

ABSTRACT
The association between the physio-pathological variables of type 2 diabetes (T2D) and gut microbiota composition suggests a new avenue to track the disease and improve the outcomes of pharmacological and non-pharmacological treatments. This enterprise requires new strategies to elucidate the metabolic disturbances occurring in the gut microbiome as the disease progresses. To this end, physiological knowledge and systems biology pave the way for characterizing microbiota and identifying strategies in a move toward healthy compositions. Here, we dissect the recent associations between gut microbiota and T2D. In addition, we discuss recent advances in how drugs, diet, and exercise modulate the microbiome to favor healthy stages. Finally, we present computational approaches for disentangling the metabolic activity underlying host-microbiota codependence. Altogether, we envision that the combination of physiology and computational modeling of microbiota metabolism will drive us to optimize the diagnosis and treatment of T2D patients in a personalized way.
KEYWORDS: Gut microbiome, type 2 diabetes, diet, physical activity, anti-diabetic drugs, high-throughput data, personalized medicine, systems biology, dysbiosis
Introduction
Type 2 diabetes mellitus (T2D) is a metabolic disorder characterized by hyperglycemia as a result of insulin resistance (IR) and a relative lack of insulin in the human body.1 Notably, dysbiosis of the gut microbiome accompanies the progression of IR in T2D and the development of microvascular (retinopathy, nephropathy, and neuropathy) and macrovascular (atherosclerosis) complications of diabetes.2,3 This dysbiosis remodels the intestinal barrier and insulin signals through metabolites derived from bacteria, which interact with receptors on epithelial, fat, muscle, liver, pancreatic, and cardiac cells. Thus, metabolic signals produced by the gut microbiome can indirectly promote IR by altering the host’s metabolism. Among these metabolic changes, we highlight metabolic endotoxemia and the low rate of production of short-chain fatty acids (SCFAs) and secondary bile acids (BAs).1 Although these findings have helped to characterize the association between the gut microbiome profile and T2D, it remains a challenge to grasp the mechanisms that drive their codependence; it is also difficult to use this knowledge to modulate the metabolic crosstalk between the microbiome and host.4
Currently, there is great interest in controlling external factors to modulate host-microbiome metabolic crosstalk and to restore patients to a healthy state. Thus, classical variables associated with lifestyle, such as diet and exercise, have increased their relevance in personalized medicine.5 Moreover, new technologies, such as fecal microbiota transplantation or bacteriophage intervention (phagosome), are promising technologies for enhancing patients’ wellness and treatment.6,7 Currently, two approaches are helping to reach these objectives. On the one hand, 16S rRNA and shotgun metagenome technologies allow us to carefully monitor the physiological state and composition of the gut microbiome in patients with different degrees of T2D.8,9 This massive amount of biological data contributes to characterizing the phenotype state of patients and evaluating how these phenotypes are altered as the disease progresses. On the other hand, the development of computational modeling approaches (CMAs) with the capacity to integrate high-throughput (HT) data is a pioneering effort to elucidate the ecological interactions of the bacterial community, and to postulate the metabolic mechanism by which T2D progresses. In particular, computational models based on inference microbial interactions and genome-scale metabolic reconstructions have emerged as a remarkable scheme required to understand the metabolic activity of the gut microbiome and to track the changes that follow the emergence of T2D.9 Currently, this field is nascent, but some pioneering advances have been reported in T2D and type 1 diabetes (T1D). In this narrative review, we analyze the state of the art of the association between the gut microbiome and T2D, the factors that modulate the interaction, and some in silico strategies to reveal the underlying metabolic mechanisms. First, we discuss the cutting-edge evidence of the relationship between host-microbiota metabolism and physiological alterations associated with T2D. Then, we discuss recent publications that highlight the importance of handling microbiome composition through lifestyle, diet, and promising intervention methods to modulate the gut microbiome. Finally, the last section is devoted to presenting and discussing frontiers in computational strategies to describe the complex interactions in the bacterial community, shed light on their organization, and build testable hypotheses to modulate metabolic mechanisms. Overall, our review highlights the importance of combining physiological knowledge, HT technologies, and computational modeling of microbiome metabolisms for designing microbiome interventions in favor of a healthy phenotype. To add an original contribution on the state of the art, the structure of the review was grounded in a bibliometric analysis with the Bibliometrix R library.10 This analysis was built with the terms “type 2 diabetes”, “gut microbiota”, “gut microbiome”, “systems biology” and “bioinformatics”, as keywords to find either in the documents’ titles or abstracts. Next, we filtered this search based on the review article as a type of document. Interestingly, we did not detect any document related to the content of our manuscript (Table S1-S2). Undoubtedly, the achievements around this field will have a strong impact on precision medicine for optimizing the outcome of treatments and improving patients’ quality of life.
The gut microbiome and T2D
The gut microbiome is a complex microbial ecosystem that coexists with various biological processes and metabolic capacities in the host.11 Through interactive evolutionary processes, hosts and their microbiomes have established mutual benefits. With the abundant evidence of this relationship and its influence on health, humans and their gut microbiome can be considered holobionts, and the health of the host depends on the microbiota and cannot be seen as disconnected from it.12 Nevertheless, there are intrinsic (genetics, age, sex, and health condition) and environmental factors (diet, antibiotic consumption, and lifestyle) that affect the composition of the gut microbiome and its structural functions. These factors, known as microbial disruptors, can alter a variety of physiological mechanisms that favor the development of pathologies such as intestinal permeability, chronic low-grade inflammation, and changes in carbohydrates metabolism and its associated signaling pathways (the insulin route).13
Understanding the biochemical processes related to the interaction between microbial disruptors and the gut microbiome could explain the remarkable relationship of the gut microbiome with its hosts. From a metabolic point of view, the gut microbiome can be conceived as a bioreactor inside the host, leading to the production of bioactive compounds, and whose dysbiosis could be associated with the development of T2D.14 The set of metabolites derived from the gut microbiome serves as a source of signals that facilitate communication between the body’s organs via the nervous system (afferent and efferent autonomic pathways). Through these signals, the gut microbiota (GM) modulate the immune, endocrine, gastrointestinal, and nervous systems, forming the microbiota-gut-brain axis.15 When bacteria-host communication fails, vital functions of the host are interrupted, causing numerous dysfunctions associated with disease. In the case of T2D, several studies have characterized the composition of GM and have confirmed the existence of a particular dysbiosis depending on biogeographical variables.16,17 For instance, in European and Chinese populations, remarkable differences between T2D and healthy subjects were the low relative abundances of butyrate-producing bacteria (Roseburia intestinalis and Faecalibacterium prausnitzii) and the higher relative abundance of species such as Lactobacillus, as well as some opportunistic pathogens like Bacteroides caccae, Clostridium hathewayi, Clostridium ramosum, Clostridium symbiosum, and Escherichia coli.18,19 Previous results suggest that an interesting aspect of these findings is that GM has been associated with the immune system (IS) through immunoinflammatory signaling, which can affect insulin sensitivity (Figure 1).20
Figure 1.
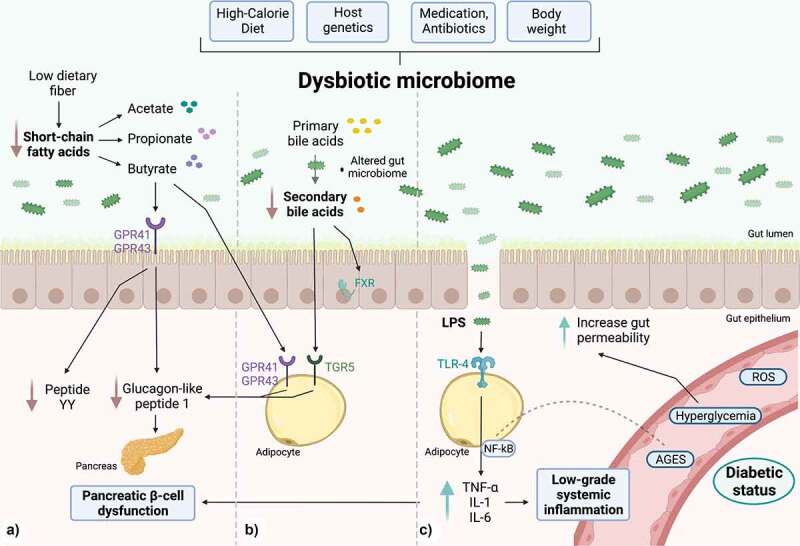
Gut microbiome-derived mechanisms are able to modulate the chronic inflammatory state in DT2. a) SCFAs, products of dietary fiber fermentation, promote GLP-1 and YY peptide secretion in L cells by activating G protein-coupled receptors such as GPR41 and GPR43. With a dysbiotic microbiome, there is an overall decline in the production of SCFAs, leading to a reduction in GLP-1 secretion, impairing pancreatic insulin secretion. b) Secondary bile acids derived from the intestinal microbiome act as mediator molecules through nuclear receptors such as the FXR receptor and the TGR5 membrane receptor, which in intestinal L cells improve glucose metabolism by stimulating GLP-1 production and promoting insulin secretion. Also, in muscle (not shown), they enhance mitochondrial activity and facilitate insulin sensitization. c) PAMPs, e.g., LPS can bind to the TLR4 receptor, and stimulate the expression of pro-inflammatory cytokines IL-6, IL-1 and TNF-α, which are characteristic of a low-grade systemic inflammatory state. There is an increase in intestinal permeability due to the direct effects of glucotoxicity and gut dysbiosis. Created with BioRender.com.
Of the most evident associations between T2D and physiological alterations, we highlight those comprising the gut microbiome and alterations of glucose metabolism through SCFA and BA secondary reduction (Figures 1A and 1b).14,21 GM typically generate SCFAs through the saccharolytic fermentation of dietary fiber. SCFA production is carried out mainly in the distal and proximal colon at a 60:20:20 molar ratio (acetate, propionate, and butyrate).22 Fifteen grams of non-digestible carbohydrates in the colon subjected to saccharolytic fermentation produce between 400 and 600 mmol/day of SCFAs.23 The effectiveness of this biochemical transformation depends on numerous factors, including pH, which influences the growing competition of bacteria in the intestine. Bacteria such as Lactobacilli, Roseburia, Faecalibacterium prausnitzii, and Bifidobacteria promote health in the host and enhance the production of SCFAs at a pH of 5.5. Alternatively, when the non-fermentable fiber is limited in the distal large intestine, the luminal pH is raised to 6.5, eliminating butyrate-producing bacteria almost entirely.24
GM uses major bacterial metabolic pathways (Embden-Meyerhof-Parnas, pentoses-phosphate, Wood-Ljungdahl, succinate, acrylate, and propanediol) to produce SCFAs.25 SCFAs play a role in insulin sensitivity in humans through incretins, which are intestinal peptides that act as hormones produced in the gastrointestinal tract by enteroendocrine cells in response to food intake. At a functional level, incretins affect the cells of the islets of Langerhans by increasing the secretion of insulin by 70% in the postprandial state and consequently decreasing glucagon secretion depending on circulating glucose. The main incretins are glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). SCFAs, products of saccharolytic fermentation, bind to G protein-coupled free fatty acid receptors in the colon mucosa, which are G-protein-coupled receptor type 41 (GPR41) and G-protein-coupled receptor type 43 (GPR43). Through the GPR41 receptor, the YY peptide is released, which reduces the energy extracted from the diet, increasing intestinal peristalsis. In contrast, GPR43 induces glucose-dependent insulin discharge, inhibits glucagon secretion, improves insulin sensitivity, reduces hepatic gluconeogenesis, and controls appetite (Figure 1a).26 Furthermore, SCFAs can prevent obesity and IR by shifting from lipogenesis to fatty acid oxidation in the liver and adipose tissue. There is evidence supported in a murine model fed a high-fat diet, where butyrate supplementation prevents an increase in body weight and increases insulin sensitivity.27 In addition, butyrate and propionate can induce intestinal gluconeogenesis, acting through a gut-brain neural circuit to improve peripheral glucose production and insulin sensitivity.28
On the other hand, several studies have reported changes in the concentration of circulating bile acid groups (BAs) in subjects with IR or T2D.29,30 These alterations were related to liver structures (primary BAs) and derivatives produced by intestinal microbes (secondary BAs).31 BAs are signaling molecules that regulate glucose and lipid metabolism. The primary BAs are secreted in the small intestine after production and glyco-taurus conjugation (N-acyl amidation with glycine or taurine substituents) in the liver. Through a bacterial hydrolase enzyme (produced by Clostridium, Bacteroides, Lactobacillus, Bifidobacterium, and Enterococcus), deconjugation is first generated.32 There is evidence pointing out that 95% of conjugated primary BAs are reabsorbed through enterohepatic circulation, while the remaining 5% escape from this mechanism, reaching the large intestine and becoming secondary BAs by the action of Firmicutes (Eubacterium spp and Clostridium spp).33
Subjects with T2D have a lower number of secondary BAs than healthy subjects. This characteristic is related to an alteration in carbohydrate metabolism since they have an insulin-sensitizing role.Thus, BAs have been proposed as metabolic integrators of energy homeostasis involved in the regulation of numerous metabolic pathways, including their synthesis and enterohepatic circulation.31 This regulation of energy metabolism would be produced through nuclear receptors, identical to the farnesoid X receptor (FXR) and the G-protein-coupled bile acid receptor (Gpbar1). Specifically, Takeda G-protein–coupled receptor 5 (TGR5) is a bile acid membrane receptor expressed in the gallbladder, ileum, colon, and brown and white adipose tissue.34 This receptor internalizes and activates a series of adenylate cyclase-dependent signals through the involvement of glucose metabolism and lipid energy in brown adipose tissue and muscle, heightened mitochondrial activity, and phosphorylation. These processes stimulate insulin sensitization in murine models of diabetic and obese conditions. Likewise, in L cells, TGR5 improves glycemic metabolism by stimulating GLP-1 production and insulin secretion (Figure 1b).33 Furthermore, secondary BAs appear to have an insulin-sensitizing role. These act as mediator molecules through nuclear receptors such as FXR receptor and the TGR5 membrane receptor expressed in myriad tissues, such as the vesicle, ileum, colon, brown adipose tissue (BAT), and white adipose tissue (WAT). In BAT and muscle, increased mitochondrial activity and phosphorylation lead to insulin sensitization in obese and diabetic mouse models.
Instead, innate immune cells (monocytes and macrophages) are considered key players in the pathogenesis of T2D, as they can recognize microbial signals such as LPS and peptidoglycan, which are considered PAMPs. In addition, they have the ability to recognize non-microbial signals released by cellular damage or stress, such as mitochondrial DNA and ATP, which are considered DAMPs.35 The signals from the gut microbiome regulate innate immunity and influence local and systemic responses.36,37 In T2D, microbiome signals can promote low-grade chronic inflammation associated with IR through toll-like receptors (TLRs). Cell surface TLRs mainly recognize microbial membrane components such as lipids, lipoproteins, and proteins. Specifically, TLR4 recognizes bacterial lipopolysaccharide (LPS).38 TLRs differentially recruit members of a set of TIR domain-containing adaptors, such as MyD88, which is utilized by all TLRs (except TLR3) and activates nuclear factor kappa-light-chain-enhancer of activated β-cells (NF-κB) and mitogen-activated protein kinases (MAPKs) for the induction of inflammatory cytokine genes.36,37 Due to a high-fat diet, these receptors are activated in response to microbial stimuli such as LPS, which is present on the membranes of gram-negative bacteria.39 TLRs, specifically TLR-4, also interact with glycosylated serum proteins called advanced glycosylation end products (AGEs)40,41 to activate the transcription of MAPKs and NF-κB. In turn, this last pathway acts as an enhancer of activated β-cells after activation by LPS.39 On the other hand, proteins (such as hemoglobin, albumin, and low-density lipoproteins [LDL]) are enzymatically glycosylated in lysine and arginine residues in the hyperglycemic state. As a consequence of this glycosylation, the production of AGEs is favored.40,41 AGEs in macrophages can bind to TLR-4, which is overexpressed in T2D. Thus, TLR activation leads to a cascade of intracellular signaling mediated by NF-κB when it is translocated to the nucleus, and activates the transcription of genes coding for cytokines and inflammatory chemokines, including tumor necrosis factor-alpha (TNF-α), interleukin 1 beta (IL-1β), and interleukin 8 (CXCL8). As a result, this process promotes the onset of the inflammatory response to hyperglycemia (Figure 1c).42
Under normal physiological conditions, insulin binds to the receptor located on the surface of myocytes, hepatocytes, and adipocytes.43 Subsequently, an intracellular signaling cascade mediated by the insulin receptor substrate (IRS) is initiated, which activates protein kinase B (Akt), and finally induces the mobilization of two isoforms of glucose transporter (GLUT-2 and GLUT-4) to introduce glucose into the cell. When β-cells are subjected to an inflammatory process, they respond through the activation of two main pathways: 1) the activation of TLR2 and TLR4 by PAMPs or DAMPs; and 2) the assembly of the inflammasome nucleotide-binding oligomerization domain (NOD)-like receptor pyrin domain-containing (NLRP3).44 This inflammatory process causes insulitis, characterized by a continuous release of IL-6, IL-8, TNF-α, and monocyte chemotactic protein 1 (MCP-1), and by the activation of insular macrophages and the recruitment of new monocytes and peripheral macrophages.43 When the inflammatory response is improperly regulated, it produces a state of damage called chronic low-grade inflammation.36 During low-grade systemic inflammation, adipose, liver, and muscle tissue endure infiltration of inflammatory macrophages producing TNF-α; this cytokine directly interferes with the tissues and reduces their ability to respond to insulin. TNF-α reduces tyrosine phosphorylation of the insulin receptor and IRS-1, and induces serine phosphorylation of IRS1, which in conjunction reduces downstream transduction of insulin signaling.45 It has recently been postulated that the presence of gut dysbiosis favors an increase in the prolonged circulation of inflammatory markers and increased intestinal permeability.46 In turn, bacterial products (PAMPs) enter the circulatory system and promote the genesis of systemic inflammation in T2D.42 Overall, the genesis of T2D is conditioned by high levels of pro-inflammatory cytokines and the composition of the GM.47,48
Given their importance, in the following sections, we describe the relationship among the gut microbiome and two variables with relevance in T2D physiopathology: IR and pancreatic β-cell dysfunction (PBD).
