent subsequent differentiation into adipocytes. Our studies indicated that PCB126 caused a proinflammatory response in preadipocytes and that maximal effects on adipogenesis occurred within 3 days of treatment (Gourronc et al. 2018). Further, the effects were found to be dependent on the AhR. We were interested in determining how PCB126 treatment leads to a cascade of transcriptional changes that cause a proinflammatory response, resulting in a block to adipogenesis. To do this, we performed RNAseq analysis on PCB126-treated preadipocytes over a time course of 9 hours, 1 day, and 3 days post-treatment. We used a high concentration of PCB126 (10 μM) that in previous studies was found not to inhibit proliferation (Gadupudi et al. 2015).
Transcripts that exhibited statistically significant (adjusted p<0.05) differences in expression with an average log2 fold change cutoff of 0.3 between PCB126-treated and vehicle controls were determined. Results were further placed into biological context with pathway analysis. The number of gene transcripts affected by PCB126 exposure increased dramatically with the treatment time, going from nearly 200 gene transcripts being altered at 9 hours to over 2,000 by day 3 post-exposure (Figure 1), a greater than 10-fold increase in the number of different gene transcripts. The magnitude of changes in the DEGs was on average much higher at the later timepoints (Figure 1, note different scales on the x-axes). Regardless of magnitude, about half (91/200) of the same DEGs at the early 9-hour time point were altered on days 1 and 3 (Figure 1). Not all the changes that occurred at early time points were DEGs in later time points, indicating that some changes were transient and/or that initial changes caused by PCB126 further activated or repressed other genes.
Figure 1:
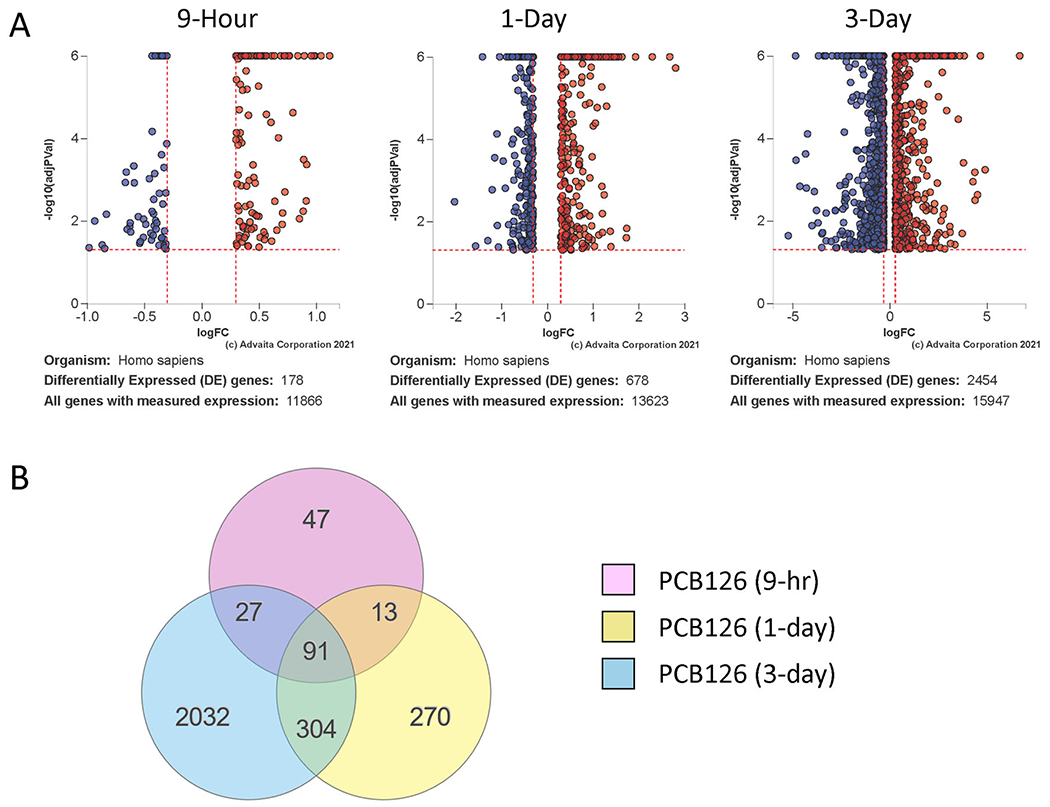
Transcript changes in PCB126-exposed preadipocytes over a time course. A. Volcano plots showing differentially expressed genes at 9-hour, 1-Day, or 3-Day post-treatment of preadipocytes with PCB126 (10 μM). Red: Upregulated; Blue: Downregulated. DE Thresholds: p-value:0.05; Log2FC: 0.3 B. Venn diagram illustrating overlap of differentially expressed genes at the different timepoints. Fdr≤0.05.
The impact analysis of the iPathwayGuide software tool allows an assessment of what upstream master regulator genes are most likely involved in causing the observed changes in gene transcript levels. AhR was the most significant upstream regulator gene at 9 hours (Table 1), even though the level of AhR gene transcript itself was not significantly altered (Figure 2). This is consistent with AhR’s known activation mechanism of binding to ligands in the cytoplasm and translocating to the nucleus to activate AhR-responsive genes rather than being upregulated itself. A progressive increase in the magnitude of change and number of AhR-responsive genes was observed over time (Figure 2). For example, an AhR-responsive gene, CYP1B1, went from an increase of log2fold of 0.988 (~2-fold) at 9 hours to a log2fold change of 1.510 (~3-fold) on day 1 and then 2.081 (~4-fold) at day 3 (Figure 2). Interestingly, while AhR was predicted to be the most significant upstream regulator at the 9-hour time point, it was not the top predicted upstream regulator on days 1 and 3, indicating a progressive shift in gene expression with time (Table 1). For example, on day 1 post-treatment, the top predicted upstream regulator was SREBF2, a transcription factor involved in sterol metabolism (Ye and DeBose-Boyd 2011) followed by NFY complex subunit genes, NFYB, NFYC, and NFYA, that code for transcription factors involved in the regulation of fatty acid synthase and other genes that act in sterol metabolism (Lu et al. 2015; Nishi-Tatsumi et al. 2017). In contrast, on day 3, the top predicted upstream regulator was IL-1β, a key factor involved in initiating a proinflammatory response in adipose tissue (Feve and Bastard 2009). IL-1β was considered a top 10 predicted upstream regulator at all time points and showed a progressive increase in the magnitude of change and number of genes involved in this pathway with time (Figure 3 and Figure S2). Thus, the earliest time point of 9 hours had a clear AhR signature. In contrast, day 1 showed changes to lipid metabolism and, by day 3, the most significant predicted upstream regulator was IL-1β, indicating a powerful shift toward a proinflammatory response at the latest time point.
