

Abstract
Accumulation of lipids and their intermediary metabolites under endoplasmic reticulum (ER) stress instigates metabolic failure, described as lipotoxicity, in the kidney. This study aimed to determine ER-stress-related sphingolipid and polyunsaturated fatty acid (PUFA) changes in human kidney cells. Tunicamycin (TM) was employed to induce ER stress and an ER stress inhibitor, tauroursodeoxycholic acid (TUDCA), was given to minimize cytotoxicity. Cell viability was determined by MTT assay. Sphingomyelin (SM), ceramide (CER), and PUFA levels were measured by LC–MS/MS. Glucose-regulated protein 78-kd (GRP78), cleaved caspase-3 and cyclooxygenase-1 (COX-1) levels were assessed by immunofluorescence. Cytosolic phospholipase A2 (cPLA2), total COX, and prostaglandin E2 (PGE2) were measured to evaluate changes in enzyme activity. Decreased cell viability was observed in TM treated cells. Administration of TUDCA following TM treatment significantly increased cell viability compared to TM treatment alone. Tunicamycin-induced ER stress was confirmed by significantly increased protein levels of GRP78. A significant increase was observed in C18-C24 CERs and caspase-3 activity, while a significant decrease occurred in sphingosine-1-phosphate (S1P) and cPLA2 activity in cells treated with TM versus controls. The decrease in cPLA2 activity was accompanied by significantly increased PUFA levels in TM treated cells. TUDCA treatment in conjunction with TM significantly decreased ER stress, C18-C24 CERs, caspase 3 activity, and increased S1P levels. Results show the buildup of long chain CERs and PUFAs in kidney cells undergoing ER stress alongside increased apoptotic activity. TUDCA administration, along with TM treatment alleviated the buildup of CERs and TM-induced apoptotic activity in kidney epithelial cells.
Keywords: kidney, tunicamycin, sphingolipid, ceramide, PUFA
1. Introduction
Changes in ER stress markers can be indicative of disease status and can be used as valuable biomarkers for different human pathologies including cancer, metabolic, degenerative, infectious, and inflammatory diseases1. Endoplasmic reticulum stress plays a role in tubular and glomerular impairment in patients with acute and chronic renal disorders. Tubular epithelial cells undergo an adaptive ER stress in hyperglycemic and proteinuric diseases. However, persistence of hyperglycemia and proteinuria may in due course lead to ER stress-induced apoptosis2. Studies have demonstrated that ER stress is implicated in the initiation of diabetic nephropathy, inflammation-induced kidney injury, renal ischemia–reperfusion and proteinuria3. Research have shown the induction of ER stress after both acute kidney ischemia4 and chronic renal tubular-interstitial injury5. Podocyte and mesangial dysfunction in the glomeruli are also associated with ER stress. In the passive Heymann nephritis model of membranous nephropathy, complement C5b-9 induces glomerular epithelial cell injury, and it is revealed that complement C5b-9 membrane attack complex increases ER stress in glomerular epithelial cells6. Preconditioning with ER stress mediators such as TM or thapsigargin improves mesangioproliferative glomerulonephritis suggesting the possibility of using therapeutic approaches targeting ER stress in certain kidney diseases7.
Kidney tubular epithelial cells are very vulnerable to chemicals, which induce ER stress such as TM, which acts by triggering ER stress through disturbing protein glycosylation8. Indeed, a single sublethal intraperitoneal injection of TM results in transient renal insufficiency in mice9. Markers of ER stress and ER-mediated cell death have also been defined in kidney tissue exposed to nephrotoxic doses of cisplatin, gentamicin, nephrotoxic metabolites of acetaminophen, and in paracetamol-induced renal tubular injury10,11.
The accumulation of lipids and their intermediary metabolites under ER stress instigates metabolic failure, described as lipotoxicity, in the kidney12. Sphingolipids are metabolically diverse lipids that have structural and signaling roles in kidney cells. Sphingolipid metabolism is altered during renal diseases, which may involve ER stress13. A recent study has demonstrated increased ceramide levels in a rodent model of renal ER stress12. Likewise, fatty acid accumulation has also been associated with podocyte ER stress14.
This study aimed to reveal TM-induced ER-stress-related sphingolipid and PUFA changes in human kidney cells. Studies commonly report that changes in sphingolipid metabolism cause intracellular signaling that triggers an unfolded protein response (UPR)15. However, it is known that this is not a unidirectional relationship and activation of UPR also affects sphingolipid metabolism15. In insulinoma cells, triggering of the UPR by thapsigargin results in increased neutral sphingomyelinase activity, leading to elevated ceramide levels16. Stimulation of the UPR response via dithiothreitol (DTT) also increases ceramide levels in insulinoma cells, via an increase in ceramide synthase expression17. In glioblastoma cells, expression of IL-24 causes activation of protein kinase R–like endoplasmic reticulum kinase (PERK), increasing ceramide levels, and causing cell death18. Expression of dominant negative PERK suppresses ceramide generation and cell death in glioblastoma cells indicating that ceramide plays a role in PERK-mediated ER stress18. Tunicamycin treatment of human head and neck squamous cell carcinoma (HNSCC) cell lines down-regulate ceramide-synthase-6-generated C16-ceramide and induces ER stress by activating transcription factor 6 (ATF6) signaling19. Likewise, thapsigargin causes an increase in ceramide, sphingosine-1-phosphate, and sphingosine levels in human keratinocytes20. Dysregulation of sphingolipid levels, particularly an increase in dihydroceramide and dihydrosphingosine, has also been reported in HEK293 cells exposed to thapsigargine and DTT21. Evidence from these studies suggests that alteration of sphingolipids plays an important role in downstream signaling in ER stress.
The ER also plays an important role in PUFA synthesis through desaturases and elongates localized in the phospholipid bilayer of the ER membrane22. Thus, ER stress is also likely to cause alterations in cellular PUFA composition and synthesis. Mammalian cells can synthesize many fatty acids except the two essential PUFAs, which include linoleic (LA, C18: 2n-6) and α-linolenic acid (ALA, C18:3n-3)23. Linoleic acid is the precursor of omega-6, while ALA is the precursor of omega-3 series of PUFAs24. Even though LA and ALA cannot be synthesized in humans, they can be metabolized to other PUFAs by the addition of double bonds and acyl chains via desaturases and elongases, respectively24. Arachidonic acid (AA, C20:4n-6) and eicosapentaenoic acid (EPA, C20:5n-3) can be metabolized to eicosanoids via COX and lipoxygenase (LOX) pathways24. The metabolism of docosahexaenoic acid (DHA, C22:6n-3) via LOX can also yield resolvins and protectins that display potent anti-inflammatory properties and are recognized in the resolution of inflammation24. Ceramide is known to activate cPLA2, which functions to break down cell membrane phospholipids and releases arachidonic acid25. Ceramide has also been reported to increase COX-2 protein levels possibly through extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) pathway26.
The hypothesis of our study was that activation of ER stress could affect both sphingolipid and polyunsaturated fatty acid metabolism in human kidney epithelial cells. The aim of our study was to investigate multiple sphingolipid types and PUFA changes in the tunicamycin-induced UPR response and to examine the effects of these changes on apoptosis activation. HEK-293 human kidney cells have epithelial morphology. Studies have described these cells as “epithelial cells”27,28 and have used these cells to study mechanisms of kidney diseases. HEK-293 cells have been studied in renal toxicity29, in kidney diseases associated with ER stress30–33 and in collagen formation linked with fibrotic kidney disease34. We combined lipidomics measurements to study ER-stress-related sphingolipid and PUFA changes in HEK-293 cell lines. The connection between ER-stress-induced lipotoxocity and inflammation were also evaluated by measuring cPLA2 activity, COX-1 protein expression, and PGE2 formation.
