

Abstract
Homocystinuria is a rare metabolic inborn disorder caused due to dysfunctional cystathionine β-synthase (CBS) enzyme activity, thus resulting in elevated levels of methionine and homocysteine in the blood and urine. The timely recognition of this rare metabolic disorder and prompt methionine-restricted diet are crucial in lessening the systemic consequences. The recalcitrant cases have a higher risk for cardiovascular diseases, neurodegenerative diseases, neural tube defects, and other severe clinical complications. This review aims to present the ophthalmic spectrum of homocystinuria and its molecular basis, the disease management, as well as the current and potential treatment approaches with a greater emphasis on preventive strategies.
Keywords: Cystathionine β-synthase, ectopia lentis, genetics, homocystinuria
Homocystinuria is a rare inborn metabolic disorder caused due to dysfunctional cystathionine β-synthase (CBS) enzyme activity, thus resulting in elevated levels of methionine and homocysteine in the blood and urine. It is more common in males and has a worldwide prevalence of 1 in 344,000.[1,2] Affected infants may have growth failure or may grow longer too fast and often have developmental delays and failure to thrive. Additionally, specific symptoms become apparent by the age of 3 years. These include ectopia lentis, iridodonesis, and high myopia. Affected individuals tend to be thin with tall stature, arachnodactyly, and Marfanoid features, because of which they are misdiagnosed as having Marfan’s syndrome.[3,4,5] This build-up could lead to several complications such as nearsightedness and dislocated eye lenses, besides a variety of neuropsychiatric disorders and vascular complications.[6]
Homocystinuria is categorized based on the site of the metabolic defect. Type 1 is due to deficiency in cystathionine β-synthase (CBS), which converts homocysteine to cystathionine. Type 2 is because of defective methylcobalamin synthesis, which converts homocysteine to methionine (Met). Type 3 is due to an abnormality in methylenetetrahydrofolate reductase, which also helps to convert homocysteine to Met [Fig. 1].
Figure 1.
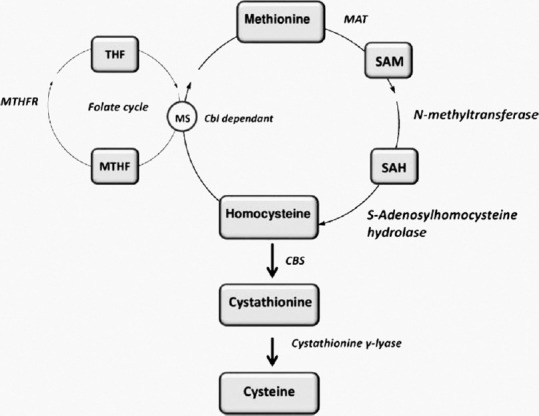
Homocystinuria pathway
The CBS gene is located on the long arm of chromosome 21.[7] More than 191 CBS mutations have been documented to date. Out of these, approximately 87% are missense mutations. However, they do not affect the CBS catalytic site.[8] Instead, they result in an unstable misfolded protein with abnormal biological function. The two most frequently reported mutations worldwide are p.G307S (31%) and p.I278T (24%).[9] The p.G307S mutation is seen more commonly in Ireland and Australia.[10] The mutation is traced to exon 8 of the CBS gene, replacing guanine at position 919 with adenine. This leads to the substitution of serine in place of glycine at position 307. Homozygous patients are usually nonresponsive to pyridoxine (B6) treatment and are severely affected.[11,12] It is also seen that this mutation is stable, inactive, and responds neither to chaperone-based therapy nor to pyridoxine treatment.[13] Similarly, the p.I278T mutation affects the catalytic domain of the CBS enzyme.[14] However, it still confers responsiveness to pyridoxine treatment.[15] The p. 1278T mutation was first identified in a pyridoxine-responsive patient with mild clinical manifestations.[16] This mutation results from incorrect excision of a 68-bp repeat polymorphism within the CBS gene.[17] Consequently, it substitutes thymine with a cytosine at position 833 of exon 8. This results in substitution of isoleucine with threonine at position 278.
Interestingly, these two most prevalent mutations found in the world were not seen in the Indian population in a study on 14 cases of homocystinuria by Kaur et al.[18] They found a total of 15 different variations, out of which seven were novel variations with a spectrum of frame-shift deletions (c.566_567del, c.402del, c.1217del), nonsense variants (c. 825C>A, c.1397C>A), missense mutation (c.362G>C), and a splicing variant (c.736+2T>C). The eight variants, c.1136G>A c.785C>T, c.430G>A c.19del, c.518_520del, c.992C>T, c.982G>A, and c.1642C>T, had already been reported from other population cohorts.
