

Abstract
Corneal endothelial dystrophies are a heterogeneous group of diseases with different modes of inheritance and genetic basis for each dystrophy. The genes associated with these diseases encode transcription factors, structural components of the stroma and Descemet membrane, cell transport proteins, and others. Congenital hereditary endothelial dystrophy (CHED) is associated with mutations in two genes, OVOL2 and SLC4A11, for dominant and recessive forms of CHED, respectively. Mutations in three genes are known to cause posterior polymorphous corneal dystrophy (PPCD). They are OVOL2 (PPCD1), ZEB1 (PPCD3), and GRHL1 (PPCD4). The PPCD2 locus involving the collagen gene COL8A2 on chromosome 1 is disputed due to insufficient evidence. Mutations in the COL8A2 gene are associated with early-onset Fuchs’ endothelial corneal dystrophy (FECD). Several genes have been associated with the more common, late-onset FECD. Alterations in each of these genes occur in a fraction of patients, and the most prevalent genetic alteration in FECD patients across the world is a triplet repeat expansion in the TCF4 gene. Knowledge of the genetics of corneal endothelial dystrophies has considerably advanced within the last decade and has contributed to better diagnosis of these dystrophies as well as opened up the possibility of novel therapeutic approaches based on the molecular mechanisms involved. The functions of genes identified to date provide insights into the pathogenic mechanisms involved in each disorder.
Keywords: CHED, corneal endothelium, endothelial dystrophy, FECD, genetics, genotype, phenotype, PPCD
The corneal endothelium develops from the migration of the first wave of mesenchyme (neural crest cells) at 5–6 weeks of gestation. The neural crest cells migrate centrally through the primary cornea formed between the lens vesicle and the surface ectoderm. These cells arrange into a sheet of overlapping cells, which forms the corneal endothelium by about 8 weeks of gestation.[1] At birth, the human cornea has 7500 endothelial cells per square (sq) millimeter (mm), while the adult cornea has about 2500 cells per sq mm due to a gradual decline in the number of endothelial cells throughout life. The endothelium actively pumps fluid out of the stroma to maintain a constant level of stromal hydration, thereby maintaining corneal transparency.[2]
Corneal endothelial dystrophies are a group of heterogeneous disorders characterized by the dysfunction and loss of the corneal endothelium. Defects in the endothelial cell layer lead to fluid accumulation within the corneal stroma, secondary changes such as scarring, epithelial bullae, vascularization, and consequent vision loss.
The major forms of endothelial dystrophies, i.e., congenital hereditary endothelial dystrophy (CHED), posterior polymorphous corneal dystrophy (PPCD), and Fuchs’ endothelial corneal dystrophy (FECD), are genetically diverse with respect to the number of associated genes and modes of inheritance [Table 1].[3] The international classification of corneal dystrophies (IC3D) laid out the parameters for systematically classifying all dystrophies by considering the various levels of evidence available for each dystrophy, including clinical, histopathological, and genetic features. The second edition of the IC3D essentially revised the anatomic basis of the corneal dystrophies laid out in the first.[4] The acellular layers of the cornea including Bowman’s layer, condensed anterior stroma, Descemet membrane (DM) and the basement membrane of the endothelium were excluded as separate entities.
Table 1.
Details of Genes and Loci for Corneal Endothelial Dystrophies
| Endothelial Dystrophy | Locus/gene | Inheritance | Gene function |
|---|---|---|---|
| CHED | CHED1/OVOL2 | AD | Transcription factor promoting MET |
| CHED2/SLC4A11 | AR | Not known; ion transporter–related | |
| PPCD | PPCD1/OVOL2 | AD | Transcription factor |
| PPCD2/COL8A2* | AD | Component of Descemet membrane | |
| PPCD3/ZEB1 | AD | Transcription factor promoting EMT | |
| PPCD4/GRHL1 | AD | Transcription factor promoting MET | |
| FECD | FECD1/COL8A2 | AD | Component of Descemet membrane |
| FECD2 (13pter-q12)/Not known | AD | Not known | |
| FECD3/TCF4 | AD | Transcription factor | |
| FECD4/SLC4A11 | Not known; ion transporter–related | ||
| FECD5 (5q33-35)/Not known | AD | Not known | |
| FECD6/ZEB1 | Complex | Transcription factor | |
| FECD7 (9p24-22)/not known | Complex | Not known | |
| FECD8/AGBL1 | AD | Glutamate decarboxylase | |
| FECD/DMPK | AD | Protein kinase |
The designations for loci FECD1–8 are as per the OMIM database (Ref. 3-available at https://omim.org/phenotypicSeries/PS136800]. *the status of the COL8A2 gene as the PPCD2 locus is questionable. AD, Autosomal dominant; EMT, Epithelial–mesenchymal transition; MET, Mesenchymal–epithelial transition. Shown above are the phenotypic details of different CEDs including associated features, and structural changes in the cornea as observed on slit lamp, specular microscopy and OCT
The common clinical features and associations are summarized in Table 1 with representative images shown in Fig. 1. There have been several recent developments concerning the genetics of endothelial dystrophies, leading to knowledge of the genes involved, particularly in the cases of autosomal dominant CHED (CHED1 locus) and PPCD (involving the PPCD1 gene) and certain forms FECD, as detailed in the following sections. These discoveries have led to a better understanding of the pathogenesis of these disorders and uncovered new molecular mechanisms involved in the disease processes. The need for the present review is, thus, to capture the current status of this field, including the substantial additions, particularly over the last few years, not covered in recent literature reviews. Herein, we attempt to present a comprehensive and updated account of the genetics of all major forms of corneal endothelial dystrophies.
Figure 1.
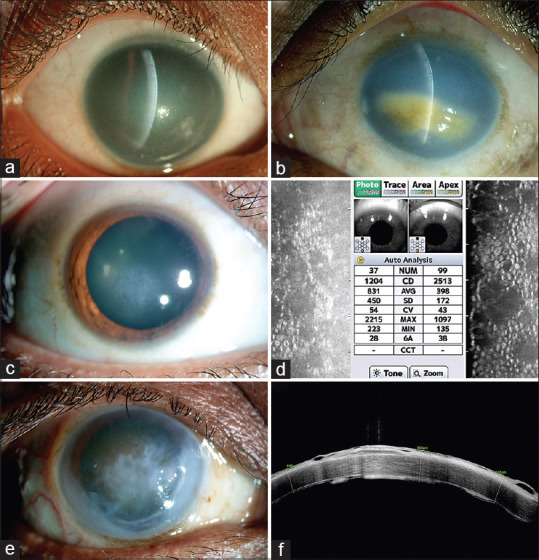
Corneal phenotypes of patients with corneal endothelial dystrophy (CED) Representative slit-lamp photographs (slit view) of the right (a) and the left (b) eyes of two different patients with CHED, showing diffuse corneal haze and thick cornea limbus to limbus throughout. Image B shows secondary spheroidal degeneration due to longstanding corneal edema; (c) slit-lamp photograph of a patient with FECD showing focal corneal edema; (d) specular microscopy of the right and left eyes of a patient with FECD showing reduced endothelial cell density and characteristic dropout areas suggestive of guttae; (e) slit-lamp photograph of a patient who has PPCD showing edema and secondary epithelial changes; (f) the optical coherence tomography of the same patient (as in e), showing deposits at the level of Descemet-endothelial complex with epithelial bullae and scarring
Congenital Hereditary Endothelial Dystrophy
Congenital hereditary endothelial dystrophy (CHED), also known as congenital hereditary corneal edema, is characterized by bilateral haze and thick corneas at birth [Table 2; Fig. 1a and b]. It may be associated with developmental glaucoma in certain cases. There are two forms of CHED recognized by their different manifestations, modes of inheritance and genetic loci: autosomal dominant CHED (as associated to the CHED1 locus) and autosomal recessive CHED (CHED2 locus). First documented in a large British family,[5] autosomal dominant CHED is characterized by a diffuse, milky opacification of the cornea, usually evident after the first year of life, stromal and epithelial edema, and an increased corneal thickness. Photophobia and epiphora are seen in the early post-natal period. The DM appears thickened on slit-lamp examination, and there is an absence of an endothelial mosaic.[5,6] Autosomal recessive CHED reportedly has a slightly earlier onset, and patients have a milky appearance of the cornea within the first few weeks of life. It is characterized by a diffuse corneal opacity having a ground-glass appearance, increased corneal thickness, lack of endothelial cells and a homogeneous thickening of the DM. Other changes in the cornea which show overlap between the two forms of CHED include epithelial atrophy, sub-epithelial fibrosis, and loss of Bowman’s layer. Spheroidal degeneration and stromal vascularization occur in association with longstanding disease.[6] Autosomal dominant CHED maps to the pericentromeric region of chromosome 20,[7] while the recessive form maps to the short arm of chromosome 20 at 20p13.[8]
Table 2.
