

Abstract
The LC3/GABARAP family of proteins is involved in nearly every stage of autophagy. Inhibition of LC3/GABARAP proteins is a promising approach to blocking autophagy, which sensitizes advanced cancers to DNA-damaging chemotherapy. Here, we report the structure-based design of stapled peptides that inhibit GABARAP with nanomolar affinities. Small changes in staple structure produced stapled peptides with very different binding modes and functional differences in LC3/GABARAP paralog selectivity, ranging from highly GABARAP-specific to broad inhibition of both subfamilies. The stapled peptides exhibited considerable cytosolic penetration and resistance to biological degradation. They also reduced autophagic flux in cultured ovarian cancer cells and sensitized ovarian cancer cells to cisplatin. These small, potent stapled peptides represent promising autophagy-modulating compounds that can be developed as novel cancer therapeutics and novel mediators of targeted protein degradation.
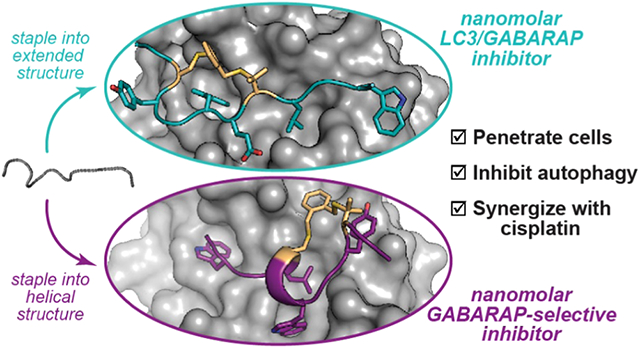
Graphical Abstract
INTRODUCTION
Autophagy is a dynamic cellular process by which cytosolic material is transported to the lysosomal compartment for degradation. Bulk autophagy recycles biomolecules into building blocks in response to starvation, while selective autophagy removes misfolded proteins, damaged organelles, and intracellular pathogens.1 Dysregulation of autophagy is a hallmark of many diseases, including neurodegenerative diseases, infectious diseases, and cancer.1 During carcinogenesis, autophagy is thought to be protective as downregulating autophagy increases oxidative stress, DNA damage, and chromosomal instability.2 However, established tumors can become overly reliant on autophagy to survive an environment with low nutrients, hypoxia, and high oxidative stress.2
There is a great deal of evidence that autophagy inhibition could be a promising therapeutic strategy for advanced-stage cancers. For example, genetic knockdown of essential autophagy genes attenuates tumor growth and extends survival in pancreatic ductal adenocarcinoma, lung cancer, and melanoma.3-5 Autophagy is induced in response to DNA-damaging chemotherapy, and autophagy has been implicated in cisplatin resistance in breast and ovarian cancers.6,7 Multiple clinical trials using the nonspecific autophagy inhibitors chloroquine and hydroxychloroquine in combination with chemotherapy have shown an overall improved response compared to chemotherapy alone for pancreatic cancer, glioblastoma, breast cancer, and other malignancies.8-11 However, hydroxychloroquine and related drugs broadly affect endolysosomal processes and are not specific to autophagy, and they can cause significant side effects at doses required for autophagy inhibition.12,13 Novel, autophagy-specific inhibitors are highly sought-after to better understand the effects of autophagy inhibition on advanced cancers and to develop therapeutics with improved efficacy and better side effect profiles.14
One strategy for specific autophagy inhibition is interruption of key protein–protein interactions.14 Members of the Atg8 protein family, including the mammalian LC3 (LC3A, LC3B, LC3C) and GABARAP (GABARAP, GABARAP-L1, GABAR-AP-L2/GATE-16) subfamilies, are involved in key protein–protein interactions at several steps in autophagy, including initiation, formation and elongation of the phagophore, cargo recruitment, autophagosome trafficking, and lysosomal fusion (Figure 1).15 Mammalian LC3 and GABARAP proteins, which we will refer to collectively as LC3/GABARAP proteins, recognize a conserved motif known as the LC3-interacting region (LIR).16,17 Proteins with LIR motifs that bind LC3 and/or GABARAP proteins include ATG4, which promotes autophagy initiation and phagophore formation, selective autophagy adapters such as p62 which recruit proteins to be degraded, and trafficking machinery including FYCO1 and the HOPS complex.18-20 Thus, disruption of protein–protein interactions involving LC3/GABARAP proteins has been recognized as a promising strategy for autophagy inhibition. For example, blocking protein–protein interactions of LC3/GABARAP proteins using engineered binding proteins sensitized acute myeloid leukemia cells to chemotherapy.21 Additionally, the application of LC3-binding peptides sensitized ovarian cancer cells to cisplatin treatment.19 GABARAP and LC3 subfamilies are important for autophagy in mammalian cells, with different yet overlapping roles (Figure 1). The different roles of GABARAPs and LC3s in cancer progression are poorly understood, so inhibitors selective to each subfamily are important for studying how they control autophagy in cancer and other pathologies.22
Figure 1.

Roles of LC3/GABARAP proteins in autophagy. Autophagy pathways proceed through a multistep process that involves protein–protein interactions of LC3/GABARAP proteins at every step. Some of the known differential roles for proteins from the LC3 and GABARAP subfamilies are noted.18-20
LIR motifs bind Atg8-family proteins in an extended conformation anchored by conserved hydrophobic side chains.16,17,23 Several studies have explored structural determinants of selectivity for LC3 and GABARAP proteins, and generally, it was observed that GABARAPs are able to accommodate a wider variety of LIR ligands.23-25 In previous work, we described structure-activity relationships for an LC3-specific LIR motif derived from FYCO1, a Rab7 effector protein that mediates autophagosome trafficking.26 We also described peptides with side-chain-to-side-chain crosslinks, or “stapled” peptides, with improved affinity, selectivity for LC3B over GABARAP, proteolytic stability, and cytosolic delivery. In this work, we report two new classes of stapled peptides that inhibit LC3/GABARAP proteins with nanomolar affinity, one GABARAP-selective class and another that inhibits both subfamilies. Notably, while most implementations of a stapled peptide strategy involve helical peptides,27 here we employ a “diversity-oriented stapling” approach that allows the synthesis and evaluation of stapled peptides with a variety of three-dimensional structures. This methodology enabled the discovery of two distinct families of stapled GABARAP ligands. We obtained crystal structures, which revealed that high affinity and selectivity were governed by small changes in the chemical structures of the staples, producing two different, high-affinity binding modes. These peptides resist biological degradation, penetrate the cytosol, inhibit autophagy, and are synergistic with DNA-damaging chemotherapy in ovarian cancer cells.