Some novel mutations identified recently are a missense change (c. 467T > C; p.L156P) and an in-frame deletion (c. 808_810del; p.E270del) reported in Pakistani children and eight novel mutations in CBS in Chinese patients. These were identified using Sanger sequencing.
Ocular Complications
Ectopia lentis
The main ocular feature associated with homocystinuria is ectopia lentis.[19] There is an alteration of self-interaction properties because of homocysteinylation of fibrillin-1 due to a reduction of disulfide-bonded C-terminal fibrillin-1 multimers.[20] This leads to weak zonules and progressive ectopia lentis. The lens dislocation is invariably bilateral, and inferonasal in 90% patients; the prevalence is 30% in infancy and increases to 80% by age 15 years [Fig. 2]. The zonules are sometimes absent compared to the stretched zonules in Marfan’s syndrome. This can lead to the pathognomonic vitreous prolapse seen in some patients.
Figure 2.

Classical ectopia lentis in homocystinuria
Mulvihill et al.,[21] in their study, have shown that ectopia lentis in homocystinuria develops after 1 year and maximum by 8 years of age.
In a study by Burke et al.,[22] 19 homocystinuria patients were studied. The ophthalmic findings in each patient were correlated concerning the biochemical control as reflected by mean plasma homocysteine and cysteine levels since the beginning of therapy. It was concluded that ectopia lentis progressed despite tight biochemical control in late detected cases.
Taylor et al.[23] divided the patients into two groups. Group A included those patients who were diagnosed by newborn screening programs. Group B patients were diagnosed by investigations of various clinical manifestations; thus, treatment was commenced following diagnosis. Group A patients had a mean age of 10.8 years. Nine were pyridoxine responsive.
Patients had 6/6 vision in each eye; eight were myopic. The mean age of group B patients was 19.4 years. Five patients were pyridoxine responsive. In five children, increasing myopia was an early abnormality. In five patients, the diagnosis was made following dislocation of the lens. Only four patients had 6/6 vision in each eye. Hua et al.[24] described a case of a 14-year-old boy with CBS deficiency presenting with recurrent dislocation of lens. A previously unreported genetic variant was identified, that is, c. 697 T > G (p. W233N). The lens was present in the anterior chamber binocularly. The intraocular pressure was 45 mmHg in the left eye with conjunctival injection and 16 mmHg in the right eye. In the right eye, the lens was present in the inferior vitreous cavity with a strand of elongated zonular fibers at 12 o’clock position to the ciliary body. Whole-exome sequencing and Sanger sequencing for the genetic variants were performed. The result showed mutations in the CBS gene at two distinct positions.
Mulvihill et al.,[21] in their study, highlighted the importance of early diagnosis and good control. They found that all 14 late-diagnosed patients had lens subluxation or dislocation at diagnosis. Six patients were in poor biochemical control, out of which two patients developed phacodonesis. Since birth, the remaining 15 patients with good biochemical control of their Homocystinuria (HCU) had no evidence of phacodonesis or lens subluxation.
Regarding the management of lens subluxation, controversy exists as to the most effective mode of treatment.[25] Prophylactic iridectomy has been suggested to prevent pupillary block glaucoma.[26] Some recommend conservative methods like mydriasis with a supine patient to redislocate the lens posteriorly. Miotics and a peripheral iridectomy follow this to avoid recurrent anterior lenticular dislocation and pupillary block, respectively.[27]
Harrison et al.[28] conducted a study to evaluate the role of conservative versus surgical treatment in ophthalmic complications of homocystinuria. The majority of ophthalmic problems are due to lenticular instability. They concluded that complications could not be prevented by conservative prophylactic treatment methods alone as majority of patients eventually required lensectomy.
High myopia
Cruysberg et al.[29] stressed the need for early diagnosis. The mean age of diagnosis was 24 years. High myopia of more than 5 D is extremely rare in children under 5 years[30] and probably affects under 2% of adults.[31] In such high myopia, an abnormality of one particular ocular refraction (i.e., axial length, corneal curvature, or the lens) is most likely.[32] Thus, if the fundus of these patients shows no clear signs of axial myopia and the corneal curvature is also normal, then lenticular myopia due to ectopia lentis should be suspected and searched for. Mulvihill et al.[21] found that all patients with lens subluxation had high degrees of myopia. The six patients with poor biochemical control were all myopic, with a mean spherical equivalent refraction of –6.4 D per eye. The remaining 15 patients had good biochemical control of their HCU since birth. Seven of these patients were emmetropic, four were hyperopic, and four were myopic. Patricia et al.[33] observed 10 patients with HCU (20 eyes). The most common abnormalities were ectopia lentis (n = 20) and myopia (n = 9).
