

Abstract
Chronic liver injury causing liver fibrosis is a major cause of morbidity and mortality worldwide. Targeting the suppression of hepatic stellate cell (HSC) activation is recognized as an effective strategy for the treatment of liver fibrosis. Ellagic acid (EA), a natural polyphenol product isolated from fruits and vegetables, possesses many biological functions. Here, EA exerts its antifibrotic activity by inducing ferroptotic cell death of activated HSCs, which is accompanied by redox-active iron accumulation, lipid peroxidation, and GSH depletion in CCl4 mice and human LX-2 cells. The specific ferroptosis inhibitor ferrostatin-1 prevented EA-induced ferroptotic cell death. Mechanistically, EA impairs the formation of vesicle-associated membrane protein 2 (VAMP2)/syntaxin 4 and VAMP2/synaptosome-associated protein 23 complexes by suppressing VAMP2 expression by enhancing its degradation in a proteasome-dependent pathway. This leads to the impairment of ferroportin (FPN, an iron exporter) translocation and intracellular iron extrusion. Interestingly, VAMP2 overexpression inhibits the role of EA in blocking FPN translocation and increasing intracellular ferritin content (an iron storage marker). In contrast, VAMP2 knockdown shows a synergistic effect on EA-mediated ferroptotic events in both HSCs. Additionally, HSC-specific overexpression of VAMP2 impaired EA-induced HSC ferroptosis in mouse liver fibrosis, and HSC-specific VAMP2 knockdown increased the inhibitory effect of EA on fibrosis. Taken together, our data suggest that the natural product EA exerts its antifibrotic effects by inducing FPN-dependent ferroptosis of HSCs by disrupting the formation of SNARE complexes, and EA will hopefully serve as a prospective compound for liver fibrosis treatment.
Keywords: Ellagic acid, Liver fibrosis, Ferroptosis, Ferroportin translocation, SNARE complexes formation
Graphical abstract
Schematic regulatory mechanism of EA action.EA suppressed FPN-dependent vesicle trafficking to plasma membrane to blocked formation SNARE complexes through induced VAMP2 degradation. This process led to FPN translocation and intercellular iron extrusion impairment. Subsequently, excessive iron produced a large amount of ROS, ultimate caused ferroptosis.

Highlights
-
•
EA exerts its antifibrotic activity by inducing ferroptotic cell death of activated HSCs in CCl4/BDL mice.
-
•
EA blocks the SNARE complexes formation by suppressing VAMP2by enhancing its degradation in a proteasome-dependent pathway.
-
•
Impairment SNARE complexes suppress FPN translocation, which in turn prevents intracellular iron extrusion.
-
•
EA induces ferroptosis of HSCs resulting from intracellular excessive iron accumulation.
1. Introduction
Liver fibrosis is a liver disease with high morbidity and mortality worldwide, and current prevention strategies are only temporarily effective [1]. Recent approaches to alleviate liver fibrosis have focused on reducing activated hepatic stellate cells (HSCs) [2], and it is worth noting that ferroptosis is considered a novel and effective method for eliminating activated HSCs. A previous study indicated that activation of autophagy by regulating beclin1 [3] and inhibition of glutathione peroxidase (GPX4) led to iron deposition and reactive oxygen species (ROS) accumulation [4], which promoted ferroptosis of HSCs and alleviated liver fibrosis [5].
Ferroptosis results from dysregulation of iron homeostasis. Iron deficiency and acquired and inherited iron overload lead to disturbances in iron metabolism [6]. Iron homeostasis is coordinated by a complex series of tightly regulated processes, and iron-regulatory proteins at the plasma membrane are indispensable in regulating iron metabolism [7]. Ferroportin (FPN) is a plasma membrane iron exporter that mediates the transport of iron from the cytoplasm to the extracellular environment [8,9]. Research shows that increased FPN expression inhibits oxidative stress-induced ferroptosis by suppressing the JNK/MTF1 pathway in nucleus pulposus cells and alleviates intervertebral disc degeneration [10], while downregulation of FPN expression induces ferroptosis by increasing iron concentrations and ROS levels in pancreatic cancer cells and attenuates the development of pancreatic cancer [11]. Additionally, deletion of FPN induces cognitive deficits by downregulating GPX4, and decreasing GSH content triggers ferroptosis in hippocampal neurons of APPswe/PS1dE9 mice, whereas restoration of FPN ameliorates ferroptosis and memory impairment, thereby alleviating Alzheimer's progression [12]. However, whether FPN regulating ferroptosis in HSCs is remain unclear.
Recently, evidence has indicated that the transport of FPN from the phagosome to the plasma membrane is accomplished by soluble NSF attachment protein receptor (SNARE)-mediated membrane fusion in macrophages, reducing iron overload by removing FPN from early phagosomes enhances host nutritional immunity to limit microbial growth [13]. SNARE-mediated membrane fusion and protein transport are accomplished by the formation of trans-SNARE complexes between vesicle (v)-SNAREs (vesicle-associated membrane proteins [VAMPs]) and SNARE proteins on target membrane (t)-SNAREs (Syntaxins and the SNAP families) [14]. VAMPs are present in distinct post-Golgi vesicular compartments, which mediate the fusion of vesicles with the plasma membrane, the trans-Golgi network and endosomes, including VAMP1, 2, 3, 4, 5, 7, and 8 [15]. (t)-SNAREs are localized on the plasma membrane and mainly include syntaxin1, syntaxin4, SNAP23 and SNAP25 [16]. (v)-SNAREs and (t)-SNAREs form a trans-SNAREs complex that drives fusion of vesicles with the plasma membrane [17]. It was found that the formation of the syntaxin4/SNAP23/VAMP2 complex promotes the transfer of the glucose transporter GLUT4 from intracellular storage to the plasma membrane of fat and muscle cells, facilitating the uptake of glucose by fat and muscle cells [18]. Disruption of VAMP2/synaxin4/SNAP23 complex formation in insulin-sensitive cell model 3T3-L1 adipocytes results in marked inhibition of insulin-dependent GLUT4 trafficking and glucose uptake [19]. Previous research has revealed that SNAP-23 single-gene deletion reduces activated HSC secretion of alpha smooth muscle actin (α-SMA) and temporarily delays the development of fibrosis in CCl4 mice and the human HSC line hTERT [20]. However, whether the SNARE mechanism mediates FPN trafficking in HSCs remains unclear.
Ellagic acid (EA) is a naturally occurring bioactive and pharmacologically active polyphenolic compound widely found in fruits, vegetables and herbs [21]. EA has displayed antioxidant, anti-inflammatory and antiproliferative properties in some disease models [[22], [23], [24]]. Research demonstrated that EA reduces pancreatic stellate cell activation by suppressing the expression of TGF-β1, which reduces pancreatic fibrosis and alleviates the development of chronic pancreatitis [25]. Moreover, EA is also able to block HSC activation by altering HSC cell morphology and further reducing α-SMA expression, thereby preventing hepatocyte degeneration and preventing further fibrosis and scarring [26,27]. However, the specific mechanism by which EA alleviates liver fibrosis remains unclear. The present study shows that EA attenuates CCl4/BDL-induced liver fibrotic injury in mice. Moreover, EA promotes FPN-dependent ferroptosis of activated HSCs by disrupting the formation of SNARE complexes. Therefore, EA is expected to be an effective treatment for liver fibrosis.
2. Materials and methods
2.1. Animal treatments
Animal experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at Taizhou University in China. The protocol was approved by the Ethics Committee for Medical Laboratory Animals of Taizhou University (The referral number for animal ethics: TZXY-2021-20211004). Male C57BL/6 mice at 5 weeks of age were purchased from Nanjing Junke Biotechnology Corporation, Ltd. (Nanjing, Jiangsu, China). The animals housed at 20 ± 2 °C with 12 h light/dark cycles and a relative humidity of 50 ± 5% under filtered, pathogen-free air, with food and water available ad libitum. Six-week-old male C57BL/6 mouse was intraperitoneally injected with vehicle (olive oil) or 2 ml/kg bodyweight CCl4 (1:9 v/v in olive oil; #1601168, Sigma, St. Louis, MO, USA) twice a week for 4 weeks. VA-Lip-Control siRNA and VA-Lip-VAMP2-siRNA/plasmid (0.75 mg/kg; Shanghai Genechem Co., Ltd, shanghai, China) were administrated intravenously 3 times a week for 2 weeks after CCl4 administrated [28]. For EA (#E2250, Sigma) treatment, mice were randomized and pretreated with EA by gavage at a dose of 50 mg/kg body weight or vehicle for 5 days (drug concentration equilibrium) prior to CCl4 administrated, and then EA treatment continued for 4 weeks. The vehicle-treated control mice received an equal volume of 0.5% carboxymethyl cellulose (CMC, 10 ml/kg bodyweight; #1096611, Sigma). Mice were killed 4 weeks after the final CCl4 injection, and serum were collected for biochemical analysis. A small portion of the liver was removed for histopathological and immunohistochemical studies by fixation with 4% paraformaldehyde (PFA; #158127, Sigma). The remaining liver was cut in pieces and rapidly frozen with liquid nitrogen for extraction of total RNA and hepatic proteins.
2.2. Liver function analysis
Alanine transaminase (ALT, #C009-2-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, PR China), aspartate aminotransferase (AST, #C010-1-1, Nanjing Jiancheng Bioengineering Institute) and lactate dehydrogenase (LDH, #MAK066, Sigma-Aldrich), and three items of liver fiber procollagen type III (PC-III; #ab285003, Abcam, Cambridge, UK), laminin (LN, #ab119572, Abcam) and collagen (#MAK322, Sigma-Aldrich) in serum of all group mice were determined by ELISA kits according to the manufacturer's instructions.
2.3. Morphological and immunostaining analyses
Liver tissues were fixed in 4% PFA for 24 h at room temperature, processed and embedded in paraffin, and sectioned at a thickness of 5 μm. The sections were deparaffinized with xylene, rehydrated in graded ethanol, and stained with hematoxylin and eosin (H&E), Sirius red and Masson. Sirius Red and Masson-stained areas from 10 random fields were quantified with ImageJ software (NIH, Bethesda, MD, USA) according to previous reported [29]. For TUNEL assays, TUNEL kit (#KGA7051, Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) was applied to identify apoptosis cells of fibrotic liver according to the manufacture's instruction.
2.4. Intracellular ferrous iron (Fe2+) measurements
The relative iron concentration in cell lysates was determined with an Iron Assay kit (#ab83366, Abcam), and the experimental method was carried out according to the instructions.