RESULTS
Peptide K1 is an Artificial, GABARAP-Selective Ligand.
The highest-affinity natural ligands for LC3B/GABARAP proteins are large, negatively charged peptides. We sought to design smaller, less charged ligands that would be more suitable as pharmacological agents, so we started with the non-natural GABARAP ligand K1, which was discovered in a phage display selection by Weiergräber et al.28 The binding mode of K1 is unique compared to all known native ligands in three respects: K1 binds with a single turn of a 310 helix instead of an extended conformation, K1 binds with the opposite N-to-C orientation compared to natural ligands, and K1 induces GABARAP to widen a key hydrophobic pocket to accommodate a Trp residue in addition to the canonical aliphatic residue (Figures 2a and S3). We tested fluorescein-tagged K1 in a direct fluorescence polarization (FP) assay with recombinant, His-tagged GABARAP and LC3B and found that it bound GABARAP with a Kd of 10 ± 1 nM and LC3B with a Kd of 1200 ± 30 nM (Figure 2b), which made it an ideal starting point for designing stapled GABARAP inhibitors.23,29
Figure 2.

Diversity-oriented stapling of K1. (a) The structure of K1 bound to GABARAP28 suggested that replacing residue 8 with Cys and then stapling it to a second Cys residue at either position 3, position 4, or position 5 could potentially stabilize the overall structure (yellow dotted lines). Cysteine side chains are depicted at all four positions, with sulfur atoms indicated by yellow spheres. (b) Cysteine-substituted K1 analogues were stapled using the linkers ortho-, meta-, or para-dimethylbenzene, and affinities for recombinant GABARAP and LC3B were measured using fluorescence polarization. Allyl-modified Cys3 peptide was tested as an unstapled control. Peptides K1 and FYCO1 were tested as GABARAP-selective and LC3B-selective controls, respectively. (c) Penicillamine-substituted analogues of peptide Cys3 were stapled, and their binding affinities for GABARAP and LC3B were measured. π denotes penicillamine. Peptides were prepared with fluorescein on their N-termini (Figure S1 and Table S1). Data are shown as the average and standard error of the mean for three or more independent trials. Primary data are shown in Figures S4-S17.
Diversity-Oriented Stapling of K1.
While K1 has a high affinity for GABARAP, applications of peptides with all-natural amino acids can be limited by their biological stability and cell penetration (see below). Covalent linking of side chains, or “peptide stapling,” is a promising strategy to minimize these limitations.2 While most stapled peptides are α-helical, we and others have shown that appropriately designed staples can improve the properties of peptides in other conformations as well.26,30-34 Based on the crystal structure of K1 bound to GABARAP,28 we designed peptides Cys5, Cys4, and Cys3, which stapled K1 in (i,i + 3), (i,i + 4), and (i,i + 5) positions, respectively (Figure 2a). We stapled each peptide using dithiol bis-alkylation34 with ortho-, meta-, and para-dimethylbenzene linkers, allowing for a “diversity-oriented stapling” approach, where the staple geometry was varied (Figure 2a).31,35,36 Stapling in (i,i + 3) positions (the Cys5 peptides) reduced binding to both GABARAP and LC3B compared to K1. Stapling in (i,i + 4) positions (the Cys4 peptides) produced stapled peptides with similar LC3B binding across staple geometries but different GABARAP binding with the best affinity afforded by the longer para linker. By contrast, for the Cys3 peptides, which were stapled in (i,i + 5) positions, the unstapled control (Cys3-allyl) and all three stapled peptides bound GABARAP with nanomolar affinity, but the staple geometry modulated LC3B binding across an order of magnitude (Figure 2b).
From these initial designs, it was clear that shorter staples were generally less compatible with GABARAP binding. We explored the effects of longer linkers by stapling (i,i + 4) positions with homocysteine instead of cysteine in either the 4th or 8th position (peptides Hcy4 and Hcy8, respectively, Figure S8). Homocysteine in either position significantly improved GABARAP binding for peptides with an ortho staple but not with meta or para staples. Homocysteine position and staple geometry had large effects on LC3B binding, with Hcy8-para having increased LC3B affinity (230 ± 30 nM) compared to other (i,i + 4) stapled peptides (Figure S8).
Because both Cys3-meta and Cys4-meta each showed only a moderate decrease in GABARAP affinity compared to K1, we hypothesized that a peptide with three cysteine substitutions stapled using a (1,3,5)-trimethylbenzene linker might also be tolerated.34,37 We found that both the unstapled Cys3.4.8-allyl and stapled Cys3.4.8-tmb peptides had GABARAP-binding affinities comparable to Cys3-meta and Cys4-meta, and the stapled Cys3.4.8-tmb maintained 35-fold binding selectivity for GABARAP over LC3B (Figure S8).
β-Branching in the Staple Modulates Binding and Selectivity.
Next, we hypothesized that β-branching in the stapled residues might modulate affinity and selectivity. Replacing Cys3 with penicillamine, a β-branched analogue of cysteine, increased binding affinity to GABARAP while maintaining selectivity over LC3B for all staple geometries and the allylated, unstapled control (Figure 2c). We confirmed that this was due to stabilization of β-strand structure by testing unstapled K1 analogues with tert-butyl glycine and tert-butyl alanine in the 3 position, which had similar affinities and selectivities (Figure S8). Unexpectedly, replacing Cys8 with penicillamine not only maintained high affinity for GABARAP but also improved LC3B binding affinity by nearly 2 orders of magnitude (compare Cys3-ortho and Pen8-ortho, Figure 2c). This effect was highly dependent on the structure of the rest of the staple, with only moderate LC3B binding observed for Pen8-para and poor LC3B binding for Pen8-meta. Ultimately, introducing β-branching into the staple produced not only Pen3-ortho, a 14 nM ligand for GABARAP with 100-fold selectivity for GABARAP over LC3B, but also Pen8-ortho, which had 12 nM affinity for GABARAP and 33 nM affinity for LC3B. Surprisingly, these two peptides differed only in the placement of two methyl groups within the staple.
