

Abstract
The efficacy of chemotherapy depends on the tumor microenvironment. This microenvironment consists of a complex cellular network that can exert both stimulatory and inhibitory effects on tumor genesis. Given the increasing interest in the effectiveness of cannabis, cannabinoids have gained much attention as a potential chemotherapy drug. Cannabinoids are a group of marker compounds found in Cannabis sativa L., more commonly known as marijuana, a psychoactive drug used since ancient times for pain management. Although the anticancer potential of C. sativa, has been recognized previously, increased attention was generated after discovering the endocannabinoid system and the successful production of cannabinoid receptors. In vitro and in vivo studies on various tumor models have shown therapeutic efficiency by modifying the tumor microenvironment. However, despite extensive attention regarding potential therapeutic implications of cannabinoids, considerable clinical and preclinical analysis is needed to adequately define the physiological, pharmacological, and medicinal aspects of this range of compounds in various disorders covered in this review. This review summarizes the key literature surrounding the role of cannabinoids in the tumor microenvironment and their future promise in cancer treatment.
Keywords: cannabinoids, angiogenic factor, hypoxia, tumor genesis, combination therapy, signaling pathways
Introduction
The interconnections between cancerous cells and their microenvironment, consisting of stromal cells (including stromal fibroblasts, endothelial, macrophages, microglia, and lymphocytes) and extracellular matrix components (ECM; fibronectin, laminin, collagen hyaluronan, etc.) components, seem to be essential to stimulate heterogeneity of cancerous cells, clonal development, and developing multi-drug resistance, eventually leading to cancer cell progression and metastasis. The tumor microenvironment enhances the activity of cellular and non-cellular aspects by using complex signaling cascades, allowing non-malignant cells to act to the tumor’s advantage. The results of this crosstalk are tumor development and maintenance and an inadequate treatment response or multi-drug resistance. 1 Cannabis sativa L. is a natural source of valuable compounds that comprise cannabinoid agonists and antagonists, which have recently been scanned for future applications as anti-tumor drugs. 2 Currently, in the UK, the use of cannabinoids has only been approved as an adjunct in moderate to severe spasticity in multiple sclerosis. 3 Cannabinoids have mostly been used as a part of palliative care to alleviate pain, relieve nausea, and stimulate appetite in cancer patients. 4 Although not yet approved for treating tumor progression, cannabinoid agonist/antagonists on the tumor microenvironment have been studied for the last 43 years. Since then, many cell lines and tumor models have been studied for the efficacy and mechanism of cannabinoids and their effect on tumors. An active component of cannabis, Δ9-tetrahydrocannabinol (THC), has been extensively studied in several in vitro and in vivo models together with other plant-derived endogenous and synthetic cannabinoids derived from plants. 5 The main natural psychoactive compound present within cannabis is THC, which has an affinity for the cannabinoid receptor (CB1) and CB2 receptors that modulate its main effects. 6 Recently, new psychoactive substances containing synthetic cannabinoids (SC) have been developed to become widely used among young adults.
Contrary to the decline in consumption of several new psychoactive substances such as the cathinones and piperazines, it appears that the number of SC users is increasing. 7 Although SC drugs are more efficacious than natural cannabinoids, their undesired adverse effects are unpredictable. One of the earliest synthetic cannabinoids that were identified as the psychoactive component is JWH-018, which has 4 times greater affinity for CB1 and 10 times for CB2 receptors than THC. 8 Side effects are associated with CB1 and CB2 expression patterns rather than with binding affinity.
Phyto-cannabinoids and synthetic cannabinoids can interact with the extracellular matrix components or other cellular pathways and thus affect the development/progression of diseases, including cancer. 9 Numerous in vitro and in vivo studies have demonstrated the anti-cancer effects of cannabinoids in various cancer types (Table 1). 10 Here, we review the literature on the effect of phytocannabinoids and synthetic cannabinoids on the tumor microenvironment, especially angiogenesis and several other factors that play a crucial role in the tumor microenvironment.
Table 1.
List of Cannabinoids and Their Effect on Various Cancer Models..
| Cannabinoid | Reported effect | Model used in the experiment | References |
|---|---|---|---|
| Δ9-THC, WIN-55,212-2 | Inhibit on VEGF, PIGF, and Ang 2 | Numerous cancer cell lines | Blázquez et al, 18 Casanova et al, 19 Portella et al 20 |
| WIN-55,212-2, HU210, AEA, Δ9-THC | Stimulate EGFR and enhance cell reproduction | NCI-H292 cells (lung) | Portella et al 20 |
| SCC-9 cells (squamous cell carcinoma) | |||
| 5637 cells (bladder) | |||
| U373-MG cells (glioblastoma) | |||
| 1321N1 cells (astrocytoma) | |||
| A494 cells (kidney) | |||
| Δ9-THC | Down-regulate TIMP-1 and MMP-2 | Glioma cell lines | Casanova et al 19 |
| JWH-133 | Human tumors from patients with recurrent glioblastoma multiforme | ||
| Nude mice xenografted with C6.9 glioma cells | |||
| Two patients with glioblastoma multiforme | |||
| CBD | |||
| Down-regulate MMP-2 | 4T1.2 cells | Seltzer et al 29 | |
| MTV-1 tumors | Δ9-THC, 2-AG | ||
| Increase cellular progression | LNCaP cells | Deryugina and Quigley, 31 Kleifeld and Overall, 32 Roomi et al 34 | |
| PC3 cells | AEA | ||
| Cause cellular regression | LNCaP cells | Deryugina and Quigley, 31 Kleifeld and Overall, 32 Roomi et al 34 | |
| PC3 cells | AEA | ||
| Decrease cell viability | DU145 cells | Blázquez et al 35 | |
| LNCaP cells | |||
| PC3 cells | 2-AG | ||
| Increase cellular proliferation | PC3 cells | Casanovas et al 36 | |
| Tumor-associated macrophages (TAM) | CBD | ||
| Decrease in F4/80-positive and Arginase-I-positive cells | Breast cancer xenografts | Deryugina and Quigley 31 | |
| Lung metastases | CBD | ||
| Produce less CCL3, MIP-2 proteins, and GM-CSF | 4T1.2 cells | Deryugina and Quigley 31 | Δ9-THC |
| Induce an apoptotic effect via p8-mediated autophagy | Glioma cells | Elbaz et al, 38 Ebos et al 39 | CBD |
| Halt glioblastoma multiforme growth | Tumor bearing mice | Mimeault et al 42 | Δ9-THC |
| Autophagic effects | Mitochondria | Handsley and Edwards 44 | WIN-55,212-2 |
| Stimulate autophagy | Mantle cell lymphoma and pancreatic, breast, glioma, and hepatocellular carcinoma cells | McKallip et al, 45 Sánchez et al 41 | Δ9-THC |
| activate p53 which triggered the apoptotic cascade | Cultured cortical neurons | Singer et al 50 | Anandamide |
| Increase cellular proliferation | Glioma stem cells (GSCs), glioma cells | Lorente et al, 51 Salazar et al 52 | SR141716A |
| CBD | |||
| Switches off Id1 and has a role in the advancement of breast cancer and metastasis in the lungs | Breast cancer | Salazar et al, 57 De Petrocellis et al 58 | |
| Metastasis in the lungs | |||
| 4T1-derived tumors | CBD | ||
| Decline migration and viability | A549 cells | Egeblad and Werb 27 | Δ9-THC, MA, JWH-133 |
| AEA, JWH-133 | |||
| Inhibit the adrenaline activated migration | SW480 cells | De Petrocellis et al 58 | |
| MDA-MB-468 cells | Met-F-AEA | ||
| Decrease in both the size and number of metastatic nodules | MDA-MB-231 cells | Sarfaraz et al 60 | CBD |
| Decrease in HIF-1α | Normoxic cells in U87-MG | Sarnataro et al 63 |
Abbreviations: Δ9-THC, delta-9-tetrahydrocannabinol; VEGF, vascular endothelial growth factor; PIGF, phosphatidylinositol glycan anchor biosynthesis class F; Ang 2, angiopoietin-2; AEA, N-arachidonoylethanolamine, arachidonoylethanolamide, anandamide; CBD, cannabidiol; MMP-2, matrix metalloproteinase-2; TIMP1, tissue inhibitor matrix metalloproteinase 1; 2-AG, 2-arachidonoylglycerol; CCL3, chemokine (C-C motif) ligand 3; GM-CSF, CSF2, granulocyte macrophage-colony stimulating factor; Met-F-AEA2, methyl-2′-F-anandamid; WIN-55,212-2, (11R)-2-methyl-11-(morpholin-4-ylmethyl)-3-[(naphthalen-1-yl)carbonyl]-9-oxa-1-azatricyclo[6.3.1.0^{4,12}]dodeca-2,4,6,8(12)-tetraene; HU210, (6aR,10aR)-9-(hydroxymethyl)-6,6-dimethyl-3-(2-methyloctan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromen-1-ol; JWH 133, (6aR,10aR)-6,6,9-trimethyl-3-(2-methylpentan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromene.
