

Abstract
RNA is dynamically modified in cells by a plethora of chemical moieties to modulate molecular functions and processes. Over 140 modifications have been identified across species and RNA types, with the highest density and diversity of modifications found in transfer RNA (tRNA). The methods used to identify and quantify these modifications have developed over recent years and continue to advance, primarily in the fields of next generation sequencing (NGS) and mass spectrometry (MS). Most current NGS methods are limited to antibody-recognized or chemically-derivatized modifications and have limitations in identifying multiple modifications simultaneously. Mass spectrometry can overcome both of these issues, accurately identifying a large number of modifications in a single run. Here we present advances in MS data acquisition for the purpose of RNA modification identification and quantitation. Using this approach, we identified multiple tRNA wobble position modifications in Arabidopsis thaliana that are upregulated in salt-stressed growth conditions and may stabilize translation of salt stress induced proteins. This work presents improvements in methods for studying RNA modifications and introduces a possible regulatory role of wobble position modifications in A. thaliana translation.
Graphical Abstract
INTRODUCTION
The RNA epitranscriptome is defined by the presence of over 140 modifications on RNA across species and RNA types1. These modifications decorate both coding and noncoding RNA and play critical roles in biological functions through modulating RNA structure, RNA-protein interactions, and nucleic acid interactions2–5. Despite RNA modifications being known to exist for over 60 years, it is likely that not all modifications have been discovered. The biological roles of known modifications have been challenging to study due to a limited number of tools to interrogate their functions. Recently, the discovery of modifications such as m6A and ac4C in mRNA and invention of novel sequencing techniques capable of localizing the marks has led to dramatically increased interest in and investigation of RNA modifications at large6,7. This surge in interest in the biological functions of RNA modifications, which have been implicated in development, cancer, and a multitude of cellular processes, has highlighted the need for additional improvements in quantitative methods of RNA modification analysis.
Much of the recent research on RNA modifications has been performed using sequencing, focused on individual modifications of mRNA8,9. These efforts have yielded important findings on mRNA stability, splicing, and translation; however, the powerful sequencing methods have significant limitations4,9,10. Sequencing provides the best data on modification locations in mRNA, but is limited to only a few modifications, and only one can be robustly analyzed at a time. Some of the techniques have high false positive rates or low specificity, such as bisulfite sequencing, and a number are not quantitative11. In addition, sequencing approaches are not as well suited for tRNA and rRNA due to the high number of diverse chemical modifications which can inhibit reverse transcription, occluding certain regions from analysis or providing false positives in RT-stop based techniques12,13. Lastly, because modification sequencing is targeted, it requires a priori knowledge of which modification is present.
Analysis of single nucleosides can provide high quality modification quantitation, produce valuable data, and inform future sequencing efforts. Mononucleoside analysis is best performed by LC-MS, which has been in use since 199014. A variety of different sample preparation techniques are commonly used in combination with mass spectrometry. Generally, a cocktail of multiple enzymes is used to ensure that modified nucleosides do not occlude cleavage of phosphoester linkages. This incubation is sometimes performed at acidic and/or basic pH at 37 °C15. In this work, we digest RNA at neutral pH and room temperature.
Mass spectrometric techniques also vary, but the implications of different approaches do not have the same burden of bias that sample preparation differences can induce16. These techniques are used largely dependent on the desired data. The common methods include targeted data acquisition, used most commonly with triple quadrupole (QQQ) mass spectrometers, and data dependent acquisition (DDA), used more commonly with instruments that can acquire full scans rapidly, such as Orbitraps17,18. As many proteomics labs lack QQQ instruments and seek to expand their analytical technologies, development of easily adopted RNA modification analysis methods for high-resolution instruments is of value. These methods come with distinct advantages; targeted QQQ acquisition provides the highest quantitative accuracy and sensitivity, and DDA allows for identification of unknowns, however, each method comes with limitations. Targeted acquisition requires prior knowledge of analytes and must be performed carefully, as QQQ instruments lack the mass accuracy to differentiate, for example, ac4C from isotopes of guanosine. Depending on acquisition parameters, DDA can acquire too few MS2 scans to use for quantitation, which can introduce more noise in measurement by requiring quantitation at the MS1 level. Decreasing dynamic exclusion can overcome this issue, however, it is at the cost of missing very low abundance nucleosides. The limitations of these methods provide an opportunity for a recent MS technique, data independent acquisition (DIA) to be used. DIA allows for both the potential detection of unanticipated molecular species and MS2 based quantitation, addressing the biggest disadvantages of targeted acquisition and DDA, respectively. In this work, we demonstrate that DIA is a viable approach for mass spectrometry-based nucleoside analysis, allowing us to better detect nucleosides and quantify their abundances.
RESULTS AND DISCUSSION
Data Independent Acquisition (DIA) for Nucleosides
Different data acquisition techniques and parameters can be used to obtain MS data, and this is mainly determined by the experiment type and downstream analysis. The three common MS techniques that can be used for nucleoside analysis are data dependent acquisition (DDA), data independent acquisition (DIA), and targeted data acquisition (Figure 1A). All three approaches have their strengths and weaknesses, some aspects of which are depicted in Figure 1A. Specifically in our studies, we compare DDA and DIA data acquisition approaches using an Orbitrap Fusion mass analyzer (Thermo Electron). In short, the ideal MS nucleoside data acquisition method needs 1) the ability to detect low abundance species, 2) sufficient MS2 scans for quantitation, 3) minimal signal interference, and 4) an unbiased approach that allows for analysis of any known and unexpected modified nucleosides of interest.
Figure 1.
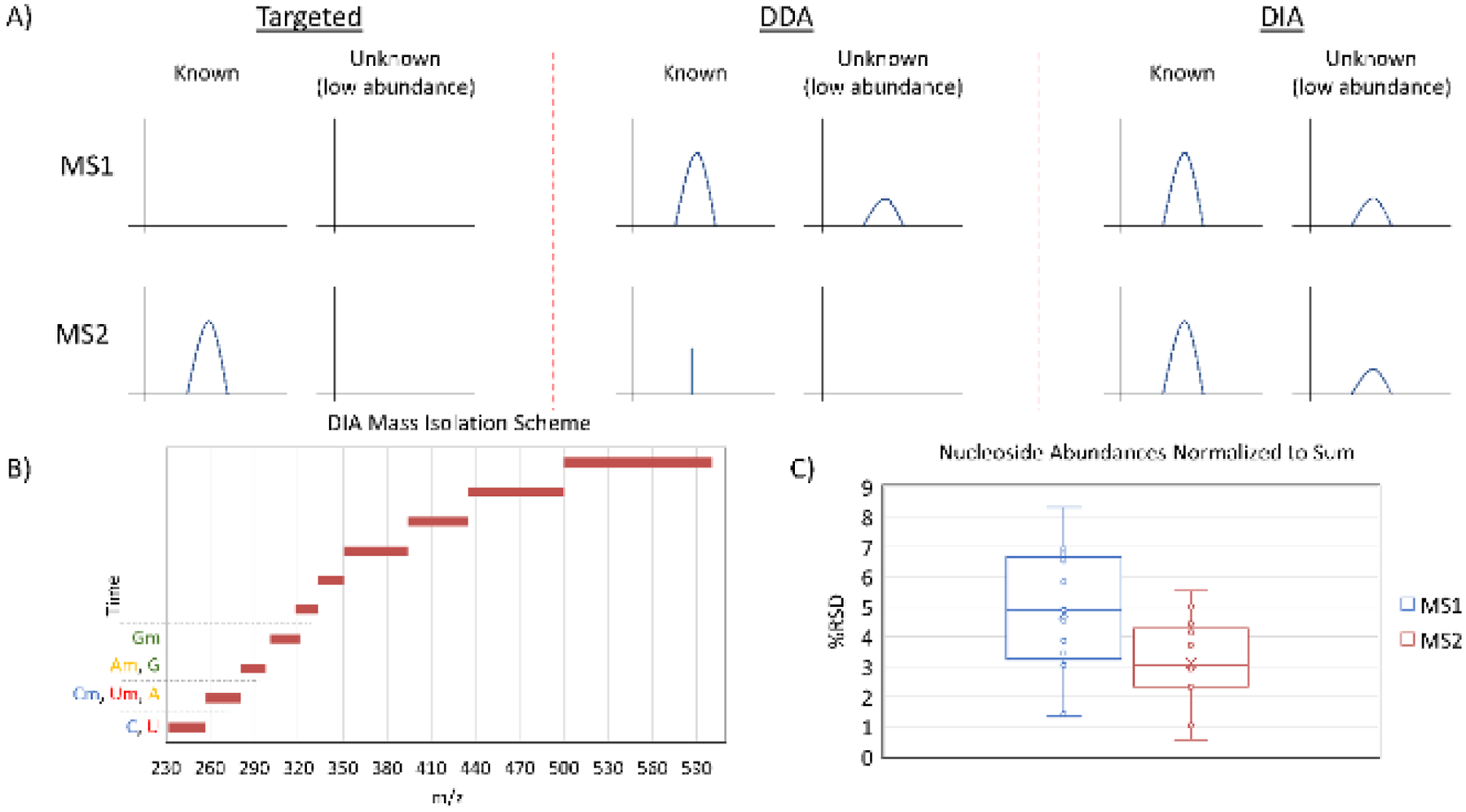
A) A comparison of MS acquisition approaches. Targeted acquisition is often performed using a QQQ mass spectrometer, which isolates a mass, fragments the molecule, and then isolates and detects a single fragment ion. DDA selects masses for fragmentation based on criteria such as abundance and often excludes previously fragmented masses from selection, resulting in few MS2 scans of abundant nucleosides and no MS2 scans of low abundance nucleosides. DIA fragments all masses in designated windows in each scan cycle, ensuring adequate points across a peak at the MS1 and MS2 levels for all nucleosides. B) The DIA mass isolation scheme designed in this work. The scan windows prevent different ribose modification states from producing the same fragment ion in the same MS2 scan from different precursors. C) A comparison of %RSD values for 5 HeLa biological replicates injected at different concentrations (1x to 5x). The nucleosides used were C, U, Ψ, A, G, m3C, m5C, Cm, I, m1A, m6A, Am, and m1acp3Ψ, and abundances were normalized to the sum of the included analytes. Within the same runs, MS2 was significantly more consistent than MS1 analysis (P < 0.05).