Crystal Structures Reveal Two Different Binding Modes.
Because Pen3-ortho and Pen8-ortho are so similar but have different selectivity, we explored their binding modes by obtaining their crystal structures bound to GABARAP. These structures revealed two completely different binding modes. Pen3-ortho, the GABARAP-selective stapled peptide, binds GABARAP with a single turn of a 310 helix in a manner highly similar to parent peptide K1 (Figures 3a and S3). Unexpectedly, Pen8-ortho binds in the opposite orientation using a more extended conformation in a manner similar to natural LIR motifs (Figures 3b and S3). These observations are consistent with the penicillamine in the eighth position disrupting the helical structure and forcing a different binding mode for Pen8-ortho. Because they bind in opposite orientations, Pen3-ortho and Pen8-ortho use different side chains to engage GABARAP’s two hydrophobic pockets. Pen8-ortho fills one pocket with Trp6 and the other with Leu9, while Pen3-ortho fills the first pocket with Trp11 and the second with both Trp6 and Leu9. This second hydrophobic pocket is expanded in the Pen3-ortho-bound structure to accommodate both side chains (Figure 3c). Notably, Trp11 in Pen8-ortho binds in a third hydrophobic pocket not typically accessed by canonical LIR motifs. This third pocket is part of a larger binding surface used by some longer natural ligands,29,38,39 but these binding interactions typically use aliphatic residues that bind as part of a longer C-terminal helix—the binding interaction with a single Trp residue has never been seen before (Figure 3d).
Figure 3.

Pen3-ortho and Pen8-ortho bind GABARAP in different binding modes. (a) Crystal structure of Pen3-ortho bound to GABARAP. (b) Crystal structure of Pen8-ortho bound to GABARAP. GABARAP is depicted in surface rendering. (c) Pen3-ortho induces a conformational change in GABARAP that expands the central hydrophobic pocket relative to other ligands, including Pen8-ortho. Residues that bind this pocket from Pen3-ortho and Pen8-ortho are shown in purple and cyan, respectively, and GABARAPs bound to Pen3-ortho and Pen8-ortho are shown in pink and cyan ribbons, respectively. (d) Trp11 of Pen8-ortho (cyan) accesses a binding pocket that has only previously been observed to be engaged with a C-terminal helix of natural ligands (ligands from AnkB, FAM134B, SEC62, RTN3, and STX17 shown in lavender).29,38-40 In all structures, some side chains are omitted for clarity.
The crystal structures offer a potential explanation for why Pen3-ortho, which binds in the artificial, K1-like conformation, is selective for GABARAP over LC3B, while Pen8-ortho, which binds in the natural, more extended conformation, binds with high affinity to both GABARAP and LC3B. We surmise that LC3B is unable to bind ligands in the K1-like conformation with high affinity due to an inability to undergo induced fit to accommodate the Trp side chain in the central hydrophobic pocket (Figure 2c). Notably, in both structures, the staple makes extended van der Waals contacts with GABARAP, implying that the staple contributes directly to the binding affinity for both ligands.
Structure-Based Design Using 4-Mercaptoproline (4MP).
Based on the dihedral angles of bound Pen3-ortho and Ramachandran plots of proline and pre-proline residues,41,42 we hypothesized that a 4-mercaptoproline (4MP) residue within the staple would favor Pen3-ortho’s GABARAP-selective binding conformation. 4MP conformationally restricts both the backbone and the staple, and we recently reported its use as a valuable building block for peptide stapling for non-α-helical peptides.32 Testing a panel of Pen3-ortho analogues with 4MP in the third position (parent peptide MP3), we found that 4MP substitution maintained high GABARAP affinity while decreasing binding affinity to LC3B for all staple geometries (Figure 4a). These data, along with the observation that 4MP substitution abolished the nanomolar LC3B affinity of Pen8-ortho (compare to MP3Pen8-ortho, Figure 4a), suggested that 4MP promotes the helical Pen3-ortho binding conformation and disfavors the extended Pen8-ortho binding conformation. The highest-affinity ligand, MP3-meta, had a binding affinity for GABARAP that was too strong to be measured by direct FP (Kd ≤ 2.5 nM; see below for measurement using an orthogonal assay) and at least 1000-fold selectivity for GABARAP over LC3B (Figure 4a).
Figure 4.

Structure-based design of improved GABARAP ligands. (a) Stapled peptides containing 4-mercaptoproline (4MP) and their affinities for recombinant GABARAP and LC3B as measured by FP. φ denotes 4MP, and π denotes penicillamine. (b) Stapled peptides with a decreased negative charge and their affinities for recombinant GABARAP and LC3B. (c) Affinities of selected peptides were measured for five human paralogs in the GABARAP/LC3 family using FP assays. For FP assays, peptides were prepared with fluorescein on their N-termini (Figure S1 and Table S1). (d) Example data (blue curves), curve fits (red curves), and binding data (table) for a biolayer interferometry (BLI) assay with selected biotinylated peptides (Figure S1 and Table S2) and recombinant GABARAP. All data are shown as the average and standard error of the mean for three or more independent trials. Primary data and additional BLI results are shown in Figures S20-S22 and Table S10.