Mulvihill et al.[34] divided the patients into three groups. With no ocular pathology (28 eyes), Group I had a mean refractive error of − 0.25 D (spherical equivalent). Group II, with phacodonesis or lens subluxation (12 eyes), had a mean refractive error of − 10.7 D. Phacodonesis was associated with simple myopia and lens subluxation with marked myopic astigmatism. Group III included patients with complete lens dislocation in at least one eye (12 eyes) and was optically aphakic with a mean refractive error of + 12.9 D. A significantly longer mean axial length was found in this group. Thus, they concluded that the axial length is significantly increased in individuals with homocystinuria and lens dislocation.
The development mechanism of such high myopia can be explained as being due to release of zonular tension from degeneration of zonular fibers, which causes spherophakia,[35] or because of the enlarged globe. In addition, patients also develop early loss of accommodation due to zonular weakness.
Retinal degeneration and detachment
Burke et al.[22] found benign peripheral cystoid retinal degeneration extending circumferentially to the globe equator in 31% of patients, although no case progressed to degenerative retinoschisis. This has been further supported based on histologic studies by Ramsey et al.[36] In addition to ectopia lentis, they suggest that peripheral cystoid retinal degeneration may also be a characteristic clinical finding in some cases of homocystinuria. Another rarely reported complication is retinal detachment.
Secondary glaucoma
The development of pupillary block glaucoma may be attributed to the anterior movement of the lens, and the mechanism of development is far from clear. The conditions for pupillary block may develop if lens with some zonular support is in a semi-fixed, but anterior position behind the iris. A second possible cause of pupillary block glaucoma is increased zonular dialysis, and the lens tends to become rounder.[28]
Larsen et al.[37] described a rare case of phacolytic glaucoma seen in a homocystinuria patient. The patient’s Intra ocular Pressure (IOP) was 38 mmHg in the right eye and 40 mmHg in the left eye. An anterior segment examination found trace conjunctival injection and 1+ anterior chamber cells in both the eyes. On slit-lamp examination, the patient appeared to be aphakic, and gonioscopy was open in both eyes. Trace cells were found in the vitreous. The optic nerve exhibited slightly enlarged cupping in both eyes. Based on the history and examination findings, phacolytic glaucoma was diagnosed.
Other ocular complications
The explanation for development of stromal opacification or bullous keratopathy seen in some patients could be the anterior movement of the lens, which may traumatize the endothelium.
The cases of iris atrophy[28] seen in 21% of eyes in the study by Harrison et al. could be attributed to embolism or thrombosis in the greater arterial circle of the iris.[34] Another report found scleral thickening, which was the result of disrupted normal collagen architecture and deposition of a mucopolysaccharide.[38]
Systemic Features
The affected individuals tend to be tall (Marfanoid habitus with arachnodactyly) and have blonde hair, osteoporosis, increased risk of fractures, pectus excavatum, and scoliosis. The individuals tend to have learning and intellectual disability (50%), seizures, and psychiatric problems. There are increased chances of cardiomegaly and platelet abnormality with hypercoagulability (thromboembolism). Also, 75% of the affected patients die by the age of 30 years. The systemic and ophthalmic features seen in homocystinuria can be distinguished from other similar clinical entities [Table 1].
Table 1.