Table 1:
Predicted upstream regulator gene pathways affected by PCB126 treatment of preadipocytes at different timepoints of 9 hours, 1 day, or 3 days. The over-representation p-value is based on the number of differentially expressed genes considered downstream of the upstream regulator gene. Fdr≤0.05
| 9-Hour | 1-Day | 3-Day | |||
|---|---|---|---|---|---|
| Gene symbol | pv_comb_p_fdr | Gene symbol | pv_comb_p_fdr | Gene symbol | pv_comb_p_fdr |
| AHR | 0.005954545 | SREBF2 | 3.56E-09 | IL1B | 5.93693E-05 |
| HGF | 0.012824388 | NFYB | 1.32184E-05 | GAST | 0.002052121 |
| GAST | 0.012824388 | NFYC | 1.32184E-05 | IL33 | 0.002052121 |
| PROK1 | 0.012824388 | NFYA | 1.94097E-05 | ATF4 | 0.002052121 |
| TNF | 0.012824388 | IL17A | 4.3981E-05 | IRF9 | 0.006077495 |
| TLR2 | 0.012824388 | IL1B | 5.65353E-05 | IL17A | 0.006552089 |
| RELA | 0.015822055 | AHR | 0.00025815 | OSM | 0.013833113 |
| IL1B | 0.015822055 | CCL2 | 0.00025815 | TNF | 0.013833113 |
| IL6 | 0.015822055 | GAST | 0.000921071 | BMP7 | 0.023533496 |
| CSF2 | 0.015822055 | TNF | 0.000988789 | NFKB1 | 0.023533496 |
Figure 2:
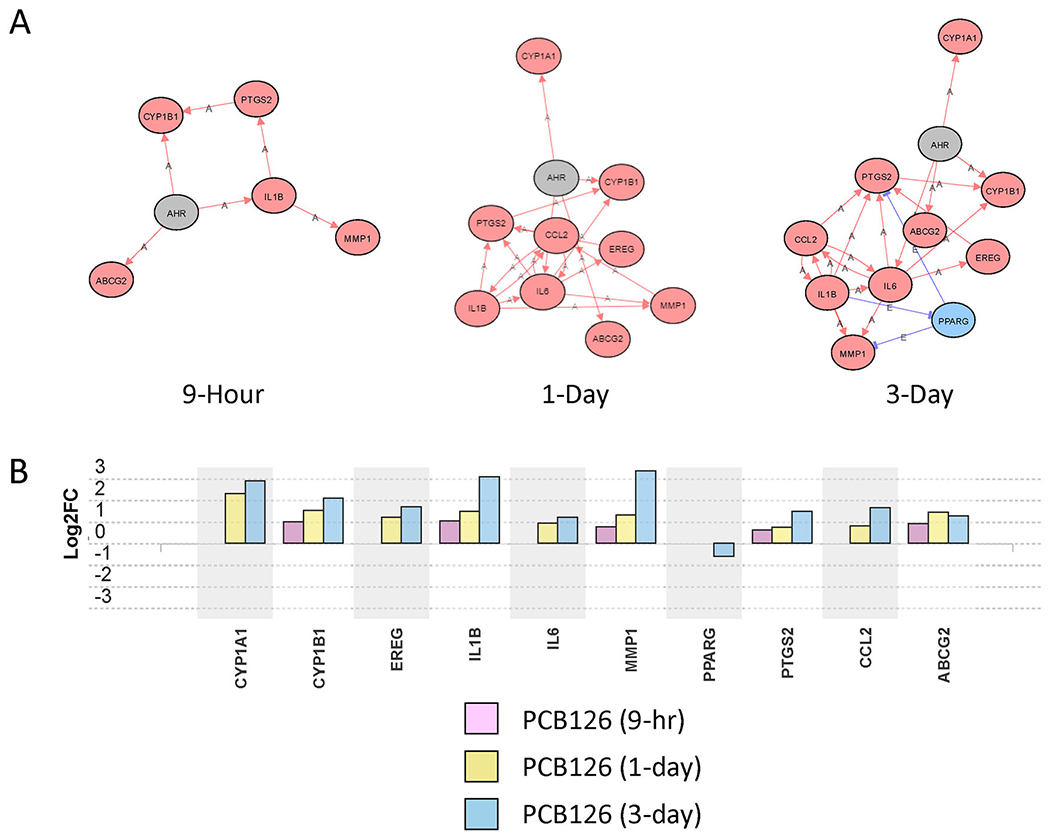
Aryl hydrocarbon receptor (AhR) pathway genes altered by PCB126 treatment of preadipocytes at different timepoints. A. Network analysis of AhR regulated genes at 9-hour, 1-day, and 3-day timepoints. Red Oval: Upregulated; Blue Oval: Downregulated; Gray Oval: Unchanged; “A”: Activated; “E”: Expression Inhibited; “I”: Inhibited; →Activation; ┤Inhibition. B. Transcript level changes (log2fold) of genes considered to be in the AhR pathway at different timepoints (9-hour, 1-day, 3-day) of PCB126-exposed preadipocytes compared to vehicle controls at the same timepoints. Fdr≤0.05.
Figure 3:
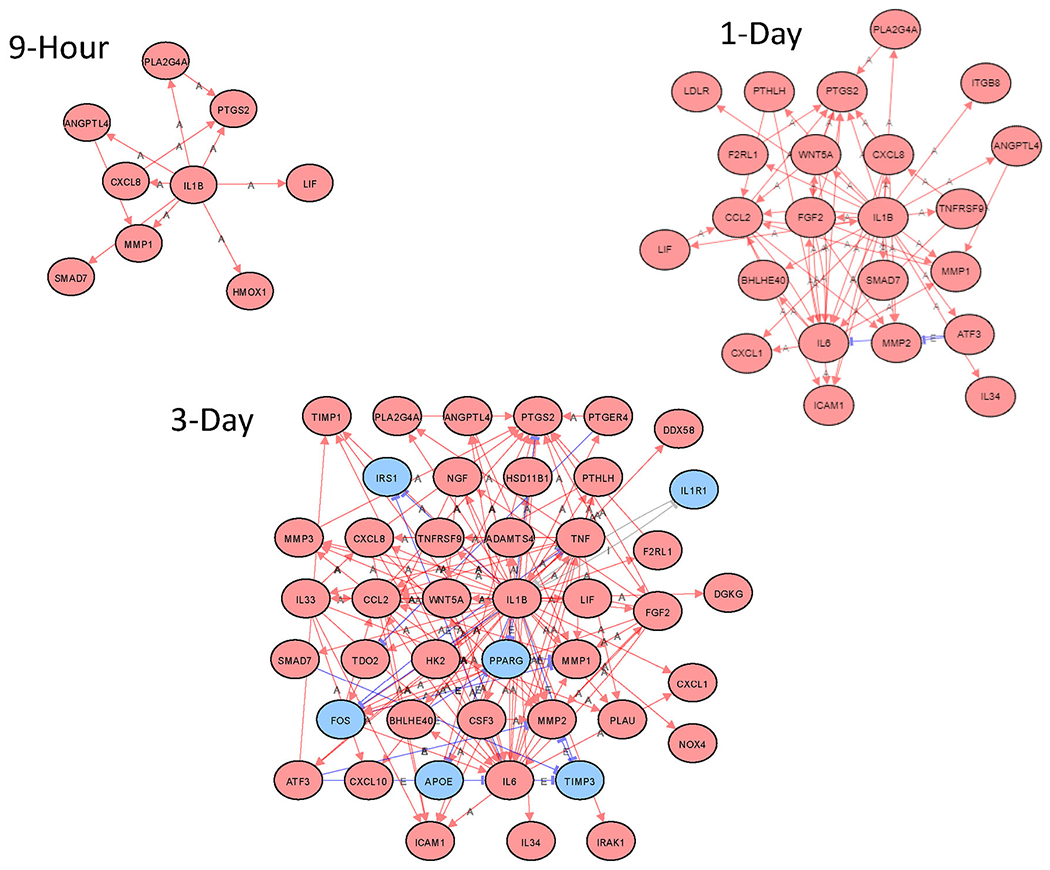
Network analysis of predicted IL-1β-regulated genes at 9-hour, 1-day, and 3-day timepoints. Red Oval: Upregulated; Blue Oval: Downregulated; Gray Oval: Unchanged; “A”: Activated; “E”: Expression Inhibited; “I”: Inhibited; →Activation; ┤Inhibition.
