

Abstract
Protein arginine methyltransferase (PRMT)5 is the major methyltransferase catalyzing symmetric dimethylation (SDM). PRMT5 regulates developmental homeostatic and disease processes in vertebrates and invertebrates, and a carcinogenic role has been observed in mammals. Recently generated tools for PRMT5 loss-of-function have allowed to demonstrate essential roles for PRMT5 in mouse and human lymphocyte biology. PRMT5 modulates CD4+ and CD8+ T cell development in the thymus, peripheral homeostasis, and differentiation into CD4+ Th17 cell phenotypes. Here, we provide a timely review of the milestones leading to our current understanding of PRMT5 in T cell biology, discuss current tools to modify PRMT5 expression/activity, and highlight mechanistic pathways.
Keywords: Arginine methylation, PRMT5, lymphocytes, autoimmunity
Methylation, the process of transferring a methyl group onto a substrate, plays crucial roles in biology. Methylation modulates developmental, homeostatic, and disease processes by providing a reversible mechanism to regulate gene expression and protein activity. The most common methylation targets are cytosines on DNA and lysine or arginine residues on proteins. The impact of DNA [1] and protein lysine methylation [2] in immune processes has been extensively reviewed elsewhere [3]. In this review, we focus on arginine methylation catalyzed by protein arginine methyltransferase (PRMT)5 in T lymphocyte biology, a field that has bloomed in recent years.
Protein arginine methylation has an important role in many processes, including RNA processing, DNA repair, and transcriptional regulation [4,5]. Arginine methylation is catalyzed by PRMT enzymes and typical targets in eukaryotes include histones and other proteins, such as small nuclear ribonucleoproteins [4]. The PRMT family comprises three types of enzymes, depending on how many methyl groups are added and where they are positioned (Fig. 1A). Type I enzymes, namely PRMT1-4, PRMT6, and PRMT8, catalyze asymmetric dimethylation (ADM) of arginine [6]. In contrast, type II enzymes PRMT5 and PRMT9 catalyze symmetric dimethylation (SDM) at the terminal amine group of arginine [7,8]. Finally, type III enzymes catalyze monomethylation, and PRMT7 is the only PRMT in this group [9]. PRMT5 has a prominent role among PRMTs, as it is responsible for the majority of type II SDM [10].
Figure 1.
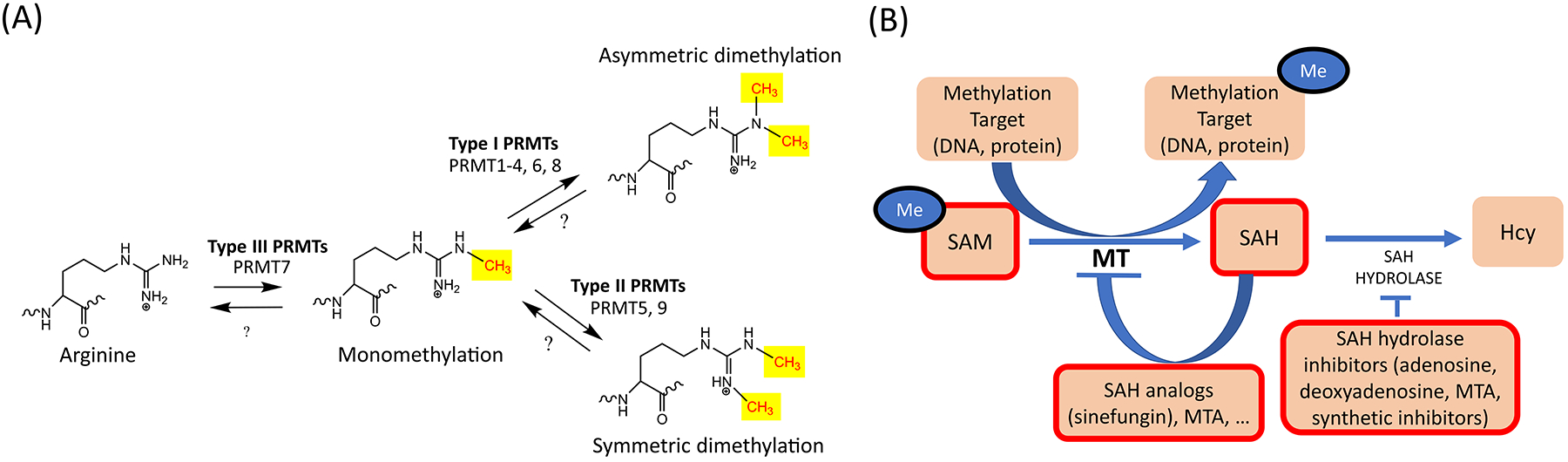
(A). Arginine methylation reactions. Arginine methylation is catalyzed by arginine methyltransferases (PRMTs), which transfer a methyl group onto the ω-nitrogen of arginine (Arg). All mammalian PRMTs modify arginine to its monomethylated state. From the monomethylated state, Type I PRMTs are able to catalyze asymmetric demethylation (ADM) of Arg [6]. Type II PRMTs are able to catalyze symmetric dimethylation (SDM) from the monomethylated state [7,8]. Type III PRMTs are solely able to catalyze monomethylation [9]. (B). S-adenosyl methionine (SAM) is the methyl donor for all methyltransferase (MT) reactions, including DNA methyltransferase (DNMT), Protein Lysine methyltransferase (PKMT), and Protein Arginine methyltransferase (PRMT) reactions. After SAM has donated its methyl group, it is converted to (S)-adenosylhomocysteine (SAH). SAH inhibits MT activity by a negative feedback loop but methylation can generally proceed if SAH is broken down by the SAH hydrolase enzyme [21]. In contrast, if SAH hydrolase is not active, SAH accumulates in the cytoplasm and inhibits SAM-dependent methylation reactions. Therefore, MT inhibitors often act by inhibiting SAH hydrolase and increasing the concentration of the natural inhibitor SAH [21]. Products with MT inhibitory activity are indicated with a thick red outline. SAM can act as an inhibitor by producing high amounts of SAH, while SAH hydrolase inhibitors do the same by inhibiting the SAH degradation [21]. Finally, SAH analogs also have general MT inhibitory activity [21].
PRMT5 plays a crucial role in the regulation of developmental and physiological processes [4,5]. This protein is highly expressed during mouse and human embryonic/fetal development and, at the steady state, in human bone marrow, lymphoid tissues, brain, gastrointestinal tract, genitourinary tract and reproductive tissuesI [11]. In addition, PRMT5 is overexpressed in many hematologic and solid human cancers [12] and, therefore, its role in disease is currently a major focus of research and therapeutic targeting. Structurally, PRMT5 regulates the symmetric dimethylation of histone H4 at R3, histone H2A, also at R3 and histone H3 at R8 [4]. In addition, it dimethylates arginine residues symmetrically in spliceosomal proteins [5]. The biological outcomes of these PRMT5-mediated modifications include the regulation of mouse and human stem cell maintenance, differentiation, and early development, cell proliferation and cell death [4,5]. Because of these activities, PRMT5 can contribute to cell transformation and oncogenesis [12]. Accordingly, the importance of PRMT5-catalyzed methylation as a cancer-driver is well established [12] and has resulted in the development of various PRMT5 inhibitor cancer therapeutics that are currently being tested for hematologic and solid cancers in five Phase I clinical trials, listed under ClinicalTrials.gov numbers NCT03573310, NCT03854227, NCT04089449, NCT02783300 and NCT03614728.II
The recognition that PRMT5 plays a crucial role in immune processes is a more recent development. Various mouse and human immune cells express PRMT5 constitutively or upon activation [13,14]. For example, whereas EBV-transformed, diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) B lymphocytes express PRMT5 [15–17], normal B lymphocytes express PRMT5 only at certain stages of B cell development in mice [13]. Moreover, mature mouse B cells express PRMT5 inducibly, following BCR engagement [13]. In non-transformed human and mouse naïve or memory helper T lymphocytes Th1 and Th2 cells, PRMT5 is induced maximally, 2–3 days post TCR engagement [18], via the NF-kB/mTOR/Myc signaling axis [19]. These findings collectively suggest a physiological and highly conserved role for this enzyme in lymphocyte biology. Recently developed PRMT5-selective inhibitors and conditional knockout (cKO) mouse tools are currently allowing to dissect the emerging role of PRMT5 in lymphocyte biology. Here, we provide a comprehensive review of the recent developments in our understanding of PRMT5 function in T lymphocytes.
A historical perspective on the role for PRMT5 in lymphocyte biology.
PRMT5-catalyzed SDM of arginine plays crucial roles in development, homeostasis and disease [3]. These functions have often been deduced from the effects of small-molecule inhibitors, or from the outcome of genetic knockdown/KO interventions. In this section, we review how natural and synthetic pan-methylation inhibitors (Table 1) and genetic strategies (Table 2) for PRMT5 loss-of-function first suggested, and then established a role for PRMT5-catalyzed arginine methylation in lymphocyte biology.
Table 1.