Angiogenesis is the process of forming blood vessels from pre-existing capillaries. It happens in both health and cancer throughout the lifetime, commencing in utero and extending until death. No metabolically active part of the body is far more than just a few 100 µm apart from a blood capillary developed by the angiogenesis process. 11 Evading an attack by the immune system is a significant event during tumor development. 12 This is achieved by dynamic interactions between different cytokines and their respective receptors in the tumor microenvironment, which attracts infiltrating cells that help tumors evade attack by the immune system. 13 Cannabinoids induce evasive resistance by inhibiting proangiogenic factors. This phenomenon highlights the need for further research on cannabinoid-induced evasive resistance and whether inhibiting proangiogenic factors enables revascularisation via alternative pathways. 14 It also highlights the need to understand the mechanisms of cannabinoid involvement in cell regression and progression, which are also highly debated. We also review how evasive resistance might be overcome by combining cannabinoids with current anti-cancer treatments. Although the therapeutic potential of cannabinoids is remarkable, there are many conflicting interpretations about these compounds. In this review, we summarize key literature surrounding the role of cannabinoids in the tumor microenvironment and their potential roles in cancer treatment.
Further factors covered in this review include the microenvironmental deprivation of oxygen supply (hypoxia distress) modulating autophagy, which facilitates cancer cell death or survival in the context of tumor development. Cancerous cells can acclimate to microenvironmental alterations more quickly than healthy cells, for example, by triggering various stress response pathways while evading anti-proliferative and inducing apoptosis. 15
Effect of Cannabinoids on Angiogenic Factors
Numerous investigations have shown that cannabinoids hinder the proliferation of many tumor cell lines and have the tendency to reduce or prevent the growth of various tumor xenograft models in laboratory animal models 16 In 1971, Judah Folkman announced that angiogenesis is crucial for solid tumors to grow beyond 1 to 2 mm3 or become metastatic. This finding provided a novel method for the anti-tumorigenic strength expressed by cannabinoids. 17 The potential effects of cannabinoids on angiogenic factors are discussed in depth in several studies; however, comprehensive information in this area is still lacking. Therefore, extensive studies surrounding angiogenesis and associated factors were conducted that play a vital role in the tumor microenvironment. New vascular pathways are developed during angiogenesis to provide tumors with sufficient nutrition, allow gaseous exchange, and facilitate waste removal. 5 Cannabinoids have significantly affected vascular endothelial growth factor (VEGF), a critical proangiogenic regulator. 18 The administration of Δ9-THC, 2-methyl-2′-F-anandamide (Met-F-AEA), WIN-55,212-2, and (6aR,10aR)-3-(1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo [b,d] pyran (JWH-133) reduced VEGF levels in numerous cancer cell lines.5,18,19 In particular, evidence shows that Δ9-THC may potentially lead to reduced VEGF production in lung cancer cell lines SW1573 and A549 and decreased vascularization of A549 cell lines in immunodeficient mice with tumor xenotransplantation. 20
The effect of cannabinoids (WIN-55,212-2 and JWH-133) on antiangiogenic factors did not influence the multidomain matrix glycoproteins thrombospondin 1 and thrombospondin 2, which naturally inhibit neovascularization in mice transplanted with melanoma carcinoma cells. 18 Similarly, in Kirsten rat sarcoma virus (K-ras) transformed skin carcinoma tumors, Met-F-AEA induces a reduction in VEGF production and VEGR1. 21 This inhibitory effect was also observed with PDV.C57 (epidermal cell lines). 18 This study injected tumorigenic epidermal cell lines (PDV.C57) into mice and left them to grow until a tumor of the desired size was achieved. Treatment for 11 days with cannabinoids showed a remarkable reduction in the 3 main angiogenic factors: VEGF, phosphatidylinositol glycan anchor biosynthesis class F (PIGF), and angiopoietin-2 (Ang 2). 18
Likewise, the effect of cannabinoids on glioma models showed a significant reduction in both VEGF and Ang 2.19,20 Similar effects were observed in a clinical trial of a cannabinoid, in which 2 patients with glioblastoma multiforme demonstrated a reduction in both VEGF and Ang218,22
Effect of Cannabinoids on Tumor Vascularization
There is sufficient evidence to suggest that suppression of VEGF does not necessarily lead to inhibition of angiogenesis. Antiangiogenic therapies targeting VEGF/VEGFR2 do not exhibit efficacy on tumor growth reduction and survival in the long term. Consequently, the initial clinical benefit is followed by a resistance to the antiangiogenic therapy and, ultimately, heightened invasion and metastasis. The experimental evidence on mouse models supports this hypothesis, where inhibition of the VEGF receptor on pancreatic islet cancer reveals primary effects, with a reduction in the vascularization and tumor metastasis, followed by regrowth due to the adaption of the tumor microenvironment. This phenomenon is often referred to as “evasive resistance.” This process was based on the upregulation of different proangiogenic factors that constitute fibroblast growth factor (FGF) ligands. 18
This observation was supported by Casanova et al 19 whose study involved using DC101 (the function-blocking antibody) in opposition to VEGFR2 in the transgene insertion 2, Douglas Hanahan (RIP1-Tag2) model of pancreatic neuroendocrine cancer (PNET). The study revealed that, in immunocompromised mice bearing RIP1-Tag2 which were treated with the neutralizing monoclonal anti-VEGFR antibody (DC101) for just 1 week, a reduction in tumor vascularization was observed; 20 following continuous 4-week treatment, a significant and more invasive phenotype was observed, deduced by histological analysis. The more aggressive tumors showed a broad front of invasion, immingled with enclosing acinar tissue compared to control tumors, which remained intact, encapsulated, and micro-invasive. 20 This theory of “evasive resistance” was tested using sunitinib, a VEGF receptor tyrosine kinase inhibitor (RTKIs), which also acts similarly to JWH-133, a C2B receptor agonist, in terms of its VEGF decreasing capabilities. Likewise, the cannabinoids Δ9-THC and WIN-55,212-2 are C2B and CB1 receptor agonists and inhibit the VEGF receptor and 2 major angiogenic factors, PIGF and Ang 2, respectively.23,24
A study on Δ9-THC effects on promoting mitogenic kinase signaling in cancer cells was demonstrated on numerous cancer cell lines.18,25 It was observed that using nanomolar strengths of Δ9-THC surprisingly enhanced cell reproduction, and this was entirely related to metalloprotease and epidermal growth factor receptor (EGFR) activity. To demonstrate cannabinoid induction of EGFR transactivation in human carcinoma cell lines, Hart et al 26 in 2004 experimented by treating the cancer cell lines, NCI-H292 (lung), SCC-9 (squamous cell carcinoma), 5637 (bladder), U373-MG (glioblastoma), 1321N1 (astrocytoma) and A494 (kidney) with the cannabinoids WIN-55,212-2, HU210, AEA, and Δ9-THC. The results were recorded using immunoblot analysis, where cannabinoids quickly caused EGFR stimulation within 3 minutes. The prior incubation of the cells with the metalloprotease inhibitor BB94 (Batimastat) or EGFR kinase-specific inhibitor AG1478, inhibited EGFR tyrosine phosphorylation due to the cannabinoid, a process that depends on metalloprotease and EGFR kinase-specific activation. Conversely, stimulation of NCI-H292 cells with arachidonyl-2′-chloroethylamide (ACEA) and BML-190, both of which are synthetic agonists of CB1 or CB2 receptors, demonstrated metalloprotease-dependent EGFR transactivation (a vital step in cell proliferation). 27
Effect of Cannabinoids on Immune Infiltration
Cannabinoids modulate significant immunological signaling pathways and receptors. 28 The understanding of the mechanism of action of the pharmacological effect of cannabidiol (CBD) highlights the therapeutic potential of CBD as an effective immunomodulator. 29 Recently, it was observed that antagonists of CB1 and CB2 receptors and TRPV1 (transient receptor potential vanilloid 1) reverse the immunomodulatory effect of CBD, which confirms that the CBD immunomodulatory inhibition is not dependent on CB1 and CB2 receptors. 30 CBD inhibits critical immune signaling pathways such as Janus Kinase/Signal transducer and activator of transcription and the nucleotide-binding oligomerization domain-like receptor signaling pathway, thus downregulating the proinflammatory cytokine production. 28 Likewise, CBD also protects against immune response-induced cytotoxicity by modulating adenosine signaling. These studies overpoweringly reinforce the immunomodulatory potential of CBD. 29
Effect of Cannabinoids on Metalloproteinases and Non-metalloproteinase
Cannabinoids have been shown to affect certain matrix metalloproteinase (MMP) family members indirectly. These zinc-dependent endopeptidases and proangiogenic factors secreted by tumor cells are involved in tumor neovascularization. 31 Inhibitors of MMP are found amongst the endogenous proteins circulating in the tumor microenvironment. This protein is a tumor inhibitor of matrix metalloproteinases-1 (TIMP-1) and proved to suppress tumor neovascularization. 32 In a study in 2014, Ramer et al attempted to merge these 2 findings to reveal whether cannabinoids that are TIMP-1 inducers can be used to inhibit angiogenesis. The experiment was initially performed using recombinant TIMP-1 to mimic cannabinoid activated TIMP-1 release from the tumor microenvironment. 33 However, cannabinoids tested on an in vitro angiogenesis model of human umbilical vein endothelial cells (HUVEC) migration, sprout, and tube formation elicited a concentration-dependent inhibition, although cellular viability remained unaltered. 34
In contrast, Blázquez et al 35 suggested that THC demonstrated downregulation of TIMP-1 in glioma cell lines and human tumors from patients with recurrent glioblastoma multiforme. This was also supported in an alternative experiment involving JWH-133, where TIMP-1 was again downregulated in nude mice xenografted with C6.9 glioma cells. Although tumor progression was not observed, it posed the question of other angiogenic factors being inhibited, thus being the main cause of tumor stasis or regression. 35 Hence, the impact of cannabinoids on the expression of TIMP-1 depends on the type of cell line used.