We used the iPathwayGuide software to elucidate what gene signatures from publicly available databases of chemical exposures of cells are most similar to the obtained data. As might be predicted, the first and second most similar top chemical signature pathways were those associated with tetrachlorodibenzodioxin (TCDD), followed by 3,4,5,3’,4’-pentachlorobiphenyl (PCB126) treatment, respectively (Table S1). These findings demonstrate that the RNAseq data and subsequent analysis are consistent with published results showing that PCB126 causes a dioxin-like response in other cell types.
We also looked at enriched biological pathways changed by PCB126 treatments. The top predicted pathway altered by PCB126 at all time points was the cytokine-cytokine receptor interaction pathway, a broad pathway involving numerous proinflammatory cytokine and chemokine genes (Table 2, Tables 3, Table 4, Table S2). The AGE-RAGE pathway in diabetic complications was another significantly altered pathway at all time points (Table S2). This pathway is of particular interest with regard to how AhR agonists such as PCB126 and dioxin might be involved in disrupting metabolism through effects on adipogenesis and adipocyte function. For many genes activated in this pathway, progressively increasing gene transcript levels occurred with lengthening exposure to PCB126 (Figure 4).
Table 2:
Pathways altered by PCB126 treatment of preadipocytes at the 9-hour timepoint (all significant, Fdr≤0.05).
| Pathway | pv_fdr |
|---|---|
| Cytokine-cytokine receptor interaction | 0.000213 |
| MAPK signaling pathway | 0.000213 |
| Rheumatoid arthritis | 0.001351 |
| Osteoclast differentiation | 0.002083 |
| AGE-RAGE signaling pathway in diabetic complications | 0.00959 |
| Pathways in cancer | 0.011362 |
| Amoebiasis | 0.014754 |
| Complement and coagulation cascades | 0.015962 |
| NF-kappa B signaling pathway | 0.017713 |
| Ovarian steroidogenesis | 0.017713 |
| Bladder cancer | 0.028612 |
| TGF-beta signaling pathway | 0.036269 |
| Ras signaling pathway | 0.037673 |
| Leishmaniasis | 0.039832 |
| MicroRNAs in cancer | 0.04307 |
| Cholesterol metabolism | 0.043771 |
| Inflammatory bowel disease | 0.043771 |
| Hematopoietic cell lineage | 0.043771 |
| Fluid shear stress and atherosclerosis | 0.04661 |
| Neuroactive ligand-receptor interaction | 0.04661 |
Table 3:
Pathways altered by PCB126 treatment of preadipocytes at the 1-Day timepoint (top 25, Fdr≤0.05).
| Pathway | pv_fdr |
|---|---|
| Cytokine-cytokine receptor interaction | 1.58E-06 |
| Viral protein interaction with cytokine and cytokine receptor | 1.58E-06 |
| AGE-RAGE signaling pathway in diabetic complications | 8.53E-05 |
| TNF signaling pathway | 0.000369 |
| Steroid biosynthesis | 0.000369 |
| Cell cycle | 0.000369 |
| Ovarian steroidogenesis | 0.000369 |
| Rheumatoid arthritis | 0.000393 |
| MAPK signaling pathway | 0.000499 |
| Neuroactive ligand-receptor interaction | 0.000775 |
| Pathways in cancer | 0.000813 |
| Malaria | 0.00103 |
| Oocyte meiosis | 0.001369 |
| NF-kappa B signaling pathway | 0.004984 |
| African trypanosomiasis | 0.005584 |
| IL-17 signaling pathway | 0.006917 |
| Amoebiasis | 0.006938 |
| Fluid shear stress and atherosclerosis | 0.010475 |
| PPAR signaling pathway | 0.011545 |
| Hematopoietic cell lineage | 0.012131 |
| Progesterone-mediated oocyte maturation | 0.012322 |
| Fanconi anemia pathway | 0.012322 |
| Rap1 signaling pathway | 0.012322 |
| Cholesterol metabolism | 0.012322 |
| Osteoclast differentiation | 0.013481 |
Table 4:
Pathways altered by PCB126 treatment of preadipocytes at the 3-Day timepoint (top 25, Fdr<0.05).
| Pathway | pv_fdr |
|---|---|
| Cytokine-cytokine receptor interaction | 3.69E-06 |
| Viral protein interaction with cytokine and cytokine receptor | 4.52E-06 |
| MAPK signaling pathway | 1.1E-05 |
| Osteoclast differentiation | 7.33E-05 |
| Neuroactive ligand-receptor interaction | 7.33E-05 |
| AGE-RAGE signaling pathway in diabetic complications | 0.000132 |
| Human papillomavirus infection | 0.000132 |
| Pathways in cancer | 0.000177 |
| Ras signaling pathway | 0.000197 |
| Complement and coagulation cascades | 0.000197 |
| Cell adhesion molecules | 0.000197 |
| Hypertrophic cardiomyopathy | 0.000197 |
| ECM-receptor interaction | 0.000197 |
| PI3K-Akt signaling pathway | 0.000197 |
| Calcium signaling pathway | 0.000197 |
| Rheumatoid arthritis | 0.000214 |
| Small cell lung cancer | 0.000337 |
| TNF signaling pathway | 0.000337 |
| Axon guidance | 0.000337 |
| Renin secretion | 0.000461 |
| IL-17 signaling pathway | 0.000501 |
| Focal adhesion | 0.000518 |
| NF-kappa B signaling pathway | 0.000553 |
| Amoebiasis | 0.00062 |
| Chemokine signaling pathway | 0.00067 |
Figure 4:
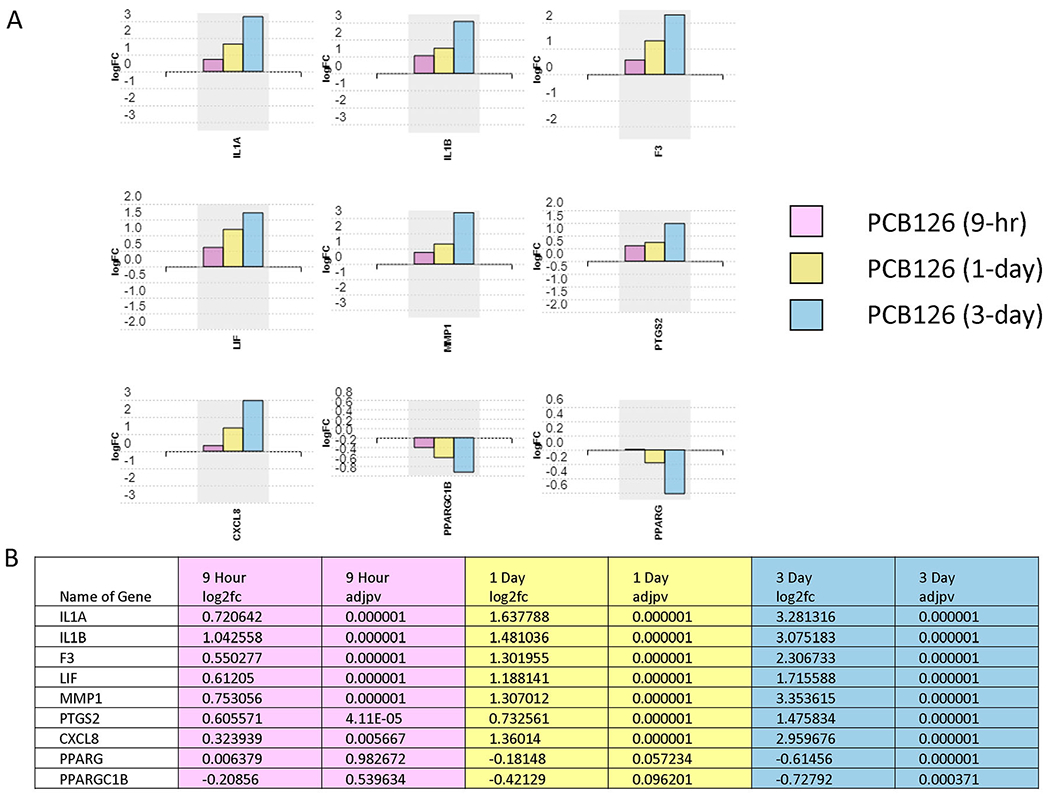
AGE-RAGE pathway gene transcript changes induced by PCB126 treatment of preadipocytes. A. Transcript level changes (log2fold) of genes the AGE-RAGE pathway at different timepoints (9-hour, 1-day, 3-day) of PCB126-exposed preadipocytes compared to vehicle controls at the same timepoints. Values represent log2fold change over vehicle control. B. Transcript level changes (log2fold) of AGE-RAGE pathway genes. Fdr<0.05. Pink: 9-hour exposure; Yellow: 1-day exposure; Blue: 3-day exposure.
