

Abstract
Although prion infections cause cognitive impairment and neuronal death, transcriptional and translational profiling shows progressive derangement within glia but surprisingly little changes within neurons. Here we expressed PrPC selectively in neurons and astrocytes of mice. After prion infection, both astrocyte and neuron‐restricted PrPC expression led to copious brain accumulation of PrPSc. As expected, neuron‐restricted expression was associated with typical prion disease. However, mice with astrocyte‐restricted PrPC expression experienced a normal life span, did not develop clinical disease, and did not show astro‐ or microgliosis. Besides confirming that PrPSc is innocuous to PrPC‐deficient neurons, these results show that astrocyte‐born PrPSc does not activate the extreme neuroinflammation that accompanies the onset of prion disease and precedes any molecular changes of neurons. This points to a nonautonomous mechanism by which prion‐infected neurons instruct astrocytes and microglia to acquire a specific cellular state that, in turn, drives neural dysfunction.
Keywords: astrocytes, neurons, prions, scrapie
Prion infection of mice that express PrPC in astrocytes leads to strong PrPSc accumulation but to little or no neuroinflammatory changes.

1. INTRODUCTION
Prion diseases are characterized by a long, largely asymptomatic incubation period. Once clinical signs and symptoms arise, the disease typically progresses very rapidly. Prion‐infected brains contain PrPSc, an aggregated and misfolded isoform of the cellular prion protein (PrPC) [1]. PrPSc seeds the nucleation of prions by recruiting PrPC; accordingly, ablation of PrPC abrogates prion propagation [2] and toxicity [3, 4].
The clinical manifestation of prion diseases, both in humans and in animal models, consists of progressive neurological signs including deterioration of cortical functions. The anatomical correlates of the disease are spongiosis (a highly characteristic form of extensive neuronal vacuolation), activation and proliferation of microglia, and astrogliosis. The cortex of patients with terminal prion diseases can show an almost total depletion of neurons [5, 6], which suggests that neurons may be the primary target of the disease. But what is the connection between prion replication and neuronal demise? In a neurografting paradigm, Prnp‐ablated neurons survive long‐term exposure to prions [4], implying that resident PrPC is necessary for the development of damage. Also, quenching neuronal PrPC expression was found to delay prion disease [7], adding to the evidence that neuronal PrPC is required for neurotoxicity.
However, recent molecular studies are painting a starkly different picture. Transcriptomic analysis performed in prion‐infected mice over the course of disease has revealed dramatic aberrations of glia‐enriched genes coinciding with the onset of clinical signs, whereas neuronal changes were less pronounced and were only detected at the terminal stage of the disease [8]. Similarly, a quantitative analysis of mRNA translation during the course of prion diseases has found that almost all changes during the progression of prion disease occur in non‐neuronal cells, except very late in disease [9]. These findings suggest that it is the glia which experiences initial dysfunction, whereas the neuronal demise is a consequence thereof.
Here we have tested the above hypothesis by systematically investigating the impact of cell type‐specific PrPC in disease. We generated transgenic mice line expressing a conditionally expressed PrP transgene, and mated them with mice expressing the Cre recombinase driven by cell type‐specific promoters. The resulting mice expressed PrPC in a cell type‐specific manner. We found that neuron‐restricted PrPC sufficed to induce neurodegeneration upon prion infection. Conversely, mice with astrocyte‐restricted PrPC expression did not experience clinical signs of scrapie after prion infection. However, we found that these mice had conspicuous PrPSc accumulation.
2. MATERIAL AND METHODS
2.1. Mice
Animal welfare and experimental procedure on the mice were performed according to the “Swiss Ethical Principles and Guidelines for Experiments on Animals” and approved by the Veterinary office of the Canton of Zurich (permit 90/2013). All efforts were made to minimize the suffering and reduce the number of animals used for the experiments. Mice were bred and housed in special hygienic grade facilities and housed in small groups (max 5 per cage) under a 12 h light/12 h dark cycle with sterilized food (Kilba No.3431, Provimi Kilba Kaiseraugst, Switzerland) and water ad libitum. Prion inoculated mice were regularly monitored according to the standard operating procedures approved by the Veterinary office and mice were humanely sacrificed once the termination criteria were reached.
2.1.1. Generation of CAG‐CAT‐PrP transgenic mice
For the generation of mice carrying a loxP‐flanked stop cassette followed by the mouse Prnp coding sequence under the CAG promoter, we used the pCAG–loxP–CAT–loxP vector (kind gift of Dr. Kimi Araki, Kumamoto University, Japan), where CAG is the CMV immediate early enhancer‐chicken β‐actin hybrid promoter and CAT the chloramphenicol acetyl transferase gene [10]. Cloning by T4 DNA ligase (New England Biolaboratories, Ipswich, MA, United States) was performed after EcoRV (New England Biolaboratories, Ipswich, MA, United States) digestion of both the PCR‐amplified Prnp cDNA and the pCAG–loxP–CAT–loxP vector. The KpnI‐SacI (New England Biolaboratories, Ipswich, MA, United States) linearized CAG‐CAT‐Prnp transgene was purified and microinjected at the transgenic facility of the University Hospital Zurich into one cell‐stage fertilized embryos from Prnp ZH1/ZH1 mice. Transgenic founders were identified by PCR using the following primers: Forward: AAC GCC AAT AGG GAC TTT CC; Reverse: ATG GGG AGA GTG AAG CAG AA (actin primers used as internal control for PCR: fwd ‐TGT TAC CAA CTG GGA CGA CA; rev‐ GAC ATG CAA GGA GTG CAA GA). Following assessment of germline transmission and establishment of the colonies, the only expresser line (Line 211) was selected based on the levels of CAT expression which were determined in brain tissue homogenates of 2‐month‐old CAG‐CAT‐Prnp mice using the CAT ELISA kit (Roche, Basel, Switzerland) according to the manufacturer's instructions. Selected line was backcrossed into C57BL6/J background and Prnp ZH3/ZH3 for nine generations.
To generate mice expressing Cre recombinase under the control of cell type‐specific promoters in ZH3 background: Synapsin1 Cre (B6.Cg‐Tg(Syn1‐Cre)671Jxm/J(#003966)) and GFAP Cre mice (B6.Cg‐Tg(Gfap‐Cre)77.6Mvs/2J(#024098)) were bred with ZH3. Cell type‐specific Cre expressors in ZH3 background were then crossed with Line 211 to generate mice lines expressing PrPC exclusively in a subset of neurons (SynCre;loxPrP) and astrocytes (GFAPCre;loxPrP). Other mice lines used in the current study are: C57BL6/6J and tga20 mice (B6.Cg‐Tg(Prnp)a20Cwe).
2.1.2. Prion inoculations
RML6 brain homogenates were prepared from the brains of terminally sick CD1 mice infected with RML6 prions. Brain homogenates were prepared in PBS (+5%BSA). For control inoculations, brain homogenates from healthy CD1 mice were used and they are referred to as noninfectious brain homogenate (NBH). Thirty microliters of RML6 (dose corresponding to 3 × 105 LD50) or NBH lysate was injected intracerebrally into 6‐ 8‐week‐old mice. Scrapie was diagnosed according to clinical criteria (ataxia, kyphosis, priapism, and hind leg paresis). Mice were sacrificed on the day of onset of terminal clinical signs of scrapie and NBH inoculated mice were sacrificed approximately at the same time. In case of mice that did not manifest prion disease, they were sacrificed approximately after 550–630 days post inoculations.
2.2. RNA sequencing and data analysis
RNA extraction from cerebella of mice was performed as described previously [8]. Data analysis and visualizations were performed using Sushi data analysis framework provided by Functional genomics center Zurich (FGCZ) from University of Zurich. Quality control and data analysis were performed as described previously [11]. Differential gene expression was performed using Edge R (version 3.0) [12] and any gene with Log2FC >0.5 and p‐value <0.05 was considered to be differentially expressed. Data intersection with genes dysregulated during the course of prion infection was performed using Multiple list comparator from www.molbiotools.com.
