

Abstract
Rhabdomyosarcomas comprise the single largest category of soft tissue sarcomas in children and adolescents in the United States, occurring in 4.5 million persons aged less than 20 years. Based on the clinic-pathologic features and genetic abnormalities identified, the rhabdomyosarcomas are classified into embryonal, alveolar, spindle cell / sclerosing and pleomorphic subtypes. Each subtype shows distinctive morphology and has characteristic genetic abnormalities. This review discusses the evolution of the classification of rhabdomyosarcoma to the present day, along with a discussion of key histomorpholgic and genetic features of each subtype and the diagnostic approach to these tumors.
Keywords: Rhabdomyosarcoma, Embryonal, Alveolar, Spindle cell / Sclerosing, Pleomorphic, MYOD1, FOXO1
HISTORICAL CLASSIFICATION OF RHABDOMYOSARCOMA
The first reports of rhabdomyosarcoma were in the mid-nineteenth century when Guersant described a polypoid tumor arising in the vagina of a young girl. 1 Due to the resemblance to a bunch of grapes, this tumor was termed as sarcoma botryoides. The next variant of rhabdomyosarcoma to be described was the embryonal rhabdomyosarcoma in the late nineteenth century by Berard, a french pathologist.1 In 1946, Stout described another variant of rhabdomyosarcoma and termed it Pleomorphic rhabdomyosarcoma.2 In 1956, Riopelle and Theriault described 6 cases of alveolar variant of rhabdomyosarcoma, with a morphologic emphasis on the alveolar histology with intervening fibrous septa.3
The first attempt at classification of rhabdomyosarcomas was by Horn and Enterline in 19584, when they collected 39 cases (23 from Hospital of University of Pennsylvania, 7 from the Children’s Hospital of Pennsylvania, 5 from Henry Ford Hospital, and 4 from other sources) and classified them into 4 distinct morphologic variants – Botryoid, Embryonal, Alveolar and Pleomorphic. (Figure 1)This was a classification based on clinical and pathological characteristics. They acknowledged that the botryoid and embryonal were closely related, differing chiefly in the distinctive gross appearance and location of the botryoid variant. The alveolar variant was recognized to occur in adolescents and young adults and involving extremities. The pleomorphic variant were those that occurred in the extremities of older age groups. This system was the most utilized classification system for much of the latter half of the twentieth century.
Figure 1:
Timeline of Classification of Rhabdomyosarcoma.
In 1989, the French group of Caillaud et al.5 published the SIOP (International Society of Pediatric Oncology) rhabdomyosarcoma classification based on over 500 soft tissue tumors in pediatric patients reviewed at 30 European centers between 1975 and 1983. The main subtypes were Embryonal, Alveolar and Pleomorphic Rhabdomyosarcomas. The embryonal rhabdomyosarcomas (ERMS) were classified into A. Loose and B. Dense types. The Loose ERMS were further subclassified into 1. Botryoid and 2. Non-botryoid; and the Dense ERMS were subclassified into 1. Poorly differentiated and 2. Well-differentiated. There was also a Not otherwise specified (NOS) subtype. (Figure 1)
In 1990, Tsokos et al.6 published an NCI classification of rhabdomyosarcomas using 159 cases, which differed from the conventional classification by taking into account both the pattern of growth and cytologic features of the tumor cells. This was the first time the group of solid tumors with compact round cells, which were previously classified under the embryonal rhabdomyosarcoma group, were classified under the alveolar subtype despite the lack of an alveolar architecture. These tumors were referred to as ‘solid alveolar rhabdomyosarcoma’. They noted that many rhabdomyosarcomas exhibiting a solid pattern of growth show an alveolar pattern in their metastatic sites or on extensive sampling of the primary tumor. They further classified all of the embryonal subgroups under the ‘favorable’ category and all forms of ‘alveolar’ subgroups under the ‘unfavorable’ category. (Figure 1)
Newton and colleagues from the Intergroup Rhabdomyosarcoma Study, using a large number of 800 rhabdomyosarcomas, published an IRS classification of rhabdomyosarcoma in 1995.7 In this study, the authors proposed a new system with the subtypes based on prognostic significance. The classification included tumors with I. Superior prognosis (Botryoid and Spindle cell rhabdomyosarcoma) II. Intermediate prognosis (Embryonal rhabdomyosarcoma) III. Poor prognosis (Alveolar rhabdomyosarcoma and undifferentiated sarcoma) and IV. Subtype where prognosis was not evaluable (Rhabdomyosarcoma with rhabdoid features).
Role of Immunohistochemistry and Molecular testing in the classification of Rhabdomyosarcoma.
The increasing use of immunohistochemistry in the 1990s lead to usage of markers specific to skeletal muscle phenotype, including desmin, Myogenin and MYOD1. This led to more accurate and reliable diagnosis of rhabdomyosarcoma. The pleomorphic variant, over the years, included spindle cell pleomorphic sarcomas which were morphologically classified as pleomorphic rhabdomyosarcomas since they involved the skeletal muscle. The usage of immunohistochemical markers helped to exclude the morphologic mimics including malignant fibrous histiocytoma (now termed undifferentiated pleomorphic sarcoma). It was also noticed that there were some differences with respect to Myogenin staining among the embryonal and alveolar subtypes. The alveolar rhabdomyosarcomas tended to stain diffusely for Myogenin while the staining was patchy in embryonal rhabdomyosarcomas.8
Karyotyping studies conducted in the 1990s showed that alveolar rhabdomyosarcomas have chromosomal translocations involving the t(2;13) and less commonly t(1;13). 9–12 Further studies using molecular methods led to the identification of the PAX3-FOXO1 and PAX7-FOXO1 gene fusions corresponding to these chromosomal translocations.13, 14 The finding of gene fusions and application of molecular ancillary testing in rhabdomyosarcomas further refined the classification by grouping the fusion-positive tumors into the alveolar subtype irrespective of the cytomorphology. Subsequent studies classifying the alveolar rhabdomyosarcomas into fusion-positive and fusion-negative subgroups showed that failure-free survival of the fusion-positive group was worse than the fusion-negative group, irrespective of histologic features. There was no difference noted between the fusion-negative alveolar rhabdomyosarcomas and the embryonal rhabdomyosarcomas.15
Evolution of the Spindle cell / Sclerosing Rhabdomyosarcoma subtype.