Prior to moving forward with biological assays, we sought to further lessen the overall negative charge. We noticed that residues Asp1 and Glu7 appear to make much more extensive polar contacts in the Pen8-ortho structure compared to the Pen3-ortho structure, as is commonly observed for the binding of canonical LIR motifs.23,39 For example, GABARAP Arg67 forms a salt bridge with Glu7 in Pen8-ortho but not Pen3-ortho (Figure S3). Thus, we hypothesized that we could reduce charge and improve selectivity by truncating the N-terminal Asp and/or replacing Glu7 with Ala. Both substitutions only moderately affected K1’s binding affinity for GABARAP, and Pen3-ortho analogues with either substitution showed 2- to 6-fold decreases in GABARAP affinity while maintaining specificity for GABARAP over LC3B (Figure 4b). Strikingly, both substitutions had larger effects on Pen8-ortho, decreasing GABARAP affinity by 20- to 30-fold while also losing all measurable binding affinity to LC3B. These results confirm the prediction that these negatively charged residues play important roles in binding LC3B but less so for GABARAP, especially when ligands bind in the non-natural, K1-like conformation. For the highest-affinity ligand, MP3-meta, either or both substitutions maintained high-affinity GABARAP binding and at least 200-fold selectivity over LC3B (Figure 4b).
Selectivity Across Human LC3/GABARAP Analogues.
We next sought to characterize binding affinities of the stapled peptides for additional human LC3/GABARAP paralogs. We set up similar FP assays with recombinant LC3A, GABARAP-L1, and GABARAP-L2 and tested selected stapled peptides and controls (Figure 4c). K1, Pen3-ortho, and MP3-meta all share a similar selectivity pattern, binding GABARAP and GABARAPL1 in the low-to-mid nanomolar range, and GABARAP-L2, LC3A, and LC3B mostly in the micromolar range. By contrast, we found that Pen8-ortho bound the five paralogs tested with Kd values between 12 and 110 nM. In fact, stapled peptide Pen8-ortho bound LC3B and LC3A with similar affinity as the natural, LC3-selective FYCO1 peptide (Figure 4c).
Orthogonal Binding Assay: Biolayer Interferometry.
The binding affinity measured for K1 in FP assays is different than the Kd in the original report by Weiergräber et al., which used surface plasmon resonance.28 These results suggested that the fluorescein attached to peptides for the FP assay may improve binding to GABARAP. To control for these effects, we used biolayer interferometry (BLI) as an orthogonal binding assay that employed biotinylated peptides (Figure S1 and Table S2). Kd values measured by BLI were, in general, 3- to 10-fold higher than Kd values measured by FP, consistent with N-terminal fluorescein contributing to binding (Figure 4d and Table S10). Importantly, the binding affinities of the highest-affinity ligands were at the titration limit of the FP assay, so BLI offered a more accurate method for comparing their binding affinities. The GABARAP-binding affinities of K1, Pen3-ortho, Pen8-ortho, and MP3-meta were measured by BLI at 55 ± 8, 39 ± 7, 14 ± 0.5, and 20 ± 1 nM, respectively. BLI data also provided insight into binding kinetics, revealing that slower off-rates appear to be primary drivers for affinity improvements for stapled analogues of K1. Pen8-ortho was the only stapled peptide with enhanced on-rate, which may be due to its canonical LIR binding mode not requiring induced fit of GABARAP (Figure 3c).
Resistance to Biological Degradation.
Proteolytic degradation is a liability for peptides, but artificial amino acids and staples can provide resistance. We measured the stability of selected GABARAP ligands following 1, 3, and 5 h incubation in freshly prepared HeLa cell lysate, a high-stringency assay for measuring susceptibility to biological degradation.26,32,43 K1 was degraded rapidly, with only 9% remaining after 1 h. Incorporating artificial amino acids such as tert-butyl alanine conferred some protease resistance, with 34% remaining at 1 h and 4% at 5 h (Figure S28). Stapling conferred even more resistance. At 5 h, between 35 and 80% of stapled peptides remained intact (Figure 5a). Interestingly, the bicyclic peptide Cys3.4.8-tmb (see above) had less peptide remaining at 5 h than most of the stapled peptides (Figure S28)–in this case, bicyclic stapling did not confer an added advantage compared to single staples with respect to biological degradation.
Figure 5.

Biological stability and cytosolic penetration of GABARAP inhibitors. (a) Chloroalkane-tagged peptides (Figure S1 and Table S3) were incubated with HeLa cell lysates for 1, 3, and 5 h, and HPLC was used to quantitate the fraction of peptide remaining. (b) Cytosolic penetration after 24 h incubation of chloroalkane-tagged peptides with HeLa cells, as measured using the chloroalkane penetration assay (CAPA).44,45 CAPA measures cytosolic penetration by monitoring the extent to which cytosolic HaloTag protein is blocked by chloroalkane-tagged peptide, with 100% fluorescence indicating no cytosolic penetration and 0% fluorescence indicating penetration sufficient to block all cytosolic HaloTag. CP50 values are derived from the curve fits shown.44,45 Chloroalkane-tagged polyarginine is shown as a positive control.44,45 All data are shown as the average and standard error of at least three independent trials. Results of individual trials and additional data at a 4 h time point are shown in Figures S24 and S25 and Table S11.
Penetration to the Cytosol.
In many cases, peptide stapling also increases cytosolic penetration.26,27,31 We examined the cytosolic penetration of our stapled GABARAP-binding peptides using the chloroalkane penetration assay (CAPA), a robust assay that measures penetration to the cytosol without interference from the material trapped at the cell surface or in endosomes.44 Importantly, while linear peptide K1 was the most prone to degradation, it was among the least cell-penetrant peptides tested (Figure 5b). This result helps to rule out degradation artifacts and supports the CAPA data report on the internalization of intact peptides.45,46 We found that, with the exception of MP3-meta, all stapled peptides had significantly improved cytosolic penetration compared to K1 (Figure 5b). Removing negative charges had modest effects, improving cytosolic penetration by 2–3 fold for some but not all stapled peptides. Overall, the CAPA data showed that stapled GABARAP ligands effectively access the cytosol when incubated for 24 h at concentrations as low as 1 to 2 micromolar.
Synergy with DNA-Damaging Agents and Effects on Autophagy.