Due to the inability to establish a rapid QQQ based DIA method, the difference between DIA and targeted or DDA methodology is best compared based on Orbitrap technology. Indeed, when designing a targeted method, careful consideration of which modifications are of interest is important, as not all of the ~160 known marks can be included in an optimized single rapid scan cycle. Many of the modifications are known to be restricted to certain species or RNA types, which can inform method design, but does not completely resolve the issue19. Additionally, preliminary work can establish retention times for known modifications, allowing a method to have segments for targeted analyses which increase the number of targets that can be included in a single method. To perform each of these steps and design a method idealized for the modified nucleosides of interest, and additional nucleosides of possible interest, is a burden that can be overcome trivially with DIA. Moreover, DIA has the benefit of potential modification discovery, and of re-mining old data after modifications are discovered. For example, in HeLa extracted RNA, we were able to identify hm5Cm, a modification that was described shortly after our data were acquired (Figure S1)20. In this run, the hm5Cm precursor was never more than the 452nd most abundant m/z, making it very unlikely to have been selected for fragmentation in high resolution DDA scans21. Thus, DIA occupies a space between targeted acquisition and DDA as, in this application, DIA retains practically none of the limitations of the other two methods (Figure 1B).
In order to facilitate discovery of modifications and accurate quantitation simultaneously, we sought to design a DIA instrument method that would provide unbiased mononucleoside data. The DIA isolation window scheme was designed to minimize interference and misidentifications even in poor chromatographic conditions: no more than 16 known unique nucleoside masses were included in a single window (326 – 351 m/z), and methylribose nucleoside fragmentation windows were separated from their respective canonical nucleoside fragmentation windows as many of their fragment ions are identical (Figure 1B). This is due to the lability of the glycosidic bond; with the exception of pseudouridine, nucleosides fragment favorably into intact bases corresponding to ribose losses22. DIA scan window optimization was performed without accounting for retention time effects, making the isolation window scheme applicable for any stationary phase and any chromatographic gradient runtime. This approach differs from a very recently published DIA method by using larger, non-uniform isolation windows designed to account for potential isobaric fragment ions23. Compared to small, uniform isolation windows, this allows for greater points across a peak and/or higher AGC targeting, enhancing quantitative precision and/or sensitivity.
To further compare DIA and DDA, we highlight the difficulty of DDA for discovery of post-transcriptional modifications and analysis of very low abundance modifications, as they can be lost in noise at the MS1 level. Moreover, DDA suffers the limitation of lacking MS2 quantitation unless dynamic exclusion is short enough to obtain several measurements across a peak (typically eight or more). Short dynamic exclusion exacerbates the discovery problem by limiting the number of low abundance peaks that can be selected for fragmentation. DIA resolves both issues by fragmenting all ions in each scan cycle regardless of abundance and ensuring that sufficient points across a peak will be obtained for quantitation. Uridine and pseudouridine demonstrate the benefits of performing DIA, as the mass of these nucleosides has higher noise than other common nucleosides. We quantitated C, U, Ψ, A, G, m3C, m5C, Cm, I, m1A, m6A, Am, and m1acp3Ψ using MS1 and MS2 scans from the same five biological replicate runs of extracted total HeLa RNA. Our MS2 quantitated results showed m5C and Cm at 0.32% and 0.56% of C respectively, and m6A and Am at 0.10% and 1.3% of A respectively, in the range of previously reported values24–27. Compared to MS1 quantitation, MS2 analysis of all listed nucleosides yielded significantly lower relative standard deviation (RSD) (Figure 1C). The greatest difference in %RSD was observed in the uridine analysis. This can be observed qualitatively in chromatograms, which show decreased noise in MS2 scans when compared to MS1 scans. In particular, uridine, inosine, and m6Am have substantially less relative noise in their respective MS2 scans (Figure 2).
Figure 2.