The RNAseq data indicated that several other pathways were altered by PCB126 treatment at all three time points (Table S2). These include the MAP kinase pathway, osteoclast differentiation, complement and coagulation cascades, rheumatoid arthritis, and the NF-κB pathway. These pathways likely play a significant role in altering adipogenesis or subsequent preadipocyte function. Other pathways (e.g. neuroactive ligand-receptor interaction, amoebiasis, hematopoietic cell lineage, and fluid shear stress and atherosclerosis) were also altered by PCB126 treatment.
While some pathways were predicted to be activated at all three time points, others were activated only at later time points. Activation of the IL-17A pathway at the later time points of days 1 and 3 (Tables 3 and 4) is of particular interest as it was recently found to be a key regulator of adipogenesis (Teijeiro et al. 2021). It is interesting to note that transcript levels of IL-17A ligand gene or the IL-17 receptor were not altered by PCB126 treatment, but it is rather downstream genes in the IL-17A pathway that are predicted to be activated (Figure 5). This finding suggests that PCB126 treatment activates the IL-17A pathway through a mechanism that does not involve the upregulation of IL-17A ligand or IL-17 receptor proteins.
Figure 5:
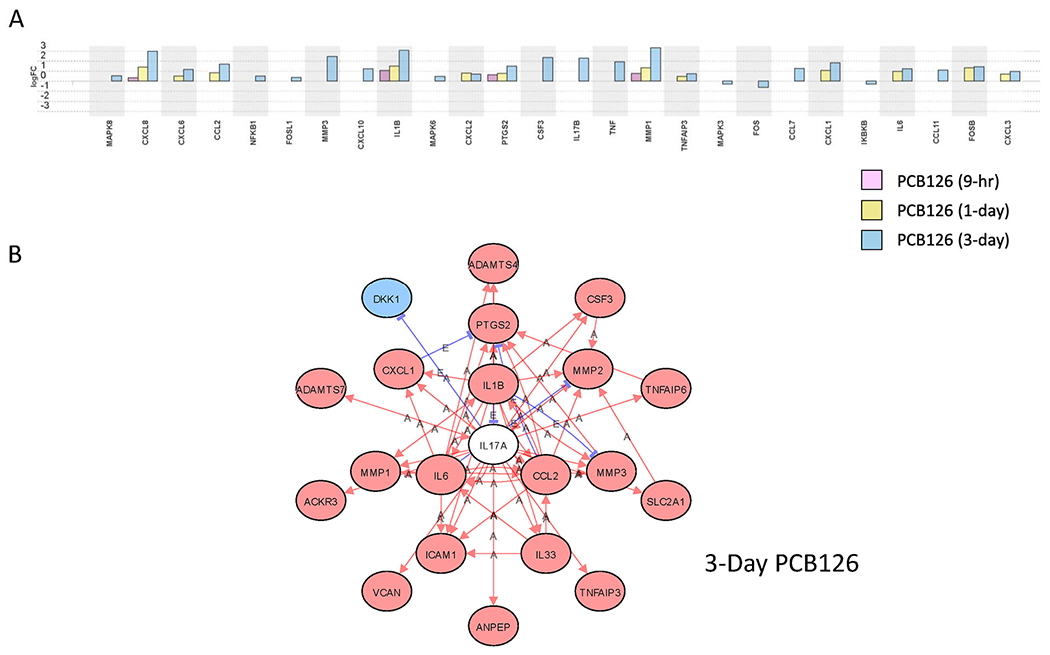
PCB126-induced changes in IL-17A pathway genes. A. Transcript level changes (log2fold) of genes in the IL-17A pathway at different timepoints (9-hour, 1-day, and 3-day) of PCB-exposed preadipocytes compared to vehicle controls at the same timepoints. B. Network analysis of IL-17A pathway genes altered by 3 days of PCB126 treatment of preadipocytes. Red Oval: Upregulated; Blue Oval: Downregulated; White Oval: Unchanged; “A”: Activated; “E”: Expression Inhibited; “I”: Inhibited; →Activation; ┤Inhibition. Fdr≤0.05.
Many of the pathways (e.g. cytokine-cytokine-receptor, rheumatoid arthritis, IL-17) that were significantly affected by PCB126 treatment, particularly at later time points, have genes known to be regulated by NF-κB. Accordingly, the NF-κB signaling pathway was predicted by the iPathwayGuide software to be activated at all time points (Table 2, Tables 3, Table 4, and Table S2).
DISCUSSION
Alterations in gene expression by PCB126 are widespread and increase with time
PCB126 is considered one of the most biologically active of the 209 congeners. It is a prevalent and persistent compound that accounts for a significant fraction of Toxicity Equivalency Factor (TEF) of dioxin-like compounds that humans are exposed to in diet, air, and the environment (Patterson et al. 2008). We have shown that PCB126 acts through AhR in preadipocytes to significantly inhibit adipogenesis and adipocyte function (Gourronc et al. 2019; Gourronc et al. 2018). In the present study, we were interested in determining the breadth of genes and pathways affected by PCB126 beyond AhR over a time course. Our results demonstrate the enormity of the changes in genes and pathways that occur over a 3-day time course. Many of these changes go beyond what are considered classic AhR responsive genes and illustrate how AhR agonists initiate a process that is long-term and consequential. The following sections address the major changes that we observed in our studies and put them in the context of how PCB126 affects preadipocyte function.
Activation of AhR-responsive genes
Many studies, including those from our group, have shown that nearly all of the effects of PCB126 on toxicity, changes in cell function, and pathology are dependent on the ability of PCB126 to activate the AhR. Our results support previous findings using preadipocytes, liver, and endothelial cells that PCB126 activates AhR responsive genes such as the cytochrome P450s, including CYP1A1 and CYPB1 (Eti et al. 2021; Gourronc et al. 2018; Liu et al. 2016; Nault et al. 2013). In most cases, classical AhR-responsive genes increased in magnitude over the time course measured. Also, as time went on, more and more genes considered to be regulated by AhR were upregulated in preadipocytes. At the early timepoint, AhR is considered the most significant upstream regulator (Table 1). On 1 day, while not the most significant upstream regulator, AhR is still considered a highly significant regulator, but is pushed down in the list because other predicted upstream regulators have lower p-values. Although AhR was not a significant upstream regulator at day 3, it is still obvious from the network analysis and transcript level changes of AhR responsive genes that it is active (Figure 2). Overall, our results are consistent with the idea that PCB126 strongly activates AhR responsive genes and that the magnitude of changes in levels of these genes increases with time of exposure. However, it is apparent that secondary responses become more active with time. These secondary responses likely contribute to the biological consequences caused by PCB126 and, likely, other AhR agonists.