Prior work indicated that autophagy is associated with cisplatin resistance in ovarian cancer and that autophagy inhibition can be synergistic with DNA-damaging chemotherapy in several solid tumors.6,21,30 To determine whether GABARAP-binding stapled peptides have similar applications, we exampled the effects of peptides alone and with cisplatin on cell growth in the OVCAR8 ovarian cancer cell line. Peptides alone showed limited effects on tumor cell growth up to 10 μM. However, all peptides enhanced cisplatin-induced growth inhibition at concentrations as low as 2.5 μM (Figures 6a and S31). Synergistic effects between peptides and cisplatin were confirmed by calculating combination indices, which were well below 1 for all peptides tested, with MP3.2-meta having the highest degree of synergy (Figure S31).
Figure 6.

Effects of stapled peptides on ovarian cancer cells. (a) Relative cell proliferation after treatment of OVCAR8 cells with peptide alone (1.25–10 μM), cisplatin alone (0.6–5 μM), or cisplatin plus peptide (1:2 ratio of cisplatin to peptide). ns denotes not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, one-way ANOVA with multiple comparisons. All peptides showed synergistic effects with cisplatin; combination indices are shown in Figure S31. Data are shown as the average and standard error of four technical replicates, and data are representative of two biological replicates (Figure S31). (b) Western blots of OVCAR8 cells treated with 25 μM peptide and/or 10 μM cisplatin for 48 h. Blot is representative of two biological replicates (Figure S32).
Because LC3B and GABARAP are involved in every step of autophagy (Figure 1), it was not clear precisely how LC3B/GABARAP inhibitors would affect autophagy. We treated OVCAR8 cells with cisplatin and/or stapled peptides and analyzed the autophagy markers p62, LC3B, and GABARAP by Western blotting (Figures 6b and S32). MP3.1-meta showed apparent cytotoxicity at 25 μM, as evidenced by low intensity in the GAPDH loading control. In all other samples, p62 levels were relatively unchanged, implying that cisplatin and cisplatin plus peptide had minimal effects on selective autophagy mediated by p62. However, as previously shown in OVCAR8,30 when cells are treated with cisplatin at sub-lethal doses, cisplatin decreases LC3B-I and GABARAP-I, which indicates induction of non-p62-dependent autophagy. When cotreated with sub-lethal doses of cisplatin and stapled peptides Pen8-ortho, Pen3.2-ortho, MP3-meta, or MP3.2-meta, OVCAR8 cells had increased LC3B-I compared to cells treated with cisplatin alone (Figure 6b). Treating cells with cisplatin and Pen3.2-ortho or MP3.2-meta also reversed the decrease in GABARAP-I observed in cells treated with cisplatin alone. These results suggest that K1-derived stapled peptides can partially block autophagy induction by cisplatin, which is a plausible mechanism for their synergistic effects on cell proliferation.
DISCUSSION
Despite decades of work on stapled α-helices, the structural and functional consequences of stapling nonhelical peptides remain difficult to predict. In this case, a diversity-oriented stapling strategy allowed for development of two classes of high-affinity GABARAP ligands: Pen8-ortho, which binds both GABARAPs and LC3s with nanomolar affinity, and the more GABARAP-selective Pen3-ortho and its analogues, including MP3-meta. The striking difference in binding modes for Pen8-ortho and Pen3-ortho, despite a difference of the locations of only two methyl groups, underscores the ability of a diversity-oriented stapling approach to broadly access conformational space for peptides, especially peptides lacking extended α-helices. These stapled peptides are among the most potent binders of LC3/GABARAP proteins described to date. Notably, native ligands with similar or higher affinity have an extended C-terminal helix (Figure 3d), are twice the size of the stapled peptides reported here, and have eight or more negative charges, making them unlikely to be useful for pharmacological inhibition.23,29 For Pen8-ortho, binding affinity appears to derive from the ability to access three hydrophobic pockets, and for Pen3-ortho, binding affinity appears to derive from engagement of a large, cryptic hydrophobic pocket that is not observed for native ligands. Both inhibitors also make extensive contact with GABARAP via their hydrophobic staples.
Our applications of stapled peptides to inhibit autophagy add to the growing literature showing that interfering with the protein-protein interactions of LC3/GABARAP proteins is a promising avenue for anti-cancer therapeutics.21,30 Inhibiting autophagy can also subvert immune system evasion by solid tumors. For example, Yamamoto and co-workers found that in pancreatic ductal adenocarcinoma cells, MHC-I is degraded by NBR1-mediated autophagy, resulting in a lower antigen presentation and decreased CD8+ T cell activation compared to autophagy-inhibited cells.47 Inhibiting autophagy has also been found to promote an antitumor immune response in high mutational burden tumors and can improve response to PD-1/PD-L1 therapy.48-51 This may explain why a prior LC3B ligand showed unusually potent antitumor effects in mice compared to its in vitro activity.30 In this work, we developed several stapled peptides with selectivity for GABARAP, including Pen3.2-ortho, MP3-meta, and MP3.2-meta, as well as one stapled peptide that inhibited both LC3 and GABARAP proteins, Pen8-ortho. These ligands all inhibited autophagy and potentiated cisplatin toxicity in OVCAR8 cells with similar potency. These results imply that blocking GABARAP may be at least as critical as blocking LC3B for efficient autophagy inhibition. Prior work which tested the effects of LC3B ligands and inhibitors did not examine their binding to GABARAP, leaving open the possibility that their biological effects were mediated in part (or perhaps even in full) by GABARAP inhibition.8,30,52 Moving forward, we expect that GABARAP-selective ligands and ligands that bind both LC3 and GABARAP proteins will be useful classes of ligands for exploring potential combination therapies with DNA-damaging agents and potentially with checkpoint inhibitors as well.47
Autophagy, and specifically the LC3/GABARAP family of proteins, is also an emerging avenue for targeted protein degradation.53 Autophagy-targeting chimeras (called AUTACs, among other acronyms)52-55 link a compound that binds a protein-of-interest to another compound that promotes recruitment to the autophagosome. AUTACs could have several advantages over proteolysis-targeting chimeras (PROTACs) because autophagy components such as LC3/GABARAP are broadly expressed in all tissue types and because autophagosome recruitment requires only physical tethering, not catalysis within a ternary complex. While peptides would likely get degraded in the lysosome, the stapled peptides reported here represent promising starting points for this exciting modality. Further, our data directly inform efforts to develop small-molecule AUTACs. For example, while LC3B is the more well-studied mammalian paralog, our data suggest GABARAP may be a better target for continued work in AUTAC development. GABARAP’s central hydrophobic pocket can widen to accommodate a larger hydrophobic ligand (Figure 3c), which is not seen in structures of LC3B, and it has several adjacent hydrophobic pockets that can be exploited to improve binding affinity (Figure 3a,b). Thus, we expect GABARAP to be a key target for recruiting proteins-of-interest for degradation via autophagy.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by NIH GM125856 and NSF 2003010 for J.A.K., a Beckman Scholars award from the Arnold O. and Mabel Beckman Foundation for M.C., Emerson Collective and MD Anderson Specialized Program of Research Excellence in Ovarian Cancer (P50 CA 217685, NCI) and NCI R01 CA 135354 for R.C.B. Jr. and Z.L., and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 267205415-SFB 1208, project B02 for D.W. The authors acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities, and they would like to thank Antoine Royant and Estelle Mossou for assistance in using beamlines ID23-2 and ID30B, respectively.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c04699.