Small molecule inhibitors of pan-methyltransferase reactions, PRMTs and PRMT5.
| Compound | Refs | Immune Effect | Species | Refs | ||
|---|---|---|---|---|---|---|
| Pan-MT inhibitors | Natural Inhibitors | SAM/SAH | [93] | Significantly inhibit the proliferation of both B and T cells | B10.BR, MRL/Mp-Fas lpr (MR L/lpr) mice and α/β TCR (Vα11+, Vβ3+) tg mice | [45] |
| Sinefungin | Significant inhibition of the proliferation of both B and T cells | BALB/c mice | [37] | |||
| MTA | Inhibition of T cell signaling, T cell proliferation, suppression of interleukin 2 (IL-2) secretion, reducing the amount of interferon gamma (IFN-γ) and interleukin 4 (IL-4) producing cells production. | Sprague Dawley rats, humans, Balb/c mice, B10.BR, MRL/Mp-Fas lpr (MR L/lpr) mice, α/β TCR (Vα11+, Vβ3+) tg mice and AKR mice | [38,39,42, 43,45,51] | |||
| Adenosine/Deoxyadenosine | Build-up occurs in ADA and results in severe combined immunodeficiency with B/T cell lymphopenia and particularly severe T cell defects. | Human, Tg-m1/m1 mice | [28,29,31, 32,35,36] | |||
| Synthetic Inhibitors | MDL 28842 | Selective inhibition of the generation of cytotoxic T cells during transplantation rejection, reduced T cell proliferation and IL-2 production. | Sprague Dawley rats, DBN1 LacJ mice, CD- 1 mice, C3H/He J mice | [25,26,42,94,95] | ||
| DHCaA | Reduced T cell proliferation and IL-2 production [26] | [26] | ||||
| DZ2002 | Suppression of CD4+ T cell activation resulting in blocked EAE induction and amelioration of established EAE. A follow-up study found treatment of isolated CD4+ T cells reduced Th1 and Th17 polarization. Isolated B cells that were activated with DZ2002 displayed reduced proliferation | C57BL/6 mice, mouse T cell hybridoma T8.1-Vav1, BALB/c mice, MRL-Faslpr mice and BXSB mice | [27,46,51, 96,97] | |||
| PRMT inhibitors | PRMT1, PRMT3, PRMT4 and 6 inhibition (PRMT5 at high concentrations) | AMI-1 | [52] | Reduction in IFN-γ secretion from Th1 cells and IL-4 secretion from Th2 cells | BALB/c mice | [43] |
| PRMT1, PRMT3, PRMT4, and PRMT5 inhibition | Compound 4 | Reduces IFN-γ secretion from Th1 cells and IL-4 secretion from Th2 cells more effectively than AMI-1 | BALB/c mice | [43] | ||
| PRMT5 Inhibitors | Substrate and SAM binding inhibitors | HLCL65 | Suppression of inflammatory Th1 cells | (MBP)Ac1 –11– specific TCR-transgenic (Tg) mice and B20.PL mice | [18] | |
| CMP5/BLL1 [15] | [15] | Suppression of IL-2 and inflammatory Th1 cells and, at higher concentrations, Th2 and naïve T cells | hu-PBL-SCID mouse model and Prmt5flox/Δ mice | [15,19] | ||
| HLCL61 | Decreased cell proliferation AML cells | Cell lines | [61] | |||
| Substrate competitive inhibitors | EPZ015666 | Significant inhibition of tumor growth in mantle cell lymphoma | Cell line | [55] | ||
| GSK3326595 | Anti-proliferative effect on human MCL cell line, currently being used in clinical trials for lymphoma. | Cell line | [56] | |||
| DC_C01 | DC_C01 was found to have antiproliferative effects in the MCL lines of Z-138, Maver-1, and Jeko-1 | Cell line | [57] | |||
| SAM competitive inhibitors | DS-437[61] | [61] | Inhibits both PRMT5 and PRMT7. FOXP3 inhibition, resulting in inhibited Treg function and tumor immunity | Foxp3Creyfp (B6.129(Cg)-Foxp3tm4(YFP/Cre)Ayr/J, 016959) and CD4cre (Tg (Cd4-cre)1Cwi/BfluJ, 017336) mice | [67] | |
| LLY-283 | Anti-tumor effect in hematological cell lines. | Cell line | [60] | |||
| Not defined | PJ-68 | Treatment of chronic myelogenous leukemia resulted in decreased numbers of disease- and PRMT5-associated CD38+ cells | Cell lines and peripheral blood or bone marrow from human patients | [58] | ||
| P5i-6 | Treatment results in inhibition of growth HT-29 and DLD-1. In DLD-1 cells, cell cycle arrest was observed in the S/G2 phase. Similar inhibition of growth of hepatic cancer cell line HepG2 was also observed. | Human colorectal cancer cell lines DLD-1 and Hep2G | [59] |
Abbreviations: ADA (Adenosine deaminase deficiency), EAE (Experimental autoimmune encephalomyelitis), AML (Acute myeloid leukemia), MCL (Mantle cell lymphoma)
Table2.
Tools for genetic PRMT5 loss-of-function.
| Method | Types | Target cell/s | Immune Effect | Refs |
|---|---|---|---|---|
| Knockdown (RNA interference) | siRNA | transformed Jurkat T cells | Suppression of IL-2 production | [42] |
| Antisense lentivirus | Transformed Mino and JeKo B cell lines | Reducing PRMT5 expression affects proliferation of transformed B cells (1.8-fold decrease in JeKo cell growth after PRMT5 knockdown), low PRMT5 expression needed for normal growth. | [16] | |
| shRNA, siRNA | Human primary Th1 and Th2 cells | Suppression of human Th1 and Th2 cell proliferation | [18] | |
| Cre driver | ||||
| Germline Full body KO mice | Homologous recombination – Cre was not used | All cells | A germline full-body knockout of Prmt5 has proved to be embryonic-lethal. This demonstrates that Prmt5 is essential in development. | [65] |
| Conditional knockout mice | Mx1-cre | Early hematopoietic progenitors | Early loss of Prmt5 in hematopoietic cell development results in drastic loss of hematopoietic stem cells, resulting in bone marrow failure and death within three weeks. Prmt5 heterozygous mice had normal bone marrow, spleen, and thymus cell populations along with normal peripheral blood count | [68] |
| CD4-cre | CD4+CD8+ double-positive stage (DP) of T cell development (deletion in all T cell subsets, including CD4+, CD8+ and iNK T cells) | No defects in thymocyte development were observed in a CD4crePRMT5fl/Δ model. Impaired thymocyte development, with loss of DP, CD4+ SP and Treg cells as well as resistance to EAE development are observed in one CD4crePrmt5fl/fl model. A third model, CD4crePrmt5fl/fl reproduced similar results of robust loss of NK T cells and requirement of Prmt5 for peripheral T cell maintenance | [14,66,69] | |
| CD4-creERT2 | Peripheral CD4+ T cells | Reduced proliferative, Th17 and Th1 recall responses as well as severely reduced numbers of memory T cell infiltration in the CNS. Resistance to EAE development. Impaired Th17 differentiation | [69] | |
| Foxp3-cre | Foxp3-expressing cells (Tregs) | Foxp3-selective Prmt5 deletion results in scurfy-like autoimmunity; normal numbers of Tregs but deficient suppressive function | [67] |
Abbreviations: IL (Interleukin), DP (Double positive), SP (Single positive)
Early studies establish a role for methylation in immune responses.
One of the earliest indications that methylation could play a key role in lymphocyte activation came from studies showing that the turnover rate of S-adenosylmethionine (SAM) was higher in stimulated than resting human peripheral blood mononuclear cells (PBMCs) [20]. Further support for the conclusions drawn from these experiments was provided by studies on the functional characterization of naturally occurring inhibitors of methylation, such as (S)-adenosyl-L-homocysteine (SAH), methylthioadenosine (MTA) and sinefungin (Fig. 1B). These inhibitors impact all methyltransferases (MTs), including DNA MTs (DNMTs), protein Lysine MTs (PKMTs) and PRMTs [21].
High concentrations of S-adenosyl methionine (SAM), the methyl-donor for all methylation reactions, also has indirect inhibitory activity via its end product SAH (Fig. 1B) [21]. The accumulation of SAH in the course of the reaction triggers an inhibitory feedback loop, which limits the methylation of the target [22]. The fungal product sinefungin is a potent SAH analog with enhanced protein methyltransferase inhibitory activity [23]. Another potent naturally occurring pan-MT inhibitor, the nucleoside MTA, is commonly used as research tool compound [24]. MTA has been reported to directly inhibit methyltransferases, although some studies suggest that it may function by inhibiting SAH hydrolase [24]. MTA bears the advantage of enhanced cell permeability relative to SAM and SAH, making it a pan-MT inhibitor of choice [21]. In addition to the naturally-occurring synthetic pan- or broad spectrum MT inhibitors, synthetic inhibitors have also been developed. These include the irreversible SAH hydrolase inhibitors MDL28842 and DHCaA [25,26], and the reversible SAH hydrolase inhibitor DZ2002 [27]. Synthetic pan-MT inhibitors are significantly toxic, by comparison with natural pan-MT inhibitors [21]. Thus, their toxicity limits their usefulness and makes natural inhibitors the inhibitors of choice.
Other naturally-occurring indirect MT inhibitors include adenosine and deoxyadenosine, which accumulate in cells of adenosine deaminase-deficient (ADA) patients [28] and inhibit the SAH hydrolase [29,30]. In humans, ADA deficiency causes severe combined immunodeficiency [31], characterized by B/T cell lymphopenia and severe T cell dysfunction [32]. CD4+ T cells from these patients proliferate poorly and exhibit defects in cytokine production following stimulation [33]. The immunological defects of ADA [33,34] have therefore suggested a link between methylation and lymphocyte biology. Studies in human peripheral blood lymphocytes where ADA was inhibited while exogenously restoring the pyrimidine product of ADA provided evidence that the antiproliferative effects of ADA deficiency were indeed due, at least in part, to inhibition of SAH hydrolase and accumulation of SAH that inhibits methylation [35]. Further support to this conclusion was provided by a mouse model of ADA deficiency (Ada−/−) that recapitulates the T/B lymphopenic phenotype of ADA patients, which was accompanied by marked inhibition of SAH hydrolase in thymic and spleen lymphoid compartments [36].