2. Materials and methods
2.1. Cell culture and treatment conditions
HEK-293 (ATCC CRL-1573) kidney cell line was originally obtained from American Type Culture Collection (ATCC; Manassas, Virginia, USA). Cells were cultured in Dulbecco’s Modified Eagle’s Medium: Nutrient Mixture F-12 (DMEM/ F-12; Gibco, Life Technologies Limited, Paisley, UK) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco, Life Technologies Corporation, Paisley, UK), and 1% penicillin–streptomycin (Gibco, Life Technologies Corporation, Grand Island, USA). Amphotericin-B (Gibco, Life Technologies Limited, Paisley, UK) was added at 200 μl per 1 L medium. The prepared medium was sterilized by passing through a .22-μm bottle-top filter and stored at 4 °C for use. Cultures were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2. After the cell culture reached 80% confluency, the cells were passaged by trypsinization (.05% Trypsin–EDTA; Gibco, Life Technologies, Paisley, UK). Cells from passages third to tenth were used in the experiments and were counted by Micro Counter 1300 device (Celeromics Technologies, Grenoble, France).
Endoplasmic reticulum stress was induced by TM (Sigma-Aldrich, St. Louis, MO, USA) prepared in dimethyl sulfoxide (DMSO; Fisher Scientific, Fair Lawn, New Jersey, USA) at a stock concentration of 10 mg/ml. Doses of 1, 5, and 10 μg/ml TM were used to determine the optimum dose of TM in creating ER stress. In most of the experiments, cells were treated with 5 μg/ml TM for 24 h to induce ER stress. The dose and duration of TM used to treat cells were based on results of cell viability studies.
The ER stress inhibitor TUDCA (EMD Millipore Corp. Billerica, MA USA) was dissolved in saline at a stock concentration of 10 mM. TUDCA was administered to HEK-293 cells at a dose range of .156–5 mM, and the effect of TUDCA administration was evaluated by cell viability assays.
In most experiments. HEK-293 cells were incubated with either 1 μl/ml DMSO or 5 μg/ml TM for 24 h. .156 mM TUDCA was applied for 12 h. In the TM + TUDCA group, TUDCA (.156 mM) was given 12 h after 5 μg/ml TM application, with a total TM incubation time of 24 h.
2.2. Cell viability assay
Cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Gold Biotechnology Inc., St. Louis, MO, USA). MTT is a yellow tetrazolium dye that turns purple when reduced to an insoluble formazan. This reduction is accomplished in living cells by NADH and NADPH-dependent oxidoreductase enzymes. After dissolution of formazan produced from the cell, absorbance is measured and compared with a control group.
Cells were grown to confluence in 96-well plates and incubated with 1–10 μg/ml TM for 12–36 h. Dose-dependent effect of TUDCA (.156–5 mM) on cell viability was assessed for 12–36 h. Control cells were prepared in plates containing only medium. After the incubation period, the MTT protocol was initiated. At least six repetitions were performed per group. The incubation medium was withdrawn, and 200 μl of 10% MTT solution was added to each well. The 96-well plate was left in the dark for 2 h at 37 °C in a 5% CO2 incubator. After the incubation period, it was observed that purple colored formazan crystals were formed on the bottom. The liquid on top was carefully aspirated and 100 μl of DMSO was added to each well. Formazan crystals were well dissolved in DMSO on the orbital shaker for 5 minutes. The absorbances at 570 and 690 nm were measured colorimetrically in a microplate reader (MicroQuant Plate Reader, Bio-Tek Instruments Inc. Vermont, USA). The 570 nm absorbance values were subtracted from the 690 nm absorbance values. The amount of formazan formation in experimental groups was expressed as % viability compared to control cells.
2.3. Immunofluorescent staining
HEK-293 cells were plated at a density of 100,000 cells per chamber in an 8 chamber-slide (Merck Millipore, Cork, Ireland). The chamber slide was kept in an incubator at 37 °C, 5% CO2 overnight in order to achieve 70% confluency. At the end of the incubation period, the medium was removed and cells were washed twice with .01 M cold PBS. Cells were fixed for 10 min at room temperature in 4% freshly prepared paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) followed by three washes in cold PBS. Cells were permeabilized in PBS with .2% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) for 10 min. After five washes in cold PBS, cells were incubated for 30 min in blocking solution [5% normal goat serum (NGS; Vector Laboratories, Burlingame, CA, USA) in PBS] and treated with either anti-GRP78 (1:200, # ab21685 Abcam, Cambridge, MA, USA), anti-cleaved caspase-3 (1:100, # 9664, Cell Signaling Technology, Danvers, MA, USA), and anti-COX-1 (1:200, # ab227513, Abcam, Cambridge, MA, USA) overnight at 4 °C. The secondary antibody, Alexa Fluor-488 conjugated goat anti-rabbit (1:300, # A11008 life Technologies, USA) was applied for 45 min at room temperature, and nuclei are counterstained with DAPI (Vector Laboratories Inc.,Burlingame, CA, USA) in all experiments. Slides were viewed under a fluorescence microscope (Olympus IX81 fully automated, Tokyo, Japan) and fluorescence intensity was quantified using NIH ImageJ 1.53e software. The corrected total cell fluorescence (CTCF) was calculated as; CTCF = Integrated Density—(area of selected cell × mean fluorescence of background readings).
2.4. Polyunsaturated fatty acid and sphingolipid measurements by LC–MS/MS
Optimized multiple reaction monitoring (MRM) methods were set up for quantifying PUFAs and sphingolipids via ultrafast liquid chromatography (UFLC; LC-20 ad UFLC XR, Shimadzu Corporation, Japan) coupled with tandem mass spectrometry (MS/MS; LCMS-8040, Shimadzu Corporation, Japan). Cellular levels of PUFAs were measured as previously described35. Standards for arachidonic acid (AA, C20:4n6), dihomogamma-linolenic acid (DGLA, C20:3n6), eicosapentaenoic acid (EPA, C20:5n3), and docosahexaenoic acid (DHA, C22:6n3) were purchased from Sigma-Aldrich (St. Louis MO, USA) and AAd8 (Deuterium labeled AA-d8 internal standard) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Samples were prepared for LC–MS/MS analysis as previously described36.
Cellular levels of sphingomyelins and ceramides were measured as previously described37. Standards for sphingosine-1-phosphate (S1P), N-palmitoyl-D-erythro-sphingosylphosphorylcholine (C16 SM), N-stearoyl-D-erythro sphingosylphosphorylcholine (C18 SM), N-lignoceroyl-D-erythro sphingosylphosphorylcholine (C24 SM) N-palmitoyl-D-erythro-sphingosine (C16 CER), N-stearoyl-D-erythro-sphingosine (C18 CER), N-arachidoyl-D-erythro-sphingosine (C20 CER), N-behenoyl-D-erythro-sphingosine (C22 CER), and N-lignoceroyl-D-erythro-sphingosine (C24 CER) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Labeled C16 CER d18:1/16:0 (Palmitoyl-U-13C16) internal standard was obtained from Cambridge Isotope Laboratories (Andover, MA, USA). Samples were prepared for LC–MS/MS analysis as previously described38.
2.5. Caspase activity
Caspase-3 activity was measured from cell lysates using a fluorometric assay kit (Catalog # K105-200 BioVision Milpitas, CA, USA). The analysis is based on detecting the cleavage of DEVD (Asp-Glu-Val-Asp)-7-amino-4-trifluoromethyl coumarin (DEVD-AFC) substrate. Free AFC emits at 500 nm, which can be measured by a fluorometer. The change in caspase-3 activity was determined by comparing AFC fluorescence from the samples with the control group. HEK-293 cells were incubated with either 1 μl/ml DMSO or 5 μg/ml TM for 24 h. About, .156 mM TUDCA was applied for 12 h. In the TM + TUDCA group, TUDCA (.156 mM) was given 12 h after 5 μg/ml TM application, with a total TM incubation time of 24 h. Control cells were incubated with only medium. At the end of the incubation period, cells were washed with PBS, 4–5 × 106 cells were resuspended in lysis buffer and incubated on ice for 10 min. Caspase-3 was determined by using 200 μg cell lysate protein in each group.