Insulin resistance
The gut microbiome plays an important role in the progression of IR to T2D.20 An imbalance between potentially pathogenic and nonpathogenic bacteria can induce crucial metabolic alterations in the entire organism, which in turn can disturb the physiological parameters in multiple body compartments.49 This perturbation has at least two complex consequences. First, IR promotes alterations in different metabolic pathways, such as lipids, amino acids, and bile acids. Second, these alterations have substantial implications for the modulation of insulin sensitivity.14 In this way, metabolites produced by GM may regulate insulin sensitivity through several components of the insulin signaling pathway, such as insulin receptor substrates (IRS) and the enzyme kinase AKT (Figure 2a).50 Furthermore, some of these IRSs can indirectly affect the flow of substrates through lipogenesis, lipid oxidation, protein synthesis and degradation, and hepatic gluconeogenesis.
Figure 2.
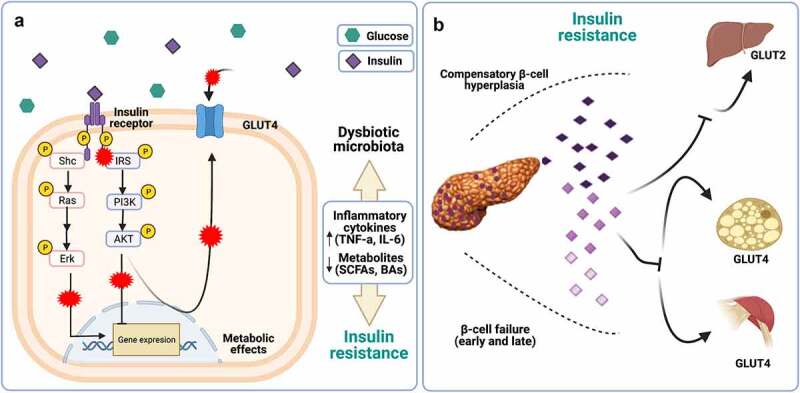
Defects in glucose transport in IR. a) Increased expression of inflammatory cytokines (TNF-a, IL −1, IL6) dependent on TLR4/MYD88 activation are negatively involved in downstream insulin signaling (marked with red crosses). Furthermore, an imbalance in the production of microbiota-derived metabolites, including SCFAs and BAs, is indirectly related to insulin resistance through the modulation of their receptors. b) Hyperglycemia is a consequence of over-demand of insulin requirements; indeed, β-cells improve the restoration of glucose homeostasis through increased insulin biosynthesis. Over time, apoptosis exceeded the rate of replication, resulting in loss of β-cells and a reduction in β-cell mass. Created with BioRender.com.
On the other hand, branched-chain amino acids (BCAAs) have recently attracted significant scientific attention because elevated blood concentrations of BCAAs are associated with states of IR, such as obesity and T2D.51 The main species associated with T2D and BCAA biosynthesis are Prevotella copri and Bacteroides vulgatus in the Danish population.52 These bacteria decreased the expression of BCAAs through catabolizing enzymes in adipose tissue and liver, and promoted an increase in inflammation in WAT, stress on the endoplasmic reticulum (ER), and an alteration in insulin signaling at the hypothalamic level.14 Additionally, BCAAs increase lipid oxidation in muscle, accumulating acylcarnitines and generating mitochondrial dysfunction. In adipose tissue, the alteration of BCAA catabolism reduces substrate flow to lipogenesis, contributing to metabolic dysfunction in IR and linking it to the progressive loss of pancreatic β-cell function.14 Ultimately, these changes generate an acute secretory response in the β-cell, resulting in a progressive dysfunction characterized by an abnormal secretory process due to the pancreatic β-cell trying to adapt and compensate for the IR, as described in the following section.
Pancreatic β-cell dysfunction
Glucose homeostasis is the product of metabolic, hormonal, neural, and microbial signals whose regulation determines the degree of glucose-dependent insulin release. The progression from normoglycemia to glucose intolerance and later to T2D occurs due to a deterioration in these signals, which gradually decreases insulin sensitivity and β-cell functionality (Figure 2b).2 This dysfunction associated with GM can be induced by two effects: 1) the increase in intestinal permeability; and 2) the chronically elevated glucose levels in the host (islet glucotoxicity).53 This physiological imbalance causes an increase in intestinal permeability, which in turn promotes the translocation of some bacteria (Proteus mirabilis and Escherichia coli) and their metabolites.54 To respond to intestinal permeability in the host, two main detection systems continuously scan for bacteria capable of translocating the intestinal mucosa or adhering to the epithelium: 1) NOD-like receptors (NLRs), which detect the presence of intracellular microbes; and 2) TLRs.38
The first detection system involves NLRs, specifically the NLR family pyrin domain containing (NLRP) subfamily, which can oligomerize into a macromolecular complex known as an inflammasome. Inflammasomes are found in the cytosol and mediate the activation of inflammatory caspases.55 Activation of the inflammasome depends on two pathways: first, NF-κB expression upon sensing PAMPs or DAMPs by their respective receptors, e.g., TLR4 binds to LPS. This process alerts the cell to express inflammasome-related genes such as inactive NLRP3, Pro-IL1, and Pro-IL-18. The second signal involves the recognition of bacterial toxins (e.g., cytosolic bacterial peptidoglycan) or host metabolites (potassium efflux) by inflammasome sensor proteins (NLRP3, NLR Family CARD Domain Containing 4 [NLRC4]).56 This recognition system allows recruitment of the adaptor protein ASC, which binds to pro-caspase-1, leading to its self-processing and activation. Caspase-1 cleaves the inactive precursor proteins of IL-1β and IL-18 into their bioactive fragments. In addition, this protease induces an inflammatory cell death known as pyroptosis.53,55 The second detection system for bacterial permeability corresponds to TLRs. These are pattern recognition receptors (PRRs) capable of recognizing PAMPs and inducing the expression of pro-inflammatory cytokines (IL-1B, IL-6, and TNF-α), mediated by the adapter molecules MyD88 and NF-κB. Taken together, these mechanisms directly affect the function of β-cells.53 Spranger et al. reported that the combined elevation of IL-6 and IL-1β, products of the activation of the two detection systems, was related to a threefold increased risk of developing T2D.57
Concerning islet glucotoxicity, chronically elevated glucose levels have been reported to impair islet function and proliferation, and induce apoptosis. In addition, the intra-islet expression of IL-1β may contribute to T2D pathogenesis by inducing the loss of mass and β-cell function. Consequently, hyperglycemia and glucotoxicity result from pancreatic dysfunction, increase intestinal permeability, and activate the inflammatory response.53,58
Factors that modulate the microbiome in patients with T2D
GM have great plasticity, which implies the ability to adapt their populations to fluctuating environmental conditions.59 Currently, their composition and diversity are modulated through classical factors such as diet, exercise, drugs, and more recently through interventions such as fecal microbiota transplantation (FMT), bacteriophage intervention through phageome studies, antibiotics, and bariatric surgery. All of these modulator methods can generate beneficial changes in the structure and function of the GM and restore them temporarily or permanently.60 In the next section, we will analyze the effect of these modulators on microbiome composition (Figure 3).
Figure 3.
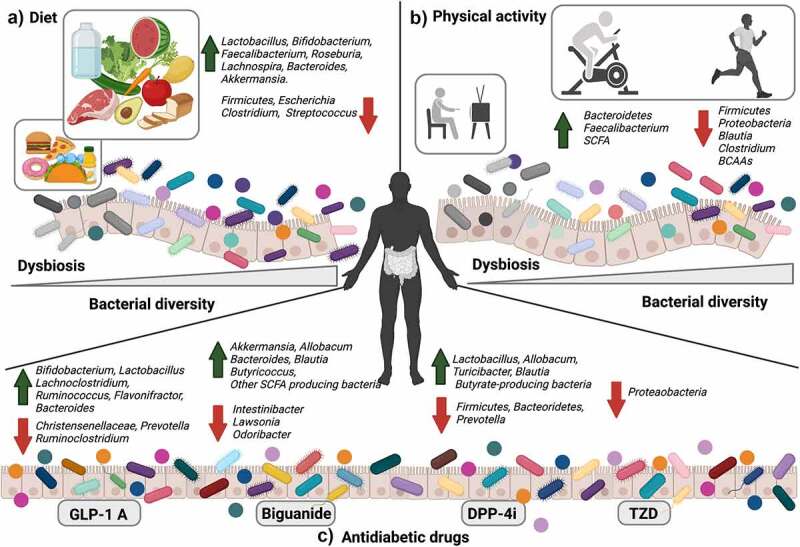
Factors that modulate the microbiome. a) Diet: The correct diet modifies the structure, and GM function increases bacterial diversity and SCFA producing species that contribute to better glycemic control in T2D patients. b) Physical activity: In sedentary subjects with T2D, physical activity increases bacterial diversity and SCFA-producing species. These changes reduce endotoxemia and increase the degradation of SCFAs and BCAAs. c) Antidiabetic drugs change the composition, diversity, and SCFA-producing bacteria in the GM, favoring glucose homeostasis through their mechanisms of action. Created with BioRender.com.
Diet
Treatment strategies for T2D include lifestyle changes, such as dietary interventions with routine physical exercise.61 Diet is a fundamental environmental contributor that interferes with the structure and function of the GM, playing a key role in modulating the benefits of human health.60 Studies on diet and microbiome associations have shown that the consumption of prebiotics and dietary fibers increases the abundance of SCFA-producing bacteria in the gut.62 At present, this modulator has been extended to include dietary patterns, eating behaviors, diet quality, and food preparation methods.63 Therefore, an unbalanced diet causes unfavorable changes in energy balance and GM composition. This dysbiosis results in weight gain and a higher risk of developing metabolic illnesses. On the other hand, a balanced diet favors appropriate changes in the microbiome composition and can promote weight loss and a healthy metabolic transformation.16 Therefore, nutrient ratios in meals affect digestive secretions, absorption, and transit time. These factors also affect human GM.63 Recent studies have estimated that for an average diet, 40 g of carbohydrates, 12 to 18 g of protein, and 2 to 10 g of fat reach the colon undigested every day and serve as substrates for microbial metabolism.64 Based on recent estimations, the gut microbiome contains 16,000 active carbohydrate enzymes and 9,000 genes involved in the complex metabolism of carbohydrates and polysaccharides, while the human genome contains only 17 of these genes.63 The superiority of gut microbes to metabolize a wide range of non-digestible nutrients is remarkable. In this way, dietary interventions modulate the GM, and these changes might contribute to better glycemic control in T2D patients.65
Advances in microbiome research have revealed the importance of diet variability and its contribution to GM composition in supporting health. Because diet influences intestinal transit time, pH, and macronutrient intake, possible approaches to achieving a healthy microbiota are to directly manage beneficial bacteria through probiotics, a Mediterranean diet, diet restrictions, a content-modified diet, and a diet high in non-digestible polysaccharides.66 The shift in diet has a dynamic effect on the composition of the GM in the short and long term and is dependent on the type of food eaten.67 Table 1 summarizes studies with statistically significant differences in the composition after dietary intervention for T2D patients.68–70 Furthermore, the differences in clinical parameters are closely related to changes at various levels of taxa (phylum, family, genus, species, and Firmicutes: Bacteroidetes relationship) (Table 1 and Table S3).
Table 1.
Descriptive characteristics and assessment of diet, physical activity, and antidiabetic drugs in GM of T2D patients.
| Nutritional/dietary interventions | ||||||||
|---|---|---|---|---|---|---|---|---|
| Country year |
Participants | Sample size | Intervention implemented | Treatment duration | Aims/Outcomes | Gut-microbiota significant differences | Clinical effects | Ref |
| Portugal 2021 |
Male and female. 40–80 years old. T2D. |
9 | Intervention: The Mediterranean diet. | 12 weeks | Primary: This study was to evaluate the modulation role of the Mediterranean diet in gut microbiota. | The effect size induced by the Mediterranean diet on the Firmicutes to Bacteroidetes ratio was considered clinically relevant after 12 weeks (p = .846; Cohen d = −0.83). Prevotella to Bacteroides ratio tended to increase right after 4 weeks of the intervention (p = .438; 4 weeks Cohen d = 0.74 and 12 weeks Cohen d = 0.74). | Twelve weeks after the intervention, HbA1c decreased by 0.67% (7.53 ± 1.07% to 6.86 ± 0.85%, p < .05; Cohen d = −0.70) and HOMA-IR decreased from 3.79 ± 2.98 to 2.76 ± 2.05 (p < .05; Cohen d = −0.41), with no accompanying changes in lipid profile. | 68 |
| China 2020 |
Male and female. ≥ 18 years old. T2D. |
45 |
Intervention: a-LCD + 84 g/day almond. Control: LFD + 56 g/day almond. |
3 months |
Primary: This study was to determine the effect of an a-LCD on depression and glycometabolism. Secondary: This study was to determine the effect in the gut microbiota and fasting GLP-1 in patients with T2D. |
At the phylum level, Firmicutes in the a-LCD group was significantly lower than that in the LFD group by the third month. At the genus level, Roseburia and Ruminococcus in the a-LCD group were significantly higher than those in the LFD group by the third month; compared to the baseline: Eubacterium, Roseburia increased significantly and Bacteroides decreased significantly in the a-LCD group. | Compared to the baseline, HbA1c levels in both groups decreased significantly (p < .01, p < .05) during the study period. At the third month, the HbA1c level in the a-LCD group decreased more than that in the LFD group (p < .01). | 69 |
| Iran 2020 |
Male and Female. Prediabetes. |
120 |
Intervention: Consume 6 g/d multispecies probiotic or inulin-based symbiotic. Control: Consume 6 g/d of either a placebo containing maltodextrin. |
6 months | Primary: This study was to determine the effects of probiotics and symbiotic supplementation on intestinal microbiome modifying in adults with prediabetes. | Six months’ supplementation with probiotics resulted in a statistically significant increase in abundance of Bacteroides fragilis to E. coli ratio. In the probiotic group, the relative proportion of Firmicutes to Bacteroidetes repressive was decreased significantly. | They do not report clinical parameters. | 70 |
| Physical exercise | ||||||||
| China 2020 |
Male and female. 40–46.5 years old. BMI between 27–30 Kg/m2. T2D. |
14 | Intervention: High-intensity exercise training. | 12 weeks |
Primary: This study was to determine the efficacy of high-intensity exercise in the prevention of diabetes. Secondary: This study was to determine the change in the gut microbiota through the intervention of exercise in all participants. |
The relative abundances of 6 species, belonging to Firmicutes, Bacteroidetes, and Proteobacteria, respectively, were significantly altered after exercise. Species falling into the Bacteroides genus and Clostridiales order, most of which are involved in the production of SCFAs, underwent a significant strain-level genomic variation by exercise. | The responders showed a remarkable 42.70% and 49.60% decrease in fasting insulin and HOMA-IR index, respectively, as well as a striking 116.29% increase of Matsuda index (a comprehensive evaluation of both hepatic and peripheral insulin sensitivity derived from oral glucose tolerance test. | 71 |
| Finland 2020 |
Male and female. 45–53 years old. BMI between 27.5–33.5 Kg/m2. |
49 |
Intervention 1: SIT (session consisted of 30-s exercise bouts). Intervention 2: MICT (40 to 60 min of moderate-intensity (60% of V˙O2peak intensity). |
2 weeks | Primary: This study was to determine the effects of SIT and MICTn intestinal metabolism and microbiota in subjects with insulin resistance. | Both training modes decreased the ratio of Firmicutes/Bacteroidetes (time, P = .04), and the abundance of Blautia spp. (time P = .051) and Clostridium spp. (time P = .04). Mainly due to the increase in the relative abundance of Bacteroidetes phyla (time, P = .03). Lachnospira genus was present in higher abundance after SIT compared with baseline (P = .025) and significantly higher abundance of Veillonella genus. Interestingly, the abundance of the Faecalibacterium genus (F. prausnitzii) was increased after MICT (P = .057) while no change after SIT was found. | Improved HBA1c (P = .003). Both training modes significantly reduced systemic inflammatory marker TNF α (p = .03) and tended to reduce C-reactive protein (p = .08) and intestinal inflammatory marker lipopolysaccharide-binding protein (p = .02). Lipopolysaccharide-binding protein correlated positively with HBA1c (r = 0.54; P = .02). | 73 |
| Italy 2019 |
Male and female. T2D. |
30 | Intervention: Program of endurance, resistance, and flexibility training. | 6 months | Primary: This study was to evaluate the role of chronic exercise on gut flora composition and leaky gut in T2D stable patients. | Chronic exercise modified the composition of the gut microbiota, reduced intestinal mycelial overgrowth, leaky gut, and systemic inflammation. | They do not report clinical parameters. | 76 |
| Canada 2015 |
Six-week-old male type 2 diabetic db/db (C57BL/KsJ-leprdb/leprdb) and db/+ (heterozygote; control) littermates. | 19 |
Intervention: Exercise (Exercise consisted of a low-intensity treadmill running 5 days/week for 60 min /session. Control: Sedentary. |
6 weeks | Primary: The purpose of this study was to determine if exercise influences the gut microbial profile in normal and diabetic mice. | This study revealed a main effect of exercise, with a greater abundance of select Firmicutes species and lower Bacteroides/Prevotella spp. in both normal and diabetic exercised mice compared to sedentary counterparts. Conversely, Bifidobacterium spp. was greater in exercised normal but not diabetic mice (exercise for diabetes interaction). | The diabetic mice had higher blood glucose (P < .001). However, there was a trend toward an interaction between diabetic state and exercise training (P = .070), such that glucose was higher in Ex-db/+ than Sed-db/+ (12.9 ± 1.9 vs. 8.1 ± 0.6 mmol/L), but similar in Ex-db/db compared to Sed-db/db (20.4 ± 2.4 vs. 21.9 ± 1.7 mmol/L). | 77 |