2.5. Total GSH and GSH/GSSG ratio measurements
Using a GSH Assay Kit (#BB-4711; BestBio, shanghai, China) to detect the intracellular total GSH and glutathione disulphide (GSSG) levels according to the manufacturers' protocols. The relative levels were analyzed on the microplate reader (BioRad, Hercules, CA, USA).
2.6. MDA measurement
To detect the lipid peroxidation product MDA content in cells using a Lipid Oxidation Detection Kit (#BB4709; BestBio) according to the manufacturers' protocols. In addition, we used another C11-BODIPY581/591 probe as a lipid peroxide indicator. After different treatments, cells were stained with 5 μM C11-BODIPY581/591 probe (#D3861; Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer's instructions. Observed of C11-BODIPY581/591 fluorescence was performed by Olympus fluorescence microscope and then evaluated with ImageJ (NIH).
2.7. Hydroxyproline analysis
Hydroxyproline content was measured according to the manufacturer's protocol (#MAK008, Sigma) in liver tissue from CCl4 and vehicle mice.
2.8. ROS measurement
2′, 7′-dichlorodihydrofluorescin diacetate (DCFH-DA; #S0033, Beyotime Institute of Biotechnology, Beijing, China) was diluted in serum-free medium at 1:1000 to a final concentration of 10 mM. The cells were seed in a 24-well plate with cover slides at the bottom and cultured to adhere to the wall. Remove the cell culture medium and add 1 ml diluted DCFH-DA for 20 min at 37 °C. Then remove the cover slides and use Olympus fluorescence microscope to observe and take images. Additionally, oxidative stress was also detected by CellROX™ Deep Red Reagent (#C10422, Molecular Probes, Life Technologies, Invitrogen, Eugene, OR, USA). After different treatment, the cells were stained with 5 μM CellROX™ Deep Red Reagent and Hoechst 33342 (#14533, Sigma-Aldrich) by adding the probe to the complete medium and incubating the cells at 37 °C for 30 min. The cells were then washed with PBS and analyzed on a Cellomics ArrayScan Infinity (Thermo Fisher Scientific).
2.9. Cell culture conditions and drug treatment
The human liver stellate cell HSC-LX-2 cells were purchased from the Shanghai Institute of Biological Science (Shanghai, China). The cells were grown in plastic culture flasks under standard conditions (37 °C with 5% CO2 in a completely humidified atmosphere) using dulbecco's modified essential medium (DMEM; #11995065, Gibco, Carlsbad, CA, USA) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin (#15640055, Gibco). Cells detachment was achieved by rinsing with 0.05% trypsin/0.02% EDTA solution. After 24 h of attachment, the cells were cultured in serum-free DMEM for 24 h and then treated with EA at an IC50 concentration or with TGF-β1 (#ab50036, Abcam) at dose of 2 ng/ml for 24 h to assay the hepatic fibrogenesis genes. EA was dissolved in dimethyl sulfoxide (DMSO; #D2650, Sigma) to form a 20 mM stock solution, which was stored at −20 °C and diluted to the desired final concentration in DMEM at the time of use. Ferrostatin-1 (#HY-100579, MCE, Taiwan, ROC), ZVAD-FMK (#HY-16658B, MCE), deferoxamine (#D9533, Sigma), proteasome inhibitor MG132 (# HY-13259, MCE), protein synthesis inhibitor cycloheximide (CHX, #HY-12320, MCE) were dissolved in DMSO at a concentration of 10 mM and stored in a dark-colored bottle at −20 °C. The stock was diluted to the required concentration with DMSO when needed. Cells grown in a medium containing an equivalent amount of DMSO without drugs served as a control.
2.10. Isolation and cultures of primary mouse HSC and hepatocyte
Primary mouse HSC and hepatocyte were isolated from the liver of 6-week-old C57BL/6 male mice. After intubation in the portal vein, the livers were perfused in situ with Ca2+-free Hank's balanced saline solution at 37 °C for 15 min and then perfused with the solution containing 5 mg/ml collagenase IV (#17104019, Gibco) and Ca2+ for 15 min at a flow rate of 10 ml/min. After a few minutes of perfusion, the livers were removed, and the digested hepatic cells were dispersed in cold DMEM-free. The cell suspension generated was filtered through a sterile 70 μm pore size nylon cell strainer (#CLS431751, Sigma) and spun 3 times at 30×g for 4 min. The pellets were suspended in DMEM/F12 medium containing 15% FBS and 1% penicillin/streptomycin supplemented with insulin, transferrin, selenium and dexamethasone for primary hepatocyte culture.
For primary HSCs isolation, after liver removed, DNase enzymes (#D4263, Sigma) were added into the digested hepatic cells to prevent filamentous gelatinous material, and the undigested debris was removed through a filter. The filtrates were centrifuged at 50×g for 5 min at 4 °C. The supernatant was collected following gradient centrifugation with 25% Nycodenz (#D2158, Sigma) to isolate primary HSCs. The number of live cells was determined with a cell viability analyzer (Epics XL flow cytometer; Beckman Coulter, Brea, CA, USA). Cells were washed and seed on 60-mm diameter tissue culture dishes, and cells morphology were assessed using an inverted microscope with an Olympus CX40 system.
2.11. Hepatocytes treatment and cell viability assay
Primary hepatocytes were incubated for 24 h in DMEM/F12/10% FBS medium. After attachment, the cells were cultured in serum-free DMEM/F12 for 24 h and then treated with 20 mM CCl4 (in 0.1% DMSO) for 48 h in the presence or absence of EA for the last 24 h. For assess for cell viability, the 10 μl CCK8 reagents (#C0042, Beyotime Institute of Biotechnology) were added to cells and incubated at 37 °C in 5% CO2 for 4 h, and then the plates were measured at 450 nm using the Tecan Safire Multi-detection Microplate Reader (Morrisville, NC, USA).
2.12. Quantitative PCR (qPCR) analysis
Total RNA was extracted from liver tissue or primary HSCs by homogenization in TRIzol reagent (#15596018, Thermo, Fisher Scientific). cDNA was synthesized by Revert Aid reverse transcriptase (#K1691, Thermo, Fisher Scientific). Realtime PCR was performed on a Bio-Rad CFX384™ real-time PCR detection system using iTaq™ Universal SYBR® Green Super mix (#1725125, Bio-Rad Laboratories, Shanghai, China). The following genes were probed with quantitative PCR using β-actin gene as loading control: α-Sma, Col1a1, Tgf-β1, Mmp-2, Mmp-9, hepcidin, VAMP2, VAMP3 and VAMP8. Primer sequences were as follows: (Human) β-actin, 5ʹ-CTCTTCCAGCCTTCCTTCCTG-3ʹ and 5ʹ-CAGCACTGTGTTGGCGTACAG; α-Sma, 5ʹ-GACTCCGACATCTCTAGCTCTT-3ʹ and 5ʹ-TGCCTTTTCCTGCACATCTGTC; Col1a1, 5ʹ-AACATGACCAAAAACCAAAAGTG-3ʹ and 5ʹ-CATTGTTTCCTGTGTCTTCTGG; Tgf-β1, 5ʹ-GGCAGTGGTTGAGCCGTGGA-3ʹ and 5ʹ-TGTTGGACAGCTGCTCCACCT; Mmp-2, 5ʹ-GTATTTGATGGCATCGCTCA-3ʹ and 5ʹ-CATTCCCTGCAAAGAACACA; Mmp-9, 5ʹ-CACTGTCCACCCCTCAGAGC-3ʹ and 5ʹ-GCCACTTGTCGGCGATAAGG; hepcidin, 5ʹ-AGCTGGATGCCCATGTTC-3ʹ and 5ʹ-CAGCACATCCCACACTTTGA; VAMP2, 5ʹ-ATGAGGGTGAACGTGGACAA-3ʹ and 5ʹ-CGGGGATTTAAGTGCTGAAGTA, VAMP3, 5ʹ-GAAAGAAAACAGGCAAGGAGG-3ʹ and 5ʹ-CATTTCAGCGATGTTAAGGGA, VAMP8, 5ʹ-TGGAACATCTCCGCAACAAG-3ʹ and 5ʹ-GTGGCAAAGAGCACAATGAAG; (Mouse) β-actin, 5′-TGTTACCAACTGGGACGA‘CA-3′ and 5′-GGGGTGTTGAAGGTCTCAAA-3′; α-Sma, 5′-GCGTGGCTATTCCTTCGTTA C-3′ and 5′-CATAGTGGTGCCCCCTGATAG -3′; Col1a1, 5′-ATTCCAGTTCGAGTATGGCGG -3′ and 5′-GTTGCTTGTCTGTTTCCGGGT -3′; Tgf-β1, 5ʹ-GCCCTGGATACCAACTATTGC-3ʹ and 5ʹ-GCAGGAGCGCACAATCATGTT-3ʹ; Mmp-2, 5ʹ-TGAAGGTCGGTGTGAACGGA-3ʹ and 5ʹ-CATGTAGCCATGAGGTCCACCAC; Mmp-9, 5ʹ-TTGACAGCGACAAGAAGTGG-3ʹ and 5ʹ-GGCACAGTAGTGGCCGTAG; hepcidin, 5ʹ-CTCCTGCTTCTCCTCCTTGC-3ʹ and 5ʹ-GCAATGTCTGCCCTGCTTTC-3ʹ; VAMP2, 5ʹ-CTGCACCTCCTCCAAACCTTAC-3ʹ and 5ʹ-CCACCAGTATTTGCGCTTGAG-3ʹ; VAMP3, 5ʹ-GGCAGTAATCGAAGACTCCAGC-3ʹ and 5ʹ-GACACTGATCCCTATCGCCCA-3ʹ; VAMP8, 5ʹ-CCGAGTTAGGAACCTGCAGAGT-3ʹ and 5ʹ-ACCTTCTGGGACGTTGTCTTGA-3ʹ. β-actin was used as a reference gene. Results are reported as normalized and calibrated ratios calculated using the 2−ΔΔCT method.