2.6. Measurement of cytosolic phospholipase A2
Activity of cPLA2 was measured via a PLA2 assay kit (Abcam, Cat No: ab133090, Cambridge, MA, USA). HEK-293 cells (4–5 × 106) were sonicated in ice-cold 50 mmol/L sodium phosphate buffer (pH 7.4) containing 1 mM EDTA. Homogenates were centrifuged (10,000 g for 15 min at 4 °C), and supernatants were stored at −80 °C. Arachidonoyl thio-PC synthetic substrate was used to detect PLA2 activity. Hydrolysis of the arachidonoyl thioester bond releases a free thiol, which was detected by 5,5′-dithiobis-2-nitrobenzoic acid (DTNB). The reaction rate at 414 nm was determined using the DTNB extinction coefficient of 10.66 mM−1 (adjusted for the path length of the solution in the well). One unit of enzyme activity was defined as the amount of enzyme that hydrolyzes one μmol of arachidonoyl thio-PC per minute at 25 °C. cPLA2 activity in the samples was reported as nmol/min/mg protein.
2.7. Measurement of cyclooxygenase activity
Cyclooxygenase activity was measured using a fluorometric assay kit (Cayman Chemical Cat No: 700200, Cat Ann Arbor, MI, USA) according to manufacturer’s instructions. The analysis uses the peroxidase component of COX. The reaction between hydroperoxy endoperoxide (PGG2) and ADPH (10-acetyl-3,7 dihydroxyphenoxazine) resulting from peroxidase COX activity produces highly fluorescent resorufine. Resorufine fluorescence is analyzed at 530 nm excitation and 585 nm emission wavelengths. HEK-293 cells (4–5 × 106) were sonicated in ice-cold PBS containing protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Cellular lysates were centrifuged at 10,000 g for 15 minutes at 4 °C and supernatants were kept at −80 °C until assayed. One unit of enzyme activity was defined as the amount of enzyme that caused the formation of 1 nmol of fluorophore per minute at 22 °C. Total COX activity in the samples was reported as nmol/min/mg protein.
2.7. Measurement of prostaglandin E2
Quantitative determination of PGE2 in HEK 293 culture media was done by an enzyme immunoassay test kit (Cayman Chemical, Cat No: 514010 Ann Arbor, MI, USA) according to the manufacturer’s instructions. A standard curve of absorbance values of known PGE2 standards was plotted as a function of the logarithm of PGE2 standard concentrations (pg/ml) using the GraphPad Prism Software program for windows version 8.4.3. (GraphPad Software Inc). PGE2 concentrations in the samples were calculated from their corresponding absorbance values via the standard curve.
2.8. Protein measurements
Protein concentrations were measured at 595 nm by a modified Bradford assay using Coomassie Plus reagent with bovine serum albumin as a standard (Pierce Chemical Company, Rockford, IL).
2.9. Statistical analysis
Statistical analysis was performed using SigmaStat statistical software version 3.5 (Sigma, St. Louis, MO, USA) or GraphPad Prism Software program for windows version 5.03. Statistical analysis for each measurement is described in figure and table legends. To compare the groups via the SigmaStat statistical software, we first performed a normality test. A test that passed indicated that the data matched the pattern expected if the data wer drawn from a population with a normal distribution. If the sample data were not normally distributed, the normality test failed. In such case, the software performed a nonparametric test. The experimental groups were compared by either one-way ANOVA (analysis of variance), two-way ANOVA, or by Kruskal–Wallis one-way ANOVA on ranks. When there was a statistically significant difference, we used multiple comparison procedures also known as post hoc tests to determine exactly, which groups were different. All pairwise multiple comparison procedures were done by either Tukey or Dunnett’s T3 multiple comparison tests.
3. Results
3.1. Cell viability analysis and evaluation of the protective efficacy of TUDCA
Dose-dependent 36-h time-course response analysis of TM-induced HEK 293 cell death is shown in Fig. 1A. Twelve-hour 10, 5, and 1 μg/ml TM administration showed no cytotoxicity, compared to control and DMSO groups. Incubation of cells with 10, 5, and 1 μg/ml TM for 24 and 36 h significantly reduced cell viability compared to control and DMSO groups in the same time frames. Likewise, 10, 5, and 1 μg/ml TM incubations for 24 and 36 h significantly reduced cell viability compared to 12-h incubations within the same dose. Cell viability analysis of TUDCA doses (.156–5 mM) is shown in Fig. 1B. It was observed that applied TUDCA doses had no significant effect on cell viability at 12- and 36-h time frames. Cells treated with higher concentrations of TUDCA (1.25–5 mM) for 24 h showed a pronounced decrease in cell viability compared to controls.
Fig. 1.
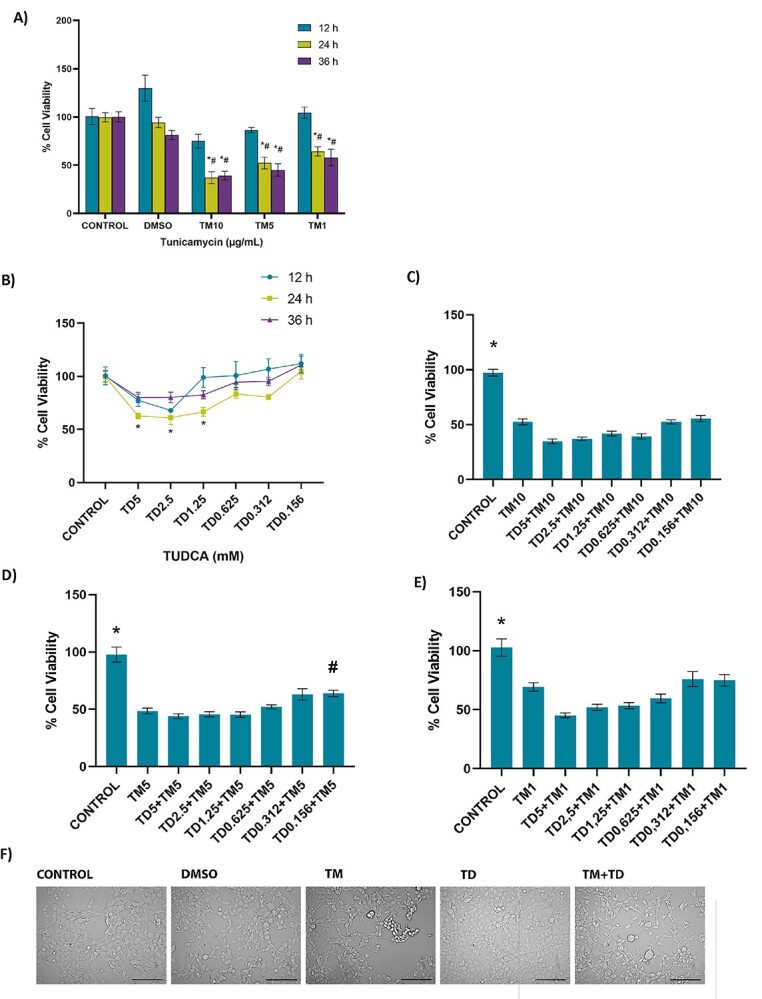
Cell viability assessed by MTT assay in HEK-293 cells. A) Dose-dependent 36-h time-course response analysis of tunicamycin-induced cell death. DMSO, cells treated with dimethyl sulfoxide (1 μl/ml); TM10, TM5, and TM1, cells treated with 10, 5, and 1 μg/ml tunicamycin. Data shown are representative of 7–8 separate experiments, and values are shown as mean ± SEM. Statistical analysis was performed by two-way ANOVA with all pairwise multiple comparison procedures done by Tukey multiple comparison test. *P < 0.05 vs. control and DMSO within the same incubation time. #P < .05, vs. 12 h within the same dose. No toxicity was observed at 12-h incubation in any TM doses. B) Dose-dependent effect of TUDCA (TD) on cell viability at 12–36-h time course. Data shown are representative of 7–8 separate experiments, and values are shown as mean ± SEM. Statistical analysis was performed by two-way ANOVA with all pairwise multiple comparison procedures done by Tukey multiple comparison test. *P < 0.05, vs. control within the 24-h incubation time. No toxicity was observed in cell viability at 12- and 36-h time-course compared to control. C) No protective effect was observed at 12-h TUDCA (TD) administration in 24 h 10 μg/ml TM toxicity. Control cells were incubated with only medium. HEK-293 cells were incubated with 10 μg/ml TM for 24 h. In the TM + TUDCA groups, TUDCA (.156–5 mM) was given 12 h after 10 μg/ml TM application, with a total TM incubation time of 24 h. Data shown are representative of 11–12 separate experiments, and values are shown as mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Dunnett’s T3 multiple comparison test. *P < 0.05 vs. all other groups. D) Protective effect of TUDCA (TD) was observed at 12 h .156 mM TD administration in 24 h 5 μg/ml TM toxicity. Control cells were incubated with only medium. HEK-293 cells were incubated with 5 μg/ml TM for 24 h. In the TM + TUDCA groups, TUDCA (.156–5 mM) was given 12 h after 5 μg/ml TM application, with a total TM incubation time of 24 h. Data shown are representative of 11–12 separate experiments, and values are shown as mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Dunnett’s T3 multiple comparison test. *P < 0.05, vs. all other groups. #P < 0.05, vs. TM5. E) No protective effect of TUDCA (TD) was observed at 12-h TD administration in 24 h 1 μg/ml TM toxicity. Control cells were incubated with only medium. HEK-293 cells were incubated with 1 μg/ml TM for 24 h. In the TM + TUDCA groups, TUDCA (.156–5 mM) was given 12 h after 1 μg/ml TM application, with a total TM incubation time of 24 h. Data shown are representative of 11–12 separate experiments, and values are shown as mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Dunnett’s T3 multiple comparison test. *P < 0.05, vs. all other groups. F) Morphological changes of TM and TUDCA administration on HEK-293 cells observed under an inverted light microscope (20X magnification) bar, 100 μm. Cells treated with 1 μl/ml DMSO for 24 h or .156 mM TUDCA for 12 h showed no significant morphological changes. Cells treated with 24 h, 5 μg/ml TM showed changes such as shrinkage, aggregation, detachment, and rounding. A decrease in cell population was also noted in the TM group. Administration of .156 mM TUDCA after 12 h 5 μg/ml TM showed a decrease in TM-related morphological deterioration.