| Antidiabetic drugs | ||||||||
| Biguanide (metformin) | ||||||||
| Colombia 2017 |
Male and female. 18–62 years old. BMI ≥18,5 kg/m2. T2D. |
98 |
Intervention: Metformin. Control: Subjects without T2D. |
5 months | Primary: This study determines the influence of metformin on the association of T2D and gut dysbiosis in an adult population. | Compared with participants without T2D, participants with diabetes taking metformin had a higher relative abundance of Akkermansia muciniphila. | Compared with ND participants, T2D-met+ participants had higher fasting glucose, HbA1c, and insulin resistance than ND participants and lower levels of the insulin-sensitizing hormone adiponectin (P < .05). | 85 |
| Sweden 2017 |
Male and female. 50–58 years old. T2D. |
40 |
Intervention: Metformin. Control: Placebo. |
4 months | Primary: This study was to determine the effects of metformin in treatment-naïve adults with DM2. | Increased abundance of Akkermansia muciniphila in individuals who received metformin for 4 months. Bifidobacterium adolescentis, fecal propionate, and butyrate concentrations were significantly increased by the metformin group. | Significant decreases in % hemoglobin A1c (HbA1c) and fasting blood glucose were observed only in the group randomized to metformin treatment. | 86 |
| Korea 2017 |
Five-week-old male C57BL/6 mice. | 18 |
Intervention: HFD + metformin. Control: HFD and regular diet. |
16 weeks | Primary: This study was to investigate the effect of metformin on the gut microbiota in elderly mice. | The abundance of the genera Akkermansia, Bacteroides, Butyricimonas, and Parabacteroides was significantly increased by metformin in mice fed an HFD. | Metformin administration for 16 weeks to mice fed an HFD significantly decreased the serum glucose level compared to mice fed only an HFD. Metformin also significantly improved glucose tolerance. | 87 |
| China 2019 |
Male and female. 40–75 years old. T2D. |
180 |
Intervention: Antidiabetic drugs (Metformin, insulin, α- glucosidase inhibitor). Control: Non-therapeutic and health subject. |
3 months | Primary: This study was to explore the interaction between the gut microbiome and T2D or hypoglycemics in the Chinese population. | Metformin increased the abundance of Spirochete, Turicibacter, Fusobacterium, and Ruminococcus (butyrate-producing bacteria), Ruminococcus was related to the impaired glucose control with regard to T2D. | They do not report clinical parameters. | 88 |
| GLP-1 analogue | ||||||||
| China 2017 |
Five-week-old male Sprague-Dawley. | 60 |
Intervention: Liraglutide 0.2 g/kg and Liraglutide 0.4 g/kg. Control: Normal and diabetic conditions. |
12 weeks | Primary: This study was to determine the influence of liraglutide on fecal microbiota in diabetic male rats. | SCFA-producing bacterias including Bacteroides, Lachnospiraceae, and probiotic bacteria, Bifidobacterium, were selectively enhanced in liraglutide-treated diabetic male rats. Lactobacillus was negatively correlated with fasting blood glucose. | Liraglutide significantly decreased serum insulin level, HOMA-IR, and IL-6 (P < .01). | 90 |
| China 2016 |
10 weeks old male ApoE -/- mice with a C57BL/6 genetic background. | 60 | Intervention: Hyperglycemia + liraglutide and Hyperglycemia + saxagliptin. Control: Normal glucose control, Normal glucose + liraglutide, and normal glucose + saxagliptin. |
8 weeks | Primary: This study determines the structural modulation of the gut microbiota and the relationship with body weight, compared evaluation of liraglutide and saxagliptin treatment. | The enriched phylotypes were the genera Allobaculum and Turicibacter within the family Erysipelotrichaceae, the genera Anaerostipes, and Blautia within the family Lachnospiraceae, the genus Lactobacillus within the family Lactobacillaceae, genus Butyricimonas within the family Porphyromonadaceae, and the genus Desulfovibrio (phylum Proteobacteria, class Deltaproteobacteria). | The mean blood glucose level was significantly lower in liraglutide-treated mice compared with the control mice, who were fed ad libitum (6.70 ± 0.43 mmol/L vs. 7.62 ± 0.68 mmol/L, p = 9.0e-6). There were no substantial differences in the LPS concentrations. | 96 |
| Dipeptidyl peptidase-4 inhibitor (DPP-4i) | ||||||||
| China 2016 |
Four-week-old male Sprague-Dawley (SD) rats (induce TD2). | 15 | Intervention: Sitagliptin. | 4 weeks | Primary: This study was on determining the effects of sitagliptin in rats (induce T2D). | At the level of genus, SCFA-producing bacteria, Blautia, Roseburia, and Clostridium, and probiotics Lactobacillus, Bifidobacterium, and so forth were identified as significantly different (T2D vs T2D-sitagliptin conditions). | Sitagliptin resulted in a significant reduction in blood glucose (p < .05). | 94 |
| China 2017 |
Five-week-old male Sprague-Dawley SD rats. | 30 |
Intervention: Vildagliptin 0.01 g/kg + HFD and Vildagliptin 0.02 g/kg + HFD. Control: Control, control + Vildagliptin 0.02 g/kg and HFD + STZ. |
12 | Primary: This study was to identify whether vildagliptin modifies the gut microbiota structure during T2D treatment. | At the phylum level, a higher relative abundance of Bacteroidetes, lower abundance of Firmicutes, and reduced ratio of Firmicutes/Bacteroidetes were observed in the vildagliptin-treated group. Moreover, vildagliptin treatment increased butyrate-producing bacteria, including Bacteroides and Erysipelotrichaeae, in the diabetic rats. | Both doses of vildagliptin treatment reduced the fasting blood glucose and HbA1c levels (P < .01). Vildagliptin treatment reduced the blood glucose levels before and after glucose load, and the area under the curve of blood glucose (P < .01). Serum insulin levels, HOMA-IR, and IL-6 levels in the diabetic rats were higher than that in the normal controls (P < .01), Vildagliptin reduced the serum insulin and IL-6 levels, alleviated insulin resistance, and increased serum GLP-1 in diabetic rats (P < .05). | 95 |
| Thiazolidinediones | ||||||||
| Denmark 2021 |
Eight weeks-old male B6.BKS(D)-Leprdb/J (db/db) mice. | 24 |
Intervention: Rosiglitazone. Control: Placebo. |
8 weeks | Primary: This study was to characterize local gut microbiome and intestinal transcriptome responses in diabetic db/db mice following rosiglitazone treatment. | Lactobacillaceae and Lachnospiraceae were predominant in the small and large intestines, respectively. While Lactobacillus was the most relatively abundant genus (>75% of all genera) in the small intestine, the large intestine was characterized by a more diverse genus composition predominantly composed of Kineothrix, Lactobacillus, and Blautia. | Plasma insulin levels remained stable throughout the entire dosing period in vehicle controls (baseline: 6999 ± 327 pg/mL; termination: 7628 ± 1076 pg/mL, p = .570) and rosiglitazone-treated db/db mice (baseline: 5988 ± 295 pg/mL; termination: 5718 ± 841 pg/mL, p = .741). Rosiglitazone significantly improved glucose excursions in two successive OGTTs performed on treatment day 28 (p < .001) and 49 (p < .001). Rosiglitazone also improved weekly fasting blood glucose levels and terminal HbA1c levels (p < .001). | 79 |
| Sulfonylureas | ||||||||
| Netherlands 2020 |
Male and female (postmenopausal). 35–75 years old. BMI scores > 25 kg/m2. T2D. |
44 |
Intervention1: Dapagliflozin. Intervention2: Gliclazide. |
12 weeks | Primary: This study was to evaluate the effects of 12-week treatment with the SGLT2 inhibitor dapagliflozin and sulfonylurea gliclazide on gut microbiome composition in T2D patients treated with metformin. | Differential abundance analysis yielded no significant shift in specific microbes. | Both dapagliflozin and gliclazide similarly improved glycemic control (p < .001), while dapagliflozin reduced gliclazide slightly increased fasting insulin (p = .011 and p = .569, respectively). | 81 |
| Antibiotics | ||||||||
| Netherlands 2022 |
Male with PreT2D and BMI was 31.0 ± 0.5 kg/m2. Male C57BL/6 J mice and 6-week-old male mice. |
57 |
Intervention: amoxicillin, vancomycin. Control: placebo. |
7 days | Primary: This study was to explore the effects of the gut microbiota on the function of the exocrine pancreas and the gut endocrine system. | Gut microbiota alters host intestinal proteome. Proteolysis-related proteins, such as elastase and dipeptidase 1, were increased by the HFD, whereas proteins involved in iron homeostasis, such as ferritin heavy chain, were reduced with the HFD. Notably, serine protease inhibitors (serpin) proteins, which have been previously linked to metabolic diseases, were one of the main groups affected by diet and gut microbiota. | In the donor mice, the HFD caused a decrease in circulating GLP-1 levels, and this was reversed with vancomycin treatment. Transferring microbiota from antibiotic-treated mice to GF mice was sufficient to transfer significant changes in GLP-1 secretion. | 101 |
| Netherlands 2016 |
Male with overweight and obese. 35–70-year-old Caucasian. |
57 |
Intervention: amoxicillin, vancomycin. Control: placebo. |
7 days | Primary: This study was to investigate how gut microbiota manipulation by antibiotics (7-day administration of amoxicillin, vancomycin, or placebo) affects host metabolism in 57 obese, prediabetic men. | The fecal microbiota composition showed that 7-day vancomycin markedly decreased microbial diversity (p < .001), whereas this was not affected by amoxicillin (p = .42) as compared to placebo. vancomycin decreased the relative abundance of mainly Gram-positive bacteria of the Firmicutes phylum. Among the most strongly affected groups were genus-like groups that contain known butyrate-producing species from Clostridium clusters IV and XIVa, such as Coprococcus eutactus, Faecalibacterium prausnitzii, and Anaerostipes caccae, as well as species involved in BA dehydroxylation such as Clostridium leptum. Conversely, Gram-negative Proteobacteria, members of Clostridium cluster IX, and vancomycin resistant Gram-positive Bacilli such as Lactobacillus plantarum and Enterococcus, showed increased relative abundance after vancomycin treatment. | Antibiotic treatment did not significantly alter Rd as compared to PLA. Additionally, no effects were found on hepatic and adipose tissue insulin sensitivity, as determined by the insulin-mediated suppression of endogenous glucose production (EGP) and plasma-free fatty acid (FFA) concentrations. In accordance, antibiotic treatment neither altered whole-body insulin sensitivity (HOMA-IR) immediately after cessation of treatment nor at 8 weeks follow-up. | 102 |
| Bariatric surgery | ||||||||
| France 2022 |
Male and female T2D with Roux-en-Y gastric bypass. | 136 | Intervention: Roux-en-Y gastric bypass. | 5 years | Primary: This study was to decipher the participation of the GM in the long-term improvement of T2D after bariatric surgery, as well as its implication in the severity of the persisting cases of T2D. | The more severe cases of unresolved T2D were associated with a major increase in the class Bacteroidia, including 12 species comprising Phocaeicola dorei, Bacteroides fragilis, and Bacteroides caecimuris. A key observation is that patients who underwent major metabolic improvements do not harbor this enrichment in Bacteroidia, as those who presented mild cases of T2D at all times. | The prevalence of patients in the Severe cluster decreased from 55% at baseline to 30% at 5 years, confirming an overall decrease in T2D severity after RYGB. | 109 |
| Portugal 2021 |
Male and female, age ≥ 20 and ≤ 65 years old; BMI ≥ 30 and < 35 kg/m2; previous diagnosis of T2D. | 20 | Intervention: Roux-en-Y gastric bypass surgery. Control: Standard medical therapy. |
12 months | Primary: This study was to evaluate gut microbiota changes after metabolic surgery versus standard medical therapy in diabetic adult patients with class 1 obesity. | Ruminococcus, unclassified_Lachnospiraceae_ family, and Faecalibacterium significantly decreased, while Klebsiella, Gammaproteobacteria, Enterobacter, unclassified_Gammaproteobacteria, unclassified_Veillonellaceae increased after 12 months of RYGB. In the medical arm, unclassified_Lachnospiraceae and Sutterella significantly decreased, while unclassified_Clostridiales and unclassified_Bacteria increased, when comparing baseline with M12. | The fasting glucose, insulinemia, C-peptide, and HOMA-IR were significantly lower in the surgical arm (p = .007, p = .020, p = .020, and p = .027, respectively), and HDL-C was higher (p = .004). The surgical arm, 5 participants (62.5%) experienced remission from their diabetes (p = .007 for comparison with the medical arm. | 108 |
T2D: Diabetes Mellitus type 2; a-LCD: almond-based low carbohydrate diet; LFD: Low-fat diet; GLP-1: glucagon-like peptide 1; BMI: body mass index; SCFA: short-chain fatty acids; SIT: sprint Interval; MICT: moderate-intensity continuous training; HFD: high-fat diet; STZ: streptozotocin; db/db: diabetic; SGLT2: sodium-glucose co-transporter-2.
Exercise
Physical activity is an effective strategy to control diabetes; however, its benefits for metabolic homeostasis remain poorly understood.71 Recently, a modulating effect of exercise on the GM was reported in humans and animals, both with T2D.72 For example, murine models subjected to physical training simultaneously promoted an increase in the abundance of Bacteroidetes and a reduction in Firmicutes and Proteobacteria. Furthermore, individuals with physical activity had greater diversity in microbiota composition and better metabolic capacity than sedentary subjects.73
At the physiological level, the effect of physical exercise on GM composition has been associated with reducing inflammatory markers and metabolic endotoxemia, increasing the production of SCFAs, and degrading BCAAs.74 Notably, Y. Liu et al. reported that the relative abundance of Firmicutes, Bacteroidetes, and Proteobacteria was significantly altered after 12 weeks of exercise in a human cohort. In addition, the species belonging to the genus Bacteroides (producers of SCFAs) increased significantly, suggesting that physical exercise for short periods of time exerts differential modulating effects on microbial composition.75 Moreover, chronic physical activity modifies GM composition and reduces intestinal permeability and systemic inflammation.76 On the other hand, Motiani et al. reported the effects of continuous training of different intensities (moderate intensity and speed interval training on intestinal metabolism) on the GM in subjects with T2D and pre-T2D. Remarkably, the authors concluded that the composition of the GM changed due to physical exercise in two weeks. Both forms of training decreased the Firmicutes/Bacteroidetes ratio, Blautia spp., and Clostridium spp. Faecalibacterium increased only in continuous moderate-intensity training. Notably, these changes in microbiome abundances were correlated with clinical parameters. For example, a lower abundance of Blautia was associated with better insulin sensitivity. Overall, regular physical activity provides metabolic benefits, even in short periods of time73,77 (see Table 1).
Drugs
Patients with T2D are clinically treated with various anti-diabetic drugs78 that normalize blood glucose by targeting different organs and through different mechanisms. For example, GLP-1 analogues stimulate insulin secretion and keep pancreatic β-cells healthy and proliferating. Inhibitors of intestinal hormones, such as dipeptide-4 (DPP-4), suppress appetite in the brain. On the other hand, sodium-dependent glucose transport-2 inhibitors (SGLT2) block renal reabsorption of glucose. Metformin (a biguanide) reduces hepatic gluconeogenesis. Meanwhile, thiazolidinediones (TZD) agonists of PPAR-γ increase glucose uptake in skeletal muscles and adipose tissues.79 Last, sulfonylureas (SUs) increase pancreatic insulin secretion.78,80,81 The effect between the GM and anti-diabetic drugs is bidirectional; the drug influences microbiota composition, and in turn, the metabolism of the microbiota may have a positive effect in the host.80 One effect of these drugs in the host is to modify the gut microbiome composition by increasing the bacteria that produce SCFAs.82 In turn, the GM and SCFAs exert effects on anti-diabetic agents, influencing their pharmacogenetics and bioavailability.80 Understanding the bidirectional drug-microbiome interaction and how it influences clinical outcomes in T2D patients is necessary to identify possible modulating mechanisms of the GM.80 Given their current application, we discuss and present some evidence of the interactions between the GM and three anti-diabetic drugs (biguanides, GLP-1 receptor antagonists, and DPP-4 inhibitors).