Furthermore, direct inhibition of a subtype of the family of metalloproteinases (not via TIMP-1 induction), MMP-2 (over-expressed in tumor cells) showed a remarkable decline in MMP-2 expression in 2 patients who were given the cannabinoid Δ9-THC. Immunomicroscopic analysis of the biopsies in 2 patients with glioblastoma multiforme that were administered local Δ9-THC showed MMP-2 levels to be lower in both patients while levels of “other” MMP expression remained unchanged in patient 1. In the case of patient 2, sufficient samples could not be taken for Western blotting analysis. The cannabinoid JWH-133 also showed a reduction in MMP-2 and tumor growth which was confirmed by immunomicroscopic analysis. Nevertheless, the levels of the “other” MMPs remained unchanged again. 36
The use of cannabinoid induction or inhibition of these MMP or TIMP-1 remains undetermined. Once again, more research is required to confirm whether cannabinoid-stimulated inhibition of TIMP-1 expression or cannabinoid induction of TIMP-1 expression is based on cell lines or dependent on the type of cannabinoid. 37 Furthermore, an experiment by Elbaz et al 38 proposed that MMP-2 expression was considerably lower in breast cancer in 4T1.2 and MTV-1 tumors when CBD was administered, supporting the anti-metastatic capabilities of cannabinoids in reducing tumor survival. Additionally, Δ9-THC and 2-AG increased cellular progression, whereas the endocannabinoid N- arachidonoylethanolamide (AEA) caused cellular regression. Sánchez et al40,41 suggested that AEA decreased cell reproduction of LNCaP and PC3 cells, whereas Δ9-THC (50 nM) has an opposite effect (increased proliferation).
This was also highlighted by Mimeault et al, 42 who showed that AEA application in DU145, LNCaP, and PC3 cell lines decreased cell viability at concentrations above 2 µM. Moreover, Nithipatikom et al 43 showed a decrease in PC3 cellular proliferation at concentrations above 1 µM, but caused an increase in cellular proliferation with the cannabinoid 2-AG at similar concentrations. Despite the role of cannabinoids in affecting angiogenic factors, tumor-associated macrophages (TAM) were investigated to ascertain if they had a role in tumor angiogenesis, cell proliferation, and invasive properties. 44 Breast cancer xenografts were examined for F4/80-positive and Arginase-I-positive macrophages by immunohistochemistry (IHC). Tumors treated with CBD showed decreased F4/80-positive and Arginase-I-positive cells compared to those untreated. This experiment was repeated in lung metastases, and a reduced F4/80-positive and Arginase-I-positive was identified. A more recent analysis in 2015 by Elbaz et al 38 hypothesized that CBD has a role in modulating cytokine production in cancer cells via the reduction of macrophages. Using cytokine array analysis, it was found that 4T1.2 cells (an aggressive breast cancer cell line) treated with CBD produced less CCL3, MIP-2 protein, and GM-CSF compared to the untreated 4T1.2 cells. The experiment highlights another mechanism of cannabinoids in modulating cytokine production in tumor cells by allowing the reduction in macrophage recruitment in tumor sites. These results imply cannabinoids have a degree of influence on tumor angiogenic factors, even though the exact mechanism and their effects on tumor progression or regression are yet to be well understood. Different cannabinoids have different effects on angiogenic factors both directly and indirectly. Simply reducing proangiogenic factors does not cause decreased cell proliferation. Reduction in one type of proangiogenic factor may lead to the expression of other proangiogenic factors, further inducing a more invasive malignant progression of tumors. As studied by McKallip et al 45 in 2005, cannabinoids must be administered at varying concentrations to determine their potential pharmacological effects on cultured cell lines. This phenomenon of evasive resistance, sometimes known as adaptive resistance, could also be used to describe cannabinoid sensitivity to cell lines. 20
A recent study also highlighted that Δ9-THC induced an apoptotic effect on glioma cells by the activation of cannabinoids receptors via p8-mediated autophagy. However, there was a significant contrast in the sensitivity of Δ9-THC-stimulated cell death. This correlates with an enhancement in the activity of certain genes in Δ9-THC-resistant glioma cells rather than the overexpression of cannabinoid receptors.40,46 One gene which showed upregulation was Midkine (MDK), also known as neurite growth-promoting factor 2 (NEGF2). This gene has previously been associated with a rise in malignancy and resistance to chemotherapeutic effects. Although this mechanism is still not fully understood, it is thought that MDK has a role in stimulating anaplastic lymphoma kinase (ALK), which ultimately blocks Δ9-THC- controlled cell death. 41
Based on this understanding, ALK inhibitors are used to control non-small cell lung carcinoma and other tumors. 47 Other growth factors stimulated by carcinoma cells are involved in cannabinoid-triggered proliferation, and this highlights the importance of recognizing and understanding all involving factors and signaling pathways. Notably, EGFR ligands have been shown to suppress cellular proliferation. 13 In 2009, Wondrak 48 experimented with investigating the major resistance system used by cancerous and normal cells to counteract oxidative stress, the NRF2 transcriptionally-regulated program.
Effect of Cannabinoids on the Oxidative Stress
Anti-cancer drugs often produce oxidative stress through the production of elevated intracellular reactive oxygen species (ROS), which is also produced as a natural by-product of aerobic metabolism. ROS is important in normal cell reproduction via the activation of growth-related pathways. The effects of ROS are mainly dependent on the basal metabolic rate of the cell. Cancer cells possess a high basal metabolic rate, making them more susceptible to redox-directed therapeutics compared to non-transformed cells. Consequently, redox-directed therapeutics were developed as cancer inhibitors or sensitizers to tumors as first-line agents. CBD, a ROS modulator and an inhibitor of antioxidant response genes, has shown promising effects on halting glioblastoma multiforme (GBM) growth. 49 Despite this, an experiment carried out by Singer et al 50 using bioluminescence measurements to monitor tumor growth showed prolonged survival in tumor bearing mice (intracranial tumors); however, the intracranial tumors resumed a rapid growth profile on day 22 despite the continuous administration of CBD (Figure 1).
Figure 1.
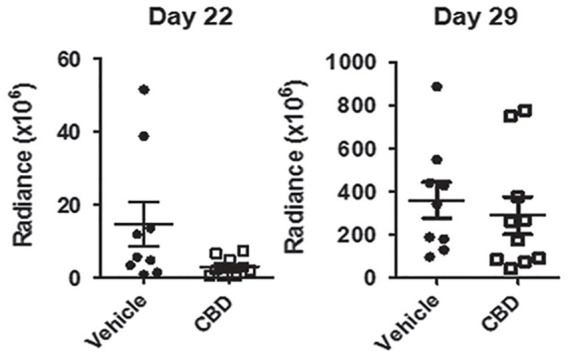
Bio-layer interferometry (BLI) measurements demonstrating an initial response to CBD (decrease in tumor after 3 weeks) and subsequent tumor resistance to treatment 1 week later (after 4 weeks) 50 (republished after obtaining permission from the author and journal).
The underlying resistance mechanism was explained by the confirmation that CBD triggered NRF2 stimulation in a dose-dependent method, which resulted in the induction of antioxidant response genes. Immunohistochemistry (IHC) analysis of mice bearing glioma and treated with CBD demonstrated an upregulation of cellular components such as NRF2 and xCT/SLC7A11when compared to untreated mice, suggesting a resistance pathway associated with redox-directed therapeutics.
Cannabinoids and Their Potential Mechanisms of Action in Cell Growth Inhibition
The endocannabinoid system is profoundly affected by a complicated set of signaling pathways through numerous receptors and CB1, CB2, TRPV1, and GRP55 and receptor-independent pathways. However, the type of receptors involved is yet to be investigated. The countless results in the field of mechanisms of cell survival and cell death remain debatable, with some researchers suggesting autophagy playing an important role rather than direct apoptosis, and others are adamant that apoptosis is directly stimulated. Autophagy involves encapsulating organelles into a double membrane vesicle for destruction and recycling by a process involving the action of lysosomes. The cannabinoid Δ9-THC exerted its autophagic effects by degradation of the key organelle, mitochondria. 51 Velasco et al and Shrivastava et al reported that WIN-55,212-2 stimulates autophagy in a dose dependent manner in mantle cell lymphoma and is prevalent in pancreatic, breast, glioma, and hepatocellular carcinoma cells. 52 Evidence suggests that cannabinoid receptor activation modulates the MAPK and Akt signaling cascades, causing cellular activity changes and stimulating the autophagy process.