The term spindle cell RMS was first introduced by the German-Italian Cooperative Sarcoma Study16 to distinguish it from the more common ERMS group, based on its distinctive clinicopathologic features and favorable outcome. Spindle cell RMS was defined as a bland spindle cell proliferation, with eosinophilic, fibrillary cytoplasm, resembling true smooth muscle differentiation. The cells were typically arranged in intersecting long fascicles, reminiscent of the ‘‘herring bone’’ pattern of adult-type fibrosarcoma. In all tumors, evidence of rhabdomyoblasts with cross-striations were noted, as well as occasional areas of classic ERMS, composed of a mixture of spindle, stellate, and round cells embedded in a myxoid stroma. The findings were then confirmed in a larger study of 800 RMS tumors from the Intergroup RMS Study (IRS).17 Patients with spindle cell RMS of non-paratesticular sites had more extensive disease compared with patients with tumors in paratesticular location. Spindle cell RMS in adults was initially reported by Rubin et al.18 and, subsequently by other authors19, 20, and described as cellular proliferation of spindle cells with morphology closely resembling fibrosarcoma, leiomyosarcoma or MPNST. In contrast with the favorable behavior reported in the pediatric age group, spindle cell RMS in adults was shown to follow an aggressive course and had a predilection for head and neck location.
Sclerosing RMS in adults was first recognized in small case series by Mentzel and Katenkamp21 and Folpe et al.22 It was defined as a ‘sclerosing, pseudovascular RMS in adults’, showing an extensive hyalinized matrix, which may mimic osteosarcoma or angiosarcoma. Although the focal alveolar architecture and the primitive round cytologic appearance suggested that it may be closely related to ARMS, the occasional presence of strap cells, the low level of myogenin expression and the absence of FOXO1-related fusions, pointed more toward an ERMS variant. The anatomic location from these combined series showed an equal distribution for head and neck and limbs, with 3 cases each.21, 22 Subsequently, sclerosing RMS was investigated in a large cohort of children and adolescents from the Intergroup RMS Study (IRS)/Children Oncology Group.23 The authors studied 13 sclerosing RMS patients, showing a 1:1 gender ratio and being equally distributed between head and neck and extremities. The clinical behavior of sclerosing RMS in pediatric age group was quite favorable, reminiscent of the outcome reported in the pediatric spindle cell RMS.16 Subsequently, it was suggested that due to overlapping morphologic features, spindle cell RMS and sclerosing RMS may represent a morphological spectrum of a distinct variant of RMS, separate from ERMS and ARMS, although unifying genetic abnormalities are still to be discovered. 24
Based on these findings, Spindle cell / sclerosing rhabdomyosarcomas were classified as a separate entity for the first time in the WHO 2013 classification of soft tissue and bone tumors.25 Subsequently, numerous molecular studies have shed light on the various subsets in this group of tumors.
Recent molecular studies on SpRMS have shed light on the genetic alterations of this entity and to molecularly classify it into three categories. (a) SpRMS occurring in the infantile/congenital setting which show recurrent NCOA2 and VGLL2 associated gene fusions including VGLL2-CITED2, VGLL2-NCOA2, TEAD1-NCOA2 and SRF-NCOA2. 26, 27 (b) MYOD1 p.L122R gene mutations have been identified as a frequent event in adult and pediatric spindle cell RMS.28–30 A subset (up to one-third) of the MYOD1-mutant SpRMS have co-existent mutations in the PIK3CA gene mutations.29, 31More recent studies have shown that a small subset of MYOD1-mutatnt RMS have associated deep deletions of CDKN2A gene.32 (c) SpRMS with no known recurrent abnormalities. Recent studies note that MYOD1 mutated SpRMS are aggressive tumors and show a worse prognosis in both pediatric and adult patients. 29, 31
2020 WHO Classification
In the most recent 2020 WHO classification33, the four subtypes of the 2013 WHO classification - Embryonal rhabdomyosarcoma, alveolar rhabdomyosarcoma (ARMS), pleomorphic rhabdomyosarcoma (PRMS) and spindle cell / sclerosing rhabdomyosarcoma (SRMS) are retained with additional mention of a few novel subtypes.
Newer subtypes of Rhabdomyosarcoma
More recently, a novel subtype of rhabomyosarcoma with a predilection for intraosseous locations and genetically characterized by EWSR1 / FUS – TFCP2 gene fusions were described.34, 35 These tumor show a spindled and epithelioid morphology and affect patients of all age groups. They have a predilection for the craniofacial bones.36 Interestingly, they also show positivity for epithelial markers (cytokeratin and / or EMA) and show overexpression of ALK. In the most recent 2020 WHO classification, this group has been tentatively classified under the spindle cell / sclerosing rhabdomyosarcoma.
A second novel group of rhabdomyosarcomas with the MEIS1-NCOA2 gene fusion have recently been described.35 They also show predilection for osseous locations and seem associated with an aggressive course. Additional studies with larger numbers of these cases will be helpful to shed light on the biology of these tumors and as to where they will be grouped in subsequent classifications.
HISTOMORPHOLOGY AND GENETICS OF RHABDOMYOSARCOMA SUBTYPES
The key features of each subtype are listed in Table 1.
Table 1.
Clinico-pathologic features and genetic alterations of Rhabdomyosarcoma subtypes.
| RMS Subtype | Age (at presentation) | Location | Morphology | Genetic alterations | Prognosis |
|---|---|---|---|---|---|
| Embryonal RMS | Pediatric (0-5) | H&N, GU, GYN tract | primitive to small blue round cells, scattered rhabdomyoblasts, botryoid pattern | Aneuploidy with chromosomal gains and losses; Alterations of the RAS family genes (HRAS, NRAS, KRAS), FGFR4, PIK3CA, NF1 and FBXW7 | Favorable |
| Alveolar RMS | Pediatric (10-20) | Extremities | alveolar pattern, sheets of medium sized cells, scattered giant cells | PAX3-FOXO1 gene fusion; PAX7-FOXO1 gene fusion | Poor |
| Spindle cell / sclerosing RMS | Infantile, Pediatric and Adults | H&N, paratesticular, extremities | spindle cells in fascicles; sclerosing ‘pseudovascular’ pattern | VGLL2 / NCOA2 gene fusions (infantile) ; MYOD1 gene mutation | Infantile – favorable; MYOD1 mutant - poor |
| Pleomorphic RMS | Adults | Extremities | pleomorphic rhabdomyoblasts | Complex genetic alterations | Poor |
Embryonal Rhabdomyosarcoma (ERMS)
ERMS is a primitive, malignant soft tissue tumor that recapitulates the phenotypic and biological features of embryonic skeletal muscle. It is the most common subtype, occurring in 2.6 per million children ages less than 15 years in the United States, with children less than 5 years of age being most commonly affected. They are more common in males than females with a ratio of 1.4:1.25 The tumors occur in equal proportion in the head and neck and the genitourinary system. Common locations in the genitourinary tract include the urinary bladder, prostate, vulva / vagina, cervix and paratesticular soft tissues. 25, 37 Besides these two general regions, ERMS occur in the biliary tract, retroperitoneum, pelvis, perineum, and abdomen and have been reported in various visceral organs, such as the liver, kidney, heart, and lungs. The botryoid variant of ERMS arise beneath a mucosal epithelial surface, limiting it to organs such as the urinary bladder, biliary tract, vulva / vagina, cervix, pharynx, conjunctiva, or auditory canal.