Among the biguanides, metformin is used as the first-line treatment in patients with T2D.83 Various studies on mouse and human models suggest that metformin increases the abundance of Akkermansia and other bacteria that produce SCFAs (Allobacum, Bacteroides, Blautia, Butyricoccus, and Phascolarctobacterium). Simultaneously, with this dysbiosis, metformin improves glucose concentrations in the patient.84 Forslund et al. in their study with T2D patients stratified by treatment regimens, found that the subjects treated with metformin changed their GM composition. They observed that patients treated with metformin presented with a higher production of butyrate and propionate than untreated patients due to the enrichment of bacteria producing SCFAs (Blautia, Bacteroides, Butyricicoccus, Bifidobacterium, Prevotella, Megasphaera, and Butyrivibrio).85 On the other hand, Shin et al. showed statistically significant differences in the abundance of Firmicutes and Bacteroidetes between mice fed a high-fat diet and treated with and without metformin.86 At the genus level, an increase in Escherichia and a decrease in Intestinibacter were recognized in mice treated with metformin.86 In addition, Na-Ri Shin et al. explored the relationship of the anti-diabetic effect of metformin and GM composition in obese mice with T2D. They found that the effect of metformin may be mediated by a specific subset of bacterial taxa associated with an increased abundance of Anaerotruncus, Lactococcus, Akkermansia, Parabacteroides, Odribacter, Alistipes, Lawsonia, Blautia, and Lactonifacter, of which Akkermansia was responsible for the greatest phylum Verrucomicrobia whose abundance was observed in T2D-induced mice treated with metformin compared to control mice with p < .05. Similarly, the authors concluded that the number of goblet cells producing mucin and mucin-degrading bacteria was higher in the group of mice with T2D and treatment with metformin vs the control group (9.5 ± 0.5 vs 6.6 ± 0.3, p > .001). Another outstanding conclusion is that the modification of GM composition was associated with greater glucose tolerance in mice treated with metformin than in control mice (p < .05).86 The previous results coincide with those found by de la Cuesta-Zuluaga et al. who studied 14 subjects with a diagnosis of T2D treated with metformin, and 42 healthy individuals with similar clinical conditions.87 They concluded that Akkermansia muciniphila and Butyrivibrio increased their abundance in subjects with T2D respect to the control. In addition, they reported a non significant change in abundance for Roseburia, Subdoligranulum, and Faecalibacterium, all of which are butyrate-producing bacteria.87 Treatment with metformin, in addition to increasing Akkermansia muciniphila in the colon, also increases the abundance of Lactobacillus in the upper small intestine, which may contribute to the anti-diabetic effect of metformin.88,89 Furthermore, metformin increases the population of SCFA-producing bacteria (Alobacum, Bacteroides, Blautia, Butyricoccus, and Phascolarctobacterium) in the gut.90 Taking into account all these findings in animal models and humans, the drug’s ability to modify bacterial diversity becomes evident by selectively increasing the abundance of specific bacteria and altering multiple metabolic pathways in the GM, such as those involved in the metabolism of glucose.
A second frequent drug clinically applied in T2D is the GLP-1 analogue. These activate GLP-1 receptors and increase resistance to inactivation by the DPP-4 enzyme. Currently, six drugs are clinically approved in this category: exenatide, liraglutide, semaglutide, albiglutide, lixisenatide, and dulaglutide.37 GLP-1 receptor agonists (liraglutide) can modulate the GM.91 Zhang et al. in a murine model, determined the effect of liraglutide (0.4 mg/kg/day) on GM composition. In this study, 58 bacteria changed significantly between the normoglycemic, diabetes-induced, and liraglutide groups (p < .05). Of these microorganisms, 11 showed statistically significant differences between the treatment and control groups. The genera Flavonifractor, Lachnoclostridium, Ruminococcus_gnavus, Flavonifractor_plautii, and Bacteroides_acidi-faciens were significantly elevated in the liraglutide treated group compared with the diabetic group. The bacteria that significantly reduced their abundance were the genera Christensenellaceae_R_7_group, Ruminococcaceae_UCG_010, Ruminoclostridium_6, Prevotella_9, and the class Mollicutes. The Bifidobacterium and Lactobacillus genera were exclusively identified in the liraglutide group compared with the healthy group.92 Complementarity, Charpentier et al. evaluated the effect of liraglutide (intervention), exendin (control), and saline solution (control) in a situation of mouse model-induced hyperglycemia. Liraglutide significantly improved insulin secretion, and this effect was associated with changes in GM composition. The frequency of the Bacteroidetes/Firmicutes phyla relationship increased in response to both GLP-1 receptor agonists. Additionally, Porphyromonadaceae and Lactobacillaceae significantly increased, while Lachnospiraceae and Bacteroidaceae decreased in abundance. Exendina modified the families Lachnospiraceae and Porphyromonadaceae, the genus Odoribacter, and Lactobacillaceae and liraglutide modified Escherichia and Shigella.91
Finally, dipeptidyl peptidase-4 inhibitor (DPP-4i) is a proteolytic enzyme found in the cell membrane of most cells in the body whose main function is to inactivate GLP-1.83 Inhibiting DPP-4 prolongs the circulating half-life of GLP-1, thereby improving its insulinotropic and glucoregulatory capabilities.93 Five drugs have so far been approved in this class: sitagliptin, saxagliptin, vildagliptin, linagliptin, and alogliptin.93 In particular, the composition of the GM can be modulated with DPP-4 inhibitors.80 Xinleng Yan et al. in their study using an animal model, determined the effect of sitagliptin on the composition of GM and its relationship with glucose intolerance. The results obtained are conclusive; treatment with sitagliptin for 12 weeks modified the GM composition, reduced Firmicutes (63.19% vs 83.56%, p < .01), and increased Bacteroidetes (32.46% vs 16.06%, p < .01). Additionally, serum glucose was reduced in comparison to mice with T2D without pharmacological treatment, showing statistical significance (p < .01).94 Also, Zhan et al. determined the effect of vildagliptin on the increase in butyrate-producing bacteria in mice with T2D. Vildagliptin was administered in two doses; high (0.02 g/kg) and low (0.01 g/kg). Statistically significant differences were found in comparison to control rats in the number of operational taxonomic units (OTU), Shannon’s index, and Chao’s index, all with p < .01. Vildagliptin modified the characteristic Firmicutes/Bacteroidetes index in the GM in mice with T2D, reduced Firmicutes, and increased Bacteroidetes, butyrate-producing bacteria, and Lactobacillus. In addition, vildagliptin treatment enriched the phyla Streptococcaceae (p < .01) and Bacteroides (p < .01), but decreased Ruminococcaceae oscillibacter (p < .05), Ruminiclostridium (p < .05), Anaerotruncus (p < .01), Eubacterium (p < .05), and Prevotellaceae (p < .05). It was determined that the animal model treated with vildagliptin at any dose reduced fasting, postprandial glucose, HbA1c, HOMA-IR, and IL-6 compared to T2D and normoglycemic mice, with statistical significance of p < .01 for each variable.95
On the other hand, Lin-Wang et al. compared the GM structural modulation of body weight and serum glucose with two treatments: liraglutide (GLP-1 receptor agonist) and saxagliptin (DPP-4 inhibitor). The authors found that mice treated with liraglutide enriched the genera Allobacum (p = .004), and Turicibacter (p = 1.77e-8), the family Erysipelotrichaceae, specifically the genera Anaerostipes (p = 5.51e-5) and Blautia (p = .039), and the family Lachnospiraceae genus Lactobacillus (p = .013). These changes in GM composition were associated with a reduction in body weight in mice treated with liraglutide. In contrast, the GM composition in treatment with saxagliptin increased the abundance of some bacteria from the class Erysipelotrichaceae, such as Lactobacillus (p = .023), Allobaculum (p = .017), and Turicibacter (p = .001). Furthermore, the abundance of Bacteoridetes decreased, specifically the genera Bacteroides (p = .003) and Prevotella (p = .018). The random glucose concentration was lower in linagliptin-treated mice, p < .05. Liraglutide and saxagliptin were shown to enrich Lactobacillus and Turicibacter. Furthermore, Lactobacillus showed inhibitory activity against DPP-4, through increased incretins and glucose homeostasis.96
Antibiotics
Antibiotics are administered to combat pathogens. However, their use disturbs the microbial composition of some important genera participating in immune, endocrinological, and metabolic functioning.97 The use of antibiotics has been associated with remarkable metabolic alterations, mainly when the application comprises broad-spectrum antibiotics and their use occurs in the first years of life. It has been estimated that antibiotics impact the abundance of 30% of the GM, producing a rapid and significant decline in richness, diversity, gene expression, and protein and metabolic activity.98 Despite the GM metabolically responding under this perturbation, the initial state is not fully recovered, and antibiotic-induced microbial alterations can remain for months or even years.99
Recently, murine model studies and human clinical trials have shown that antibiotics can modulate the GM with antibiotic treatment, which in turn, could reduce glucose intolerance, adiposity, and adipose tissue inflammation. Using a murine model, Fujisaka et al. determined the effect of antibiotics on the GM and host metabolism. In this study, cB6J, 129 T, and 129 J mice were treated with a placebo, vancomycin, or metronidazole in their drinking water. Vancomycin treatment lowered the relative abundance of Firmicutes in B6J mice to 37% (p = .009) and in 129 T mice to 50% of untreated HFD levels (p= .003). This dysbiosis was associated with a rise in the relative abundance of Proteobacteria.100 In 129 J mice, metronidazole and vancomycin markedly diminished Verrucomicrobia from 66 to 0% (p = .002) and 23% (p = .007), respectively, favoring the predominance of Proteobacteria and Firmicutes. HFD B6J mice treated with vancomycin and metronidazole exhibited reduced blood glucose levels and improved glucose tolerance during the OGTT.100 Furthermore, Gridhar et al. evaluated the effect of antibiotic treatment and an HFD on the metabolism and function of the pancreas. The C57BL/6 J mice were divided into four groups: 1) fed a standard chow diet, 2) an HFD for six weeks, 3) fed a standard diet plus oral vancomycin, and 4) metronidazole in the last two weeks of the experiment. Vancomycin treatment significantly increased glucose tolerance and insulin sensitivity in HFD-exposed mice.101 On the other hand, Reijnders et al. investigated how GM manipulation by antibiotics (a 7-day administration of amoxicillin, vancomycin, or placebo) affects host metabolism in 57 obese and pre-diabetic men. This study was a double-blind controlled clinical trial in which the population was randomized into three groups: the placebo, amoxicillin, and vancomycin. Vancomycin at seven days decreased the diversity of intestinal microbiota compared to the placebo (p < .001); however, amoxicillin did not affect diversity (p = .42). Likewise, the group treated with vancomycin decreased the relative abundance of butyrate producing bacteria, such as Coprococcus eutactus, Faecalibacterium prausnitzii, and Anaerostipes caccae, as well as species involved in BA dehydroxylation, like Clostridium leptum. In contrast, patients treated with amoxicillin did not experience a change in the composition of the microbiota after seven days of treatment.102 The data obtained in this clinical trial were in contrast with several previous studies on rodents, which indicated that antibiotic treatment can improve glucose homeostasis and metabolic alterations.103 In human species, the effects of antibiotics on glycemic control or insulin sensitivity remain inconclusive.102
Bariatric surgery
Bariatric surgery (BS) refers to surgical procedures designed to achieve weight loss and long-term glycemic control in patients with T2D and obesity. Importantly, it can achieve better outcomes than non-surgical interventions (medication and diet).104 The two most common procedures in BS are sleeve gastrectomy (SG) and Roux-en-Y gastric bypass (RYGB), both with comparable efficacy. According to the American Diabetes Association (ADA) and the International Diabetes Federation (IDF) guidelines, BS is recommended in individuals with T2D and body max index (BMI) of at least 35 kg/m2, it can be an option for individuals with mild obesity (BMI of 30–34.9 kg/m2) who have inadequate glycemic control despite optimal medical management.104
Noticeably, BS can achieve a remission of T2D in 23 to 60% of patients, defined by normalization of blood glucose (FPG below 126 mg/dL or estimated HbA1c below 6.5%) without the need for normoglycemic medications.104 After BS, an improvement in metabolism occurs even before weight loss begins. Although the mechanisms underlying these favorable responses are not fully understood, the gut neuroendocrine system, gut hormones, bile acids, and GM have been proposed as key mediators.105
Remarkable changes in GM composition were observed after BS. In general, patients treated with BS experience an early increase in the richness of the GM, which may reflect an attempt to restore intestinal homeostasis. There is also a profound shift in certain genera associated with an improvement in glucose metabolism and a reduction in systemic inflammation markers. A comparative study of GM composition in T2D patients before and after BS showed an increase in the relative abundance of Escherichia, Klebsiella, and Akkermansia muciniphila and a decline in the relative abundance of Faecalibacterium prausnitzii, Lactobacillus acidophilus, and Coprococcus.106 Different BS procedures affect GM differently, with the most notable change occurring in RYGB. Sanchez-Alcoholado et al. found that patients after SG experienced an increase in the relative abundance of Akkermansia, Eubacterium, Haemophilus and Blautia. In contrast, patients after RYGB saw a preferential increase in the relative abundance of Veillonella and Granucatiella.107 In addition, the microbiota are altered in individuals with mild obesity treated with RYGB, with a rise in the relative abundance of Klebsiella, Gammaproteo-bacteria, Enterobacter, Gammaproteobacteria_un-classified, and Veillonellaceae_unclassified.108
The role of the GM in the prognosis of T2D remission after BS has also been studied. J. Debédat et al. demonstrated that an inadequate response to RYGB in T2D patients is associated with an increase in the Bacteroida class (such as Bacteroides fragilis species, Bacteroides vulgatus, Phocaicola dorei, and Bacteroides caecimuris) before and five years after surgery compared to post-RYGB patients with adequate remission. In addition, they investigated the causal link between GM and BS by performing human-to-mouse (free mice) FMT. They found that the phenotype of IR can be induced in a recipient mouse by receiving a transplantation of fecal microbiota from patients with inadequate remission after RYGB.109
Several microbiota-mediated mechanisms have been proposed to induce glycemic control and improve insulin sensitivity after BS. One of them is the crosstalk between the BAs and the GM. After BS, there is an increase in the blood concentrations of primary BAs and secondary BAs in T2D patients. Altogether, the overproduction of these BAs impacts lipid and glucose metabolism, and seems to be associated with the remission and improvements of T2D patients treated with BS.110 However, these outcomes are in contrast to those reported by Ilhan et al. who characterized the fecal metabolome of an American cohort with severe obesity and T2D after BS (the majority of subjects had resolution of diabetes and other comorbidities 12 months after treatment). They found a decline in the concentration of secondary BAs at 12 months after surgery compared to the non-surgical controls.111 Overall, these results imply that GM and BS have an important codependence in the improvement of T2D patients. Consequently, the study on the differences in GM composition is critical for understanding the pathways underlying metabolic improvement after surgery.