Summary of Phenotypes of Corneal Endothelial Dystrophies
| Dystrophy | Clinical features on slit lamp | Other associations | Features observed by specular microscopy | Features on OCT |
|---|---|---|---|---|
| CHED | Diffuse corneal haze, | Developmental glaucoma | Not visible due to stromal haze | Thickened cornea |
| Thick corneas, abnormalities in corneal shape and size | Amblyopia | Epithelial hyperreflectivity in those with longstanding edema | ||
| Edema in longstanding/severe cases/those with raised IOP | Nystagmus | |||
| Secondary corneal changes such as spheroidal degeneration | Sensorineural deafness (Harboyan syndrome) | |||
| PPCD | Vesicles, bands, and placoid lesions at the level of DM | Meridional amblyopia | Dropout areas corresponding to the vesicles/bands | Rail-track appearance of DM- endothelial complex, |
| Thickened DM, iris changes | Keratoconus | Low endothelial cell density | Epithelial hyperreflectivity in longstanding edema | |
| Thick and steep cornea with edema | ||||
| Epithelial changes in longstanding edema | ||||
| Corneal vascularization & secondary degenerative changes | ||||
| FECD | Guttae typically more in central versus peripheral cornea | Glaucoma (open-angle and angle-closure) | Guttae | Thickened DM in some cases, Epithelial hyperreflectivity in those with edema |
| Pigments on the endothelial surface | ||||
| Epithelial changes in longstanding edema corneal vascularization can occur | Areas between guttae may show endothelial cells parameters such as increased coefficient of variation and mean cell area, correlating with endothelial health |
Table 2 shows the major phenotypic changes for each endothelial dystrophy as observable under slit lamp, specular microscopy and OCT. Other associations denote pathologies not involving the endothelium
Autosomal dominant CHED
Pearce et al. initially reported the dominant form of CHED as congenital endothelial corneal dystrophy in a British family of 78 individuals, half of whom were affected[5]. The same family was further elaborated upon,[6] and the disease locus was mapped to chromosome 20 by Toma et al.[7] Although two other families were reported as autosomal dominant CHED (CHED1), the clinical and ultrastructural features in these cases suggested that the phenotype corresponded to PPCD.[9] Electron microscopy of the cornea in one of the patients was noted to consist of fibroblast-like cells two–three layers thick, replacing the endothelium, microvilli facing the anterior chamber, hemidesmosomes, and cytoplasmic filaments: These were all characteristic of PPCD.[10,11] Another distinguishing feature of dominant CHED from PPCD is that in the former case, corneal haze is evident within the first year of life.[12] The very rare occurrence of dominant CHED and the absence of any further reports in the literature to support its existence apart from the original pedigree mentioned above led to its being eliminated as a unique entity by the IC3D.[3] However, the British index family reported by Pearce et al.,[5] later extending to seven generations with 36 affected individuals, was revisited and subjected to a detailed analysis. These studies led to the CHED1 gene being mapped to a more refined interval on chromosome 20, thus facilitating its identification.[12] The affected members in this family presented with photophobia, epiphora, and corneal haze, developing by one year of age. Whole-genome sequencing of affected members identified the pathogenic change, consisting of a duplication (-339-361dup) in the promoter of the Ovo-like 2 (OVOL2) gene in the family. Cloning and heterologous expression of the mutant OVOL2 promoter showed greater activation of a reporter gene than the wild-type promoter in cell lines,[12] suggesting that the CHED1 mutation deregulates and thereby increases promoter activity. Thus, the putative mechanism of the disease involves an aberrant and ectopic expression of the OVOL2 gene in endothelial cells of patients with dominant CHED.
Ovo-like 2 is a transcription factor belonging to the Ovo family, homologous to the Drosophila Ovo. These proteins contain four C2H2-type zinc fingers at their C-termini. The mammalian OVOL2 gene is expressed in several embryonic and post-natal tissues and regulates the process of mesenchymal–epithelial transition (MET). In skin keratinocytes, OVOL2 induces proliferation but suppresses differentiation.[13] It induces MET in dermal fibroblasts cooperatively with other epithelial reprogramming factors by enhancing the expression of epithelial genes and suppressing fibroblast-associated genes.[14] It is expressed in the corneal epithelium but absent in corneal endothelium and stromal fibroblasts.[12] Its overexpression in the presence of mutations in its promoter may thus lead to a phenotypic alteration of corneal endothelial cells due to changes in their gene expression program during development.
Autosomal recessive CHED
The gene for autosomal recessive CHED (CHED2) was mapped onto chromosome 20p13 in a family from Myanmar, and mutations in the solute carrier family 4 member 11 (SLC4A11) gene were identified in the original family and a series of other CHED2 families from Pakistan and India.[15,16] The SLC4A11 gene encodes a transmembrane protein implicated in the transport of ions across the membrane, although its exact function is not understood as yet. Apart from the reports mentioned above, mutational heterogeneity is evident from the types and distribution of mutations in SLC4A11 detected in families from different regions to date.[17,18,19,20,21,22] In addition to CHED, Harboyan syndrome, a disorder consisting of CHED and progressive perceptive deafness, is also associated with mutations in SLC4A11.[23]
The SLC4A11 gene was isolated as a bicarbonate-transporter-related protein (BTR1) expressed in several glandular tissues[24] and is homologous to the borate transporter Arabidopsis thaliana, known as AtBor. In the absence of borate ions, BTR1 conducts Na+ and OH-, while in the presence of borate, it functions as a Na-coupled borate cotransporter (hence named NaBC1). NaBC1 has been characterized as an electrogenic, voltage-regulated transporter, important in mediating borate homeostasis in plants and animal cells.[25] Based on its similarity to the erythrocyte anion exchanger AE1 protein, SLC4A11 is proposed to have fourteen transmembrane segments with amino and carboxy termini facing the cytosol.[26] Knockout of the Slc4a11 gene in mice is associated with a corneal phenotype resembling CHED. The mutant mice show a decreased density of endothelial cells, increased cell size, loss of hexagonal shape of the cells, thickening of the DM disorganization of stromal collagen, and an overall increase in corneal thickness.[27] CHED-associated mutants of SLC4A11 expressed in cell lines display defects in translocation to the cell membrane and increased oxidative stress with apoptotic cell death.[28]
Posterior Polymorphous Corneal Dystrophy
Posterior polymorphous corneal dystrophy (PPCD) manifests as a diverse spectrum of clinical features [Table 2; Fig. 1e and f]. A vast majority of patients with PPCD may be asymptomatic and may present with a refractive error or amblyopia when one eye is involved. There are others that present with gradual loss of vision with variable age at onset of symptoms ranging from childhood to adulthood. Common features include corneal edema, thickened DM with band-like or vesicular lesions, iridocorneal adhesions and normal or raised intraocular pressure.[29,30] The condition is known to be associated with steep corneal curvatures.[31] The essential change in the endothelial cells is metaplasia with transformation to a multi-layered stratified epithelium having tonofilaments, desmosomes, and microvilli.[32] PPCD is an autosomal dominant disorder and has four associated genes, designated as PPCD1 to PPCD4. Three of the genes are critical regulators of differentiation, inducing changes in cell type, either from epithelial to mesenchymal or the reverse.