Botryoid tumors have a characteristic polypoid appearance with clusters of small, sessile or pedunculated nodules that abut an epithelial surface.
Histologically, ERMS show primitive oval to spindle cells with minimal cytoplasm. The background can be loose myxoid or the cells can be compactly arranged in sheets. (Figure 2A–B) Some areas show small blue round cell morphology. As these cells differentiate, they progressively acquire more cytoplasmic eosinophilia and elongate shapes, varyingly described as “tadpole”, “strap”, and “spider” cells and deemed to be evidence of rhabdomyoblastic differentiation. Differentiation tends to be more evident following chemotherapy, as differentiated elements become the predominant cell population, separated by therapy induced necrosis and fibrosis. The botryoid variant of ERMS contains linear aggregates of tumor cells (cambium layer) that tightly abut an epithelial surface. This variant also shows variable numbers of polypoid nodules, often with an abundant, loose, myxoid stroma. The anaplastic variant of ERMS is characterized by the presence of enlarged, atypical cells with hyperchromatic nuclei. Anaplastic features can be focal or diffuse and bizarre, multipolar, mitoses are also often present. (Figure 2C)
Figure 2:
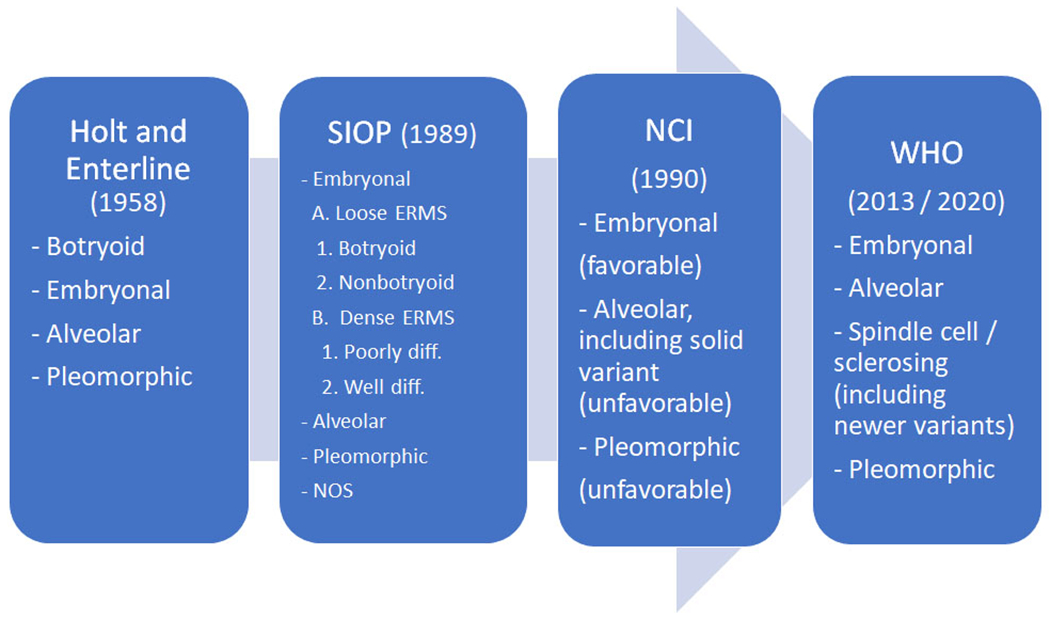
Embryonal Rhabdomyosarcoma. A. Low power image showing cellular neoplasm with hyper and hypocellular areas. (H&E, 200x) B. High power image showing primitive spindle cells with scattered rhabdomyoblasts. (H&E, 400x). C. High power image of ERMS with scattered large anaplastic cells. (H&E, 400x). D. Immunohistochemical stain for Myogenin showing scattered nuclear positivity.
Immunohistochemically, markers of skeletal muscle differentiation are positive in ERMS. Desmin is the most frequently used diagnostic marker and shows diffuse staining. ERMS typically shows patchy positivity for Myogenin and MYO-D1. (Figure 2D)
Genetics
ERMS do not show any gene fusions. Molecular analyses typically show aneuploidy with multiple copy number gains and losses noted. Whole chromosome gains with polysomy of chromosome 8 are common. Whole chromosome losses of 10 and 15 are noted.38 The specific genes associated with ERMS include RAS family genes (HRAS, NRAS, KRAS), FGFR4, PIK3CA, NF1 and FBXW7. 32, 39, 40
Prognosis
Prognosis can be determined by stage, histological classification, age, and site of origin. Staging, in children, is accomplished by clinical evaluation (Intergroup Rhabdomyosarcoma Study Group (IRSG) Stage) and / or surgicopathological evaluation (IRSG Group).41 The IRSG subdivides RMS into low, intermediate and high risk groups for purposes of protocol based therapy. Younger patients (1-9 yrs) tend to have a more favorable prognosis than infants and adolescents. 42 ERMS have a better prognosis than ARMS and botryoid variants have a superior outcome as a group.43 ERMS with diffuse anaplasia may have a worse outcome than the other subsets of embryonal rhabdomyosarcoma. 44
In adults, the reported range of ERMS is from 21 to 54%. Although historical data indicate that ERMS in adults fare poorly and have a higher relapse rate, recent studies have shown that using IRSG groups and staging for adult ERMS and treating them with pediatric protocols significantly improves the survival of adult patients.45, 46
Alveolar Rhabdomyosarcoma
Alveolar rhabdomyosarcoma (ARMS) is a cellular malignant neoplasm composed of a monomorphous population of primitive cells with round nuclei and features of arrested myogenesis. ARMS occur at all ages, more often in adolescents and young adults than in younger children. The median ages of occurrence is between 6.8 and 9.0 years.37 They occur less frequently than ERMS and comprise about 20% of all pediatric RMS. The male: female ratio is approximately even, and no geographic or racial predilection is reported. Alveolar rhabdomyosarcomas commonly arise in the extremities. Additional sites of involvement include the paraspinal and the perineal regions and the paranasal sinuses. Clinically, ARMS typically present as rapidly growing extremity masses. Tumors at other sites such as paranasal, perirectal and paraspinal mainly cause symptoms of compression of the surrounding structures. ARMS tend to be high stage lesions at presentation and form expansile, rapidly growing soft tissue tumors.