Fecal microbiota transplantation
FMT has become an outstanding research topic with potential applications in clinical medicine and biomedicine.112 Recently, FMT has been proven to be an effective method for treating and preventing the recurrence of gastrointestinal disorders through host-microbiota interactions; for example, FMT treatment against Clostridium difficile infection.113 Furthermore, FMT has been suggested as a therapeutic approach to modulate chronic and metabolic conditions such as T2D.114 Vrieze et al. established the effects of infusing GM from lean donors to male recipients with metabolic syndrome on the recipients’ microbiota composition and glucose metabolism. Six weeks after microbiota infusion, receptor insulin sensitivity increased along with butyrate-producing bacteria levels.115 Likewise, the authors demonstrated that FMT with or without lifestyle changes increased butyrate-producing bacteria in subjects with obesity and T2D. A recent study highlighted that the combination of lifestyle changes and FMT increased Bifidobacterium and Lactobacillus compared to FMT alone.116 In addition, Kootte et al. studied the effect of allogeneic FMT (from lean donors) on metabolism in relation to GM composition at 6 and 18 weeks after treatment. Moreover, the authors obtained microbiome composition in autologous FMT as the control treatment (placebo) in the donor subjects. In their clinical trial, the authors observed a statistically significant increase in insulin sensitivity after 6 weeks of allogeneic transplantation, accompanied by an altered composition of the GM. Further, using the GM composition, it was possible to classify the status of responders versus non-responders to the allogeneic FMT (recipient operating characteristics [ROC] AUC 0.88). Metabolic responders to FMT were characterized by lower baseline GM diversity and a higher abundance of Subdoligranulum variabile and Dorea longicatena than non-responders, while the abundance of Eubacterium ventriosum and Ruminococcus torques was lower in the baseline fecal samples. Although FMT has been proposed as a candidate to treat T2D, the molecular mechanisms underlying the therapeutic benefits are not yet understood.116
On the other hand, in a murine model with T2D, FMT increased the number of species and the alpha diversity indices (Shannon and Simpson). In addition, the level of HbA1c decreased and improved pancreatic β-cell function (measurement performed with HOMA-B) in the FMT group. Notably, this study suggests that GM transplantation reverses IR and damages islets.117 Additionally, Zhang et al. determined the effect of transplanted fecal bacteria from Kazakhs (the Kazakh Chinese ethnic group) with normal glucose tolerance on male db/db mice with T2D. In these recipient mice, the levels of Desulfovibrio and Clostridium coccoides in the intestine were significantly reduced, but the abundance of Akkermansia muciniphila and the expression of colonic protein histone deacetylase-3 (HDAC3) in the colon samples were increased. These results indicate that Akkermansia muciniphila could have affected the metabolism of the murine model by regulating the expression of HDAC3. This is because it can activate brown fat cells to oxidize lipids, increase metabolism and promote weight loss to counter T2D. Furthermore, in this study, HDAC3 was positively correlated with glycolipid levels, suggesting that Akkermansia muciniphila may be the main intestinal probiotic that improves metabolism in T2D.118 Based on previous evidence, FMT has been recognized as a therapeutic strategy that in combination with lifestyle can be potentially effective in the treatment of T2D.119 However, more evidence in humans needs to be generated, and we suggest that for future interventions, determining the baseline fecal microbiota composition may help predict treatment efficacy.112
Phageome
Obesity and T2D are associated with changes in gut bacterial composition, but little is known about the role of the virome in T2D disease development. Yang et al. reported the results of viral-bacterial transkingdom correlation for a Chinese cohort of 101 lean controls and 128 obese subjects (74 diagnosed with T2D). The authors found a decreased number of correlations between the relative abundance of the virome and bacteriome in obese subjects compared to the lean controls. Furthermore, obese subjects with T2D displayed an increased number of negative correlations and a lower number of positive correlations compared to the lean controls.120
Given the extensive evidence that phages can shape the composition and function of bacterial communities, the phageome of the human gut has been studied in T2D patients. For example, Yingfei et al. conducted a computational study of associations (with SparCC, a tool to infer correlation networks) between bacterial and phage abundance in a Chinese cohort of 74 healthy patients and 71 T2D patients.121 Their results imply that the number of phages in the intestinal tract of diabetes subjects was significantly increased, especially in the group with seven phage OTUs (pOTUs) (Siphoviridae phage family for Lactobacillus, Listeria and Staphylococcus). However, they identified pOTUs belonging to the Caudovirales order, which has several limitations of taxonomic annotation. Despite these incipient achievements providing little insight into the mechanisms by which the phageome participates in T2D, they supply evidence of their involvement in the disease and pave the way for further research to discover human gut phage functions in the development of T2D.121
Systems biology: In silico modeling of metabolism in gut microbial communities
High-throughput technologies (HTs) are powerful tools, as they offer new insight into the functioning and behavior of microbial communities. However, data is not sufficient, and the generation of knowledge requires computational schemes with the capacity to integrate data and reach conclusions at a systemic level. As depicted in Figure 4, data obtained from HT can be used as input in several systems biology (SB) tools to provide insight into the structure and function of microbial communities.122 For example, proteomic studies and 16S rRNA data suggest a lower abundance of the Lachnospiraceae family in obese diabetic mice than in the control group.123 Additionally, Reeves et al. suggested that inoculation with Lachnospiraceae in germ-free mice is associated with suppression of Clostridium difficile colonization.124 Altogether, these findings suggest that an increase in the abundance of Lachnospiraceae can be considered beneficial and is highly associated with the maintenance of gut homeostasis and health.123 On the other hand, integration of whole metagenome shotgun (WMS) with metabolomics pointed out that a high fiber diet was associated with a decrease in the abundance of sulfate reducer bacteria and a rise in SCFA producers, which in turn is related to better levels of HbA1c and increased GLP-1 in the host.125 Moreover, an integrative analysis with WMS and targeted metabolomics performed on a T2D-Chinese cohort supplies evidence that SCFA metabolism enriches its activity after metformin application. Simultaneously, this last finding was associated with higher levels of butyrate and propionate metabolites.88
Figure 4.
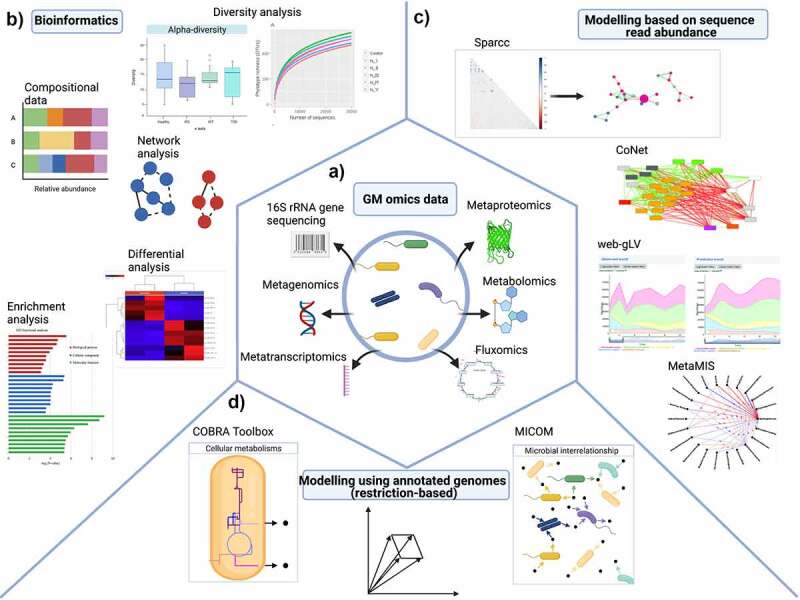
SB approaches used with omics datasets from T2D. a) Data collection by HT technologies, b) Bioinformatics, c) Modeling based on sequence read abundance, and d) Modeling by using annotated genomes (restriction-based). Adapted from116–121 Created with BioRender.com.
In terms of the development and progression of patients with T2D, the information obtained through HT is in constant growth. For example, Zhou et al. carried out deep profiling of the transcriptomes, metabolomes, cytokines, proteome, and GM of a cohort of 106 subjects (healthy and pre-T2D). This report revealed two main conclusions about GM and individuals with prediabetes. First, GM is differ between healthy and IR subjects. Second, the GM (conceived as a holobiont) in subjects with IR responded differently than those associated with healthy subjects when the immune system was stimulated (immunization and viral respiratory infections). Moreover, the global co-association analysis revealed precise host-microbe interactions that differed between healthy and IR subjects. In this way, the correlations between GM and host circulating cytokines revealed several interactions in IR subjects (p < .05). For example, IL-1β was positively correlated with Barnesiella, and TNF-α was inversely correlated with Faecalibacterium.126 Given that the host’s IL-1 activity127 and IFN-γ128 signaling are related to Barnesiella and Faecalibacterium, they are associated with TNF-α, which might enhance IL-17 activity.124 The absence of these correlations in IR subjects suggests that IR could affect associations between GM and cytokines.126 Moreover, the genera Clostridium-XIVb and Phascolarctobacterium were significantly correlated (p < .05) with Holdemania in healthy participants, and Holdemania was correlated with Clostridium-XlVa, Clostridium-XVII and Collinsella. Thus, IR and healthy associations can indicate different patterns of GM interactions between the two groups.126 These findings support the idea that systemic analyses offer novel insight into the role of GM in the development and course of T2D disease.
An effort to discover new insights about the function, structure, and design of GM has been triggered by the development of qualitative and quantitative computational models.129 Due to their relevance to elucidate regulatory and metabolic mechanisms, we discuss the main computational approaches that have been used to explore the relationship between GM and T2D, which are summarized in Figure 4.
Bioinformatic and functional studies
The generation of extended comprehensive studies for GM requires massive high-quality data and bioinformatic methods to derive a proper biological interpretation. For example, one of the first and most fundamental questions belongs to the taxonomic composition of the GM and the phylogenetic connections of its members. An approach for taxonomic classification is based on the counts of specific marker genes., i.e., 16S rRNA, 18S rRNA, and internal transcribed spacer. These approaches have enabled the analysis of different and complex habitats in the GM. In particular, 16S rRNA has been frequently used due to its capacity to analyze large numbers of samples, i.e., multiple patients and longitudinal studies.130
At the same time, several bioinformatics tools have been developed to examine the reads obtained from 16S rRNA technologies, most of which rely on three main steps: preprocessing and quality control, taxonomic assignment, and ecological analyses.131 After the primary analysis, it is possible to perform more specific analyses, (such as those focused on enrichment, differential abundance, and association networks), at several taxonomic levels (e.g., genus, family).132
The taxonomic assignment is a key step in the 16S rRNA sequencing data analysis pipeline. Currently, three representative tools have been successfully and widely applied in 16S analysis: Quantitative Insights Into Microbial Ecology (QIIME2), Mothur, and DADA2. In general, these tools have reliable and comparable results in terms of performance for 16S analysis under different next generation sequencing technologies.133
Although 16S analysis allows us to obtain information on the taxonomic and phylogenetic community structure at the genus level, these methods cannot provide information about the functional genes in the bacterial community. Despite this limitation, there are bioinformatic strategies to fill this gap. Phylogeny is actively correlated with functional gene patterns;134 thus, we can predict functional genetic information through diverse bioinformatics tools, such as the phylogenetic investigation of communities by reconstructing unobserved states (PICRUSt2) and Tax4Fun.135
However, we need to consider that HT data (e.g., 16S rRNA reads) fall into a class of data termed “closed” or “compositional”, which includes particular geometric and statistical properties; this makes establishing microbial taxa associations between communities challenging.136 Hence, the following bioinformatic tools have been developed for statistical analysis of microbiome data, such as LEfSe and MaAsLin2.137 They provide several methods for data normalization and transformation. Additionally, SparCC, and SPEIC-EASI address the compositional problem by assuming that few species are correlated, and BAnOCC makes no assumptions about the microbial data.138 In addition, several supervised learning algorithms have been proposed to identify a subset of highly predictive taxa from the different stages of the disease. Due to the high complexity of the data, it is necessary that these algorithms be able to model the complex interactions and non-linear effects between microbial communities. As such, the most commonly used methods are support vector machine, random forest, and multilayer perceptron with variable predictive accuracy.139
Modeling based on sequence read abundance: Inferencing of microbial ecological rules
Among the computational approaches available to analyze the microbiome, we highlight those associated with reconstructing the ecological structure from HT, such as 16S rRNA and WMS, SparCC,140 MENA,141 LSA,141 CoNet,142 SPIEC-EASI,140 MetaMIS,143 and Web-gLV,144 among others fall in this category. These bioinformatic tools integrate correlation networks and include methods for OTU pre-processing and microbial associations. The most common output from all of these tools is the inference of a microbial association network and the estimation of its robustness through their topological parameters, such as cluster coefficient, connectivity, and modularity.145
By following these strategies, some remarkable studies about T2D and microbiome association have been recently reported (Table 2). For instance, Ross et al. explored the association between GM composition and T2D in a cohort of 63 Mexican-American subjects from Cameron County (CCHC) (Texas, US).146 This geographic location was selected due to the high rate of obesity and T2D cases that emerge there each year.147 According to this report, the authors identified more reads in Lachnospiraceae and Roseburia (Firmicutes) in CCHC subjects that in the Human Microbiome Project (HMP). Moreover, the authors deduced that Lachnospiraceae plays a key role in metabolic functions in the progression of T2D in CCHC subjects.146
Table 2.
T2D analysis reported by modeling based on sequence read abundance approach.
| SB/Bioinformatic Tool | Seq- Tech | Population | Associated genera with T2D | Reference |
|---|---|---|---|---|
| SparCC | Roche pyroseq 454 (V1-V3) 16S |
63 patients with T2D and obesity | Lachnospiraceae | 146 |
| SparCC | Illumina MiSeq (V3-V4) 16S 300 bp | 450 patients with T2D and hyperlipidemia | Blautia and Faecalibacterium spp. | 148 |
| SparCC | Illumina MiSeq WMS |
71 patients with T2D | Siphoviridae** | 121 |
| SparCC | Illumina MiSeq (V1-V3) 16S 300 bp |
106 patients with preT2D |
Barnesiella and Faecalibacterium |
126 |
| DESeq2 | Illumina MiSeq (V4) 16S 250 bp | 427 patients with IFG, IGT, IFG+IGT, T2D and NG* | Escherichia, Veillonella, Blautia and Anaerostipes | 170 |
| CoNet | Illumina HiSeq (V3-V4) 16S 250 bp |
83 patients with T2D and diabetic retinopathy | Anaerobiospirillum, Gardnerella, Cloacibacillus and Leptotrichia | 154 |
| SparCC | Illumina NovaSeq 6000 WMS and VLP |
74 patients with T2D | Escherichia phage, Geobacillus phage,and Lactobacillus phage | 120 |
*Normal Glucose Tolerance (NG), **A virus family.
In another study using SparCC, it was found that alterations in GM are involved in the treatment of T2D with hyperlipidemia in a Chinese cohort of 450 subjects exposed to two clinical interventions: metformin and AMC (a Chinese herbal formula of Rhizoma Anemarrhenae, Momordica charantia, Coptis chinensis, Aloe vera, and red yeast rice).148,149 In the metformin-treated group, the authors noted a remarkable increase in Blautia spp, (SCFA producer), which is in line with other studies conducted on animals.90 Also, they observed a decline in Akkermansia in T2D Chinese subjects with metformin treatment. However, these findings do not agree with those of other studies on humans, where Akkermansia increases after metformin is consumed.84,118,150,151 This discrepancy might be explained when we take into account strain-specific functions; thus, the authors suggested further studies (such as molecular analysis the full ribosomal 16S gene and phylogenetic trees) to clarify this controversy.148,149 On the other hand, treatment with AMC increased the abundance of two genera related to butyrate production (i.e., Faecalibacterium and Roseburia).152 Interestingly, Faecalibacterium prausnitzii has been reported as a functionally important bacterium to prevent physiological damage through the production of butyrate and anti-inflammatory metabolites.153 Moreover, AMC had a stronger modulatory effect on the GM than metformin treatment in terms of improving IR and triglyceride levels. This can be explained by the synergistic effects of the multiple phytochemicals present in AMC.148 Concerning other diseases caused by T2D, the information is limited.154 Das et al. published an interesting study on the association between GM dysbiosis in T2D and diabetic retinopathy (DR) with an Indian cohort of 30 subjects. Regarding T2D subjects, the authors found positive associations between a chronic low-grade inflammatory state and pathogenic genera such as Gardnerella, Atopobium, Fusobacterium, Gemella, Halomonas, and Vagococcus. Moreover, and with DR participants, a reduction in the abundance of anti-inflammatory and probiotic bacteria in comparison to other genera was observed. However, in terms of the GM of T2D and DR, the authors did not report significant differences at the genus level.154
Last, related to modeling based on sequence read abundance, generalized Lotka-Volterra modeling enhances the classical predator-prey models of the two species, which are broadly used in ecology.144 The main advantage of these models is their capacity to estimate the native growth and interaction constraints of uncultured microbes in a given environment from temporal data.155 Once the parameters are delimited, we can analyze the variations in GM composition over time with unidentified initial conditions.
Modeling using annotated genomes (constrained-based)
Genome-scale metabolic reconstruction and constraint-based modeling is a paradigm in systems biology to quantitatively explore the metabolic activity of a bacterium or a group of bacteria constrained in a specific metabolic environment. This approach is characterized by three global elements: the use of genome-scale metabolic reconstruction for microorganisms, their mathematical representation to simulate their metabolic phenotypes, and their integrative description with HT technologies.156 A variety of computational tools based on genome-scale metabolic models (GEMS) has been developed in recent years with the purpose of quantitatively examining metabolic activity in the microbiome. Among these methods, we highlight the COBRA Toolbox and cobrapy, computational frameworks developed to model genome-scale metabolic reconstructions through constraint-based modeling.157 Based on this approach, there have been some efforts to characterize the microbial metabolic community at small, medium, and large scales. Among the efforts to model metabolic activity and species-level interactions in GM, we list computational strategies such as OptCom,158 cFBA,159 CASINO,160 BacArena,161 Dynamic OptCom,162 bialSim,163 and MICOM.9 These schemes differ from each other in several ways: 1) the amount of genome-scale metabolic reconstruction capable of being included in the simulation, 2) the dynamic condition constraining the state of the system (steady or unsteady state), and 3) the computational procedure to define and calculate the community objective function in the microbial community.164
In terms of T2D and metabolic modeling of microbiota through annotated genomes, the information is scarce but continues to growth with several articles.165 For example, Rosario et al. used the COBRA Toolbox to model the contribution of four bacteria (Escherichia spp, Akkermansia muciniphila, Subdoligranulum variabile, and Intestinibacter bartletti) to the physiology of T2D patients undergoing metformin treatment.166 To characterize the metabolic alterations produced by this dysbiosis, they applied flux balance analysis (FBA) coupled with synthetic lethality analysis interactions to identify patterns of growth. Their results suggest that the metabolism of Escherichia sp., A. muciniphila, S. variabile, and I. bartlettii may explain the features observed in T2D-metformin patients, which are related to commensal and competing behavior through extracellular compounds, including SCFAs, H2, and amino acids.
From the other side, Diener et al. (2020) reported MICOM,9 an OptCom based framework capable of simulating the GM metabolism of almost 850 instances of genome-scale metabolic reconstruction simultaneously, starting from the relative abundance of bacteria obtained from 16S or metagenome technologies. Remarkably, the authors were able to derive the growth rates that correspond directly to the observed replication rates. Moreover, they integrated several constraints, such as taxon abundance and adjustable dietary input, to prepare personalized metabolic models for individual GM samples. With T2D, T1D, and the healthy control data of 186 subjects, the authors showed that the community-level production of SCFAs was heterogeneous and mostly distinctive at the individual level. Also, their model output showed complex cross-feeding associations among bacteria, which mirrors the complex community structure that is difficult to measure in vivo.9 In addition, MICOM was able to predict reduced SCFA (butyrate and propionate) production levels in T2D participants, with a consecutive restoration of these rates found in subjects with metformin treatment. In general, they reported that changes in taxon abundance or diet have highly personalized effects.9 Based on these findings, the in silico modeling of the metabolic activity of microbiota has started to be a fundamental and useful approach to obtain quantitative mechanistic explanations that complement association studies. This last point involves genome-scale metabolic reconstruction of bacterial communities in the gut microbiome and diet; all of these are placed in the context of a personalized background.