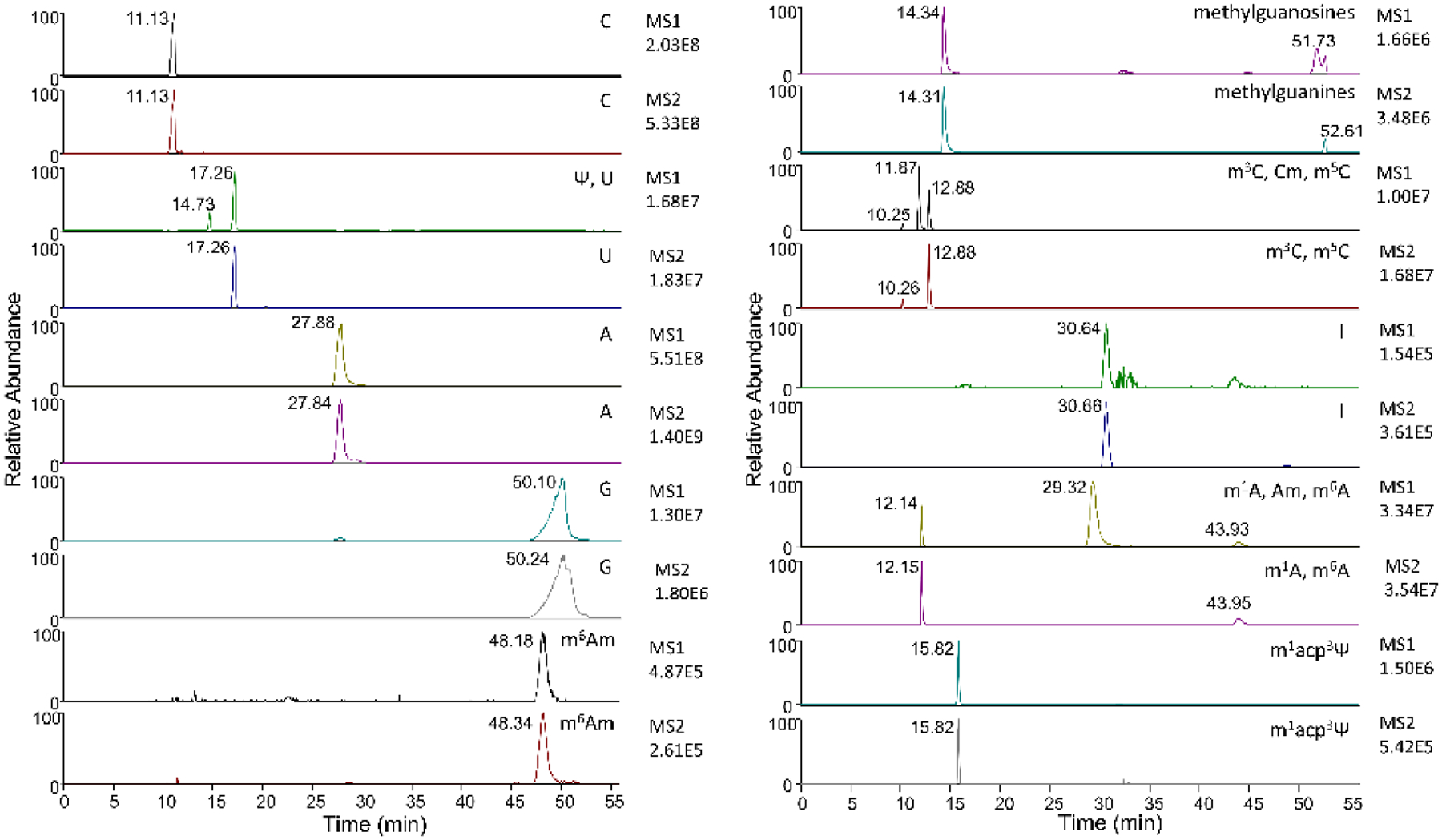
MS1 and MS2 chromatograms of HeLa nucleosides using DIA. Modifications are listed in order of appearance, with retention times denoting which peaks correspond to the listed modifications, ordered respectively. A reduction in noise can be observed in the MS2 scans, most clearly for U, m6Am, and I. MS2 peaks for Ψ, Cm, and Am are not present as their fragment masses differ from the nucleobases of U and the base modifications of C and A.
We then generated calibration curves for A, U, G, C, io6A, i6A, mcmo5U, and t6A in a background of 13C6 glucose treated HeLa RNA using MS1 and DIA MS2 quantitation (Figure 3). The DIA method was modified to ensure that any heavy ribose containing HeLa nucleosides without at least one 13C atom in the nucleobase were fragmented separately from the standards. Peaks were integrated in Skyline, manually validated, and exported for processing28. The calibration curves consistently yielded better linearity at the MS2 level, and lower limit of quantitation values (Table S1) as determined by the curves. In particular, MS2 quantitation allowed for inclusion of lower levels of uridine, guanosine, io6A, and mcmo5U in the linear range of the curves. This was most pronounced for mcmo5U, in which the lowest level injection was not observable at the MS1 level, and for uridine, in which the lowest four injections were not observable. These data show that DIA is capable of high quality MS2 quantitation without the need for targeted fragmentation or low dynamic exclusion and has the potential to identify nucleosides that are not observable at the MS1 level. Combined with Skyline data processing, this workflow simplifies MS nucleoside analysis from method design to results and can accommodate the preferred LC method of the user.
Figure 3.
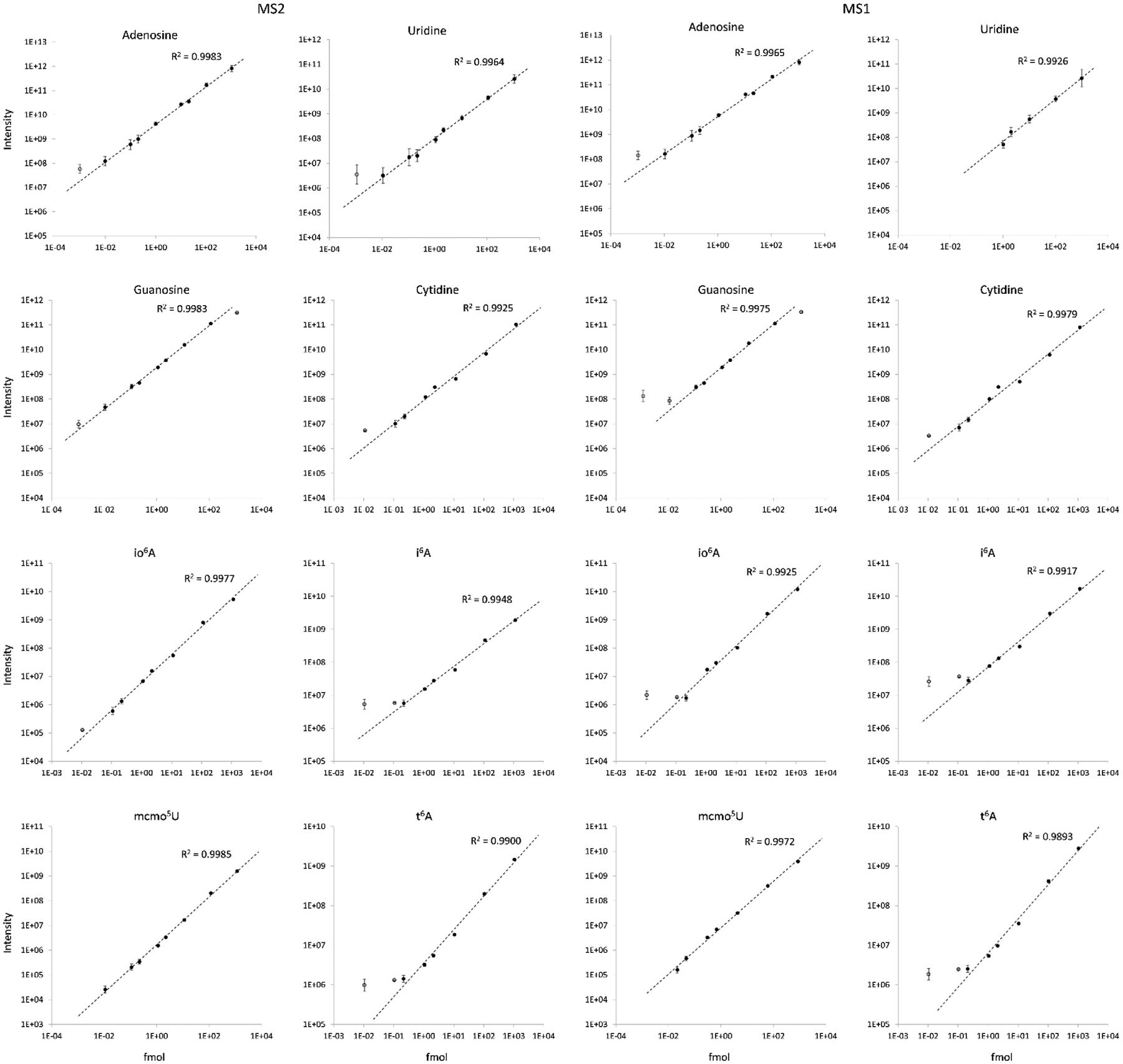
Calibration curves of nucleoside standards in a matrix of 13C-glucose treated HeLa RNA. Unfilled points were not included in linear curves. All MS2 curves yielded superior R2 values to MS1 curves and lower limits of quantitation (Table S1). Some data are absent from uridine and mcmo5U MS1 curves but were present in the MS2 scans.
When using new LC methods, it is not always necessary to know expected nucleoside retention times beforehand, as many nucleosides have unique precursor and fragment ion m/z values, and even some isobaric species have unique fragments. Notably, HCD has previously been used to generate unique fragments from isobaric nucleosides, and these fragments were recently characterized in-depth29. Using HCD fragmentation for DIA provided some fragments matching the literature at low levels (Table S2). In an hour-long method, high energy HCD or targeted MS3 can be used for isobaric nucleosides without resulting in fewer than eight MS2 scans of the targeted isomers. These experiments were performed using standards to validate methylcytidine isomer retention times and fragmentation spectra (Figure S2). A direct comparison to QQQ methodology cannot be easily performed, but DIA is an attractive method for quantitation in Orbitrap instruments, as it is similar to targeted Orbitrap methods but has fewer limitations30.