Materials and methods, chemical structures, compound characterization, additional data on binding, degradation, cell penetration, and effects in cell culture, full data sets for binding assays and cell penetration assay, and crystallographic data tables (PDF)
The authors declare no competing financial interest.
Contributor Information
Hawley Brown, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
Mia Chung, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
Alina Üffing, Institute of Biological Information Processing, Structural Biochemistry (IBI-7), Forschungszentrum Jülich, 52425 Jülich, Germany; Institut für Physikalische Biologie, Heinrich-Heine-Universität Düsseldorf, 40225 Düsseldorf, Germany.
Nefeli Batistatou, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
Tiffany Tsang, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
Samantha Doskocil, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, United States.
Weiqun Mao, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, United States.
Dieter Willbold, Institute of Biological Information Processing, Structural Biochemistry (IBI-7), Forschungszentrum Jülich, 52425 Jülich, Germany; Institut für Physikalische Biologie, Heinrich-Heine-Universität Düsseldorf, 40225 Düsseldorf, Germany.
Robert C. Bast, Jr., Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, United States
Zhen Lu, Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, United States.
Oliver H. Weiergräber, Institute of Biological Information Processing, Structural Biochemistry (IBI-7), Forschungszentrum Jülich, 52425 Jülich, Germany
Joshua A. Kritzer, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
REFERENCES
- (1).Levine B; Kroemer G Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).White E The Role for Autophagy in Cancer. J. Clin. Invest 2015, 125, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Karsli-Uzunbas G; Guo JY; Price S; Teng X; Laddha SV; Khor S; Kalaany NY; Jacks T; Chan CS; Rabinowitz JD; White E Autophagy Is Required for Glucose Homeostasis and Lung Tumor Maintenance. Cancer Discovery 2014, 4, 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Xie X; Koh JY; Price S; White E; Mehnert JM Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discovery 2015, 5, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yang S; Wang X; Contino G; Liesa M; Sahin E; Ying H; Bause A; Li Y; Stommel JM; Dell’antonio G; Mautner J; Tonon G; Haigis M; Shirihai OS; Doglioni C; Bardeesy N; Kimmelman AC Pancreatic Cancers Require Autophagy for Tumor Growth. Genes Dev. 2011, 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jiang Y; Ji F; Liu Y; He M; Zhang Z; Yang J; Wang N; Zhong C; Jin Q; Ye X; Chen T Cisplatin-Induced Autophagy Protects Breast Cancer Cells from Apoptosis by Regulating Yes-Associated Protein. Oncol. Rep 2017, 38, 3668–3676. [DOI] [PubMed] [Google Scholar]
- (7).Wang J; Wu GS Role of Autophagy in Cisplatin Resistance in Ovarian Cancer Cells. J. Biol. Chem 2014, 289, 17163–17173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).AlMasri SS; Zenati MS; Desilva A; Nassour I; Boone BA; Singhi AD; Bartlett DL; Liotta LA; Espina V; Loughran P; Lotze MT; Paniccia A; Zeh HJ III; Zureikat AH; Bahary N Encouraging Long-Term Survival Following Autophagy Inhibition Using Neoadjuvant Hydroxychloroquine and Gemcitabine for High-Risk Patients with Resectable Pancreatic Carcinoma. Cancer Med. 2021, 10, 7233–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Anand K; Niravath P; Patel T; Ensor J; Rodriguez A; Boone T; Wong ST; Chang JC A Phase II Study of the Efficacy and Safety of Chloroquine in Combination With Taxanes in the Treatment of Patients With Advanced or Metastatic Anthracycline-Refractory Breast Cancer. Clin. Breast Cancer 2021, 21, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Compter I; Eekers DBP; Hoeben A; Rouschop KMA; Reymen B; Ackermans L; Beckervordersantforth J; Bauer NJC; Anten MM; Wesseling P; Postma AA; De Ruysscher D; Lambin P Chloroquine Combined with Concurrent Radiotherapy and Temozolomide for Newly Diagnosed Glioblastoma: A Phase IB Trial. Autophagy 2021, 17, 2604–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Xu R; Ji Z; Xu C; Zhu J The Clinical Value of Using Chloroquine or Hydroxychloroquine as Autophagy Inhibitors in the Treatment of Cancers. Medicine 2018, 97, No. e12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Leung L-SB; Neal JW; Wakelee HA; Sequist LV; Marmor MF Rapid Onset of Retinal Toxicity From High-Dose Hydroxychloroquine Given for Cancer Therapy. Am. J. Ophthalmol 2015, 160, 799–805. [DOI] [PubMed] [Google Scholar]
- (13).Mauthe M; Orhon I; Rocchi C; Zhou X; Luhr M; Hijlkema K-J; Coppes RP; Engedal N; Mari M; Reggiori F Chloroquine Inhibits Autophagic Flux by Decreasing Autophagosome-Lysosome Fusion. Autophagy 2018, 14, 1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kocak M; Erdi SE; Jorba G; Maestro I; Farrés J; Kirkin V; Martinez A; Pless O Targeting Autophagy in Disease: Established and New Strategies. Autophagy 2022, 18, 473–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lee Y-K; Lee J-A Role of the Mammalian ATG8/LC3 Family in Autophagy: Differential and Compensatory Roles in the Spatiotemporal Regulation of Autophagy. BMB Rep. 2016, 49, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Birgisdottir ÅB; Lamark T; Johansen T The LIR Motif – Crucial for Selective Autophagy. J. Cell Sci 2013, 126, 3237–3247. [DOI] [PubMed] [Google Scholar]