Differentiating features of homocystinuria from other similar clinical entities
| Disease | Systemic features | Ocular features |
|---|---|---|
| Marfan’s syndrome | Joint hypermobility | Ectopia lentis, myopia, retinal detachment, cataract, glaucoma, amblyopia, strabismus |
| Pectus excavatum, Pes planus, scoliosis, Mitral Valve Prolapse (MVP), pneumothorax, aortic root dilation, increased arm span: height ratio, skin striae | ||
| Ehlers-Danlos syndrome | Skin hyperextensibility, fragility, joint hypermobility, scoliosis, hip dislocation, cigarette paper scars on the skin, spontaneous rupture of medium- and large-sized arteries | Ectopia lentis, myopia, blue sclera, retinal detachment, globe rupture secondary to trauma |
| Weill-Marchesani syndrome | Short stature, brachydactyly, joint stiffness | Microspherophakia and lens dislocation |
| Hyperlysinemia | Intellectual disability, behavioral problems, cognitive impairment | Ectopia lentis |
| Sulfite oxidase deficiency | Encephalopathy, seizures, opisthotonus, microcephaly, dystonia, ataxia, developmental regression | Ectopia lentis, elongated palpebral fissures, cortical blindness |
| Aniridia | Behavioral problems, developmental delays, glucose intolerance | Nystagmus, glaucoma, cataract |
| Congenital glaucoma | Watering, photophobia, blepharospasm, scleral thinning, buphthalmos, Haab’s striae, subluxated lens, disk cupping | |
| Ectopia lentis et pupillae | Lens dislocation, iris abnormalities, refractive error, cataract, raised IOP, Retinal Detachment (RD) |
Diagnostic Tests
Two indices of plasma homocysteine levels have been widely used, free homocysteine (fHcy) and total homocysteine (tHcy); the latter is the sum of free and bound homocysteine. The amino acid (AA) analyzer is used to measure plasma fHcy and is found to be an insensitive marker as it only becomes detectable once the tHcy exceeds approximately 50–60 μmol/L.[39] Thus, determination of plasma fHcy is no longer recommended. Detection of urinary disulfide homocysteine (called the cyanide nitroprusside test) is even less sensitive as this only becomes detectable once the plasma tHcy exceeds approximately 150 μmol/L.[39]
Further biochemical support for CBS deficiency can be obtained by analyzing plasma Met and cystathionine. Patients with CBS deficiency have low cystathionine (reference range 0.05–0.08 and 0.35–0.5 µµol/L) and high Met concentrations (reference range 12–15 and 40–45 μmol/L) with a grossly abnormal ratio of these two metabolites.
The intake of pyridoxine is the major confounder that may alter the test results. Decreases in tHcy concentration occur after administration of pharmacological doses of pyridoxine in a substantial proportion of CBS-deficient patients.[40,41] It is important to avoid intake of any pyridoxine supplements for at least 2 weeks before sampling plasma for tHcy measurement.[42]
The gold standard test to confirm CBS deficiency is by using radioactive or deuterium-labeled substrates and measuring cystathionine production from homocysteine and serine in cultured fibroblasts.[43,44]
Genetic testing
Genetic testing is important in homocystinuria. Because this condition results from a genetic mutation, it is not possible to prevent it. Genetic testing gives people an idea of their risk of passing on a gene mutation that can lead to homocystinuria.
The gold standard in molecular diagnostics is CBS gene sequencing. However, there are certain technical pitfalls, like the polymerase chain reaction (PCR) may amplify only one of the parental alleles, fail to detect larger rearrangements, or sequence portions of the gene inadequately.
Interpreting the results of mutation analysis is straightforward if the genetic variant is already known. However, interpretation of novel variants detected in genomic DNA may be difficult. Targeted mutation analysis is done to detect heterozygotes among individuals in whom genetic variants are segregating in the family.
Some of the genetic tests available in India are MGM084, a homocystinuria gene panel, MGM272, a clinical exome of 26 MB (80–100×), and MGM274, which carries out whole-exome sequencing (80–100×). The method used is next-generation sequencing, with Illumina being the sequencing platform. The type of sample taken can be peripheral blood or purified genomic DNA or chorionic villus sample or amniotic fluid or dried blood spots or product of conception.
Prenatal diagnosis
Targeted mutation analysis in native or cultured chorionic villi or amniocytes can be performed if the mutations are already known in the family and both parents. If the mutations are unknown (or known in only one parent), CBS activity is determined in cultured amniocytes.
Newborn screening
The median Met concentration of 103 μmol/L in CBS-deficient patients is substantially higher than the median of 20 μmol/L in healthy neonates.[45] The sensitivity of Met for detecting CBS deficiency is not precisely known, and 20%–50% of pyridoxine nonresponsive cases may be missed.[2,46] In fact, it is suggested that screening detects even fewer pyridoxine-responsive cases. Reducing the Met cut-off concentrations increases the detection rate. Thus, to increase sensitivity, Met cut-off values of 39 and 50 μmol/L are now proposed.[47]
Elevated blood Met concentrations occur not only in CBS deficiency, but also in liver disease, methionine adenosyltransferase I/III deficiency, and several other inborn errors of metabolism.[48] The low specificity of Met as the first-tier analyte can be substantially improved by quantifying tHcy as a second-tier marker.[47,48] This approach can reduce the false-positive rate fivefold.[47] In addition, calculating the Met/tHcy ratio may discriminate between CBS and Methionine Adenosyl Transferase (MAT)I/III deficiency.
Treatment
Generally, treatment of homocystinuria patients with varying missense mutations is based on a combined high dose of cofactors of homocysteine metabolism and a lifelong Met-restricted diet.