As nearly all known nucleosides fragment into the intact base, searching data can be performed by attempting to identify ribose loss, methylribose loss, or canonical nucleobases in the event a modification is fragmented as well. This allows for post-acquisition data searches for neutral losses and nucleobases, both which can be performed rapidly in MS-DIAL, and the latter of which can easily be applied in Thermo Xcalibur Qual Browser. For example, searching for adenine across different DIA MS2 windows can identify fragment ions for which the respective precursor can be identified by process of elimination within the range of the fragmentation window (Figure S3). DIA is not without limitations, as there is a higher potential for artefacts or coelutions to appear to be bona fide nucleosides31. This issue only differs from DDA in that precursor ions can be misattributed to product ions, therefore, one of the first validations steps to perform in the event of modification discovery is targeted fragmentation. Internal standards can also be used with this DIA method provided that stable isotopes are present on the base. An alternative fragmentation scheme can also be used to ensure all modifications and their standards fragment in the same window (Table S3), ensuring AGC target matched quantitation. Overall, DIA decreases MS2 cycle time, ensures adequate points across each peak, fragments nucleosides regardless of abundance, and enables discovery of previously unobserved modifications in studies that may otherwise have missed new findings.
Wobble Position Modifications in Arabidopsis thaliana
RNA in Arabidopsis thaliana has previously been reported to undergo modification changes in salt stress. Previous analyses have reported modification changes in mRNA that altered structure and transcription, however, not all translational changes could be attributed to mRNA modification and structure32. To analyze another potential translation regulation mechanism, we used our DIA method with MS2 level nucleobase quantitation to examine RNA modifications, focusing on wobble position uridine modifications, in salt stressed A. thaliana (Table S4). Our results showed significant increases in 5-aminomethyluridine (nm5U), 5-carbamoylmethyluridine (ncm5U), 5-carbamoylhydroxymethyluridine (nchm5U), and 5-methoxycarbonylmethyluridine (mcm5U) (Figure 4), but no significant changes in tRNA modifications D and io6A. Common rRNA modification ac4C also did not show changes, nor did mRNA cap modification m7G. The significantly altered ncm5U modification is found at the wobble position of tRNAPro(UGG) in multiple species33–37. Additionally, it has been reported that ncm5U or a modification in the same deposition pathway is likely be modified at U34 in A. thaliana tRNAPro(UGG) as well, however, it has not been proven38–40.
Figure 4.
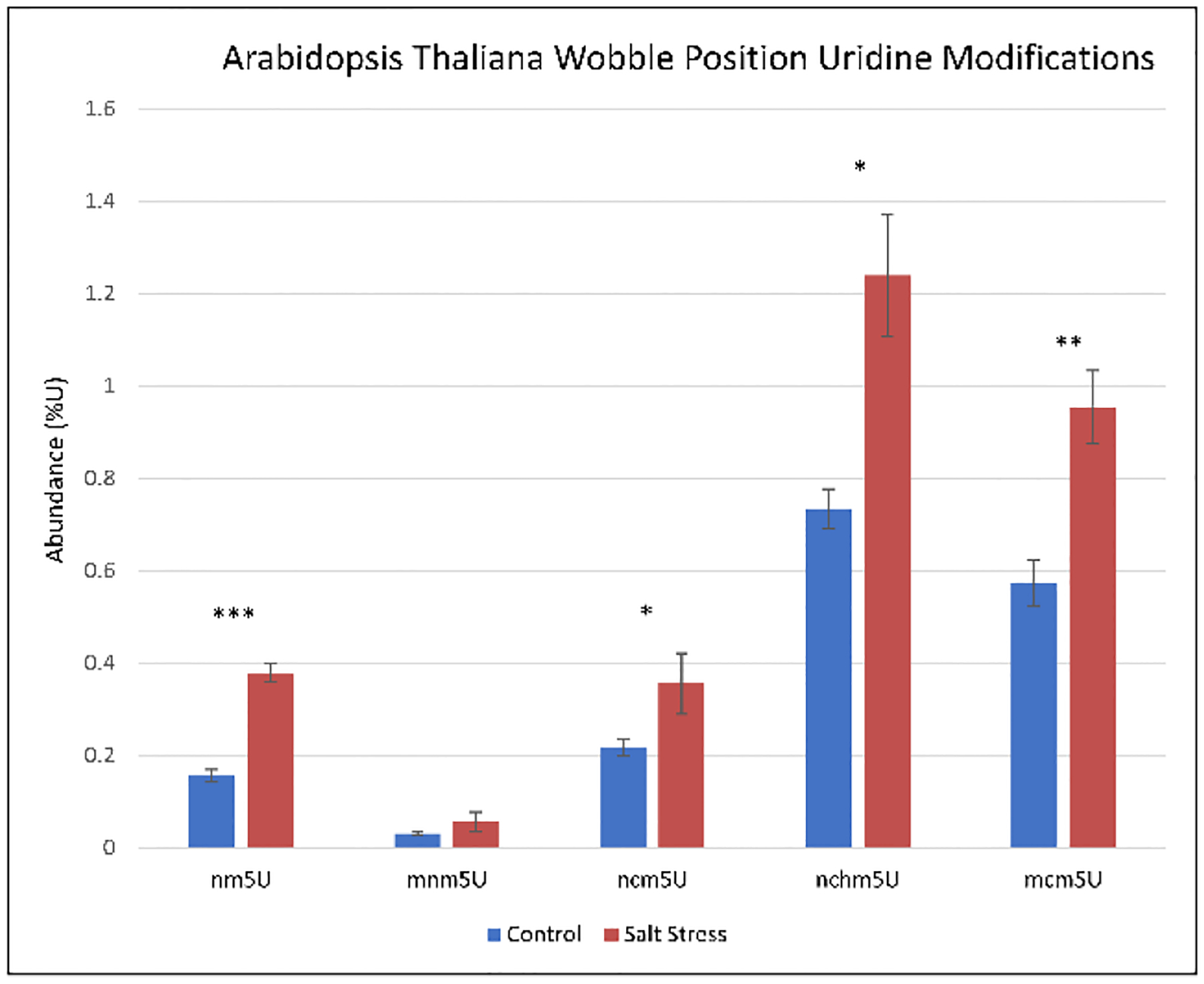
Uridine modifications upregulated in salt stressed A. thaliana. All modifications have been previously observed in only the wobble position of different organisms. ***P < 0.001, **P < 0.01, *P < 0.05
The biogenesis of ncm5U, nchm5U, and mcm5U have previously been shown to be TRM9 and TRM11 dependent, and ncm5U also required AtELP1 activity. Consistent with our RNA modification data, TRM11 was shown to be upregulated in transient salt stress in A. thaliana41. Although our previously published proteomics experiments did not detect TRM9 or TRM11 due to dynamic range limitations, our previously published RNA-seq data did show increases in AtELP1 and in two isoforms each of TRM9 and TRM11 in salt stress (Table 1)32.
Table 1:
mRNA Levels of tRNA Modifiersa.
| Gene | Control (RPM) | Salt (RPM) |
|---|---|---|
| TRM11 Isoform 1: AT3G26410 | 5.60 | 8.85 |
| TRM11 Isoform 2: AT4G23020 | 5.14 | 7.00 |
| TRM9 Isoform 1: AT1G31600 | 10.28 | 14.55 |
| TRM9 Isoform 2: AT1G36310 | 24.05 | 25.57 |
| AtELP1: AT3G52850 | 69.49 | 119.35 |
Table 1: mRNA Levels of tRNA Modifiers. A. thaliana mRNA reads per million for RNA modifying enzymes. The listed modifications catalyze deposition of identified upregulated wobble position uridine modifications. All mRNA transcripts were upregulated in salt stress.