2.3. Cerebellar organotypic cultured slices (COCS)
About 350‐µm‐thick COCS were prepared from 9‐ to 12‐day‐old mice pups as described previously [13]. COCS cultures were maintained in a standard incubator (37°C, 5% CO2, 95% humidity) and the medium was replenished three times per week.
2.3.1. Prion inoculations of COCS
Freshly prepared COCS were inoculated with Rocky mountain laboratory strain 6 prions (RML6) or as a control with either noninfectious brain homogenate (NBH) as described previously. Slices were maintained for a further 56 days followed by fixation with 4% paraformaldehyde and staining with NeuN (to label cerebellar granule neurons), calbindin (to label Purkinjee cells), and DAPI (to label nuclei). Slices were imaged at 4x magnification on a fluorescence microscope (BX‐61, Olympus). NeuN and calbindin morphometry was analyzed using analySISvc5.0 software and neurotoxicity was defined as significant loss of NeuN or calbindin‐positive neuronal layer loss over NBH treatment.
2.3.2. POM1 treatment of COCS
Toxicity in COCS was induced by treatment with anti‐PrPC antibody (POM1) targeting the globular domain of the protein as described previously [14]. COCS were treated with either POM1 (67nM) or as control with IgG for 10 days after a 14‐day recovery period of initial gliosis due to tissue preparation. COCS were fixed, imaged, and analyzed as described for the prion inoculated slices. Antibody treatment was randomly assigned to individual wells.
2.4. Western blots
Mice brains (RML6 and NBH inoculated) were 3wsx3esxfor 5 min in 10 vol of lysis buffer (0.5% Nonidet P‐40, 0.5% 3‐[(3‐cholamidopropyl)dimethylammonio]‐1‐propanesulfonate (CHAPS)), protease inhibitors (complete Mini, Roche), phosphatase inhibitors (PhosphoSTOP, Roche) in PBS, and centrifuged at 1000 g for 5 min at 4°C to remove debris prior to analysis by SDS–PAGE (Novex NuPAGE 10% Bis‐Tris Gels). After electrophoresis, gel was transferred to iBlot I (Invitrogen) and transferred onto nitrocellulose membranes. Membranes were blocked in 5% Sureblock for 1 h at room temperature followed by incubation with primary antibody overnight. Membrane was washed 3x (15 min each) with PBS‐Triton (0.2%) followed by incubation with HRP‐tagged secondary antibody (Peroxidase‐Goat Anti‐Mouse IgG (H + L) (#62‐6520) or Peroxidase‐Goat Anti‐Rabbit IgG (H + L) (#111.035.045); 1h at room temperature) and further washes (3x, 10 min). Membrane was developed with Luminata Crescendo (Millipore) and images were acquired using LAS‐3000 Imaging system from FUJI. Densitometry analysis was performed using Quantity One software (BioRAD) and data were plotted using Graphpad software.
2.5. Immunohistochemistry
Brain tissues were fixed in formalin and treated with concentrated formic acid to inactivate prions. About 2µm thick sections were prepared from these brains and deparaffinized using graded alcohols and then subjected to antigen retrieval using 10mM citrate buffer (pH 6). Astrogliosis, microgliosis, and the presence of protease‐resistant prion deposits were visualized by staining brain sections with GFAP (1∶1000, Agilent technologies), IBA1 (1∶2500, WAKO), and the SAF84 antibody (1∶200, SPI bio), respectively on a NexES immunohistochemistry robot (Ventana instruments) using an IVIEW DAB Detection Kit (Ventana). Sections were also counterstained with hematoxylin and eosin when appropriate. Images were acquired using NanoZoomer scanner (Hamamatsu Photonics) and visualized using NanoZoomer digital pathology software (NDPview; Hamamatsu Photonics). Images were acquired using Olympus BX61 Upright fluorescent microscope. Quantifications of IBA1, GFAP staining was performed on the entire brain section from the acquired images using Image J.
2.6. Antibodies
The following antibodies are used in the current study.
| Antibody | Source | Cat. No |
|---|---|---|
| anti‐SNAP25 antibody | Abcam | ab5666 |
| anti‐GFAP | Agilent technologies | Z03344 |
| anti‐Iba1 | WAKO | 019‐19741 |
| anti‐SAF84 | Bertin bioreagent | A03208 |
| anti‐NeuN | Millipore | MAB377X |
| anti‐calbindin | Abcam | ab108404 |
| anti‐calnexin | Enzo Life sciences | ADI‐SPA‐865‐D |
| anti IL1β | Abcam | ab9722 |
| anti‐PrPC antibodies (POM1, POM2, POM19) | Aguzzi Lab | (15) |
| anti‐actin | Millipore | MAB1501R |
2.7. Statistical analysis
Details of the type of statistical tests performed are described in the figure legends. Results are represented as mean of replicates (µSEM). Data were analyzed using GraphPad software.
3. RESULTS
3.1. Generation of mice with cell type‐restricted PrPC expression
We developed a versatile transgenic mouse model that directs expression to specific cell types in a robust and controllable manner. We used a murine Prnp cDNA under the transcriptional control of the cytomegalovirus/chicken beta actin/rabbit beta‐globin gene (CAG) promoter and a loxP‐stop‐loxP (LSL) cassette flanking the chloramphenicol acetyl transferase (CAT) gene [10]. When crossed to mice expressing the Cre recombinase tissue specifically, the CAT gene and the transcriptional stop cassette flanked by LoxP sites are excised, permitting activation of PrPC expression (Figure 1A).
FIGURE 1.
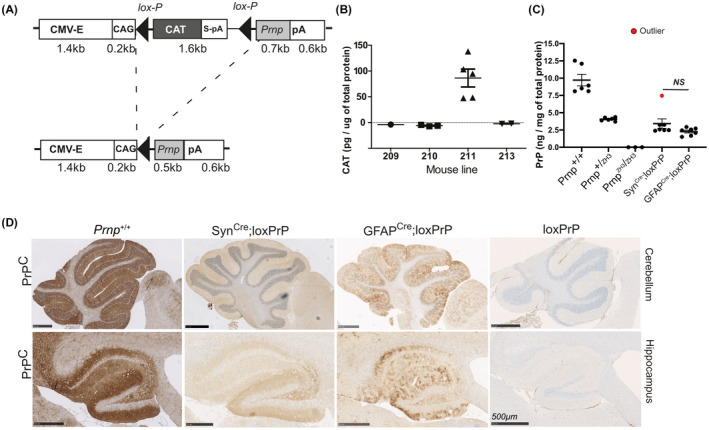
Generation and phenotyping of mice expressing PrP in defined brain compartments. (A) Schematic of the CAG‐CAT‐Prnp transgene: The transgene is driven by the CMV enhancer followed by chicken beta actin (CAG) promoter. The chloramphenicol acetyl transferase (CAT) expression cassette contains an SV40 polyadenylation signal (S‐pA) and is flanked by lox‐P sites, followed by the Prnp coding sequence and a rabbit β‐globin polyadenylation signal (pA). The Prnp transgene is expressed only upon excision of the CAT stop sequence by the Cre recombinase. (B) Transgenic CAT expression in brain homogenates, assessed by ELISA. Assays here and in panel C were performed as triplicates; plots represent mean ± SEM. Line 211 was the only line that displayed CAT expression, and was selected for crossing with Prnp ZH3/ZH3 mice. (C) The resulting Prnp ZH3/ZH3;CAG‐CAT‐PrP mice were crossed with Cre driver lines. Brain PrPC expression was compared with Prnp +/+, Prnp +/ZH and Prnp ZH3/ZH3 mice by ELISA. Crosses to SynCre and GFAPCre mice yielded mice expected to selectively express PrPC in a subset of neurons and astrocytes, respectively. Dots: ELISA on individual mouse brain. Red dot: outlier SynCre;loxPrP mouse with elevated expression of PrPC. Grubbs test was performed to identify the outlier. Statistics: one‐way ANOVA followed by Bonferroni's post‐hoc test. NS: not significant. (D) Immunohistochemistry of brain sections of SynCre;loxPrP and GFAPCre;loxPrP mice with anti‐PrPC antibody POM2. Negative and positive controls: loxPrP and wild‐type mice brain sections. PrPC was found mainly in the hippocampus of SynCre;loxPrP mice, whereas GFAPCre;loxPrP mice showed prominent staining in both hippocampus and cerebellum
Pronuclear injection of the CAG‐CAT‐PrP construct into one‐cell stage fertilized embryos from mice ablated of cellular PrPC (Prnp ZH1/ZH1) resulted in the generation of seven transgenic lines, four of which transmitted the transgene to their F1 offspring (Figure S1A). We next monitored the expression of CAT in brain lysates from offspring of all four transgenic lines using quantitative enzyme‐linked immunosorbent assay (ELISA). Only line 211 showed sustained expression of CAT (Figure 1B). Line 211 was further bred to co‐isogenic C57BL/6 Prnp‐ablated mice (Prnp ZH3/ZH3) for nine successive generations in order to eliminate any potential genetic confounder effects on the phenotypes observed [16].