These researchers suggested that autophagy precedes apoptosis. It is linked to activation of CB1 and CB2, inducing serine palmitoyltransferase, an enzyme that regulates de novo ceramide generation. 46 Ceramide activates p8 (nuclear protein 1) by stimulating endoplasmic reticulum-associated eIF2α, which initiates a signaling cascade through tribble homolog 3, AKT rapamycin complex 1 to activate autophagy. 53 A slightly different mechanism was proposed by Carracedo et al, 54 who suggested that ceramide-dependent p8 upregulation (de novo-synthesized ceramide) and it is downstream (ATF-4, CHOP, and TRB3) targets like activating transcription factor-4, homologous protein, and TRB3 (cell death-inducible) are the mechanisms for its anti-tumorigenic actions (Figure 2). The study utilized real-time quantitative PCR; researchers demonstrated that THC increases p8 mRNA expression in C6.9 cells but not in C6.4 cells. In addition, THC enhances p8 protein expression in C6.9 cells but not in C6.4 cells.
Figure 2.

Schematic representation of the endocannabinoid system as suggested by Carracedo et al, 54 2006 (reproduced using Biorender).
There are no indications of autophagy in melanoma cell lines, leukemia, rhabdomyosarcoma, or prostate cancer. Endogenous cannabinoids play a significant role in regulating the de novo synthesis of ceramides (lipid-based components of the phospholipid bilayer involved in structure and cellular signaling). Also, ceramide autophagy has a crucial role in signaling regarding the control of apoptosis. The link between ceramide signaling (by involving cannabinoids) and some cancers is becoming ever more prevalent in the research to alter tumor microenvironment for cell apoptosis. 55 In glioma cells, either CB1 or CB2 is related to increased ceramide levels. The upregulation of ceramide leads to activating of the ERK (extracellular signal-regulated kinase) pathway via Raf-1, p38 MAPK stimulation, which ultimately activates caspase (a family of cysteine proteases, which have significance roles in apoptosis). 56
To highlight this pathway further, Δ9-THC activated p53 in cultured cortical neurons by binding to the CB1 receptor. This process triggered the apoptotic cascade involving Bcl-2 (B-cell lymphoma 2) and Bcl-2 associated X protein (Bcl-2-like protein 4), suggesting that the endocannabinoid system causes cell destruction via the process of apoptosis rather than autophagy. Both signaling pathways involve the stimulation of ceramide levels, and in addition to this, cannabinoids exhibit their direct effects on cAMP (Cyclic adenosine monophosphate) through adenylate cyclase inhibition of protein kinase A and gene transcription. 57 De Petrocellis et al proposed an entirely different mechanism and studied the effects of cannabinoids on hormone-responsive cancers. The initial experiment was conducted on anandamide targeting tumor cells expressing estrogenic and prolactin receptors. Therefore, it was hypothesized that anandamide could exert anti-proliferative effects by interfering with their receptors or agonists. This hypothesis was later declined when the breast cancer cell line MCF-7 showed no affinity between cannabimimetic compounds and estrogenic receptors. 58
In contrast, prolactin-Stat5 signaling (produced in high amounts in most breast cancer cell lines, including human breast carcinomas at approximately 0.35 µg/mL daily) has been proposed to have accelerating effects on the G1/S phase of the mitotic cycle. Another experiment carried out by De Petrocellis et al 58 highlighted the effects of prolactin on cellular proliferation of EFM-19 cells upon administering 50 ng/mL of prolactin exogenously. In the case of prolactin receptor antagonists or prolactin monoclonal antibodies (mAb), as expected, a complete blockade of cellular reproduction was observed in MCF-7 cell lines. Furthermore, cellular proliferation was also reduced in EFM-19, BT-474, and T-47D cells. 59
The question arises, whether anandamide exerts its effects on prolactin agonists or prolactin receptors? To answer this, results from the experiment by De Petrocellis et al 58 suggested that cell lines most receptive to anandamide such as MCF-7, EFM-1, and T-47D cells responded considerably to antibody treatment (70%-98% inhibition of growth with 20 µg/mL of antibody). In contrast, in cell lines that had the lowest sensitivity to anandamide (BT-474), only 29.1% of cellular proliferation inhibition was observed when exposed to the antibody. This indicates that the potency of anandamide appears similar to the degree of dependency of human breast cancer cell (HBC) reproduction on endogenous prolactin.
However, additional cellular proliferation inhibition was not observed when the submaximal concentration of anandamide and submaximal concentration of the prolactin antibody was administered. 60 This finding suggests that endogenous cannabinoids, such as anandamide do not directly affect prolactin levels but rather affect prolactin receptors. This theory of the cannabinoid effect on different receptors is also supported because the decrease in prolactin caused by the antibody did not show any further reduction in cellular proliferation.61,62 Administration of a lower dose of anandamide (0.1-0.5 µM), along with a cannabinoid receptor antagonist (SR 141716A, 0.5 µM), showed an increase in cellular proliferation. 63
The down-regulating effect on the prolactin receptor is thought to be mediated by a CB1 receptor as the anti-proliferative effect of anandamide was reversed when co-incubated with SR 141716A (an inverse agonist of CB1 receptor). In another case, the presence of SR 141716A caused inhibition of cellular proliferation through the effect of extracellular signal-regulated kinase ½ (ERK ½) co-localized inside membrane lipid rafts. These rafts, comprising glycosphingolipids and protein receptors organized in glycolipoprotein microdomains, have a crucial role in breast tumors and metastasis. Rinaldi-Carmona et al proposed that SR 121716A, besides its antagonistic characteristics, also possesses opposite agonist effects. This allows it to block mitogen-activated protein kinase (MAPK) and adenylate cyclase, thus having significant anti-tumor effects. 64 The key requirements in SR 141616A-induced antiproliferation signaling are lipid raft integrity on the cell membrane and overly expressed CB1 receptors. 51 It was proposed by Moffett et al 65 in 2000 that this feature (lipid raft integrity) increases the efficiency and specificity of signal transduction by facilitating interactions between proteins to increase the efficiency and specificity of signal transduction by facilitating crosstalk between pathways.
The evidence suggests that SR 141716A requires the raft integrity to exercise its antireproductive effects by signaling. When MCD, a chemical used to extract lipid components from cell membranes, was administered, the antiproliferative effect of SR 141716A was diminished. The proposed mechanism shows further promise in controlling cellular growth in human breast cancer cell lines. It is unsurprising to indicate that this is just one of the many possible tumor stress mechanisms during cell transformation. The mechanisms of action of cannabinoids on cellular proliferation or inhibition are not just influenced by their microenvironment but also by the different features of tumors such as prolactin-producing pituitary carcinoma; overexpression of CBD receptors; the presence of prolactin receptors; or the presence of a lipid raft component. 66 A more recent mechanism suggested relates to the effect of cannabinoids on intercellular adhesion molecule 1 (ICAM-1), an essential molecule within the signaling transduction pathway and with a major role in regulating lymphocyte-activated killer (LAK) cells. LAKs have a role in deactivating potential cancer cells by their cytotoxic action. The identification of CBD in controlling ICAM-1 was supported by the fact that the LAK cytolytic function was reversed when ICAM-1 neutralizing antibody was administered. LAK function was also reversed via the blockade of CB1, CB2, and TRPV1 receptors, suggesting the role of cannabinoids in modulating LAK via the activation of ICAM-1 67
Cannabis and Tumor Migration
Several studies have linked the ability of cannabinoids to control tumor survival, which is considered a pivotal finding in the field of oncology.68-70 Tumors possess a heightened proliferation rate, breaking through basement membranes and invading extracellular space through tissue and organs. 71 Growth beyond a certain distance from normal tissue blood vessels is a characteristic of all tumors. The ability of tumors to secrete growth factors imparts them to survive hypoxic conditions by angiogenesis, developing new blood from vascular structures. Endothelial cells grow toward the source of chemo-attractants (tumor) and develop a disorganized tumor vasculature. Tumor cells may survive and grow by 3 survival mechanisms: migration, invasion, and sprout formation. Combining all 3 results in an increased spreading of cancerous cells to other parts of the body. The term used to describe this is metastasis, which involves an initial break away from the main tumor attachment and the degradation of matrix metalloproteinases (MMP) that makes up the surrounding extracellular matrix, which separates the tumor from the tumor adjoining tissues. By degenerating these proteins, carcinoma cells can disregard the extracellular matrix and release it. Metastasis is regarded as a fundamental hallmark of cancer and is often very difficult to treat clinically 72
Migration is defined as a unified multistep process that plays a significant role in cancer progression while invasion encompasses this and spreads cancerous cells through the extracellular matrix into the neighboring tissue. This leads to degradation of the extracellular matrix, and proteolysis occurs (Cell Biolabs, Inc, n.d.). Id1, an important gene in metastasis (Figure 3) is “switched off” when CBD is administered. It has a role in advancing breast cancer and metastasis in lungs. 73
Figure 3.