Patients are said to be pyridoxine responsive if, after initiation of treatment, the plasma tHcy becomes less than 50 µµol/L or fHcy is not detected or is less than 10 µµol/L or total fHcy (i.e., twice the homocysteine concentration plus homocysteine–cysteine disulfide concentration) is less than 20 μmol/L.
Pyridoxine doses of 10–40 mg can achieve biochemical targets in some patients with the p.P49L and p.I278T mutations.[49] Pyridoxine doses of 200 mg/day or less can achieve the biochemical targets in many other patients. Higher doses and additional treatment are required in partially pyridoxine-responsive patients. There is no evidence to support that pyridoxine has beneficial effects independent of lowering Homocysteine (Hcy) concentrations in patients unresponsive to pyridoxine.
Dietary management of CBS deficiency can be highly successful and should be considered for all pyridoxine-unresponsive and partially pyridoxine-responsive patients.[41] Restricting intake of the essential AA, Met, reduces the precursor load on the trans-sulfuration pathway, thereby reducing Hcy production.
Discussion
Homocystinuria, a rare metabolic disorder, causes a variety of ocular complications. Often, it goes unnoticed at birth, and the individuals first present to the ophthalmologist with high myopia. The role of ophthalmologists in such a scenario increases manifold to find the cause of abnormally high myopia and to correlate other systemic findings with the help of a physician to reach a final diagnosis of homocystinuria. In addition, the relevant diagnostic tests along with genetic testing should be carried out. This is essential as it can prevent the life-threatening complication of thromboembolic events in such individuals. As we have seen, the most common ocular complication seen in homocystinuria is ectopia lentis. The importance of good dietary control and prompt surgical management has already been stressed to prevent permanent vision-threatening complications like corneal opacity, optic atrophy, and so on. Early surgical intervention with lens extraction and aphakic correction with spectacles or secondary Intra Ocular Lens (IOLs) goes a long way in providing a reasonably good quality of life to these patients. Table 2 illustrates few important studies done in the past regarding ocular manifestations of homocystinuria [Table 2].
Table 2.
Previous studies on ocular manifestations in homocystinuria
| Previous studies | Author | Conclusion |
|---|---|---|
| Seven novel genetic variants in a North Indian cohort with classical homocystinuria | Kaur et al. | Identified the spectrum of variants prevailing in the CBS gene responsible for classical homocystinuria from India |
| Ocular findings among patients with late-diagnosed or poorly controlled homocystinuria compared with a screened, well-controlled population | Mulvihill et al. | Compared the ocular features in HCU patients who had late diagnosis or were noncompliant to treatment and found worse prognosis compared to a control group of early-diagnosed and well-controlled subjects |
| Ocular complications in homocystinuria - Early and late treated | Burke et al. | Dietary treatment and early diagnosis prevented ocular complications associated with homocystinuria |
| Homocystinuria due to cystathionine beta-synthase deficiency in Ireland: 25 years’ experience of a newborn screened and treated population with reference to clinical outcome and biochemical control | Naughten et al. | Newborn screening, early commencement of dietary treatment, and low lifetime free homocysteine levels were associated with significantly reduced probability of developing complications compared to the untreated HCU patients |
| Ophthalmic abnormalities in homocystinuria: The value of screening | Taylor et al. | Stresses upon the role of early screening and prompt treatment |
| Management of ophthalmic complications of homocystinuria | Harrison et al. | Surgical management of ocular complications is associated with good prognosis compared to conservative management |
| Delay in diagnosis of homocystinuria: Retrospective study of consecutive patients | Cruysberg et al. | Early recognition of unusual high myopia, ectopia lentis, systemic association, and biochemical testing are associated with good prognosis |
To date, more than 191 CBS mutations have been identified. This eventually leads to impairment of conversion of homocysteine to cystathionine, resulting in elevation of both Met and homocysteine levels in blood and urine, leading to hyperhomocysteinemia, homocystinuria, hypermethioninemia, and homocysteinemia. The two most frequently reported mutations worldwide are p.G307S (31%) and p.I278T (24%). The p.G307S mutation produces severe effects in the homozygous individuals, and it is also found to be minimally responsive to nonresponsive to pyridoxine treatment. On the other hand, the p. 1278T mutation is pyridoxine responsive with mild clinical manifestations.
Diagnosis of homocystinuria has been traditionally made using a qualitative urinary cyanide nitroprusside test that detects the presence of urinary disulfide homocysteine. Blood tests also measure fHcy and tHcy levels. Further diagnosis can be made by analyzing plasma Met and cystathionine. The gold standard test to confirm CBS deficiency is taking the cultured fibroblasts and determining cystathionine production from homocysteine and serine.