To evaluate the robustness of CAG‐CAT system in generating cell type‐specific PrPC expressors, we crossed Line 211 with mice expressing Cre under the control of the synapsin‐1 (Syn) or glial fibrillary acidic protein (GFAP) promoters in order to direct PrPC expression selectively to neurons (SynCre;loxPrP) or astrocytes (GFAPCre;loxPrP).
Mice were tested by ear‐biopsy PCR for the CAG‐CAT‐PrP and the appropriate Cre transgene, and brain lysates from double‐positive mice were subjected to ELISA. There were no significant differences in the expression levels of PrPC between SynCre;loxPrP and GFAPCre;loxPrP mice in their brains. As expected, the overall protein levels of PrPC were lower in brain lysates of mice expressing cell type‐specific PrPC than the wild‐type mice or hemizygous Prnp ZH3/+ mice (Figure 1C). We next sought to investigate the expression pattern of PrPC in various brain regions of the three transgenic lines. Immunohistochemistry of entire brain sections from SynCre;loxPrP and GFAPCre;loxPrP mice using the anti‐PrP antibody POM19 revealed differential expression of PrPC. SynCre;loxPrP mice expressed PrPC predominantly in the hippocampus, whereas GFAPCre;loxPrP mice expressed PrPC in both cerebellum and hippocampus (Figure 1D, Figure S1B).
To further assess the cell type‐restricted expression of PrPC, we performed immunofluorescence stainings on cerebellar brain sections. Mice lacking PrPC (Prnp ZH3/ZH3) and wild‐type mice were used as negative and positive controls. In cerebellar sections of SynCre;loxPrP mice, PrPC expression was exclusively observed in Purkinje cells. Unlike SynCre;loxPrP mice, co‐staining with MAP‐2, which labels mature neuronal population, revealed that PrPC expression in wild‐type mice showed a diffuse neuronal staining. Prnp ZH3/ZH3 mice did not show any staining of PrPC (Figure 2A). While several glia drivers were shown to also label neuronal subpopulations [17], the GFAP‐Cre driver used here was shown to exclusively label astrocytes [17]. Co‐staining with GFAP in cerebellar sections of GFAPCre;loxPrP mice revealed astrocytic localization of PrPC. Finally, no expression of PrPC was seen in the absence of Cre (Figure 2B).
FIGURE 2.
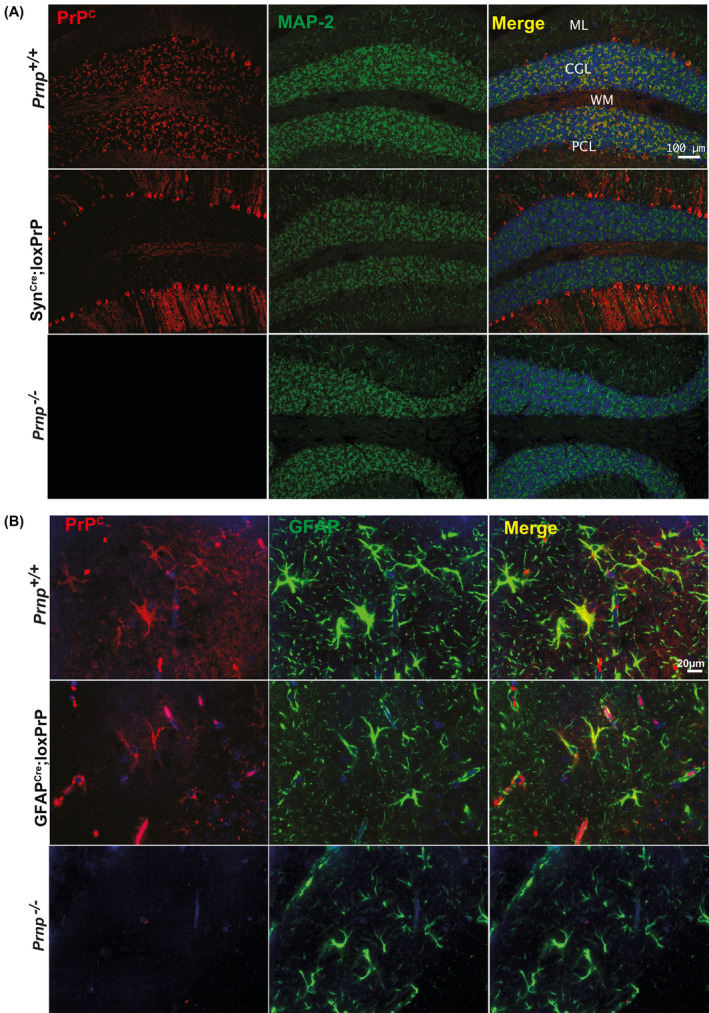
PrPC expression patterns in CAG‐CAT‐PrP mice. (A and B) Cerebellar sections of wild‐type and transgenic mice (as indicated) were immunostained for PrPC (red) as well as for MAP2 (microtubule‐associated protein 2), GFAP (glial fibrillary acidic protein), which were used as neuronal and astrocytic markers, respectively (green). As controls cerebellar sections from Prnp ablated mice were used. Blue: cell nuclei (DAPI). In Prnp +/+ mice, PrPC is detected in the cerebellar granule cell layer (CGL), Purkinje cells (PCL), and molecular layer (ML). In SynCre;loxPrP cerebella (A), PrPC was mostly detected in Purkinje cells. In GFAPCre;loxPrP, PrPC was exclusively detected in astrocytes colocalizing with GFAP. Prnp ablated mice revealed absence of any unspecific staining. Each staining was performed on sections from at least three individual mice
3.2. Role of neuronal and astrocytic PrPC in the manifestation of prion disease
We sought to investigate the kinetics and the manifestation of prion disease in the newly generated mice expressing PrPC in a subset of neurons [18]. Five SynCre;loxPrP mice were intracerebrally inoculated with prions (Rocky Mountain Laboratory strain, passage #6, henceforth termed RML6). For control, we used wild‐type C57BL6/J mice, mice transgenic for SynCre but not for loxPrP, and vice versa. SynCre;loxPrP mice developed clinical scrapie, albeit with significantly longer incubation times than wild‐type mice (435 vs 196 days after inoculation (dpi), respectively, p = 0.002) (Figure 3A). Hence neuronal PrPC expression suffices to confer susceptibility to prion disease. We next assessed proteinase K (PK)‐resistant prion protein (termed PrPSc) in brain lysates of terminally scrapie‐sick wild‐type and SynCre;loxPrP mice, as well as age‐matched loxPrP, SynCre or Prnp ZH3/ZH3 mice. Western blotting of PK‐treated lysates (25 µg/ml, 37°C, 1h) revealed PrPSc only in wild‐type and SynCre;loxPrP lysates (Figure 3B). Next, five GFAPCre;loxPrP transgenic mice were inoculated intracerebrally with RML6 prions. For control we used wild‐type mice, GFAPCre mice, loxPrP mice, and Prnp ZH3/ZH3 mice. Over a period of 627 dpi, neither GFAPCre;loxPrP mice nor Prnp ZH3/ZH3 mice developed any signs of scrapie, whereas Prnp +/+ mice (with or without Cre transgenes) developed terminal scrapie at 167 ± 5 dpi (Figure 3C). Western blots showed similar amounts of PrPSc in terminally scrapie‐sick wild‐type mice and in 2‐year‐old prion‐infected GFAPCre;loxPrP mice (627 dpi) (Figure 3D).