2.13. Western blot analysis and immunoprecipitation
The liver tissue of mice or primary HSC was extracted using RIPA Lysis and Extraction Buffer (#89901; Thermo Fisher Scientific) and protein concentration was determined by BCA. Samples containing equal amounts of protein (15–20 μg) were separated by 8–12% SDS-PAGE and transferred to PVDF membranes. Membranes were blocked with 5% BSA for 2 h at room temperature and incubated overnight at 4 °C with primary antibodies against Desmin (#5332, CST; 1:1000), Col1a1(#72026, CST; 1:2000), glutathione peroxidase 4 (GPX4; #52455, CST; 1:1000), transferrin (TFN; #ab278498, Abcam; 1:1000), divalent metal ion transporter (DMT1; #15083, CST; 1:1000), ferroportin (FPN; #NBP1-21502, Novus, Minneapolis, MN, USA; 1:1000), ferritin (FTH1; #4393, CST; 1:1000), hepcidin (#MAB9505, R&D; 1:1000), syntaxin 4 (sc-101301, Santa Cruz Biotechnology, CA, USA; 1:1000), synaptosome-associated protein of 23 kDa (SNAP23; sc-166244, Santa Cruz Biotechnology; 1:1000), VAMP2 ((#13508, CST; 1:1000), VAMP3 (#13640, CST; 1:1000), VAMP8 (#13060, CST; 1:1000), α-SMA (#ab7817, Abcam; 1:1000), TGF-β (#ab215715, Abcam; 1:1000), and GAPDH (#10494-1-AP, Proteintech; 1:2000). Secondary horseradish peroxidaseconjugated anti-rabbit (#ab6721, Abcam; 1:2000) or anti-mouse antibody (#ab6728, Abcam; 1:2000) was applied. Enhanced chemiluminescence (#1705060, Bio-Rad, Hercules, CA, USA) was used to visualize bands, which were quantified by Image J 5.0 software.
For immunoprecipitation, cells were lysed with RIPA buffer for 30 min on ice. Then cell lysates were adjusted to equal amounts of protein (500 μg) and immunoprecipitated with VAMP2, syntaxin 4, SNAP23 antibodies at 4 °C overnight. Rabbit IgG control antibody (#3423, CST; 1:50) was used as a negative control. Immunoprecipitates were pulled down with Protein PLUS A/G Agarose (#sc-2003, Santa Cruz Biotechnology) followed by western blot analysis.
2.14. Plasmid transfection and adenovirus infection
Cells were infected with recombinant adenovirus expressing a Flag-tagged full-length mouse VAMP2 gene (AdVAMP2) or a shRNA targeting VAMP2 (AdshVAMP2; Shanghai GeneChem Co., Ltd) (MOI 20-50) for 24 h. The Lacz adenovirus (AdLacz) was used as negative control. Additionally, in EA regulates FPN translocation experiment, FPN shRNA, Control shRNA, FPN plasmid and Control vector were ordered from Shanghai GeneChem Co., Ltd. Subsequent Western blot was performed to analyze transfection efficiency.
2.15. Statistical analyses
All experiments and analyses were conducted with the experimenter blinded to the drug treatment and were performed in triplicate. All data in this study were expressed as mean ± standard error of the mean (SEM). Significant differences were determined by t-test or one-way analysis of variance (ANOVA). All statistical analyses were performed using GraphPad Prism software (Version 6.0; GraphPad Software, San Diego, CA, USA). P < 0.05 was considered significant throughout this study.
3. Results
3.1. EA ameliorates CCl4-induced hepatic fibrotic injury in mice
Fig. 1A shows the chemical structure of EA. In our preliminary study, EA was used at doses of 25, 50, and 100 mg/kg body weight for daily oral administration in mice for 8 weeks. No changes in sizes, gross anatomical features or body weights were observed. Moreover, hematoxylin and eosin (H&E) staining showed normal liver morphology in mice treated with EA at three doses for 8 weeks (Fig. 1B), suggesting that EA had little toxicity in normal mice. As such, the dose of 50 mg/kg body weight EA was used for our studies, and this dose was considered one tenth of the proposed oral LD50 mg/kg body weight. EA, as a natural polyphenolic compound, has a hepatoprotective effect [30]. We evaluated the impact of EA on hepatic injury in an animal model induced by CCl4 with intraperitoneal injection. Macroscopic examination showed that pathological morphological changes occurred in the livers of CCl4 mice compared with the control livers, but EA treatment significantly improved the pathological changes in the livers (Fig. 1C). The liver index (liver weight/body weight) was positively associated with the degree of liver damage. As expected, EA treatment reduced the liver index compared to CCl4 treatment alone (Fig. 1D). H&E staining was used to perform histological pathological examinations in mouse livers. EA treatment markedly improved the CCl4-induced disordered arrangement of hepatocytes and decreased the false leaves of the livers, fiber connective tissue and inflammatory cell infiltration (Fig. 1C). In addition, in blood biochemistry analyses, alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) activities were all significantly diminished by EA treatment, indicative of improvement of liver function in CCl4 mice (Fig. 1E). Collectively, EA exerted a hepatoprotective effect.
Fig. 1.

EA treatment attenuated hepatic injury and fibrosis induced by CCl4 in mice
(A) The chemical structure of EA. (B) Body weights of mice receiving either control treatment or EA treatment with EA at doses of 25, 50 and 100 mg/kg body weight for 8 weeks (n = 40), and liver sections were subjected to H&E staining (n = 10). (C) Representative photographs show the pathological changes in the livers observed by macroscopic examination; liver sections were stained with H&E, Masson and Sirius Red. The liver fibrosis stage was scored in a double-blind method. The Masson and Sirius red staining areas of the mice and the liver/body weight ratio (D) were calculated. n = 10. (E) Liver function was assessed by ALT, AST and LDH activities, and (F) three items of liver fiber (PC-III, LN and collagen) in the serum of mice were measured by commercial kits or ELISA kits. n = 10. (G) Western blot and (H) qPCR assays were used to detect the protein and mRNA expression levels of α-SMA, Col1a1, TGF-β1 and desmin in isolated primary HSCs from liver tissue and quantify them. n = 5. (I) Collagen content was measured by biochemical determination of hydroxyproline (per mg of liver) in the livers of mice. n = 10. (J) Immunohistochemical staining of α-SMA and TGF-β1 was determined in liver sections. Brown arrow indicates positive area. n = 5. Data are expressed as the mean ± SEM of three independent experiments; *P < 0.05, **P < 0.01 between the indicated groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Then, the role of EA in liver fibrosis was examined. The levels of serum liver fibrosis indicators, including PC-III, LN, and collagen, were detected using ELISA kits. EA treatment greatly decreased these indices of liver fibrosis in CCl4 mice (Fig. 1F). The excessive accumulation of ECM is the main cause of hepatic fibrogenesis [31]. Sirius Red and Masson's trichrome staining revealed that EA treatment reduced the CCl4-induced degree of collagen deposition in mouse livers (Fig. 1C). Consistently, EA treatment notably attenuated increases in the levels of α-SMA, collagen type 1 (Col1a1) and TGF-β1, the most abundant ECM proteins in fibrotic liver tissue, and HSC activation-associated proteins, including desmin (Fig. 1G). Similarly, α-Sma, Col1a1 and Tgf-β1 mRNA levels were decreased in CCl4 mice treated with EA (Fig. 1H). EA treatment also prevented the increase in hydroxyproline content (Fig. 1I). We further determined the regulatory effects of EA on key proteins in the fibrotic pathway. Immunostaining of mouse livers showed that the protein levels of α-SMA and TGF-β1 were significantly decreased by EA treatment (Fig. 1J).
To further assess the antifibrotic effect of EA, we generated a bile duct ligation (BDL) mouse model, another classical murine liver fibrosis model. EA treatment mitigated the development of liver fibrosis in the BDL mice at 4 weeks, as evidenced by histopathological improvement and the changes in Sirius Red staining (Fig. 2A). Repression of α-SMA and TGF-β1 gene expression was verified by the results of immunohistochemistry, western blot and qPCR assays (Fig. 2A–C). Similarly, hepatic hydroxyproline content was lessened by EA treatment (Fig. 2D). The above results demonstrate that EA has the capability to inhibit liver fibrosis development and progression.
Fig. 2.

EA treatment suppresses BDL-induced liver fibrosis and inhibits the activation of HSCs
(A) Representative images of liver sections from control and BDL mice treated with or without EA are shown. Liver sections (4 μm) were stained with H&E and Sirius Red for histopathological study and IHC for α-SMA or TGF-β1 were performed on the liver sections of mice. The Sirius Red, α-SMA and TGF-β1 staining areas of the mice were calculated. n = 10. (B) Western blot and (C) qPCR assays were used to detect the protein and mRNA expression levels of α-SMA and TGF-β1 in isolated primary HSCs from liver tissue and quantify them. n = 5. (D) Liver hydroxyproline levels were determined using a hydroxyproline assay kit in mice. n = 10. (E) Western blot and (F) qPCR assays were used to detect the protein and mRNA expression levels of α-SMA, Col1a1 and desmin in primary HSCs and LX-2 cells treated with TGF-β1 or/with EA and quantify them. n = 5. (G) qPCR assays were used to detect the mRNA expression levels of Mmp-2 and Mmp-9 in cells as described above. n = 5. (H) The inhibition ratio of cell growth was calculated in primary HSCs and LX-2 cells treated with EA and the apoptosis inhibitor ZVAD-FMK for 24 h n = 5. Data are expressed as the mean ± SEM of three independent experiments; *P < 0.05, **P < 0.01 between the indicated groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. EA treatment suppresses the activation of HSCs in vitro
On the basis of the decrease in hepatic fibrosis induced by treatment with EA in CCl4 mice, we next assessed whether the same antifibrotic effects could be seen in vitro. It is well known that the activation of HSCs plays an important role in fibrogenesis. TGF-β1 is the key profibrogenic cytokine in HSC activation and liver fibrogenesis [32]. In the present study, TGF-β1 (2 ng/ml) was used to stimulate HSC activation. LX-2, a human HSC line, and mouse primary HSCs isolated from wild-type C57BL/6 mouse livers were incubated with EA at doses of 40 or 45 μM (LX-2 cells: IC50 ~ 37 μM, primary HSCs: IC50 ~ 44 μM) and TGF-β1 for 24 h. As expected, EA prevented LX-2 cells and primary HSCs from TGF-β1-induced α-SMA, col1a1 and desmin expression (Fig. 2E). The same results could be seen in qPCR analysis (Fig. 2F). Moreover, EA treatment significantly lessened the levels of TGF-β1-induced target gene transcripts, including matrix metalloproteinase (MMP)-2 and MMP-9 (Fig. 2G). Interestingly, the treatment of activated HSCs with EA and the apoptosis inhibitor ZVAD-FMK (10 μM) for 24 h showed that EA can induce activated HSC death but not through inducing cell apoptosis (Fig. 2H), indicating that another factor is involved in the suppression of HSC activation. These results confirmed the inhibitory effect of EA on fibrosis in mice.