Posterior polymorphous corneal dystrophy 1
The posterior polymorphous corneal dystrophy 1 (PPCD1) locus was mapped by linkage analysis of a large family to a region of 30 cM on the long arm of chromosome 20,[33] encompassing the genetic locus of CHED1. One of the genes investigated for its association with PPCD is the visual system homeobox 1 (VSX1) gene, which maps within this genetic interval. It encodes a transcription factor expressed in ocular tissues. However, the examination of two Czech families excluded this gene from the PPCD1 locus.[34] Further investigation of a large American family of 29 members[35] and multiple Czech families with PPCD confirmed the PPCD1 genomic interval on chromosome 20. Common ancestry in the latter families helped refine the locus for PPCD1.[36] Analysis of genes in the mapped PPCD1 interval in affected individuals led to the detection of a common mutation in the promoter of the OVOL2 gene consisting of a change of −370T>C in sixteen Czech PPCD families. Mutations of − 307T>C and − 274T >G of OVOL2 were identified in British families with PPCD.[12] The mutation of −307T > C was also detected in the family that was originally mapped to the PPCD1 locus.[37] These observations established OVOL2 as the gene for dominant CHED and PPCD, indicating that they are allelic disorders.
Posterior polymorphous corneal dystrophy 2
The posterior polymorphous corneal dystrophy 2 (PPCD2) locus was attributed to the collagen 8 alpha 2 (COL8A2) gene on chromosome 1p34-p32 to study one family of two affected individuals. This locus was earlier mapped in a family with FECD, and the COL8A2 gene in the critical region was considered a suitable candidate gene for both FECD and PPCD due to its expression in the DM. Mutation in the COL8A2 gene of Gln455Lys was identified in one family with a reported diagnosis of PPCD comprising two affected individuals, in addition to two families with early-onset FECD.[38] Curiously, two families with early-onset FECD and one family with PPCD, all from Northern England, were reported to have a common founder haplotype for five markers at the disease locus. It is questionable that families with two different clinical conditions can share a founder haplotype, even if the same gene is involved. One possibility is that a diagnosis of a disease as PPCD in the concerned family is incorrect. This idea is contradicted by the statement that ultrastructural analysis of the patients’ corneas confirmed the characteristic features of PPCD, such as multilayering of the corneal endothelium and presence of desmosomes and microvilli.[38] Possible explanations for a shared haplotype between the families could be that the five markers used to determine haplotypes around the COL8A2 gene are not sufficiently polymorphic or that the particular haplotypes reported are present at a high frequency in the population studied. In the absence of data on haplotypes of normal controls from the same region, one cannot ascertain the reasons for shared haplotypes between the families. Furthermore, the involvement of the COL8A2 gene in PPCD is not supported by other studies, which found no pathogenic alterations in PPCD cases, consisting of five[39] and fourteen probands tested.[40] Sequence changes that were identified in patients were also found in normal controls. Thus, the association of the COL8A2 gene with PPCD is questionable.
Posterior polymorphous corneal dystrophy 3 (PPCD3)
The posterior polymorphous corneal dystrophy 3 (PPCD3) locus was mapped to the short arm of chromosome 10 in a large family in which affected members presented with vesicular, geographic, or band-like lesions in the DM. The zinc finger E-box binding homeobox 1 (ZEB1) gene is located in the mapped region for PPCD3 and encodes a homeodomain zinc finger transcription factor that represses the epithelial phenotype. Analysis of the ZEB1 gene in the family revealed a frameshift mutation consisting of a two base-pair deletion (the cDNA change is c.2916_2917del).
Pathogenic mutations were detected in four families upon screening of ten additional families in this study,[41] with all of the mutations involving frameshifts or stop codons. Similar types of mutations in ZEB1 have been found in other populations of PPCD patients as well.[42,43,44,45,46] Individuals with ZEB1 mutations also have other non-ocular phenotypes such as hernias and hydroceles in males, occasional hernias in women, and skeletal defects in some families.[41,42,47] Hypoplasia or agenesis of the corpus callosum has been observed in various patients with PPCD3. Two probands with defects in the corpus callosum and PPCD had a single base deletion (c. 449delG) and a two base pair deletions (c.1913-1914delCA), respectively.[48] Interstitial deletion of about 2 Mb encompassing the ZEB1 gene was detected in another patient with agenesis of the corpus callosum and PPCD.[49] One of the targets regulated by ZEB1 is the collagen gene COL4A3. It has ZEB1-binding sites in its promoter and is repressed by ZEB1.[41] Thus, the cornea of a patient with a mutation in ZEB1 showed ectopic expression of COL4A3, supporting the idea that altered gene expression contributes to an abnormal Descemet membrane. The phenotypes associated with ZEB1 mutations in humans are recapitulated in mouse models having a knockout of ZEB1, though the homozygous knockout mice die shortly after birth. The knockout mice show corneal defects such as ectopic expression of epithelial genes in the endothelium, abnormal proliferation of keratocytes and endothelium, iridocorneal adhesions and numerous skeletal defects.[50,51]
ZEB1 is a transcription factor that regulates the process of EMT (epithelial–mesenchymal transition), the opposite of MET (mesenchymal–epithelial transition). EMT is a transition of cell state that can occur during development, wound healing, fibrosis and disease, in which epithelial cells lose their characteristic features such as gene expression profile involving repression of epithelial gene expression and activation of mesenchymal genes, changes in cell polarity and cell–cell adhesion. A key event in the transition is the switch from E-cadherin to N-cadherin gene expression, which alters the adhesion properties of the cells.[52] These changes bring about a mesenchymal phenotype. As mentioned above, haploinsufficiency of ZEB1 in a PPCD3 patient was associated with a decreased endothelial expression of COL4A3, but changes in other collagens appear less conclusive. The overall transcriptomic profile of corneal endothelium in PPCD3 patients displayed a significant loss of endothelial-specific gene expression, ectopic expression of epithelial cadherin 1, and loss of ZEB1 expression.[53]
Posterior polymorphous corneal dystrophy 4
The fourth locus for PPCD, designated as PPCD4, was mapped in a nine-generation Czech family with an autosomal dominant transmission of PPCD onto chromosome 8q22-24. Whole genome sequencing and analysis of sequences in the mapped PPCD4 region showed a mutation of c.20+544G>T, located within the intron of the grainy head-like 2 (GRHL2) gene, in the family.[54] The GRHL2 gene encodes a transcription factor that represses both ZEB1 activity and the process of EMT. Though located within the first intron of the gene, in silico analysis of the intron sequence suggested that it regulates transcription of the gene. The c.20+544G>T mutation was detected in three additional PPCD families of Czech origin. Two more mutations in the GRHL2 promoter were identified in British subjects, also located in the intron (c.20+257delT and c.20+133delA). The GRHL2 mutations were associated with intra- and interfamilial variability in phenotypes, with onset of loss of vision ranging from childhood until adulthood in different individuals. Ages at surgery and endothelial cell phenotypes also showed a wide variation. Asymmetric disease was noted in some cases. Similar to OVOL2, the expression of GRHL2 is undetectable in fetal or adult corneal endothelium but found at high levels in corneal epithelial cells. Analysis of mutant promoters in cell lines suggests an increased activity of the mutants compared with the normal sequence.[54] Furthermore, levels of GRHL2 are found to be increased in PPCD1 corneas as well as other corneal tissues without a mutation in any of the known PPCD loci.[55] These observations imply a mechanism of ectopic expression of the GRHL2 gene in the corneal endothelium of PPCD patients carrying the mutations mentioned above, leading to an epithelial-like transformation of these cells.