Histologically, they are highly cellular neoplasms and composed of primitive cells with round nuclei, with fibrovascular septa that separate the tumor cells into discrete nests. These nests contain central clusters of cells with loss of cohesion around the periphery, giving an ‘alveolar’ appearance. (Figure 3A–B) Giant cells with rhabdomyoblastic differentiation are common. Solid variant of ARMS lack the fibrovascular stroma and form sheets of round cells with variable rhabdomyoblastic differentiation.6(Figure 3C)
Figure 3:
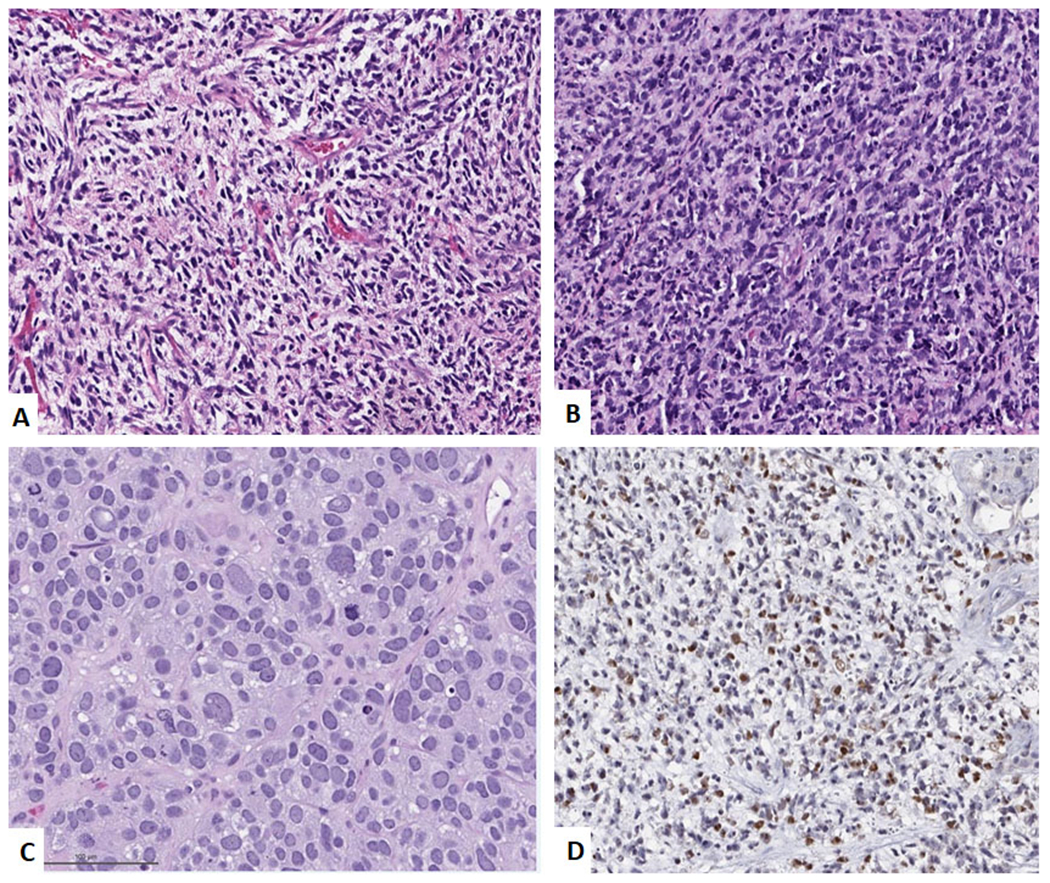
Alveolar Rhabdomyosarcoma. A. Low power image showing a nested alveolar pattern of arrangement of the tumor cells with the nests separated by fibrous septa. (H&E, 100x) B. High power image showing medium-sized tumor cells with scattered giant cells. (H&E, 400x) C. High power image showing solid variant with sheets of medium-sized tumor cells. (H&E, 400x) and D. Immunohistochemical stain for Myogenin showing diffuse positivity.
Immunohistochemically, ARMS stain strongly for desmin. Myogenin and MyoD1 typically show a diffuse, strong nuclear staining pattern. 8(Figure 3D) The diffuse positivity for myogenin is unlike the focal patchy staining seen in ERMS and can provide a clue to the subtype on small biopsy material. About one-third to one-half of the ARMS in the head and neck have been reported to show positivity for epithelial markers. 47
Genetics
ARMS are characterized by recurrent translocations. A t(2;13)(q35;q14) is found in the majority of cases and a t(1;13)(p36;q14) is seen in a smaller subset of cases.48 These translocations juxtapose the PAX3 or PAX7 genes on chromosomes 2 and 1, respectively, with the FOXO1 gene on chromosome 13, to generate fusion genes which encode PAX3-FOXO1 and PAX7-FOXO1 fusion proteins.14, 49 PAX3 and PAX7 are related members of the paired box family of transcription factors whereas FOXO1 is a member of the forkhead transcription factor family. The PAX3- FOXO1 and PAX7-FOXO1 fusion products contain the PAX3/PAX7 DNA binding domain and the FOXO1 transcriptional activation domain, and function as potent transcriptional activators.50 In addition to this functional change, the translocations generate high levels of these chimeric proteins which activate downstream transcriptional targets, and are postulated to exert oncogenic effects by altering control of proliferation, apoptosis, and differentiation. The most frequent amplifications seen in ARMS, involve chromosomal regions 12q13-15 and 2p24, 51 regions that contains many growth-related genes such as the GLI, CDK4, MDM2, and MYCN genes. The PAX7-FOXO1 fusion gene is amplified in the majority of ARMS cases with the t(1;13) translocation.52
Prognosis
ARMS are high grade neoplasms that are more aggressive than ERMS.26 IRSG grouping is predictive of outcome. Although some studies postulate that PAX7-FOXO1 positive tumors behave in a more benign fashion than PAX3-FOXO1 tumors, 53 the prognostic value of the different type of gene fusion is uncertain. Unlike ERMS, ARMS is one of the few sarcomas that can metastasize to the regional lymph nodes. Imaging of the regional lymph nodes should be part of the work up for these patients. In some institutions, sentinel lymph node mapping is used. In adults, the reported range of ARMS is 18 – 33%. Similar to the pediatric population, ARMS in adults seem to have a worse prognosis when compared to ERMS.45, 46
Pleomorphic Rhabdomyosarcoma
Pleomorphic rhabdomyosarcoma (PRMS) is a high grade sarcoma occurring almost exclusively in adults and consisting of bizarre polygonal, round, and spindle cells which display evidence of skeletal muscle differentiation with no evidence of embryonal or alveolar component. These lesions are more common in men and present at a median age in the 6th to 7th decade. 54 These tumors usually occur in the deep soft tissues of the lower extremities but have been reported in a wide variety of other locations. 55, 56 Clinically, most patients present with a rapidly- growing painful swelling. Tumors are usually large (5-15 cm), well circumscribed, and often surrounded by a pseudocapsule. The cut surface is tan and fleshy with variable hemorrhage and necrosis. Morphologically, these are pleomorphic sarcomas composed of undifferentiated round to spindle cells and an admixture of polygonal cells with densely eosinophilic cytoplasm in spindle, tadpole, and racquet-like contours. Cross striations are not usually seen. (Figure 4 D–E) Immunohistochemically, PRMS, like other rhabdomyosarcoma subtypes, express desmin, with MyoD1 and Myogenin showing focal positivity. (Figure 4F) Genetically, PRMS show a complex karyotype with copy number alterations and unbalanced structural alterations.57 The prognosis for these tumors is poor.