3.3. EA treatment induces ferroptosis in activated HSCs
We then investigated the mechanism of fibrosis inhibition by EA treatment. The effect of EA on global gene expression patterns of CCl4 mice was assessed at the transcriptome level using RNA sequencing (RNA-seq) experiments. Primary HSCs were isolated from CCl4 mice treated with or without EA, and total RNA was extracted for sequencing (RNA-Seq). Using >1.5-fold change for upregulation and <0.5-fold change for downregulation as cutoffs, 419 mRNAs were upregulated and 286 mRNAs were downregulated in CCl4 mice treated with EA, as shown in the heatmap (Fig. 3A) and volcano plot (Fig. 3B). As expected, some established fibrosis-related genes, such as COL1A1 (0.71-fold), MMP9 (0.82-fold), FN1 (0.54-fold), TIMP1 (0.62-fold) and MMP2 (0.65-fold), were found among these downregulated genes. Notably, several recognized ferroptosis-related genes, such as PTGS2 (3.77-fold), GPX4 (2.51-fold) and ferritin1 (FTH1) (4.73-fold), were found among these upregulated genes. Gene set enrichment analysis (GSEA) was further performed to determine the effects of EA-induced transcriptomic changes on biological functions and pathways. Bioinformatics analysis predicted ferroptosis regulators whose activities were positively related to EA treatment in CCl4 mice (Fig. 3C). The data indicate that ferroptosis is induced in activated HSCs.
Fig. 3.

EA treatment suppresses fibrosis via ferroptosis induced by the activated HSC pathway. Primary HSCs were further isolated from CCl4 mice treated with or without EA, and total RNA was extracted for RNA-Seq.
(A) Microarray heatmap illustrating genes with at least a 2-fold change in transcript levels between isolated primary HSCs treated with EA and control cells. Bright blue, underexpression; gray, no change; bright red, overexpression (CCl4, n = 3; CCl4+EA, n = 3). (B) Volcano plot demonstrates clustering of isolated primary HSCs. Hierarchical cluster analysis of significantly differentially expressed mRNAs: bright blue, underexpression; gray, no change; bright red, overexpression (CCl4, n = 3; CCl4+EA, n = 3). (C) Gene set enrichment analysis (GSEA) indicated a significant change in ferroptosis signaling induced by EA treatment. NES, normalized enrichment score. (D) The levels of total GSH, GSH/GSSG ratio, (E) intracellular ferrous iron, (F) MDA and (G) the fluorescence intensity of C11-BODIPY581/591 were assayed in isolated primary HSCs from control and CCl4 mice treated with or without EA (n = 8–10). (H) Representative western blot and summary data of PTGS2 and GPX4 in control and CCl4 mice. Data were normalized to GAPDH. n = 5. (I) Representative images of PTGS2 (green) and α-SMA (red) costained liver sections from control and CCl4 mice collected at 4 weeks. Cell nuclei were stained with DAPI (blue). (J) The morphological mitochondrial changes in control and CCl4 mice treated with or without EA were determined by transmission electron microscopy analysis. Representative photographs are shown. n = 3. The black arrow indicates mitochondria. Data are expressed as the mean ± SEM of three independent experiments; *P < 0.05, **P < 0.01 between the indicated groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
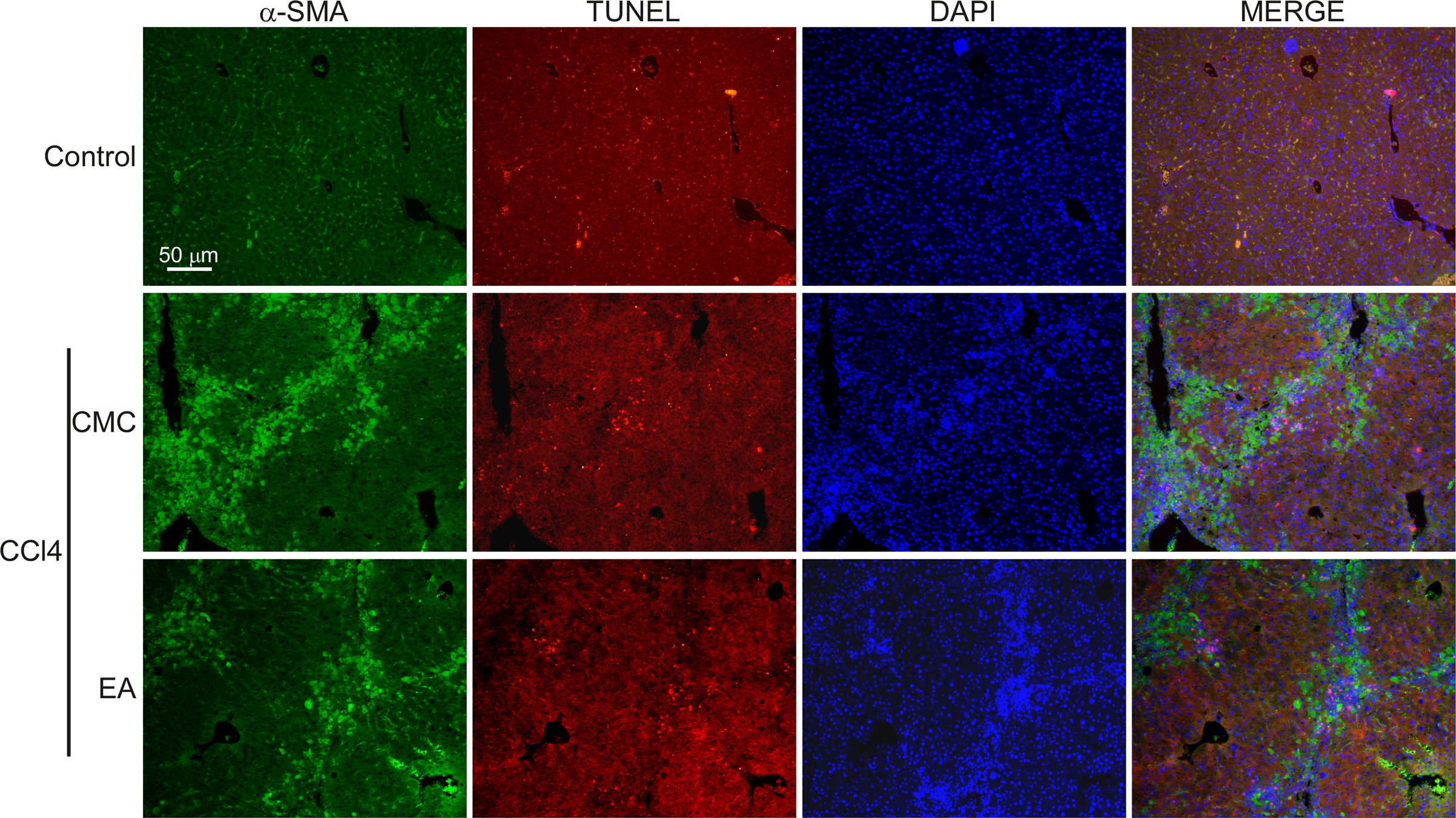
Reports have shown that targeting HSCs activated by ferroptosis can inhibit hepatic fibrosis [33], and whether EA treatment can induce ferroptosis in activated HSCs was investigated. Ferroptosis is characterized by redox-active iron accumulation, glutathione (GSH) depletion, and lipid peroxidation. Thus, we examined the levels of intracellular redox-active iron, total GSH, GSH/GSSG ratio, malondialdehyde (MDA) and the fluorescence intensity of C11-BODIPY581/591 in isolated primary HSCs from control and CCl4 mice. Consistent with the in vitro results, EA treatment induced ferroptotic events, including GSH depletion (Fig. 3D), redox-active iron accumulation (Fig. 3E), and lipid peroxidation (Fig. 3F–G), in CCl4 mice. Moreover, we also detected the protein levels of the ferroptosis markers prostaglandin-endoperoxide synthase 2 (PTGS2) and glutathione peroxidase 4 (GPX4, a central regulator of ferroptosis) by western blotting. The results showed that EA treatment significantly increased the expression of PTGS2 while decreasing GPX4 expression in CCl4 mouse livers (Fig. 3H). In addition, immunostaining for PTGS2 and the activated HSC marker α-SMA showed that EA treatment induced the expression of PTGS2 in activated HSCs (Fig. 3I). Moreover, electron microscopy observation showed that CCl4-treated primary HSCs exhibited more swollen mitochondria and a reduced number of cristae with a more lamellar phenotype than control HSCs. However, these changes were significantly alleviated by EA treatment (Fig. 3J). Additionally, we also examined if EA treatment reduced HSCs activation was through induced apoptosis of HSCs. We preformed co-immunostaining for apoptosis and activated HSCs using TUNEL and α-SMA antibody in CCl4 mice liver. Results showed that EA treatment greatly inhibited activation of HSCs induced by CCl4. Moreover, CCl4 greatly increased the number of TUNEL-positive cells compared with control mice, but EA treatment did not significantly change the increase TUNEL-positive cells (Fig. S1), suggesting that EA is not involved in inducing apoptosis of HSCs during fibrosis.
Next, we further observed the impact of EA on ferroptosis in HSCs. The results showed that EA-mediated growth inhibition in isolated primary HSCs and LX2 cells by TGF-β1 stimulants was prevented by the ferroptosis inhibitor ferrostatin-1 (1 μM) but not the apoptosis inhibitor ZVAD-FMK (Fig. 4A). Moreover, propidium iodide (PI) staining showed that EA treatment resulted in a reduction in live cells and an increase in dead cells compared with the control cells, whereas ferrostain-1, but not ZVAD-FMK, completely reversed the effect of EA on cell death (Fig. 4B). Moreover, we examined the levels of intracellular redox-active iron, total GSH, GSH/GSSG ratio, MDA and C11-BODIPY581/591 in HSCs and LX-2 cells. As expected, EA treatment induced ferroptotic events, including GSH depletion (Fig. 4C), redox-active iron accumulation (Fig. 4D), and lipid peroxidation (Fig. 4E–F), in HSCs. Ferrostain-1 completely blocked GSH depletion, MDA production and redox-active iron accumulation in the induction of ferroptosis (Fig. 4C–F). Additionally, the cytotoxic effect of EA on HSCs was also assessed. HSCs were incubated with EA (20, 40, 80 μM in primary HSCs; 22.5, 45, 90 μM in LX-2 cells) without TGF-β1 for 24 and 48 h. The results showed that the growth of primary HSCs and LX-2 cells was not inhibited by EA treatment, suggesting that EA was safe and nontoxic for HSCs, not activated HSCs (Fig. 4G).
Fig. 4.

EA treatment induces ferroptosis in activated HSCs.