Exploration of the role of methylation on lymphocyte activation revealed that MT inhibition by both naturally-occurring (sinefungin, MTA and SAM) [37–41] and synthetic (MDL28842 and DHCaA) [25,26] inhibitors interfered with mouse and/or human T lymphocyte proliferation and mouse B cell proliferation. MT inhibition has also been shown to impair activation of NFAT, the interleukin 2 (IL2) promoter, and secretion of IL-2 in PMA-activated human Jurkat CD4+ T cells [42]. In addition, MT inhibition has blunted the secretion of Th1 cytokines (IFN-γ, IL-2, TNF-α, GM-CSF) and Th2 cytokines (IL-4, IL-5, IL-6, IL-10) from primary mouse Th1 and Th2 cells, respectively [43–45].
The lymphocyte suppressive activities of MT inhibitors, and the beneficial MTA treatment of lupus-prone mice (MRL/lpr) have led to the idea that MT inhibition might be beneficial in human autoimmune systemic lupus erythematosus (SLE), although this remains to be assessed [45]. Similarly, the pan-MT inhibitors MTA and DZ2002 have robustly suppressed the clinical symptoms of experimental autoimmune encephalomyelitis (EAE), the mouse model of Multiple Sclerosis (MS) [41,46]. High doses of MTA were found to be more effective in reducing the severity of EAE than the MS therapies approved at the time, glatiramer acetate and IFN-β; a combination of MTA with either of these therapies resulted in the highest reduction of disease severity compared to controls [47]. In tumors with low expression or mutations of the MTA-degrading enzyme MTA phosphorylase (MTAP), MTA accumulates and can impact other cells in the tumor microenvironment. For example, tumor-derived MTA has been shown to accumulate in MTAP-deficient melanoma tumor cells and inhibit human T cell proliferation in a mixed lymphocyte reaction, revealing a novel immune evasion strategy employed by tumor cells [48]. Overall, early studies with pan-MT inhibitors and other models provided strong evidence of the importance of MT in T lymphocyte biology and provided the rationale to further explore the specific role of arginine methylation in T cells.
Later studies establish a role for arginine methylation in lymphocyte biology.
Another round of studies provided evidence that the T cell immunomodulatory effects of the pan-MT inhibitors were due primarily to protein arginine methyltransferase (PRMT) inhibition [49]. For example, human Jurkat T cell activation has been linked to PRMT activity and arginine methylation of Vav1 and other proteins [50,51]. Vav-1 is a signaling molecule downstream of the TCR that is methylated upon CD28 costimulation in human and mouse transformed T cells [51]. Vav1, an important effector of CD28, was found to undergo CD28-dependent symmetric arginine dimethylation in Jurkat T cells activated with SEB-treated B5-3.1 cells, suggesting that Vav1 might be a PRMT5 target [51]. This possibility was confirmed via immunoprecipitation (IP) experiments [51]. The functional significance of Vav1 arginine methylation was supported by experiments showing that loss of Vav1 methylation correlated with suppressed IL-2 production in human Jurkat T cells treated with the pan-MT inhibitor DZ2002 relative to untreated cells [51]. In other human Jurkat T-cell experiments, symmetric arginine dimethylation was detected on proteins bound to the IL2 promoter during Jurkat T cell activation via PMA [42]. Loss of this modification in Jurkat T cells treated with PRMT5 siRNA indicated that the enzyme responsible for the SDM of these proteins was PRMT5 [42].
The first specific arginine methyltransferase inhibitor, AMI-1, was reported in 2004 [52]. AMI-1 selectively inhibits type I arginine methyltransferases PRMT1, PRMT3, PRMT4 and PRMT6, and, at higher concentrations, type II arginine methyltransferase PRMT5. Compound 4, a less polar derivative of AMI-1, is more selective than AMI-1 toward PRMT1, PRMT3, and PRMT4. In addition, it has higher inhibitory activity toward PRMT5 [43]. Compound 4 has reduced IFN-γ secretion from primary murine Th1 cells and IL-4 secretion from murine Th2 cells in vitro, more effectively than AMI-1 [43].
Although all, or at least most PRMTs may have immunomodulatory activities, PRMT5 has been the most conclusively shown to be immunomodulatory [42,49–51]. This link, combined with the discovery that PRMT5 may be a promising therapeutic target for hematologic malignancies [16,17], breast cancer [53] and lung cancer [54] has led to parallel efforts by several academic and industry groups to develop selective PRMT5 inhibitors (Box 1). Such efforts have led to the discovery of a series of selective PRMT5 inhibitors, including CMP5/BLL1III, HLCL61IV and HLCL65IV [15,18,55]. These inhibitors bind reversibly to both the methyl donor SAM and the substrate binding sites [18]. The PRMT5 selective inhibitor EPZ015666 interacts only with the substrate binding site [55]. Other inhibitors include compound GSK3326595 [56] and compounds DC_P33, and DC_C01 [57]. Other SAM competitive PRMT5 inhibitors include PJ-68 [58], P5i-6 [59], and LLY-283 [60].
Most of these compounds have been tested in in vitro cancer models, demonstrating anti-proliferative activity: GSK3326595, has inhibited the proliferation of human mantle cell lymphoma (MCL) cell lines [56]. GSK3326595-related, but more potent compounds DC_P33 and DC_C01 have also displayed antiproliferative activities against transformed human cell lines [57]. CMP5 and HLCL61 decreased human B cell transformation and the proliferation of acute myeloid leukemia (AML) cells, respectively [15,61]. PJ-68 treatment of chronic myelogenous leukemia cells led to a decrease in the number of CD38+ cells [58]. P5i-6 arrested the human colorectal cancer cell lines HT-29 and DLD-1 in the S/G2 phase of the cell cycle, inhibiting their proliferation [59]. Finally, LLY-283 had similar anti-oncogenic effects than the inhibitors above in human leukemia cell lines [60]. An impact of selective PRMT5 inhibitors on normal mouse T lymphocytes was first demonstrated with inhibitors CMP5 and HLCL65, which showed anti-proliferative activity on memory Th1 cells and, to a lesser extent, naïve and memory Th2 cells [18,19]. These suppressive effects on proliferation were conserved from mouse to human primary T cells and were also observed with PRMT5 siRNA knockdown in human primary CD4+ Th cells, demonstrating the role of PRMT5 in T cell proliferation [18,19]. Recently, additional selective PRMT5 inhibitors EPZ015666, PF-9927 and C220 have had similar in vitro anti- proliferative effects on human CD4+ or CD8+ T cells [62–64].
Genetic mouse models conclusively demonstrate a role for PRMT5 in lymphocyte biology.
Genetic mouse models addressing the role of PRMT5 in hematopoiesis and T cell development have the advantage of eliminating the possibility of off-target effects, a common problem with small molecule inhibitors. The constitutive ablation of Prmt5 in mice is embryonic-lethal [65]. As a result, all studies addressing the phenotypic outcome of the genetic inactivation of Prmt5 in mice have been carried out on Cre-lox cKO mouse models, which allow temporal- and/or site-specific Prmt5 ablation [66–69]. Recent studies revealed that the homozygous ablation of Prmt5 in hematopoietic stem and progenitor cells (HSPCs) in PRMT5fl/fl-Mx1-cre mice resulted in a dramatic reduction in the abundance of HSPCs in the bone marrow (BM), in addition to BM failure and animal death within three weeks [68]. These mice also exhibited defects in early thymocyte development, as evidenced by the increased abundance of CD4+CD8+ double positive (DP) and the decreased abundance of CD4−CD8− double negative (DN) and CD8+ single positive (SP) thymocytes [68]. Given that Mx1-Cre inactivates Prmt5 in all hematopoietic lineages, the design of these experiments did not address the question of whether the observed effects on thymocyte development were cell-autonomous or whether they were due to the modulation of thymocyte differentiation by other lineages of Prmt5-deficient hematopoietic cells.
The preceding data and the effects of PRMT5 inhibitors on T cell function raised the question of what would be the impact of the targeted ablation of Prmt5 in T cells. The development of inducible and/or cell-specific cKO PRMT5 models [66,69] has now allowed to provide insights into the roles of Prmt5 in mouse T cell biology (Fig. 2, Key Figure). Models from two laboratories, namely CD4crePRMT5fl/Δ and CD4crePRMT5fl/fl provide constitutive T cell-specific Prmt5 deletion during thymic development [66,69]. Both models report using the same CD4cre transgene [70], but a notable difference is that the homozygous deletion of Prmt5 in T cells is harbored either within a full-body heterozygous PRMT5 KO (initially driven at an earlier generation via β-actin cre [66]) or a wildtype WT [69] background, respectively. While no defects in thymocyte development were observed in the CD4crePRMT5fl/Δ model [66], the CD4crePRMT5fl/fl model from our group found decreased numbers of CD4+ SP and regulatory T cells (Treg)in the thymus [69]. The reason for this discrepancy is unclear, but could stem from the different exons deleted in the models (exon 4 vs exon 7), or the ubiquitous heterozygote Prmt5 KO background in the CD4crePRMT5fl/Δ model. The latter supposition is intriguing since heterozygote T cell-specific cKO (CD4crePrmt5fl/WT) controls showed a similar decrease in thymic CD4+ SP cell numbers than homozygote T cell-specific cKO (CD4crePRMT5fl/fl) mice in our study [69]. This would imply that full wildtype (WT) controls are necessary to fully capture Prmt5 deletion effects on thymocyte development. Nonetheless, thymic total CD4+ and Treg number defects appear to stem from a defect in expansion, rather than at the thymocyte development level, given that all evaluated thymic populations were present at percentages similar to those of control mice. At the peripheral level, both models showed that deletion of Prmt5 at the CD4+CD8+ thymocyte stage severely impaired peripheral CD4+, CD8+ and iNKT peripheral T cell compartments [66,69]. This effect was linked to reduced transcript and protein expression and altered transcript splicing of the IL-2R common gamma chain (γc) and JAK3 in naïve and anti-CD3/CD28 stimulated CD4+, CD8+ and in iNK T cells from CD4crePrmt5fl/Δ mice [66]. Ultimately, this resulted in decreased strength of γc receptor-associated cytokine signaling relative to controls [66]. In the course of revising this manuscript, an additional report on CD4crePrmt5fl/fl mice also confirmed loss of iNK T cells in thymus and spleen of these animals, in addition to a requirement for Prmt5 in peripheral CD4+ and CD8+ T cell maintenance [14].