FIGURE 3.
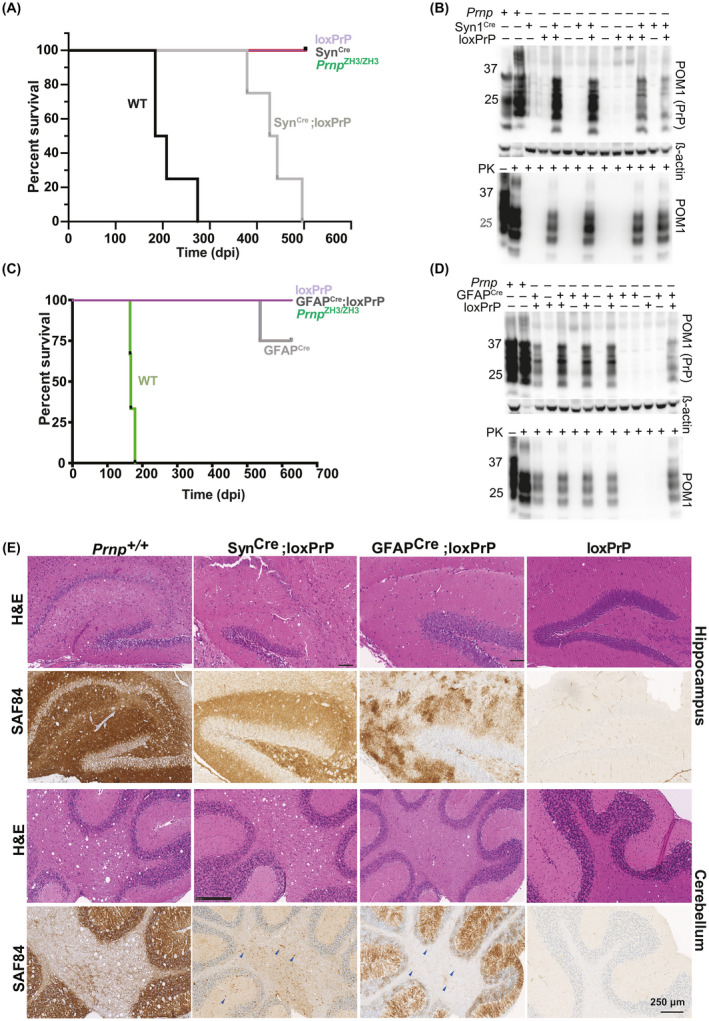
Neuron‐selective, but not astrocyte‐selective, PrPC expression confers susceptibility to prion disease. (A) Survival of SynCre;loxPrP, loxPrP, SynCre, Prnp ZH3/ZH3 and wild‐type mice inoculated intracerebrally with RML6 prions. The median incubations for SynCre−;loxPrP (n = 5) and wild‐type mice (n = 4) were 435 and 195 days post inoculation (dpi), respectively. Survival curves were compared by a log‐rank (Mantel‐Cox) test. (B) Total PrP (upper panel) and PK‐resistant PrPSc (lower panel) in brains of prion‐infected, terminally sick mice. SynCre;loxPrP and wild‐type brains, but not loxPrP brains, contained PrPSc. β‐actin: loading control. Lane #2 (upper panel) was intentionally underloaded to avoid overexposure. (C) Survival curves of GFAP1Cre;loxPrP, loxPrP, Prnp ZH3/ZH3 and wild‐type mice inoculated with RML6 prions intracerebrally. GFAPCre;loxPrP (n = 5) did not develop clinical signs of prion disease. Wild‐type mice (n = 4): 167 dpi (median incubation time). One of the GFAPCre mice was sacrificed (544 dpi) because of acute dermatitis and did not exhibit scrapie signs or PrPSc accumulation. (D) Western blot of total PrP and PK‐digested PrPSc in brain homogenates of prion‐infected GFAPCre;loxPrP (627 dpi) and control mice. GFAPCre;loxPrP harbor copious PrPSc. (E) Hippocampal and cerebellar histology of prion‐infected SynCre;loxPrP, GFAPCre;loxPrP, loxPrP, and Prnp +/+ mice. Slices were stained with hematoxylin and eosin (H&E) and anti‐PrP antibody SAF84. Both astrocyte‐ and neuron‐restricted PrP transgenic mice accumulated PrPSc, but their deposition patterns differed profoundly. Blue arrows in cerebellar regions of SynCre;loxPrP and GFAPcre;loxPrP: PrPSc deposits
Histological analysis of hippocampal and cerebellar brain sections revealed vacuolation (spongiosis) in SynCre;loxPrP mice, albeit less pronounced than in wild‐type mice, whereas GFAPCre;loxPrP did not show any vacuolation (Figure 3E). Immunohistochemistry with anti‐PrP antibody, SAF84 [19] in hippocampal sections of SynCre;loxPrP mice revealed the presence of PrP deposits, albeit at lower amounts than in wild‐type mice, while mice lacking PrP or Cre‐recombinase did not show any staining (Figure 3E). SynCre;loxPrP mice, which expressed PrPC predominantly in Purkinje cells, exhibited fewer PrP deposits than wild‐type mice (Figure 3E). SAF84 immunostaining in prion inoculated GFAPCre;loxPrP mice however revealed PrP deposits in both cerebellum and hippocampus (Figure 3E). Taken together, these results suggest that mice with neuron‐restricted PrPC develop clinical and histological features of prion disease whereas PrPC expression in astrocytes alone does not lead to prion disease, yet it supports PrPSc accumulation.
3.3. Astrocyte‐restricted PrPC does not induce neurodegeneration in COCS
We next generated cerebellar organotypic cultured slices (COCS) from 8‐day‐old SynCre;loxPrP mice and inoculated them with RML6 prions or with non‐infectious brain homogenate (NBH). At 56 dpi, these COCS were immunostained with NeuN (staining cerebellar granule cells) and calbindin (staining Purkinje cells) [20].
Neuronal loss in COCS was measured by morphometric assessment of cerebellar granule layer (CGL) area immunoreactive to the antibodies against NeuN and calbindin. COCS generated from SynCre;loxPrP mice showed a significant decrease in calbindin staining, confirming neurodegeneration of PrP expressing Purkinje cells in the SynCre;loxPrP mice. Surprisingly, there was also a significant loss of afferent NeuN+ cerebellar granule neurons, possibly due to secondary effects arising from Purkinje cell death [21]. In contrast, control COCS from loxPrP mice did now show any neurodegeneration (Figure 4A).
FIGURE 4.