(A) Cell viability of primary HSCs, LX-2 cells and primary hepatocytes was assayed. n = 5. (B) Primary HSCs and LX-2 cells were treated with EA (LX-2 cells: 40 μM; primary HSCs: 45 μM), ferrostatin-1 (1 μM) and ZVAD-FMK (10 μM), and cells were stained with PI (red fluorescence) to examine the dead cells. (C) Total GSH, GSH/GSSG ratio, (D) intracellular ferrous iron, (E) MDA and (F) C11-BODIPY581/591 levels were assayed in primary HSCs, LX-2 cells and primary hepatocytes treated with TGF-β1 and/or EA. n = 5. (G) The cytotoxicity of EA was observed in primary HSCs and LX-2 cells using a cell viability assay. Primary HSCs and LX-2 cells were treated with EA (20, 40, 80 μM in primary HSCs; 22.5, 45, 90 μM in LX-2 cells) for 24 and 48 h n = 5. Data are expressed as the mean ± SEM of three independent experiments; *P < 0.05, **P < 0.01 between the indicated groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Additionally, we also examined whether EA had a similar effect on hepatocytes. Mouse primary hepatocytes were isolated from C57BL/6 mouse livers and incubated with EA (IC50 ~ 64 μM) at doses of 32.5, 65 and 130 μM or/and TGF-β1 for 24 and 48 h. However, the cell viability analysis showed that EA treatment could not suppress the growth and viability of the hepatocytes. Furthermore, to assess whether EA treatment affect hepatocyte growth in injured condition, in vitro experiments were first conducted on primary hepatocytes that had been exposed to CCl4 (20 mM) or/and EA at doses of 32.5, 65 and 130 μM for 48 h. Whereas hepatocyte growth was inhibited by CCl4, this outcome was not reversed in cells to which EA at any dose was added for the final 24 h (Fig. S2). Moreover, treatment with EA did not trigger GSH depletion, redox-active iron overload or MDA production in primary hepatocytes (Fig. 4C–F) or HepG2 cells (data not shown), suggesting that EA has a selective effect on HSC growth inhibition and ferroptosis induction.
These data suggest that EA treatment activates HSCs to undergo ferroptosis.
3.4. EA prevents iron extrusion and FPN translocation in HSCs
Iron overload can trigger ferroptosis in HSCs. To unveil the mechanism behind EA-enhanced iron accumulation in activated HSCs, we measured iron regulatory proteins, including transferrin (TFN), divalent metal ion transporter (DMT1) and FPN, in activated HSCs. Consistent with previous studies, reduced expression of TFN and DMT1 and increased FPN expression were seen in primary HSCs isolated from CCl4 mouse livers; however, although there was no significant alteration in the total protein level of FPN by EA treatment, a marked decrease was seen in plasma membrane FPN levels in isolated HSCs, and no change was observed in TFN and DMT1 levels in either the total or plasma membrane (Fig. 5A). Similarly, EA treatment also reduced the FPN level of the plasma membrane in TGF-β1-treated primary HSCs and LX-2 cells (Fig. 5B). These data indicate that EA may regulate FPN trafficking.
Fig. 5.

EA prevents iron extrusion and FPN translocation in HSCs
(A) Representative western blots and summary data showing the expression of the iron transport-related proteins DMT1, TFN and FPN in total protein and membrane protein extracts of isolated primary HSCs from control and CCl4 mice treated with or without EA. Data were normalized to GAPDH. n = 5. (B) Representative western blots and summary data showing the expression of the iron transport proteins DMT1, TFN and FPN in total protein and membrane protein extracts of primary HSCs and LX-2 cells treated with or without TGF-β1 and/or EA. Data were normalized to GAPDH. n = 5. (C) Hepatic hepcidin mRNA and protein levels in primary HSCs of control and CCl4 mice treated with or without EA for 4 weeks. Data were normalized to β-actin or GAPDH. n = 3–5. (D) Representative western blots and summary data showing the expression of FTH1 in isolated primary HSCs from control and CCl4 mice treated with or without EA. Data were normalized to GAPDH. n = 5. (E) Representative image of ROS probe labeling with DCFH-DA, (F) ROS content analysis and (G) CellROX™ Deep Red staining in primary HSCs and LX-2 cells treated with TGF-β1 or/and EA and an iron chelating agent, DFO. Cell nuclei were stained with Hoechst 33342 (blue). (H) Quantitation and statistical analysis of oxidative stress based on CellROX™ Deep Red Reagent staining. Data are expressed as the mean ± SEM of three independent experiments; *P < 0.05, **P < 0.01 between the indicated groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Hepcidin, the master regulator of iron homeostasis, mediates iron through the internalization and degradation of FPN [34]. We next considered the possibility that EA treatment decreased the expression of FPN on the plasma membrane through a hepcidin-dependent pathway. To address this possibility, hepcidin expression levels were detected. In CCl4 mice, hepcidin protein and mRNA levels were both decreased in primary HSCs. However, EA treatment did not alter the level of hepcidin expression (Fig. 5C), and a similar result was observed in TGF-β1-treated HSCs, suggesting that EA regulates the levels of FPN on the plasma membrane independent of the ability of FPN to bind hepcidin.
We then determined whether the EA-induced reduction in FPN expression in the plasma membrane was responsible for ferroptosis in activated HSCs. Since FPN acts as an iron exporter, if reduced in the plasma membrane could lead to iron accumulation in the cytoplasm, the iron content in HSCs was assessed. EA treatment greatly increased the intracellular ferritin FTH1 content (a main iron storage protein) in activated HSCs isolated from CCl4 mice (Fig. 5D). Excess iron produces a large amount of ROS, and intracellular ROS are the ultimate cause of ferroptosis. We examined the level of ROS in activated HSCs. ROS probe DCFH-DA staining showed that the fluorescence intensity was significantly enhanced by EA treatment in TGF-β1-treated primary HSCs and LX-2 cells (Fig. 5E). Additionally, we used CellROX™ Deep Red Reagent as oxidative stress indicator to detect ROS level. The results we observed were consistent with DCFH-DA staining (Fig. 5G). However, deferoxamine (DFO), an iron chelating agent, rescued the potentiated effect of EA treatment on ROS levels (Fig. 5E–G). Similar results were observed in the ROS content analysis (Fig. 5F, H).
Next, to directly examine if EA-induced ferroptosis is associated with FPN in activated HSCs, primary HSCs and LX-2 cells were pretreated with FPN shRNA or FPN plasmid, followed TGF-β1 or/and EA treatment. Western blot analysis confirmed that FPN shRNA significantly decreased FPN protein level in primary HSCs and LX-2 cells whereas FPN plasmid markedly increased it (Fig. S3A). Then we examined the levels of intracellular redox-active iron, total GSH, GSH/GSSG ratio, MDA and C11-BODIPY581/591by assay kits and fluorescence probe. As expected, FPN knockdown greatly increased in GSH depletion, redox-active iron accumulation, lipid peroxidation in TGF-β1-treated HSCs. The similar results were seen in EA treatment combined with FPN shRNA. In contrast, FPN plasmid suppressed ferroptosis events (Fig. S3B). Interestingly, the effect of FPN plasmid on ferroptosis events was blocked by EA treatment (Fig. S3B). Additionally, intracellular iron content was also assessed the in HSCs treated with FPN shRNA or FPN plasmid. Western blot analysis showed that knockdown of FPN significant increased the FTH1 level compared to control shRNA in activated HSCs (Fig. S3C). EA treatment did not alter the effect of FPN shRNA on increased of FTH1 level. However, EA treatment greatly abolished the effect of FPN plasmid on the FTH1 expression (Fig. S3C), suggesting that EA inducing ferroptosis is FPN dependent.
These data suggest that EA treatment regulates FPN trafficking and iron transport in HSCs.
3.5. VAMP2 is involved in EA-regulated FPN translocation
The membrane fusion machinery is involved in the exocytosis of activated HSCs and integral membrane protein FPN translocation in macrophages [13]. To determine whether SNAREs are involved in EA-regulated FPN translocation, we first examined the vesicle (v)-SNAREs (vesicle-associated membrane proteins, VAMPs) and target membrane (t)-SNAREs (syntaxin4, synaptosome-associated protein of 23 kDa [SNAP23] and SNAP25) levels. We found that Vamp2, 3, 8, syntaxin4 and Snap23 mRNA levels were expressed in primary HSCs, but we did not find SNAP25 expression. Similar results were observed in LX-2 cells (Fig. 6A). TGF-β1 stimulation greatly increased the expression of VAMP2 and VAMP3 in activated HSCs. However, EA treatment suppressed only the increase in VAMP2 expression but not the increase in VAMP3 expression at the protein level (Fig. 6B). Notably, EA treatment did not change the TGF-β1-induced increase in Vamp2 and Vamp3 mRNA levels (Fig. 6A). In addition, EA treatment did not affect the expression of VAMP8, syntaxin4 or SNAP23 at the mRNA and protein levels (Fig. 6A, B). Additionally, the inhibitory effect of EA on VAMP2 protein expression was reversed by the proteasome inhibitor MG-132 but not the lysosome blocker chloroquine (Fig. 6C). By using the protein synthesis inhibitor cycloheximide (CHX), the degradation of VAMP2 was markedly enhanced by EA treatment (Fig. 6D), suggesting that the attenuation of VAMP2 is a posttranslational modification and that EA inhibits VAMP2 expression by enhancing VAMP2 degradation through a proteasome-dependent pathway. We further examined the role of EA in SNARE assembly in activated HSCs. EA treatment markedly limited the formation of VAMP2/syntaxin4 and VAMP2/SNAP23 complexes, as shown by the immunoprecipitation assay (Fig. 6E), suggesting that degradation of VAMP2 by EA contributes to the impairment of SNARE formation.
Fig. 6.

VAMP2 is involved in EA-regulated FPN translocation.
(A) mRNA expression levels of Vamp (Vamp2, Vamp8 and Vamp8), SNAP23 and syntaxin 4 in primary HSCs from C57BL/6 mice and LX-2 cells. n = 4. (B) Protein expression of VAMP2, VAMP3, VAMP8, syntaxin 4, and SNAP23 in primary HSCs and LX-2 cells treated with TGF-β1 and/or EA. n = 5. (C) VAMP2 expression in primary HSCs and LX2 cells pretreated with MG-132 (10 μM) or CQ (1 μM) for 30 min followed by TGF-β1 and/or EA treatment. n = 5. (D) Following TGF-β1 treatment, VAMP2 expression was analyzed in primary HSCs and LX2 cells pretreated with CHX (100 μg/mL) for the indicated durations. n = 5. (E) Immunoblotting analysis of syntaxin 4 and SNAP23 in VAMP2 immunoprecipitates from primary HSCs and LX2 cells treated with TGF-β1 and/or EA. n = 4. (F) Primary HSCs and LX-2 cells were infected with different concentrations of AdshVAMP2, AdVAMP2 or control adenovirus (AdLacz) for 24 h. VAMP2 protein expression was examined. n = 5. (G) After HSCs were infected with AdshVAMP2 or AdVAMP2 followed by TGF-β1 treatment, the cells were incubated in EA for 24 h, and then the distribution of FPN and (H) FTH1 expression in those cells was examined by western blotting. n = 5. Data were normalized to GAPDH and are expressed as the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 between the indicated groups.