Due to the common identification of these uridine modifications in the anticodon loop, we sought to investigate the possibility that they stabilize salt stress response translation42,43. Proline tRNAs were selected for analysis due to the similarity between A. thaliana tRNAPro(UGG) and the same tRNA in modification annotated sequences. Codon enrichment for tRNAPro(UGG) was calculated as the quantity of CCA codons in an mRNA transcript divided by sum of CCT, CCC, and CCG codons, each of which encodes a different proline tRNA. This enrichment was compared to a measure of translation efficacy, normalized fold change in protein abundance over normalized fold change in mRNA transcript abundance, ΔMS/ΔmRNA (Figure 5). The results show that highly CCA enriched proteins more often had increases in ΔMS/ΔmRNA than decreases. Two of the most CCA enriched proteins, FQR1 and PBB1 have previously been reported to be upregulated in salt stress conditions44–47. These two mRNA transcripts were stabilized; however, the stability was not m6A induced, suggesting an alternative mechanism. The transcript with the most CCA codons, At4G22485, had a moderate CCA enrichment but yielded a ΔMS/ΔmRNA value of 1.44 and was previously reported to be upregulated in salt stress48. Finally, we performed the same analysis for tRNAPro(AGG) (Figure S4), which did not show a positive trend in ΔMS/ΔmRNA at high CCT codon enrichment. These data provide an example of how RNA modification analysis can be integrated with RNA-seq and proteomic datasets to identify correlations in biology. However, additional work is required to determine if the wobble position uridine modifications are causative in salt-stress response translation stabilization.
Figure 5.
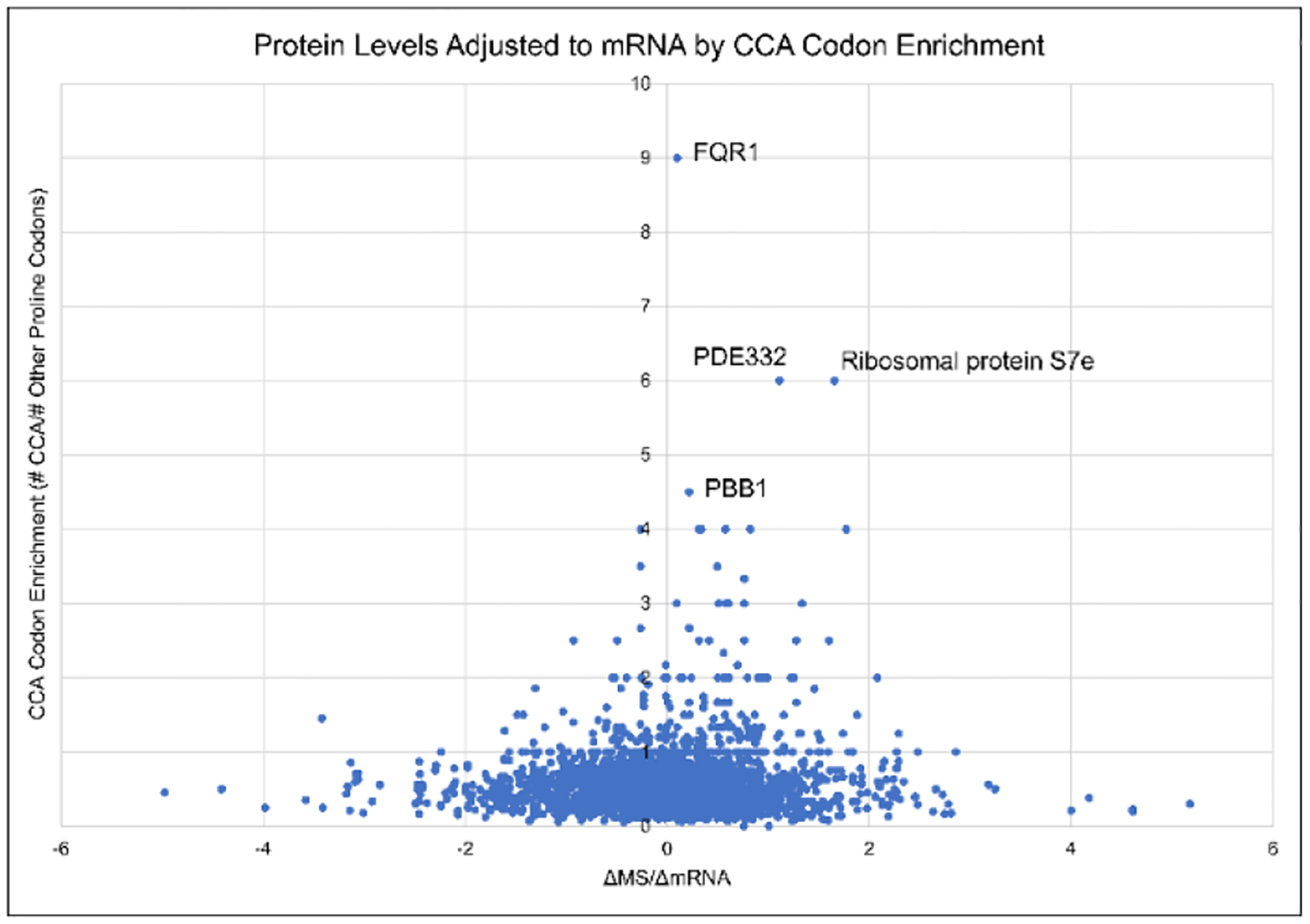
Log2 normalized ΔMS/ΔmRNA values plotted based on CCA-enrichment of the mRNA transcript. The highest CCA-enriched transcripts all show positive ΔMS/ΔmRNA values, suggesting translation stabilization for these proteins. Both FQR1 and PBB1 have been reported to be upregulated in salt stress conditions.
Conclusions
In this work, we present DIA as an improvement to Orbitrap MS based RNA mononucleoside analysis. The fragmentation windows were designed to ensure that different precursor ions with isobaric fragment ions are isolated in different MS2 scans. For any chromatographic method, this is a simple approach that guarantees fragmentation of all knowns and unknowns while simultaneously allowing for MS1 and MS2 based quantitation. We used 13C glucose labeling to generate MS1 and MS2 quantitated calibration curves in a relevant matrix, demonstrating the advantages of MS2 quantitation. Using this DIA method, we established that tRNA modifications in Arabidopsis thaliana undergo changes in response to salt stress and suggest a possible regulatory mechanism for salt stress protein translation. We also demonstrate that DIA is superb at identifying modifications without a priori knowledge and showcase an example of mining old data with hm5Cm. These efforts represent a step forward in advancing RNA mononucleoside modification detection by mass spectrometry.
Methods
Plant Growth and Harvesting
Plant growth took place in a chamber with a controlled cycle of 16 light hours and 8 dark hours at 22 °C. The ecotype of Arabidopsis thaliana was UPQ10:NTF/ACT2p:BirA Columbia-0. Salt concentrations were based on fresh weight decreases as previously determined32.
Rosette leaves were crushed in liquid N2 and RNA was extracted using Qiazol lysis reagent. QIAshredders (QIAGEN) were used to homogenize samples and miRNeasy mini columns (QIAGEN) were used to extract RNA. Samples were treated with RNase-free DNase (QIAGEN) for 30 minutes at room temperature and then RNA was ethanol precipitated.
RNA Purification
HeLa RNA was purified using TRIzol as previously described49. In short, cells were suspended in TRIzol/TriPure and centrifuged. The supernatant was mixed with chloroform and centrifuged. The aqueous phase was collected, and RNA was precipitated by adding isopropanol. RNA was washed with ethanol and then dried in a Savant SpeedVac.
Cell Culture
HeLa cells were grown in Dulbecco’s Modified Essential Medium (DMEM) lacking glucose, supplemented with 10% FBS, 1% glutamax, 1% penicillin/streptomycin, 1% sodium pyruvate, and 4.5 g/L 13C6 glucose. Cells were cultured until the ribose moieties were completely 13C labeled and the bases contained at least one 13C. RNA was extracted using TRIzol as previously described.