Three of the four PPCD genes encode transcription factors that determine the status of differentiation of the endothelium. The pathogenic mechanisms in PPCD are thus consistent with a lack of regulation of cell fate due to mutations in the ZEB1, OVOL2, and GRHL2 genes, leading to the altered epithelial-like morphology of the corneal endothelium in this disease. Loss of activity in the case of ZEB1 mutations and deregulated or ectopic activity in the case of OVOL2 and GRHL2 mutations are implicated in the pathogenesis of various types of PPCD. An aspect that is yet to be understood is the extent and nature of the regulatory region of the GRHL2 gene since mutations identified in PPCD4 include sequences downstream of the transcription start site in the intron of the gene. Knowledge of the specific intronic sequences of GRHL2 that regulate its expression, and their mode of action, may help in understanding the factors which interact with the promoter and regulate MET. Apart from the known PPCD genes, there appears to be further genetic heterogeneity in the disease. Since screening of several families for mutations in the relevant regions, all the known PPCD genes did not reveal any mutations in them.[55] There may be other loci involved in the disease or further mutational heterogeneity involving other regions in the same genes that may explain unresolved cases.
Fuchs’ Endothelial Corneal Dystrophy
Fuchs’ endothelial corneal dystrophy (FECD) is common in many parts of the world, particularly in the older population. It commonly manifests with early morning symptoms of decreased vision, corneal haze and poor night vision. It has a female preponderance and has a prevalence ranging from about 3% to 11% in different parts of the world, being much more common in Western countries than in Asia.[56] Characteristic signs include reduced endothelial cell density, abnormal size and shape of endothelial cells, corneal edema, and formation of excrescences (guttae) on the DM, which is abnormally thickened [Table 2; Fig. 1c and d]. It is graded according to clinical severity on the Krachmer grading scale from grade 0 to grade 6, depending on the extent of guttae and the presence of corneal edema.[57] FECD presents as early- and late-onset forms. The early-onset form is rare and develops within the first decade,[58] progressing through the second to third decades.
In contrast, the more typical late-onset form has its onset in the second to third decades and manifests after the fifth decade. The details of loci and genes associated with FECD are summarized in Table 1; loci are designated as FECD1–FECD8. Three loci are mapped (FECD2, FECD5, and FECD7), the genes of which are unknown.
Early-onset FECD
Collagen 8 alpha 2 gene
Early-onset FECD is associated with mutations in the collagen 8 alpha 2 (COL8A2) gene. Linkage mapping of a three-generation family with early-onset FECD assigned the locus to chromosome 1p34-32.[38] The COL8A2 gene in this locus encodes a component of the DM and thus represents a strong candidate for FECD and PPCD. A missense change of glutamine-455 to lysine (c.1363C>A; Gln455Lys) was identified in the original family; the same variant was observed in two more families with FECD and a family with PPCD. Another mutation of lysine-450 to tryptophan (c.1349T>G; Lys450Trp) was reported for early-onset FECD in a six-generation family. It was associated with much greater severity of the disease when compared with other familial FECD patients.[59] Other mutations in COL8A2 reported in patients with late-onset FECD such as arginine-155 to glutamine (c.464G>A; Arg155Gln), arginine-304 to glutamine (c.911G>A; Arg304Gln), arginine-434 to histidine (c.1300G>A; Arg434His), and glutamine-455 to lysine (c.1363C>A; Gln455Lys), have not been confirmed to be specific to the disease since they were also found in normal individuals.[39,60] Overall, COL8A2 mutations involve amino acid substitutions that are associated with early-onset FECD. The role of the COL8A2 gene in late-onset disease is not confirmed from existing data. Knock-in mouse models for COL8A2 mutations lysine-450 to tryptophan (Lys450Trp) and glutamine-455 to lysine (Gln455Lys) have been generated by replacing the normal mouse Col8a2 gene with the selected mutant genes. These mice show changes similar to the human phenotypes with a gradual decline in endothelial cell density, increase in the number of guttae and loss of hexagonal shape of the endothelial cells.[61,62]
Late-onset FECD
Zinc finger E-box-binding protein 1 gene
The zinc finger E-box-binding protein 1 (ZEB1) gene was investigated for association with FECD based on its already established role in the pathogenesis of a related disorder, PPCD. A missense change of asparagine-696 to serine (Asn696Ser) in a conserved protein region was found in one out of 74 unrelated patients with the late-onset disease.[63] A larger study involving 384 cases of late-onset FECD in Caucasian patients reported missense mutations in the ZEB1 gene in less than 2% of patients. A locus on chromosome 9p mapped in one multiplex family with FECD is postulated to interact with a missense mutation (glutamine-840 to proline (Gln840Pro)) in ZEB1 based on phenotypic severity of individuals having the risk haplotype in this locus as well as the Gln840Pro mutation.[64] A later study of Indian patients reported mutations in ZEB1, including one nonsense and four missense mutations in 6 out of 82 patients with FECD.[65]
Transcription factor 4 gene
The Transcription factor 4 (TCF4) gene encodes a helix–loop–helix transcription factor E2-2. Association of the TCF4 gene with FECD was obtained through a genome-wide study of FECD cases of European origin. Significant associations with the disease were obtained with several markers in this region, and the strongest association was obtained with a single nucleotide polymorphism (SNP) rs613872, suggesting an increased risk of about five-fold for each allele.[66] This effect was replicated through other association studies of FECD cases that are possibly mostly Caucasian and also confirmed by linkage analysis of multiplex families.[67,68] A marginal association of FECD with rs613872 was seen in Indian subjects with a sample size of forty-four subjects.[69] On the other hand, a different SNP in the same locus, rs17089887, showed significant association with FECD in this study of Indian FECD cases.