To clarify the pathophysiological relevance of VAMP2 in EA-regulated FPN translocation, HSCs were infected with VAMP2 shRNA adenovirus (AdshVAMP2) or VAMP2 adenovirus (AdVAMP2) and their respective control adenovirus (Fig. 6F). EA and VAMP2 shRNA markedly decreased the plasma membrane FPN level in primary HSCs and LX2 cells following TGF-β1 stimulation, whereas the VAMP2 plasmid abrogated the inhibitory effect of EA treatment on the plasma membrane FPN level (Fig. 6G). Since FPN acts as an iron exporter, the effect of iron content on SNARE assembly in HSCs was assessed. EA and VAMP2 shRNA treatment increased the intracellular ferritin content (an iron storage marker) in primary HSCs and LX2 cells, while the VAMP2 plasmid had the opposite effect (Fig. 6H), reflecting the ability of EA to regulate intracellular iron. Collectively, these findings indicate that EA treatment blocks FPN translocation, leading to ferroptosis of HSCs by promoting VAMP2 degradation and the subsequent impairment of SNARE formation.
3.6. VAMP2 is indispensable due to the role of EA on ferroptosis induced in liver fibrosis
To investigate whether VAMP2 is required for the regulatory effect of EA on ferroptosis, we examined ferroptosis-associated indicators in TGF-β1-treated primary HSCs and LX2 cells treated with AdshVAMP2 or AdVAMP2. VAMP2 knockdown markedly potentiated the EA-induced increase in redox-active iron accumulation, lipid peroxidation, GSH depletion, and ROS content in TGF-β1-treated HSCs, while the enhanced effect of EA on ferroptosis was blocked by AdVAMP2 (Fig. 7A–D). Additionally, knockdown of VAMP2 further enhanced the reduction in EA-induced α-Sma and col1a1 expression and Mmp-2 and Mmp-9 mRNA levels. However, VAMP2 upregulation offset the effects of EA treatment on the inhibition of collagen deposition (Fig. 7E).
Fig. 7.

VAMP2 is indispensable for the role of EA on ferroptosis induced in liver fibrosis.
(A) Intracellular ferrous iron, (B) MDA, the relative fluorescence intensity of C11-BODIPY581/591, (C) Total GSH, GSH/GSSG ratio and (D) ROS content with DCFH-DA and CellROX™ Deep Red staining were assayed in AdshVAMP2- and/or AdVAMP2-infected primary HSCs and LX-2 cells treated with TGF-β1 and/or EA. n = 5. (E) The expression of the fibrosis-related genes α-Sma, Col1a1, Mmp2 and Mmp9 were analyzed in AdshVAMP2- and/or AdVAMP2-infected primary HSCs and LX-2 cells treated with TGF-β1 and/or EA. n = 5. (F) Representative images of liver sections from control and CCl4 mice injected with VA-Lip-VAMP-siRNA/plasmid are shown. Thin sections (4 μm) were stained with H&E, Sirius Red, and Masson for histopathological study. The liver fibrosis stage was scored in a double-blind method. n = 10. (G) Liver hydroxyproline levels were determined using a hydroxyproline assay kit in mice as described above. n = 10. (H) IHC for α-SMA or TGF-β1 was performed on the liver sections of mice as described above. (I) α-Sma and Tgf-β1 mRNA expression was assayed in liver tissues of mice as described above. n = 3–5. Data are expressed as the mean ± SEM of three independent experiments; *P < 0.05, **P < 0.01 between the indicated groups. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To further prove the important role of VAMP2 in EA-mediated anti-hepatic fibrosis, according to a reported method [28], vitamin A-coupled liposomes carrying VAMP2 siRNA/plasmid (VA-Lip-VAMP-siRNA/plasmid) were constructed to knock down/infect VAMP2 expression in HSCs. We assessed the impact of HSC-specific VAMP2 knockdown and overexpression on liver fibrosis in vivo. H&E, Masson, and Sirius red staining showed that although VAMP2 knockdown reduced liver fibrosis, EA treatment combined with VA-Lip-VAMP2-siRNA significantly alleviated hepatic fibrosis characterized by decreased collagen deposition compared with CCl4 mice, whereas the improvement of EA treatment on liver fibrosis was completely abrogated by overexpression of VAMP2 (Fig. 7F–G). Moreover, immunohistochemistry revealed that EA treatment combined with VAMP2-siRNA greatly reduced the expression of α-SMA and TGF-β1, whereas overexpression of VAMP2 almost completely abolished the inhibitory effect of EA on α-SMA and TGF-β1 expression (Fig. 7H). qPCR also showed that EA treatment combined with VAMP2-siRNA caused a decrease in α-Sma and Tgf-β1 mRNA expression, whereas overexpression of VAMP2 abrogated the effects of EA treatment on α-Sma and Tgf-β1 mRNA expression (Fig. 7I). Taken together, these results show that suppression of VAMP2 enhances EA-induced HSC ferroptosis in mouse liver fibrosis.
4. Discussion
In this study, we used EA to evaluate the protection against liver fibrosis in classical mouse models. The main manifestations are as follows: 1) EA mitigated liver fibrotic injury by inducing ferroptosis of activated HSCs in CCl4 and BDL mice and primary HSCs and human LX2 cells; 2) EA prevented FPN translocation of the plasma membrane, leading to disordered intercellular iron homeostasis in HSCs; and 3) EA reduced VAMP2 expression in a posttranslational modification manner and promoted VAMP2 degradation through a proteasome-dependent pathway, which impaired assembly of the SNARE complex (VAMP2/Syntaxin4 and VAMP2/SNAP23), ultimately blocking FPN trafficking.
EA, as a natural polyphenolic compound, has been shown to have many biological functions in various animal disease models [[35], [36], [37]]. Moreover, in vivo and in vitro studies in rodents show that EA is safe and nontoxic [37]. This was consistent with our result. In a recent study, EA was considered to have anti-fibrotic effects due to its ability to inhibit the transformation of HSCs into myofibroblasts [26] and can reduce the secretion of MMPs and TIMP of HSC activation in an alcohol-induced liver disease model [38]. Here, EA treatment also significantly improved liver injury both in CCl4 and BDL mice and mitigated liver fibrosis by inhibiting HSC activation and collagen deposition. Likewise, EA showed a similar antifibrotic effect in vitro. This result is consistent with other reports [36]. However, the further mechanism by which EA inhibits liver fibrosis is unclear.
A growing number of recent studies suggest that ferroptosis may be an attractive strategy for the prevention and treatment of liver fibrosis. In BDL-induced hepatic fibrosis, the activated BRD7/p53/SLC25A28 axis promotes mitochondrial iron accumulation and lipid peroxidation, resulting in HSC ferroptosis [39]. Upregulation of NCOA4 expression triggers HSC ferroptosis through the autophagy pathway to alleviate liver fibrosis [40]. To further explore the mechanism by which EA reduces liver fibrosis, we performed whole-transcriptome sequencing analysis using HSCs from CCl4 mice. Bioinformatics analysis showed that EA downregulated fibrotic-related genes and upregulated ferroptotic-related genes, indicating that EA mitigated fibrosis by inducing HSC ferroptosis. As we expected, EA treatment triggered ferropotic events, as evidenced by iron overload, GSH depletion, and MDA accumulation in HSCs. Moreover, immunofluorescence costaining verified that α-SMA-positive cells undergo ferroptotic processes. A study revealed that EA can inhibit colon cancer and esophageal cancer development by inducing cell cycle arrest and apoptosis [41,42]. In our studies, the ferroptosis inhibitor ferrostatin-1 relieved the growth inhibition of HSCs induced by EA, while ZVAD-FMK (an apoptosis inhibitor) could not. Moreover, EA treatment could not further increase the CCl4-induced cell apoptosis, clearly illustrating that EA enhanced the death of activated HSCs via the ferroptotic pathway rather than the apoptosis pathway. Interestingly, EA-induced HSC ferroptosis has a selective effect; EA neither inhibited growth nor induced ferroptotic events in both primary hepatocytes and human HepG2 cells in normal condition or under injury condition, which is consistent with previous findings that no growth inhibition was observed when hepatocytes and HepG2 cells were treated with 50 μM EA [43,44], suggesting that EA has a selective effect on ferroptosis induction.
The iron regulatory system involves multiple proteins coregulating iron import, neutralization, storage, and export, including TFN, DMT1, ferritin, and FPN [45]. TFN-mediated iron transport into cells is the most common iron uptake pathway; it decreases TFN expression via endocytosis, reduces iron uptake, and blocks mitochondrial ROS formation to protect neuronal cells from ferroptosis [46]. Additionally, DMT1 has been shown to be involved in the iron absorption process. Targeting DMT1 expression can induce ferroptosis of glioblastoma cells, whereas inhibition of DMT1 reduces cellular ROS levels, iron deposition, and MDA levels and thus inhibits ferroptosis [47]. In contrast, in our study, EA did not affect the expression of TFN or DMT1 in either the total or the plasma membrane. This finding revealed that EA-induced HSC ferroptosis may not be mediated by iron import disorder. Evidence reveals that ferroptosis can be induced by FPN but not TFN or DMT1 in human nucleus pulposus cells treated with TBHP [48]. FPN is indispensable in iron output, and downregulating FPN expression induces iron accumulation in breast cancer cells, leading to ferroptosis and delaying the development of breast cancer, while FPN overexpression reduces ROS levels and inhibits ferroptosis [49]. Here, EA only downregulated the FPN expression level of the plasma membrane, but the total expression level and mRNA level did not change. This suggests that FPN did not undergo posttranscriptional and translational modification, which might be a barrier to trafficking. Evidence has shown that knockdown of FPN triggers ferroptosis by increasing the accumulation of iron and lipid ROS in neuroblastoma cells [50]. Consistently, EA-induced reduction of plasma membrane FPN resulted in the accumulation of intracellular ferritin and further increased ROS levels in activated HSCs. As expect, DFO (an iron chelator) almost completely blocked the enhancing effect of EA on ROS levels. Moreover, our experiments also confirmed that FPN knockdown induced ferroptosis events of activated HSCs, whereas FPN overexpression could inhibit them. However, EA treatment blocked the effect of FPN plasmid on ferroptosis events. This finding suggests that FPN-regulated iron transport is involved in EA-induced ferroptosis of HSCs. The plasma membrane expression of FPN is negatively regulated by the hepatic iron-regulating hormone hepcidin, and inhibition of hepcidin increases FPN expression to reduce intracellular free iron levels in macrophages, thereby reducing atherosclerosis [51,52]. However, in our studies, EA did not affect the expression of hepcidin, suggesting that the EA-induced reduction in plasma membrane FPN was independent of hepcidin. This allows us to focus on FPN trafficking. FPN trafficking may be related to the vesicular transport network in the macrophage membrane [53], but it is unknown whether FPN transport is also regulated by vesicle trafficking in HSCs.