Supplementary table 1 shows cell viability when TUDCA (.156–5 mM) and TM (1–10 μg/ml) were applied together for 24 h. Treatment of cells with TM and TUDCA together did not increase cell viability compared to TM treatment alone. Supplementary table 2 shows cell viability when TUDCA (.156–5 mM) was applied 12 h before 24 h TM (1–10 μg/ml) treatment. Pretreatment of cells with TUDCA for 12 h did not increase cell viability compared to TM treatment alone. Figure 1C–E show cell viability when TUDCA (.156–5 mM) was given 12 h after TM (1–10 μg/ml) application, with a total TM incubation time of 24 h. Results confirm that low dose of TUDCA (.156 mM) is protective when employed 12 h following the initiation of ER stress via 5 μg/ml TM treatment, with a total TM incubation time of 24 h. Figure 1F shows morphological changes of TM and TUDCA administration on HEK-293 cells. Cells treated with 1 μl/ml DMSO for 24 h or .156 mM TUDCA for 12 h showed no significant morphological changes while cells treated with 24 h, 5 μg/ml TM showed changes such as shrinkage, aggregation, detachment, and rounding. A decrease in cell population was also apparent in the TM group. Administration of .156 mM TUDCA after 12 h 5 μg/ml TM resulted in a diminution of TM-related morphological deterioration.
3.2. Induction of ER stress
Glucose-regulated protein-78 kd levels increase in cells undergoing ER stress, therefore, GRP78 immunofluorescence staining was performed on HEK-293 cells to demonstrate the presence of ER stress. Figure 2A and B shows GRP78 immunostaining and quantitation, respectively. It was observed that GRP78 levels significantly increased in TM-treated cells, compared to other groups. TUDCA application 12 h after the initiation of ER stress significantly decreased GRP78 protein levels in TM + TUDCA-treated cells.
Fig. 2.

Levels of GRP78 in HEK-293 cells. A) Representative immunofluorescent staining of GRP78 in HEK-293 cells incubated with either 1 μl/ml DMSO or 5 μg/ml TM for 24 h. .156 mM TUDCA (TD) was applied for 12 h. In the TM + TD group, TUDCA (.156 mM) was given 12 h after 5 μg/ml TM application, with a total TM incubation time of 24 h. Bar, 100 μm. B) Quantitation of GRP78 fluorescence staining was done by ImageJ software. Data shown are representative of 15 separate experiments, and values are shown as mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Dunnett’s T3 multiple comparison test. *P < 0.001, vs. all other groups. #P < 0.001, vs. control, DMSO, and TD groups.
3.3. Sphingolipid levels
Endogenous sphingolipid levels measured in HEK-293 cells are given in Table 1. There was no significant difference in sphingomyelin levels between the groups. Likewise, there was no statistically significant difference between the groups for C16, C20, and C22 ceramide levels. C18 and C24 ceramide levels were significantly increased in cells treated with TM compared to other groups. Sphingosine-1-phosphate levels decreased significantly in the TM-treated group compared to the control. Treatment with TUDCA 12 h after the initiation of ER stress decreased C18 and C24 ceramide and increased S1P levels in TM + TUDCA-treated cells.
Table 1.
Sphingolipid levels in kidney cells.
| Sphingolipids (ng/mg protein) | CONTROL (n = 6) | DMSO (n = 8) | TM (n = 6) | TD (n = 6) | TM + TD (n = 6) |
|---|---|---|---|---|---|
| 16:0 SM (d18:1/16:0) | 1038.82 ± 59.49 | 948.98 ± 96.67 | 962.59 ± 192.72 | 1069.71 ± 165.35 | 1033.01 ± 71.09 |
| 18:0 SM (d18:1/18:0) | 306.69 ± 27.31 | 312.16 ± 30.36 | 301.41 ± 22.85 | 311.65 ± 33.75 | 296.59 ± 26.16 |
| 24:0 SM (d18:1/24:0) | 339.12 ± 55.49 | 360.78 ± 19.52 | 364.16 ± 16.93 | 381.11 ± 26.91 | 353.19 ± 15.50 |
| C16 Ceramide (d18:1/16:0) | 182.90 ± 20.90 | 176.60 ± 20.54 | 185.41 ± 19.76 | 183.44 ± 24.79 | 186.66 ± 8.43 |
| C18 Ceramide (d18:1/18:0) | 119.42 ± 8.29 | 113.06 ± 14.52 | 161.33 ± 9.65* | 117.69 ± 14.49 | 104.66 ± 7.33 |
| C20 Ceramide (d18:1/20:0) | 41.28 ± 1.42 | 38.26 ± 4.98 | 39.63 ± 1.92 | 40.19 ± 1.89 | 41.87 ± 6.90 |
| C22 Ceramide (d18:1/22:0) | 105.55 ± 7.45 | 111.11 ± 2.40 | 115.06 ± 9.81 | 108.28 ± 10.92 | 109.48 ± 4.88 |
| C24 Ceramide (d18:1/24:0) | 367.18 ± 71.27 | 378.16 ± 15.95 | 600.08 ± 15.30* | 359.77 ± 20.50 | 377.26 ± 6.56 |
| S1P | 4.84 ± 1.72 | 4.21 ± 0.60 | 2.77 ± 0.68** | 4.02 ± 0.76 | 3.97 ± 1.30 |
All values are mean ± SD. SM, sphingomyelin; S1P, sphingosine-1-phosphate; DMSO, cells treated with dimethyl sulfoxide (1 μl/ml); TM, cells treated with 5 μg/ml tunicamycin within the 24 h incubation time; TD, cells treated with 0,156 mM TUDCA within the 12 h incubation time; TM + TD, 12 h .156 mM TD administration in 24 h, 5 μg/ml TM toxicity. Cells were treated with .156 mM TD after 12 h 5 μg/ml TM administration. Total incubation time was 24 h. Statistical analysis was by one-way analysis of variance or Kruskal–Wallis one-way analysis of variance on ranks, and all pairwise multiple comparison procedures were done by Tukey test. *, P < 0.001 vs. control, DMSO, TD, and TM + TD. **, P < 0.05 vs. control.