Another polymorphic marker in the TCF4 gene linked to the rs613872 SNP is a trinucleotide repeat sequence made up of CTG repeats in an intron of the gene. Significant expansions of triplet repeats at this locus are associated with FECD. Repeat numbers of above a threshold of 50 repeats are reported in 79% of cases and about 3% of controls.[70] The repeat expansion shows a strong association with FECD, and a repeat number greater than 40 repeats displayed complete penetrance in 52% of cases and incomplete penetrance in an additional 10% of the families of Caucasian origin.[71] This association was reported in different populations, including Chinese,[72] Japanese,[73] Indian,[74] German,[75] and Australian.[76] The disease mechanism in cases of expansion of triplet repeats in TCF4 is that the repeat sequences are transcribed into expanded CUG repeat RNA. The repeat RNA binds and sequesters a splicing factor known as MBNL (muscleblind-like protein), causing widespread mis-splicing of several other genes in the endothelium. The mutant CUG repeat RNA accumulates and is visible as nuclear foci in the affected corneal endothelial cells using fluorescence in situ hybridization.[77,78] A quantitative analysis of the MBNL proteins in endothelial tissues and cell lines suggests that the levels of the protein are lower in human corneal endothelial tissue and localized mostly in the cytoplasm. Furthermore, the MBNL proteins are required for stabilization of foci. These data may explain the presence of RNA foci as well as the pathologic effect of the triplet expansion in TCF4 gene in the corneal endothelium.[79,80]
Solute carrier family 4 member 11 gene
The association of solute carrier family 4 member 11 (SLC4A11) gene mutations in autosomal recessive CHED makes it a suitable candidate gene for FECD as well, and it has therefore been screened in patients with FECD. A study of 89 Asian patients found four patients with mutations in SLC4A11.[81] Analysis of the mutant proteins in vitro showed that they had defects in localizing to the cell surface. A study of 45 Indian patients with late-onset FECD found four missense changes in five patients.[82] In contrast, no significant associations of SLC4A11 mutations were detected in another study of 80 Indian patients with FECD[83] or African American patients.[84] A different approach to studying the association of SLC4A11 mutations with FECD was employed to investigate a series of eight families of patients with CHED2. Here, the heterozygous parents of CHED-affected children with SLC4A11 mutations showed early signs of FECD, though they were asymptomatic.[85]
Other genes associated with late-onset FECD
Other genes have been associated with FECD through studies on large families and a series of cases. These genes are found to be involved in 1%–2% of FECD patients. The AGBL1 gene encodes a carboxypeptidase that modifies target proteins by deglutamylation. It is expressed in the corneal endothelium, and the wild-type protein has a cytoplasmic localization. Mutations in the ATP-GTP binding protein-like 1 (AGBL1) gene are associated with FECD in both familial and sporadic forms of the late-onset disease.[86] Another FECD locus mapped to chromosome 18q in three large families is ascribed to the lipoxygenase homology domain 1 (LOXHD1) gene, though a variant was found in only one of the families. Based on available data, the association of LOXHD1 variants with FECD must be interpreted cautiously due to the high percentage of novel variants detected at this locus in control populations and patients.[87]
A CTG trinucleotide repeat expansion in the 3’-untranslated region of the DMPK gene results in myotonic dystrophy type 1 (DM1). The DM1 protein kinase (DMPK) gene encodes for dystrophia myotonica protein kinase. While nearly all DM1 patients develop bilateral iridescent cataracts, studies have shown that 36%–46% of DM1 probands develop FECD.[88,89] In a screen of 317 FECD probands, one patient harbored a DMPK expansion without a prior diagnosis of DM1, suggesting that DMPK mutations contribute to the genetic burden of FECD but are rare. CUG repeat RNA foci co-localizing with MBNL1 have been observed in the corneal endothelial cells of a DM1 subject with FECD.[88]
Genome-wide association studies of large cohorts of FECD cases of European ancestry have shown significant associations with polymorphisms in other genes, in addition to a strong association with TCF4.[90] Apart from the various risk alleles associated with FECD, analysis of mitochondrial variants has suggested associations with mitochondrial markers having protective effects for FECD.[91]
Thus, from all the studies done so far, late-onset FECD appears to be highly genetically heterogeneous, and a small percentage (no more than 5%–10%) of patients have mutations at each of the known FECD loci such as ZEB1 and SLC4A11. The largest pathogenic impact among the known FECD loci is that of the TCF4 intronic repeat sequence, found in a substantial fraction of patients from different populations across the world. Since this repeat expansion is associated with gross mis-splicing and the presence of RNA foci, CRISPR-Cas approaches are being developed to target and block the repeat RNA[92] or cut the repeat DNA sequences by gene editing as well as by using antisense oligonucleotides to bind to and block the mutant RNA.[93,94]
A form of X-linked endothelial dystrophy has been reported in one family. The locus for endothelial dystrophy in a family of 60 members studied maps to chromosome Xq25.[95] The affected individuals displayed variable phenotypes, including corneal clouding, band keratopathy, and endothelial cells appearing like “moon craters”. Microscopy showed degeneration of the endothelium and thickening of the DM.
Summary and Clinical Relevance
Many recent advances in the genetics of corneal endothelial dystrophies have brought to light pathways and mechanisms underlying the development of these diseases and pointed to correlations between genotype and phenotype. Despite a high degree of genetic heterogeneity, particularly for PPCD and FECD, the prevalence of mutations in the existing genes are rapidly being defined in patients from different regions. The application of genome sequencing may further facilitate the identification of new loci or novel types of pathogenic changes in existing genes in the near future and enhance our understanding of the underlying genetics of these diseases. A significant corollary of the new developments in the field lies in the possibility of developing suitable new therapies for these disorders based on their known genetic and molecular mechanisms. Proof of concept has already been obtained for using specific approaches to inhibit the triplet repeat expansion–mediated disease pathways in FECD. Genetic screening may aid in establishing a genotype–phenotype correlation for patients. In a majority of cases, a meticulous slit-lamp examination and histological analysis wherever available help in determining the exact nature of the endothelial disease, although a diagnosis in patients with unusual manifestations can be supported by genetic testing. In familial forms of PPCD, knowing the mutation can aid in early screening and detection of affected but asymptomatic individuals. Developments in genetics have improved our knowledge of the corneal endothelial dystrophies and reduced the inaccuracies in their nomenclature. An accurate diagnosis of the specific type of endothelial dystrophy assists in planning the most appropriate management strategy and prognostication of the clinical condition. Furthermore, with the advent of alternatives such as pharmacotherapy and targeted molecular therapy in the management of endothelial dystrophies, a precise diagnosis of the clinical phenotype has become increasingly paramount.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