Discussion and perspectives
The prevalence of T2D has become a serious public health problem worldwide.167 Genetic components, a sedentary lifestyle, and dietary habits (low dietary fiber and high fat consumption) are etiological factors that contribute to the development of T2D.168 Recently, GM dysbiosis has been integrated as a factor associated with the rapid progression from IR to T2D. Notably, this dysbiosis can remodel functions of the intestinal barrier and metabolic pathways in the host. In particular, these alterations are closely associated with SCFA production, bile acid transformation, adipose tissue inflammation, and chronic low-grade systemic inflammation.169 In this review, we have discussed in detail how the physiological variables in T2D are associated with a disruption of microbiota in the host, as well as how lifestyle, drugs, and new promising interventions like FMT can control and reshape their profiles to benefit the health of the host. Altogether, these findings offer innovative possibilities for preventing and treating T2D. However, their implementation still faces some challenges that need to be addressed in the coming years. Let us review some of them.
First, inter-individuality variability is an inherent factor that underlines the need to define microbiome profiles at the ethnic, geographic, and sociocultural levels. While in Chinese treatment-naive individuals with T2D Akkermansia muciniphila, Faecalibacterium prausnitzii and Roseburia intestinalis are relevant to the phenotype,151 Escherichia-Shigella, Veillonella, Blautia and Anaerostipes were associated with T2D progression in a Mexican cohort.170 These and other reports provide evidence of the need to characterize the local microbiome, and reinforce the need to generate an individualized intervention to enhance glycemic control through the synergistic effects of lifestyle, diet, probiotics, drugs, or surgical interventions.
Second, dietary components orchestrate and modulate GM composition and thus alter the host’s metabolism.171 However, the response to diet differs among individuals, with some being non-responders to dietary interventions,172 since what can be a good diet for one person might not work for someone else. Personalized nutrition requires frequent evaluation of anthropometric, biochemical, and clinical variables for designing optimal interventions that produce a favorable change in the patient’s metabolism. Although the idea is clear, the practical implementation of this dietary plan in conjunction with other adjuvant factors, such as drugs and changes in lifestyle, remains a challenge in many countries. Some questions with inconclusive answers should be addressed in the future. For instance, the long-term effect of diet on the patient, and how to design it in terms of socio-cultural factors. Although some contributions have been reported in this aspect,173 we consider the implementation of these strategies to represent the vanguard of personalized nutrition.
Third, antidiabetic treatments positively influence the host through changes in GM and the host’s metabolism. For example, metformin can facilitate a healthy state by increasing the abundance of the Lactobacillus and Akkermansia muciniphila strains.87 Other drugs, such as DPP-4 inhibitors, decrease the abundance of Oscillibacter and increase the abundance of Lactobacillus. These compositional changes reduce TLR ligands and pro-inflammatory cytokines and increase propionate. Although metformin is the first-line treatment in most T2D patients, there is a spectrum of drugs that are administered according to clinical variables defining the individual (such as comorbidities, patient preferences, tolerability, and cost). One challenge here is to define the role of the microbiome in selecting the treatment, which is unclear, because there is a specific pattern of microbiome phenotypes that can increase the success of the treatment. To this end, longitudinal studies that integrate the different omics are necessary to verify the effect on the long-term composition and help clarify the role of microbial ecology in the host. Remarkably, this information can generate valuable information to help decide on precision medical treatments that slow down the progression and long-term vascular complications of the disease, and which increase quality of life for patients with T2D.
Fourth, FMT could be used as a favorable modulator of insulin resistance, showing potential in the treatment of T2D patients. However, the Food and Drug Administration (FDA) has issued several warnings on FMT regarding the potential risk of serious or life-threatening infections. In June 2019, approximately two immunocompromised adults developed invasive infections caused by ESBL-producing Escherichia coli, and consequently one died.174 Additionally, in March 2020, the FDA reported that two patients developed enteropathogenic Escherichia coli (EPEC) and Shiga toxin-producing Escherichia coli (STEC) infection after receiving FMT.175 Therefore, FMT as an experimental therapy requires a well-defined and standardized methodology to design properly conducted clinical trials in the next few years. Despite their practical implications, information on the mechanisms underlying its modulatory effect on microbiota is currently scarce.
Fifth, beyond HT technologies, SB offers new approaches to mechanistically understand T2D development, physiopathology, and its relationship with GM through the constant development of integrative computational approaches. For example, in the framework of modeling based on sequence read abundance, SparCC (a tool to infer correlation networks) has proven to be an outstanding procedure for estimating correlations in microbiota composition, and it has become the gold standard for association studies related to GM.
On the other hand, constrained-based modeling allows for the versatility needed to simulate bacterial communities in numerous conditions that cannot be performed in vivo, such as Clostridium difficile infection. In this way, computational modeling of metabolism in microbiota, through software such as MICOM, offers a computational framework to infer the growth rates of selected bacteria and the metabolic interactions into GM. Moreover, it provides a high-throughput platform for generating mechanistic hypotheses and testing them in clinical assays. We suggest that the most useful application of metabolic models in bacterial communities is to offer detailed functional metabolic inferences that can serve as a means for novel hypothesis testing.170 Despite these computational themes, which have been applied in GM, their use is pioneering in exploring the metabolic consequences of T2D. The outcomes of such approaches comprise the foundations for developing more accurate models by including information about the GM and their ecological relationships with the host (as a holobiont). For example, Thiele et al. (2020) investigated a complete sex-specific model of humans that involved 26 organs and six cell types in the blood to study inter-organ metabolic fluxes and to explore host-microbe cometabolism.176 The accuracy of the hypothesis, generated with genome-scale metabolic reconstruction, the reproducibility, and reuse should be a challenge to face in the future of T2D.
Finally, in recent years, there has been a real revolution in the field of big data. In this sense, real-world data and real-world evidence play a more relevant role in medical care decisions.177 A vast amount of health-related data is now being collected and stored due to the increased use of computers, mobile devices, wearable devices, and other biosensors.178 These data have the potential to allow us to better design and conduct clinical trials and studies in health care to answer questions that were previously unfeasible. In addition, with the development of new and sophisticated analytical capabilities, we can better scrutinize data and apply the results of the analysis to the development and approval of medical products. For example, in the study of GM, it is necessary to design studies with a sufficient number of subjects of both sexes. In addition, information on lifestyle, diet, mental health, comorbidities, and metadata should be included that allow for comprehensive interpretation of the GM. To this end, it is necessary to have electronic databases at different levels (state, national, and world) for multicenter data analysis. Likewise, the capacity of omics sciences should be increased with shotgun studies, meta-transcriptomics, meta-metabolomics, meta-proteomics, and medium-term fluxomic studies to grasp the dynamics of metabolism in the microbial community and the host.122 In conclusion, we now have more measurable variables and are able to add more “data layers” to systems biology studies. Undoubtedly, we are only at the tip of the iceberg of understanding host-microbiome interactions and their specific mechanisms of modulation, one of the frontiers in the medicine of this century.
Supplementary Material
Acknowledgments
The authors thank the financial support from CONACYT (Grant Ciencia de Frontera 2019, FORDECYT-PRONACES/425859/2020), PAPIIT-UNAM (IA202720), and an internal grant from the National Institute of Genomic Medicine (INMEGEN, México). YEM-L is a doctoral student. This paper is part of the requirements to obtain the degree of Doctor in Science program in Ciencias Médicas, Odontológicas y de la Salud by Universidad Nacional Autónoma de México (UNAM) and received CONACyT fellowship 629384. DAE-H is a postdoctoral research associate at INMEGEN and received CONACyT fellowship to CVU 420693. JPS-C is a student from the Master in Sciences program in Ciencias Bioquímicas, UNAM and received CONACyT fellowship to CVU 1005702. DN-R is a student from the Master in Sciences program in Ciencias Bioquímicas, UNAM and received CONACyT fellowship to CVU 1083211.
Funding Statement
The authors thank the financial support from CONACYT (Grant Ciencia de Frontera 2019, FORDECYT-PRONACES/425859/2020), PAPIIT-UNAM (IA202720), and an internal grant from the National Institute of Genomic Medicine (INMEGEN, México).
Author Contributions
All authors wrote, discussed, and reviewed the manuscript.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2111952
References
- 1.Sharma S, Tripathi P.. Gut microbiome and type 2 diabetes: where we are and where to go? J Nutr Biochem. 2019;63:101–35. doi: 10.1016/j.jnutbio.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H, Martín C. Pathophysiology of Type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):6275. doi: 10.3390/ijms21176275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iatcu CO, Steen A, Covasa M. Gut Microbiota and Complications of Type-2 Diabetes. Nutrients. 2021:14. doi: 10.3390/nu14010166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao L, Lou H, Peng Y, Chen S, Zhang Y, Li X. Comprehensive relationships between gut microbiome and faecal metabolome in individuals with type 2 diabetes and its complications. Endocrine. 2019;66(3):526–537. doi: 10.1007/s12020-019-02103-8. [DOI] [PubMed] [Google Scholar]
- 5.Moore JB. From personalised nutrition to precision medicine: the rise of consumer genomics and digital health. Proc Nutr Soc. 2020;79:300–310. doi: 10.1017/S0029665120006977. [DOI] [PubMed] [Google Scholar]
- 6.Napolitano M, Covasa M. Microbiota transplant in the treatment of obesity and diabetes: current and future perspectives. Front Microbiol. 2020;11:590370. doi: 10.3389/fmicb.2020.590370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Łusiak-Szelachowska M, Weber-Dąbrowska B, Żaczek M, Borysowski J, Górski A. The Presence of Bacteriophages in the Human Body: good, Bad or Neutral? Microorganisms. 2020;8:2012. doi: 10.3390/microorganisms8122012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allaband C, McDonald D, Vázquez-Baeza Y, Minich JJ, Tripathi A, Brenner DA, Loomba R, Smarr L, Sandborn WJ, Schnabl B, et al. Microbiome 101: studying, analyzing, and interpreting gut microbiome data for clinicians. Clin Gastroenterol Hepatol. 2019;17:218–230. doi: 10.1016/j.cgh.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diener C, Gibbons SM, Resendis-Antonio O. MICOM: metagenome-scale modeling to infer metabolic interactions in the gut microbiota. mSystems. 2020:5. doi: 10.1128/mSystems.00606-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aria M, Cuccurullo C. bibliometrix: an R-tool for comprehensive science mapping analysis. J Informetr. 2017;11(4):959–975. doi: 10.1016/j.joi.2017.08.007. [DOI] [Google Scholar]
- 11.Thursby E, Juge N. Introduction to the human gut microbiota . Biochemical Journal. 2017;474:1823–1836. doi: 10.1042/bcj20160510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van de Guchte M, Blottière HM, Doré J. Humans as holobionts: implications for prevention and therapy. Microbiome. 2018;6:81. doi: 10.1186/s40168-018-0466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vos Wm, Nieuwdorp M. Genomics: a gut prediction. Nature. 2013;498:48–49. [DOI] [PubMed] [Google Scholar]
- 14.Yang Q, Vijayakumar A, Kahn BB. Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol. 2018;19:654–672. doi: 10.1038/s41580-018-0044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akpinar O. The gut-brain axis: interactions between microbiota and nervous systems. J Cell Neurosci Oxidative Stress. 2018;10:783. doi: 10.37212/jcnos.610103. [DOI] [Google Scholar]
- 16.Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MC. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms. 2019:7. doi: 10.3390/microorganisms7010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dwiyanto J, Hussain MH, Reidpath D, Ong KS, Qasim A, Lee SWH, Lee SM, Foo SC, Chong CW, Rahman S. Ethnicity influences the gut microbiota of individuals sharing a geographical location: a cross-sectional study from a middle-income country. Sci Rep. 2021;11:2618. doi: 10.1038/s41598-021-82311-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 19.Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99–103. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- 20.Larsen N, Vogensen FK, van den Berg Fwj, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sørensen SJ, Hansen LH, Jakobsen M, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakaroun RM, Massier L, Kovacs P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: perpetrators or Bystanders? Nutrients. 2020;12:1082. doi: 10.3390/nu12041082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin-Gallausiaux C, Marinelli L, Blottière HM, Larraufie P, Lapaque N. SCFA: mechanisms and functional importance in the gut. Proc Nutr Soc. 2021;80(1):37–49. doi: 10.1017/S0029665120006916. [DOI] [PubMed] [Google Scholar]
- 23.Tungland B . Short-Chain Fatty Acid Production and Functional Aspects on Host Metabolism. Hum Microbiota Health Dis. 2018:37–106. doi: 10.1016/b978-0-12-814649-1.00002-8. [DOI] [Google Scholar]
- 24.Rios-Covian D, Salazar N, Gueimonde M, de Los Reyes-gavilan CG. Shaping the Metabolism of Intestinal Bacteroides Population through Diet to Improve Human Health. Front Microbiol. 2017;8:376. doi: 10.3389/fmicb.2017.00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hino S, Mizushima T, Kaneko K, Kawai E, Kondo T, Genda T, Yamada T, Hase K, Nishimura N, Morita T. Mucin-derived o-glycans act as endogenous fiber and sustain mucosal immune homeostasis via short-chain fatty acid production in rat cecum. J Nutr. 2020;150(10):2656–2665. doi: 10.1093/jn/nxaa097. [DOI] [PubMed] [Google Scholar]
- 26.Muñoz-Garach A, Diaz-Perdigones C, Tinahones FJ. Microbiota y diabetes mellitus tipo 2. Endocrinol Nutr. 2016;63(10):560–568. doi: 10.1016/j.endonu.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 27.G DB, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk Th, Oosterveer MH, Jonker JW, Groen AK, Reijngoud DJ, et al. Short-Chain Fatty Acids Protect Against High-Fat Diet–Induced Obesity via a PPARγ-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes. 2015;64(7):2398–2408. doi: 10.2337/db14-1213. [DOI] [PubMed] [Google Scholar]
- 28.De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Bäckhed F, Mithieux G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. 2014;156(1–2):84–96. doi: 10.1016/j.cell.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 29.Lee S-G, Lee Y-H, Choi E, Cho Y, Kim J-H. Fasting serum bile acids concentration is associated with insulin resistance independently of diabetes status. Clin Chem Lab Med. 2019;57(8):1218–1228. doi: 10.1515/cclm-2018-0741. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Chen C, Xie C, Huang W, Young RL, Jones KL, Horowitz M, Rayner CK, Sun Z, Wu T. Serum bile acid response to oral glucose is attenuated in patients with early type 2 diabetes and correlates with 2-hour plasma glucose in individuals without diabetes. Diabetes Obes Metab. 2022;24(6):1132–1142. doi: 10.1111/dom.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao X, Fritsche J, Wang J, Chen J, Rittig K, Schmitt-Kopplin P, Fritsche A, Häring H-U, Schleicher ED, Xu G, et al. Metabonomic fingerprints of fasting plasma and spot urine reveal human pre-diabetic metabolic traits. Metabolomics. 2010;6(3):362–374. doi: 10.1007/s11306-010-0203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, Nicholson JK, Holmes E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4523–4530. doi: 10.1073/pnas.1006734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palau-Rodriguez M, Tulipani S, Isabel Queipo-Ortuño M, Urpi-Sarda M, Tinahones FJ, Andres-Lacueva C. Metabolomic insights into the intricate gut microbial-host interaction in the development of obesity and type 2 diabetes. Front Microbiol. 2015;6:1151. doi: 10.3389/fmicb.2015.01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pols TWH, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5: a valuable metabolic target. Dig Dis. 2011;29(1):37–44. doi: 10.1159/000324126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sircana A, Framarin L, Leone N, Berrutti M, Castellino F, Parente R, De Michieli F, Paschetta E, Musso G. Altered Gut Microbiota in Type 2 Diabetes: just a Coincidence? Curr Diab Rep. 2018;18:98. doi: 10.1007/s11892-018-1057-6. [DOI] [PubMed] [Google Scholar]