Activation of proinflammatory pathways by PCB126
Our previous studies and those of others have demonstrated that PCB126 activates a proinflammatory response in preadipocytes, liver, and endothelial cells (Chapados and Boucher 2017; Gourronc et al. 2018; Kim et al. 2012; Liu et al. 2016). In our studies with preadipocytes, maximum activation of proinflammatory response genes didn’t occur until 2 to 3 days after PCB126 treatment of preadipocytes (Gourronc et al. 2018). The RNAseq results presented here support these findings. Many proinflammatory cytokine and chemokine genes were activated partially by the 9-hour time point but increased further in magnitude over the 3-day time course.
Several proinflammatory pathways significantly affected by PCB126 exposure are of interest. One pathway that was considered to be significantly altered at later time points is the IL-17A pathway. AhR agonists such as kynurenine have been linked to the activation of Th17 T cells, specifically (Esser et al. 2009; Van der Leek et al. 2017). While numerous genes purportedly downstream of IL-17A were upregulated, the IL-17A gene was not affected by PCB126, nor were IL-17 receptors upregulated. IL-17A is mainly expressed and secreted by T cells, with some evidence that myeloid cells such as macrophages, neutrophils, and mast cells also express IL-17A (Bechara et al. 2021). Thus, since these cells are not present in the experiments performed in this study, PCB126 is likely activating this pathway through a mechanism that does not involve IL-17A ligand, specifically. Activation of the IL-17A pathway leads to activation of NF-κB (Goldminz et al. 2013). Activation of NF-κB subsequently upregulates numerous cytokines and chemokines such as IL-6, IL-8, IL-1β , and others, all of which were found to be upregulated by PCB126 in our studies. It is of interest that the IL-17A axis has been shown to play an important role in diet-induced obesity and metabolic syndrome and, specifically, that IL-17A inhibits adipogenesis (Bechara et al. 2021; Li et al. 2019; Teijeiro et al. 2021). Whether this is the mechanism by which PCB126 inhibits adipogenesis is unknown and will be the subject of future studies.
AGE-RAGE pathway activation
One of the top signature pathways was the AGE-RAGE pathway of diabetic complications. AGEs are a heterogeneous group of macromolecules produced by the glycation of proteins, lipids, and nucleic acids (Strieder-Barboza et al. 2019; Wu et al. 2011; Yamagishi et al. 2009). Endogenous AGEs are produced by interactions between glucose and proteins. Glycated proteins are known to interact with a pattern recognition receptor called RAGE. Activation of RAGE has been shown to cause a proinflammatory response and oxidative stress. Activation of RAGE is associated with the development of chronic diseases such as atherosclerosis, obesity, insulin resistance, and cardiac disease (Egana-Gorrono et al. 2020; Lopez-Diez et al. 2016). The RAGE pathway is complex and consists of activation of the MAP kinase pathway, the NF-κB pathway, TGFβ1, and other pathways resulting in the induction of numerous proinflammatory cytokines and chemokines (e.g., IL-6, IL-8, IL-1β, TNFα), as well as genes involved in tissue remodeling (e.g. Matrix Metalloproteinases, MMPs), coagulation (e.g., Tissue Factor, F3), and apoptosis.
As noted, many AGE-RAGE pathway genes were activated by PCB126. These effects may be mainly implemented through activation of the NF-κB pathway, but our results indicate that the MAP kinase pathway and TGFβ1 pathways are also affected by PCB126. In fact, the MAP kinase pathway was consistently activated across all three time points. Activation of the MAPK pathway by AhR agonists, including PCB126, has been previously reported (Song and Freedman 2005). The exact mechanisms of activation of this pathway by AhR are unclear. Still, it is possible that the upregulation of genes involved in the MAPK signaling pathway is not through the binding of AhR to classic xenobiotic response elements (XREs)(Weiss et al. 2005). The regulatory mechanisms of AhR on MAPKs are multifactorial and include phosphorylation by various kinases (Henklova et al. 2008). How the MAPK signaling pathway is activated by PCB126 and its role in mediating the biological effects on adipogenesis and adipocyte function are questions that require further investigation.
There is limited information in the literature linking exposure to environmental persistent organic pollutants, specifically dioxin-like compounds, to AGE-RAGE activation. A recent study showed that PCB126 caused activation of the AGE-RAGE pathway in HepaRG liver cells (Mesnage et al. 2018). Another study in which several persistent organic pollutants, including pp-dichlorodiphenyldichloroethylene (DDE), pp-dichlorodiphenyldichloroethane (DDD), hexachlorobenzene, dieldrin, transnonachlor, PCB153, PCB138, PCB118, PCB77, and PCB126 were gavaged into mice demonstrated activation of the AGE-RAGE pathway specifically in cardiac tissue (Coole et al. 2019). Thus, PCB exposure affects the AGE-RAGE pathway in the liver and cardiac cells. Activation of AGE-RAGE has been associated with inflammation and insulin resistance in adipose tissue (Strieder-Barboza et al. 2019; Wu et al. 2011; Yamagishi et al. 2009). The strong association of AGE-RAGE with diabetes would suggest that activation of AGE-RAGE in preadipocytes, as we have seen here, would be associated with the development of diabetes. Further studies will be needed to tease out how AGE-RAGE is activated, what this activation means regarding the disruption of adipose function, and how this can lead to the development of diabetes.
The effects of PCB126 on adipogenesis and adipocyte function
Our previous studies demonstrated a clear inhibition of adipogenesis by exposure of preadipocytes to PCB126 (Gadupudi et al. 2015). This inhibition was associated with subsequent disruption of adipocyte function, including blockage of the thermogenic response (Gourronc et al. 2019). All of these alterations were dependent on the AhR (Gourronc et al. 2019; Gourronc et al. 2018). The findings presented in the current study shed light on the many changes caused by PCB126 exposure in preadipocytes. Our results demonstrate that AhR genes are activated at the 9-hour time point. However, the most significant predicted upstream regulator gene at day 1 is SREBF2 (aka SREBP2) (Table 1). SREBF2 is followed on the list by NFY complex subunit genes, NFYA, NFYB, and NFYC. Thus, the changes observed on day 1 indicate a shift in expression of genes involved in sterol and fatty acid metabolism. Such alterations would be expected to directly affect lipogenesis and adipocyte gene expression (Lu et al. 2015; Nishi-Tatsumi et al. 2017; Ye and DeBose-Boyd 2011). Changes on day 3, on the other hand, point to massive changes in inflammatory response genes. Along with the upregulation of cytokine and chemokine genes, it is of interest to note that PPARγ, the master regulator of adipogenesis, was downregulated at the day 3 time point. This finding is consistent with our previous studies indicating downregulation in PPARγ in differentiated adipocytes treated with PCB126 (Gadupudi et al. 2015). That PPARγ is already downregulated in preadipocytes (before differentiation) by PCB126 suggests that this is a key factor in blocking subsequent adipogenesis.
Considering the number and magnitude of changes that occur upon PCB126 treatment, it is not surprising that adipogenesis is significantly disrupted. Preadipocytes are an under-appreciated cell type with regard to their role in responses to environmental toxins. Our results demonstrate that they are highly proinflammatory. In previous studies, inflammation in adipose tissue has been mainly attributed to infiltrating macrophages (Rosen and Spiegelman 2014). It is possible that preadipocytes, which are thought to make up 5-10% of adipose tissue, also contribute to this response. Adipose inflammation is strongly linked to obesity and the development of insulin resistance. As PCBs and other POPs accumulate in adipose tissue, the likelihood of these compounds causing a proinflammatory response is high, leading to cardiometabolic disease.