This work of C.K, S.C., and M.R. was supported by grants from the Science and Engineering Research Board and the Department of Biotechnology (Government of India). The authors (C.K, S.C., and M.R.) also acknowledge support from the Hyderabad Eye Research Foundation.
References
- 1.Tuft SJ, Coster DJ. The corneal endothelium. Eye. 1990;4:389–424. doi: 10.1038/eye.1990.53. [DOI] [PubMed] [Google Scholar]
- 2.Bourne WM. Biology of the corneal endothelium in health and disease. Eye. 2003;17:912–8. doi: 10.1038/sj.eye.6700559. [DOI] [PubMed] [Google Scholar]
- 3.McKusick-Nathans Institute of Genetic Medicine. Johns Hopkins University; Baltimore, MD: [Last accessed on 2022 Mar]. Online Mendelian Inheritance in Man, OMIM®. Available from:https://omim.org/ [Google Scholar]
- 4.Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, et al. IC3D classification of corneal dystrophies--edition 2. Cornea. 2015;34:117–59. doi: 10.1097/ICO.0000000000000307. [DOI] [PubMed] [Google Scholar]
- 5.Pearce WG, Tripathi RC, Morgan G. Congenital endothelial corneal dystrophy. Clinical, pathological, and genetic study. Br J Ophthalmol. 1969;53:577–91. doi: 10.1136/bjo.53.9.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkness CM, McCartney A, Rice NS, Garner A, Steele AD. Congenital hereditary corneal edema of Maumenee:Its clinical features, management, and pathology. Br J Ophthalmol. 1987;71:130–44. doi: 10.1136/bjo.71.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toma NM, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA, Bhattacharya SS. Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet. 1995;4:2395–98. doi: 10.1093/hmg/4.12.2395. [DOI] [PubMed] [Google Scholar]
- 8.Hand CK, Harmon DL, Kennedy SM, FitzSimon JS, Collum LM, Parfrey NA. Localization of the gene for autosomal recessive congenital hereditary endothelial dystrophy (CHED2) to chromosome 20 by homozygosity mapping. Genomics. 1999;61:1–4. doi: 10.1006/geno.1999.5920. [DOI] [PubMed] [Google Scholar]
- 9.Levenson JE, Chandler JW, Kaufman HE. Affected asymptomatic relatives in congenital hereditary endothelial dystrophy. Am J Ophthalmol. 1973;76:967–71. doi: 10.1016/0002-9394(73)90090-1. [DOI] [PubMed] [Google Scholar]
- 10.Kanai A, Waltman S, Polack FM, Kaufman HE. Electron microscopic study of hereditary corneal edema. Invest Ophthalmol. 1971;10:89–99. [PubMed] [Google Scholar]
- 11.Kanai A, Kaufman HE. Further electron microscopic study of hereditary corneal edema. Invest Ophthalmol. 1971;10:545–54. [PubMed] [Google Scholar]
- 12.Davidson AE, Liskova P, Evans CJ, Dudakova L, Nosková L, Pontikos N, et al. Autosomal-dominant corneal endothelial dystrophies CHED1 and PPCD1 are allelic disorders caused by non-coding mutations in the promoter of OVOL2. Am J Hum Genet. 2016;98:75–89. doi: 10.1016/j.ajhg.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wells J, Lee B, Cai AQ, Karapetyan A, Lee WJ, Rugg E, et al. Ovol2 suppresses cell cycling and terminal differentiation of keratinocytes by directly repressing c-Myc and Notch 1. J Biol Chem. 2009;284:29125–35. doi: 10.1074/jbc.M109.008847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe K, Liu Y, Noguchi S, Murray M, Chang JC, Kishima M, et al. OVOL2 induces mesenchymal-to-epithelial transition in fibroblasts and enhances cell-state reprogramming towards epithelial lineages. Sci Rep. 2019;9:6490. doi: 10.1038/s41598-019-43021-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2) Nat Genet. 2006;38:755–57. doi: 10.1038/ng1824. [DOI] [PubMed] [Google Scholar]
- 16.Jiao X, Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Gangopadhyay N, et al. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet. 2007;44:64–8. doi: 10.1136/jmg.2006.044644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramprasad VL, Ebenezer ND, Aung T, Rajagopal R, Yong VH, Tuft SJ, et al. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2) Mutation in brief #958 Online. Hum Mutat. 2007;28:522–3. doi: 10.1002/humu.9487. [DOI] [PubMed] [Google Scholar]
- 18.Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Kannabiran C. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Mol Vis. 2007;13:1327–32. [PubMed] [Google Scholar]
- 19.Hemadevi B, Veitia RA, Srinivasan M, Arunkumar J, Prajna NV, Lesaffre C, et al. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch Ophthalmol. 2008;126:700–8. doi: 10.1001/archopht.126.5.700. [DOI] [PubMed] [Google Scholar]
- 20.Aldahmesh MA, Khan AO, Meyer BF, Alkuraya FS. Mutational spectrum of SLC4A11 in autosomal recessive CHED in Saudi Arabia. Invest Ophthalmol Vis Sci. 2009;50:4142–5. doi: 10.1167/iovs.08-3006. [DOI] [PubMed] [Google Scholar]
- 21.Paliwal P, Sharma A, Tandon R, Sharma N, Titiyal JS, Sen S, et al. Congenital hereditary endothelial dystrophy-mutation analysis of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Mol Vis. 2010;16:2955–63. [PMC free article] [PubMed] [Google Scholar]
- 22.Kodaganur SG, Kapoor S, Veerappa AM, Tontanahal SJ, Sarda A, Yathish S, et al. Mutation analysis of the SLC4A11 gene in Indian families with congenital hereditary endothelial dystrophy 2 and a review of the literature. Mol Vis. 2013;19:1694–706. [PMC free article] [PubMed] [Google Scholar]
- 23.Desir J, Moya G, Reish O, Van Regemorter N, Deconinck H, David KL, et al. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J Med Genet. 2007;44:322–6. doi: 10.1136/jmg.2006.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker MD, Ourmozdi EP, Tanner MJ. Human BTR1, a new bicarbonate transporter superfamily member and human AE4 from kidney. Biochem Biophys Res Commun. 2001;282:1103–9. doi: 10.1006/bbrc.2001.4692. [DOI] [PubMed] [Google Scholar]
- 25.Park M, Li Q, Shcheynikov N, Zeng W, Muallem S. NaBC1 is a ubiquitous electrogenic Na+-coupled borate transporter essential for cellular boron homeostasis and cell growth and proliferation. Mol Cell. 2004;16:331–41. doi: 10.1016/j.molcel.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 26.Vilas GL, Morgan PE, Loganathan SK, Quon A, Casey JR. A biochemical framework for SLC4A11, the plasma membrane protein defective in corneal dystrophies. Biochemistry. 2011;50:2157–69. doi: 10.1021/bi101887z. [DOI] [PubMed] [Google Scholar]
- 27.Han SB, Ang HP, Poh R, Chaurasia SS, Peh G, Liu J, et al. Mice with a targeted disruption of Slc4a11 model the progressive corneal changes of congenital hereditary endothelial dystrophy. Invest Ophthalmol Vis Sci. 2013;54:6179–89. doi: 10.1167/iovs.13-12089. [DOI] [PubMed] [Google Scholar]
- 28.Roy S, Praneetha DC, Vendra VP. Mutations in the corneal endothelial dystrophy-associated gene SLC4A11 render the cells more vulnerable to oxidative insults. Cornea. 2015;34:668–74. doi: 10.1097/ICO.0000000000000421. [DOI] [PubMed] [Google Scholar]
- 29.Krachmer JH. Posterior polymorphous corneal dystrophy:A disease characterized by epithelial-like endothelial cells which influence management and prognosis. Trans Am Ophthalmol Soc. 1985;83:413–75. [PMC free article] [PubMed] [Google Scholar]
- 30.Chaurasia S, Mittal R, Bichappa G, Ramappa M, Murthy SI. Clinical characterization of posterior polymorphous corneal dystrophy in patients of Indian ethnicity. Int Ophthalmol. 2017;37:945–52. doi: 10.1007/s10792-016-0360-y. [DOI] [PubMed] [Google Scholar]
- 31.Aldave AJ, Ann LB, Frausto RF, Nguyen CK, Yu F, Raber IM. Classification of posterior polymorphous corneal dystrophy as a corneal ectatic disorder following confirmation of associated significant corneal steepening. JAMA Ophthalmol. 2013;131:1583. doi: 10.1001/jamaophthalmol.2013.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7. doi: 10.1186/1750-1172-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Héon E, Mathers WD, Alward WL, Weisenthal RW, Sunden SL, Fishbaugh JA, et al. Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol Genet. 1995;4:485–8. doi: 10.1093/hmg/4.3.485. [DOI] [PubMed] [Google Scholar]
- 34.Gwilliam R, Liskova P, Filipec M, Kmoch S, Jirsova K, Huckle EJ, et al. Posterior polymorphous corneal dystrophy in Czech families maps to chromosome 20 and excludes the VSX1 gene. Invest Ophthalmol Vis Sci. 2005;46:4480–4. doi: 10.1167/iovs.05-0269. [DOI] [PubMed] [Google Scholar]