3.4. Caspase-3 activity
Figure 3A and B shows cleaved caspase-3 immunostaining and quantitation, respectively. Incubation of HEK-293 cells with TM for 24 h significantly increased cleaved caspase-3 levels compared to other groups. Significantly increased caspase-3 activity in TM treated cells, compared with other groups, confirmed immunofluorescence staining (Fig. 3C). Treatment of cells with low-dose TUDCA after the initiation of ER stress significantly decreased caspase-3 activity and cleaved caspase-3 level.
Fig. 3.

Caspase-3 in kidney cells. A) Representative immunofluorescent staining of cleaved caspase-3 in HEK-293 cells incubated with either 1 μl/ml DMSO or 5 μg/ml TM for 24 h. .156 mM TUDCA (TD) was applied for 12 h. In the TM + TD group, TUDCA (.156 mM) was given 12 h after 5 μg/ml TM application, with a total TM incubation time of 24 h. Bar, 200 μm. B) Quantitation of cleaved caspase-3 fluorescence staining was estimated by ImageJ software. Data shown are representative of 15 separate experiments and values are shown as mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Tukey’s multiple comparison test. *P < 0.001, vs. all other groups. C) Caspase-3 activity. Data shown are representative of 6 separate experiments and values are shown as mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Tukey’s multiple comparison test. *P < 0.001, vs. all other groups.
3.5. Cytosolic phospholipase A2, cyclooxygenase, and prostaglandin E2 levels
Treatment of HEK-293 cells with .5 μg/ml TM for 24 h significantly decreased cPLA2 activity (mean ± SEM) compared to control, DMSO, and TUDCA groups. Treatment of cells with low-dose TUDCA after the initiation of ER stress had no significant effect on cPLA2 activity compared with the TM group. Measured enzyme activity in control, DMSO, TM, TUDCA, and TM + TUDCA treated cells were 15.91 ± .60; 15.13 ± .74; 12.79 ± .46; 15.17 ± .67; and 12.75 ± .75 nmol/min/mg protein, respectively (Fig. 4A).
Fig. 4.

Cytosolic phospholipase A2 (cPLA2), cyclooxygenase (COX) and prostaglandin E2 (PGE2) in HEK-293 cells. Cells were incubated with either 1 μl/ml DMSO or 5 μg/ml TM for 24 h. .156 mM TUDCA (TD) was applied for 12 h. In the TM + TD group, TUDCA (.156 mM) was given 12 h after 5 μg/ml TM application, with a total TM incubation time of 24 h. A) cPLA2 activity. Data shown are representative of 7–8 separate experiments and values are mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Tukey’s multiple comparison test. *P < 0.001 vs. control, DMSO, and TD groups. B) Representative immunofluorescent staining of COX-1 in HEK-293 cells. Bar, 100 μm. C) Quantitation of COX-1 fluorescence staining was estimated by ImageJ software. Data shown are representative of 15 separate experiments and values are shown as mean ± SEM. Statistical analysis was performed by Kruskal–Wallis analysis with all pairwise multiple comparison procedures done by Dunn’s multiple comparison test. No significant difference was observed among the groups. D) Total COX activity. Data shown are representative of 4 separate experiments and values are shown as mean ± SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Tukey’s multiple comparison test. No significant difference was observed among the groups. E) PGE2 levels in cell culture media. Data shown are representative of 5 separate experiments and values are shown as mean ± -SEM. Statistical analysis was performed by one-way ANOVA with all pairwise multiple comparison procedures done by Dunnett’s T3 multiple comparison test. No significant difference was observed among the groups.
Cyclooxygenase-1 immunofluorescence staining was performed in HEK-293 cells and total COX activity was measured (Fig. 4B-D). Immunofluorescence imaging and quantitation of COX-1 showed no noticeable difference among the different treatment groups (Fig. 4B and C, respectively). Likewise, no significant difference was observed among treatment groups with regard to COX activity. Measured enzyme activity in control, DMSO, TM, TUDCA, and TM + TUDCA treated cells were; .35 ± .02; .32 ± .01; .32 ± .02; and .37 ± .01 nmol/min/mg protein, respectively (Fig. 4D).
PGE2 levels (mean ± SEM) also showed no significant difference among the different treatment groups (Fig. 1E). Measured PGE2 levels in control, DMSO, TM, TUDCA, and TM + TUDCA-treated cells were: 77,04 ± 2,79; 75,41 ± 4,62; 70,46 ± 2,39; 75,37 ± 6,04; and 71,48 ± 3,46 pg/ml, respectively.
3.6. Polyunsaturated fatty acid levels
Endogenous PUFA levels measured in HEK-293 cells are given in Table 2. All measured PUFAs were significantly increased in TM and TM + TUDCA groups compared to control, DMSO, and TUDCA groups.
Table 2.
Analysis of polyunsaturated fatty acids in kidney cells.
| Group (n = 8) | DGLA (C20:3n6) | AA (C20:4n6) | EPA (C20:5n3) | DHA (C22:6n3) |
|---|---|---|---|---|
| CONTROL | 9.04 ± 2.81 | 5.54 ± 1.53 | 0.93 ± 0.04 | 4.56 ± 0.76 |
| DMSO | 8.55 ± 3.59 | 5.01 ± 1.70 | 0.85 ± 0.07 | 3.96 ± 0.64 |
| TM | 13.73 ± 2.46* | 15.02 ± 2.32* | 2.76 ± 0.37* | 9.48 ± 1.34* |
| TD | 9.34 ± 3.20 | 4.82 ± 1.47 | 0.79 ± 0.24 | 3.79 ± 0.45 |
| TM + TD | 15.24 ± 2.77* | 13.96 ± 2.27* | 2.14 ± 0.34* | 9.04 ± 1.26* |
Values are expressed as μg fatty acid /5 × 106 cell. Data are reported as mean ± SD. DGLA, dihomogamma-linolenic acid; AA, arachidonic acid; EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid. DMSO, cells treated with dimethyl sulfoxide (1 μl/ml); TM, cells treated with 5 μg/ml tunicamycin within the 24 h incubation time; TD, cells treated with 0,156 mM TUDCA within the 12 h incubation time; TM + TD, 12 h .156 mM TD administration in 24 h, 5 μg/ml TM toxicity. Cells were treated with .156 mM TD after 12 h 5 μg/ml TM administration. Total incubation time was 24 h. Statistical analysis was by one-way analysis of Variance or Kruskal–Wallis one-way analysis of variance on ranks and all pairwise multiple comparison procedures were done by Tukey test. *, P < 0.05 vs. control, DMSO, and TD.
4. Discussion
This study reports the noteworthy observation that TM treatment in human kidney epithelial cells causes increased ceramide production that can be regulated by an ER stress inhibitor, TUDCA. Data are also presented showing down regulation of caspase-3 activity in response to ER stress inhibition through TUDCA. Tunicamycin-induced cPLA2 inhibition and consequential PUFA accumulation can also give rise to cellular toxicity in renal cells, as described herein.
Treatment of HEK-293 kidney cells with 5 μg/ml TM significantly decreased cell viability as reported previously39. Data demonstrated that TM specifically damaged HEK-293 cell cultures. These results are consistent with previous reports showing renal toxicity of TM in both in vivo and in vitro experimental models40,41. Tunicamycin acts by triggering ER stress through disturbing protein glycosylation8. Cellular toxicity in our experimental model is likely to be a consequence of ER-stress-related cell death signaling, since treatment with an ER stress inhibitor, TUDCA, resulted in a marked increase in cell viability (Fig. 1). TUDCA acts as a chaperone in the treatment of ER-stress-related diseases by restraining the UPR42. TUDCA holds back the induction of the initial signaling steps in the three major pathways of UPR. It suppresses the synthesis and binding of activated transcription factors to the GRP78 promoter43. In addition, TUDCA has also been reported to decrease oxidative stress, inhibit apoptosis and reduce inflammation in several in-vitro and in-vivo models of various diseases44. Apart from TUDCA, different modulators also target UPR protein pathways. Potent PERK inhibitors such as GSK2656157 target PERK kinase and can be orally administered45. Apigenin targets upregulation of ATF6 expression46 while kaempferol leads to down regulation of ATF647. Inositol-requiring kinase 1 (IRE1) is dimerized and autophosphorylated during ER stress. Activated IRE1 gains RNAase activity. Salicylaldimines are examples of IRE1αRNase active-site inhibitors48.