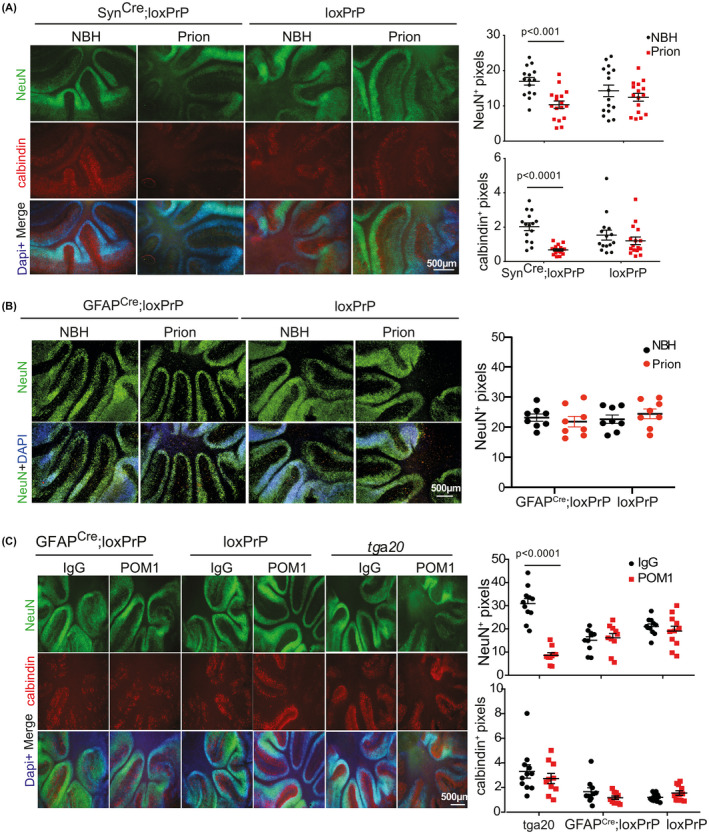
COCS from astrocyte‐restricted PrPC mice are resistant to neurodegeneration induced by prion mimetics. (A) Fluorescence micrographs of cerebellar organotypic cultured slices (COCS) of SynCre;loxPrP and loxPrP COCS infected with prions (56 dpi) or exposed to noninfectious brain homogenate (NBH). Purkinje cells were identified by calbindin immunostaining. Calbindin and NeuN morphometry (right) shows degeneration of granule and Purkinje cells in prion‐infected SynCre;loxPrP COCS. Each dot represents a cerebellar slice. Here and henceforth: one‐way ANOVA was used for statistical analysis. (B) Fluorescence micrographs of GFAPCre;loxPrP and loxPrP COCS infected with prions or exposed to non‐infectious brain homogenate (NBH). Slices were cultured for 56 days and stained with anti‐NeuN antibody. There was no discernible loss of NeuN signal in prion‐infected COCS. For control, we stained COCS from loxPrP mice. Right panel: quantification. Each dot represents a cerebellar slice. One‐way ANOVA was used for statistical analysis; NS: not significant. Nuclei were stained with DAPI. (C) Fluorescence micrographs of GFAPCre;loxPrP, loxPrP, and Tga20 COCS exposed to the neurotoxic anti‐PrP antibody POM1 or IgG control and cultured for 10 days were stained for NeuN (green), calbindin (red), and DAPI (blue). POM1 did not trigger neuronal loss in GFAPCre;loxPrP COCS, whereas neurodegeneration was extensive in Tga20 COCS. Each dot represents a cerebellar slice
To further challenge the conclusion that astrocyte‐restricted PrPC expression does not restore prion‐dependent neurodegeneration, we generated COCS from 9‐day‐old pups of GFAPCre;loxPrP and for control from loxPrP mice. COCS were inoculated with RML6 prions or NBH. At 56dpi COCS were subjected to immunostaining with NeuN. COCS from GFAPCre;loxPrP did not show any signs of neuronal loss (Figure 4B). We next investigated if COCS from GFAPCre;loxPrP are also resistant to prion mimetics. COCS from 9–day‐old GFAPCre;loxPrP mice were treated with the prion‐mimetic antibody POM1 or with pooled mouse IgG, fixed after 10 days of treatment, and immunostained with NeuN and calbindin. As controls, COCS were generated from tga20 (mice overexpressing PrPC) [22] and loxPrP mice were used. COCS from tga20 mice, but not from GFAPCre;loxPrP and loxPrP mice, showed conspicuous neuronal loss (Figure 4C).
3.4. Mice with neuron or astrocyte restricted PrPC expression do not activate microglia upon prion infection.
Prion diseases typically feature extreme activation and proliferation of microglia to an extent rarely seen in any other brain diseases. We assessed the status of microglia and astrocytes by immunohistochemistry for Iba1 and GFAP on the cortical and hippocampal brain sections of terminally scrapie‐sick wild‐type mice and SynCre;loxPrP mice. For control, we used loxPrP mice. While wild‐type mice showed a high microglia density, we were surprised to find that SynCre;loxPrP mice did not show more microglial activation and astrogliosis than control mice (Figure 5A–C, Figure S2A) despite being terminally scrapie‐sick. We performed the same analysis in prion‐infected GFAPCre;loxPrP mice. Also here, the staining of cortical and hippocampal sections with anti‐Iba1 and anti‐GFAP did not reveal any microglia activation or astrogliosis beyond the baseline of control mice (GFAPCre−;loxPrP, GFAPCre;loxPrP−), whereas wild‐type mice showed a brisk enhancement of Iba1 and GFAP immunoreactivity (Figure 5A‐C, Figure S2A).
FIGURE 5.
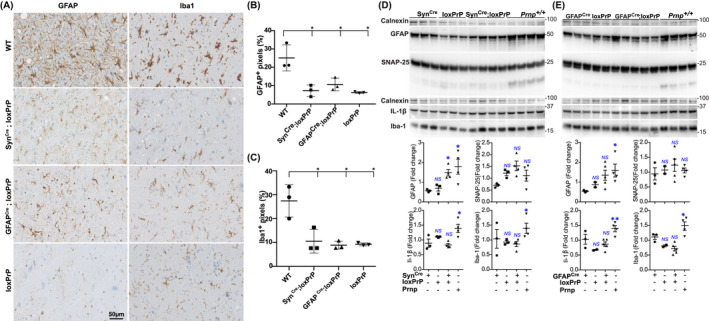
Prion infection of neuron and astrocyte‐specific PrP expressors does not induce microglia activation. (A) Microglia (Iba1) and astrocyte (GFAP) immunostains of the cortical sections of prion‐inoculated SynCre;loxPrP (379 dpi), GFAPCre;loxPrP (627 days), loxPrP (504 dpi), and wild‐type (184 dpi) mice. (B and C) Quantification of GFAP+ (B) and Iba1+ (C) pixels in a cross‐section of complete mouse brain. Expression of both markers was conspicuously increased in Prnp +/+ mice, whereas SynCre;loxPrP and GFAPCre;loxPrP revealed significantly lower amounts of GFAP and Iba1. Each dot represents quantification performed on brain sections obtained from an individual mouse. Statistics: one‐way Anova test. (D and E) Western blot analysis of brain lysates generated from the enrire hemispheres and related quantification of GFAP, SNAP‐25, IL1‐Beta, and Iba‐1 in prion inoculated SynCre;loxPrP mice (D) and GFAPCre;loxPrP mice (E) versus controls. Calnexin was used for loading control. Only SNAP‐25 showed elevated expression in prion‐infected SynCre;loxPrP mice, whereas all other markers of microglial activation remained unaltered. Each dot represents data from lysate obtained from an individual mouse. Statistics: one‐way Anova test
3.5. Molecular changes associated with microglial activation remain unaltered in mice with cell type restricted PrPC upon prion infection
Microglial changes typically precede the onset of the clinical signs of the disease, and are accompanied by the expression of pro‐inflammatory cytokines such as TNFa, IL1α, and IL1β. Hemispheric brain lysates from the same prion‐infected mice as above were then subjected to western blotting to monitor the expression of SNAP25 (a presynaptic protein engulfed by microglia previously shown to be reduced in prion infections before the onset of motor defects [23]), GFAP, IL‐1β, and Iba‐1. Terminally scrapie‐sick wild‐type mice showed a substantial increase in the expression of GFAP, Iba‐1, and IL‐1β, whereas SynCre;loxPrP and GFAPCre;loxPrP lysates were similar to the negative controls. Only SNAP‐25 was slightly elevated in SynCre;loxPrP mice (Figure 5D,E). These results indicate that microglial activation is not induced by prion infection in mice expressing PrPC only in neurons or astrocytes.