Genetic testing can also be done by sequencing the CBS gene. The gene can be detected easily, and the results of molecular genetics are interpreted easily if the genetic variant is already known. This becomes difficult in the case of novel mutations.
Newborn screening can be done by measuring plasma Met concentration, which is found to be exceptionally high in newborns with homocystinuria. Tandem mass spectrometry can also be used for newborn screening.
The mainstay of treatment remains pyridoxine supplementation, the dose of which is 10–40 mg that may be sufficient in pyridoxine-responsive patients. In those patients who are partially responsive, a dose of 200 mg/day can achieve the biochemical targets. The problem arises for the pyridoxine-unresponsive patients, for whom dietary restriction of Met has been suggested. Treatment for CBS deficiency must be continued throughout life, as loss of biochemical control in later life is associated with serious complications.
The importance of early diagnosis by clinical examination along with confirmation using a battery of tests available, including blood tests and genetic tests, is well known, since early diagnosis helps in initiating the dietary modifications that arrest the progression of various complications associated with homocystinuria, as seen in previous studies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Yap S, Naughten E. Homocystinuria due to cystathionine betasynthase deficiency in Ireland:25 years'experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J Inherit Metab Dis. 1998;21:738–47. doi: 10.1023/a:1005445132327. [DOI] [PubMed] [Google Scholar]
- 2.Naughten ER, Yap S, Mayne PD. Newborn screening for homocystinuria:Irish and world experience. Eur J Pediatr. 1998;157(Suppl 2):S84–7. doi: 10.1007/pl00014310. [DOI] [PubMed] [Google Scholar]
- 3.Boers GHJ, Polder TW, Cruysberg JRM, Schoonderwaldt HC, Peetoom JJ, van Ruyven TWJ, et al. Homocystinuria versus Marfan's syndrome:The therapeutic relevance of the differential diagnosis. Neth J Med. 1984;27:206–12. [PubMed] [Google Scholar]
- 4.McKusick VA. Homocystinuria. In: McKusick VA, editor. Heritable Disorders of Connective Tissue. St Louis: Mosby; 1972. pp. 224–81. [Google Scholar]
- 5.Brenton DP, Dow CJ, James JIP, Ray RL, Wynne-Davies R. Homocystinuria and Marfan's syndrome:A comparison. J Bone Joint Surg Br. 1972;54:277–98. [PubMed] [Google Scholar]
- 6.Cross HE, Jensen AD. Ocular manifestations in the Marfan syndrome and homocystinuria. Am J Ophthalmol. 1973;75:405–20. doi: 10.1016/0002-9394(73)91149-5. [DOI] [PubMed] [Google Scholar]
- 7.The Human Gene Mutation Database Cystathionine-Beta- Synthase. [Last accessed on 2020 Jan 25];2019 [Google Scholar]
- 8.Kraus JP, Janosík M, Kozich V, Mandell R, Shih V, Sperandeo MP, et al. Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat. 1999;13:362–75. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 9.Garland J, Prasad A, Vardy C, Prasad C. Homocystinuria:Challenges in diagnosis and management. Paediatr Child Health. 1999;4:557–62. doi: 10.1093/pch/4.8.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallagher PM. High frequency (71%) of cystathionine b-synthase mutation G307S in Irish homocystinuria patients. Hum Mutat. 1995;6:177–80. doi: 10.1002/humu.1380060211. [DOI] [PubMed] [Google Scholar]
- 11.de Franchis R. Four novel mutations in the cystathionine b-synthase gene:Effect of a second linked mutation on the severity of the homocystinuric phenotype. Hum Mutat. 1999;13:453–7. doi: 10.1002/(SICI)1098-1004(1999)13:6<453::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 12.Kelly PJ, Furie KL, Kistler JP, Barron M, Picard EH, Mandell R, et al. Stroke in young patients with hyperhomocysteinemia due to cystathionine beta-synthase deficiency. Neurology. 2003;60:275–9. doi: 10.1212/01.wnl.0000042479.55406.b3. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S. Mouse modeling and structural analysis of the p. G307S mutation in human cystathionine b-synthase (CBS) reveal effects on CBS activity but not stability. J Biol Chem. 2018;293:13921–31. doi: 10.1074/jbc.RA118.002164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majtan T, Pey AL, Gimenez-Mascarell P, Martínez-Cruz LA, Szabo C, Kožich V, et al. Potential pharmacological chaperones for cystathionine beta-synthase-deficient homocystinuria. Handb Exp Pharmacol. 2018;245:345–83. doi: 10.1007/164_2017_72. [DOI] [PubMed] [Google Scholar]