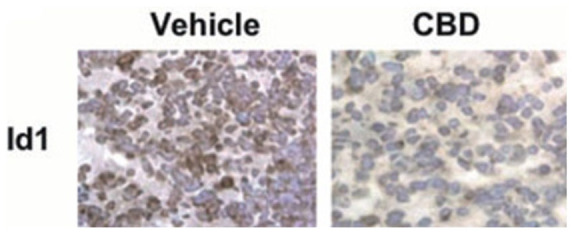
CBD-induced inhibition of Id-1. Immunohistochemical detection of Id1 on lung tissue (left) using vehicle and CBD treated 4T1-derived tumors (right) 73 (Republished after obtaining permission from the author and journal).
Ramer et al 33 conducted an experiment to tackle a probable indirect antiangiogenic performance of cannabinoids by modulating the tumor cell microenvironment. The human alveolar epithelial cell line A549 was initially treated with a vehicle or different cannabinoids for 48 hours. The conditioned media, collected and centrifuged, were subsequently used to prepare cell suspensions of HUVEC at a final density of 1 × 10 5 cells per sample. HUVEC in conditioned medium were evaluated for migration utilizing Boyden chamber analysis, 48-well plates to determine survivability, or Matrigel-coated 48-well plates to assess tubular formation after just 1 day of incubation. 74 Finally, to measure the indirect effects of three-dimensional sprout formation of HUVEC in fibrin gels, the A549 cells underwent EGM-2 treatment 3 times every 48 hours, in parallel. As with the previous methods, the conditioned media were centrifuged and transferred onto the fibrin gels containing HUVEC-coated codex 3 microcarrier beads. Another study suggested that conditioned media acquired from A549 cells treated with vehicle for 2 days enhanced the viability, migration, and sprout formation of HUVEC compared to HUVEC incubated with non-conditioned EGM-2 medium for fibrin bead assays. By contrast, HUVEC suspended in conditioned media of A549 cells treated with 3 µM CBD for 48 hours showed a considerable decline in migration and viability compared to conditioned media from vehicle-treated A549 cells. These results were mirrored with the use of other cannabinoids at the same concentrations, specifically, Δ9-THC, MA, or JWH-133. 33 Concerning sprout formation, conditioned media acquired from cannabinoid-treated A549 cells also elicited a decrease in sprout number compared to HUVECS incubated with non-cannabinoid A549 cells. 25 Other evidence supporting cannabinoid association with metastasis and invasion of cancer include specific markers related to migration, adhesion, invasion, and metastasis. 75 An experiment conducted in 2004 suggested that the adrenalin-activated migration of SW480 and MDA-MB-468 cells was again inhibited with cannabinoids AEA (40 nM) and JWH-133 (10 nM). 76
Cannabidiol administered at doses greater than 3 μM inhibited the migration of U87 glioma cells in a Boyden chamber analysis. However, the effect of cannabinoid-induced migration was not inhibited when the antagonists SR 141716 or SR 1445289 of CB1 or CB2, respectively, were incubated together. This experiment shows that cannabinoid-induced inhibition is mediated by cannabinoid receptors and other mechanisms. 77
Effect of Cannabinoids on Cellular Adhesion
Cannabinoids also exert a direct effect on cellular adhesion. This process is essential for maintaining the integrity of the extracellular matrix and any dysfunction that may cause tumor metastasis. An experiment highlighting metastatic spread using MDA-MB-231 cells and the cannabinoid MDA-F-AEA (0.5 mg/kg every 3 days for 3 weeks) demonstrated a decrease in the size and number of metastatic nodules. To confirm the direct effects of cannabinoids on metastasis, the effect was antagonized by SR 141716. 58 Unfortunately, despite the clear evidence that cannabinoids may inhibit metastasis, the long-term effects and potential mechanisms of inducing resistance remain unclear. Although cannabinoids demonstrate tumor regression, experiments have only been carried out in vivo for no longer than 21 days.
Hypoxia
Another characteristic of tumors is their ability to survive in severe hypoxic conditions. 78 Tumor cells activate transcriptional factors such as hypoxia-inducible- factor 1 (HIF-1), a known transcriptional factor called a regulator of hypoxic conditions. HIF-1 works by induction, invasion, angiogenesis, glycolytic metabolism, and upregulation of other cell survival molecules. 60 Solinas et al 79 experimented on 2 different brain carcinoma cell lines, U87-MG and T98G, derived from malignant gliomas and human glioblastoma multiforme tumors, respectively. Both cell lines were exposed to 50 µM CoCl2 or 1% pO2 for 24 hours to mimic hypoxic conditions. Under these hypoxic conditions, CBD at 5 and 9 µM induced a considerable decrease in HIF-1α compared to normoxic cells in U87-MG. Unlike U87-MG, T98G did not activate any substantial changes in HIF-1α levels when CBD was administered. 62 The study highlights the distinction between different cell lines and thus different effects of CBD on each cell line. T98G is already present with high amounts of HIF-1α levels in normoxic conditions, whereas, in U87-MG, it is barely detectable. A distinct difference in the upstream pathways of HIF-1α activation could be responsible for the difference in the 2 cell lines. T98G cells are very insensitive to CBD treatment compared to U87-MG.
Moreover, T98G insensitivity was also highlighted with Δ9-THC, and is thus considered cannabinoid resistant. The difference must be due to the origin of the brain tumors, where U87-MG cell lines were derived from glioblastoma/astrocytoma. However, T98G was derived from glioblastoma multiforme. 80
Combination Therapy
Recently, the use of combination therapies has suggested numerous theoretical advantages compared to the “cannabinoid only” treatment. Adding an adjunct agent allows a multiple-killing strategy whereby several mechanisms reduce tumor growth. It also provides the advantage of minimizing the effects of adaptive resistance often associated with cancerous tissues. In agreement with this phenomenon, research conducted in 2011 by Torres et al 81 further supported this hypothesis. The team experimented with glioblastoma multiforme (GBM), one of the most frequent and aggressive forms of cancer often associated with death within 12 to 15 months of diagnosis.61,62 The alkylating agent temozolomide (TMZ), first-line treatment in the management of GBM, was combined with Δ9-THC, which produced profound results. Submaximal doses of TMZ and Δ9-THC were selected after being initially treated on 3 different cell lines as single constituents. The in vivo relevance of combining these 2 agents was determined by subcutaneous injection of U87MG in immunodeficient mice and was allowed to reach 250 mm3. 60
The results highlighted a synergistic effect when Δ9-THC and TMZ are combined in their tumor regressing capabilities compared to treatment as single agents. To further elaborate on the effects of combined therapy, Torres et al 81 investigated the combination of 2 cannabinoids (Δ9-THC and CBD) on glioma cells. Similar to the effects witnessed with Δ9-THC and TMZ, the combined effect of these 2 cannabinoids at submaximal concentrations of 7.5 mg/kg/day of each agent reduced tumor growth of U87MG cell-derived subcutaneous xenografts. The effects were more significant in comparison to the treatment of this cell line than individual agents. 81
Adjunct therapy in cancer allows tumor growth to be targeted by interfering with multiple tumor survival mechanisms and enhancing their ability to further induce cell death by minimizing the survival strategies adopted by tumor cells. Δ9-THC works through the ceramide pathway, in contrast to CBD, which works through ROS production, ultimately causing both autophagy and apoptosis or simply apoptosis.46,63 Other potential benefits of combining 2 cannabinoids, such as the non-psychoactive CBD with psychoactive Δ9-THC, would be to enhance anti-tumoral activity and reduce Δ9-THC associated psychoactive side effects. 60
Torres et al 81 further investigated the potential for triple therapy. When used as a single agent, the experiment combined Δ9-THC, CBD, and TMZ to counteract the effects of resistance to TMZ. A major contributor to the poor prognosis of GBM is drug resistance because of overexpression of O 6 -methylguanine methyltransferase (MGMT), which is responsible for catalyzing the removal of methyl groups on the O 6 position of guanine bases on DNA, ultimately neutralizing the chemotherapeutic activity 82 The results of the experiment, when administered at a submaximal dose of each agent, reduced the growth of U87MG and T98G glioma cells. 60 A triple combined therapy may be necessary for specific cell lines where resistance is prevalent instead of U87MG cells, where only a single agent would be sufficient. 83
Cannabinoids as Adjuvant
Adjuvant therapy is delivered after primary treatment to destroy the remaining cancerous cells 84 cannabinoid CBD was used as an adjunct alongside bortezomib (BORT) in multiple myeloma (MM) cell lines. MM is a B-cell-derived hematological malignancy that causes clonal proliferation of plasma cells and accumulation in the bone marrow. 85 BORT, a proteasome inhibitor, is currently used to treat refractory and relapsed MM patients. Like most chemotherapeutic agents, drug resistance develops over time. 86 Morelli et al 87 analyzed samples of a particular protein often associated with the regulation of tumor growth, progression, invasion, and angiogenesis. It has previously been demonstrated that overexpression of this protein, transient receptor potential vanilloid type-2 (TRPV2), resulted in an increase in drug intake and enhanced sensitivity to chemotherapeutic agents by the inhibition of the Ras/Raf/MEK/ERK in glioblastoma. 88
In the case of MM, gene mutations in TRPV2 have been detected, and SNP analysis has shown a 5-Mb 17p11.2-p12 amplification in KMS-26 myeloma cells. Despite this, no functional role has been deduced for TRPV2 in MM. 69 CBD has shown a particular affinity to this receptor in glioma cell lines, including peroxisome proliferator-activated receptor gamma (PPARγ), and has been shown to increase its activity by increasing drug uptake cytotoxic activity.71-73 The obtained results suggested a positive outcome by using a combination of BORT and CBD. A BrdU incorporation assay has shown that CBD or BORT treatment reduced proliferation in MM cell lines, with a major reduction observed in cells presenting TRPV2 receptors compared to those without TRPV2 receptors. Although single agents reduced proliferation (Figure 4), a combination of BORT and CBD suggested a more profound reduction in cell proliferation (Figure 5).