Figure 4:
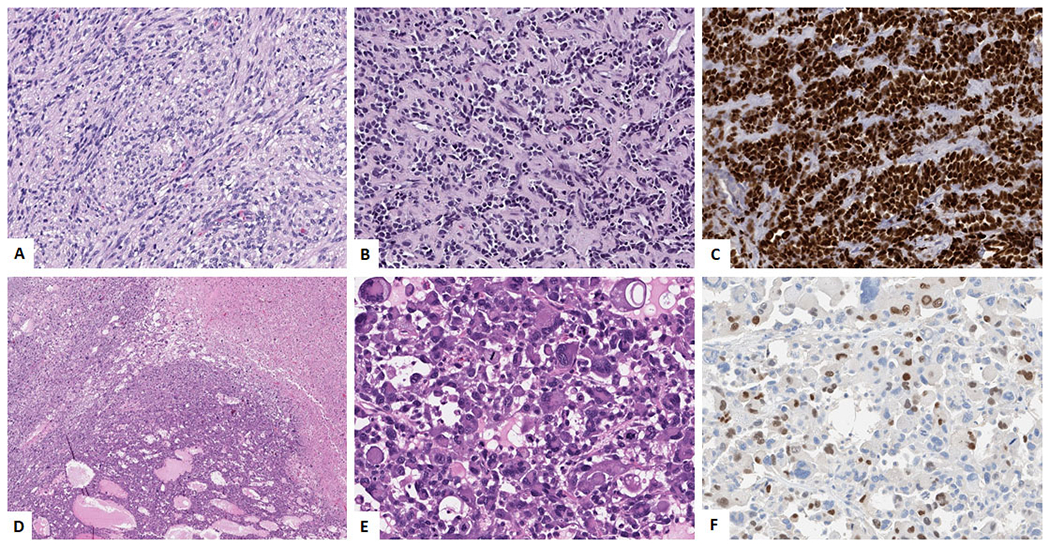
Spindle cell / Sclerosing Rhabdomyosarcoma (A-C). A. Spindle cell morphology showing uniform spindle cells in intersecting fascicles. (H&E, 200x). B. Sclerosing ‘pseudovascular’ morphology composed of small nests of tumor cells with central clearing, in a sclerosing collagenous background.(H&E, 200x) C. Immunohistochemical stain for MYOD-1 showing diffuse positivity; Pleomorphic Rhabdomyosarcoma (D-F). D. Low power image showing sheets of tumor cells with areas of necrosis (H&E, 100x). E. High power image showing large cells with rhabdoid morphology and large pleomorphic nuclei (H&E, 400x). F. Immunohistochemical stain for Myogenin showing focal positivity.
The morphologic differential diagnoses include other high grade tumors in adults including dedifferentiated liposarcoma and high grade undifferentiated pleomorphic sarcoma Immunohistochemical stains and ancillary studies help to make the right diagnosis.
Spindle Cell / Sclerosing Rhabdomyosarcoma (SpRMS)
SpRMS can occur as painless masses or can have symptoms due to compression. Grossly they are usually well circumscribed with sections showing a grey white whorled cut surface. Microscopically, SpRMS can show a varying morphology. Some tumors show a bland spindle cell proliferation, with eosinophilic, fibrillary cytoplasm, resembling true smooth muscle differentiation. The cells are typically arranged in intersecting long fascicles, reminiscent of the “herring bone” pattern of adult-type fibrosarcoma. (Figure 4A) Evidence of rhabdomyoblasts with cross-striations can be seen in some tumors. Other patterns that can be seen in SpRMS is that of a cellular proliferation of spindle cells with morphology closely resembling fibrosarcoma, leiomyosarcoma, or MPNST. The sclerosing variant of SpRMS shows tumor cells in an extensive hyalinized matrix with focal alveolar architecture giving a pseudovascular appearance. (Figure 4B) Some sclerotic areas may mimic osteosarcoma. Immunohistochemically, SpRMS show diffuse positivity for desmin. Myogenin usually shows rare focal positivity with some cases being negative. MyoD1 stain is positive in the tumor cells. (Figure 4C)
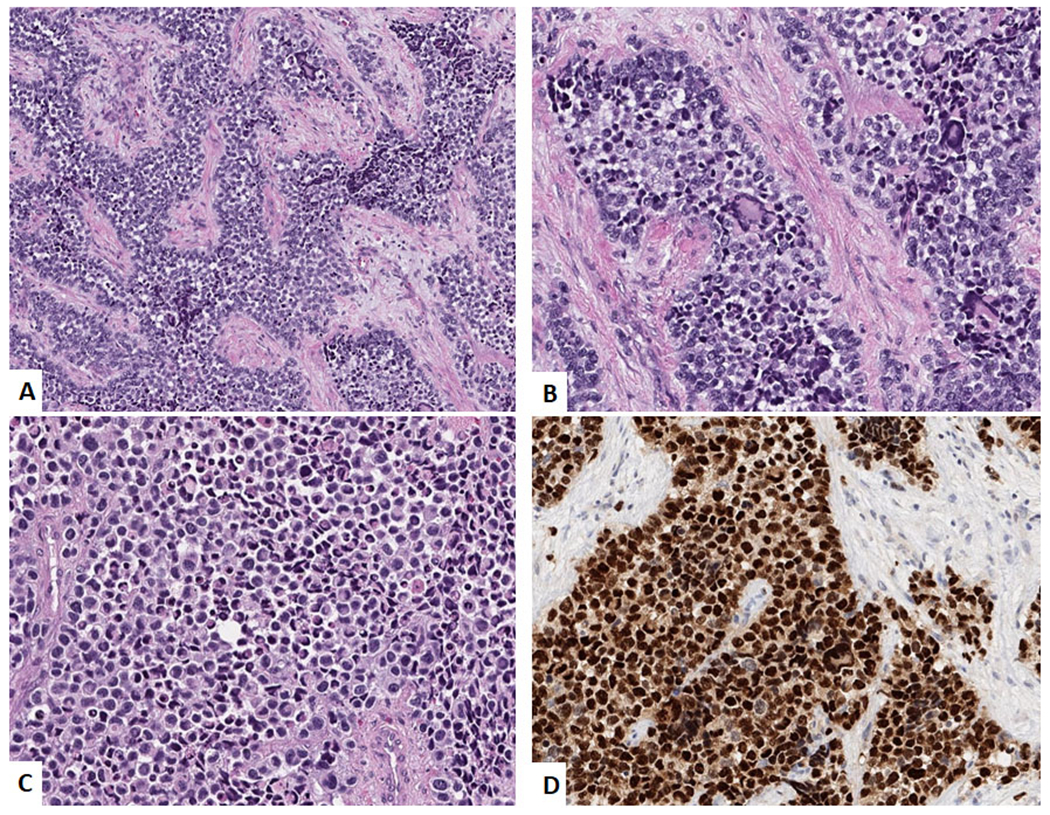
The recently described subtype of rhabomyosarcoma, with a predilection for intraosseous locations and genetically characterized by EWSR1 / FUS – TFCP2 gene fusions, has been tentatively classified under the spindle cell / sclerosing rhabdomyosarcoma.34, 35 They have a predilection for the craniofacial bones. These tumors morphologically show a spindled and epithelioid morphology. (Figure 5A–B) Interestingly, apart from positivity for desmin, Myogenin and MYOD1 (Figure 5C–D), they also show positivity for epithelial markers (cytokeratin and / or EMA) and show overexpression of ALK. (Figure 5E–F)