- (17).Noda NN; Ohsumi Y; Inagaki F Atg8-Family Interacting Motif Crucial for Selective Autophagy. FEBS Lett. 2010, 584, 1379–1385. [DOI] [PubMed] [Google Scholar]
- (18).Weidberg H; Shvets E; Shpilka T; Shimron F; Shinder V; Elazar Z LC3 and GATE-16/GABARAP Subfamilies Are Both Essential yet Act Differently in Autophagosome Biogenesis. EMBO J. 2010, 29, 1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nguyen TN; Padman BS; Usher J; Oorschot V; Ramm G; Lazarou M Atg8 Family LC3/GABARAP Proteins Are Crucial for Autophagosome–Lysosome Fusion but Not Autophagosome Formation during PINK1/Parkin Mitophagy and Starvation. J. Cell Biol 2016, 215, 857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Johansen T; Lamark T Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol 2020, 432, 80–103. [DOI] [PubMed] [Google Scholar]
- (21).Putyrski M; Vakhrusheva O; Bonn F; Guntur S; Vorobyov A; Brandts C; Dikic I; Ernst A Disrupting the LC3 Interaction Region (LIR) Binding of Selective Autophagy Receptors Sensitizes AML Cell Lines to Cytarabine. Front. Cell Dev. Biol 2020, 8, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jacquet M; Guittaut M; Fraichard A; Despouy G The Functions of Atg8-Family Proteins in Autophagy and Cancer: Linked or Unrelated? Autophagy 2021, 17, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wirth M; Zhang W; Razi M; Nyoni L; Joshi D; O’Reilly N; Johansen T; Tooze SA; Mouilleron S Molecular Determinants Regulating Selective Binding of Autophagy Adapters and Receptors to ATG8 Proteins. Nat. Commun 2019, 10, No. 2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rogov VV; Stolz A; Ravichandran AC; Rios-Szwed DO; Suzuki H; Kniss A; Löhr F; Wakatsuki S; Dötsch V; Dikic I; Dobson RC; McEwan DG Structural and Functional Analysis of the GABARAP Interaction Motif (GIM. EMBO Rep. 2017, 18, 1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huber J; Obata M; Gruber J; Akutsu M; Löhr F; Rogova N; Güntert P; Dikic I; Kirkin V; Komatsu M; Dötsch V; Rogov VV An Atypical LIR Motif within UBA5 (Ubiquitin like Modifier Activating Enzyme 5) Interacts with GABARAP Proteins and Mediates Membrane Localization of UBA5. Autophagy 2020, 16, 256–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cerulli RA; Shehaj L; Brown H; Pace J; Mei Y; Kritzer JA Stapled Peptide Inhibitors of Autophagy Adapter LC3B. ChemBioChem 2020, 21, 2777–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Walensky LD; Bird GH Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem 2014, 57, 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Weiergräber OH; Stangler T; Thielmann Y; Mohrlüder J; Wiesehan K; Willbold D Ligand Binding Mode of GABAA Receptor-Associated Protein. J. Mol. Biol 2008, 381, 1320–1331. [DOI] [PubMed] [Google Scholar]
- (29).Li J; Zhu R; Chen K; Zheng H; Zhao H; Yuan C; Zhang H; Wang C; Zhang M Potent and Specific Atg8-Targeting Autophagy Inhibitory Peptides from Giant Ankyrins. Nat. Chem. Biol 2018, 14, 778–787. [DOI] [PubMed] [Google Scholar]
- (30).Gray JP; Uddin MN; Chaudhari R; Sutton MN; Yang H; Rask P; Locke H; Engel BJ; Batistatou N; Wang J; Grindel BJ; Bhattacharya P; Gammon ST; Zhang S; Piwnica-Worms D; Kritzer JA; Lu Z; Bast RC; Millward SW Directed Evolution of Cyclic Peptides for Inhibition of Autophagy. Chem. Sci 2021, 12, 3526–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Peraro L; Zou Z; Makwana KM; Cummings AE; Ball HL; Yu H; Lin Y-S; Levine B; Kritzer JA Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy. J. Am. Chem. Soc 2017, 139, 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Pace JR; Lampkin BJ; Abakah C; Moyer A; Miao J; Deprey K; Cerulli RA; Lin Y-S; Baleja JD; Baker D; Kritzer JA Stapled β-Hairpins Featuring 4-Mercaptoproline. J. Am. Chem. Soc 2021,143, 15039–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sawyer N; Arora PS Hydrogen Bond Surrogate Stabilization of β-Hairpins. ACS Chem. Biol 2018, 13, 2027–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Timmerman P; Beld J; Puijk WC; Meloen RH Rapid and Quantitative Cyclization of Multiple Peptide Loops onto Synthetic Scaffolds for Structural Mimicry of Protein Surfaces. ChemBioChem 2005, 6, 821–824. [DOI] [PubMed] [Google Scholar]
- (35).Siegert TR; Bird MJ; Makwana KM; Kritzer JA Analysis of Loops That Mediate Protein–Protein Interactions and Translation into Submicromolar Inhibitors. J. Am. Chem. Soc 2016, 138, 12876–12884. [DOI] [PubMed] [Google Scholar]
- (36).Peraro L; Siegert TR; Kritzer JA Conformational Restriction of Peptides Using Dithiol Bis-Alkylation. Methods Enzymol. 2016, 580, 303–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Heinis C; Rutherford T; Freund S; Winter G Phage-Encoded Combinatorial Chemical Libraries Based on Bicyclic Peptides. Nat. Chem. Biol 2009, 5, 502–507. [DOI] [PubMed] [Google Scholar]