Treatment of HEK-293 cells with the ER stressor TM, caused a significant increase in GRP78 protein levels (Fig. 2). Increased protein levels of GRP78 in the kidney, has become a well-known marker for the presence of ER stress49. GRP78 acts as a chaperone within the ER lumen and some can be attached to PKR-like ER kinase (PERK). Increased formation of unfolded proteins, results in detachment of GRP78 from PERK and binding of GRP78 to unfolded proteins, thereby challenging to preserve homeostasis in the ER. Detachment of GRP78 from PERK, allows PERK phosphorylation50. We found that inhibition of ER stress in TM + TUDCA treated cells significantly decreased protein levels of GRP78, indicating that ER stress was alleviated by TUDCA treatment in HEK-293 cells.
A considerable increase was noted in cellular levels of C18 and C24 CERs in HEK-293 cells treated with TM compared to other groups (table 1). We found no significant difference between the groups for C16, C20 and C22 ceramide levels. S1P levels decreased significantly in TM treated cells compared to control. De novo biosynthesis of ceramides occurs by ceramide synthases (CerS)51 while the salvage pathway also supplies ceramides mainly via the activation of sphingomyelinases52. Ceramidases catalyze hydrolysis of ceramides to generate sphingosine, which is phosphorylated to form S1P53. Ceramidases poses glycosylation sites which are required for normal protein function53. Tunicamycin treatment is known to down-regulate CerS-6-generated C16-ceramide in HNSCC cell lines19 and decrease neutral sphingomyelinase activity in retinal pigment epithelial cells38. A similar effect of tunicamycin in HEK-293 may, to some extent, explain unaltered C16, C20, and C22 ceramide levels observed herein. The accumulation of C18 and C24 ceramides in the presence of tunicamycin could be a result of decreased ceramidase activity due to blockade of glycosylation which may also explain the decreased S1P levels observed in tunicamycin treated cells. Sphingosine-1-phosphate is well documented to be a pro-survival molecule53 and thus its reduction can further promote the apoptotic signaling of ceramide. Our results confirm recent studies that have shown the buildup of ceramide in a rodent model of renal ER stress and in human retinal pigment epithelial cells subjected to TM-induced ER stress12,38. To our knowledge, this is the first study to show increased levels of long chain fatty acid containing ceramides in HEK-293 cells treated with TM.
Previous work have revealed that ceramide inhibits sarcoplasmic/endoplasmic reticulum Ca2+- ATPase (SERCA) resulting in [Ca2+] depletion in the ER54. This outcome of ceramide is comparable to thapsigargin, a typical inducer of ER stress. Thapsigargin selectively inhibits SERCA, causing Ca2+ depletion from the ER lumen and activation of ER stress55. The successive increase of Ca2+ concentration in the cytoplasm and mitochondrial matrix is an important factor in ceramide-induced apoptosis56. TUDCA therapy produced a significant drop in long chain CER levels in cells treated with TM compared to TM treatment alone. The observed decrease in C18 and C24 CERs after TUDCA treatment may offer a new mechanism by which TUDCA lessens ER stress.
To our knowledge, this is the first study to report decreased levels of S1P in HEK-293 cells treated with TM. Sphingosine-1-phosphate, similar to ceramide, can be expected to function as both structural and/or cell signaling molecule in the kidney57. Kidney epithelial cells can generate S1P which operates as an opponent of ceramide to maintain cell proliferation and protect cells from various injuries58. Sphingosine-1-phosphate can be dephosphorylated to sphingosine by S1P phosphatase or irreversibly degraded by S1P lyase leading to the formation of ethanolamine-1-phosphate and C16 fatty aldehyde. Intriguingly, in the kidney, S1P lyase has been reported to take part in the occurrence of proteinuria in mice59, while genetic mutations in the gene coding for S1P lyase are connected with nephrotic syndrome in humans60. Our results support the protective role of S1P in kidney epithelial cells and further demonstrate that inhibition of ER stress via TUDCA treatment can increase levels of S1P. Most recent substantial progress in the understanding of biologically active sphingolipid synthesis, particularly within ceramide and S1P-mediated pathways, has resulted in the acknowledgement of the fundamental function of these molecules in cell survival. Ceramide, a principal molecule in sphingolipid metabolism, activates antiproliferative and apoptotic responses, while S1P triggers reactions that make this lipid a cell proliferating molecule61. The finding of increased C18-C24 CERs and decreased S1P suggests that the balance of ceramide/S1P determines ceramide-induced caspase-3 activation in our cell model.
Caspase-3 activity was increased in TM treated HEK-293 cells (Fig. 3). Caspases are major enzymes involved in apoptosis, which operates through two main pathways. The extrinsic pathway, requires binding of Fas ligand and tumor necrosis factor-α (TNF-α) to membrane receptors initiating caspase-8 or caspase-10 activation. The intrinsic pathway is activated by stress-induced mitochondrial cytochrome c release62. Cytochrome c binds to apoptotic protease activating factor-1 (Apaf-1) and procaspase-9, which activates caspase-9. Both pathways congregate via caspase-3 activation, causing morphological transformations such as blebbing and nuclear degradation62. Transcription factors produced under ER stress, such as C/EBP-homologous protein (CHOP), have been implicated in ER stress-induced apoptosis via decreasing the expression of B-cell lymphoma/leukemia-2 (Bcl-2) and initiating cytochrome c release63. Furthermore, CHOP-insufficiency generates resistance to ER stress-induced cell death64.
Secretory (sPLA2), cPLA2, and calcium independent PLA2 are major phospholipase A2 enzymes which hydrolyze the phospholipid bond at the sn-2 position and liberate arachidonic acid. Incubation of HEK-293 cells with TM significantly decreased cPLA2 activity compared to control, DMSO, and TUDCA groups (Fig. 4). We observed that treatment with TUDCA had no major effect on cPLA2 activity in cells exposed to TM. Previous work have reported that ceramide activates cPLA2 by binding to the CaLB domain of the enzyme, making possible its membrane docking and arachidonic acid release25. Tunicamycin-induced ER stress in HEK-293 cells increased ceramide formation but did not lead to ceramide-induced cPLA2 activation. The observed decrease of cPLA2 activity in our cell model is likely caused by decreased N-glycosylation of cPLA2. Tunicamycin decreases N-glycosylation in HEK-293 cells, and this regulation is not dependent on changes in protein expression levels65. N-glycosylation of PLA2 is required for catalytic activity of the enzyme66 which is present in HEK-293 cells67,68.
Ceramide is also known to increase COX-2 transcription by activating extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK)26. In agreement with decreased cPLA2 activity, we have found no significant change in total COX activity, COX-1 protein and PGE2 levels following TM treatment (Fig. 4). Decreased availability of free arachidonic acid is probable to cause no major change in COX activity and PGE2 synthesis.
To the best of our knowledge, this study is the first to measure endogenous PUFA levels in HEK-293 cells following TM treatment. Tunicamycin incubation of HEK-293 cells resulted in significantly increased omega-3 and omega-6 -PUFA levels (Table 2). Increase in HEK-293 kidney cell PUFA levels suggests that TM leads to PUFA accumulation as a consequence of cPLA2 inhibition, causing a buildup of PUFA pools. We observed that treatment with TUDCA had no major effect on cPLA2 activity in cells exposed to TM. Our results support a recent study which has shown that cPLA2 inhibition by diclofenac results in significantly increased PUFA levels in kidney epithelial cells36. Increased PUFA levels in HEK-293 cells following TM treatment is likely to enhance the development of cellular injury by destructing mitochondrial membrane potential (MMP)69. Mitochondrial membrane potential, evaluated as a measure of mitochondrial membrane injury, is distorted in cells treated with high PUFA concentrations69. PUFAs also increase reactive oxygen species generation70, which may also be connected with mitochondrial malfunction.