Figure 5:
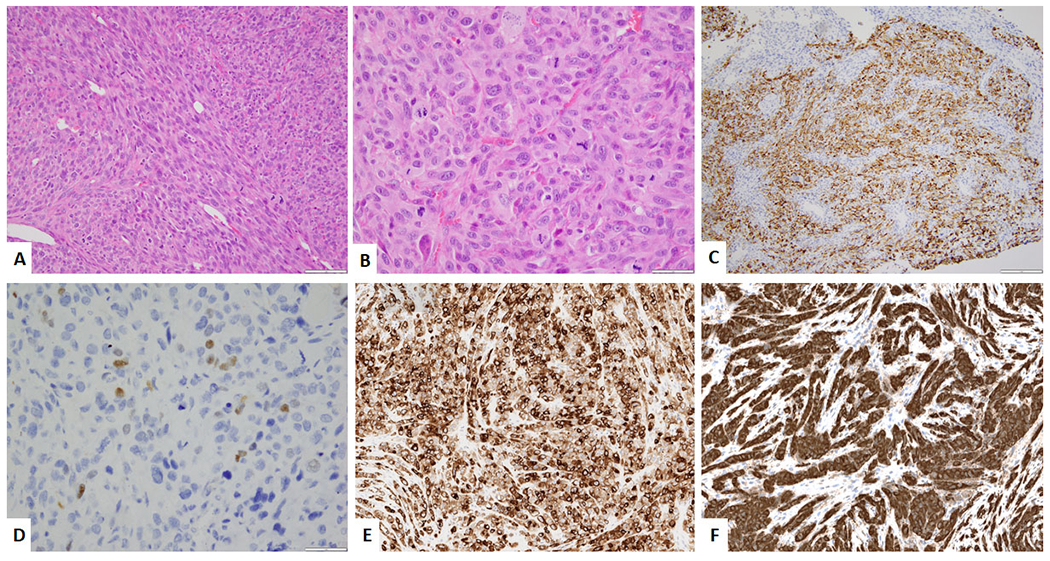
Rhabdomyosarcoma with EWSR1-TFCP2 gene fusion. A. Low power image showing spindled and epithelioid morphology. (H&E, 200x). B. High power image of the epithelioid areas. (H&E, 400x) C. Immunohistochemical stain for desmin showing relatively diffuse positivity. D. Immunohistochemical stain for Myogenin showing focal positivity. E. Immunohistochemical stain for Pan-Cytokeratin showing relatively diffuse positivity. F. Immunohistochemical stain for ALK showing diffuse positivity.
Prognosis
The recent genetic findings in SpRMS have significantly increased our understanding of the biology of SpRMS. The infantile SpRMS with the VGLL2 associated gene fusions appear to have a favorable prognosis. 26 In contrast, the MYOD1 mutated SpRMS seem to be aggressive tumors and have a significantly worse prognosis, despite aggressive chemoradiation therapy, in both the pediatric and adult age groups. 29, 31 Tumors with EWSR1/FUS-TFCP2 gene fusions have also been shown to behave aggressively and have a poor prognosis.58
Diagnostic Approach To Rhabdomyosarcoma
Rhabdomyosarcomas that occur in pediatric age group include embryonal, alveolar and spindle cell / sclerosing subtypes. In adults, pleomorphic rhabdomyosarcoma should also be included as part of the differential diagnosis. The subtypes of rhabdomyosarcoma in pediatric and adult age groups have variable prognosis. (Figure 6) Based on the clinical presentation and imaging findings, the initial approach for a tissue diagnosis is to get a surgical core biopsy. A core biopsy is preferable to fine needle aspiration biopsy to assess the morphologic architecture as well as to obtain adequate tissue for cytogenetic and molecular analysis. The immunohistochemical panel used for diagnosis depends on the morphology of the tumor. If the tumor shows morphology of a small blue round cell tumor with no hint of rhabdomyoblastic differentiation, immunohistochemical stains for desmin, myogenin and MyoD1 should be included along with a larger panel of stains performed. If the tumor shows distinctive rhabdomyoblastic differentiation, immunohistochemical stains for desmin, myogenin and MyoD1 should be performed. Although the morphology and immunohistochemical stains suggest a subtype of RMS, it is standard practice to perform ancillary fluorescence-in-situ-hybridization or molecular studies for the detection of FOXO1 gene rearrangement on all newly diagnosed cases to exclude the possibility of fusion-positive ARMS. If the morphology is suggestive of SpRMS, DNA- and RNA-based molecular studies should be performed to detect presence of MYOD1 mutation, a finding that predicts aggressive behavior of the disease, or possible gene fusions.