Key Figure, Figure 2. PRMT5 can modulate multiple facets of T cell development and function in humans and mice.
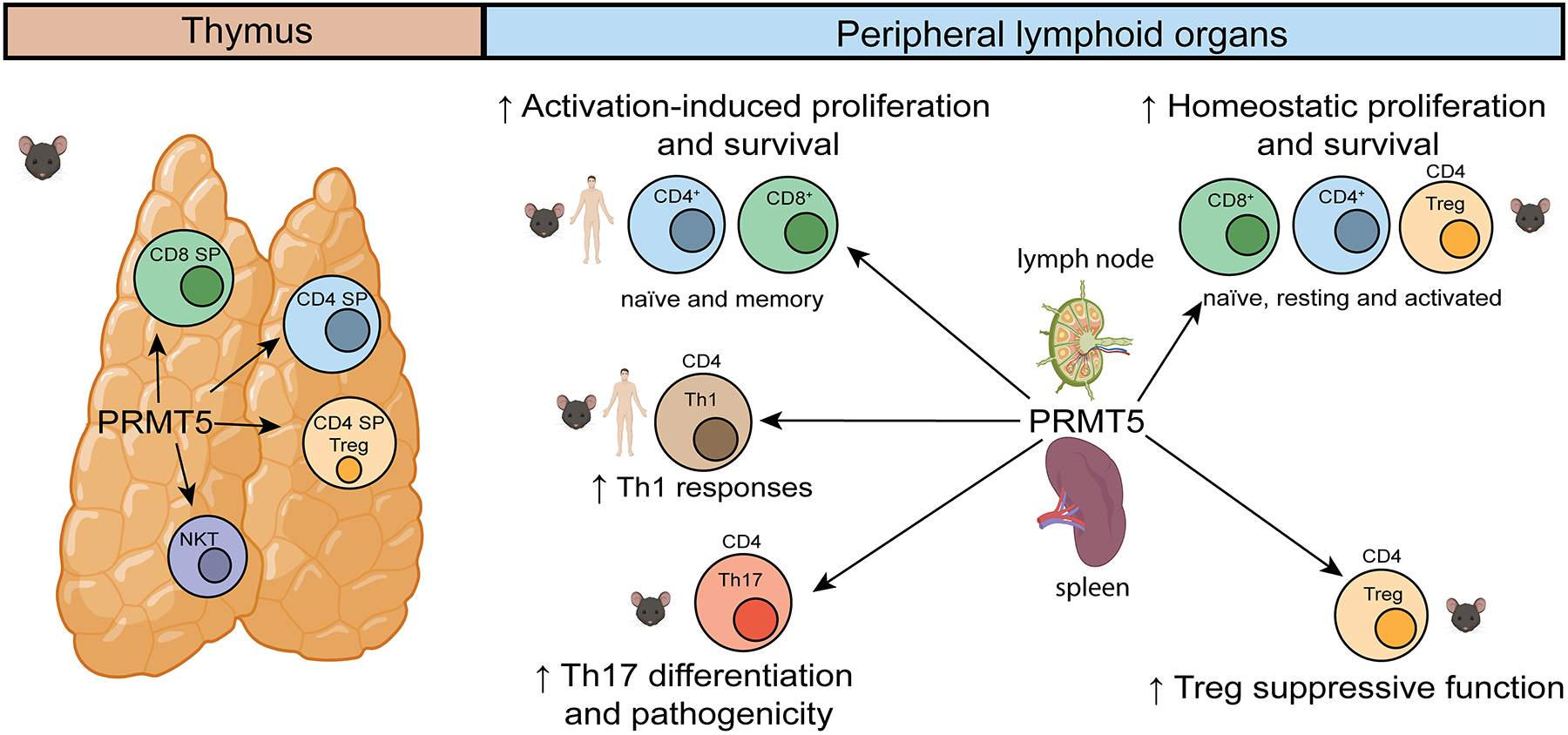
T cell specific, CD4cre-driven, Prmt5 deletion during thymic development in CD4crePrmt5fl/fl or CD4cre-Prmt5fl/Δ mice results in robust defects in thymic numbers of NKT cells[14,66,69]. Additional defects in CD4+ , CD8+ and Treg total thymic numbers, but not percentages, is observed in comparison to defined control mice in CD4crePrmt5fl/fl mice [69]. In spleen and lymph nodes of CD4crePrmt5fl/fl or CD4cre-Prmt5fl/Δ mice, reduced CD4+, CD8+, Treg and iNK T cells indicate a role in homeostatic proliferation [14,66,69]. Studies with human and mouse naïve and memory T cells treated with PRMT5 inhibitors or via siRNA, shRNA or mouse cKO strategies, demonstrate deficient activation and proliferative responses [14,22,66,69]. Tregs in mice with a Treg-specific deletion of Prmt5 (Foxp3crePrmt5fl/fl) have reduced Treg function and develop scurfy-like autoimmunity, consistent with a role for Prmt5 in Treg function [67]. Arrows represent the links between PRMT5 and various immune cells and/or functions. This figure was created using BioRender (https://biorender.com/).
An additional mouse model (CD4creERT2PRMT5fl/fl) used tamoxifen-inducible Prmt5 deletion in CD4+ Th cells in mice to dissect the role of Prmt5 in peripheral immune Th cell responses and disease; this model allowed normal T cell development prior to CD4-creERT2 driven deletion upon tamoxifen treatment [69]. The peripheral CD4+ Th cell Prmt5 cKO mice (short-term tamoxifen-treated CD4creERT2PRMT5fl/fl mice) were completely resistant to EAE development. Moreover, the central nervous system (CNS) of these mice failed to develop in vivo myelin oligodendrocyte glycoprotein (MOG)-specific pathogenic Th17 and Th1 cell responses after MOG immunization, as evidenced from abrogated MOG-splenocyte T cell proliferation and IFN-γ or IL-17+ T cell detection via flow cytometry, as well as impaired recruitment of memory T cells into the CNS, as observed via CNS infiltrating cell quantification at the peak of EAE disease [69]. In addition, a severe defect in the differentiation of naïve T cells towards the Th17 phenotype was observed -- deemed to be mediated by impaired cholesterol metabolism and ROR-γt activation (see below). These results conclusively established that mouse CD4+ T cell Prmt5 was necessary to drive pathogenic Th cell-mediated EAE autoimmunity [69]. This has important implications for autoimmune therapy, although the possibility of global immunosuppressive effects will need to be carefully evaluated.
Mechanisms of PRMT5-dependent control of cellular function.
PRMT5 regulates cell biology and function through a number of mechanisms and methylation targets, resulting in gene expression control and modulation. Various molecules have been identified as direct targets of PRMT5 methylation in vertebrates, invertebrates and/or plants, including histones [71], splicing proteins [72], DNA damage response components such as p53 protein [73,74] as well as signaling pathway components and transcription factors such as NF-kB [75,76], Vav1 [51], SREBP [77] and hnRNPA1 [78] (Fig. 3). The majority of these targets and mechanisms have been identified in cancer or transformed cell studies, and these findings may or may not reflect what occurs in non-transformed lymphocytes. Much is yet unknown regarding the specific mechanisms by which PRMT5 methylation controls lymphocyte development, activation, and differentiation. However, a few breakthroughs have recently provided substantial insights into how PRMT5 methylation promotes T cell proliferation, peripheral homeostasis and Th17 differentiation. In the following section, we discuss these mechanisms and highlight remaining outstanding questions (Outstanding Questions) in the field.
Figure 3. General PRMT5 methylation targets and mechanisms.