- 15.Scriver CR. New York, NY, USA: McGraw-Hill; 2001. The Metabolic &Molecular Bases of Inherited Disease. [Google Scholar]
- 16.Kozich V, Kraus JP. Screening for mutations by expressing patient cDNA segments in E.coli:Homocystinuria due to cystathionine beta-synthase deficiency. Hum Mutat. 1992;1:113–23. doi: 10.1002/humu.1380010206. [DOI] [PubMed] [Google Scholar]
- 17.Sperandeo MP, de Franchis R, Andria G, Sebastio G. A 68-bp insertion found in a homocystinuria patient is a common variant and is skipped by alternative splicing of the cystathionine beta-synthase mRNA. Am J Hum Genet. 1996;59:1391–3. [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur R, Attri SV, Saini AG, Sankhyan N, Singh S, Faruq M, et al. Seven novel genetic variants in a North Indian cohort with classical homocystinuria. Sci Rep. 2020;10:17299. doi: 10.1038/s41598-020-73475-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spaeth GL, Barber GW. Homocystinuria- Its ocular manifestations. J Pediatr Ophthalmol. 1996;3:42–8. [Google Scholar]
- 20.Hubmacher D, Cirulis JT, Miao M, Keeley FW, Reinhardt DP. Functional consequences of homocysteinylation of the elastic fiber proteins fibrillin-1 and tropoelastin. J Biol Chem. 2010;285:1188–98. doi: 10.1074/jbc.M109.021246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulvihill A, Yap S, O'Keefe M, Howard PM, Naughten ER. Ocular findings among patients with late-diagnosed or poorly controlled homocystinuria compared with a screened, well- controlled population. J AAPOS. 2001;5:311–5. doi: 10.1067/mpa.2001.118219. [DOI] [PubMed] [Google Scholar]
- 22.Burke JP, O'Keefe M, Bowell R, Naughten ER. Ocular complications in homocystinuria—early and late treated. Br J Ophthalmol. 1989;73:427–31. doi: 10.1136/bjo.73.6.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor RH, Burke J, O'Keefe M, Beighi B, Naughton E. Ophthalmic abnormalities in homocystinuria:The value of screening. Eye. 1998;12:427–30. doi: 10.1038/eye.1998.100. [DOI] [PubMed] [Google Scholar]
- 24.Hua N, Ning Y, Zheng H, Zhao L, Qian X, Wormington C, et al. Recurrent dislocation of binocular crystal lenses in a patient with cystathionine beta-synthase deficiency. BMC Ophthalmol. 2021;21:212. doi: 10.1186/s12886-021-01974-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen AD, Cross HE. Surgical treatment of dislocated lenses in the Marfan syndrome and homocystinuria. Trans Am Acad Ophthalmol Otolaryngol. 1972;76:1491–9. [PubMed] [Google Scholar]
- 26.Cross HE. Differential diagnosis and treatment of dislocated lenses. Birth Defects Orig Artic Ser. 1976;12:335–46. [PubMed] [Google Scholar]
- 27.Bloch RS. Homocystinuria. In: Fraunfelder FT, Roy FH, editors. Current Ocular Therapy 4. Philadelphia: Saunders; 1995. pp. 156–7. [Google Scholar]
- 28.Harrison DA, Mullaney PB, Mesfer SA, Awad AH, Dhindsa H. Management of ophthalmic complications of homocystinuria. Ophthalmology. 1998;105:1886–90. doi: 10.1016/S0161-6420(98)91035-1. [DOI] [PubMed] [Google Scholar]
- 29.Cruysberg JR, Boers GH, Trijbels JM, Deutman AF. Delay in diagnosis of homocystinuria:Retrospective study of consecutive patients. BMJ. 1996;313:1037–40. doi: 10.1136/bmj.313.7064.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohindra I, Held R. Refraction in humans from birth to five years. Doc Ophthalmol Proc Ser. 1981;28:17–29. [Google Scholar]
- 31.Sorsby A, Sheridan M, Leary GA, Benjamin B. Vision, visual acuity, and ocular refraction of young men. Findings in a sample of 1033 subjects. Br Med J. 1960;1:1394–8. doi: 10.1136/bmj.1.5183.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorsby A, Leary GA, Richards MJ. Correlation ametropia and component ametropia. Vision Res. 1962;2:309–13. [Google Scholar]