- (38).Li Y; Cheng X; Li M; Wang Y; Fu T; Zhou Z; Wang Y; Gong X; Xu X; Liu J; Pan L Decoding Three Distinct States of the Syntaxin17 SNARE Motif in Mediating Autophagosome–Lysosome Fusion. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 21391–21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhao J; Li Z; Li J The Crystal Structure of the FAM134B–GABARAP Complex Provides Mechanistic Insights into the Selective Binding of FAM134 to the GABARAP Subfamily. FEBS Open Bio 2021, 12, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mochida K; Yamasaki A; Matoba K; Kirisako H; Noda NN; Nakatogawa H Super-Assembly of ER-Phagy Receptor Atg40 Induces Local ER Remodeling at Contacts with Forming Autophagosomal Membranes. Nat. Commun 2020, 11, No. 3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ho BK; Brasseur R The Ramachandran Plots of Glycine and Pre-Proline. BMC Struct. Biol 2005, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).MacArthur MW; Thornton JM Influence of Proline Residues on Protein Conformation. J. Mol. Biol 1991, 218, 397–412. [DOI] [PubMed] [Google Scholar]
- (43).Partridge AW; Kaan HYK; Juang Y-C; Sadruddin A; Lim S; Brown CJ; Ng S; Thean D; Ferrer F; Johannes C; Yuen TY; Kannan S; Aronica P; Tan YS; Pradhan MR; Verma CS; Hochman J; Chen S; Wan H; Ha S; Sherborne B; Lane DP; Sawyer TK Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties. Molecules 2019, 24, 2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Peraro L; Deprey KL; Moser MK; Zou Z; Ball HL; Levine B; Kritzer JA Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J.Am. Chem. Soc 2018, 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Deprey K; Kritzer JA Quantitative Measurement of Cytosolic Penetration Using the Chloroalkane Penetration Assay. Methods Enzymol. 2020, 641, 277–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Deprey K; Batistatou N; Debets MF; Godfrey J; VanderWall KB; Miles RR; Shehaj L; Guo J; Andreucci A; Kandasamy P; Lu G; Shimizu M; Vargeese C; Kritzer JA Quantitative Measurement of Cytosolic and Nuclear Penetration of Oligonucleotide Therapeutics. ACS Chem. Biol 2022, 17, 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yamamoto K; Venida A; Yano J; Biancur DE; Kakiuchi M; Gupta S; Sohn ASW; Mukhopadhyay S; Lin EY; Parker SJ; Banh RS; Paulo JA; Wen KW; Debnath J; Kim GE; Mancias JD; Fearon DT; Perera RM; Kimmelman AC Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature 2020, 581, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Deng J; Thennavan A; Dolgalev I; Chen T; Li J; Marzio A; Poirier JT; Peng DH; Bulatovic M; Mukhopadhyay S; Silver H; Papadopoulos E; Pyon V; Thakurdin C; Han H; Li F; Li S; Ding H; Hu H; Pan Y; Weerasekara V; Jiang B; Wang ES; Ahearn I; Philips M; Papagiannakopoulos T; Tsirigos A; Rothenberg E; Gainor J; Freeman GJ; Rudin CM; Gray NS; Hammerman PS; Pagano M; Heymach JV; Perou CM; Bardeesy N; Wong K-K ULK1 Inhibition Overcomes Compromised Antigen Presentation and Restores Antitumor Immunity in LKB1-Mutant Lung Cancer. Nat. Cancer 2021, 2, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Poillet-Perez L; Sharp DW; Yang Y; Laddha SV; Ibrahim M; Bommareddy PK; Hu ZS; Vieth J; Haas M; Bosenberg MW; Rabinowitz JD; Cao J; Guan J-L; Ganesan S; Chan CS; Mehnert JM; Lattime EC; White E Autophagy Promotes Growth of Tumors with High Mutational Burden by Inhibiting a T-Cell Immune Response. Nat. Cancer 2020, 1, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Noman MZ; Parpal S; Van Moer K; Moer KV; Xiao M; Yu Y; Arakelian T; Viklund J; Viklund J; Milito AD; De Milito A; Hasmim M; Hasmim M; Andersson M; Andersson M; Amaravadi RK; Amaravadi RK; Martinsson J; Martinsson J; Berchem G; Berchem G; Janji B Inhibition of Vps34 Reprograms Cold into Hot Inflamed Tumors and Improves Anti–PD-1/PD-L1 Immunotherapy. Sci. Adv 2020, 6, No. eaax7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wang X; Wu WKK; Gao J; Li Z; Dong B; Lin X; Li Y; Li Y; Gong J; Qi C; Peng Z; Yu J; Shen L Autophagy Inhibition Enhances PD-L1 Expression in Gastric Cancer. J. Exp. Clin. Cancer Res 2019, 38, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Li Z; Wang C; Wang Z; Zhu C; Li J; Sha T; Ma L; Gao C; Yang Y; Sun Y; Wang J; Sun X; Lu C; Difiglia M; Mei Y; Ding C; Luo S; Dang Y; Ding Y; Fei Y; Lu B Allele-Selective Lowering of Mutant HTT Protein by HTT–LC3 Linker Compounds. Nature 2019, 575, 203–209. [DOI] [PubMed] [Google Scholar]
- (53).Alabi SB; Crews CM Major Advances in Targeted Protein Degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem 2021, 296, No. 100647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Takahashi D; Moriyama J; Nakamura T; Miki E; Takahashi E; Sato A; Akaike T; Itto-Nakama K; Arimoto H AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810. [DOI] [PubMed] [Google Scholar]
- (55).Pei J; Pan X; Wang A; Shuai W; Bu F; Tang P; Zhang S; Zhang Y; Wang G; Ouyang L Developing Potent LC3-Targeting AUTAC Tools for Protein Degradation with Selective Autophagy. Chem. Commun 2021, 57, 13194–13197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.