- 36.Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30:492–506. doi: 10.1038/s41422-020-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran KL, Park YI, Pandya S, Muliyil NJ, Jensen BD, Huynh K, Nguyen QT. Overview of Glucagon-Like Peptide-1 Receptor Agonists for the Treatment of Patients with Type 2 Diabetes. Am Health Drug Benefits. 2017;10:178–188. [PMC free article] [PubMed] [Google Scholar]
- 38.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley RE. Obesity and the human microbiome. Curr Opin Gastroenterol. 2010;26(1):5–11. doi: 10.1097/MOG.0b013e328333d751. [DOI] [PubMed] [Google Scholar]
- 40.Briceno Noriega D, Zenker HE, Croes C-A, Ewaz A, Ruinemans-Koerts J, Savelkoul HFJ, van Neerven Rjj, Teodorowicz M. Receptor Mediated Effects of Advanced Glycation End Products (AGEs) on Innate and Adaptative Immunity: relevance for Food Allergy. Nutrients. 2022:14. doi: 10.3390/nu14020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chawla D, Tripathi AK. Role of advanced glycation end products (AGEs) and its receptor (RAGE)-mediated diabetic vascular complications. Integr Food, Nutr Metab. 2019:6. doi: 10.15761/ifnm.1000267. [DOI] [Google Scholar]
- 42.Jialal I, Huet BA, Kaur H, Chien A, Devaraj S. Increased toll-like receptor activity in patients with metabolic syndrome. Diabetes Care. 2012;35:900–904. doi: 10.2337/dc11-2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.León-Pedroza JI, González-Tapia LA, Del Olmo-Gil E, Castellanos-Rodríguez D, Escobedo G, González-Chávez A. Inflamación sistémica de grado bajo y su relación con el desarrollo de enfermedades metabólicas: de la evidencia molecular a la aplicación clínica. Cir Cir. 2015;83:543–551. doi: 10.1016/j.circir.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 44.Imai Y, Dobrian AD, Weaver JR, Butcher MJ, Cole BK, Galkina EV, Morris MA, Taylor-Fishwick DA, Nadler JL. Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes Metab. 2013;15(Suppl 3):117–129. doi: 10.1111/dom.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sethi JK, Hotamisligil GS. Metabolic Messengers: tumour necrosis factor. Nat Metab. 2021;3:1302–1312. doi: 10.1038/s42255-021-00470-z. [DOI] [PubMed] [Google Scholar]
- 46.Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–522. doi: 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Q, Vijayakumar A, Kahn BB. Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol. 2018;19:654–672. doi: 10.1038/s41580-018-0044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chakaroun RM, Massier L, Kovacs P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: perpetrators or Bystanders? Nutrients. 2020;12:1082. doi: 10.3390/nu12041082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaike AH, Paul D, Bhute S, Dhotre DP, Pande P, Upadhyaya S, Reddy Y, Sampath R, Ghosh D, Chandraprabha D, et al. The gut microbial diversity of newly diagnosed diabetics but not of prediabetics is significantly different from that of healthy nondiabetics. mSystems. 2020;5(2). doi: 10.1128/msystems.00578-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang HR, Lee H-Y. Mechanisms linking gut microbial metabolites to insulin resistance. World J Diabetes. 2021;12:730–744. doi: 10.4239/wjd.v12.i6.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gojda J, Cahova M. Gut Microbiota as the Link between Elevated BCAA Serum Levels and Insulin Resistance. Biomolecules. 2021:11. doi: 10.3390/biom11101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedersen C, Gallagher E, Horton F, Ellis RJ, Ijaz UZ, Wu H, Jaiyeola E, Diribe O, Duparc T, Cani PD, et al. Host-microbiome interactions in human type 2 diabetes following prebiotic fibre (galacto-oligosaccharide) intake. Br J Nutr. 2016;116:1869–1877. doi: 10.1017/S0007114516004086. [DOI] [PubMed] [Google Scholar]
- 53.Böni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, Pattou F, Halban PA, Weir GC, Donath MY. Increased Interleukin (IL)-1β Messenger Ribonucleic Acid Expression in β-Cells of Individuals with Type 2 Diabetes and Regulation of IL-1β in Human Islets by Glucose and Autostimulation. J Clin Endocrinol Met. 2008;93:4065–4074. doi: 10.1210/jc.2008-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez-Alcoholado L, Castellano-Castillo D, Jordán-Martínez L, Moreno-Indias I, Cardila-Cruz P, Elena D, Muñoz-Garcia AJ, Queipo-Ortuño MI, Jimenez-Navarro M. Role of gut microbiota on cardio-metabolic parameters and immunity in coronary artery disease patients with and without type-2 diabetes mellitus. Front Microbiol. 2017;8:1936. doi: 10.3389/fmicb.2017.01936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cordero MD, Alcocer-Gómez E. Inflammasomes: clinical and Therapeutic Implications. Sevilla, Spain: Springer; 2018. [Google Scholar]
- 56.Malaguarnera R, Scamporrino A, Filippello A, Di Mauro S, Minardo A, Purrello F, Piro S. The entero-insular axis: a journey in the physiopathology of diabetes. Explor Med. 2020:1. doi: 10.37349/emed.2020.00025. [DOI] [Google Scholar]
- 57.Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, Boeing H, Pfeiffer AFH. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 58.Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, Braverman S, Tengeler AC, Barak O, Elazar M, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018;359:1376–1383. doi: 10.1126/science.aar3318. [DOI] [PubMed] [Google Scholar]
- 59.Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. 2019;16:35–56. doi: 10.1038/s41575-018-0061-2. [DOI] [PubMed] [Google Scholar]
- 60.Hasan N, Yang H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ. 2019;7:e7502. doi: 10.7717/peerj.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makki K, Deehan EC, Walter J, Bäckhed F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe. 2018;23:705–715. doi: 10.1016/j.chom.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 62.Liu X, Cao S, Zhang X. Modulation of gut microbiota–brain axis by probiotics, prebiotics, and diet. J Agric Food Chem. 2015;63:7885–7895. doi: 10.1021/acs.jafc.5b02404. [DOI] [PubMed] [Google Scholar]
- 63.Holscher HD. Diet Affects the Gastrointestinal Microbiota and Health. J Acad Nutr Diet. 2020;120:495–499. doi: 10.1016/j.jand.2019.12.016. [DOI] [PubMed] [Google Scholar]
- 64.Shortt C, Hasselwander O, Meynier A, Nauta A, Fernández EN, Putz P, Rowland I, Swann J, Türk J, Vermeiren J, et al. Systematic review of the effects of the intestinal microbiota on selected nutrients and non-nutrients. Eur J Nutr. 2018;57:25–49. doi: 10.1007/s00394-017-1546-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Durganaudu H, Kunasegaran T, Ramadas A. Dietary glycaemic index and type 2 diabetes mellitus: potential modulation of gut microbiota. Prog Biophys Mol. 3. doi: 10.36877/pmmb.a0000082. [DOI] [Google Scholar]
- 66.Hills R, Pontefract B, Mishcon H, Black C, Sutton S, Theberge C. Gut Microbiome: profound Implications for Diet and Disease. Nutrients. 2019;11:1613. doi: 10.3390/nu11071613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leeming ER, Johnson AJ, Spector TD, Le Roy CI. Effect of Diet on the Gut Microbiota: rethinking Intervention Duration. Nutrients. 2019;11:2862. doi: 10.3390/nu11122862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ismael S, Silvestre MP, Vasques M, Araújo JR, Morais J, Duarte MI, Pestana D, Faria A, Pereira-Leal JB, Vaz J, et al. A pilot study on the metabolic impact of mediterranean diet in type 2 diabetes: is gut microbiota the key? Nutrients. 2021;13. doi: 10.3390/nu13041228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ren M, Zhang H, Qi J, Hu A, Jiang Q, Hou Y, Feng Q, Ojo O, Wang X. An almond-based low carbohydrate diet improves depression and glycometabolism in patients with type 2 diabetes through modulating gut microbiota and glp-1: a randomized controlled trial. Nutrients. 2020:12. doi: 10.3390/nu12103036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kassaian N, Feizi A, Rostami S, Aminorroaya A, Yaran M, Amini M. The effects of 6 mo of supplementation with probiotics and synbiotics on gut microbiota in the adults with prediabetes: a double blind randomized clinical trial. Nutrition. 2020;79-80:110854. doi: 10.1016/j.nut.2020.110854. [DOI] [PubMed] [Google Scholar]
- 71.Liu Y, Wang Y, Ni Y, Cheung CKY, Lam KSL, Wang Y, Xia Z, Ye D, Guo J, Tse MA, et al. Gut Microbiome Fermentation Determines the Efficacy of Exercise for Diabetes Prevention. Cell Metabolism. 2020;31:77–91.e5. doi: 10.1016/j.cmet.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 72.Clauss M, Gérard P, Mosca A, Leclerc M. Interplay Between Exercise and Gut Microbiome in the Context of Human Health and Performance. Front Nutr. 2021;8:637010. doi: 10.3389/fnut.2021.637010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Motiani KK, Carmen Collado M, Eskelinen -J-J, Virtanen KA, Löyttyniemi E, Salminen S, Nuutila P, Kalliokoski KK, Hannukainen JC. Exercise training modulates gut microbiota profile and improves endotoxemia. Med Sci Sports Exerc. 2020;52:94–104. doi: 10.1249/mss.0000000000002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Utzschneider KM, Kratz M, Damman CJ, Hullarg M. Mechanisms linking the gut microbiome and glucose metabolism. J Clin Endocrinol Metab. 2016;101:1445–1454. doi: 10.1210/jc.2015-4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Y, Wang Y, Ni Y, Cheung CKY, Lam KSL, Wang Y, Xia Z, Ye D, Guo J, Tse MA, et al. Gut Microbiome Fermentation Determines the Efficacy of Exercise for Diabetes Prevention. Cell Metabolism. 2020;31:77–91.e5. doi: 10.1016/j.cmet.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 76.Pasini E, Corsetti G, Assanelli D, Testa C, Romano C, Dioguardi FS, Aquilani R. Effects of chronic exercise on gut microbiota and intestinal barrier in human with type 2 diabetes. Minerva Med. 2019;110:3–11. doi: 10.23736/S0026-4806.18.05589-1. [DOI] [PubMed] [Google Scholar]
- 77.Lambert JE, Myslicki JP, Bomhof MR, Belke DD, Shearer J, Reimer RA. Exercise training modifies gut microbiota in normal and diabetic mice. Appl Physiol Nutr Metab. 2015;40:749–752. doi: 10.1139/apnm-2014-0452. [DOI] [PubMed] [Google Scholar]
- 78.Chaudhury A, Duvoor C, Reddy Dendi VS, Kraleti S, Chada A, Ravilla R, Marco A, Shekhawat NS, Montales MT, Kuriakose K, et al. Clinical Review of Antidiabetic Drugs: implications for Type 2 Diabetes Mellitus Management. Front Endocrinol. 2017;8:6. doi: 10.3389/fendo.2017.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Madsen MSA, Grønlund RV, Eid J, Christensen-Dalsgaard M, Sommer M, Rigbolt K, Madsen MR, Jelsing J, Vrang N, Hansen HH, et al. Characterization of local gut microbiome and intestinal transcriptome responses to rosiglitazone treatment in diabetic db/db mice. Biomed Pharmacother. 2021;133:110966. doi: 10.1016/j.biopha.2020.110966. [DOI] [PubMed] [Google Scholar]
- 80.Whang A, Nagpal R, Yadav H. Bi-directional drug-microbiome interactions of anti-diabetics. EBioMedicine. 2019;39:591–602. doi: 10.1016/j.ebiom.2018.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Bommel Ejm, Herrema H, Davids M, Kramer MHH, Nieuwdorp M, van Raalte DH. Effects of 12-week treatment with dapagliflozin and gliclazide on faecal microbiome: results of a double-blind randomized trial in patients with type 2 diabetes. Diabetes Metab. 2020;46:164–168. doi: 10.1016/j.diabet.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 82.Montandon S, Jornayvaz F. Effects of Antidiabetic Drugs on Gut Microbiota Composition. Genes. 2017;8:250. doi: 10.3390/genes8100250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chaudhury A, Duvoor C, Reddy Dendi VS, Kraleti S, Chada A, Ravilla R, Marco A, Shekhawat NS, Montales MT, Kuriakose K, et al. Clinical Review of Antidiabetic Drugs: implications for Type 2 Diabetes Mellitus Management. Front Endocrinol. 2017;8:6. doi: 10.3389/fendo.2017.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Q, Hu N. Effects of Metformin on the Gut Microbiota in Obesity and Type 2 Diabetes Mellitus. Diabetes, Metabolic Syndrome and Obesity: targets and Therapy. 2020;13:5003–5014. doi: 10.2147/dmso.s286430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Pedersen HK, et al. Erratum: corrigendum: disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2017;545:116. doi: 10.1038/nature22318. [DOI] [PubMed] [Google Scholar]
- 86.Shin N-R, Lee J-C, Lee H-Y, Kim M-S, Whon TW, Lee M-S, Bae J-W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63:727–735. doi: 10.1136/gutjnl-2012-303839. [DOI] [PubMed] [Google Scholar]
- 87.de la Cuesta-zuluaga J, Mueller NT, Corrales-Agudelo V, Velásquez-Mejía EP, Carmona JA, Abad JM, Escobar JS. Metformin is associated with higher relative abundance of mucin-degrading akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care. 2017;40:54–62. doi: 10.2337/dc16-1324. [DOI] [PubMed] [Google Scholar]
- 88.Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, Ståhlman M, Olsson LM, Serino M, Planas-Fèlix M, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23:850–858. doi: 10.1038/nm.4345. [DOI] [PubMed] [Google Scholar]
- 89.Lee H, Lee Y, Kim J, An J, Lee S, Kong H, Song Y, Lee C-K, Kim K. Modulation of the gut microbiota by metformin improves metabolic profiles in aged obese mice. Gut Microbes. 2018;9:155–165. doi: 10.1080/19490976.2017.1405209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang X, Zhao Y, Xu J, Xue Z, Zhang M, Pang X, Zhang X, Zhao L. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci Rep. 2015:5. doi: 10.1038/srep14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Charpentier J, Briand F, Lelouvier B, Servant F, Azalbert V, Puel A, Christensen JE, Waget A, Branchereau M, Garret C, et al. Liraglutide targets the gut microbiota and the intestinal immune system to regulate insulin secretion. Acta Diabetologica. 2021;58:881–897. doi: 10.1007/s00592-020-01657-8. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, Wang T, Wang X. Featured article: structure moderation of gut microbiota in liraglutide-treated diabetic male rats. Exp Biol Med. 2018;243:34–44. doi: 10.1177/1535370217743765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karagiannis T, Boura P, Tsapas A. Safety of dipeptidyl peptidase 4 inhibitors: a perspective review. Ther Adv Drug Saf. 2014;5:138–146. doi: 10.1177/2042098614523031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yan X, Feng B, Li P, Tang Z, Wang L. Microflora disturbance during progression of glucose intolerance and effect of sitagliptin: an animal study. J Diabetes Res. 2016;2016:1–10. doi: 10.1155/2016/2093171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang Q, Xiao X, Li M, Yu M, Ping F, Zheng J, Wang T, Wang X. Vildagliptin increases butyrate-producing bacteria in the gut of diabetic rats. PLoS One. 2017;12:e0184735. doi: 10.1371/journal.pone.0184735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang L, Li P, Tang Z, Yan X, Feng B. Structural modulation of the gut microbiota and the relationship with body weight: compared evaluation of liraglutide and saxagliptin treatment. Sci Rep. 2016:6. doi: 10.1038/srep33251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Francino MP. Antibiotics and the Human Gut Microbiome: dysbioses and Accumulation of Resistances. Front Microbiol. 2015;6:1543. doi: 10.3389/fmicb.2015.01543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Francino MP. Antibiotics and the Human Gut Microbiome: dysbioses and Accumulation of Resistances. Front Microbiol. 2015;6:1543. doi: 10.3389/fmicb.2015.01543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fujisaka S, Ussar S, Clish C, Devkota S, Dreyfuss JM, Sakaguchi M, Soto M, Konishi M, Softic S, Altindis E, et al. Antibiotic effects on gut microbiota and metabolism are host dependent. J Clin Invest. 2016;126(12):4430–4443. doi: 10.1172/jci86674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Girdhar K, Soto M, Huang Q, Orliaguet L, Cederquist C, Sundaresh B, Hu J, Figura M, Raisingani A, Canfora EE, et al. gut microbiota regulate pancreatic growth, exocrine function, and gut hormones. Diabetes. 2022;71(5):945–960. doi: 10.2337/db21-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reijnders D, Goossens GH, Hermes GDA, Neis EPJG, van der Beek CM, Most J, Holst JJ, Lenaerts K, Kootte RS, Nieuwdorp M, et al. Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: a randomized double-blind placebo-controlled trial. Cell Metab. 2016;24:341. doi: 10.1016/j.cmet.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 103.Vrieze A, de Groot Pf, Kootte RS, Knaapen M, van Nood E, Nieuwdorp M. Fecal transplant: a safe and sustainable clinical therapy for restoring intestinal microbial balance in human disease? Best Pract Res Clin Gastroenterol. 2013;27:127–137. doi: 10.1016/j.bpg.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 104.Schauer PR, Nor Hanipah Z, Rubino F. Metabolic surgery for treating type 2 diabetes mellitus: now supported by the world’s leading diabetes organizations. Cleve Clin J Med. 2017;84:S47–56. doi: 10.3949/ccjm.84.s1.06. [DOI] [PubMed] [Google Scholar]
- 105.Jin Z-L, Liu W. Progress in treatment of type 2 diabetes by bariatric surgery. World J Diabetes. 2021;12(8):1187–1199. doi: 10.4239/wjd.v12.i8.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Graessler J, Qin Y, Zhong H, Zhang J, Licinio J, Wong M-L, Xu A, Chavakis T, Bornstein AB, Ehrhart-Bornstein M, et al. Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: correlation with inflammatory and metabolic parameters. Pharmacogenomics J. 2013;13:514–522. doi: 10.1038/tpj.2012.43. [DOI] [PubMed] [Google Scholar]