3.6. Prion infection does not elicit its transcriptional signature in mice expressing astrocyte‐restricted PrPC
Previous studies have longitudinally mapped transcriptional changes associated with prion disease in mice, and revealed changes in gene expression from as early as 4 weeks post prion infection [8]. We asked if GFAPCre;loxPrP mice, which do not develop clinical signs after prion infection, exhibit transcriptional changes upon prion infection. Three GFAPCre;loxPrP mice were intracerebrally inoculated with RML6 prions; for control we inoculated two GFAPCre;loxPrP mice with non‐infectious brain homogenate (NBH). Prion‐infected mice did not manifest prion disease and were humanely euthanized along with the NBH‐treated mice at approximately 627 dpi. RNA was isolated from cerebella of these mice and processed for RNA sequencing (Figure 6A). Unsupervised clustering of the 100 genes with highest variance across all the samples did not reveal any separation between the prion‐infected and NBH‐treated samples (Figure 6B). Data analysis to identify differentially expressed genes (DEGs) in prion inoculated vs NBH‐treated mice revealed absence of any DEGs when a filter of Log2FC >0.5 and FDR <0.05 was applied (Figure 6A). We then relaxed the stringency by applying a filter of Log2FC >0.5 and p‐value <0.05. This revealed 168 DEGs in prion inoculated GFAPCre;loxPrP mice. Of the 168 DEGs, 104 genes were upregulated, and 64 genes were downregulated (Figure 6C, Table S1). Functional gene ontology studies revealed upregulated genes were enriched in biological processes associated with hormonal response pathways (Figure S2B), whereas the downregulated genes were not enriched in any pathway. A total of 3723 genes were previously shown to have their expression altered at least at one measured time point during the course of prion disease in C57BL6/J mice inoculated with RML6 prions. We next tested if any of the DEGs from the prion‐inoculated GFAPCre;loxPrP mice overlapped with the DEGs observed during the progression of prion disease in C57BL6/J mice. Intersection plots revealed that 43 genes with altered expression in GFAPCre;loxPrP mice were also differentially expressed during the prion disease (Figure 6D, Table S2). These 43 DEGs however did not correlate with any particular time point during the progression of prion disease. We deduce that PrPSc produced by astrocytes does not induce the transcriptional changes typical of prion diseases.
FIGURE 6.
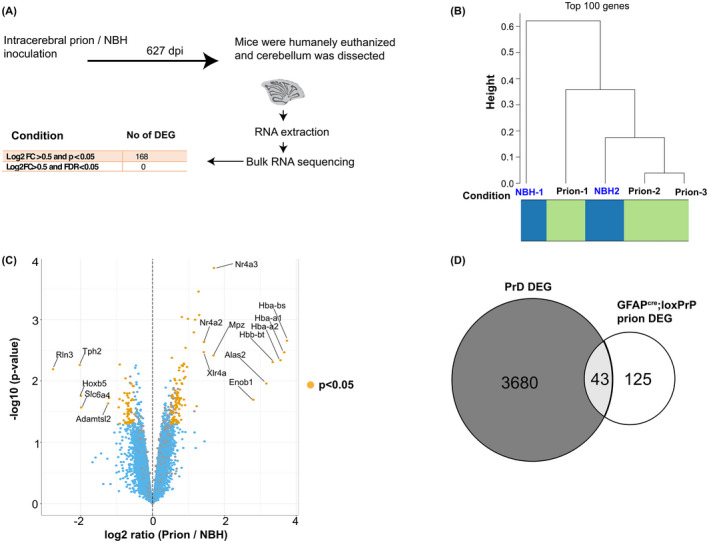
Prion disease‐specific transcriptional signature is absent in astrocyte‐specific PrPC expressors infected with prions. (A) Schematic representation of the sample collection (cerebellum) and bulk RNA sequencing after prion or NBH inoculation of GFAPCre;loxPrP mice. The number of differentially expressed genes (DEGs) and the filtering criteria used to derive them are indicated. (B) Hierarchical clustering analysis based on the 100 genes with highest variance across all the samples did not reveal a separation between NBH and prion‐infected mice (n = 3 for prion inoculations and n = 2 for NBH injections). (C) Volcano plot showing differentially expressed genes in the prion inoculated GFAPCre;loxPrP mice compared with the NBH‐injected counterparts. Genes with Log2FC >0.5 and p‐value <0.05 were considered as DEGs. The identities of the top 10 upregulated and top 5 downregulated genes are reported in the plot. Log2FC: log twofold change. (D) Intersection between the DEGs observed during the progression of prion disease in C57BL6/J mice (3723 genes) and DEGs in prion inoculated GFAPCre;loxPrP mice (168 genes). The intersection consists of only 43 genes, suggesting that prion‐infected GFAPCre;loxPrP mice do not exhibit any prion‐specific transcriptomic signature
4. DISCUSSION
While many transgenic mouse lines with tissue‐specific PrPC expression have been generated in the past two decades, we still lack a flexible generic model system allowing for expression in any given cell type under highly controlled conditions. What is worse, several of the transgenic models alleged to display cell‐specific PrPC expression in reality exhibit illegitimate expression in ectopic compartments: the widely used neuron‐specific enolase (NSE) promoter can be expressed in glial cells [24] and earlier versions of a GFAP‐driven construct are expressed in neurons [25, 26]. This situation has motivated us to generate the lines described in this study. We opted to use the CAG‐CAT system [10] and in its native state, the CAG‐CAT transgene leads to sustained and ubiquitous expression of chloramphenicol acetyl transferase (CAT), whose presence can be easily monitored with an enzyme activity assay. Upon CRE‐mediated recombination, the loxP‐flanked CAT minigene and its polyadenylation signal are excised, and transcription of PrPC is enabled. We tested the system with two CRE‐expressor crossings directing PrPC expression to neurons and astrocytes. We chose the Synapsin‐1 promoter as it is selectively, but broadly expressed in many types of neurons. Conversely, the mGFAP line 77.6 used in the current study has previously been shown to express Cre exclusively in astrocytes [17]. Although expressed in different cell types, PrPC expression in these lines remained comparable.
The data reported here enable new insights into two aspects of prion pathology: cell type‐specific replication and cell type‐specific toxicity. The expression of PrPC is a necessary but insufficient prerequisite to prion replication, and multiple cell types replicate prions during their journey from the periphery to the brain. However, many tissues do not replicate prions despite expression of PrPC. While the replication competence of neurons is well established, the data on astrocytes have remained controversial. One of us (AA) reported that mice expressing hamster PrPC off a GFAP promoter would replicate prions and develop disease [27]. However, a similar construct was found to be active ectopically in certain populations of neurons, raising the possibility of low‐level expression of the transgene and thereby [25] raising doubts on the validity of the previous report. The GFAP‐Cre line used in the current study, unlike the previous GFAP promoter‐derived constructs has no expression in the postnatal or adult neural stem cells or their progeny [17]. The specificity of transgenic mice described in the current study is arguably more stringent. Although these mice did not develop any histological signs of neuroinflammation and clinical disease, we confirmed that astrocytes are capable of prion replication and PrPSc deposition in vivo.
The terminal stage of prion diseases is typically characterized by extensive neuronal loss. This observation has led to the implicit assumption that neurons are the primary targets, and possibly the primary driver, of prion diseases. An impressive panoply of evidence has accumulated that neuronal damage is crucially dependent on expression of PrPC by the neurons targeted by prions. The first hint came from grafting PrPC‐expressing neural tissue into PrPC‐deficient mice. After prion infection, grafts developed all the pathological features of prion disease, yet no damage was observed in the adjoining tissue lacking PrPC despite PrPSc deposition loss [28, 29]. An additional line of evidence came from partial depletion of neuronal PrPC, which decelerated the development of disease despite deposition of PrPSc [7]. Furthermore, transgenic mice expressing a hamster prion protein in neurons became susceptible to prion disease after exposure to hamster prion, suggesting that mere expression of PrPC in neurons suffices to develop prion disease [30].