Evidence has revealed that SNARE-mediated membrane fusion and trafficking are involved in FPN transport, and NSF, an inhibitor of SNARE, prevents FPN transport to the plasma membrane, but the exact mechanism remains unknown [13]. SNAP23, SNAP25, syntaxin4 and VAMP2 are considered major components of the SNARE protein complex. Notably, we found that SNAP25 is not expressed in primary HSCs, which is consistent with previous studies [20]. The SNAP23/Syntaxin4/VAMP2 complex is involved in mediating cAMP-stimulated renin release and paraglomerular exocytosis [54] and can also improve insulin resistance by regulating glucose receptor transport [55]. Moreover, SNAP23/VAMP2 mediates exocytosis in circulating vesicles containing transferrin receptors, whereas reduced VAMP2 expression affects complex assembly and directly inhibits exocytosis [56]. We reveal that EA reduces VAMP2 expression at the protein level, not the mRNA level, suggesting that EA affects the posttranslational modification of VAMP2. However, EA has no regulatory role in the expression of VAMP3 and VAMP8 in HSCs. Protein ubiquitination is a mechanism of VAMP2 degradation [57]. However, here, the downregulation of VAMP2 protein expression by EA was reversed by MG-132, a proteasome inhibitor, but not by the lysosomal blocker chloroquine. This suggests that EA reduced VAMP2 expression by enhancing VAMP2 degradation through a proteasome-dependent pathway. Iron treatment can reduce VAMP2 levels in hippocampal neurons, leading to iron-induced memory impairment by affecting synaptic transmission and vesicle fusion [58]. Similarly, EA blocked the formation of the VAMP2/Syntaxin4 and VAMP2/SNAP23 complexes by reducing VAMP2 expression. Disruption of the SNARE complex affects FPN transport [13], and knockdown of VAMP2 results in further downregulation of FPN. This finding suggests that EA impairs FPN translocation by inhibiting VAMP2 and further blocking the assembly of the SNARE complex. A study showed that the downregulation of FPN increases the cellular ferritin concentration and oxidative stress response and activates ferroptosis in a rat model of LPS-induced endotoxemia [48]. Consistent with this, we revealed that ferritin was significantly increased in HSCs after combined treatment with EA and VAMP2 knockdown. This finding suggests that VAMP2 inhibition by EA leads to intercellular iron overload by regulating FPN trafficking. In addition, VAMP2 knockdown enhanced the inhibition of liver fibrosis by EA in vivo and in vitro. This finding indicates that VAMP2 is indispensable in EA-induced HSC ferroptosis and suppression of liver fibrosis.
In conclusion, we demonstrate that EA exerts an antifibrotic effect by inducing FPN-dependent ferroptosis of activated HSCs by promoting VAMP2 degradation and blocking the formation of SNARE complexes in CCl4 mice (Fig. 8). Thus, EA will hopefully serve as a prospective compound for liver fibrosis treatment.
Fig. 8.

Schematic regulatory mechanism of EA action
EA suppressed FPN-dependent vesicle trafficking to plasma membrane to blocked formation SNARE complexes through induced VAMP2 degradation. This process led to FPN translocation and intercellular iron extrusion impairment. Subsequently, excessive iron produced a large amount of ROS, ultimate caused ferroptosis of HSC.
Author contribution
The authors alone are responsible for the content and writing of the paper. Lihua Li, Kunpeng Wang, Rongjun Jia, Jing Xie, Liman Ma, Zhiqing Hao and Weiwei Zhang performed the research, Lihua Li, Jinggang Mo and Fu Ren collected and analyzed the data, Lihua Li, Jinggang Mo and Fu Ren designed the research study. All authors contributed equally and have read and approved the final manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This study was supported by the National Natural Science Foundation of China [No. 31571184] and Taizhou Science and Technology Planning Project [No. 21ywb78].
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102435.
Contributor Information
Lihua Li, Email: lilihua1018@sina.com.
Kunpeng Wang, Email: wkpeng101@126.com.
Rongjun Jia, Email: 1085881994@qq.com.
Jing xie, Email: 347584368@qq.com.
Liman Ma, Email: limanma1986@163.com.
Zhiqing Hao, Email: 546573578@qq.com.
Weiwei Zhang, Email: 2793353146@qq.com.
Jinggang Mo, Email: mojg@tzzxyy.com.
Fu Ren, Email: rf@symc.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Effect of EA on apoptosis of activated HSCs in CCl4 mice liver
Co-immunostaining was performed for apoptosis and activated HSCs using TUNEL assay kit and α-SMA antibody in CCl4 mice liver. Representative images of α-SMA (green) and TUNEL (red) co-stained liver sections from control and CCl4 mice collected at 4 weeks. Cell nuclei were stained with DAPI (blue). n = 3.
{kind=link}
Effect of EA on primary hepatocytes during fibrotic injury
Primary hepatocytes were serum-starved for 12 h and then exposed for 48 h to 20 mM CCl4 (in 0.1% DMSO) or DMSO carrier (0.1%), with or without EA at doses of 32.5, 65, 130 μM for the final 24 h after which cell viability was assessed by CCK8 assay. n = 5. Data are expressed as the mean ± SEM of three independent experiments.
{kind=link}
FPN reduction contributes to EA-induced ferroptosis in activated HSCs
(A) Primary HSCs and LX-2 cells were infected with different concentrations of Control shRNA, FPN shRNA, Control vector and FPN plasmid for 24 h. FPN protein expression was examined by Western bolt analysis. n = 3. (B) Total GSH, GSH/GSSG ratio, intracellular ferrous iron, MDA and C11-BODIPY581/591 levels were assayed in FPN shRNA- and/or FPN plasmid-infected primary HSCs and LX-2 cells treated with TGF-β1 and/or EA. n = 5. (C) After HSCs were infected with FPN shRNA or FPN plasmid followed by TGF-β1 treatment, the cells were incubated in EA for 24 h, and then FTH1 expression in those cells was examined by Western blotting. n = 4. Data were normalized to GAPDH and are expressed as the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 between the indicated groups.
{kind=link}
Data availability
No data was used for the research described in the article.
References
- 1.Roehlen N., Crouchet E., Baumert T.F. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. 2020;9(4) doi: 10.3390/cells9040875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisseleva T., Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021;18(3):151–166. doi: 10.1038/s41575-020-00372-7. [DOI] [PubMed] [Google Scholar]
- 3.Shen M., Li Y., Wang Y., et al. N(6)-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021;47 doi: 10.1016/j.redox.2021.102151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan S., Wei C., Liu G., et al. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1α/SLC7A11 pathway. Cell Prolif. 2022;55(1) doi: 10.1111/cpr.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang D., Chen X., Kang R., et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner A., Alan B., Yilmaz D., et al. Despite genetic iron overload, Hfe-Hemochromatosis mice do not show bone loss. JBMR plus. 2019;3(9) doi: 10.1002/jbm4.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon S.J., Lemberg K.M., Lamprecht M.R., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drakesmith H., Nemeth E., Ganz T. Ironing out ferroportin. Cell Metabol. 2015;22(5):777–787. doi: 10.1016/j.cmet.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gammella E., Correnti M., Cairo G., et al. Iron availability in tissue microenvironment: the key role of ferroportin. Int. J. Mol. Sci. 2021;22(6) doi: 10.3390/ijms22062986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu S., Song Y., Luo R., et al. Ferroportin-dependent iron homeostasis protects against oxidative stress-induced nucleus pulposus cell ferroptosis and ameliorates intervertebral disc degeneration in vivo. Oxid. Med. Cell. Longev. 2021;2021 doi: 10.1155/2021/6670497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song Z., Xiang X., Li J., et al. Ruscogenin induces ferroptosis in pancreatic cancer cells. Oncol. Rep. 2020;43(2):516–524. doi: 10.3892/or.2019.7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao W.D., Pang P., Zhou X.T., et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell Death Differ. 2021;28(5):1548–1562. doi: 10.1038/s41418-020-00685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flannagan R.S., Farrell T.J., Trothen S.M., et al. Rapid removal of phagosomal ferroportin in macrophages contributes to nutritional immunity. Blood Adv. 2021;5(2):459–474. doi: 10.1182/bloodadvances.2020002833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sutton R.B., Fasshauer D., Jahn R., et al. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395(6700):347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 15.Hasan N., Corbin D., Hu C. Fusogenic pairings of vesicle-associated membrane proteins (VAMPs) and plasma membrane t-SNAREs--VAMP5 as the exception. PLoS One. 2010;5(12) doi: 10.1371/journal.pone.0014238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu C., Hardee D., Minnear F. Membrane fusion by VAMP3 and plasma membrane t-SNAREs. Exp. Cell Res. 2007;313(15):3198–3209. doi: 10.1016/j.yexcr.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Hughson F.M. Chaperoning SNARE folding and assembly. Annu. Rev. Biochem. 2021;90:581–603. doi: 10.1146/annurev-biochem-081820-103615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brandie F.M., Aran V., Verma A., et al. Negative regulation of syntaxin4/SNAP-23/VAMP2-mediated membrane fusion by Munc18c in vitro. PLoS One. 2008;3(12) doi: 10.1371/journal.pone.0004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawaguchi T., Tamori Y., Kanda H., et al. The t-SNAREs syntaxin4 and SNAP23 but not v-SNARE VAMP2 are indispensable to tether GLUT4 vesicles at the plasma membrane in adipocyte. Biochem. Biophys. Res. Commun. 2010;391(3):1336–1341. doi: 10.1016/j.bbrc.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 20.Eubanks H.B., Lavoie E.G., Goree J., et al. Reduction in SNAP-23 alters microfilament organization in myofibrobastic hepatic stellate cells. Gene Expr. 2020;20(1):25–37. doi: 10.3727/105221619x15742818049365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta A., Singh A.K., Kumar R., et al. Neuroprotective potential of ellagic acid: a critical review. Adv. Nutr. 2021;12(4):1211–1238. doi: 10.1093/advances/nmab007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang I., Espín J.C., Carr T.P., et al. Raspberry seed flour attenuates high-sucrose diet-mediated hepatic stress and adipose tissue inflammation. J. Nutr. Biochem. 2016;32:64–72. doi: 10.1016/j.jnutbio.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Diao M., Liang Y., Zhao J., et al. Complexation of ellagic acid with α-lactalbumin and its antioxidant property. Food Chem. 2022;372 doi: 10.1016/j.foodchem.2021.131307. [DOI] [PubMed] [Google Scholar]