- 107.Sánchez-Alcoholado L, Gutiérrez-Repiso C, Gómez-Pérez AM, García-Fuentes E, Tinahones FJ, Moreno-Indias I. Gut microbiota adaptation after weight loss by Roux-en-Y gastric bypass or sleeve gastrectomy bariatric surgeries. Surg Obes Relat Dis. 2019;15(11):1888–1895. doi: 10.1016/j.soard.2019.08.551. [DOI] [PubMed] [Google Scholar]
- 108.Lau E, Belda E, Picq P, Carvalho D, Ferreira-Magalhães M, Silva MM, Barroso I, Correia F, Vaz CP, Miranda I, et al. Gut microbiota changes after metabolic surgery in adult diabetic patients with mild obesity: a randomised controlled trial. Diabetol Metab Syndr. 2021;13:56. doi: 10.1186/s13098-021-00672-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Debédat J, Le Roy T, Voland L, Belda E, Alili R, Adriouch S, Bel Lassen P, Kasahara K, Hutchison E, Genser L, et al. The human gut microbiota contributes to type-2 diabetes non-resolution 5-years after Roux-en-Y gastric bypass. Gut Microbes. 2022;14:2050635. doi: 10.1080/19490976.2022.2050635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu H, Hu C, Zhang X, Jia W. Role of gut microbiota, bile acids and their cross-talk in the effects of bariatric surgery on obesity and type 2 diabetes. J Diabetes Investig. 2018;9(1):13–20. doi: 10.1111/jdi.12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ilhan ZE, DiBaise JK, Dautel SE, Isern NG, Kim Y-M, Hoyt DW, Schepmoes AA, Brewer HM, Weitz KK, Metz TO, et al. Temporospatial shifts in the human gut microbiome and metabolome after gastric bypass surgery. NPJ Biofilms Microbiomes. 2020;6(1):12. doi: 10.1038/s41522-020-0122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hanssen NMJ, de Vos WM, Nieuwdorp M. Fecal microbiota transplantation in human metabolic diseases: from a murky past to a bright future? Cell Metabolism. 2021;33(6):1098–1110. doi: 10.1016/j.cmet.2021.05.005. [DOI] [PubMed] [Google Scholar]
- 113.Hanssen NMJ, de Vos WM, Nieuwdorp M. Fecal microbiota transplantation in human metabolic diseases: from a murky past to a bright future? Cell Metabolism. 2021;33:1098–1110. doi: 10.1016/j.cmet.2021.05.005. [DOI] [PubMed] [Google Scholar]
- 114.Huda MN, Nazmul Huda M, Kim M, Bennett BJ. Modulating the Microbiota as a Therapeutic Intervention for Type 2 Diabetes. Front Endocrinol. 2021:12. doi: 10.3389/fendo.2021.632335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JFW, Dallinga–Thie GM, Ackermans MT, Serlie MJ, Oozeer R, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–6.e7. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 116.Ng SC, Xu Z, Mak JWY, Yang K, Liu Q, Zuo T, Tang W, Lau L, Lui RN, Wong SH, et al. Microbiota engraftment after faecal microbiota transplantation in obese subjects with type 2 diabetes: a 24-week, double-blind, randomised controlled trial. Gut. 2022;71:716–723. doi: 10.1136/gutjnl-2020-323617. [DOI] [PubMed] [Google Scholar]
- 117.Wang H, Lu Y, Yan Y, Tian S, Zheng D, Leng D, Wang C, Jiao J, Wang Z, Bai Y. Promising treatment for type 2 diabetes: fecal microbiota transplantation reverses insulin resistance and impaired islets. Front Cell Infect Microbiol. 2019;9:455. doi: 10.3389/fcimb.2019.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang L, Zhou W, Zhan L, Hou S, Zhao C, Bi T, Lu X. Fecal microbiota transplantation alters the susceptibility of obese rats to type 2 diabetes mellitus. Aging. 2020;12(17):17480–17502. doi: 10.18632/aging.103756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Han J-L, Lin H-L. Intestinal microbiota and type 2 diabetes: from mechanism insights to therapeutic perspective. World J Gastroenterol. 2014;20(47):17737–17745. doi: 10.3748/wjg.v20.i47.17737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang K, Niu J, Zuo T, Sun Y, Xu Z, Tang W, Liu Q, Zhang J, Ng E, Wong SKH, et al. Alterations in the gut virome in obesity and type 2 diabetes mellitus. Gastroenterology. 2021;161:1257–69.e13. doi: 10.1053/j.gastro.2021.06.056. [DOI] [PubMed] [Google Scholar]
- 121.Ma Y, You X, Mai G, Tokuyasu T, Liu C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome. 2018:6. doi: 10.1186/s40168-018-0410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang X, Li L, Butcher J, Stintzi A, Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. 2019:7. doi: 10.1186/s40168-019-0767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Garcia-Mazcorro JF, Pedreschi R, Chew B, Dowd SE, Kawas JR, Noratto GD. Dietary supplementation with raspberry extracts modifies the fecal microbiota in obese diabetic db/db mice. J Microbiol Biotechnol. 2018;28:1247–1259. doi: 10.4014/jmb.1803.03020. [DOI] [PubMed] [Google Scholar]
- 124.Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun. 2012;80:3786–3794. doi: 10.1128/IAI.00647-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, Fu H, Xue X, Lu C, Ma J, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359:1151–1156. doi: 10.1126/science.aao5774. [DOI] [PubMed] [Google Scholar]
- 126.Zhou W, Reza Sailani M, Contrepois K, Zhou Y, Ahadi S, Leopold SR, Zhang MJ, Rao V, Avina M, Mishra T, et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature. 2019;569(7758):663–671. doi: 10.1038/s41586-019-1236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rogier R, Ederveen THA, Boekhorst J, Wopereis H, Scher JU, Manasson J, Frambach SJC, Knol J, Garssen J, van der Kraan PM, et al. Aberrant intestinal microbiota due to IL-1 receptor antagonist deficiency promotes IL-17- and TLR4-dependent arthritis. Microbiome. 2017;5. doi: 10.1186/s40168-017-0278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, ter Horst R, Jansen T, Jacobs L, Bonder MJ, et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell. 2016;167:1125–36.e8. doi: 10.1016/j.cell.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kumar M, Ji B, Zengler K, Nielsen J. Modelling approaches for studying the microbiome. Nat Microbiol. 2019;4:1253–1267. doi: 10.1038/s41564-019-0491-9. [DOI] [PubMed] [Google Scholar]
- 130.Miossec MJ, Valenzuela SL, Mendez KN, Castro‐Nallar E. Computational Methods for Human Microbiome Analysis. Curr Protoc Microbiol. 2017:47. doi: 10.1002/cpmc.41. [DOI] [PubMed] [Google Scholar]
- 131.Marizzoni M, Gurry T, Provasi S, Greub G, Lopizzo N, Ribaldi F, Festari C, Mazzelli M, Mombelli E, Salvatore M, et al. Comparison of bioinformatics pipelines and operating systems for the analyses of 16s rrna gene amplicon sequences in human fecal samples. Front Microbiol. 2020;11:1262. doi: 10.3389/fmicb.2020.01262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Briefings Bioinf. 2021;22:178–193. doi: 10.1093/bib/bbz155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Prodan A, Tremaroli V, Brolin H, Zwinderman AH, Nieuwdorp M, Levin E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One. 2020;15:e0227434. doi: 10.1371/journal.pone.0227434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Akerman NH, Butterfield DA, Huber JA. Phylogenetic diversity and functional gene patterns of sulfur-oxidizing subseafloor Epsilonproteobacteria in diffuse hydrothermal vent fluids. Front Microbiol. 2013;4:185. doi: 10.3389/fmicb.2013.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Greenacre M. Compositional data analysis. Ann Rev Stat Appl. 2021;8(1):271–299. doi: 10.1146/annurev-statistics-042720-124436. [DOI] [Google Scholar]
- 137.Ghosh A, Firdous S, Saha S. Bioinformatics for Human Microbiome. Adv Bioinf. 2021;333–350. doi: 10.1007/978-981-33-6191-1_17. [DOI] [Google Scholar]
- 138.Schwager E, Mallick H, Ventz S, Huttenhower C. A Bayesian method for detecting pairwise associations in compositional data. PLoS Comput Biol. 2017;13:e1005852. doi: 10.1371/journal.pcbi.1005852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Topçuoğlu BD, Lesniak NA, Ruffin MT, Wiens J, Schloss PD. A framework for effective application of machine learning to microbiome-based classification problems. mBio. 2020:11. doi: 10.1128/mbio.00434-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, Gonzalez A, Kosciolek T, McCall L-I, McDonald D, et al. Best practices for analysing microbiomes. Nat Rev Microbiol. 2018;16:410–422. doi: 10.1038/s41579-018-0029-9. [DOI] [PubMed] [Google Scholar]
- 141.Deng Y, Jiang Y-H, Yang Y, He Z, Luo F, Zhou J. Molecular ecological network analyses. BMC Bioinf. 2012:13. doi: 10.1186/1471-2105-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Faust K, Raes J. CoNet app: inference of biological association networks using Cytoscape. F1000Res. 2016;5:1519. doi: 10.12688/f1000research.9050.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shaw GT-W, Pao -Y-Y, Wang D. MetaMIS: a metagenomic microbial interaction simulator based on microbial community profiles. BMC Bioinf. 2016:17. doi: 10.1186/s12859-016-1359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kuntal BK, Gadgil C, Mande SS. Web-gLV: a Web Based platform for lotka-volterra based modeling and simulation of microbial populations. Front Microbiol. 2019;10:288. doi: 10.3389/fmicb.2019.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Peng G-S, Wu J. Optimal network topology for structural robustness based on natural connectivity. Phys A: Stat Mech Appl. 2016;443:212–220. doi: 10.1016/j.physa.2015.09.023. [DOI] [Google Scholar]
- 146.Ross MC, Muzny DM, McCormick JB, Gibbs RA, Fisher-Hoch SP, Petrosino JF. 16S gut community of the Cameron County Hispanic Cohort. Microbiome. 2015:3. doi: 10.1186/s40168-015-0072-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bastida E, Cuellar I, Villas P. Prevalence of diabetes mellitus and related conditions in a south texas mexican american sample. J Commu Health Nurs. 2001;18:75–84. doi: 10.1207/s15327655jchn1802_01. [DOI] [PubMed] [Google Scholar]
- 148.Tong X, Xu J, Lian F, Yu X, Zhao Y, Xu L, Zhang M, Zhao X, Shen J, Wu S, et al. Structural alteration of gut microbiota during the amelioration of human type 2 diabetes with hyperlipidemia by metformin and a traditional chinese herbal formula: a multicenter, randomized, open label clinical trial. mBio. 2018;9. doi: 10.1128/mbio.02392-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yan J, Sheng L, Li H. is it the Holy Grail for ameliorating metabolic diseases? Gut Microbes. 2021;13:1984104. doi: 10.1080/19490976.2021.1984104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.de la Cuesta-zuluaga J, Mueller NT, Corrales-Agudelo V, Velásquez-Mejía EP, Carmona JA, Abad JM, Escobar JS. Metformin is associated with higher relative abundance of mucin-degrading akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care. 2017;40:54–62. doi: 10.2337/dc16-1324. [DOI] [PubMed] [Google Scholar]
- 151.Zhang J, Ni Y, Qian L, Fang Q, Zheng T, Zhang M, Gao Q, Zhang Y, Ni J, Hou X, et al. Decreased abundance of akkermansia muciniphila leads to the impairment of insulin secretion and glucose homeostasis in lean type 2 diabetes. Adv Sci. 2021;8:2100536. doi: 10.1002/advs.202100536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Duncan SH, Hold GL, Barcenilla A, Stewart CS, Flint HJ. Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int J Syst Evol Microbiol. 2002;52:1615–1620. doi: 10.1099/00207713-52-5-1615. [DOI] [PubMed] [Google Scholar]
- 153.Quévrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, Miquel S, Carlier L, Bermúdez-Humarán LG, Pigneur B, et al. Identification of an anti-inflammatory protein fromFaecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut. 2016;65:415–425. doi: 10.1136/gutjnl-2014-307649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Das T, Jayasudha R, Chakravarthy S, Prashanthi GS, Bhargava A, Tyagi M, Rani PK, Pappuru RR, Sharma S, Shivaji S. Alterations in the gut bacterial microbiome in people with type 2 diabetes mellitus and diabetic retinopathy. Sci Rep. 2021:11. doi: 10.1038/s41598-021-82538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bucci V, Xavier JB. Towards predictive models of the human gut microbiome. J Mol Biol. 2014;426:3907–3916. doi: 10.1016/j.jmb.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lieven C, Beber ME, Olivier BG, Bergmann FT, Ataman M, Babaei P, Bartell JA, Blank LM, Chauhan S, Correia K, et al. MEMOTE for standardized genome-scale metabolic model testing. Nat Biotechnol. 2020;38:272–276. doi: 10.1038/s41587-020-0446-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. COBRApy: constraints-based reconstruction and analysis for python. BMC Syst Biol. 2013;7:74. doi: 10.1186/1752-0509-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zomorrodi AR, Maranas CD. OptCom: a multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol. 2012;8:e1002363. doi: 10.1371/journal.pcbi.1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Khandelwal RA, Olivier BG, Röling WFM, Teusink B, Bruggeman FJ. Community flux balance analysis for microbial consortia at balanced growth. PLoS One. 2013;8:e64567. doi: 10.1371/journal.pone.0064567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, de Wouters T, Juste C, Rizkalla S, Chilloux J, et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015;22:320–331. doi: 10.1016/j.cmet.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 161.Bauer E, Zimmermann J, Baldini F, Thiele I, Kaleta C. BacArena: individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput Biol. 2017;13:e1005544. doi: 10.1371/journal.pcbi.1005544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zomorrodi AR, Islam MM, Maranas CD. D-Optcom: Dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth Biol. 2014;3:247–257. doi: 10.1021/sb4001307. [DOI] [PubMed] [Google Scholar]
- 163.Popp D, Centler F. μBialSim: constraint-based dynamic simulation of complex microbiomes. Front Bioeng Biotechnol. 2020;8:574. doi: 10.3389/fbioe.2020.00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bengtsson-Palme J. Microbial model communities: to understand complexity, harness the power of simplicity. Comput Struct Biotechnol J. 2020;18:3987–4001. doi: 10.1016/j.csbj.2020.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dillard LR, Payne DD, Papin JA. Mechanistic models of microbial community metabolism. Mol Omics. 2021;17:365–375. doi: 10.1039/D0MO00154F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rosario D, Benfeitas R, Bidkhori G, Zhang C, Uhlen M, Shoaie S, Mardinoglu A. Understanding the representative gut microbiota dysbiosis in metformin-treated type 2 diabetes patients using genome-scale metabolic modeling. Front Physiol. 2018;9:775. doi: 10.3389/fphys.2018.00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Guasch-Ferré M, Hruby A, Toledo E, Clish CB, Martínez-González MA, Salas-Salvadó J, Hu FB. Metabolomics in prediabetes and diabetes: a systematic review and meta-analysis. Diabetes Care. 2016;39:833–846. doi: 10.2337/dc15-2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sami W, Alabdulwahhab KM, Ab Hamid MR, Alasbali TA, Alwadani FA, Ahmad MS. Dietary Knowledge among Adults with Type 2 Diabetes—Kingdom of Saudi Arabia. Int J Environ Res Public Health. 2020;17(3):858. doi: 10.3390/ijerph17030858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Diener C, de Lourdes Reyes-escogido M, Jimenez-Ceja LM, Matus M, Gomez-Navarro CM, Chu ND, Zhong V, Elizabeth Tejero M, Alm E, Resendis-Antonio O, et al. Progressive Shifts in the gut microbiome reflect prediabetes and diabetes development in a treatment-Naive Mexican Cohort. Front Endocrinol. 2021;11. doi: 10.3389/fendo.2020.602326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vernocchi P, Del Chierico F, Putignani L. Gut microbiota metabolism and interaction with food components. Int J Mol Sci. 2020;21(10):3688. doi: 10.3390/ijms21103688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Díaz-Rizzolo DA, Kostov B, López-Siles M, Serra A, Colungo C, González-de-paz L, Martinez-Medina M, Sisó-Almirall A, Gomis R. Healthy dietary pattern and their corresponding gut microbiota profile are linked to a lower risk of type 2 diabetes, independent of the presence of obesity. Clin Nutr. 2020;39(2):524–532. doi: 10.1016/j.clnu.2019.02.035. [DOI] [PubMed] [Google Scholar]
- 173.Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, Ben-Yacov O, Lador D, Avnit-Sagi T, Lotan-Pompan M, et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell. 2015;163(5):1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 174.Wilcox MH, McGovern BH, Hecht GA. The efficacy and safety of fecal microbiota transplant for recurrent clostridiumdifficile infection: current understanding and gap analysis. Open Forum Infect Dis. 2020:7. doi: 10.1093/ofid/ofaa114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zellmer C, Sater MRA, Huntley MH, Osman M, Olesen SW, Ramakrishna B. Shiga toxin–producing escherichia coli transmission via fecal microbiota transplant. Clin Infect Dis. 2021;72(11):e876–80. doi: 10.1093/cid/ciaa1486. [DOI] [PubMed] [Google Scholar]
- 176.Thiele I, Sahoo S, Heinken A, Hertel J, Heirendt L, Aurich MK, Fleming RM. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol Syst Biol. 2020;16(5):e8982. doi: 10.15252/msb.20198982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lipworth W. Real-world data to generate evidence about healthcare interventions: the application of an ethics framework for big data in health and research. Asian Bioeth Rev. 2019;11:289–298. doi: 10.1007/s41649-019-00095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vijayan V, Connolly JP, Condell J, McKelvey N, Gardiner P. Review of wearable devices and data collection considerations for connected health. Sensors. 2021;21:5589. doi: 10.3390/s21165589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.