On the other hand, a growing number of observations is challenging the perception that neurons are the sole, or even the dominant, cellular actor in prion pathology. Recently concluded longitudinal transcriptomic study mapping the changes in gene expression during the course of prion infection has revealed that glial perturbations occur simultaneously to the onset of clinical disease. Surprisingly, no changes in the expression of neuron‐enriched transcript were detectable at this time point. Instead, suppression of neuronal transcripts was only observed at the terminal stage of disease [8]. The onset of changes associated with actively translating genes during the course of prion infection also revealed glial perturbation as the major contributors in driving the course of prion disease [9]. More importantly, disease‐associated microglia (DAM) genes and A1 astrocytes, both of which are renowned glia signatures in several neurodegenerative diseases were upregulated in prion disease. Overall, these studies revealed an important role for non‐neuronal cell types in the manifestation of prion disease.
4.1. Contribution of cell type‐specific PrPC to prion disease
Prion inoculated SynCre;loxPrP mice displayed all the characteristic histopathological and clinical features of prion disease. The incubation time was much longer than that of wild‐type mice (median survival: 435 vs 196 days), which may be explained by the lower total brain PrPC concentration.
Although the mechanisms responsible for neuronal death are yet to be elucidated, it is evident that PrPSc deposits from non‐neuronal counterparts could accelerate the conversion of neuronal PrPC and thereby aggravating neuronal loss. Interestingly, these mice showed no activation of microglia and astrocytes and this further confirms the hypothesis that glial activation drives the progression of the disease and in its absence the onset of the disease follows a much longer time course. One interesting question that arises is whether the glial activation in prion disease requires the presence of PrPC in both neurons and astrocytes and what is the interplay between neurons and astrocytes? Studies have pointed out toward an important role for neurons and neuronal activity not only in determining astrocytic fate but also how they can be potentially altered in neurodegeneration [31]. The SynCre;loxPrP mice line delinks glial activation from neuronal death and could be potentially used as a model system to study the mechanisms associated with neuronal death in prion disease.
Among non‐neuronal cells, astrocytes are the cell type producing highest amount of PrPC [32] followed by oligodendrocytes. Previous studies have shown astrocytes are not only capable of replicating and propagating prions but also can deposit PrPSc aggregates [27], whereas PrPC expression restricted to myelinating cells failed to accumulate PrPSc or develop prion disease [33]. Transgenic mice expressing PrPC exclusively in astrocytes (GFAPCre;loxPrP) do not develop prion disease and do not show any of the classical pathological features apart from deposition of PrPSc. These mice also fail to recapitulate glia activation or upregulate any of the inflammation markers. RNA sequencing data corroborated the absence of molecular markers of disease. Differentially expressed genes became identifiable only when the stringency of the analysis was substantially relaxed, and even then they did not appear to be enriched in any specific cell types or pathways. We conclude that astrocyte‐selective prion infection has remarkably bland effects on the brain.
In a previous study, mice expressing PrPC under the expression of GFAP promoter were shown to replicate prions but did not develop scrapie [34]. The capability of astrocytes to replicate prions was however questioned because the GFAP promoter fragment used to generate the mice also exhibited partial neuronal expression [25, 26]. With the newly generated mouse models described here, we have confirmed that astrocytes are indeed capable of replicating prions. These findings vindicate a report that cultured iPSC‐derived astrocytes can replicate CJD prions [35]. Furthermore, a recent study has documented the transport of PrPSc from astrocytes to neurons, suggesting a noncell‐autonomous mechanism of toxicity [36]. The results presented here argue that this is not the case and that accumulation and transport of astrocytic PrPSc does not suffice to induce neurotoxicity.
In the future, these newly generated mice lines can help answering some of the long‐standing questions in the prion field and may represent ideal tools to delineate the role and contribution of each of the cell types in manifestation of prion disease.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Adriano Aguzzi initiated and supervised the project, and wrote the manuscript. Asvin K. K. Lakkaraju performed RNA sequencing experiment, analyzed all the data, and wrote the manuscript. Assunta Senatore performed slice cultures, immunohistochemistry, ELISA, western blots, and analyzed the data. Silvia Sorce contributed to generation of mice lines, immunohistochemistry, ELISA, and analyzed the data. Mario Nuvolone contributed to generation of mice lines. Jingjing Guo, Petra Schwarz, Rita Moos provided the technical help in performing all the experiments in the manuscript. Pawel Pelczar performed microinjections and contributed to the generation of mice lines. All authors have read and approved the final version of the manuscript.
MATERIAL AVAILABILITY
All unique reagents used for this study will be available from the lead contact upon request with a material transfer agreement.
Supporting information
Fig S1
FIGURE S1 (A) Polymerase chain reaction (PCR) performed on tail biopsies of seven transgenic founder mice revealed successful integration of CAT‐PrP transgene. For control, we used CAT‐PrP cDNA and DNA extracted from wild‐type C57BL6/J mouse tail biopsies. Actin was simultaneously amplified as a positive control. Samples were migrated on 2% agarose in Tris–EDTA buffer by electrophoresis. Lines 208, 214, and 215 failed to transmit the transgene to F1 generation. (B) Immunohistochemistry of brain sections of SynCre;loxPrP and GFAPCre;loxPrP mice with anti‐PrPC antibody POM2. Left panel: Whole brain section montage. Right panel: Magnified images of cerebellum and hippocampal regions stained with POM2. Images reveal differential PrPC distribution in SynCre;loxPrP and GFAPCre;loxPrP mice
{kind=link}
Fig S2
FIGURE S2 (A) Microglia (Iba1) and astrocyte (GFAP) immunostaining of the hippocampal sections of prion‐inoculated SynCre;loxPrP (379 dpi), GFAPCre;loxPrP (627 days), loxPrP (504 dpi), and wild‐type (184 dpi) mice. Expression of GFAP and Iba1 was upregulated in Prnp +/+ mice, whereas SynCre;loxPrP and GFAPCre;loxPrP revealed significantly lower expression levels of the two markers. (B) Biological process enrichment obtained by gene ontology analysis on the upregulated genes in prion‐infected GFAPCre;loxPrP mice revealed that most upregulated genes were involved in pathways associated with hormone response
{kind=link}
Fig S3
FIGURE S3 Uncropped western blots. All western blots pertaining to this study are shown here in their original format as generated by the blot imager software. Sequence of staining in case multiple antibodies were used is indicated by numbering. No editing was performed
{kind=link}
Table S1
TABLE S1 Table including detailed RNA Sequencing results including the differentially expressed genes, p‐values, FDR values in prion‐infected GFAPCre;loxPrP mice
Table S2
TABLE S2 Table showing differentially expressed genes that are common to prion disease (PrD) and prion‐infected GFAPCre;loxPrP mice (GFAPCre;loxPrP prion)
ACKNOWLEDGMENTS
The authors thank Mirzet Delic and Paulina Pawlak for animal husbandry, and Merve Avar for help with data analysis of RNA sequencing data. AA is the recipient of an Advanced Grant of the European Research Council (ERC 670958), the Swiss National Foundation (SNF: 179040), the Nomis Foundation and SystemsX.ch. AS, SS, and AKKL are recipients of a grant from the Synapsis Foundation.
Lakkaraju AKK, Sorce S, Senatore A, Nuvolone M, Guo J, Schwarz P, et al. Glial activation in prion diseases is selectively triggered by neuronal PrPSc . Brain Pathol. 2022;32:e13056. 10.1111/bpa.13056
Asvin K. K. Lakkaraju, Silvia Sorce, and Assunta Senatore contributed equally to this work.
DATA AVAILABILITY STATEMENT
All original data have been included in the article. Uncropped western blots from the entire manuscript are included in Figure S3. No cropping was performed on any of the microscopy images. Any additional information/data required will be made available by the corresponding author upon reasonable request. No source code was generated in this study.