There are several limitations of our study. Studies based on cell culture and the data they provide are limited to cell-specific pharmacodynamics, biochemical pathways, and genetic variations. Cell culture focuses on isolated cells other than tissues or organisms. For this reason, chemical, hormonal, and physiological outcomes that may occur specific to the in vivo environment cannot be examined. Pharmacokinetic effects that can occur in cell cultures may differ from tissues and organisms. Thus, animal models are also needed to evaluate the in vivo effects of ER stress-induced lipotoxicity in the kidney.
In conclusion, treatment with TM increased ceramide and GRP78 protein levels while decreasing S1P concentrations, leading to caspase-3 activation in human HEK-293 cells. Inhibition of ER stress by TUDCA decreased ceramide/S1P levels, reduced caspase-3 activity, caused a significant reduction of GRP 78 expression in HEK-293 cells. Our data suggests that inhibition of ER stress and ceramide formation can potentially be a pharmaceutical target in the treatment of ER-stress-related kidney disease.
Authors’ contributions
T.Ç. and Ç.Y. participated in the investigation, formal analysis, and validation of the study. E.K., participated in the conceptualization and methodology of the study. M.A. participated in the conceptualization, methodology, writing, reviewing, and editing of the study.
Funding
This work was supported by a grant from Akdeniz University Research Foundation (TYL-2019-5107).
Conflict of interest statement
The authors declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and material
The data that support the findings of this study are available from the corresponding author, (MA), upon reasonable request.
Supplementary Material
Contributor Information
Tuğçe Çeker, Department of Medical Biochemistry, Akdeniz University, Faculty of Medicine, Antalya 07070, Turkey.
Çağatay Yılmaz, Department of Medical Biochemistry, Akdeniz University, Faculty of Medicine, Antalya 07070, Turkey.
Esma Kırımlıoglu, Department of Histology and Embryology, Akdeniz University, Faculty of Medicine, Antalya 07070, Turkey.
Mutay Aslan, Department of Medical Biochemistry, Akdeniz University, Faculty of Medicine, Antalya 07070, Turkey; Department of Gene and Cell Therapy, Akdeniz University, Faculty of Medicine, Antalya 07070, Turkey.
References
- 1. Almanza A, Carlesso A, Chintha C, et al. . Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019:286(2):241–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Inagi R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp Nephrol. 2009:112(1):e1–e9. [DOI] [PubMed] [Google Scholar]
- 3. Taniguchi M, Yoshida H. Endoplasmic reticulum stress in kidney function and disease. Curr Opin Nephrol Hypertens. 2015:24(4):345–350. [DOI] [PubMed] [Google Scholar]
- 4. Kuznetsov G, Bush KT, Zhang PL, Nigam SK. Perturbations in maturation of secretory proteins and their association with endoplasmic reticulum chaperones in a cell culture model for epithelial ischemia. Proc Natl Acad Sci U S A. 1996:93(16):8584–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kimura K, Jin H, Ogawa M, Aoe T. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem Biophys Res Commun. 2008:366(4):1048–1053. [DOI] [PubMed] [Google Scholar]
- 6. Cybulsky AV, Takano T, Papillon J, Khadir A, Liu J, Peng H. Complement C5b-9 membrane attack complex increases expression of endoplasmic reticulum stress proteins in glomerular epithelial cells. J Biol Chem. 2002:277(44):41342–41351. [DOI] [PubMed] [Google Scholar]
- 7. Inagi R, Kumagai T, Nishi H, et al. . Preconditioning with endoplasmic reticulum stress ameliorates mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 2008:19(5):915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Price NP, Tsvetanova B. Biosynthesis of the tunicamycins: a review. J Antibiot (Tokyo). 2007:60(8):485–491. [DOI] [PubMed] [Google Scholar]
- 9. Zinszner H, Kuroda M, Wang X, et al. . CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998:12(7):982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lorz C, Justo P, Sanz A, Subirá D, Egido J, Ortiz A. Paracetamol-induced renal tubular injury: a role for ER stress. J Am Soc Nephrol. 2004:15(2):380–389. [DOI] [PubMed] [Google Scholar]
- 11. Peyrou M, Hanna PE, Cribb AE. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol Sci. 2007:99(1):346–353. [DOI] [PubMed] [Google Scholar]
- 12. Aslan M, Elpek Ö, Akkaya B, Balaban HT, Afşar E. Organ function, sphingolipid levels and inflammation in tunicamycin induced endoplasmic reticulum stress in male rats. Hum Exp Toxicol. 2021:40(2):259–273. [DOI] [PubMed] [Google Scholar]
- 13. Ueda N. Sphingolipids in genetic and acquired forms of chronic kidney diseases. Curr Med Chem. 2017:24(12):1238–1275. [DOI] [PubMed] [Google Scholar]
- 14. Sieber J, Lindenmeyer MT, Kampe K, et al. . Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol. 2010:299(4):F821–F829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bennett MK, Wallington-Beddoe CT, Pitson SM. Sphingolipids and the unfolded protein response. Biochim Biophys Acta Mol Cell Biol Lipids. 2019:1864(10):1483–1494. [DOI] [PubMed] [Google Scholar]
- 16. Lei X, Zhang S, Bohrer A, Bao S, Song H, Ramanadham S. The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry. 2007:46(35):10170–10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Epstein S, Kirkpatrick CL, Castillon GA, et al. . Activation of the unfolded protein response pathway causes ceramide accumulation in yeast and INS-1E insulinoma cells. J Lipid Res. 2012:53(3):412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yacoub A, Hamed HA, Allegood J, et al. . PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res. 2010:70(3):1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Senkal CE, Ponnusamy S, Manevich Y, et al. . Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J Biol Chem. 2011:286(49):42446–42458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park K, Elias PM, Shin KO, et al. . A novel role of a lipid species, sphingosine-1-phosphate, in epithelial innate immunity. Mol Cell Biol. 2013:33(4):752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tam AB, Roberts LS, Chandra V, et al. . The UPR activator ATF6 responds to Proteotoxic and Lipotoxic stress by distinct mechanisms. Dev Cell. 2018:46(3):327–343.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rioux V, Pédrono F, Legrand P. Regulation of mammalian desaturases by myristic acid: N-terminal myristoylation and other modulations. Biochim Biophys Acta. 2011:1811(1):1–8. [DOI] [PubMed] [Google Scholar]
- 23. Spector AA, Kim HY. Discovery of essential fatty acids. J Lipid Res. 2015:56(1):11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wall R, Ross RP, Fitzgerald GF, Stanton C. Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev. 2010:68(5):280–289. [DOI] [PubMed] [Google Scholar]
- 25. Huwiler A, Johansen B, Skarstad A, Pfeilschifter J. Ceramide binds to the CaLB domain of cytosolic phospholipase A2 and facilitates its membrane docking and arachidonic acid release. FASEB J. 2001:15(1):7–9. [DOI] [PubMed] [Google Scholar]
- 26. Subbaramaiah K, Chung WJ, Dannenberg AJ. Ceramide regulates the transcription of cyclooxygenase-2. Evidence for involvement of extracellular signal-regulated kinase/c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways. J Biol Chem. 1998:273(49):32943–32949. [DOI] [PubMed] [Google Scholar]
- 27. Cusick JK, Mustian A, Goldberg K, Reyland ME. RELT induces cellular death in HEK 293 epithelial cells. Cell Immunol. 2010:261(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ashokkumar B, Vaziri ND, Said HM. Thiamin uptake by the human-derived renal epithelial (HEK-293) cells: cellular and molecular mechanisms. Am J Physiol Renal Physiol. 2006:291(4):F796–F805. [DOI] [PubMed] [Google Scholar]
- 29. Chen YC, Andrew Lin KY, Chen KF, Jiang XY, Lin CH. In vitro renal toxicity evaluation of copper-based metal-organic framework HKUST-1 on human embryonic kidney cells. Environ Pollut. 2021:273:116528. [DOI] [PubMed] [Google Scholar]