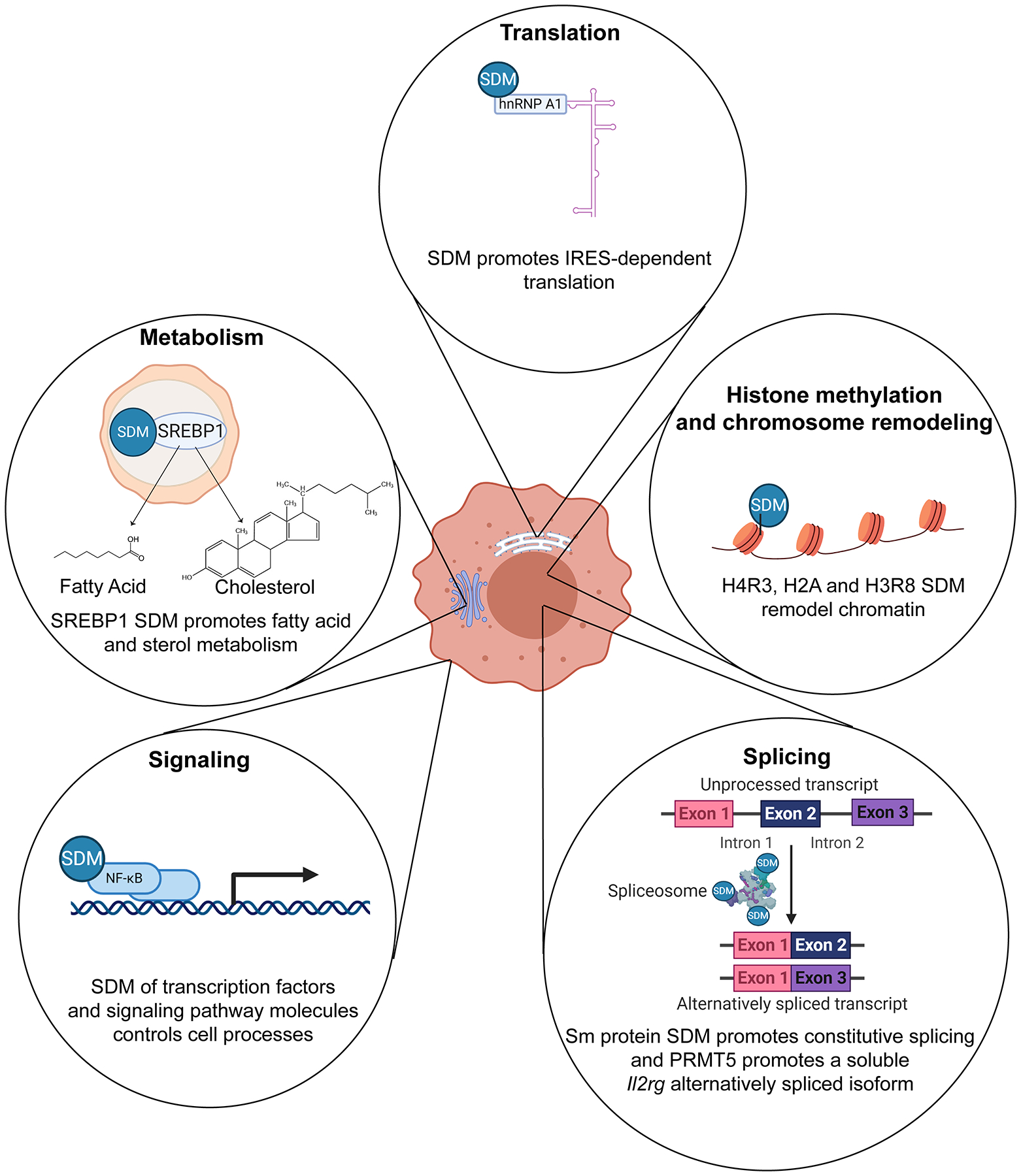
Studies in animal and plant cells show that PRMT5 symmetrically dimethylates (SDM) multiple targets in cytoplasmic and nuclear compartments, thereby influencing key cellular processes. Histone methylation: in the nucleus, SDM of histones H4R3, H3R8 and H2A generally results in chromatin repression via direct histone effects and recruitment of repressor complexes [8,89,90]. Most of the chromatin remodeling effects have been shown in non-T cell models [12]. Splicing: splicing proteins, such as SmD3 and SmB,B’, which can be found in the cytoplasm and nucleus, are also methylated by PRMT5 [82]. SDM of Sm proteins PRMT5 enhances spliceosome assembly and promotes alternative splicing switches [91]. SmD and SmB methylation has been observed in mouse T cells and is thought to increase Il2rg and Jak3 alternative splicing and expression [66]. Signaling: multiple cytoplasmic proteins, such as NF-kB and Vav-1, involved in receptor signaling pathways are methylated by PRMT5 [51,75]. Vav-1 methylation has been observed in Jurkat T cells and is believed to promote T cell costimulation [51]. Metabolism: SREBP is a lipid biogenesis transcription factor located in the endoplasmic reticulum and Golgi. Upon cleavage in the Golgi, SREBP is activated and translocates to the nucleus, where it transactivates fatty acid and cholesterol biosynthesis enzyme expression [92]. PRMT5 has been shown methylate SREBP and promote cholesterol metabolism in mouse T cells, thereby promoting Th17 differentiation [69]. Translation: PRMT5 has been shown to SDM ribonucleoprotein hnRNPA1 in cancer cells, thereby promoting translation of internal ribosomal entry site (IRES)-containing transcripts [78]. This figure was created using BioRender (https://biorender.com/).
Outstanding Questions Box.
Which T cell relevant PRMT5 targets modulate T cell biology and through which mechanisms?
What is the specific role of PRMT5 in the normal biology of other lymphocytes (CD8+ T cells, B cells, NK cells,..)?
Does PRMT5 play a role in other immune cells, such as neutrophils, macrophages, dendritic cells, innate lymphoid cells?
Are PRMT5 alterations linked to human multiple sclerosis or other T cell-mediated diseases?
What is an appropriate benefit/risk ratio of PRMT5 inhibition on immune cell compartments during PRMT5 inhibitor-based cancer treatments?
PRMT5 can promote T cell signaling, IL-2 production and T cell proliferation.
To our knowledge, the first evidence that PRMT5 and SDM played a role in T cell activation was generated in human CD4+ Jurkat T cells, an acute T cell leukemia cell line often used as an in vitro model. In this study, MTA treatment and PRMT5 knockdown suppressed two key surrogates of Jurkat T cell activation, namely IL-2 and NFAT-dependent transcription [42]. Although PRMT5 was not induced after Jurkat T cell activation, this effect was linked to PRMT5 activity when increased methylation in the IL2 locus was observed relative to controls [42]. IL-2 is an essential driver of T cell proliferation, but the impact of PRMT5 on such a process required studies in non-transformed primary T cells. Subsequently, selective PRMT5 inhibition with CMP5 and HLCL65 strongly suppressed IL-2 production and T cell proliferation in mouse and human Th1 and Th2 primary T cells [18]. PRMT5 expression and SDM were induced in these cells in vitro, reaching a maximum peak 2–3 days following TCR engagement with anti-CD3/CD28. Maximal PRMT5 induction coincided with maximal proliferation following TCR-induced activation [18]. The requirement for PRMT5 in mouse and human primary naïve CD4+ Th and differentiated Th1 and Th2 cell proliferation after anti-CD3/CD28 activation has been conclusively validated at the genetic level via Prmt5/PRMT5 shRNA or siRNA knockdown and in CD4creERT2PRMT5fl/fl cKO mouse primary T cell experiments [18,66,69]. Furthermore, suppressed IL-2 production due to PRMT5 inhibitor HLCL65 treatment appeared to mediate -- at least partially -- these CD4+ Th cell proliferation defects, as evidenced from restored Th1 cell proliferation following exogenous IL-2 supplementation [18]. Of note, chromatin immunoprecipitation using an anti-pan-SDM antibody resulted in PMA activation-dependent detection of SDM proteins bound to the IL2 promoter in human Jurkat T cells [42]. Together with the PRMT5-dependence of IL-2 production, this may imply a positive role for SDM by PRMT5 on proteins close to the IL2 locus. However, the nature of these IL2 promoter associated proteins has not been elucidated. While this may be unrelated, probing PMA-activated Jurkat T cells with the pan-SDM antibody after PRMT5 siRNA knockdown showed detectable decreases of SDM on bands corresponding to SmD3 and SmB,B’ spliceosome proteins [42]. Control of TCR signaling by PRMT5 might also potentially mediate some of the proliferative effects of PRMT5 in T cells. Since Vav-1 is thought to interact with NF-kB and NFAT complexes [79], the methylation of Vav-1 is presumed to lead to activation NFAT- and NF-κB-dependent genes during T cell activation [51], although this remains to be robustly tested. Nevertheless, this body of work establishes a crucial role for PRMT5 in T cell expansion and activation and, although mechanistic details are not fully elucidated, these effects may possibly be mediated by promoting TCR signaling and growth factor cytokine and cytokine receptor expression.
PRMT5 can promote γ-chain signaling, controlling NK cell development and homeostatic T cell expansion and survival.
Lymphocyte development, differentiation and survival require signaling through cytokine receptors. Multiple receptors, namely, IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21 receptors, share the γc subunit [80]. Congenital mutations in the γc result in severe combined immunodeficiency (SCID) generally characterized by absent T and NK cells, and dysfunctional B cells in mice and humans [81]. Signaling through γ-chain cytokine receptors is mediated via the JAK3 kinase, and γc mutations result in other types of human SCID disease [81]. Prmt5 promotes normal mouse T and NK cell lymphocyte development and peripheral survival by controlling γc and JAK3 expression [66]. These effects were linked to Prmt5 methylation of splicing proteins and enhanced pre-mRNA splicing of the IL-2 receptor γ-chain, as evidenced from decreased Sm protein SDM, decreased Il2rg and Jak3 expression and changes in il2rg and Jak3 alternatively spliced isoforms in CD4crePRMT5fl/Δ mice CD4+ , CD8+ and iNK T cells [66]. Of note, PRMT5 methylation of Sm proteins has been shown to be essential for spliceosome formation, which is expected to increase constitutive splicing efficiency and gene expression, as well as alter the pattern of alternative splicing in several non-T cell models [72,82]. In mouse CD4+ and CD8+ T cells, at least one Sm protein, SmD3, has been shown to be methylated by Prmt5 [66]. Although a direct causative link between altered Sm methylation/splicing and in vivo effects in CD4crePRMT5fl/Δ has not yet been demonstrated, CD4crePRMT5fl/Δ mice suffered from loss of iNK T cells and a reduction in peripheral lymphoid organ survival of CD4+ Th and CD8+ T cells was observed in mice [66]. Other mouse models where Prmt5 is deleted at thymic or earlier stages of development have also been linked to a robust loss of iNK T cells and reduced total numbers of CD4+ SP, Treg and CD8+ SP thymocytes relative to control mice [68,69], possibly due to reduced thymic expansion. However, the most dramatic loss of CD4+ and CD8+ T cells has been observed in spleen and lymph nodes of CD4crePRMT5fl/fl mice [66,69], consistent with γ-chain and JAK3 signaling roles in peripheral homeostasis and survival [83,84].
PRMT5 can promote cholesterol biosynthesis and Th17 differentiation.