Figure 6:
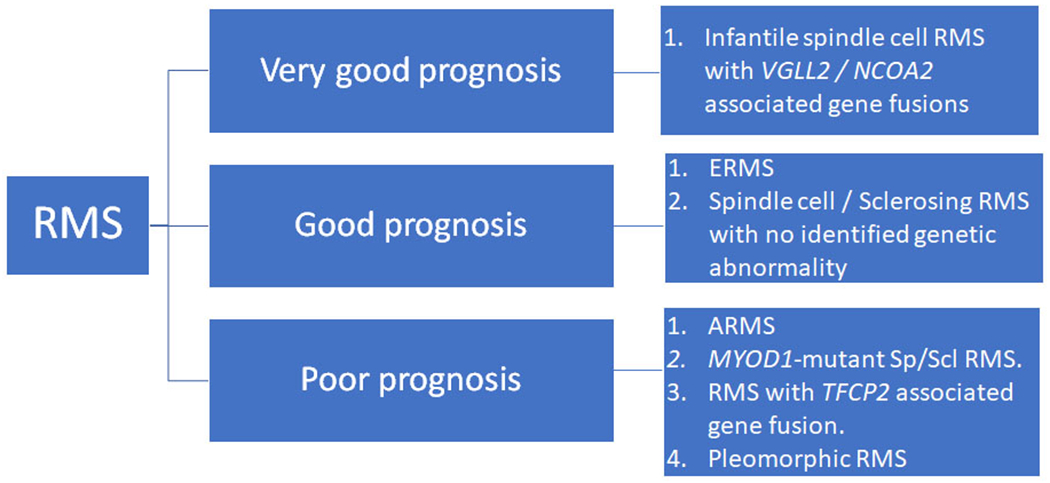
Schematic showing the prognostic classification of rhabdomyosarcoma based on the current prognostic information.
Footnotes
DISCLOSURE STATEMENT: No disclosures.
REFERENCES
- 1.Parham DM, Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol, 2001. 14(5): p. 506–14. [DOI] [PubMed] [Google Scholar]
- 2.Stout AP, Rhabdomyosarcoma of the Skeletal Muscles. Ann Surg, 1946. 123(3): p. 447–72. [PMC free article] [PubMed] [Google Scholar]
- 3.Riopelle JL and Theriault JP, [An unknown type of soft part sarcoma: alveolar rhabdomyosarcoma]. Ann Anat Pathol (Paris), 1956. 1(1): p. 88–111. [PubMed] [Google Scholar]
- 4.Horn RC Jr. and Enterline HT, Rhabdomyosarcoma: a clinicopathological study and classification of 39 cases. Cancer, 1958. 11(1): p. 181–99. [DOI] [PubMed] [Google Scholar]
- 5.Caillaud JM, Gerard-Marchant R, Marsden HB, et al. , Histopathological classification of childhood rhabdomyosarcoma: a report from the International Society of Pediatric Oncology pathology panel. Med Pediatr Oncol, 1989. 17(5): p. 391–400. [DOI] [PubMed] [Google Scholar]
- 6.Tsokos M, Webber BL, Parham DM, et al. , Rhabdomyosarcoma. A new classification scheme related to prognosis. Arch Pathol Lab Med, 1992. 116(8): p. 847–55. [PubMed] [Google Scholar]
- 7.Newton WA Jr., Gehan EA, Webber BL, et al. , Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer, 1995. 76(6): p. 1073–85. [DOI] [PubMed] [Google Scholar]
- 8.Dias P, Chen B, Dilday B, et al. , Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol, 2000. 156(2): p. 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Douglass EC, Valentine M, Etcubanas E, et al. , A specific chromosomal abnormality in rhabdomyosarcoma. Cytogenet Cell Genet, 1987. 45(3-4): p. 148–55. [DOI] [PubMed] [Google Scholar]
- 10.Turc-Carel C, Lizard-Nacol S, Justrabo E, et al. , Consistent chromosomal translocation in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet, 1986. 19(3-4): p. 361–2. [DOI] [PubMed] [Google Scholar]
- 11.Wang-Wuu S, Soukup S, Ballard E, et al. , Chromosomal analysis of sixteen human rhabdomyosarcomas. Cancer Res, 1988. 48(4): p. 983–7. [PubMed] [Google Scholar]
- 12.Biegel JA, Meek RS, Parmiter AH, et al. , Chromosomal translocation t(1;13)(p36;q14) in a case of rhabdomyosarcoma. Genes Chromosomes Cancer, 1991. 3(6): p. 483–4. [DOI] [PubMed] [Google Scholar]
- 13.Barr FG, Galili N, Holick J, et al. , Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet, 1993. 3(2): p. 113–7. [DOI] [PubMed] [Google Scholar]
- 14.Davis RJ, D’Cruz CM, Lovell MA, et al. , Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res, 1994. 54(11): p. 2869–72. [PubMed] [Google Scholar]
- 15.Williamson D, Missiaglia E, de Reynies A, et al. , Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol, 2010. 28(13): p. 2151–8. [DOI] [PubMed] [Google Scholar]
- 16.Cavazzana AO, Schmidt D, Ninfo V, et al. , Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol, 1992. 16(3): p. 229–35. [DOI] [PubMed] [Google Scholar]
- 17.Leuschner I, Newton WA Jr., Schmidt D, et al. , Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol, 1993. 17(3): p. 221–30. [DOI] [PubMed] [Google Scholar]
- 18.Rubin BP, Hasserjian RP, Singer S, et al. , Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol, 1998. 22(4): p. 459–64. [DOI] [PubMed] [Google Scholar]
- 19.Nascimento AF and Fletcher CD, Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol, 2005. 29(8): p. 1106–13. [PubMed] [Google Scholar]
- 20.Mentzel T and Kuhnen C, Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch, 2006. 449(5): p. 554–60. [DOI] [PubMed] [Google Scholar]
- 21.Mentzel T and Katenkamp D, Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch, 2000. 436(4): p. 305–11. [DOI] [PubMed] [Google Scholar]
- 22.Folpe AL, McKenney JK, Bridge JA, et al. , Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol, 2002. 26(9): p. 1175–83. [DOI] [PubMed] [Google Scholar]
- 23.Chiles MC, Parham DM, Qualman SJ, et al. , Sclerosing rhabdomyosarcomas in children and adolescents: a clinicopathologic review of 13 cases from the Intergroup Rhabdomyosarcoma Study Group and Children’s Oncology Group. Pediatr Dev Pathol, 2004. 7(6): p. 583–94. [DOI] [PubMed] [Google Scholar]
- 24.Mentzel T, [Spindle cell rhabdomyosarcoma in adults: a new entity in the spectrum of malignant mesenchymal tumors of soft tissues]. Pathologe, 2010. 31(2): p. 91–6. [DOI] [PubMed] [Google Scholar]
- 25.Fletcher C, Bridge JA, Hogendoorn PC, et al. , WHO Classification of Tumours of Soft Tissue and Bone. 2013: IARC: Lyon. [Google Scholar]
- 26.Alaggio R, Zhang L, Sung YS, et al. , A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. Am J Surg Pathol, 2016. 40(2): p. 224–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mosquera JM, Sboner A, Zhang L, et al. , Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer, 2013. 52(6): p. 538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szuhai K, de Jong D, Leung WY, et al. , Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol, 2014. 232(3): p. 300–7. [DOI] [PubMed] [Google Scholar]