- 35.Yellore VS, Papp JC, Sobel E, Khan MA, Rayner SA, Farber DB, et al. Replication and refinement of linkage of posterior polymorphous corneal dystrophy to the posterior polymorphous corneal dystrophy 1 locus on chromosome 20. Genet Med. 2007;9:228–34. doi: 10.1097/gim.0b013e31803c4dc2. [DOI] [PubMed] [Google Scholar]
- 36.Liskova P, Gwilliam R, Filipec M, Jirsova K, Reinstein Merjava S, et al. High prevalence of posterior polymorphous corneal dystrophy in the Czech Republic;linkage disequilibrium mapping and dating an ancestral mutation. PLoS One. 2012;7:e45495. doi: 10.1371/journal.pone.0045495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung DD, Frausto RF, Cervantes AE, Gee KM, Zakharevich M, Hanser EM, et al. Confirmation of the OVOL2 promoter mutation c.-307T>C in posterior polymorphous corneal dystrophy 1. PLoS One. 2017;12:e0169215. doi: 10.1371/journal.pone.0169215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R, Cousin P, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10:2415–23. doi: 10.1093/hmg/10.21.2415. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi A, Fujiki K, Murakami A, Kato T, Chen LZ, Onoe H, et al. Analysis of COL8A2 gene mutation in Japanese patients with Fuchs'endothelial dystrophy and posterior polymorphous dystrophy. Jpn J Ophthalmol. 2004;48:195–8. doi: 10.1007/s10384-003-0063-6. [DOI] [PubMed] [Google Scholar]
- 40.Yellore VS, Rayner SA, Emmert-Buck L, Tabin GC, Raber I, Hannush SB, et al. No pathogenic mutations identified in the COL8A2 gene or four positional candidate genes in patients with posterior polymorphous corneal dystrophy. Invest Ophthalmol Vis Sci. 2005;46:1599–603. doi: 10.1167/iovs.04-1321. [DOI] [PubMed] [Google Scholar]
- 41.Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA, et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005;77:694–708. doi: 10.1086/497348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aldave AJ, Yellore VS, Yu F, Bourla N, Sonmez B, Salem AK, et al. Posterior polymorphous corneal dystrophy is associated with TCF8 gene mutations and abdominal hernia. Am J Med Genet A. 2007;143A:2549–56. doi: 10.1002/ajmg.a.31978. [DOI] [PubMed] [Google Scholar]
- 43.Liskova P, Tuft SJ, Gwilliam R, Ebenezer ND, Jirsova K, Prescott Q, et al. Novel mutations in the ZEB1 gene identified in Czech and British patients with posterior polymorphous corneal dystrophy. Hum Mutat. 2007;28:638. doi: 10.1002/humu.9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liskova P, Evans CJ, Davidson AE, Zaliova M, Dudakova L, Trkova M, et al. Heterozygous deletions at the ZEB1 locus verify haploinsufficiency as the mechanism of disease for posterior polymorphous corneal dystrophy type 3. Eur J Hum Genet. 2016;24:985–91. doi: 10.1038/ejhg.2015.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bakhtiari P, Frausto RF, Roldan AN, Wang C, Yu F, Aldave AJ. Exclusion of pathogenic promoter region variants and identification of novel nonsense mutations in the zinc finger E-box binding homeobox 1 gene in posterior polymorphous corneal dystrophy. Mol Vis. 2013;19:575–80. [PMC free article] [PubMed] [Google Scholar]
- 46.Lechner J, Dash DP, Muszynska D, Hosseini M, Segev F, George S, et al. Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation. Invest Ophthalmol Vis Sci. 2013;54:3215–23. doi: 10.1167/iovs.13-11781. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen DQ, Hosseini M, Billingsley G, Héon E, Churchill AJ. Clinical phenotype of posterior polymorphous corneal dystrophy in a family with a novel ZEB1 mutation. Acta Ophthalmol. 2010;88:695–9. doi: 10.1111/j.1755-3768.2009.01511.x. [DOI] [PubMed] [Google Scholar]
- 48.Jang MS, Roldan AN, Frausto RF, Aldave AJ. Posterior polymorphous corneal dystrophy 3 is associated with agenesis and hypoplasia of the corpus callosum. Vision Res. 2014;100:88–92. doi: 10.1016/j.visres.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaudhry A, Chung BH, Stavropoulos DJ, Araya MP, Ali A, Heon E, et al. Agenesis of the corpus callosum, developmental delay, autism spectrum disorder, facial dysmorphism, and posterior polymorphous corneal dystrophy associated with ZEB1 gene deletion. Am J Med Genet A. 2017;73:2467–71. doi: 10.1002/ajmg.a.38321. [DOI] [PubMed] [Google Scholar]
- 50.Takagi T, Moribe H, Kondoh H, Higashi Y. DeltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development. 1998;125:21–31. doi: 10.1242/dev.125.1.21. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Peng X, Tan J, Darling DS, Kaplan HJ, Dean DC. Zeb1 mutant mice as a model of posterior corneal dystrophy. Invest Ophthalmol Vis Sci. 2008;49:1843–9. doi: 10.1167/iovs.07-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chung DD, Frausto RF, Lin BR, Hanser EM, Cohen Z, Aldave AJ. Transcriptomic profiling of posterior polymorphous corneal dystrophy. Invest Ophthalmol Vis Sci. 2017;58:3202–14. doi: 10.1167/iovs.17-21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liskova P, Dudakova L, Evans CJ, Rojas Lopez KE, Pontikos N, Athanasiou D, et al. Ectopic GRHL2 expression due to non-coding mutations promotes cell state transition and causes posterior polymorphous corneal dystrophy 4. Am J Hum Genet. 2018;102:447–59. doi: 10.1016/j.ajhg.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chung DD, Zhang W, Jatavallabhula K, Barrington A, Jung J, Aldave AJ. Alterations in GRHL2-OVOL2-ZEB1 axis and aberrant activation of Wnt signaling lead to altered gene transcription in posterior polymorphous corneal dystrophy. Exp Eye Res. 2019;188:107696. doi: 10.1016/j.exer.2019.107696. doi:10.1016/j.exer.2019.107696. [DOI] [PubMed] [Google Scholar]
- 56.Soh YQ, Kocaba V, Pinto M, Mehta JS. Fuchs endothelial corneal dystrophy and corneal endothelial diseases:East meets west. Eye (Lond) 2020;34:427–41. doi: 10.1038/s41433-019-0497-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krachmer JH, Purcell JJ, Jr, Young CW, Bucher KD. Corneal endothelial. A study of 64 families. Arch Ophthalmol. 1978;96:2036–9. doi: 10.1001/archopht.1978.03910060424004. [DOI] [PubMed] [Google Scholar]
- 58.Chaurasia S, Ramappa M. Fuchs endothelial corneal dystrophy in a child. Cornea. 2017;36:e17–8. doi: 10.1097/ICO.0000000000001188. [DOI] [PubMed] [Google Scholar]
- 59.Gottsch JD, Sundin OH, Liu SH, Jun AS, Broman KW, Stark WJ, et al. Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of Fuchs corneal dystrophy. Invest Ophthalmol Vis Sci. 2005;46:1934–39. doi: 10.1167/iovs.04-0937. [DOI] [PubMed] [Google Scholar]
- 60.Aldave AJ, Rayner SA, Salem AK, Yoo GL, Kim BT, Saeedian M, et al. No pathogenic mutations identified in the COL8A1 and COL8A2 genes in familial Fuchs corneal dystrophy. Invest Ophthalmol Vis Sci. 2006;47:3787–90. doi: 10.1167/iovs.05-1635. [DOI] [PubMed] [Google Scholar]
- 61.Jun AS, Meng H, Ramanan N, Matthaei M, Chakravarti S, Bonshek R, et al. An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis. Hum Mol Genet. 2012;21:384–93. doi: 10.1093/hmg/ddr473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meng H, Matthaei M, Ramanan N, Grebe R, Chakravarti S, Speck CL, et al. L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy. Invest Ophthalmol Vis Sci. 2013;54:1887–97. doi: 10.1167/iovs.12-11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehta JS, Vithana EN, Tan DT, Yong VH, Yam GH, Law RW, et al. Analysis of the posterior polymorphous corneal dystrophy 3 gene, TCF8, in late-onset Fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2008;49:184–8. doi: 10.1167/iovs.07-0847. [DOI] [PubMed] [Google Scholar]
- 64.Riazuddin SA, Zaghloul NA, Al-Saif A, Davey L, Diplas BH, Meadows DN, et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010;86:45–53. doi: 10.1016/j.ajhg.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta R, Kumawat BL, Paliwal P, Tandon R, Sharma N, Sen S, et al. Association of ZEB1 and TCF4 rs613872 changes with late onset Fuchs endothelial corneal dystrophy in Northern India. Mol Vis. 2015;21:1252–60. [PMC free article] [PubMed] [Google Scholar]