- 30. Fan Y, Xiao W, Li Z, et al. . RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun. 2015:6:7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuo CY, Lin CH, Hsu T. VHL inactivation in precancerous kidney cells induces an inflammatory response via ER stress-activated IRE1α Signaling. Cancer Res. 2017:77(13):3406–3416. [DOI] [PubMed] [Google Scholar]
- 32. Park SJ, Kim Y, Chen YM. Endoplasmic reticulum stress and monogenic kidney diseases in precision nephrology. Pediatr Nephrol. 2019:34(9):1493–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peyrou M, Cribb AE. Effect of endoplasmic reticulum stress preconditioning on cytotoxicity of clinically relevant nephrotoxins in renal cell lines. Toxicol in Vitro. 2007:21(5):878–886. [DOI] [PubMed] [Google Scholar]
- 34. Hennemeier I, Humpf HU, Gekle M, Schwerdt G. Role of microRNA-29b in the ochratoxin A-induced enhanced collagen formation in human kidney cells. Toxicology. 2014:324:116–122. [DOI] [PubMed] [Google Scholar]
- 35. Aslan M, Özcan F, Aslan I, Yücel G. LC-MS/MS analysis of plasma polyunsaturated fatty acids in type 2 diabetic patients after insulin analog initiation therapy. Lipids Health Dis. 2013:12:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aslan M, Kırımlıoğlu E, Afşar E, Çeker T, Yılmaz Ç. Increased PUFA levels in kidney epithelial cells in the course of diclofenac toxicity. Toxicol in Vitro. 2020:66:104836. [DOI] [PubMed] [Google Scholar]
- 37. Özer H, Aslan İ, Oruç MT, et al. . Early postoperative changes of sphingomyelins and ceramides after laparoscopic sleeve gastrectomy. Lipids Health Dis. 2018:17(1):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Afşar E, Kırımlıoglu E, Çeker T, Yılmaz Ç, Demir N, Aslan M. Effect of ER stress on sphingolipid levels and apoptotic pathways in retinal pigment epithelial cells. Redox Biol. 2020:30:101430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chiappisi E, Ringseis R, Eder K, Gessner DK. Effect of endoplasmic reticulum stress on metabolic and stress signaling and kidney-specific functions in Madin-Darby bovine kidney cells. J Dairy Sci. 2017:100(8):6689–6706. [DOI] [PubMed] [Google Scholar]
- 40. Carlisle RE, Brimble E, Werner KE, et al. . 4-Phenylbutyrate inhibits tunicamycin-induced acute kidney injury via CHOP/GADD153 repression. PLoS One. 2014:9(1):e84663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hammad AS, Ravindran S, Khalil A, Munusamy S. Structure-activity relationship of piperine and its synthetic amide analogs for therapeutic potential to prevent experimentally induced ER stress in vitro. Cell Stress Chaperones. 2017:22(3):417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berger E, Haller D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem Biophys Res Commun. 2011:409(4):610–615. [DOI] [PubMed] [Google Scholar]
- 43. Kusaczuk M. Tauroursodeoxycholate-bile acid with chaperoning activity: molecular and cellular effects and therapeutic perspectives. Cells. 2019:8(12):1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vang S, Longley K, Steer CJ, Low WC. The unexpected uses of Urso- and Tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob Adv Health Med. 2014:3(3):58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Atkins C, Liu Q, Minthorn E, et al. . Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013:73(6):1993–2002. [DOI] [PubMed] [Google Scholar]
- 46. Chen D, Landis-Piwowar KR, Chen MS, Dou QP. Inhibition of proteasome activity by the dietary flavonoid apigenin is associated with growth inhibition in cultured breast cancer cells and xenografts. Breast Cancer Res. 2007:9(6):R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim DS, Ha KC, Kwon DY, et al. . Kaempferol protects ischemia/reperfusion-induced cardiac damage through the regulation of endoplasmic reticulum stress. Immunopharmacol Immunotoxicol. 2008:30(2):257–270. [DOI] [PubMed] [Google Scholar]
- 48. Volkmann K, Lucas JL, Vuga D, et al. . Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011:286(14):12743–12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhong Y, Wang B, Hu S, et al. . The role of endoplasmic reticulum stress in renal damage caused by acute mercury chloride poisoning. J Toxicol Sci. 2020:45(9):589–598. [DOI] [PubMed] [Google Scholar]
- 50. Zhu G, Lee AS. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J Cell Physiol. 2015:230(7):1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ben-David O, Futerman AH. The role of the ceramide acyl chain length in neurodegeneration: involvement of ceramide synthases. NeuroMolecular Med. 2010:12(4):341–350. [DOI] [PubMed] [Google Scholar]
- 52. Zeidan YH, Hannun YA. The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr Mol Med. 2010:10(5):454–466. [DOI] [PubMed] [Google Scholar]
- 53. Mao C, Obeid LM. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta. 2008:1781(9):424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu Z, Xia Y, Li B, et al. . Induction of ER stress-mediated apoptosis by ceramide via disruption of ER ca(2+) homeostasis in human adenoid cystic carcinoma cells. Cell Biosci. 2014:4:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Doan NT, Paulsen ES, Sehgal P, et al. . Targeting thapsigargin towards tumors. Steroids. 2015:97:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001:20(11):2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Merscher S, Fornoni A. Podocyte pathology and nephropathy - sphingolipids in glomerular diseases. Front Endocrinol (Lausanne). 2014:5:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mitrofanova A, Drexler Y, Merscher S, Fornoni A. Role of sphingolipid Signaling in glomerular diseases: focus on DKD and FSGS. J Cell Signal. 2020:1(3):56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schümann J, Grevot A, Ledieu D, et al. . Reduced activity of Sphingosine-1-phosphate Lyase induces podocyte-related glomerular proteinuria, skin irritation, and platelet activation. Toxicol Pathol. 2015:43(5):694–703. [DOI] [PubMed] [Google Scholar]
- 60. Prasad R, Hadjidemetriou I, Maharaj A, et al. . Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest. 2017:127(3):942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ueda N. Ceramide-induced apoptosis in renal tubular cells: a role of mitochondria and sphingosine-1-phoshate. Int J Mol Sci. 2015:16(3):5076–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. D'Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019:43(6):582–592. [DOI] [PubMed] [Google Scholar]
- 63. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004:11(4):381–389. [DOI] [PubMed] [Google Scholar]
- 64. Marciniak SJ, Yun CY, Oyadomari S, et al. . CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004:18(24):3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang X, Wang Z, Guo L, Zhu Z, Zhang Y. Proteome-wide analysis of N-glycosylation stoichiometry using SWATH technology. J Proteome Res. 2017:16(10):3830–3840. [DOI] [PubMed] [Google Scholar]
- 66. Hefner Y, Borsch-Haubold AG, Murakami M, et al. . Serine 727 phosphorylation and activation of cytosolic phospholipase A2 by MNK1-related protein kinases. J Biol Chem. 2000:275(48):37542–37551. [DOI] [PubMed] [Google Scholar]
- 67. Pavicevic Z, Leslie CC, Malik KU. cPLA2 phosphorylation at serine-515 and serine-505 is required for arachidonic acid release in vascular smooth muscle cells. J Lipid Res. 2008:49(4):724–737. [DOI] [PubMed] [Google Scholar]
- 68. Hiraoka M, Okamoto K, Ohguro H, Abe A. Role of N-glycosylation of human lysosomal phospholipase A2 for the formation of catalytically active enzyme. J Lipid Res. 2013:54(11):3098–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu X, Yu H, Ma Q, Shen S, Das UN. Linoleic acid suppresses colorectal cancer cell growth by inducing oxidant stress and mitochondrial dysfunction. Lipids Health Dis. 2010:9:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Das UN. Arachidonic acid and other unsaturated fatty acids and some of their metabolites function as endogenous antimicrobial molecules: a review. J Adv Res. 2018:11:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, (MA), upon reasonable request.