PRMT5 has also been recently recognized as an important modulator of cellular metabolism [69,77,85]. Effects on overall energy metabolism [85], as well as on lipid metabolism [69,77] have been observed in human cancer cells. Specifically, PRMT5 has been shown to promote Warburg anaerobic glycolytic metabolism in human breast and pancreatic cancer cells [85,86], as evidenced from decreased glucose consumption or decreased Extracellular Acidification Rate (ECAR), respectively, following PRMT5 knockdown. Presumably, these metabolic impacts might potentially be mediated by gene expression control exerted by PRMT5 at the chromatin, splicing or translation levels. However, PRMT5 also regulates cellular metabolism by direct methylation of the key lipid metabolism transcription factor sterol response binding protein (SREBP1) [69,77]. SREBP1 is retained in the ER by INSIG-1 interaction, preventing its pro-lipid biogenesis nuclear activity. When cholesterol and lipid concentrations in cellular membranes decrease, due to reduced lipidic/cholesterol availability, SREBP1 is proteolytically cleaved to its active nuclear form [87]. PRMT5 methylation of SREBP on its C-terminal domain increases SREPB stability and nuclear activity relative to unmethylated SREBP and has been linked to high lipid and/or cholesterol biosynthesis [77]. This suggests that PRMT5 methylation may promote lipid and/or cholesterol biosynthesis.
Recently, similar metabolic effects were observed in murine and /or human T lymphocytes[69]. PRMT5 knockdown in human transformed human Jurkat T cells [69] led to reduced glycolytic metabolism-- measured by ECAR—and reduced oxidative phosphorylation --- measured by Oxygen Consumption Rate (OCR), metabolism [69]. These results should also be studied in primary murine and human T cells since Jurkat T cells are derived from a T cell leukemia line, and constitute a highly artefactual model. Of note, PRMT5 has also been recently implicated in lipid metabolism and Th17 cell differentiation. Specifically, CD4crePRMT5fl/fl Th0 and Th17 cells were found to be deficient in the expression of enzymes in the cholesterol biosynthetic pathway in activated primary murine T lymphocytes [69]. This pathway yields cholesterol pathway intermediates (CPI) that directly activate the Th17 transcription factor ROR-γt, as evidenced from induced Th17 differentiation in mouse CD4+ Th cells treated with various cholesterol pathway intermediates [88]. Also, PRMT5 knockdown in Jurkat T cells resulted in loss of SREBP methylation and RORγt activity [69]. Accordingly, PRMT5 expression was found to be essential for Th17 differentiation, as evidenced from the lack of Th17 differentiation in CD4crePRMT5fl/fl CD4 T cells [69]. Also, Th17 differentiation was restored in CD4crePRMT5fl/fl CD4 T cells supplemented with the CPI desmosterol during Th17 differentiation [69]. Finally, in a mouse model of EAE, as discussed above, induced Prmt5 deletion in the peripheral CD4+ Th cell compartment completely protected the animals from disease development relative to WT control mice, revealing the importance of this pathway in this model of autoimmune disease.
Concluding remarks
The importance of PRMT5-catalyzed methylation as a cancer-driver is well established and has resulted in the development of PRMT5 inhibitor cancer therapeutics currently being assessed in various clinical trials. Recent studies have now conclusively established a role for PRMT5 in CD4+, CD8+ and iNK T cell immune responses [18,66,69]. This field therefore constitutes an exciting novel area of Immunology research. Although some mechanistic insights are starting to emerge, many questions remain (Outstanding Questions), particularly with regard to our understanding of the mechanisms by which PRMT5 controls lymphocyte biology. Many of the PRMT5 methylation targets that have been identified in cancer or transformed cell lines are also expressed in mouse and human lymphocytes. Hence, future studies identifying specific PRMT5 methylation targets and the mechanisms they control in non-transformed lymphocytes should address a portion of this knowledge gap. Increased mechanistic insight relevant to the role of PRMT5 in the immune system and across species may help address fundamental basic Immunology questions and possibly reveal additional candidate therapeutic targets to treat certain autoimmune and inflammatory diseases.
Highlights Box.
PRMT5 promotes thymic CD4+ SP, CD8+ SP, Treg and iNK murine T cell production.
PRMT5 promotes activation-induced proliferation in mouse and human T cells.
PRMT5 promotes peripheral homeostatic CD4+ and CD8+ T cell proliferation and survival via modulation of γc cytokine receptor splicing.
PRMT5 promotes Th17 differentiation via enhancement of cholesterol biosynthesis and production of ROR-γt agonistic cholesterol pathway intermediates.
T cell PRMT5 is required for T cell mediated autoimmunity in EAE mouse models of Multiple Sclerosis.
Acknowledgments
MGA is supported by funds from the NIH National Institute of Allergy and Infectious Diseases grants R01AI121405, 1R21AI127354, 1R03AI151769 and The Ohio State University School of Health and Rehabilitation Sciences start-up funds. PT is supported by the funds from the NIH National Cancer Institute grant 01-CA186729 (to PNT). GL is supported by the Pelotonia Postdoctoral Fellowship. For purposes of conflict of interest disclosure, MGA is listed as an inventor in a patent of PRMT5 inhibitors.
Glossary
- DNA damage response
biological network of pathways that detects lesions in DNA and promotes their repair.
- Extracellular Acidification Rate (ECAR)
measure of the cell culture medium acidification that results from lactate production through glycolysis. It is used as a method of quantifying glycolytic activity.
- iNKT cell
type of NKT cell characterized by the expression of an invariant aβ T-cell receptor (TCR) and classical NK cell markers. iNKT cells recognize glycolipid antigens such as α-galactosylceramide presented by the MHC class 1-like molecule CD1d.
- Myelin Oligodendrocyte Glycoprotein (MOG)
glycoprotein present on the myelin sheath. It is a prototypical target antigen in the EAE animal model of multiple sclerosis CNS autoimmunity.
- Oxygen Consumption Rate (OCR)
Quantitative measurement of the loss of culture media oxygen resulting from consumption during tri-carboxylic acid cycle/oxidative phosphorylation. It is used as a measure of oxidative phosphorylation activity.
- ROR-γt
signature transcription factor of inflammatory Th17 cells.
- Scurfy like autoimmunity
Autoimmune condition in the scurfy mouse model caused by mutations in the transcription factor FoxP3 that abrogate Treg development. These mice develop multi-organ inflammation and die at around four weeks of age.
- Th1 cell
An inflammatory T helper cell lineage that produces IFN-γ and characterized by the transcription factor T-bet. Th1 differentiation is triggered by polarizing cytokine interleukin 12 (IL-12).
- Th17 cell
inflammatory T helper cell lineage producing interleukin 17 (IL-17) and characterized by the transcription factor ROR-γt. Th17 differentiation is triggered by transforming growth factor beta (TGF-β), interleukin 6 (IL-6), interleukin 21 (IL-21) and interleukin 23 (IL-23).
- Warburg anaerobic glycolytic metabolism
type of glycolytic metabolism that does not require oxygen. In contrast to classical aerobic glycolysis that provides pyruvate and fuels the TCA cycle/mitochondria oxidative phosphorylation, Warburg glycolysis diverts pyruvate to lactate. Warburg glycolysis is observed in cancer cells and certain types of activated immune cells.