Figure 4.

CBD mediated reduction in proliferation in RPMI (RPMI8226) and RPMITRPV2 cells. Assessment based on CBD (20 μM) and BORT (3 ng/mL) alone or together 67 (republished after obtaining permission from the author and journal). Results showing the effects of CBD (20 μM) and BORT (3 ng/mL) alone or together on their cytotoxic capabilities were also analyzed.
Figure 5.
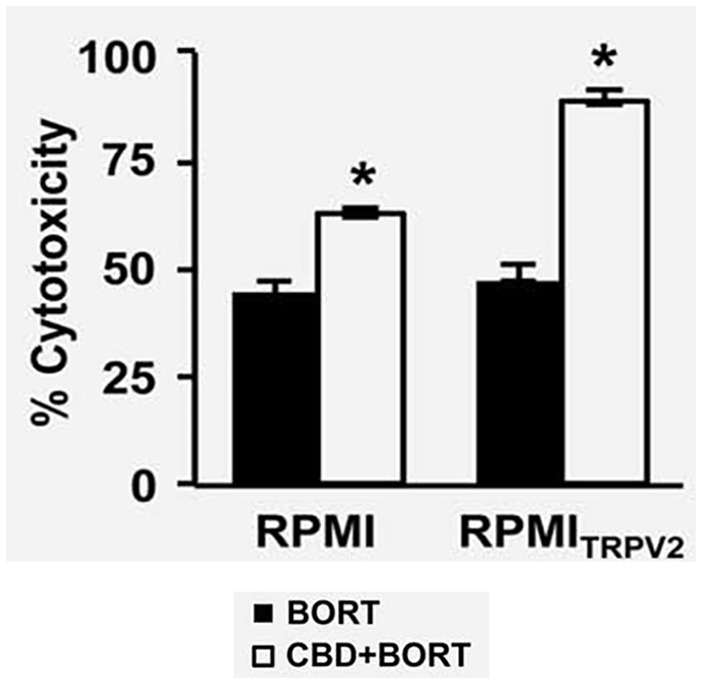
The cytotoxic effect of BORT (3 ng/mL) alone or together with CBD (20 μM) 67 (republished after obtaining permission from the author and journal).
These results again support the hypothesis of combining current anticancer agents alongside cannabinoids in combating drug resistance often associated with single agents. CBD is just one of many examples where the chemotherapeutic activity of current anti-cancer drugs can be potentiated synergistically by modifying specific receptors that can be targeted on tumor cells to allow drugs to maximize their activity. Given the role of MEK/ERK and AKT pathways in MM, the rationale behind this particular combination is that BORT induces cell cycle arrest and death of MM cells by many mechanisms such as interference with the phosphorylation of AKT synergism with ERK inhibitors.85-87 The combined effect of CBD-BORT reduced ERK and AKT signaling and switched off ERK and AKT phosphorylation in MM cells treated with this dual therapy, with the effect being maximized in those cells presenting TRPV2 receptors. 89
Other potential roles of this chosen combination are its effects on nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), an important protein complex essential for DNA transcription, cytokine production, and maintaining cell survival. NF-κB signaling is said to have a role in MM pathogenesis via the “classical” p50/p65 and “alternative” p52/RelB pathways. Suppression of the NF-κB pathway has been shown to induce MM cell death in active CD138+.76-78 The combination effect of CBD and BORT also affected this particular pathway by reducing the nuclear binding capabilities of p65, suggesting the role of TRPV2 expression in MM and an increase in sensitivity to CBD. Furthermore, the potential to reduce the dose of BORT and minimize potential side effects or adverse drug reactions associated with it, providing more reasons to rationalize dual therapy alongside cannabinoids, was also explored. 89
Combination therapy has not always produced promising results. Jacobsson et al 90 experimented with showing whether the concomitant use of Δ9-THC and tamoxifen (a drug often used in breast cancer) affected cell viability and whether the synergistic effect of both agents provided a new therapeutic strategy in the treatment of glioma. An initial experiment using AEA and 10% fetal bovine serum (FBS) to determine any significant effects on cell viability produced no significant data. Another experiment using cannabinoids (Δ9-THC or CBD) alongside tamoxifen to C6 glioma cells and with 10% FBS produced no significant effect on cell viability. Similar experiments with the administration of cannabinoids (Δ9-THC or CBD) alongside tamoxifen on C6 glioma cells and with FBS Free and 0.4% FBS produced significant effects on cell viability in a dose-dependent manner (Figure 6). 90
Figure 6.

Effects of different cannabinoids combined with tamoxifen in reducing cell viability in C6 glioma cells over 6 days 90 (republished after obtaining permission from the author and journal).
The mechanism for the increased sensitivity to cannabinoid tamoxifen therapy at reduced FBS is yet to be understood. The results suggest a need for further investigations into the potential therapeutic uses of cannabinoid-chemotherapeutic combination therapy and the effects of FBS on cell line sensitivity. 91 Moreover, although cell lines provide us with the basis for whether a particular agent can inhibit growth or change tumor characteristics, they are not always a complete representation of the underlying disease. Other GBM cell lines, A172, GB-1, U251-MG, and U373-MG, or MM cell lines, which possess different tumor characteristics, must be investigated to represent the disease accurately. Typical cell culture media are deficient in metabolites, growth molecules, cytokines, and other factors representing the tumor microenvironment’s true nature. In other words, the numerous multicellular interfaces within which all tumor cells interact in vivo are not replicated for cells grown in cell culture plates and, although tumor growth is reduced, these cell lines do not represent the actual events that occur inside a human.92-100
Currently, there is minimal published research on the pharmacokinetic profile of cannabinoids when administered orally regarding an anti-tumor activity or sensitizing of tumors to first line-agents. Unfortunately, the available data from contemporaneous research suggest numerous gaps in the knowledge base. Toxicity profiles of cannabinoids have still not been established; however, although some side effects of cannabinoids have been established, their long-term effects on modifying the tumor microenvironment in vivo are yet to be explored. Systematically determining the major pathways regarding treatment efficacy with non-psychoactive cannabinoids would allow us to determine which cannabinoids could target specific tumors or which cannabinoid-drug combinations could be used to help reduce or potentially eradicate tumor growth.
Conclusion and Future Perspectives
A significant gap remains open for further research to determine the effects of cannabinoids on the tumor microenvironment over a more extended period and if or when resistance does eventually appear. In addition, in vitro testing is not an accurate representation of in vivo; in other words, cells grown in a growth medium are not representative of a tumor cell in vivo and thus illustrate the problems associated when screening for pharmaceutical ingredients. The anti-glioma efficacy of cannabinoids was reported from a clinical trial that involved only 2 patients; more detailed studies are required to determine the actual role of cannabinoids in the microenvironment. 27 Research on cannabinoids and their potential therapeutic function has been ongoing since 1971, and their proposed mechanisms in reducing/increasing cell growth have been changed and debated. As research in this field continues, understanding cannabinoid effects on signaling in tumor growth will improve, and thus discrepancies and outdated information on the mechanism of action on cannabinoids will become updated and more credible.