- 66.Baratz KH, Tosakulwong N, Ryu E, Brown WL, Branham K, Chen W, et al. E2-2 protein and Fuchs's corneal dystrophy. N Engl J Med. 2010;363:1016–24. doi: 10.1056/NEJMoa1007064. [DOI] [PubMed] [Google Scholar]
- 67.Li YJ, Minear MA, Rimmler J, Zhao B, Balajonda E, Hauser MA, et al. Replication of TCF4 through association and linkage studies in late-onset Fuchs endothelial corneal dystrophy. PLoS One. 2011;6:e18044. doi: 10.1371/journal.pone.0018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riazuddin SA, McGlumphy EJ, Yeo WS, Wang J, Katsanis N, Gottsch JD. Replication of the TCF4 intronic variant in late-onset Fuchs corneal dystrophy and evidence of independence from the FCD2 locus. Invest Ophthalmol Vis Sci. 2011;52:2825–9. doi: 10.1167/iovs.10-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nanda GG, Padhy B, Samal S, Das S, Alone DP. Genetic association of TCF4 intronic polymorphisms, CTG18.1 and rs17089887, with Fuchs'endothelial corneal dystrophy in an Indian population. Invest Ophthalmol Vis Sci. 2014;55:7674–80. doi: 10.1167/iovs.14-15297. [DOI] [PubMed] [Google Scholar]
- 70.Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012;7:e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mootha VV, Gong X, Ku HC, Xing C. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs'endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014;55:33–42. doi: 10.1167/iovs.13-12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xing C, Gong X, Hussain I, Khor CC, Tan DT, Aung T, et al. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs'corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci. 2014;55:7073–8. doi: 10.1167/iovs.14-15390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakano M, Okumura N, Nakagawa H, Koizumi N, Ikeda Y, Ueno M, et al. Trinucleotide repeat expansion in the TCF4 gene in Fuchs'endothelial corneal dystrophy in Japanese. Invest Ophthalmol Vis Sci. 2015;56:4865–9. doi: 10.1167/iovs.15-17082. [DOI] [PubMed] [Google Scholar]
- 74.Rao BS, Tharigopala A, Rachapalli SR, Rajagopal R, Soumittra N. Association of polymorphisms in the intron of TCF4 gene to late-onset Fuchs endothelial corneal dystrophy:An Indian cohort study. Indian J Ophthalmol. 2017;65:931–5. doi: 10.4103/ijo.IJO_191_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Foja S, Luther M, Hoffmann K, Rupprecht A, Gruenauer-Kloevekorn C. CTG18.1 repeat expansion may reduce TCF4 gene expression in corneal endothelial cells of German patients with Fuchs'dystrophy. Graefes Arch Clin Exp Ophthalmol. 2017;255:1621–31. doi: 10.1007/s00417-017-3697-7. [DOI] [PubMed] [Google Scholar]
- 76.Kuot A, Hewitt AW, Snibson GR, Souzeau E, Mills R, Craig JE, et al. TGC repeat expansion in the TCF4 gene increases the risk of Fuchs'endothelial corneal dystrophy in Australian cases. PLoS One. 2017;12:e0183719. doi: 10.1371/journal.pone.0183719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Du J, Aleff RA, Soragni E, Kalari K, Nie J, Tang X, et al. RNA toxicity and missplicing in the common eye disease Fuchs endothelial corneal dystrophy. J Biol Chem. 2015;290:5979–90. doi: 10.1074/jbc.M114.621607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mootha VV, Hussain I, Cunnusamy K, Graham E, Gong X, Neelam S, et al. CF4 Triplet repeat expansion and nuclear RNA foci in Fuchs'endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2015;56:2003–11. doi: 10.1167/iovs.14-16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rong Z, Hu J, Corey DR, Mootha VV. Quantitative studies of Muscleblind proteins and their interaction with TCF4 RNA foci support involvement in the mechanism of Fuchs'dystrophy. Invest Ophthalmol Vis Sci. 2019;60:3980–91. doi: 10.1167/iovs.19-27641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chu Y, Hu J, Liang H, Kanchwala M, Xing C, Beebe W, et al. Analyzing pre-symptomatic tissue to gain insights into the molecular and mechanistic origins of late-onset degenerative trinucleotide repeat disease. Nucleic Acids Res. 2020;48:6740–58. doi: 10.1093/nar/gkaa422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH, Venkataraman D, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008;17:656–66. doi: 10.1093/hmg/ddm337. [DOI] [PubMed] [Google Scholar]
- 82.Soumittra N, Loganathan SK, Madhavan D, Ramprasad VL, Arokiasamy T, Sumathi S, et al. Biosynthetic and functional defects in newly identified SLC4A11 mutations and absence of COL8A2 mutations in Fuchs endothelial corneal dystrophy. J Hum Genet. 2014;59:444–53. doi: 10.1038/jhg.2014.55. [DOI] [PubMed] [Google Scholar]
- 83.Hemadevi B, Srinivasan M, Arunkumar J, Prajna NV, Sundaresan P. Genetic analysis of patients with Fuchs endothelial corneal dystrophy in India. BMC Ophthalmol. 2010;10:3. doi: 10.1186/1471-2415-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Minear MA, Li YJ, Rimmler J, Balajonda E, Watson S, Allingham RR, et al. Genetic screen of African Americans with Fuchs endothelial corneal dystrophy. Mol Vis. 2013;19:2508–16. [PMC free article] [PubMed] [Google Scholar]
- 85.Chaurasia S, Ramappa M, Annapurna M, Kannabiran C. Coexistence of congenital hereditary endothelial dystrophy and Fuchs endothelial corneal dystrophy associated with SLC4A11 mutations in affected families. Cornea. 2020;39:354–7. doi: 10.1097/ICO.0000000000002183. [DOI] [PubMed] [Google Scholar]
- 86.Riazuddin SA, Vasanth S, Katsanis N, Gottsch JD. Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am J Hum Genet. 2013;93:758–64. doi: 10.1016/j.ajhg.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Riazuddin SA, Parker DS, McGlumphy EJ, Oh EC, Iliff BW, Schmedt T, et al. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am J Hum Genet. 2012;90:533–9. doi: 10.1016/j.ajhg.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mootha VV, Hansen B, Rong Z, Mammen PP, Zhou Z, Xing C, et al. Fuchs'endothelial corneal dystrophy and RNA foci in patients with myotonic dystrophy. Invest Ophthalmol Vis Sci. 2017;58:4579–85. doi: 10.1167/iovs.17-22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Winkler NS, Milone M, Martinez-Thompson JM, Raja H, Aleff RA, Patel SV, et al. Fuchs'endothelial corneal dystrophy in patients with myotonic dystrophy, type 1. Invest Ophthalmol Vis Sci. 2018;59:3053–7. doi: 10.1167/iovs.17-23160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Afshari NA, Igo RP, Jr, Morris NJ, Stambolian D, Sharma S, Pulagam VL, et al. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat Commun. 2017;8:14898. doi: 10.1038/ncomms14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y-J, Minear MA, Qin X, Rimmler J, Hauser MA, Allingham RR, et al. Mitochondrial polymorphism A10398G and haplogroup I are associated with Fuchs'endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014;55:4577–84. doi: 10.1167/iovs.13-13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rong Z, Gong X, Hulleman JD, Corey DR, Mootha VV. Trinucleotide repeat-targeting dCas9 as a therapeutic strategy for Fuchs'endothelial corneal dystrophy. Trans Vis Sci Tech. 2020;9:47. doi: 10.1167/tvst.9.9.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu J, Rong Z, Gong X, Zhou Z, Sharma VK, Xing C, et al. Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs'. dystrophy Hum Mol Genet. 2018;27:1015–26. doi: 10.1093/hmg/ddy018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zarouchlioti C, Sanchez-Pintado B, Hafford Tear NJ, Klein P, Liskova P, Dulla K, et al. Antisense therapy for a common corneal dystrophy ameliorates TCF4 repeat expansion-mediated toxicity. Am J Hum Genet. 2018;102:528–39. doi: 10.1016/j.ajhg.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmid E, Lisch W, Philipp W, Lechner S, Göttinger W, Schlötzer-Schrehardt U, et al. A new, X-linked endothelial corneal dystrophy. Am J Ophthalmol. 2006;141:478–87. doi: 10.1016/j.ajo.2005.10.020. [DOI] [PubMed] [Google Scholar]