References
- [1].Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet 2018;19:81–92. [DOI] [PubMed] [Google Scholar]
- [2].Luo M. Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem Rev 2018. 10.1021/acs.chemrev.8b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Blanc RS, Richard S. Arginine Methylation: The Coming of Age. Mol Cell 2017;65:8–24. 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- [4].Bedford MT, Richard S. Arginine methylation: An emerging regulator of protein function. Mol Cell 2005;18:263–72. 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- [5].Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol 2019;20:642–57. [DOI] [PubMed] [Google Scholar]
- [6].Gary JD, Clarke S. RNA and protein interactions modulated by protein arginine methylation. Prog Nucleic Acid Res Mol Biol 1998;61:65–131. [DOI] [PubMed] [Google Scholar]
- [7].Yang Y, Hadjikyriacou A, Xia Z, Gayatri S, Kim D, Zurita-Lopez C, et al. PRMT9 is a Type II methyltransferase that methylates the splicing factor SAP145. Nat Commun 2015;6:6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Branscombe TL, Frankel A, Lee JH, Cook JR, Yang Z, Pestka S, et al. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem 2001;276:32971–6. 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- [9].Zurita-Lopez CI, Sandberg T, Kelly R, Clarke SG. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming ω-NG-monomethylated arginine residues. J Biol Chem 2012;287:7859–70. 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bedford MT, Clarke SG. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol Cell 2009;33:1–13. 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science 2015;347:1260419–1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- [12].Shailesh H, Zakaria ZZ, Baiocchi R, Sif S. Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 2018. 10.18632/oncotarget.26404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Litzler LC, Zahn A, Meli AP, Hébert S, Patenaude AM, Methot SP, et al. PRMT5 is essential for B cell development and germinal center dynamics. Nat Commun 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tanaka Y, Nagai Y, Okumura M, Greene MI, Kambayashi T. PRMT5 Is Required for T Cell Survival and Proliferation by Maintaining Cytokine Signaling. Front Immunol 2020;11. 10.3389/fimmu.2020.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Alinari L, Mahasenan KV, Yan F, Karkhanis V, Chung JH, Smith EM, et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015;125:2530–43. 10.1182/blood-2014-12-619783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif SS. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J 2007;26:3558–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chung J, Karkhanis V, Tae S, Yan F, Smith P, Ayers LW, et al. Protein Arginine Methyltransferase 5 (PRMT5) Inhibition Induces Lymphoma Cell Death through Reactivation of the Retinoblastoma Tumor Suppressor Pathway and Polycomb Repressor Complex 2 (PRC2) Silencing. J Biol Chem 2013;288:35534–47. 10.1074/jbc.M113.510669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Webb LM, Amici SA, Jablonski KA, Savardekar H, Panfil AR, Li L, et al. PRMT5-Selective Inhibitors Suppress Inflammatory T Cell Responses and Experimental Autoimmune Encephalomyelitis. J Immunol 2017;198:1439–51. 10.4049/jimmunol.1601702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Webb LM, Narvaez Miranda J, Amici SA, Sengupta S, Nagy G, Guerau-de-Arellano M. NF-κB/mTOR/MYC Axis Drives PRMT5 Protein Induction After T Cell Activation via Transcriptional and Non-transcriptional Mechanisms. Front Immunol 2019;10:524. 10.3389/fimmu.2019.00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].German DC, Bloch CA, Kredich NM. Measurements of S-adenosylmethionine and L-homocysteine metabolism in cultured human lymphoid cells. J Biol Chem 1983;258:10997–1003. [PubMed] [Google Scholar]
- [21].Zhang J, Zheng YG. SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases. ACS Chem Biol 2016;11:583–97. 10.1021/acschembio.5b00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Webb LM, Guerau-de-Arellano M. Emerging Role for Methylation in Multiple Sclerosis: Beyond DNA. 2017. 10.1016/j.molmed.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Borchardt RT, Eiden LE, Wu BS, Rutledge CO. Sinefungin, a potent inhibitor of S-adenosylmethionine: Protein O-methyltransferase. Biochem Biophys Res Commun 1979. 10.1016/0006-291X(79)91866-7. [DOI] [PubMed] [Google Scholar]
- [24].Clarke SG. 16 Inhibition of mammalian protein methyltransferases by 5’-methylthioadenosine (MTA): A mechanism of action of dietary same? The Enzymes 2006;24:467–93. 10.1016/S1874-6047(06)80018-1. [DOI] [PubMed] [Google Scholar]
- [25].Wolos JA, Frondorf KA, Babcock GF, Stripp SA, Bowlin TL. Immunomodulation by an inhibitor of S-adenosyl-L-homocysteine hydrolase: inhibition of in vitro and in vivo allogeneic responses. Cell Immunol 1993;149:402–8. 10.1006/cimm.1993.1165. [DOI] [PubMed] [Google Scholar]
- [26].Saso Y, Conner EM, Teegarden BR, Yuan C-S. S-Adenosyl-l-homocysteine Hydrolase Inhibitor Mediates Immunosuppressive Effects in Vivo: Suppression of Delayed Type Hypersensitivity Ear Swelling and Peptidoglycan Polysaccharide-Induced Arthritis. J Pharmacol Exp Ther 2001;296. [PubMed] [Google Scholar]
- [27].Wu Q-L, Fu Y-F, Zhou W-L, Wang J-X, Feng Y-H, Liu J, et al. Inhibition of S-adenosyl-L-homocysteine hydrolase induces immunosuppression. J Pharmacol Exp Ther 2005;313:705–11. 10.1124/jpet.104.080416. [DOI] [PubMed] [Google Scholar]
- [28].Donofrio J, Coleman MS, Hutton JJ, Daoud A, Lampkin B, Dyminski J. Overproduction of adenine deoxynucleosides and deoxynucleotides in adenosine deaminase deficiency with severe combined immunodeficiency disease. J Clin Invest 1978. 10.1172/JCI109201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hershfield MS, Kredich NM, Ownby DR, Ownby H, Buckley R. In vivo inactivation of erythrocyte S-adenosylhomocysteine hydrolase by 2’-deoxyadenosine in adenosine deaminase-deficient patients. J Clin Invest 1979. 10.1172/JCI109367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hershfield MS. Apparent suicide inactivation of human lymphoblast S-adenosyl-homocysteine hydrolase by 2’-deoxyadenosine and adenine arabinoside. A basis for direct toxic effects of analogs of adenosine. J Biol Chem 1979;254:22–5. [PubMed] [Google Scholar]
- [31].Meuwissen HJ, Pollara B, Pickering RJ. Combined immunodeficiency disease associated with adenosine deaminase deficiency. Report on a workshop held in Albany, New York, October 1, 1973. J Pediatr 1975;86:169–81. 10.1016/s0022-3476(75)80463-x. [DOI] [PubMed] [Google Scholar]
- [32].Buckley RH, Schiff RI, Schiff SE, Markert ML, Williams LW, Harville TO, et al. Human severe combined immunodeficiency: Genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr 1997. 10.1016/S0022-3476(97)70199-9. [DOI] [PubMed] [Google Scholar]
- [33].Cassani B, Mirolo M, Cattaneo F, Benninghoff U, Hershfield M, Carlucci F, et al. Altered intracellular and extracellular signaling leads to impaired T-cell functions in ADA-SCID patients. Blood 2008;111:4209–19. 10.1182/blood-2007-05-092429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA-SCID: Initial trial results after 4 years. Science 1995. 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- [35].Johnston JM, Kredich NM. Inhibition of methylation by adenosine in adenosine deaminase-inhibited, phytohemagglutinin-stimulated human lymphocytes. J Immunol Baltim Md 1950 1979;123:97–103. [PubMed] [Google Scholar]
- [36].Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem 1998. 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- [37].Kim Bok Ryang, Yang Kyu Hwan. Effects of sinefungin and 5′-deoxy-5′-S-isobutyl-adenosine on lipopolysaccharide-induced proliferation and protein N-methylation of arginyl residues in murine splenic lymphocytes. Toxicol Lett 1991. 10.1016/0378-4274(91)90061-A. [DOI] [PubMed] [Google Scholar]
- [38].Wolford RW, MacDonald MR, Zehfus B, Rogers TJ, Ferro AJ. Effect of 5”-methylthioadenosine and its analogs on murine lymphoid cell proliferation. Cancer Res 1981;41:3035–9. [PubMed] [Google Scholar]
- [39].Christa L, Thuillier L, Perignon JL. 5’-Deoxy-5’-methylthioadenosine inhibition of rat T lymphocyte phosphodiesterase: correlation with inhibition of Con A induced proliferation. Biochem Biophys Res Commun 1983;113:425–32. 10.1016/0006-291x(83)91743-6. [DOI] [PubMed] [Google Scholar]
- [40].Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013;13:37–50. [DOI] [PubMed] [Google Scholar]
- [41].Moreno B, Hevia H, Santamaria M, Sepulcre J, Muñoz J, García-Trevijano ER, et al. Methylthioadenosine reverses brain autoimmune disease. Ann Neurol 2006;60:323–34. 10.1002/ana.20895. [DOI] [PubMed] [Google Scholar]
- [42].Richard S, Morel M, Eroux PC. Arginine methylation regulates IL-2 gene expression: a role for protein arginine methyltransferase 5 (PRMT5). Biochem J 2005;388:379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bonham K, Hemmers S, Lim Y-H, Hill DM, Finn MG, Mowen KA. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production. FEBS J 2010;277:2096–108. 10.1111/j.1742-4658.2010.07623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lawson BR, Manenkova Y, Ahamed J, Chen X, Zou J-P, Baccala R, et al. Inhibition of Transmethylation Down-Regulates CD4 T Cell Activation and Curtails Development of Autoimmunity in a Model System. J Immunol 2007. 10.4049/jimmunol.178.8.5366. [DOI] [PubMed] [Google Scholar]
- [45].Yang M-L, Gee AJP, Gee RJ, Zurita-Lopez CI, Khare S, Clarke SG, et al. Lupus autoimmunity altered by cellular methylation metabolism. Autoimmunity 2013;46:21–31. 10.3109/08916934.2012.732133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fu Y-F, Zhu Y-N, Ni J, Zhong X-G, Tang W, Re Y-D, et al. A reversible S-adenosyl-L-homocysteine hydrolase inhibitor ameliorates experimental autoimmune encephalomyelitis by inhibiting T cell activation. J Pharmacol Exp Ther 2006;319:799–808. 10.1124/jpet.106.107185. [DOI] [PubMed] [Google Scholar]
- [47].Moreno B, Fernandez-Diez B, Penta A Di, Villoslada P. Preclinical studies of methylthioadenosine for the treatment of multiple sclerosis n.d 10.1177/1352458510375968. [DOI] [PubMed] [Google Scholar]
- [48].Henrich FC, Singer K, Poller K, Bernhardt L, Strobl CD, Limm K, et al. Suppressive effects of tumor cell-derived 5′-deoxy-5′-methylthioadenosine on human T cells. OncoImmunology 2016. 10.1080/2162402X.2016.1184802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Parry RV, Ward SG. Protein arginine methylation: a new handle on T lymphocytes? Trends Immunol 2010;31:164–9. 10.1016/j.it.2010.01.006. [DOI] [PubMed] [Google Scholar]