REFERENCES
- 1. Aguzzi A. Cell biology: Beyond the prion principle. Nature. 2009;459(7249):924–5. [DOI] [PubMed] [Google Scholar]
- 2. Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73(7):1339–47. [DOI] [PubMed] [Google Scholar]
- 3. Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, et al. Normal host prion protein necessary for scrapie‐induced neurotoxicity. Nature. 1996;379(6563):339–43. [DOI] [PubMed] [Google Scholar]
- 4. Brandner S, Raeber A, Sailer A, Blattler T, Fischer M, Weissmann C, et al. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci U S A. 1996;93(23):13148–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aguzzi A, Nuvolone M, Zhu C. The immunobiology of prion diseases. Nat Rev Immunol. 2013;13(12):888–902. [DOI] [PubMed] [Google Scholar]
- 6. Budka H. Neuropathology of prion diseases. Br Med Bull. 2003;66:121–30. [DOI] [PubMed] [Google Scholar]
- 7. Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302(5646):871–4. [DOI] [PubMed] [Google Scholar]
- 8. Sorce S, Nuvolone M, Russo G, Chincisan A, Heinzer D, Avar M, et al. Genome‐wide transcriptomics identifies an early preclinical signature of prion infection. PLoS Pathog. 2020;16(6):e1008653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scheckel C, Imeri M, Schwarz P, Aguzzi A. Ribosomal profiling during prion disease uncovers progressive translational derangement in glia but not in neurons. eLife. 2020;9:e62911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Araki K, Araki M, Miyazaki J, Vassalli P. Site‐specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc Natl Acad Sci U S A. 1995;92(1):160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Henzi A, Senatore A, Lakkaraju AKK, Scheckel C, Muhle J, Reimann R, et al. Soluble dimeric prion protein ligand activates Adgrg6 receptor but does not rescue early signs of demyelination in PrP‐deficient mice. PLoS One. 2020;15(11):e0242137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falsig J, Aguzzi A. The prion organotypic slice culture assay–POSCA. Nat Protoc. 2008;3(4):555–62. [DOI] [PubMed] [Google Scholar]
- 14. Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501(7465):102–6. [DOI] [PubMed] [Google Scholar]
- 15. Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, et al. The POM monoclonals: a comprehensive set of antibodies to non‐overlapping prion protein epitopes. PLoS One. 2008;3(12):e3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nuvolone M, Hermann M, Sorce S, Russo G, Tiberi C, Schwarz P, et al. Strictly co‐isogenic C57BL/6J‐Prnp‐/‐ mice: a rigorous resource for prion science. J Exp Med. 2016;213(3):313–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gregorian C, Nakashima J, Le Belle J, Ohab J, Kim R, Liu A, et al. Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J Neurosci. 2009;29(6):1874–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15(7):859–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demart S, Fournier JG, Creminon C, Frobert Y, Lamoury F, Marce D, et al. New insight into abnormal prion protein using monoclonal antibodies. Biochem Biophys Res Commun. 1999;265(3):652–7. [DOI] [PubMed] [Google Scholar]
- 20. Weyer A, Schilling K. Developmental and cell type‐specific expression of the neuronal marker NeuN in the murine cerebellum. J Neurosci Res. 2003;73(3):400–9. [DOI] [PubMed] [Google Scholar]
- 21. Doughty ML, De Jager PL, Korsmeyer SJ, Heintz N. Neurodegeneration in Lurcher mice occurs via multiple cell death pathways. J Neurosci. 2000;20(10):3687–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, et al. Prion protein (PrP) with amino‐proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15(6):1255–64. [PMC free article] [PubMed] [Google Scholar]
- 23. Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, et al. Sustained translational repression by eIF2alpha‐P mediates prion neurodegeneration. Nature. 2012;485(7399):507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kugler S, Kilic E, Bahr M. Human synapsin 1 gene promoter confers highly neuron‐specific long‐term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 2003;10(4):337–47. [DOI] [PubMed] [Google Scholar]
- 25. Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53‐null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14(8):994–1004. [PMC free article] [PubMed] [Google Scholar]
- 26. Zhuo L, Theis M, Alvarez‐Maya I, Brenner M, Willecke K, Messing A. hGFAP‐cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31(2):85–94. [DOI] [PubMed] [Google Scholar]
- 27. Raeber AJ, Race RE, Brandner S, Priola SA, Sailer A, Bessen RA, et al. Astrocyte‐specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J. 1997;16(20):6057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brandner S, Klein MA, Frigg R, Pekarik V, Parizek P, Raeber A, et al. Neuroinvasion of prions: insights from mouse models. Exp Physiol. 2000;85(6):705–12. [DOI] [PubMed] [Google Scholar]
- 29. Glatzel M, Aguzzi A. PrP(C) expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol. 2000;81(Pt 11):2813–21. [DOI] [PubMed] [Google Scholar]
- 30. Race RE, Priola SA, Bessen RA, Ernst D, Dockter J, Rall GF, et al. Neuron‐specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron. 1995;15(5):1183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hasel P, Dando O, Jiwaji Z, Baxter P, Todd AC, Heron S, et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat Commun. 2017;8:15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hartmann CA, Martins VR, Lima FR. High levels of cellular prion protein improve astrocyte development. FEBS Lett. 2013;587(2):238–44. [DOI] [PubMed] [Google Scholar]
- 33. Prinz M, Montrasio F, Furukawa H, van der Haar ME, Schwarz P, Rulicke T, et al. Intrinsic resistance of oligodendrocytes to prion infection. J Neurosci. 2004;24(26):5974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diedrich JF, Bendheim PE, Kim YS, Carp RI, Haase AT. Scrapie‐associated prion protein accumulates in astrocytes during scrapie infection. Proc Natl Acad Sci U S A. 1991;88(2):375–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krejciova Z, Alibhai J, Zhao C, Krencik R, Rzechorzek NM, Ullian EM, et al. Human stem cell‐derived astrocytes replicate human prions in a PRNP genotype‐dependent manner. J Exp Med. 2017;214(12):3481–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Victoria GS, Arkhipenko A, Zhu S, Syan S, Zurzolo C. Astrocyte‐to‐neuron intercellular prion transfer is mediated by cell‐cell contact. Sci Rep. 2016;6:20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
FIGURE S1 (A) Polymerase chain reaction (PCR) performed on tail biopsies of seven transgenic founder mice revealed successful integration of CAT‐PrP transgene. For control, we used CAT‐PrP cDNA and DNA extracted from wild‐type C57BL6/J mouse tail biopsies. Actin was simultaneously amplified as a positive control. Samples were migrated on 2% agarose in Tris–EDTA buffer by electrophoresis. Lines 208, 214, and 215 failed to transmit the transgene to F1 generation. (B) Immunohistochemistry of brain sections of SynCre;loxPrP and GFAPCre;loxPrP mice with anti‐PrPC antibody POM2. Left panel: Whole brain section montage. Right panel: Magnified images of cerebellum and hippocampal regions stained with POM2. Images reveal differential PrPC distribution in SynCre;loxPrP and GFAPCre;loxPrP mice
Fig S2
FIGURE S2 (A) Microglia (Iba1) and astrocyte (GFAP) immunostaining of the hippocampal sections of prion‐inoculated SynCre;loxPrP (379 dpi), GFAPCre;loxPrP (627 days), loxPrP (504 dpi), and wild‐type (184 dpi) mice. Expression of GFAP and Iba1 was upregulated in Prnp +/+ mice, whereas SynCre;loxPrP and GFAPCre;loxPrP revealed significantly lower expression levels of the two markers. (B) Biological process enrichment obtained by gene ontology analysis on the upregulated genes in prion‐infected GFAPCre;loxPrP mice revealed that most upregulated genes were involved in pathways associated with hormone response
Fig S3
FIGURE S3 Uncropped western blots. All western blots pertaining to this study are shown here in their original format as generated by the blot imager software. Sequence of staining in case multiple antibodies were used is indicated by numbering. No editing was performed
Table S1
TABLE S1 Table including detailed RNA Sequencing results including the differentially expressed genes, p‐values, FDR values in prion‐infected GFAPCre;loxPrP mice
Table S2
TABLE S2 Table showing differentially expressed genes that are common to prion disease (PrD) and prion‐infected GFAPCre;loxPrP mice (GFAPCre;loxPrP prion)
Data Availability Statement
All original data have been included in the article. Uncropped western blots from the entire manuscript are included in Figure S3. No cropping was performed on any of the microscopy images. Any additional information/data required will be made available by the corresponding author upon reasonable request. No source code was generated in this study.