The 10 μM PCB126 that we used in our study is considered a high concentration compared to what might be expected for human exposure. Our previous studies have shown that this concentration causes a reproducible and large effect on adipogenesis without causing overt cytotoxicity (Gadupudi et al. 2015; Gourronc et al. 2018). The concentration used in our study was a one-time treatment over a relatively short time frame compared to what would occur in human exposure. To measure the exact dose of human exposure to AhR ligands in their lifetime is difficult. It should be noted that humans are not only exposed to PCB126 but that there are several AhR ligands in our environment (e.g. many pesticides, TCDD, PAHs, and other dioxin-like PCBs such as PCB77 and PCB118) that are likely to affect human health (Everett and Thompson 2012). Cumulative quantities of AHR ligands, particularly in fat, can reach high levels (Darbre 2017). While a high concentration of PCB126 was used in our studies, the results give insight into mechanisms that help to identify changes relevant to human health and provide important information to explore the mechanisms of AhR activation in modulating adipogenesis and adipocyte function. However, it would be interesting to perform additional studies in our model system using lower PCB126 concentrations over a longer-term treatment (e.g., several passages).
Relationship of in vitro RNAseq results to in vivo studies
The findings presented here were generated in vitro using human preadipocytes. Several in vivo studies have been done to assess the effects of PCB126 on various aspects of disease. Exposure of rats to PCB126 leads to morbidity over time, including liver steatosis and ending in wasting syndrome (Gadupudi et al. 2016). Effects were observed that are associated with toxicity on liver energy metabolism, including inhibition of AMPK-CREB signaling (Eti et al. 2021; Gadupudi et al. 2016). Other effects mediated by PCB126 include reproductive toxicity (Klenov et al. 2021). AhR knockout rats were resistant to these effects, indicating that these effects are mediated through AhR (Eti et al. 2021; Klenov et al. 2021). Other studies in mice have whown that PCB126 causes peripheral vascular disease along with steatosis (Wahlang et al. 2017). While these animal studies have not focused on adipose, specifically, and they were done using acute doses of PCB126, they illustrate how PCB126 exposure progressively leads to disease in rodents. This is not unexpected given our finding that PCB126 exposure initiates a cascade of transcriptional events that result in progressive disruption of numerous biological pathways.
Aside from PCB126, other in vivo studies have linked AhR to cardiometabolic disease particularly with regard to its effects on adipose tissue. Exposures of mice to TCDD or another coplanar PCB, PCB77, were shown to lead to the development of obesity and disruption of glucose homeostasis (Arsenescu et al. 2008; Baker et al. 2013; Brulport et al. 2017). Studies using whole body knockout of AhR or tissue-specific knockout that leads to loss of AhR in preadipocytes (by using PDGFRα-Cre) have demonstrated that loss of AhR in these conditions leads to protection against high-fat diet (HFD) induced obesity that is specifically linked to increased adipose thermogenesis (Gourronc et al. 2020; Jaeger et al. 2017; Xu et al. 2015). In contrast, a study by Baker et al. reported that knockout of AHR in mature adipocytes (by using Adiponectin-Cre) resulted in increased body weight, increased adipose mass, increased adipose inflammation, and impaired glucose tolerance when mice were fed a HFD (Baker et al. 2015). These findings illustrate the complexity of how AHR affects adipogenesis and adipocyte function. The differing results may be due to when AHR is knocked out. Not having AHR in preadipocytes or earlier precursor cells (i.e., in the whole body knock out or via PDGFRα-Cre) versus knockout in mature adipocytes (i.e., via Adiponectin-Cre) may lead to different consequences in the context of HFD with regard to the development of obesity. Other studies have shown that chemical inhibitors of AhR may offer protection against HFD-induced obesity and steatosis (Girer et al. 2020; Moyer et al. 2017; Rojas et al. 2020). These in vivo studies point to an important role for AhR in cardiometabolic disease and are relevant to our finding that PCB126 disrupts transcriptional pathways in preadipocytes.
Future studies
Our findings provide a framework for future studies on how PCB126 and other toxicants that are AhR agonists cause disease. Given our results, it is no surprise that PCB126 is so biologically active in affecting the functions of a wide variety of cell types and tissues. The RNAseq studies reported here lay the groundwork for future work on how PCB126 and AhR activation affect the different pathways to cause preadipocyte dysfunction. As the results will be publicly available, it is hoped that other laboratories will be able to utilize the information in their own studies on how dioxin-like compounds cause disease.
Supplementary Material
Acknowledgments and Funding
The RNAseq data and analyses were obtained at the Genomics and Bioinformatics Divisions of the Iowa Institute of Human Genetics which is supported, in part, by the University of Iowa Carver College of Medicine and the Holden Comprehensive Cancer Center (National Cancer Institute of the National Institutes of Health under Award Number P30 CA086862). This study was supported by NIH P42 ES013661 (AJK, JAA, LWR, HJL) and a pilot grant from the Environmental Health Research Center P30 ES005605 (AJK, JAA).
REFERENCES
- Arsenescu V, Arsenescu RI, King V, Swanson H, Cassis LA. 2008. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect. 116(6):761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Karounos M, English V, Fang J, Wei Y, Stromberg A, Sunkara M, Morris AJ, Swanson HI, Cassis LA. 2013. Coplanar polychlorinated biphenyls impair glucose homeostasis in lean c57bl/6 mice and mitigate beneficial effects of weight loss on glucose homeostasis in obese mice. Environ Health Perspect. 121(1):105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Shoemaker R, English V, Larian N, Sunkara M, Morris AJ, Walker M, Yiannikouris F, Cassis LA. 2015. Effects of adipocyte aryl hydrocarbon receptor deficiency on pcb-induced disruption of glucose homeostasis in lean and obese mice. Environ Health Perspect. 123(10):944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara R, McGeachy MJ, Gaffen SL. 2021. The metabolism-modulating activity of il-17 signaling in health and disease. J Exp Med. 218(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer A, Biziuk M. 2009. Environmental fate and global distribution of polychlorinated biphenyls. Rev Environ Contam Toxicol. 201:137–158. [DOI] [PubMed] [Google Scholar]
- Brulport A, Le Corre L, Chagnon MC. 2017. Chronic exposure of 2,3,7,8-tetrachlorodibenzo-p-dioxin (tcdd) induces an obesogenic effect in c57bl/6j mice fed a high fat diet. Toxicology. 390:43–52. [DOI] [PubMed] [Google Scholar]
- Chapados NA, Boucher MP. 2017. Liver metabolic disruption induced after a single exposure to pcb126 in rats. Environ Sci Pollut Res Int. 24(2):1854–1861. [DOI] [PubMed] [Google Scholar]