- [50].Smith-Garvin JE, Koretzky GA, Jordan MS. T Cell Activation. Annu Rev Immunol 2009;27:591–619. 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Blanchet F, Cardona A, Letimier FA, Hershfield MS, Acuto O. CD28 costimulatory signal induces protein arginine methylation in T cells. J Exp Med 2005;202:371–7. 10.1084/jem.20050176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cheng D, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small Molecule Regulators of Protein Arginine Methyltransferases. J Biol Chem 2004;279:23892–9. 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- [53].Chiang K, Zielinska AE, Shaaban AM, Sanchez-Bailon MP, Jarrold J, Clarke TL, et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep 2017;21:3498–513. 10.1016/j.celrep.2017.11.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sheng X, Wang Z. Protein arginine methyltransferase 5 regulates multiple signalling pathways to promote lung cancer cell proliferation. BMC Cancer 2016;16:567. 10.1186/s12885-016-2632-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Penebre E, Kuplast KG, Majer CR, Johnston LD, Rioux N, Munchhof M, et al. Identification of a First-in-Class PRMT5 Inhibitor with Potent in Vitro and in Vivo Activity in Preclinical Models of Mantle Cell Lymphoma. Blood 2014;124:438–438. 10.1182/blood.v124.21.438.438. [DOI] [Google Scholar]
- [56].Gerhart SV, Kellner WA, Thompson C, Pappalardi MB, Zhang X-P, Montes de Oca R, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep 2018;8:9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ye Y, Zhang B, Mao R, Zhang C, Wang Y, Xing J, et al. Discovery and optimization of selective inhibitors of protein arginine methyltransferase 5 by docking-based virtual screening. Org Biomol Chem 2017;15:3648–61. 10.1039/c7ob00070g. [DOI] [PubMed] [Google Scholar]
- [58].Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y, et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest 2016;126:3961–80. 10.1172/JCI85239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ji S, Ma S, Wang W-J, Huang S-Z, Wang T-Q, Xiang R, et al. Discovery of selective protein arginine methyltransferase 5 inhibitors and biological evaluations. Chem Biol Drug Des 2017;89:585–98. 10.1111/cbdd.12881. [DOI] [PubMed] [Google Scholar]
- [60].Bonday ZQ, Cortez GS, Grogan MJ, Antonysamy S, Weichert K, Bocchinfuso WP, et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med Chem Lett 2018;9:612–7. 10.1021/acsmedchemlett.8b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 2016;30:789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Strobl CD, Schaffer S, Haug T, Völkl S, Peter K, Singer K, et al. Selective PRMT5 Inhibitors Suppress Human CD8+ T Cells by Upregulation of p53 and Impairment of the AKT Pathway Similar to the Tumor Metabolite MTA. Mol Cancer Ther 2020;19:409–19. 10.1158/1535-7163.MCT-19-0189. [DOI] [PubMed] [Google Scholar]
- [63].Metz PJ, Ching KA, Xie T, Delgado Cuenca P, Niessen S, Tatlock JH, et al. Symmetric Arginine Dimethylation Is Selectively Required for mRNA Splicing and the Initiation of Type I and Type III Interferon Signaling. Cell Rep 2020;30:1935–1950.e8. 10.1016/j.celrep.2020.01.054. [DOI] [PubMed] [Google Scholar]
- [64].Snyder K, Zitzer NC, Gao Y, Choe HK, Sell NE, Neidemire-Colley L, et al. PRMT5 regulates T cell interferon response and is a target for acute graft-versus-host disease. JCI Insight 2020. 10.1172/jci.insight.131099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tee W-W, Pardo M, Theunissen TW, Yu L, Choudhary JS, Hajkova P, et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev 2010;24:2772–7. 10.1101/gad.606110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Inoue M, Okamoto K, Terashima A, Nitta T, Muro R, Negishi-Koga T, et al. Arginine methylation controls the strength of γc-family cytokine signaling in T cell maintenance. Nat Immunol 2018;19:1265–76. [DOI] [PubMed] [Google Scholar]
- [67].Nagai Y, Ji MQ, Zhu F, Xiao Y, Tanaka Y, Kambayashi T, et al. PRMT5 Associates With the FOXP3 Homomer and When Disabled Enhances Targeted p185erbB2/neu Tumor Immunotherapy. Front Immunol 2019;10:174. 10.3389/fimmu.2019.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Liu F, Cheng G, Hamard P-J, Greenblatt S, Wang L, Man N, et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest 2015;125:3532–44. 10.1172/JCI81749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Webb LM, Sengupta S, Edell C, Piedra-Quintero ZL, Amici SA, Miranda JN, et al. Protein arginine methyltransferase 5 promotes cholesterol biosynthesis–mediated Th17 responses and autoimmunity. J Clin Invest 2020;130:1683–98. 10.1172/jci131254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, et al. A Critical Role for Dnmt1 and DNA Methylation in T Cell Development, Function, and Survival. Immunity 2001;15:763–74. 10.1016/S1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- [71].Burgos ES, Wilczek C, Onikubo T, Bonanno JB, Jansong J, Reimer U, et al. Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J Biol Chem 2015;290:9674–89. 10.1074/jbc.M115.636894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Meister G, Eggert C, Bühler D, Brahms H, Kambach C, Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol 2001;11:1990–4. 10.1016/S0960-9822(01)00592-9. [DOI] [PubMed] [Google Scholar]
- [73].Jansson M, Durant ST, Cho E-C, Sheahan S, Edelmann M, Kessler B, et al. Arginine methylation regulates the p53 response. Nat Cell Biol 2008;10:1431–9. [DOI] [PubMed] [Google Scholar]
- [74].Scoumanne A, Zhang J, Chen X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res 2009;37:4965–76. 10.1093/nar/gkp516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang D-B, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc Natl Acad Sci U S A 2013;110:13516–21. 10.1073/pnas.1311784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Harris DP, Bandyopadhyay S, Maxwell TJ, Willard B, DiCorleto PE. Tumor necrosis factor (TNF)-α induction of CXCL10 in endothelial cells requires protein arginine methyltransferase 5 (PRMT5)-mediated nuclear factor (NF)-κB p65 methylation. J Biol Chem 2014;289:15328–39. 10.1074/jbc.M114.547349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Liu L, Zhao X, Zhao L, Li J, Yang H, Zhu Z, et al. Arginine methylation of SREBP1a via PRMT5 promotes de novo lipogenesis and tumor growth. Cancer Res 2016;76:1260–72. 10.1158/0008-5472.CAN-15-1766. [DOI] [PubMed] [Google Scholar]
- [78].Gao G, Dhar S, Bedford MT. PRMT5 regulates IRES-dependent translation via methylation of hnRNP A1. Nucleic Acids Res 2017;45:4359–69. 10.1093/nar/gkw1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Houlard M, Arudchandran R, Regnier-Ricard F, Germani A, Gisselbrecht S, Blank U, et al. Vav1 is a component of transcriptionally active complexes. J Exp Med 2002;195:1115–27. 10.1084/jem.20011701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, et al. Cutting Edge: The Common γ-Chain Is an Indispensable Subunit of the IL-21 Receptor Complex. J Immunol 2001;167:1–5. 10.4049/jimmunol.167.1.1. [DOI] [PubMed] [Google Scholar]
- [81].Kohn LA, Seet CS, Scholes J, Codrea F, Chan R, Zaidi-Merchant S, et al. Human Lymphoid Development in the Absence of Common γ-Chain Receptor Signaling. J Immunol 2014;192:5050–8. 10.4049/jimmunol.1303496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Friesen WJ, Paushkin S, Wyce A, Massenet S, Pesiridis GS, Van Duyne G, et al. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol 2001;21:8289–300. 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sohn SJ, Forbush KA, Nguyen N, Witthuhn B, Nosaka T, Ihle JN, et al. Requirement for Jak3 in mature T cells: its role in regulation of T cell homeostasis. J Immunol Baltim Md 1950 1998;160:2130–8. [PubMed] [Google Scholar]
- [84].Nakajima H, Shores EW, Noguchi M, Leonard WJ. The Common Cytokine Receptor γ Chain Plays an Essential Role in Regulating Lymphoid Homeostasis. J Exp Med 1997;185:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Qin Y, Hu Q, Xu J, Ji S, Dai W, Liu W, et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun Signal 2019;17:30. 10.1186/s12964-019-0344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Han X, Wei L, Wu B. PRMT5 Promotes Aerobic Glycolysis and Invasion of Breast Cancer Cells by Regulating the LXRα/NF-κBp65 Pathway. OncoTargets Ther 2020;13:3347–57. 10.2147/OTT.S239730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun Lond Engl 2018;38:27. 10.1186/s40880-018-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hu X, Wang Y, Hao L-Y, Liu X, Lesch CA, Sanchez BM, et al. Sterol metabolism controls T(H)17 differentiation by generating endogenous RORγ agonists. Nat Chem Biol 2015;11:141–7. [DOI] [PubMed] [Google Scholar]
- [89].Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004;24:9630–45. 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Pei Y, Niu L, Lu F, Liu C, Zhai J, Kong X, et al. Mutations in the Type II Protein Arginine Methyltransferase AtPRMT5 Result in Pleiotropic Developmental Defects in Arabidopsis. Plant Physiol 2007;144:1913–23. 10.1104/pp.107.099531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 2013;27:1903–16. 10.1101/gad.219899.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bertolio R, Napoletano F, Mano M, Maurer-Stroh S, Fantuz M, Zannini A, et al. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat Commun 2019;10:1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Williams-Ashman HG, Seidenfeld J, Galletti P. Trends in the biochemical pharmacology of 5′-deoxy-5′-methylthioadenosine. Biochem Pharmacol 1982. 10.1016/0006-2952(82)90171-X. [DOI] [PubMed] [Google Scholar]
- [94].Wolos JA, Frondorf KA, Esser RE. Immunosuppression mediated by an inhibitor of S-adenosyl-L-homocysteine hydrolase. Prevention and treatment of collagen-induced arthritis. J Immunol Baltim Md 1950 1993;151:526–34. [PubMed] [Google Scholar]
- [95].Wolos JA, Frondorf KA, Davis GF, Jarvi ET, McCarthy JR, Bowlin TL. Selective inhibition of T cell activation by an inhibitor of S-adenosyl-L-homocysteine hydrolase. J Immunol Baltim Md 1950 1993;150:3264–73. [PubMed] [Google Scholar]
- [96].Lawson BR, Manenkova Y, Ahamed J, Chen X, Zou J-P, Baccala R, et al. Inhibition of Transmethylation Down-Regulates CD4 T Cell Activation and Curtails Development of Autoimmunity in a Model System. J Immunol 2007. 10.4049/jimmunol.178.8.5366. [DOI] [PubMed] [Google Scholar]
- [97].Tardif V, Manenkova Y, Berger M, Hoebe K, Zuo J-P, Yuan C, et al. Critical role of transmethylation in TLR signaling and systemic lupus erythematosus. Clin Immunol Orlando Fla 2013;147:133–43. 10.1016/j.clim.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]