- 24.Rahnasto-Rilla M., Järvenpää J., Huovinen M., et al. Effects of galloflavin and ellagic acid on sirtuin 6 and its anti-tumorigenic activities. Biomed. Pharmacother. 2020;131 doi: 10.1016/j.biopha.2020.110701. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki N., Masamune A., Kikuta K., et al. Ellagic acid inhibits pancreatic fibrosis in male Wistar Bonn/Kobori rats. Dig. Dis. Sci. 2009;54(4):802–810. doi: 10.1007/s10620-008-0423-7. [DOI] [PubMed] [Google Scholar]
- 26.Buniatian G.H. Stages of activation of hepatic stellate cells: effects of ellagic acid, an inhibiter of liver fibrosis, on their differentiation in culture. Cell Prolif. 2003;36(6):307–319. doi: 10.1046/j.1365-2184.2003.00287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baradaran Rahimi V., Ghadiri M., Ramezani M., et al. Antiinflammatory and anti-cancer activities of pomegranate and its constituent, ellagic acid: evidence from cellular, animal, and clinical studies. Phytother Res. 2020;34(4):685–720. doi: 10.1002/ptr.6565. [DOI] [PubMed] [Google Scholar]
- 28.Wu X., Wu X., Ma Y., et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat. Commun. 2016;7 doi: 10.1038/ncomms13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie J., Fan Y., Jia R., et al. Yes-associated protein regulates the hepatoprotective effect of vitamin D receptor activation through promoting adaptive bile duct remodeling in cholestatic mice. J. Pathol. 2021;255(1):95–106. doi: 10.1002/path.5750. [DOI] [PubMed] [Google Scholar]
- 30.García-Niño W.R., Zazueta C. Ellagic acid: pharmacological activities and molecular mechanisms involved in liver protection. Pharmacol. Res. 2015;97:84–103. doi: 10.1016/j.phrs.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Gandhi C.R. Hepatic stellate cell activation and pro-fibrogenic signals. J. Hepatol. 2017;67(5):1104–1105. doi: 10.1016/j.jhep.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dooley S., ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. 2012;347(1):245–256. doi: 10.1007/s00441-011-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z., Yao Z., Wang L., et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14(12):2083–2103. doi: 10.1080/15548627.2018.1503146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ross S.L., Tran L., Winters A., et al. Molecular mechanism of hepcidin-mediated ferroportin internalization requires ferroportin lysines, not tyrosines or JAK-STAT. Cell Metabol. 2012;15(6):905–917. doi: 10.1016/j.cmet.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 35.Afifi N.A., Ibrahim M.A., Galal M.K. Hepatoprotective influence of quercetin and ellagic acid on thioacetamide-induced hepatotoxicity in rats. Can. J. Physiol. Pharmacol. 2018;96(6):624–629. doi: 10.1139/cjpp-2017-0651. [DOI] [PubMed] [Google Scholar]
- 36.Nitha A., Prabha S.P., Ansil P.N., et al. Methanolic extract of Woodfordia fruticosa Kurz flowers ameliorates carbon tetrachloride-induced chronic hepatic fibrosis in rats. Toxicol. Ind. Health. 2016;32(7):1224–1236. doi: 10.1177/0748233714552120. [DOI] [PubMed] [Google Scholar]
- 37.Abdelkader N.F., Elyamany M., Gad A.M., et al. Ellagic acid attenuates liver toxicity induced by valproic acid in rats. J. Pharmacol. Sci. 2020;143(1):23–29. doi: 10.1016/j.jphs.2020.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Devipriya N., Sudheer A.R., Srinivasan M., et al. Effect of ellagic acid, a plant polyphenol, on fibrotic markers (MMPs and TIMPs) during alcohol-induced hepatotoxicity. Toxicol. Mech. Methods. 2007;17(6):349–356. doi: 10.1080/15376510601077003. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z., Guo M., Shen M., et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z., Wang X., Wang Z., et al. Dihydroartemisinin alleviates hepatic fibrosis through inducing ferroptosis in hepatic stellate cells. Biofactors. 2021;47(5):801–818. doi: 10.1002/biof.1764. [DOI] [PubMed] [Google Scholar]
- 41.Xu Y.Y., Wang W.W., Huang J., et al. Ellagic acid induces esophageal squamous cell carcinoma cell apoptosis by modulating SHP-1/STAT3 signaling. Kaohsiung J. Med. Sci. 2020;36(9):699–704. doi: 10.1002/kjm2.12224. [DOI] [PubMed] [Google Scholar]
- 42.Zhao J., Li G., Wei J., et al. Ellagic acid induces cell cycle arrest and apoptosis via the TGF-β1/Smad3 signaling pathway in human colon cancer HCT-116 cells. Oncol. Rep. 2020;44(2):768–776. doi: 10.3892/or.2020.7617. [DOI] [PubMed] [Google Scholar]
- 43.Ieda A., Wada M., Moriyasu Y., et al. Ellagic acid suppresses ApoB secretion and enhances ApoA-1 secretion from human hepatoma cells, HepG2. Molecules. 2021;26(13) doi: 10.3390/molecules26133885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Girish C., Koner B.C., Jayanthi S., et al. Hepatoprotective activity of picroliv, curcumin and ellagic acid compared to silymarin on paracetamol induced liver toxicity in mice. Fundam. Clin. Pharmacol. 2009;23(6):735–745. doi: 10.1111/j.1472-8206.2009.00722.x. [DOI] [PubMed] [Google Scholar]
- 45.Hentze M.W., Muckenthaler M.U., Galy B., et al. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142(1):24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 46.Clemente L.P., Rabenau M., Tang S., et al. Dynasore blocks ferroptosis through combined modulation of iron uptake and inhibition of mitochondrial respiration. Cells. 2020;9(10) doi: 10.3390/cells9102259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song Q., Peng S., Sun Z., et al. Temozolomide drives ferroptosis via a DMT1-dependent pathway in glioblastoma cells. Yonsei Med. J. 2021;62(9):843–849. doi: 10.3349/ymj.2021.62.9.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang J., Kong B., Shuai W., et al. Ferroportin-mediated ferroptosis involved in new-onset atrial fibrillation with LPS-induced endotoxemia. Eur. J. Pharmacol. 2021;913 doi: 10.1016/j.ejphar.2021.174622. [DOI] [PubMed] [Google Scholar]
- 49.Ma S., Henson E.S., Chen Y., et al. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7(7) doi: 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geng N., Shi B.J., Li S.L., et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018;22(12):3826–3836. doi: 10.26355/eurrev_201806_15267. [DOI] [PubMed] [Google Scholar]
- 51.Vogt A.S., Arsiwala T., Mohsen M., et al. On iron metabolism and its regulation. Int. J. Mol. Sci. 2021;22(9):4591. doi: 10.3390/ijms22094591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saeed O., Otsuka F., Polavarapu R., et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012;32(2):299–307. doi: 10.1161/atvbaha.111.240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCreedy R.A., Fleet J.C. Forward genetics used to identify new gene Mon1a with critical role in controlling macrophage iron metabolism and iron recycling from erythrocytes. Nutr. Rev. 2009;67(10):607–610. doi: 10.1111/j.1753-4887.2009.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mendez M., Gaisano H.Y. Role of the SNARE protein SNAP23 on cAMP-stimulated renin release in mouse juxtaglomerular cells. Am. J. Physiol. Ren. Physiol. 2013;304(5):F498–F504. doi: 10.1152/ajprenal.00556.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohseni R., ArabSadeghabadi Z., Ziamajidi N., et al. Oral administration of resveratrol-loaded solid lipid nanoparticle improves insulin resistance through targeting expression of SNARE proteins in adipose and muscle tissue in rats with type 2 diabetes. Nanoscale Res. Lett. 2019;14(1):227. doi: 10.1186/s11671-019-3042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kubo K., Kobayashi M., Nozaki S., et al. SNAP23/25 and VAMP2 mediate exocytic event of transferrin receptor-containing recycling vesicles. Biology open. 2015;4(7):910–920. doi: 10.1242/bio.012146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann-Conaway S., Brockmann M.M., Schneider K., et al. Parkin contributes to synaptic vesicle autophagy in Bassoon-deficient mice. Elife. 2020;9 doi: 10.7554/eLife.56590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X., Zhang J., Zhou L., et al. Long-term iron exposure causes widespread molecular alterations associated with memory impairment in mice. Food Chem. Toxicol. 2019;130:242–252. doi: 10.1016/j.fct.2019.05.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of EA on apoptosis of activated HSCs in CCl4 mice liver
Co-immunostaining was performed for apoptosis and activated HSCs using TUNEL assay kit and α-SMA antibody in CCl4 mice liver. Representative images of α-SMA (green) and TUNEL (red) co-stained liver sections from control and CCl4 mice collected at 4 weeks. Cell nuclei were stained with DAPI (blue). n = 3.
Effect of EA on primary hepatocytes during fibrotic injury
Primary hepatocytes were serum-starved for 12 h and then exposed for 48 h to 20 mM CCl4 (in 0.1% DMSO) or DMSO carrier (0.1%), with or without EA at doses of 32.5, 65, 130 μM for the final 24 h after which cell viability was assessed by CCK8 assay. n = 5. Data are expressed as the mean ± SEM of three independent experiments.
FPN reduction contributes to EA-induced ferroptosis in activated HSCs
(A) Primary HSCs and LX-2 cells were infected with different concentrations of Control shRNA, FPN shRNA, Control vector and FPN plasmid for 24 h. FPN protein expression was examined by Western bolt analysis. n = 3. (B) Total GSH, GSH/GSSG ratio, intracellular ferrous iron, MDA and C11-BODIPY581/591 levels were assayed in FPN shRNA- and/or FPN plasmid-infected primary HSCs and LX-2 cells treated with TGF-β1 and/or EA. n = 5. (C) After HSCs were infected with FPN shRNA or FPN plasmid followed by TGF-β1 treatment, the cells were incubated in EA for 24 h, and then FTH1 expression in those cells was examined by Western blotting. n = 4. Data were normalized to GAPDH and are expressed as the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 between the indicated groups.
Data Availability Statement
No data was used for the research described in the article.