RNA-Sequencing and Mass Spectrometry Data Analysis
RNA-sequencing (GEO: GSE108852) and mass spectrometry raw data were obtained from previous work32. RNA-sequencing data were normalized to reads per million (RPM) as previously described. Mass spectrometry data were processed in MaxQuant and iBAQ values were used to normalize protein abundances relative to average abundance within runs. Both sets of data were log2 transformed for salt stress fold change over wild type. The ΔMS/ΔmRNA values were calculated by subtracting the resultant RNA log2 fold change from the resultant mass spectrometry log2 fold change.
RNA Digestion and Sample Processing
Purified RNA samples were resuspended in water, quantitated by nanodrop, and 500 ng of RNA was added to the reaction mixture: 10 μL of 1 mM ZnCl2, 30 mM sodium acetate, and digestion enzymes. As it was previously determined that basic conditions can alter modifications, digestion was performed at pH 7.215,50,51. Four enzymes were used to digest the RNA to mononucleosides: 5 mU/μL of nuclease P1, 500 μU/μL of phosphodiesterase I, 6.25 μU/μL of phosphodiesterase II, and 5 mU/μL of shrimp alkaline phosphatase. Digestion was performed at room temperature for 4 hours, 8 hours, or overnight. Because A. thaliana samples had the potential to contain excess salt, post-digestion protein precipitation and sample dilution were not considered adequate for analysis50,52,61,62,53–60. Indeed, we observed salt matrix effects in salt-added standards (Figure S5). Therefore, digested samples were desalted using in-house hypercarb PGC (Thermo Scientific) stagetips, similar to previously used commercial kits17. In short, digested RNA was loaded onto the resin in 0.1% formic acid, washed with 0.1% formic acid, and eluted in 0.1% formic acid in 80% acetonitrile. For runs using weakly acidic HPLC buffers, ammonium acetate can be used instead of formic acid.
Liquid Chromatography
Online nanoflow PGC runs were performed using 0.1% formic acid as buffer A and 0.1% formic acid in acetonitrile as buffer B at 600 nL/min. Gradients were run as 0% B to 16% B over 5 minutes, to 32% B over 50 minutes, to 55% B over 16 minutes, and finally to 100% B over 14 minutes. 100% B was held for 5 minutes, totaling a 90 minute run. The same gradient was proportionally adjusted to a 60 minute run, the only exception being that the 5 minute 100% B wash was retained. Microflow C18 runs were performed using the same buffers at 0.3 mL/min. The gradient was run as 0% B for 3 minutes followed by an increase to 2% B over 8 minutes, then to 15% B over 5 minutes, and finally, a 5 minute 100% B wash. Nanoflow C18 runs were performed using 0.1% formic acid as buffer A and 0.1% formic acid in 80% acetonitrile as buffer B with a flow rate of 300 nL/min. Separation was achieved using a 34 minute gradient from 0% B to 10% B, and then a 10 minute gradient from 10% B to 30% B.
Mass spectrometry
The DIA method used one full MS scan followed by eleven DIA MS2 scans. The full MS scan was from 200 to 800 m/z at 60k resolution with an AGC target of 400k and a maximum injection time of 50 ms. DIA fragmentation used 24% HCD energy and scan windows were, in order, 232 to 257, 257 to 271, 271 to 285, 285 to 298, 298 to 312, 312 to 326, 326 to 351, 351 to 394, 394 to 435, 435 to 500, and 500 to 600 m/z. All Orbitrap methods were paired with the nanoflow PGC LC method except for the calibration curve. Targeted QQQ methods were paired with microflow chromatography and used nucleoside monoisotopic masses for Q1 selection and corresponding nucleobase monoisotopic masses for Q3 selection except for pseudouridine, for which the nucleobase detection was at 125.035 m/z.
Nucleoside Calibration Curve
Nucleoside standards were mixed into a background of 13C labeled HeLa RNA and injected at levels of 1 pmol, 100 fmol, 10 fmol, 2 fmol, 1 fmol, 200 amol, 100 amol, 10 amol, 1 amol, and 100 zmol. Calibration curves were performed using C18 nanoflow chromatography.
Data Analysis
Mass spectrometry data were analyzed manually and by using Skyline28. Layout templates in Thermo Xcalibur Qual Browser were generated for MS1 scans using predicted m/z values based on chemical formulas in MODOMICS database with a tolerance of 10 ppm. Layouts for MS2 scans were designed based on expected ribose or methylribose loss and determined empirically for modified pseudouridines. Peaks were identified and integrated automatically, and then inspected and adjusted by manually as necessary. Nucleoside identities were confirmed by reference standards, MS2 fragmentation patterns, and/or by MS3 (when applicable).
Rapid automated data analysis was performed using Skyline with an inclusion list for all known RNA modifications (see supplement). Expected retention times were varied by stationary phase and gradients, manually adjusted as necessary. Peaks were integrated automatically and inspected manually.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge funding from NIH grants CA196539, AI118891 and NS111997, and NSF grants IOS-1849708, MCB-1623887, IOS-1758532, and IOS-1444490.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplemental figures S1 through S5 and supplemental tables S1 through S4 (PDF)
REFERENCES
- (1).Boccaletto P; Machnicka MA; Purta E; Piatkowski P; Baginski B; Wirecki TK; de Crécy-Lagard V; Ross R; Limbach PA; Kotter A; Helm M; Bujnicki JM MODOMICS: A Database of RNA Modification Pathways. 2017 Update. Nucleic Acids Res. 2018, 46 (D1), D303–D307. 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Shi H; Wei J; He C Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74 (4), 640–650. 10.1016/j.molcel.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jung Y; Goldman D Role of RNA Modifications in Brain and Behavior. Genes. Brain. Behav 2018, 17 (3), e12444. 10.1111/gbb.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Thapar R; Bacolla A; Oyeniran C; Brickner JR; Chinnam NB; Mosammaparast N; Tainer JA RNA Modifications: Reversal Mechanisms and Cancer. Biochemistry 2019, 58 (5), 312–329. 10.1021/acs.biochem.8b00949. [DOI] [PubMed] [Google Scholar]
- (5).McCown PJ; Ruszkowska A; Kunkler CN; Breger K; Hulewicz JP; Wang MC; Springer NA; Brown JA Naturally Occurring Modified Ribonucleosides. WIREs RNA 2020, n/a (n/a), e1595. 10.1002/wrna.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fu Y; Dominissini D; Rechavi G; He C Gene Expression Regulation Mediated through Reversible M6A RNA Methylation. Nat. Rev. Genet 2014, 15 (5), 293–306. 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- (7).Arango D; Sturgill D; Alhusaini N; Dillman AA; Sweet TJ; Hanson G; Hosogane M; Sinclair WR; Nanan KK; Mandler MD; Fox SD; Zengeya TT; Andresson T; Meier JL; Coller J; Oberdoerffer S Acetylation of Cytidine in MRNA Promotes Translation Efficiency. Cell 2018, 175 (7), 1872–1886.e24. 10.1016/j.cell.2018.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Safra M; Sas-Chen A; Nir R; Winkler R; Nachshon A; Bar-Yaacov D; Erlacher M; Rossmanith W; Stern-Ginossar N; Schwartz S The M1A Landscape on Cytosolic and Mitochondrial MRNA at Single-Base Resolution. Nature 2017, 551 (7679), 251–255. 10.1038/nature24456. [DOI] [PubMed] [Google Scholar]