- 29.Agaram NP, Chen CL, Zhang L, et al. , Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer, 2014. 53(9): p. 779–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohsaka S, Shukla N, Ameur N, et al. , A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet, 2014. 46(6): p. 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agaram NP, LaQuaglia MP, Alaggio R, et al. , MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: an aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod Pathol, 2019. 32(1): p. 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shern JF, Selfe J, Izquierdo E, et al. , Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. J Clin Oncol, 2021: p. JCO2003060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.WHO Classification of Tumours Editorial Board eds. World Health Organization classification of tumours: soft tissue and bone tumours. 5th ed. Vol. 3. 2020, Lyon: IARC. [Google Scholar]
- 34.Watson S, Perrin V, Guillemot D, et al. , Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol, 2018. 245(1): p. 29–40. [DOI] [PubMed] [Google Scholar]
- 35.Agaram NP, Zhang L, Sung YS, et al. , Expanding the Spectrum of Intraosseous Rhabdomyosarcoma: Correlation Between 2 Distinct Gene Fusions and Phenotype. Am J Surg Pathol, 2019. 43(5): p. 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu B, Suurmeijer AJH, Agaram NP, et al. , Head and neck rhabdomyosarcoma with TFCP2 fusions and ALK overexpression: a clinicopathological and molecular analysis of 11 cases. Histopathology, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newton WA Jr., Soule EH, Hamoudi AB, et al. , Histopathology of childhood sarcomas, Intergroup Rhabdomyosarcoma Studies I and II: clinicopathologic correlation. J Clin Oncol, 1988. 6(1): p. 67–75. [DOI] [PubMed] [Google Scholar]
- 38.Weber-Hall S, Anderson J, McManus A, et al. , Gains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridization. Cancer Res, 1996. 56(14): p. 3220–4. [PubMed] [Google Scholar]
- 39.Paulson V, Chandler G, Rakheja D, et al. , High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer, 2011. 50(6): p. 397–408. [DOI] [PubMed] [Google Scholar]
- 40.Shern JF, Chen L, Chmielecki J, et al. , Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov, 2014. 4(2): p. 216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer WH and Spunt SL, Soft tissue sarcomas of childhood. Cancer Treat Rev, 2004. 30(3): p. 269–80. [DOI] [PubMed] [Google Scholar]
- 42.Sultan I, Qaddoumi I, Yaser S, et al. , Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol, 2009. 27(20): p. 3391–7. [DOI] [PubMed] [Google Scholar]
- 43.Wexler LH and Ladanyi M, Diagnosing alveolar rhabdomyosarcoma: morphology must be coupled with fusion confirmation. J Clin Oncol, 2010. 28(13): p. 2126–8. [DOI] [PubMed] [Google Scholar]
- 44.Kodet R, Newton WA Jr., Hamoudi AB, et al. , Childhood rhabdomyosarcoma with anaplastic (pleomorphic) features. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol, 1993. 17(5): p. 443–53. [DOI] [PubMed] [Google Scholar]
- 45.Bompas E, Campion L, Italiano A, et al. , Outcome of 449 adult patients with rhabdomyosarcoma: an observational ambispective nationwide study. Cancer Med, 2018. 7(8): p. 4023–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerber NK, Wexler LH, Singer S, et al. , Adult rhabdomyosarcoma survival improved with treatment on multimodality protocols. Int J Radiat Oncol Biol Phys, 2013. 86(1): p. 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson LDR, Jo VY, Agaimy A, et al. , Sinonasal Tract Alveolar Rhabdomyosarcoma in Adults: A Clinicopathologic and Immunophenotypic Study of Fifty-Two Cases with Emphasis on Epithelial Immunoreactivity. Head Neck Pathol, 2018. 12(2): p. 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barr FG, Molecular genetics and pathogenesis of rhabdomyosarcoma. J Pediatr Hematol Oncol, 1997. 19(6): p. 483–91. [DOI] [PubMed] [Google Scholar]
- 49.Barr FG, Chatten J, D’Cruz CM, et al. , Molecular assays for chromosomal translocations in the diagnosis of pediatric soft tissue sarcomas. JAMA, 1995. 273(7): p. 553–7. [PubMed] [Google Scholar]
- 50.Bennicelli JL, Advani S, Schafer BW, et al. , PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene, 1999. 18(30): p. 4348–56. [DOI] [PubMed] [Google Scholar]
- 51.Gordon AT, Brinkschmidt C, Anderson J, et al. , A novel and consistent amplicon at 13q31 associated with alveolar rhabdomyosarcoma. Genes Chromosomes Cancer, 2000. 28(2): p. 220–6. [PubMed] [Google Scholar]
- 52.Barr FG, Nauta LE, Davis RJ, et al. , In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet, 1996. 5(1): p. 15–21. [DOI] [PubMed] [Google Scholar]
- 53.Kelly KM, Womer RB, Sorensen PH, et al. , Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol, 1997. 15(5): p. 1831–6. [DOI] [PubMed] [Google Scholar]
- 54.Furlong MA, Mentzel T, and Fanburg-Smith JC, Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol, 2001. 14(6): p. 595–603. [DOI] [PubMed] [Google Scholar]
- 55.Schurch W, Begin LR, Seemayer TA, et al. , Pleomorphic soft tissue myogenic sarcomas of adulthood. A reappraisal in the mid-1990s. Am J Surg Pathol, 1996. 20(2): p. 131–47. [DOI] [PubMed] [Google Scholar]
- 56.Stock N, Chibon F, Binh MB, et al. , Adult-type rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am J Surg Pathol, 2009. 33(12): p. 1850–9. [DOI] [PubMed] [Google Scholar]
- 57.Li G, Ogose A, Kawashima H, et al. , Cytogenetic and real-time quantitative reverse-transcriptase polymerase chain reaction analyses in pleomorphic rhabdomyosarcoma. Cancer Genet Cytogenet, 2009. 192(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 58.Le Loarer F, Cleven AHG, Bouvier C, et al. , A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol, 2020. 33(3): p. 404–419. [DOI] [PubMed] [Google Scholar]