- Cohen P, Spiegelman BM. 2015. Brown and beige fat: Molecular parts of a thermogenic machine. Diabetes. 64(7):2346–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Spiegelman BM. 2016. Cell biology of fat storage. Mol Biol Cell. 27(16):2523–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coole JB, Burr SS, Kay AM, Singh JA, Kondakala S, Yang EJ, Kaplan BLF, Howell GE 3rd, Stewart JA Jr. 2019. Persistent organic pollutants (pops) increase rage signaling to promote downstream cardiovascular remodeling. Environ Toxicol. 34(10):1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbre PD. 2017. Endocrine disruptors and obesity. Curr Obes Rep. 6(1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirinck E, Jorens PG, Covaci A, Geens T, Roosens L, Neels H, Mertens I, Van Gaal L. 2011. Obesity and persistent organic pollutants: Possible obesogenic effect of organochlorine pesticides and polychlorinated biphenyls. Obesity (Silver Spring). 19(4):709–714. [DOI] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R. 2007. A systems biology approach for pathway level analysis. Genome Res. 17(10):1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusanov S, Ruzzin J, Kiviranta H, Klemsdal TO, Retterstol L, Rantakokko P, Airaksinen R, Djurovic S, Tonstad S. 2018. Associations between persistent organic pollutants and metabolic syndrome in morbidly obese individuals. Nutr Metab Cardiovasc Dis. 28(7):735–742. [DOI] [PubMed] [Google Scholar]
- Egana-Gorrono L, Lopez-Diez R, Yepuri G, Ramirez LS, Reverdatto S, Gugger PF, Shekhtman A, Ramasamy R, Schmidt AM. 2020. Receptor for advanced glycation end products (rage) and mechanisms and therapeutic opportunities in diabetes and cardiovascular disease: Insights from human subjects and animal models. Front Cardiovasc Med. 7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C, Rannug A, Stockinger B. 2009. The aryl hydrocarbon receptor in immunity. Trends Immunol. 30(9):447–454. [DOI] [PubMed] [Google Scholar]
- Eti NA, Flor S, Iqbal K, Scott RL, Klenov VE, Gibson-Corley KN, Soares MJ, Ludewig G, Robertson LW. 2021. Pcb126 induced toxic actions on liver energy metabolism is mediated by ahr in rats. Toxicology. 153054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett CJ, Thompson OM. 2012. Associations of dioxins, furans and dioxin-like pcbs with diabetes and pre-diabetes: Is the toxic equivalency approach useful? Environ Res. 118:107–111. [DOI] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Kaller M. 2016. Multiqc: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32(19):3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feve B, Bastard JP. 2009. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 5(6):305–311. [DOI] [PubMed] [Google Scholar]
- Gadupudi G, Gourronc FA, Ludewig G, Robertson LW, Klingelhutz AJ. 2015. Pcb126 inhibits adipogenesis of human preadipocytes. Toxicology in vitro : an international journal published in association with BIBRA. 29(1):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadupudi GS, Elser BA, Sandgruber FA, Li X, Gibson-Corley KN, Robertson LW. 2018. Pcb126 inhibits the activation of ampk-creb signal transduction required for energy sensing in liver. Toxicol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadupudi GS, Klaren WD, Olivier AK, Klingelhutz AJ, Robertson LW. 2016. Pcb126-induced disruption in gluconeogenesis and fatty acid oxidation precedes fatty liver in male rats. Toxicol Sci. 149(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girer NG, Tomlinson CR, Elferink CJ. 2020. The aryl hydrocarbon receptor in energy balance: The road from dioxin-induced wasting syndrome to combating obesity with ahr ligands. Int J Mol Sci. 22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldminz AM, Au SC, Kim N, Gottlieb AB, Lizzul PF. 2013. Nf-kappab: An essential transcription factor in psoriasis. J Dermatol Sci. 69(2):89–94. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Casanova JE, Pertuz-Cruz SL, Caicedo-Ortega NH, Rojas-Gomez DM. 2020. Adipogenesis regulation and endocrine disruptors: Emerging insights in obesity. Biomed Res Int. 2020:7453786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourronc FA, Markan KR, Kulhankova K, Zhu Z, Sheehy R, Quelle DE, Zingman LV, Kurago ZB, Ankrum JA, Klingelhutz AJ. 2020. Pdgfralpha-cre mediated knockout of the aryl hydrocarbon receptor protects mice from high-fat diet induced obesity and hepatic steatosis. PloS one. 15(7):e0236741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourronc FA, Perdew GH, Robertson LW, Klingelhutz AJ. 2019. Pcb126 blocks the thermogenic beiging response of adipocytes. Environ Sci Pollut Res Int. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourronc FA, Robertson LW, Klingelhutz AJ. 2018. A delayed proinflammatory response of human preadipocytes to pcb126 is dependent on the aryl hydrocarbon receptor. Environ Sci Pollut Res Int. 25(17):16481–16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice BA, Nelson RG, Williams DE, Knowler WC, Mason C, Hanson RL, Bullard KM, Pavkov ME. 2017. Associations between persistent organic pollutants, type 2 diabetes, diabetic nephropathy and mortality. Occup Environ Med. 74(7):521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm FA, Hu D, Kania-Korwel I, Lehmler HJ, Ludewig G, Hornbuckle KC, Duffel MW, Bergman A, Robertson LW. 2015. Metabolism and metabolites of polychlorinated biphenyls. Crit Rev Toxicol. 45(3):245–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson B, Gogg S, Hedjazifar S, Jenndahl L, Hammarstedt A, Smith U. 2009. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am J Physiol Endocrinol Metab. 297(5):E999–E1003. [DOI] [PubMed] [Google Scholar]
- Heindel JJ, Newbold R, Schug TT. 2015. Endocrine disruptors and obesity. Nat Rev Endocrinol. 11(11):653–661. [DOI] [PubMed] [Google Scholar]
- Henklova P, Vrzal R, Ulrichova J, Dvorak Z. 2008. Role of mitogen-activated protein kinases in aryl hydrocarbon receptor signaling. Chem Biol Interact. 172(2):93–104. [DOI] [PubMed] [Google Scholar]
- Henriquez-Hernandez LA, Luzardo OP, Valeron PF, Zumbado M, Serra-Majem L, Camacho M, Gonzalez-Antuna A, Boada LD. 2017. Persistent organic pollutants and risk of diabetes and obesity on healthy adults: Results from a cross-sectional study in spain. Sci Total Environ. 607–608:1096-1102. [DOI] [PubMed] [Google Scholar]
- Hestermann EV, Stegeman JJ, Hahn ME. 2000. Relative contributions of affinity and intrinsic efficacy to aryl hydrocarbon receptor ligand potency. Toxicology and applied pharmacology. 168(2):160–172. [DOI] [PubMed] [Google Scholar]
- Hu X, Adamcakova-Dodd A, Lehmler HJ, Hu D, Kania-Korwel I, Hornbuckle KC, Thorne PS. 2010. Time course of congener uptake and elimination in rats after short-term inhalation exposure to an airborne polychlorinated biphenyl (pcb) mixture. Environ Sci Technol. 44(17):6893–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson E, Shoemaker R, Larian N, Cassis L. 2017. Adipose tissue as a site of toxin accumulation. Compr Physiol. 7(4):1085–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger C, Xu C, Sun M, Krager S, Tischkau SA. 2017. Aryl hydrocarbon receptor-deficient mice are protected from high fat diet-induced changes in metabolic rhythms. Chronobiol Int. 34(3):318–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SN, Vyas SM, Duffel MW, Parkin S, Lehmler HJ. 2011. Synthesis of sterically hindered polychlorinated biphenyl derivatives. Synthesis (Stuttg). 7:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Draghici S. 2005. Ontological analysis of gene expression data: Current tools, limitations, and open problems. Bioinformatics. 21(18):3587–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. Hisat: A fast spliced aligner with low memory requirements. Nat Methods. 12(4)357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with hisat2 and hisat-genotype. Nat Biotechnol. 37(8):907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Pelloux V, Guyot E, Tordjman J, Bui LC, Chevallier A, Forest C, Benelli C, Clement K, Barouki R. 2012. Inflammatory pathway genes belong to major targets of persistent organic pollutants in adipose cells. Environ Health Perspect. 120(4):508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenov V, Flor S, Ganesan S, Adur M, Eti N, Iqbal K, Soares MJ, Ludewig G, Ross JW, Robertson LW et al. 2021. The aryl hydrocarbon receptor mediates reproductive toxicity of polychlorinated biphenyl congener 126 in rats. Toxicology and applied pharmacology. 426:115639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlgruber A, Lynch L. 2015. Adipose tissue inflammation in the pathogenesis of type 2 diabetes. Curr Diab Rep. 15(11):92. [DOI] [PubMed] [Google Scholar]