- (9).Motorin Y; Helm M Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes (Basel). 2019, 10 (1), 35. 10.3390/genes10010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li X; Xiong X; Yi C Epitranscriptome Sequencing Technologies: Decoding RNA Modifications. Nat. Methods 2017, 14 (1), 23–31. 10.1038/nmeth.4110. [DOI] [PubMed] [Google Scholar]
- (11).Helm M; Motorin Y Detecting RNA Modifications in the Epitranscriptome: Predict and Validate. Nat. Rev. Genet 2017, 18, 275. [DOI] [PubMed] [Google Scholar]
- (12).Torres AG; Reina O; Stephan-Otto Attolini C; Ribas de Pouplana L Differential Expression of Human TRNA Genes Drives the Abundance of TRNA-Derived Fragments. Proc. Natl. Acad. Sci 2019, 116 (17), 8451 LP–8456. 10.1073/pnas.1821120116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shi J; Zhang Y; Tan D; Zhang X; Yan M; Zhang Y; Franklin R; Shahbazi M; Mackinlay K; Liu S; Kuhle B; James ER; Zhang L; Qu Y; Zhai Q; Zhao W; Zhao L; Zhou C; Gu W; Murn J; Guo J; Carrell DT; Wang Y; Chen X; Cairns BR; Yang X; Schimmel P; Zernicka-Goetz M; Cheloufi S; Zhang Y; Zhou T; Chen Q PANDORA-Seq Expands the Repertoire of Regulatory Small RNAs by Overcoming RNA Modifications. Nat. Cell Biol 2021, 23 (4), 424–436. 10.1038/s41556-021-00652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pomerantz SC; McCloskey JA Analysis of RNA Hydrolyzates by Liquid Chromatography-Mass Spectrometry. Methods Enzymol. 1990, 193, 796–824. 10.1016/0076-6879(90)93452-q. [DOI] [PubMed] [Google Scholar]
- (15).Miyauchi K; Kimura S; Suzuki T A Cyclic Form of N6-Threonylcarbamoyladenosine as a Widely Distributed TRNA Hypermodification. Nat. Chem. Biol 2013, 9 (2), 105–111. 10.1038/nchembio.1137. [DOI] [PubMed] [Google Scholar]
- (16).Hu A; Noble WS; Wolf-Yadlin A Technical Advances in Proteomics: New Developments in Data-Independent Acquisition. F1000Research 2016, 5, F1000 Faculty Rev-419. 10.12688/f1000research.7042.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sarin LP; Kienast SD; Leufken J; Ross RL; Dziergowska A; Debiec K; Sochacka E; Limbach PA; Fufezan C; Drexler HCA; Leidel SA Nano LC-MS Using Capillary Columns Enables Accurate Quantification of Modified Ribonucleosides at Low Femtomol Levels. RNA 2018, 24 (10), 1403–1417. 10.1261/rna.065482.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Thüring K; Schmid K; Keller P; Helm M LC-MS Analysis of Methylated RNA BT - RNA Methylation: Methods and Protocols; Lusser A, Ed.; Springer New York: New York, NY, 2017; pp 3–18. 10.1007/978-1-4939-6807-7_1. [DOI] [PubMed] [Google Scholar]
- (19).Lane BG Historical Perspectives on RNA Nucleoside Modifications. Modification and Editing of RNA. April 28, 1998, pp 1–20. 10.1128/9781555818296.ch1. [DOI] [Google Scholar]
- (20).Huber SM; van Delft P; Tanpure A; Miska EA; Balasubramanian S 2′-O-Methyl-5-Hydroxymethylcytidine: A Second Oxidative Derivative of 5-Methylcytidine in RNA. J. Am. Chem. Soc 2017, 139 (5), 1766–1769. 10.1021/jacs.6b12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Davies V; Wandy J; Weidt S; van der Hooft JJJ; Miller A; Daly R; Rogers S Rapid Development of Improved Data-Dependent Acquisition Strategies. Anal. Chem 2021, 93 (14), 5676–5683. 10.1021/acs.analchem.0c03895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wetzel C; Limbach PA Mass Spectrometry of Modified RNAs: Recent Developments. Analyst 2016, 141 (1), 16–23. 10.1039/c5an01797a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Espadas G; Morales-Sanfrutos J; Medina R; Lucas MC; Novoa EM; Sabidó E High-Performance Nano-Flow Liquid Chromatography Column Combined with High- and Low-Collision Energy Data-Independent Acquisition Enables Targeted and Discovery Identification of Modified Ribonucleotides by Mass Spectrometry. J. Chromatogr. A 2022, 1665, 462803. 10.1016/j.chroma.2022.462803. [DOI] [PubMed] [Google Scholar]
- (24).Lin X; Chai G; Wu Y; Li J; Chen F; Liu J; Luo G; Tauler J; Du J; Lin S; He C; Wang H RNA M6A Methylation Regulates the Epithelial Mesenchymal Transition of Cancer Cells and Translation of Snail. Nat. Commun 2019, 10 (1), 2065. 10.1038/s41467-019-09865-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (25).Roundtree IA; Luo G-Z; Zhang Z; Wang X; Zhou T; Cui Y; Sha J; Huang X; Guerrero L; Xie P; He E; Shen B; He C YTHDC1 Mediates Nuclear Export of N6-Methyladenosine Methylated MRNAs. Elife 2017, 6, e31311. 10.7554/eLife.31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yu Y; Zhu S-H; Yuan F; Zhang X-H; Lu Y-Y; Zhou Y-L; Zhang X-X Ultrasensitive and Simultaneous Determination of RNA Modified Nucleotides by Sheathless Interfaced Capillary Electrophoresis–Tandem Mass Spectrometry. Chem. Commun 2019, 55 (53), 7595–7598. 10.1039/C9CC03195B. [DOI] [PubMed] [Google Scholar]
- (27).Fu L; Amato NJ; Wang P; McGowan SJ; Niedernhofer LJ; Wang Y Simultaneous Quantification of Methylated Cytidine and Adenosine in Cellular and Tissue RNA by Nano-Flow Liquid Chromatography–Tandem Mass Spectrometry Coupled with the Stable Isotope-Dilution Method. Anal. Chem 2015, 87 (15), 7653–7659. 10.1021/acs.analchem.5b00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26 (7), 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jora M; Burns AP; Ross RL; Lobue PA; Zhao R; Palumbo CM; Beal PA; Addepalli B; Limbach PA Differentiating Positional Isomers of Nucleoside Modifications by Higher-Energy Collisional Dissociation Mass Spectrometry (HCD MS). J. Am. Soc. Mass Spectrom 2018, 29 (8), 1745–1756. 10.1007/s13361-018-1999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Quinn R; Basanta-Sanchez M; Rose RE; Fabris D Direct Infusion Analysis of Nucleotide Mixtures of Very Similar or Identical Elemental Composition. J. Mass Spectrom 2013, 48 (6), 703–712. 10.1002/jms.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tyagi SK; Dryhurst G Electrochemical Oxidation of 9β-D-Ribofuranosyluric Acid in Basic Solution. J. Electroanal. Chem. Interfacial Electrochem 1987, 223 (1), 119–141. 10.1016/0022-0728(87)85255-5. [DOI] [Google Scholar]
- (32).Kramer MC; Janssen KA; Palos K; Nelson ADL; Vandivier LE; Garcia BA; Lyons E; Beilstein MA; Gregory BD N6-Methyladenosine and RNA Secondary Structure Affect Transcript Stability and Protein Abundance during Systemic Salt Stress in Arabidopsis. Plant Direct 2020, 4 (7), e00239. 10.1002/pld3.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hoffman KS; Berg MD; Shilton BH; Brandl CJ; O’Donoghue P Genetic Selection for Mistranslation Rescues a Defective Co-Chaperone in Yeast. Nucleic Acids Res. 2017, 45 (6), 3407–3421. 10.1093/nar/gkw1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Huang BO; Johansson MJO; Byström AS An Early Step in Wobble Uridine TRNA Modification Requires the Elongator Complex. RNA 2005, 11 (4), 424–436. 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hayashi S; Mori S; Suzuki T; Suzuki T; Yoshihisa T Impact of Intron Removal from TRNA Genes on Saccharomyces Cerevisiae. Nucleic Acids Res. 2019, 47 (11), 5936–5949. 