Studies on cannabinoids have shown significant potential in cancer treatment (dependent on the cell line or tumor type). There is a robust set of data both in vitro (cancer cell lines) and in vivo. However, detailed human clinical trial data is required to reach a definite conclusion. Significant hurdles still need to be overcome. There are still major disagreements and gaps between academics’ knowledge regarding cannabinoids and their role in the tumor microenvironment. Current research only goes as far as showing the potential of specificity and efficacy to be precursors to clinical treatments. The resolution of the conflicting evidence can be overcome and must remain a priority for future development of cannabinoids as the next generation of cancer treatment. The IC50 (inhibitory concentration), ED50 (effective dose), GI50 (growth inhibition), and LC50 (lethal concentration) must be first determined on a range of individual cannabinoids to establish long-term effects such as resistance, toxicity, and prognosis. This will then provide the basis of NCI 60 testing to enable more accurate determination of the mechanisms of action, which is currently lacking. The use of cannabinoids as combination therapy with current chemotherapeutic agents or cannabis extracts has also been investigated by numerous researchers; however, research remains slow, and momentum must be gained as their potential is enormous and may revolutionize the way we treat cancer in the future.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs: Hamid A. Bakshi  https://orcid.org/0000-0002-1966-040X
https://orcid.org/0000-0002-1966-040X
Murtaza M. Tambuwala
https://orcid.org/0000-0001-8499-9891
References
- 1. Baghban R, Roshangar L, Jahanban-Esfahlan R, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020;18:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richter G, Hartsel H, Eades JA, Hickory JB, Makriyannis A. Cannabis sativa: an overview. In: Gupta RC, Lall R, Srivastava A, eds. Nutraceuticals. 2nd ed. Academic Press; 2021:603-624. [Google Scholar]
- 3. NHS. Cannabis fact sheet - multiple sclerosis trust. Accessed November 1, 2018. http://www.nhs.uk/ipgmedia/national/multiple%20sclerosis%20trust/assets/cannabis.pdf
- 4. Winstock A, Lynskey M, Borschmann R, Waldron J. Risk of emergency medical treatment following consumption of cannabis or synthetic cannabinoids in a large global sample. J Psychopharmacol. 2015;29:698-703. [DOI] [PubMed] [Google Scholar]
- 5. McDougall SR, Anderson AR, Chaplain MA. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. J Theor Biol. 2006;241:564-589. [DOI] [PubMed] [Google Scholar]
- 6. Ashton CH. Pharmacology and effects of cannabis: a brief review. Br J Psychiatry. 2001;178:101-106. [DOI] [PubMed] [Google Scholar]
- 7. Pertwee RG. Pharmacological actions of cannabinoids. Handb Exp Pharmacol. 2005;168:1-51. [DOI] [PubMed] [Google Scholar]
- 8. Castaneto MS, Gorelick DA, Desrosiers NA, Hartman RL, Pirard S, Huestis MA. Synthetic cannabinoids: epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014;144:12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gunderson EW, Haughey HM, Ait-Daoud N, Joshi AS, Hart CL. A survey of synthetic cannabinoid consumption by current cannabis users. Subst Abuse. 2014;35:184-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen K, Weinstein AM. Synthetic and non-synthetic cannabinoid drugs and their adverse effects-a review from public health prospective. Front Public Health. 2018;6:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ellis LM, Liu W, Ahmad SA, et al. Overview of angiogenesis: biologic implications for antiangiogenic therapy. Semin Oncol. 2001;28:94-104. [DOI] [PubMed] [Google Scholar]
- 12. Abo-Elnazar S, Moaaz M, Ghoneim H, Molokhia T, El-Korany W. Th17/Treg imbalance in opioids and cannabinoids addiction: relationship of NF-kB activation in CD4+ T cells. Egypt J Immunol. 2014;21:33-47. [PubMed] [Google Scholar]
- 13. Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400-411. [DOI] [PubMed] [Google Scholar]
- 14. Keen L, 2nd, Turner AD. Differential effects of self-reported lifetime marijuana use on interleukin-1 alpha and tumor necrosis factor in African American adults. J Behav Med. 2015;38:527-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674. [DOI] [PubMed] [Google Scholar]
- 16. Guindon J, Hohmann AG. The endocannabinoid system and cancer: therapeutic implication. Br J Pharmacol. 2011;163:1447-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-1186. [DOI] [PubMed] [Google Scholar]
- 18. Blázquez C, González-Feria L, Alvarez L, Haro A, Casanova ML, Guzmán M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004;64:5617-5623. [DOI] [PubMed] [Google Scholar]
- 19. Casanova ML, Blázquez C, Martínez-Palacio J, et al. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J Clin Investig. 2003;111:43-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Portella G, Laezza C, Laccetti P, De Petrocellis L, Di Marzo V, Bifulco M. Inhibitory effects of cannabinoid CB1 receptor stimulation on tumor growth and metastatic spreading: actions on signals involved in angiogenesis and metastasis. FASEB J. 2003;17:1771-1773. [DOI] [PubMed] [Google Scholar]
- 21. Preet A, Ganju RK, Groopman JE. Delta9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene. 2008;27:339-346. [DOI] [PubMed] [Google Scholar]
- 22. Davis ME. Glioblastoma: overview of disease and treatment. Clin J Oncol Nurs. 2016;20:S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bergers G, Hanahan D. Modes of resistance to antiangiogenic therapy. Nat Rev Cancer. 2008;8:592-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210-221. (Erratum in: Nat Rev Clin Oncol. 2011 Jun;8(6):316. Erratum in: Nat Rev Clin Oncol. 2011;8(4):221). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kogan NM, Blázquez C, Alvarez L, et al. A cannabinoid quinone inhibits angiogenesis by targeting vascular endothelial cells. Mol Pharmacol. 2006;70:51-59. [DOI] [PubMed] [Google Scholar]
- 26. Hart S, Fischer OM, Ullrich A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 2004;64:1943-1950. doi: 10.1158/0008-5472.can-03-3720 [DOI] [PubMed] [Google Scholar]
- 27. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161-174. [DOI] [PubMed] [Google Scholar]
- 28. Peyravian N, Deo S, Daunert S, Jimenez JJ. Cannabidiol as a novel therapeutic for immune modulation. ImmunoTargets Ther. 2020;9:131-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seltzer ES, Watters AK, MacKenzie D, Jr, Granat LM, Zhang D. Cannabidiol (CBD) as a promising anti-cancer drug. Cancers. 2020;12:3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lowin T, Straub RH. Cannabinoid-based drugs targeting CB1 and TRPV1, the sympathetic nervous system, and arthritis. Arthritis Res Ther. 2015;17:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9-34. [DOI] [PubMed] [Google Scholar]
- 32. Kleifeld O, Overall CM. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227-239. [DOI] [PubMed] [Google Scholar]
- 33. Ramer R, Fischer S, Haustein M, Manda K, Hinz B. Cannabinoids inhibit angiogenic capacities of endothelial cells via release of tissue inhibitor of matrix metalloproteinases-1 from lung cancer cells. Biochem Pharmacol. 2014;91:202-216. [DOI] [PubMed] [Google Scholar]
- 34. Roomi MW, Monterrey JC, Kalinovsky T, Rath M, Niedzwiecki A. Patterns of MMP-2 and MMP-9 expression in human cancer cell lines. Oncol Rep. 2009;21:1323-1333. [DOI] [PubMed] [Google Scholar]
- 35. Blázquez C, Carracedo A, Salazar M, et al. Down-regulation of tissue inhibitor of metalloproteinases-1 in gliomas: a new marker of cannabinoid antitumoral activity? Neuropharmacol. 2008;54:235-243. doi: 10.1016/j.neuropharm.2007.06.021 [DOI] [PubMed] [Google Scholar]
- 36. Casanovas O, Hager JH, Chun MG, Hanahan D. Incomplete inhibition of the Rb tumor suppressor pathway in the context of inactivated p53 is sufficient for pancreatic islet tumorigenesis. Oncogene. 2005;24:6597-6604. [DOI] [PubMed] [Google Scholar]
- 37. Pàez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of cannabidiol in breast cancer. Mol Oncol. 2015;9:906-919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sánchez MG, Sánchez AM, Ruiz-Llorente L, Díaz-Laviada I. Enhancement of androgen receptor expression induced by (R)-methanandamide in prostate LNCaP cells. FEBS Lett. 2003;555:561-566. [DOI] [PubMed] [Google Scholar]
- 41. Sánchez MG, Ruiz-Llorente L, Sánchez AM, Díaz-Laviada I. Activation of phosphoinositide 3-kinase/PKB pathway by CB1 and CB2 cannabinoid receptors expressed in prostate PC-3 cells. Involvement in raf-1 stimulation and NGF induction. Cell Signal. 2003;15:851-859. [DOI] [PubMed] [Google Scholar]
- 42. Mimeault M, Pommery N, Wattez N, Bailly C, Hénichart JP. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: implication of epidermal growth factor receptor downregulation and ceramide production. Prostate. 2003;56:1-12. [DOI] [PubMed] [Google Scholar]
- 43. Nithipatikom K, Isbell MA, Endsley MP, Woodliff JE, Campbell WB. Anti-proliferative effect of a putative endocannabinoid, 2-arachidonylglyceryl ether in prostate carcinoma cells. Prostaglandins Other Lipid Mediat. 2011;94:34-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Handsley MM, Edwards DR. Metalloproteinases and their inhibitors in tumor angiogenesis. Int J Cancer. 2005;115:849-860. [DOI] [PubMed] [Google Scholar]
- 45. McKallip RJ, Nagarkatti M, Nagarkatti PS. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J Immunol. 2005;174:3281-3289. [DOI] [PubMed] [Google Scholar]