- 33.Gus PI, Donis KC, Marinho D, Martins TF, de Souza CFM, Carloto RB, et al. Ocular manifestations in classic homocystinuria. Ophthalmic Genet. 2021;42:71–4. doi: 10.1080/13816810.2020.1821384. [DOI] [PubMed] [Google Scholar]
- 34.Mulvihill A, O'Keeffe M, Yap S, Naughten E, Howard P, Lanigan B. Ocular axial length in homocystinuria patients with and without ocular changes:Effects of early treatment and biochemical control. J AAPOS. 2004;8:254–8. doi: 10.1016/j.jaapos.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Gerding H. Ocular complications and a new surgical approach to lens dislocation in homocystinuria due to cystathionine-b-synthetase deficiency. Eur J Pediatr. 1998;157(Suppl 2):S94–101. doi: 10.1007/pl00014312. [DOI] [PubMed] [Google Scholar]
- 36.Ramsey MS, Yanoff M, Fine BS. The ocular histopathology of homocystinuria. A light and electron microscopic study. Am J Ophthalmol. 1972;74:377–85. doi: 10.1016/0002-9394(72)90895-1. [DOI] [PubMed] [Google Scholar]
- 37.Larsen C, Boysen J. Phacolytic Glaucoma and Homocystinuria. Challenging Cases, Glaucoma Today. 2015 July/Aug; [Google Scholar]
- 38.Straatsma BR, Foos RB, Femam SS. Degenerative diseases of the peripheral retina. In: Duane TD, editor. Clinical Ophthalmology. chap. 26. Vol. 3. Hagerstown: Harper and Row; 1978. pp. 8–10. [Google Scholar]
- 39.Traboulsi EI, Silva JC, Geraghty MT, Maumenee IH, Valle D, Green WR. Ocular histopathologic characteristics of cobalamin C type vitamin B12 defect with methylmalonic aciduria and homocystinuria. Am J Ophthalmol. 1992;113:269–80. doi: 10.1016/s0002-9394(14)71578-8. [DOI] [PubMed] [Google Scholar]
- 40.Moat SJ, Bonham JR, Tanner MS, Allen JC, Powers HJ. Recommended approaches for the laboratory measurement of homocysteine in the diagnosis and monitoring of patients with hyperhomocysteinaemia. Ann Clin Biochem. 1999;36:372–9. doi: 10.1177/000456329903600311. [DOI] [PubMed] [Google Scholar]
- 41.Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet. 1985;37:1–31. [PMC free article] [PubMed] [Google Scholar]
- 42.Magner M, Krupkova L, Honzik T, Zeman J, Hyanek J, Kozich V. Vascular presentation of cystathionine beta-synthase deficiency in adulthood. J Inherit Metab Dis. 2011;34:33–7. doi: 10.1007/s10545-010-9146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orendac M, Zeman J, Stabler SP, Allen RH, Kraus JP, Bodamer O, et al. Homocystinuria due to cystathionine beta-synthase deficiency:Novel biochemical findings and treatment efficacy. J Inherit Metab Dis. 2003;26:761–73. doi: 10.1023/B:BOLI.0000009963.88420.c2. [DOI] [PubMed] [Google Scholar]
- 44.Kraus JP. Cystathionine beta-synthase (human) Methods Enzymol. 1987;143:388–94. doi: 10.1016/0076-6879(87)43068-1. [DOI] [PubMed] [Google Scholar]
- 45.Smith DE, Mendes MI, Kluijtmans LA, Janssen MC, Smulders YM, Blom HJ. A liquid chromatography mass spectrometry method for the measurement of cystathionine beta-synthase activity in cell extracts. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;911:186–91. doi: 10.1016/j.jchromb.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 46.McHugh D, Cameron CA, Abdenur JE, Abdulrahman M, Adair O, Al Nuaimi SA, et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry:A worldwide collaborative project. Genet Med. 2011;13:230–54. doi: 10.1097/GIM.0b013e31820d5e67. [DOI] [PubMed] [Google Scholar]
- 47.Gan-Schreier H, Kebbewar M, Fang-Hoffmann J, Wilrich J, Abdoh G, Ben-Omran T, et al. Newborn population screening for classic homocystinuria by determination of total homocysteine from Guthrie cards. J Pediatr. 2010;156:427–432. doi: 10.1016/j.jpeds.2009.09.054. [DOI] [PubMed] [Google Scholar]
- 48.Turgeon CT, Magera MJ, Cuthbert CD, Loken PR, Gavrilov DK, Tortorelli S, et al. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin Chem. 2010;56:1686–95. doi: 10.1373/clinchem.2010.148957. [DOI] [PubMed] [Google Scholar]
- 49.Mudd SH. Hypermethioninemias of genetic and non-genetic origin:A review. Am J Med Genet C Semin Med Genet. 2011;157C:3–32. doi: 10.1002/ajmg.c.30293. [DOI] [PubMed] [Google Scholar]