10.1093/nar/gkz270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chou H-J; Donnard E; Gustafsson HT; Garber M; Rando OJ Transcriptome-Wide Analysis of Roles for TRNA Modifications in Translational Regulation. Mol. Cell 2017, 68 (5), 978–992.e4. 10.1016/j.molcel.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Johansson MJO; Esberg A; Huang B; Björk GR; Byström AS Eukaryotic Wobble Uridine Modifications Promote a Functionally Redundant Decoding System. Mol. Cell. Biol 2008, 28 (10), 3301 LP–3312. 10.1128/MCB.01542-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chen P; Jäger G; Zheng B Transfer RNA Modifications and Genes for Modifying Enzymes in Arabidopsis Thaliana. BMC Plant Biol. 2010, 10, 201. 10.1186/1471-2229-10-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Warren JM; Salinas-Giegé T; Hummel G; Coots NL; Svendsen JM; Brown KC; Drouard L; Sloan DB Combining TRNA Sequencing Methods to Characterize Plant TRNA Expression and Post-Transcriptional Modification. RNA Biol. 2021, 18 (1), 64–78. 10.1080/15476286.2020.1792089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nakai Y; Horiguchi G; Iwabuchi K; Harada A; Nakai M; Hara-Nishimura I; Yano T TRNA Wobble Modification Affects Leaf Cell Development in Arabidopsis Thaliana. Plant Cell Physiol. 2019, 60 (9), 2026–2039. 10.1093/pcp/pcz064. [DOI] [PubMed] [Google Scholar]
- (41).Jülke S; Ludwig-Müller J Response of Arabidopsis Thaliana Roots with Altered Lipid Transfer Protein (LTP) Gene Expression to the Clubroot Disease and Salt Stress. Plants (Basel, Switzerland) 2015, 5 (1), 2. 10.3390/plants5010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Endres L; Dedon PC; Begley TJ Codon-Biased Translation Can Be Regulated by Wobble-Base TRNA Modification Systems during Cellular Stress Responses. RNA Biol. 2015, 12 (6), 603–614. 10.1080/15476286.2015.1031947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gu C; Begley TJ; Dedon PC TRNA Modifications Regulate Translation during Cellular Stress. FEBS Lett. 2014, 588 (23), 4287–4296. 10.1016/j.febslet.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bach-Pages M; Homma F; Kourelis J; Kaschani F; Mohammed S; Kaiser M; van der Hoorn RAL; Castello A; Preston GM Discovering the RNA-Binding Proteome of Plant Leaves with an Improved RNA Interactome Capture Method. Biomolecules 2020, 10 (4), 661. 10.3390/biom10040661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Xu F-Q; Xue H-W The Ubiquitin-Proteasome System in Plant Responses to Environments. Plant. Cell Environ 2019, 42 (10), 2931–2944. 10.1111/pce.13633. [DOI] [PubMed] [Google Scholar]
- (46).Ditt RF; Kerr KF; de Figueiredo P; Delrow J; Comai L; Nester EW The Arabidopsis Thaliana Transcriptome in Response to Agrobacterium Tumefaciens. Mol. Plant. Microbe. Interact 2006, 19 (6), 665–681. 10.1094/MPMI-19-0665. [DOI] [PubMed] [Google Scholar]
- (47).Witzel K; Matros A; Strickert M; Kaspar S; Peukert M; Mühling KH; Börner A; Mock H-P Salinity Stress in Roots of Contrasting Barley Genotypes Reveals Time-Distinct and Genotype-Specific Patterns for Defined Proteins. Mol. Plant 2014, 7 (2), 336–355. 10.1093/mp/sst063. [DOI] [PubMed] [Google Scholar]
- (48).He Z; Li Z; Lu H; Huo L; Wang Z; Wang Y; Ji X The NAC Protein from Tamarix Hispida, ThNAC7, Confers Salt and Osmotic Stress Tolerance by Increasing Reactive Oxygen Species Scavenging Capability. Plants (Basel, Switzerland) 2019, 8 (7), 221. 10.3390/plants8070221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).He C; Bozler J; Janssen KA; Wilusz JE; Garcia BA; Schorn AJ; Bonasio R TET2 Chemically Modifies TRNAs and Regulates TRNA Fragment Levels. Nat. Struct. Mol. Biol 2020. 10.1038/s41594-020-00526-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kasai H; Murao K; Nishimura S; Liehr JG; Crain PF; McCloskey JA Structure Determination of a Modified Nucleoside Isolated from Escherichia Coli Transfer Ribonucleic Acid. Eur. J. Biochem 1976, 69 (2), 435–444. 10.1111/j.1432-1033.1976.tb10928.x. [DOI] [Google Scholar]
- (51).Dominissini D; Nachtergaele S; Moshitch-Moshkovitz S; Peer E; Kol N; Ben-Haim MS; Dai Q; Di Segni A; Salmon-Divon M; Clark WC; Zheng G; Pan T; Solomon O; Eyal E; Hershkovitz V; Han D; Doré LC; Amariglio N; Rechavi G; He C The Dynamic N(1)-Methyladenosine Methylome in Eukaryotic Messenger RNA. Nature 2016, 530 (7591), 441–446. 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Dudley E; Bond L Mass Spectrometry Analysis of Nucleosides and Nucleotides. Mass Spectrom. Rev 2014, 33 (4), 302–331. 10.1002/mas.21388. [DOI] [PubMed] [Google Scholar]
- (53).Synold T; Xi B; Wuenschell GE; Tamae D; Figarola JL; Rahbar S; Termini J Advanced Glycation End Products of DNA: Quantification of N2-(1-Carboxyethyl)-2’-Deoxyguanosine in Biological Samples by Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry. Chem. Res. Toxicol 2008, 21 (11), 2148–2155. 10.1021/tx800224y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Castleberry CM; Rodicio LP; Limbach PA Electrospray Ionization Mass Spectrometry of Oligonucleotides. Curr. Protoc. nucleic acid Chem 2008, Chapter 10, Unit 10.2. 10.1002/0471142700.nc1002s35. [DOI] [PubMed] [Google Scholar]
- (55).Cai WM; Chionh YH; Hia F; Gu C; Kellner S; McBee ME; Ng CS; Pang YLJ; Prestwich EG; Lim KS; Babu IR; Begley TJ; Dedon PC A Platform for Discovery and Quantification of Modified Ribonucleosides in RNA: Application to Stress-Induced Reprogramming of TRNA Modifications. Methods Enzymol. 2015, 560, 29–71. 10.1016/bs.mie.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).van Dongen WD; Niessen WMA Bioanalytical LC-MS of Therapeutic Oligonucleotides. Bioanalysis 2011, 3 (5), 541–564. 10.4155/bio.11.8. [DOI] [PubMed] [Google Scholar]
- (57).Thüring K; Schmid K; Keller P; Helm M Analysis of RNA Modifications by Liquid Chromatography–Tandem Mass Spectrometry. Methods 2016, 107, 48–56. 10.1016/j.ymeth.2016.03.019. [DOI] [PubMed] [Google Scholar]
- (58).Crain PF B. T.-M. in E. [42] Preparation and Enzymatic Hydrolysis of DNA and RNA for Mass Spectrometry. In Mass Spectrometry; Academic Press, 1990; Vol. 193, pp 782–790. 10.1016/0076-6879(90)93450-Y. [DOI] [PubMed] [Google Scholar]
- (59).Sakaguchi Y; Miyauchi K; Kang B; Suzuki T Nucleoside Analysis by Hydrophilic Interaction Liquid Chromatography Coupled with Mass Spectrometry. Methods Enzymol. 2015, 560, 19–28. 10.1016/bs.mie.2015.03.015. [DOI] [PubMed] [Google Scholar]
- (60).Noestheden M; Hu Q; Tonary AM; Tay L-L; Pezacki JP Evaluation of Chemical Labeling Strategies for Monitoring HCV RNA Using Vibrational Microscopy. Org. Biomol. Chem 2007, 5 (15), 2380–2389. 10.1039/B704812B. [DOI] [PubMed] [Google Scholar]
- (61).Rose RE; Pazos MA 2nd; Curcio MJ; Fabris D Global Epitranscriptomics Profiling of RNA Post-Transcriptional Modifications as an Effective Tool for Investigating the Epitranscriptomics of Stress Response. Mol. Cell. Proteomics 2016, 15 (3), 932–944. 10.1074/mcp.M115.054718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kellner S; Ochel A; Thüring K; Spenkuch F; Neumann J; Sharma S; Entian K-D; Schneider D; Helm M Absolute and Relative Quantification of RNA Modifications via Biosynthetic Isotopomers. Nucleic Acids Res. 2014, 42 (18), e142. 10.1093/nar/gku733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.