- Lee YM, Kim KS, Kim SA, Hong NS, Lee SJ, Lee DH. 2014. Prospective associations between persistent organic pollutants and metabolic syndrome: A nested case-control study. Sci Total Environ. 496:219–225. [DOI] [PubMed] [Google Scholar]
- Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. 2019. I1-17 receptor-based signaling and implications for disease. Nat Immunol. 20(12):1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XL, H.J. 2019. Authentication of 3,3’,4,4’,5-pentachlorobiphenyl. figshare. [Google Scholar]
- Liu D, Perkins JT, Hennig B. 2016. Egcg prevents pcb-126-induced endothelial cell inflammation via epigenetic modifications of nf-kappab target genes in human endothelial cells. J Nutr Biochem. 28:164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Diez R, Shekhtman A, Ramasamy R, Schmidt AM. 2016. Cellular mechanisms and consequences of glycation in atherosclerosis and obesity. Biochim Biophys Acta. 1862(12):2244–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YH, Dallner OS, Birsoy K, Fayzikhodjaeva G, Friedman JM. 2015. Nuclear factor-y is an adipogenic factor that regulates leptin gene expression. Mol Metab. 4(5):392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesnage R, Biserni M, Balu S, Frainay C, Poupin N, Jourdan F, Wozniak E, Xenakis T, Mein CA, Antoniou MN. 2018. Integrated transcriptomics and metabolomics reveal signatures of lipid metabolism dysregulation in heparg liver cells exposed to pcb 126. Arch Toxicol. 92(8):2533–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer BJ, Rojas IY, Kerley-Hamilton JS, Nemani KV, Trask HW, Ringelberg CS, Gimi B, Demidenko E, Tomlinson CR. 2017. Obesity and fatty liver are prevented by inhibition of the aryl hydrocarbon receptor in both female and male mice. Nutrition research. 44:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R, Forgacs AL, Dere E, Zacharewski TR. 2013. Comparisons of differential gene expression elicited by tcdd, pcb126, betanf, or icz in mouse hepatoma hepa1c1c7 cells and c57bl/6 mouse liver. Toxicol Lett. 223(l):52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TM, Shafi A, Nguyen T, Draghici S. 2019. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 20(1):203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi-Tatsumi M, Yahagi N, Takeuchi Y, Toya N, Takarada A, Murayama Y, Aita Y, Sawada Y, Piao X, Oya Y et al. 2017. A key role of nuclear factor y in the refeeding response of fatty acid synthase in adipocytes. FEBS Lett. 591(7):965–978. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 14(4):417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson DG Jr., Turner WE, Caudill SP, Needham LL. 2008. Total teq reference range (pcdds, pcdfs, cpcbs, mono-pcbs) for the us population 2001-2002. Chemosphere. 73(1 Suppl):S261–277. [DOI] [PubMed] [Google Scholar]
- Rojas IY, Moyer BJ, Ringelberg CS, Tomlinson CR. 2020. Reversal of obesity and liver steatosis in mice via inhibition of aryl hydrocarbon receptor and altered gene expression of cyp1b1, pparalpha, scd1, and osteopontin. Int J Obes (Lond). 44(4):948–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. 2014. What we talk about when we talk about fat. Cell. 156(1-2):20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzin J, Petersen R, Meugnier E, Madsen L, Lock EJ, Lillefosse H, Ma T, Pesenti S, Sonne SB, Marstrand TT et al. 2010. Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ Health Perspect. 118(4):465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saktrakulkla P, Lan T, Hua J, Marek RF, Thorne PS, Hornbuckle KC. 2020. Polychlorinated biphenyls in food. Environ Sci Technol. 54(18):11443–11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, Robinson MD. 2015. Differential analyses for rna-seq: Transcript-level estimates improve gene-level inferences. F1000Res. 4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MO, Freedman JH. 2005. Activation of mitogen activated protein kinases by pcb126 (3,3’,4,4’,5-pentachlorobiphenyl) in hepg2 cells. Toxicol Sci. 84(2):308–318. [DOI] [PubMed] [Google Scholar]
- Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng YH et al. 2013. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. 123(1):215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieder-Barboza C, Baker NA, Flesher CG, Karmakar M, Neeley CK, Polsinelli D, Dimick JB, Finks JF, Ghaferi AA, Varban OA et al. 2019. Advanced glycation end-products regulate extracellular matrix-adipocyte metabolic crosstalk in diabetes. Sci Rep. 9(1):19748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KW, Novak RF, Anderson HA, Birnbaum LS, Blystone C, Devito M, Jacobs D, Kohrle J, Lee DH, Rylander L et al. 2013. Evaluation of the association between persistent organic pollutants (pops) and diabetes in epidemiological studies: A national toxicology program workshop review. Environ Health Perspect. 121(7):774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijeiro A, Garrido A, Ferre A, Perna C, Djouder N. 2021. Inhibition of the il-17a axis in adipocytes suppresses diet-induced obesity and metabolic disorders in mice. Nat Metab. 3(4):496–512. [DOI] [PubMed] [Google Scholar]
- Van der Leek AP, Yanishevsky Y, Kozyrskyj AL. 2017. The kynurenine pathway as a novel link between allergy and the gut microbiome. Front Immunol. 8:1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu BG, Gourronc FA, Bernlohr DA, Schlievert PM, Klingelhutz AJ. 2013. Staphylococcal superantigens stimulate immortalized human adipocytes to produce chemokines. PloS one. 8(10):e77988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Barney J, Thompson B, Wang C, Hamad OM, Hoffman JB, Petriello MC, Morris AJ, Hennig B. 2017. Editor’s highlight: Pcb126 exposure increases risk for peripheral vascular diseases in a liver injury mouse model. Toxicol Sci. 160(2):256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Adamcakova-Dodd A, Flor S, Gosse L, Klenov VE, Stolwijk JM, Lehmler HJ, Hornbuckle KC, Ludewig G, Robertson LW et al. 2020. Comprehensive subchronic inhalation toxicity assessment of an indoor school air mixture of pcbs. Environ Sci Technol. 54(24):15976–15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss C, Faust D, Durk H, Kolluri SK, Pelzer A, Schneider S, Dietrich C, Oesch F, Gottlicher M. 2005. Tcdd induces c-jun expression via a novel ah (dioxin) receptor-mediated p38-mapk-dependent pathway. Oncogene. 24(31):4975–4983. [DOI] [PubMed] [Google Scholar]
- Wu CH, Huang HW, Huang SM, Lin JA, Yeh CT, Yen GC. 2011. Age-induced interference of glucose uptake and transport as a possible cause of insulin resistance in adipocytes. J Agric Food Chem. 59(14):7978–7984. [DOI] [PubMed] [Google Scholar]
- Xu CX, Wang C, Zhang ZM, Jaeger CD, Krager SL, Bottum KM, Liu J, Liao DF, Tischkau SA. 2015. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int J Obes (Lond). 39(8):1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi S, Nakamura K, Matsui T. 2009. Regulation of advanced glycation end product (age)-receptor (rage) system by ppar-gamma agonists and its implication in cardiovascular disease. Pharmacol Res. 60(3):174–178. [DOI] [PubMed] [Google Scholar]
- Ye J, DeBose-Boyd RA. 2011. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol. 3(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.