- 46. Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47. doi: 10.1038/nrc704 [DOI] [PubMed] [Google Scholar]
- 47. Place AE, Jin Huh S, Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 2011;13:227-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal. 2009;11:3013-3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cudaback E, Marrs W, Moeller T, Stella N. The expression level of CB1 and CB2 receptors determines their efficacy at inducing apoptosis in astrocytomas. PLoS One. 2010;5:e8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singer E, Judkins J, Salomonis N, et al. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Discov. 2015;6:e1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lorente M, Torres S, Salazar M, et al. Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell Death Differ. 2011;18:959-973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Salazar M, Carracedo A, Salanueva ÍJ, et al. TRB3 links ER stress to autophagy in cannabinoid antitumoral action. Autophagy. 2009;5:1048-1049. [DOI] [PubMed] [Google Scholar]
- 53. Velasco G, Sánchez C, Guzmán M. Towards the use of cannabinoids as anti-tumour agents. Nat Rev Cancer. 2012;12:436-444. [DOI] [PubMed] [Google Scholar]
- 54. Carracedo A, Lorente M, Egia A, et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell. 2006;9:301-312. [DOI] [PubMed] [Google Scholar]
- 55. Shrivastava A, Kuzontkoski PM, Groopman JE, Prasad A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the crosstalk between apoptosis and autophagy. Mol Cancer Ther. 2011;10:1161-1172. [DOI] [PubMed] [Google Scholar]
- 56. Perry DK, Carton J, Shah AK, Meredith F, Uhlinger DJ, Hannun YA. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J Biol Chem. 2000;275:9078-9084. [DOI] [PubMed] [Google Scholar]
- 57. Salazar M, Carracedo A, Salanueva IJ, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Investig. 2009;119:1359-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. De Petrocellis L, Melck D, Palmisano A, et al. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci USA. 1998;95:8375-8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gómez del Pulgar T, Velasco G, Sánchez C, Haro A, Guzmán M. De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem J. 2002;363:183-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sarfaraz S, Adhami VM, Syed DN, Afaq F, Mukhtar H. Cannabinoids for cancer treatment: progress and promise. Cancer Res. 2008;68:339-342. [DOI] [PubMed] [Google Scholar]
- 61. Galve-Roperh I, Sánchez C, Cortés ML, Gómez del Pulgar T, Izquierdo M, Guzmán M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6:313-319. [DOI] [PubMed] [Google Scholar]
- 62. Sarfaraz S, Afaq F, Adhami VM, Mukhtar H. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005;65:1635-1641. [DOI] [PubMed] [Google Scholar]
- 63. Sarnataro D, Pisanti S, Santoro A, et al. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Mol Pharmacol. 2006;70:1298-1306. [DOI] [PubMed] [Google Scholar]
- 64. Rinaldi-Carmona M, Barth F, Héaulme M, et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240-244. [DOI] [PubMed] [Google Scholar]
- 65. Moffett S, Brown DA, Linder ME. Lipid-dependent targeting of G proteins into rafts. J Biol Chem. 2000;275:2191-2198. [DOI] [PubMed] [Google Scholar]
- 66. Walker C, Mojares E, Del Río Hernández A. Role of extracellular matrix in development and cancer progression. Int J Mol Sci. 2018;19:3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Haustein M, Ramer R, Linnebacher M, Manda K, Hinz B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Biochem Pharmacol. 2014;92:312-325. [DOI] [PubMed] [Google Scholar]
- 68. Dariš B, Tancer Verboten M, Knez Ž, Ferk P. Cannabinoids in cancer treatment: therapeutic potential and legislation. Bosn J Basic Med Sci. 2019;19:14-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Demuth DG, Molleman A. Cannabinoid signalling. Life Sci. 2006;78:549-563. [DOI] [PubMed] [Google Scholar]
- 70. Velasco G, Sánchez C, Guzmán M. Anticancer mechanisms of cannabinoids. Curr Oncol. 2016;23:S23-S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Murase R, Kawamura R, Singer E, et al. Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. Br J Pharmacol. 2014;171:4464-4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hermanson DJ, Marnett LJ. Cannabinoids, endocannabinoids, and cancer. Cancer Metastasis Rev. 2011;30:599-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Joseph J, Niggemann B, Zaenker K, Entschladen F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol Immunother. 2004;53:723-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro cell migration and invasion assays. J Vis Exp. 2014;88:51046. doi: 10.3791/51046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vaccani A, Massi P, Colombo A, Rubino T, Parolaro D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br J Pharmacol. 2005;144:1032-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Grimaldi C, Pisanti S, Laezza C, et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp Cell Res. 2006;312:363-373. [DOI] [PubMed] [Google Scholar]
- 77. Nieder C, Adam M, Molls M, Grosu AL. Therapeutic options for recurrent high-grade glioma in adult patients: recent advances. Crit Rev Oncol Hematol. 2006;60:181-193. [DOI] [PubMed] [Google Scholar]
- 78. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. ActaNeuropathol. 2007;114:97-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Solinas M, Massi P, Cinquina V, et al. Cannabidiol, a non-psychoactive cannabinoid compound, inhibits proliferation and invasion in U87-MG and T98G glioma cells through a multitarget effect. PLoS One. 2013;8:e76918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Massi P, Valenti M, Vaccani A, et al. 5-Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor activity of cannabidiol, a nonpsychoactive cannabinoid. J Neurochem. 2008;104:1091-1100. [DOI] [PubMed] [Google Scholar]
- 81. Torres S, Lorente M, Rodríguez-Fornés F, et al. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther. 2011;10:90-103. [DOI] [PubMed] [Google Scholar]
- 82. Hegi ME, Liu L, Herman JG, et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008;26:4189-4199. [DOI] [PubMed] [Google Scholar]
- 83. Rajkumar SV. Multiple myeloma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:57-65. [DOI] [PubMed] [Google Scholar]
- 84. Lowe MC, Kudchadkar RR. Neoadjuvant therapy for melanoma. Surg Oncol Clin N Am. 2020;29:445-453. [DOI] [PubMed] [Google Scholar]
- 85. Morelli MB, Offidani M, Alesiani F, et al. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int J Cancer. 2014;134:2534-2546. [DOI] [PubMed] [Google Scholar]
- 86. Morelli MB, Nabissi M, Amantini C, et al. The transient receptor potential vanilloid-2 cation channel impairs glioblastoma stem-like cell proliferation and promotes differentiation. Int J Cancer. 2012;131:E1067-E1077. [DOI] [PubMed] [Google Scholar]
- 87. Santoni G, Farfariello V, Liberati S, et al. The role of transient receptor potential vanilloid type-2 ion channels in innate and adaptive immune responses. Front Immunol. 2013;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci. 2008;28:6231-6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. De Petrocellis L, Ligresti A, Moriello AS, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163:1479-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jacobsson SO, Rongård E, Stridh M, Tiger G, Fowler CJ. Serum-dependent effects of tamoxifen and cannabinoids upon C6 glioma cell viability. Biochem Pharmacol. 2000;60:1807-1813. [DOI] [PubMed] [Google Scholar]
- 91. Yamada T, Ueda T, Shibata Y, et al. TRPV2 activation induces apoptotic cell death in human T24 bladder cancer cells: a potential therapeutic target for bladder cancer. Urology. 2010;76:509.e1-509.e7. [DOI] [PubMed] [Google Scholar]
- 92. Chauhan D, Hideshima T, Anderson KC. A novel proteasome inhibitor NPI-0052 as an anticancer therapy. Br J Cancer. 2006;95:961-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim K, Kong SY, Fulciniti M, et al. Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br J Haematol. 2010;149:537-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Que W, Chen J, Chuang M, Jiang D. Knockdown of c-Met enhances sensitivity to bortezomib in human multiple myeloma U266 cells via inhibiting Akt/mTOR activity. APMIS. 2012;120:195-203. [DOI] [PubMed] [Google Scholar]
- 95. Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010;115:3541-3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sen R. Control of B lymphocyte apoptosis by the transcription factor NF-kappaB. Immunity. 2006;25:871-883. [DOI] [PubMed] [Google Scholar]
- 97. Lorch JH, Thomas TO, Schmoll HJ. Bortezomib inhibits cell-cell adhesion and cell migration and enhances epidermal growth factor receptor inhibitor-induced cell death in squamous cell cancer. Cancer Res. 2007;67:727-734. [DOI] [PubMed] [Google Scholar]
- 98. Dipasquale B, Colombatti M, Tridente G. Morphological heterogeneity and phenotype modifications during long term in vitro cultures of six new human glioblastoma cell lines. Tumori. 1990;76:172-178. [DOI] [PubMed] [Google Scholar]
- 99. Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;39:29-36. [DOI] [PubMed] [Google Scholar]
- 100. Pieper RO. Defined human cellular systems in the study of glioma development. Front Biosci. 2003;8:s19-s27. [DOI] [PubMed] [Google Scholar]